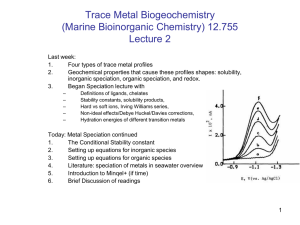
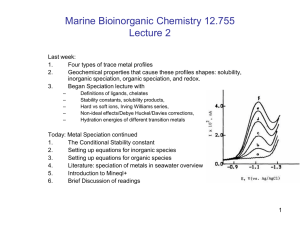
Fe Manuscript - Earth & Planetary Sciences
advertisement

SCOR - IUPAC WORKING GROUP ON IRON IN THE OCEANS CHAPTER 6: IRON: ANALYTICAL METHODS FOR THE DETERMINATION OF CONCENTRATIONS AND SPECIATION KENNETH W. BRULAND and EDEN L. RUE Institute of Marine Sciences, University of California at Santa Cruz Santa Cruz, CA 95064, USA 1. INTRODUCTION ............................................................................... 2 1.1 BACKGROUND INFORMATION - CONCENTRATIONS ................................. 4 1.2 BACKGROUND INFORMATION - SPECIATION AND AVAILABILITY ........ 6 2. THE FIRST STEP: THE DETERMINATION OF DISSOLVED AND PARTICULATE IRON .................................................................. 8 2.1 CONTAMINATION DURING SAMPLING AND ANALYSIS ............................ 8 2.2 FILTRATION ....................................................................................................... 10 2.2.1 Conventional Filtration ............................................................................. 10 2.2.2 Ultrafiltration ............................................................................................. 11 2.3 ANALYSIS OF DISSOLVED FE ......................................................................... 12 2.3.1 The Importance of Acidification ................................................................ 12 2.3.2 Extraction/Preconcentration of Dissolved Fe............................................ 13 2.3.3 Direct Electrochemical Analysis of Dissolved Fe...................................... 15 2.3.4 Operationally-defined, Labile Dissolved Fe Measurements Determined by On-line Methods .................................................................................................. 16 2.4 ANALYSIS OF PARTICULATE FE .................................................................... 17 2.5 ACIDIFICATION AND ANALYSIS OF UNFILTERED SAMPLES - AN OPERATIONALLY DEFINED MEASUREMENT OF "DISSOLVABLE" FE ........ 18 3. THE DETERMINATION OF THE CHEMICAL SPECIATION OF IRON IN THE DISSOLVED FRACTION ................................... 20 3.1 REDOX SPECIATION - FE(II) VS. FE(III).......................................................... 20 3.2 INORGANIC SPECIATION - FE(III)' AND FE(II)' ............................................. 21 3.3 COMPLEXATION OR CHELATION WITH ORGANIC LIGANDS ................. 22 4. NEW DIRECTIONS ......................................................................... 28 5. REFERENCES .................................................................................. 31 1 1. INTRODUCTION This chapter discusses analytical methods and approaches for the determination of the concentration and chemical speciation of iron in seawater. In their text book Principles and Applications of Aquatic Chemistry, Morel and Hering (1993) state “The elucidation of the chemical speciation of trace elements in natural waters is probably the greatest remaining challenge to analytical chemists; the objective is to demonstrate and quantify the existence of fractions of chemical constituents as picomolar concentrations of perhaps ephemeral species.” Of all the trace elements, the determination of iron and the elucidation of its chemical speciation present the greatest analytical challenge, due to its extremely low concentration in the ocean and its ubiquitousness as a contaminant. Despite the difficulties involved, the fact that iron is arguably the most important trace metal in seawater due to its role as an essential, and at times, biolimiting micronutrient, has stimulated the development of a variety of new analytical methodologies. Ideally, these techniques would directly determine the chemical speciation of iron among the soluble, colloidal and particulate fractions. However, many of the techniques provide only indirect measurements that are only operationally defined, and thus their interpretation can be ambiguous. The marine chemistry of iron in seawater is depicted in Figure 1, which conceptualizes the various forms and chemical species in which iron can be partitioned. The chemical form of iron is defined by physical size fractions separated on the basis of filtration methods - either with conventional membrane filters or with the use of various ultra-filtration methods. Chemical species are defined chemical constituents within a particular form or size fraction of the metal. The chemistry of iron is further complicated in that it can exist in two different redox states, Fe(III) or Fe(II), either within a variety of soluble coordination complexes with inorganic ligands (the sum of all inorganic Fe(III)hydrolysis species is Fe(III)) and organic ligands (Fe-organic ligand complexes are termed FeLi), or in a variety of colloidal and/or particulate forms. Complexation with both inorganic and organic ligands and adsorption to particle surfaces is highly pH dependent, and as a result, an assessment of the ambient chemical speciation of iron needs to be carried out at ambient pH. As various methods are developed, especially the on-line methods, it is important to better understand just what fraction of iron these methods are measuring. There is recent evidence that the bulk of the dissolved Fe in the open ocean is complexed with dissolved organic ligands (van den Berg 1995; Rue and Bruland 1995, 1997; Wu and Luther 1995). We lack knowledge at this time, however, as to the 2 chemical nature of these ligands, their sources and sinks, and their "raison d’être." In addition, we have a poor understanding of the various potential solid phases containing iron, whether colloidal in nature or particulate. Because of this lack of information, it is somewhat presumptuous of us to even use the term "chemical speciation." Figure 1. Various chemical forms and species of iron which can exist in dissolved and particulate phases. Convincing arguments can now be made to elevate iron to the same status as nitrogen, phosphorus and silicon as important nutrients influencing global biogeochemical cycles. In particular, there is now considerable evidence that the availability of iron controls the productivity, species composition, and trophic structure of planktonic communities in large regions of the ocean (Sunda, this volume). Although we increasingly recognize iron's importance in the oceans (Bruland et al. 1991; Wells et al. 1995; Hutchins 1995; Price and Morel 1998; Falkowski et al. 1998), its marine chemistry is complex and still not well understood. Factors which contribute to this obscurity 3 include: 1) the extremely low concentration in seawater due to both the low solubility of Fe(III) in oxygenated seawater (Stumm and Morgan 1996; Millero 1998) and its high biological requirement (Brand 1991; Sunda and Huntsmann 1995), 2) the high particulate abundance which varies dramatically both spatially and temporally (Gordon and Martin 1988; Wu and Luther 1996) , 3) the hydrolysis chemistry of Fe(III), which includes the uncertain role of species such as Fe(OH)03 (Hudson 1998), which in turn makes it difficult to agree on estimates of its inorganic solubility, 4) the colloid chemistry whereby both inorganic and organic colloidal forms of iron play a potentially important, but poorly understood role (Powell et al. 1995; Wells et al. 1998; Takeda et al. 1998), 5) the redox chemistry leading to both Fe(III) and Fe(II) forms being important due to an active photochemically driven redox cycle (Johnson et al. 1994; Wells et al. 1995, Sunda -this volume), 6) the coordination or organic ligation chemistry, whereby its chemical speciation appears to be dominated by Fe(III)-chelates due to the presence of a slight excess of strong Fe(III)-binding organic ligands of biological origin (Rue and Bruland 1995, 1997; Gledhill and van den Berg 1994; van den Berg 1995; Wu and Luther 1995), 7) the potential importance of Fe(II)-binding ligands which would tend to stabilize Fe(II) in surface seawater (Gledhill and van den Berg 1995), 8) the difficulties in discriminating between abiotic and biotic forms of particulate-Fe, and 9) the ease with which iron contamination can create artifacts at every step in the collection and analysis. As a result of our lack of detailed knowledge of the marine chemistry of iron and these potential contamination problems, we need to approach the analyses with great caution and a proper appreciation for potential artifacts when applying analytical methods to determine iron concentrations and its chemical speciation. 1.1 BACKGROUND INFORMATION - CONCENTRATIONS In oceanic surface waters, concentrations of dissolved Fe (defined as the iron concentration in the filtrate passing through a conventional 0.2 or 0.4 m filter) commonly range from 0.02 nM (20 pM) to 1 nM. In remote high-nutrient lowchlorophyll (HNLC) regimes such as the equatorial Pacific, subarctic Pacific, and parts of the Southern Ocean, iron can be a limiting nutrient with dissolved Fe concentrations in surface waters on the order of 0.02 to 0.05 nM (20 to 50 pM). These concentrations are low enough for dissolved Fe to be diffusion limiting for all but the smallest phytoplankton cells (Hudson and Morel 1990; Sunda and Huntsman 1995; Sunda - this volume). Characteristic vertical profiles of dissolved Fe in the upper 500 meters of the water column are presented in Figure 2. Particulate forms of iron (i.e., those retained by a 0.2 or 0.4 m pore size membrane filter) in these HNLC areas can exist at concentrations 4 higher than dissolved Fe (Price and Morel 1998) with biogenic, detrital and lithogenic particulate fractions all being important (Figure 2). In surface waters of the oligotrophic gyre of the central North Pacific, dissolved Fe exists at concentrations ranging from 0.02 to 0.4 nM and can exceed the concentration of particulate Fe (Bruland et al. 1994) (Figure 2). Dissolved Fe in open ocean deep waters is on the order of 0.6 nM (Johnson et al. 1997 and references therein). Figure 2. Vertical profiles of dissolved (o) and particulate () Fe A) the subarctic Pacific and B) the central North Pacific; from Martin et al. (1989) and Bruland et al. (1994). In coastal waters, concentrations of dissolved Fe are commonly in the range of 0.1 to 10 nM (Wu and Luther 1996; Gordon and Martin 1988; Bruland et al. in prep.), with values exceeding 1 M in the low salinity regime of estuaries (Boyle et al. 1976; Sholkovitz et al. 1977; Powell et al. 1996). The concentration of particulate Fe in coastal waters can be especially high and extremely variable due to the amount of iron-rich aluminosilicate clays of terrigenous origin suspended in these waters either supplied directly by rivers or resuspended from shelf sediments. For example, in an offshore/onshore transect in the northwest Atlantic, Wu and Luther (1996) found dissolved Fe to vary from 0.3 nM offshore to 14 nM over the inner shelf area, while the 5 particulate Fe varied from 0.8 nM to 11.1 µM, respectively. At all the stations in this northwest Atlantic transect, the particulate Fe concentration was much greater than dissolved Fe, and at the near shore station in Wu and Luther’s study, the particulate Fe was close to 3 orders-of-magnitude greater than dissolved Fe. Similar high particulate Fe concentrations are observed in the nearshore shelf areas off the West coast of the U.S. (Martin and Gordon 1988; Bruland et al. in prep.). Acetic acid-leachable particulate Fe concentrations in these near shore shelf waters can be on the order of 3 µM - a concentration close to 1000 fold greater than the dissolved Fe values (Bruland et al. in prep.). Thus, concentrations of both dissolved and weak acid-leachable particulate Fe can vary by four orders-of-magnitude in different regimes. 1.2 BACKGROUND INFORMATION - SPECIATION AND AVAILABILITY The majority of the dissolved Fe in remote, low-Fe, high-nitrate low-chlorophyll (HNLC) regimes appears to be chelated with organic ligands which resemble siderophores in their conditional stability constants and molecular weight (Rue and Bruland 1997). In HNLC areas such as the equatorial Pacific, it appears that these chelated forms of iron are primarily less than 1000 Daltons in nominal molecular weight (Rue and Bruland, 1997), with only a small fraction existing as larger, colloidal size material. Results on the chemical speciation of iron in central gyre regions of the North Pacific and North Atlantic both indicate that the bulk of the dissolved Fe exists as organic Fe(III)-chelates (Rue and Bruland 1995; Wu and Luther 1995). Although the data is very limited, it even appears that iron in the deep ocean exists primarily as Fe(III)-chelates (Rue and Bruland 1995; DeBaar et al. in prep). Dissolved Fe in coastal waters also appears to exist associated with organic ligands (Gledhill and van den Berg 1994; van den Berg 1995; Bruland et al. in prep.). In contrast to the low-iron open ocean, however, the high-iron Narragansett Bay has the bulk of what appears to be organically complexed dissolved Fe existing as a colloidal Fe fraction, with only a small amount found in the < 1000 Dalton ultrafiltrate (Rue et al. in prep.). Powell et al. (1996) used ultrafiltration to carry out a size-fractionation study of dissolved iron and dissolved organic carbon in the Ochlockonee estuary. They showed that in high-iron low-salinity regions of the estuary that the vast majority of iron was in the high molecular weight fraction (>10,000 Daltons nominal molecular weight), but that this component was only a minimal fraction in higher salinity regions. Thus, the chemical form of dissolved Fe appears to change dramatically from being primarily associated with a complex, higher molecular weight, colloidal fraction in estuarine and fresh waters, to 6 being in the form of low molecular weight, water soluble, Fe(III)-organic chelates in oceanic surface waters. Much of the current interest in the marine chemistry of iron stems from its role as a limiting micronutrient affecting plankton productivity and biological species composition. Trace metal assimilation by phytoplankton has historically been modeled using the free ion concentration model (Morel and Hering 1993), with iron uptake by phytoplankton typically correlated with free, hydrated Fe3+ concentrations (using EDTA buffered solutions). It is now realized, however, that it is the more abundant, kinetically labile, hydrolysis species comprising Fe(III) (such as Fe(OH)2 ) that actually control uptake rates of inorganic iron (Hudson and Morel, 1990; Hudson 1998). As a result, recent papers dealing with iron limitation in laboratory culture media correlate effects with [Fe(III)] rather than [Fe3+] (Sunda and Huntsman, 1995). Recent findings demonstrating that the bulk of the dissolved Fe in the ocean exists chelated to organic ligands means that labile inorganic species (Fe(III)) exist at extremely low concentrations, perhaps requiring photochemical or other redox mechanisms to provide an adequate supply of Fe (Rue and Bruland 1997; Price and Morel 1998; Sunda, this volume). At this point, we are just beginning to understand how chelated iron might be accessible by the biological community (Maldonado and Price 1999; Sunda - this volume). Prokaryotes and even unicellular eukaryotes such as diatoms appear to be able to utilize some of the chelated iron (Price et al. 1994; Price and Morel 1997; Hutchins et al. 1998, 1999). There are suggestions that eukaryotic phytoplankton such as diatoms can access this chelated Fe(III) by using cell surface-bound reductases to reduce the chelated Fe(III) to Fe(II) which then dissociates and is subsequently assimilated either as Fe(II)' or after re-oxidation, as Fe(III)' (Maldonado and Price 1999). There is some data that suggest that the protozoan grazers of the microbial community can utilize and solubilize colloidal Fe (Barbeau et al. 1996). There is also evidence that particulate Fe associated with the plankton community can be remineralized and reused as regenerated iron much in the same way as nitrogen is recycled (Hutchins et al. 1993; Hutchins and Bruland 1994). Price and Morel (1998) estimated that 83% of the iron uptake by microorganisms within the euphotic zone of the equatorial Pacific was from such biological regeneration, while the remaining 17% was supplied from external sources such as aeolian deposition. In addition, it has been suggested that various members of the phytoplankton and microplankton community can utilize phagocytosis to engulf and partially digest particulate Fe (Raven 1997), thereby rendering a fraction of the particulate Fe available over an extended time scale. Recent evidence suggests that the photosynthetic 7 phytoplankton Ochromonas can obtain iron directly in particulate form by ingesting bacteria (Maranger et al. 1998). As a result of the diversity of iron uptake strategies employed by various marine microorganisms, some of the "non-available" iron is likely recycled by various mechanisms and at various rates into biologically available forms (Wells et al. 1995). Therefore, most of the dissolved Fe and part of the particulate Fe may eventually be made available to the plankton community. 2. THE FIRST STEP: THE DETERMINATION OF DISSOLVED AND PARTICULATE IRON Over the past three decades, the development of trace metal clean techniques and sensitive analytical methods such as graphite furnace atomic absorption spectrometry have led the way in providing reliable profiles for iron in the worlds oceans (Bruland et al. 1979; Gordon et al. 1982; Landing and Bruland 1987; Martin et al. 1989; Johnson et al. 1997). A meaningful characterization of iron chemistry within a given water body begins with an accurate determination of iron in the dissolved and particulate forms or size fractions (Figure 1). Filtration is a critical first step in the analysis of iron, particularly when the details of the chemical speciation within these forms and the rates and mechanisms of transformations among the various species of iron are poorly understood. The use of conventional filtration for defining the traditional categories of “dissolved” (< 0.2 - 0.4 m) and “particulate” (> 0.4 - 0.2 m) fractions has been justified primarily from its biological importance as being necessary to effectively filter living cells such as heterotrophic and photoautotrophic bacteria from the dissolved phase. This has become particularly important in open ocean surface waters where the presence and importance of photosynthetic cyanobacteria such as Synechococcus and Prochlorococcus have recently been recognized (Chisholm et al. 1988). 2.1 CONTAMINATION DURING SAMPLING AND ANALYSIS Iron is arguably one of the two most difficult metals to determine in seawater (the other being zinc), in part due to its ubiquitous nature which increases the potential for contamination in all steps of the method, from collection, filtration and storage to analysis of seawater samples. There are numerous unpublished tales of contamination problems experienced by various laboratories attempting to measure dissolved Fe in seawater. One illustrative published example of iron (and zinc) contamination was presented by the late John Martin and his coworkers (Martin et al. 1993) in an early Joint Global Ocean Flux Study (JGOFS) study. John Martin’s research group had an excellent track record of being able to collect and determine iron in seawater without contamination problems. 8 They initiated a study designed to test for potential trace metal contamination during sample collection and handling of a primary productivity incubation study. Five separate laboratories responsible for measuring primary productivity during the JGOFS North Atlantic Bloom Experiment were provided with specially cleaned bottles from Martin’s research group. Each group was asked to fill three of the bottles directly from their water samplers which they used on a routine basis to collect “clean” seawater samples for their primary productivity measurements and to fill three other bottles after water had been kept in their incubation bottles for 24 hours. The samples were then returned to Martin’s Figure 3. Results from initial water and after 24 hours of incubation for various research groups in the North Atlantic JGOFS study from Martin et al. 1993. Laboratory #1 is Martin’s research group at MLML. For Laboratories 1, 3 and 4, the first 3 data points are 9 for the water collected initially and for laboratories 1 and 3, the last three data points are for the water from the incubation bottles after 24 hours. group for preservation and future laboratory analysis. One of the five laboratories had unusual difficulties with contamination of the samples and so their results are not included. The remaining four laboratories were identified by number only in order to maintain anonymity. Laboratory 1 was revealed to be John Martin’s research group. Results for iron, zinc, copper and nickel are presented in Figure 3. The copper and nickel results are presented as examples of potentially toxic trace elements to illustrate that all four of the laboratories performed fairly well collecting and manipulating seawater for copper measurements. Three of the laboratories faired well in keeping nickel contamination minimal (remember, however, that the Martin research group supplied them all with rigorously cleaned sample bottles). The essential micronutrient trace metals, iron and zinc, however, were found to be much more prone to contamination, and only Martin’s laboratory in this experiment was successful in keeping samples free from contamination for these two metals. All the research groups, with the exception of John Martin’s laboratory, were inadvertently carrying out high iron and zinc enrichment experiments in their productivity incubations. The minimum precautions used by most research groups doing studies on iron involve the use of special winches with KevlarTM hydrowire and TeflonTM-coated GOFloTM bottles for collecting deeper samples and/or special clean pumping systems for collecting surface samples. Research groups studying iron generally make use of cleanroom vans or containers for all sample processing including filtration, aliquoting, acidification, reagent preparation, etc. Stringent precautions with respect to the choice of materials, precleaning of all materials, preparation or choice of all reagents, etc. need to be followed (Howard and Statham 1993, and references therein). On our research cruises we find it extremely useful to routinely check sampling bottles and our surface pumping systems for iron and zinc contamination with our shipboard analyses systems prior to initiating sample collection. We find that even after careful acid cleaning, a great deal of flushing/rinsing and conditioning of the sampling system is necessary prior to collecting samples. 2.2 FILTRATION 2.2.1 Conventional Filtration Filtration with conventional membrane filters of 0.2 - 0.4 m pore size provides the widely accepted way of operationally defining particulate versus dissolved metal forms in seawater (Landing and Yeats 1991; Landing and Lewis 1991). The rationale for this pore 10 size of 0.2 to 0.4 m and the use of polycarbonate filters is to separate heterotrophic and photoautotrophic bacteria from the dissolved phase. These bacteria exist at concentrations on the order of 108 to 109 cells per liter. Photosynthetic bacteria (such as Synechococcus and Prochlorococcus sp) are approximately 1.0 and 0.6 m diameter, respectively (Chisholm, 1992), while heteterotrophic bacteria can be rod shaped with a diameter on the order of 0.5 m. Marine microbiologists generally opt for 0.2 m pore size filters to ensure a complete separation of bacteria. These requirements demand the use of absolute filters with a sharp and well-defined size cutoff such as the polycarbonate track-etched (PCTE) membrane filters provided by Nuclepore and Poretics. They are preferred over depth type filters that have poorly defined cutoffs that may allow larger particles to penetrate through the filters. Limitations for membrane filters as opposed to depth type filters include fragility, lower flow rates and decreased capacity. A common clean filtration system for trace metals makes use of 142 mm diameter PCTE membrane filters mounted in TeflonTM filter sandwiches (Bruland et al. 1979). The filters undergo lengthy acid cleaning using a series of nitric and hydrochloric acid leaches of increasing purity followed by a Q-H2O (Q = sub-boiling quartz distilled) rinse before drying on a class 100 bench. The TeflonTM filter sandwiches need a vigorous initial cleaning with hot acids and are then stored in a weak Q-grade acid bath until use. A low overpressure of less than 10 psi of 0.2 µm filtered nitrogen is used in an attempt to minimize any cell rupture and lysing. It is a common procedure to filter between 28 and 56 liters of seawater and discard the initial 5 liters of filtrate passed through the filtration system. Surface samples can also be collected using TeflonTM tubing attached to a towed fish boomed out from the research vessel and an all TeflonTM double diaphragm pump. This recent use of surface water pumping systems has allowed collection of large volumes of clean seawater either on station or along transects while underway. Filter capsules, rather than the 142 mm diameter PCTE membrane filters mounted in TeflonTM filter sandwiches, have often been used with these systems due to their higher flow rates, increased capacity and ease of use (Kozelka et al. 1998). However, due to the unavailability of absolute size cutoff type filters with appropriate materials of construction, most surface pump systems have used polypropylene depth type filter capsules (Micron Separations Inc. - MSI) that can withstand the acid cleaning necessary for trace metal sampling of seawater. There are newly available MSI filter capsules with a 0.4 m absolute TeflonTM membrane filter material. Further work needs to be performed to rigorously evaluate the performance of these cartridge filters. 2.2.2 Ultrafiltration 11 There is a marked difference between most fresh waters and open ocean seawater in the amount of the dissolved Fe and dissolved organic matter associated with the colloidal size fraction. The bulk of dissolved Fe and humic acids in fresh waters is in the colloidal form in sizes of tens to hundreds of nanometers (Buffle 1992; Davison and De Vitre 1992; Powell et al. 1996). The majority of this colloidal Fe and humic acid is, however, coagulated and removed in the low salinity mixing regimes of estuaries (Boyle et al. 1977; Sholkovitz and Copeland 1981; Mayer 1982) and never makes it to the oceans. Filterable (dissolved) Fe in inflowing rivers occurs at concentrations on the order of 1 to 25 M, but this largely colloidal-sized iron is efficiently removed in estuaries and coastal waters by salinity induced flocculation and settling. A study of size fractionated iron concentrations by Kuma et al. (1998) revealed that the portion of colloidal Fe (0.025 to 0.45 m size fraction) in the high iron (dissolved Fe concentrations of 15 to 20 nM) estuarine and coastal waters of Funka Bay, Japan was approximately 90% of the dissolved Fe. In oceanic waters (with [FeT] ~ 1 nM) they found the colloidal Fe franction to comprise only 10 % of the dissolved Fe. Similarly, ultrafiltration studies of dissolved organic carbon carried out on open ocean samples have shown that only a very small portion of the dissolved organic matter is found in the colloidal size fraction of greater than 1 nm or 1000 Daltons nominal molecular weight. The majority of dissolved organic matter in the open ocean consists of relatively low molecular weight moieties (< 1000 Daltons nominal molecular weight) such as carbohydrates (Skoog and Benner 1997). The severe problems observed in fresh water with coagulation of colloidal Fe and organic matter in the diffusion layer at the filter's surface (Buffle et al. 1992) are minimized in open ocean seawater samples. For these reasons, unlike in freshwater, conventional filtration of open ocean seawater through PCTE membrane filters is relatively free of artifacts. Results from analyses of unfiltered seawater samples are particularly difficult to interpret with respect to the biogeochemical cycling of iron. For example, slight differences in the degree of acidification can lead to significant and poorly understood differences in the amount of particulate Fe solubilized during acidification. Analyses of unfiltered samples will lead to a poorly-defined, highly-operational, measurement of iron. It is strongly recommended that immediate conventional filtration using 0.2 to 0.4 m pore size filters be the first physical separation step performed prior to any further chemical analyses. 2.3 ANALYSIS OF DISSOLVED FE 2.3.1 The Importance of Acidification 12 A key step in the analysis of total dissolved Fe in seawater is acidification. Currently, however, this step varies widely between research groups. Two research groups with a long history of making dissolved Fe measurements are the Moss Landing Marine Laboratory (MLML) (Mike Gordon, the late John Martin, Ken Johnson and Kenneth Coale) and the University of California at Santa Cruz (UCSC) (currently Geoffrey Smith, Eden Rue and Ken Bruland). Both of these research groups use 4 mL of 6 M hydrochloric acid (made using sub-boiling quartz distillation) per liter of seawater (24 mequiv/L of H+) to acidify their samples to a pH on the order of 1.7. Seawater samples are collected, immediately filtered and acidified at sea, and stored acidified for at least one month prior to analyses back at the shore based laboratories. These research groups have argued that this degree of acidification is necessary for preservation and adequate to liberate organically complexed iron and any potential colloidal Fe in the dissolved fraction and is essential in ensuring a measurement of total dissolved Fe. Two European groups with a long history of determining dissolved Fe in seawater, Westerlund and co-workers and de Baar and coworkers, acidify samples with 1 mL of concentrated (15 M) nitric acid per liter of seawater (15 mequiv/L of H+ added) to a pH of approximately 1.8-1.9. Acid concentrations and storage times less than this may well be adequate, but a rigorous evaluation of these approaches on a variety of different water types has not been undertaken. For example, acidification with microwave heating may facilitate a marked decrease in the required acidification time (Obata et al. 1993). The analysis of dissolved Fe has been carried out by two general approaches: 1) extraction/preconcentration with either solvents or solid phases followed by analyses of the concentrated extract, or 2) direct analysis of the seawater. 2.3.2 Extraction/Preconcentration of Dissolved Fe The MLML and the UCSC research groups have analyzed total dissolved Fe with dithiocarbamate/solvent extraction as a preconcentration step followed by quantification by graphite furnace atomic absorption spectrometry (GFAAS) (Bruland et al. 1979; Gordon et al. 1982; Landing and Bruland 1987). This well-characterized, quantitative method involves a pH adjustment to pH 4.0-4.5 using an ammonium acetate buffer along with the addition of pyrrolidine dithiocarbamate (PDC) and diethyl dithiocarbamate (DDC), followed by a double extraction of the Fe-dithiocarbamate neutral complexes into chloroform. The chloroform fractions are combined in quartz beakers with nitric acid added, whereupon they are slowly evaporated to dryness and subsequently reconstituted in weak nitric acid for GFAAS quantification. Westerlund and co-workers and de Baar and coworkers use a very similar approach with minor variations. They utilize the same 13 mixture of dithiocarbamate ligands (PDC and DDC), but use freon rather than chloroform as the organic solvent (Danielsson et al. 1978). Wells and Bruland (1998) have recently developed a method modified after an approach used by King and Fritz (1985) and van Geen and Boyle (1990) in which a solidphase extraction method is substituted into the extraction scheme, rather than the liquid/liquid solvent extraction. Wells and Bruland (1998) used the hydroxy-substituted dithiocarbamate ligand, bis(2-hydroxyethyl) dithiocarbamate - a more water soluble dithiocarbamate ligand. The samples are buffered to a neutral pH with maleic acid/ammonium maleate buffer and the Fe-dithiocarbamate complex is then extracted onto a polystyrene-based C-18 resin column at a flow rate of 10 mL min-1. The column is rinsed with deionized water to remove sea salts and eluted with a 1 N nitric acid/methanol solution. The eluate is gently taken to dryness and reconstituted in weak nitric acid for quantification by either inductively coupled plasma mass spectrometry (ICPMS) or GFAAS. A key feature of the above extraction methods is that a large excess of ligands is added at the same time that the samples are pH adjusted with the appropriate buffer. At this time, the large excess of dissolved dithiocarbamate ligands results in essentially a quantitative complexation of iron as dithiocarbamate complexes. This provides minimal chance for iron to re-establish equilibrium with the low concentrations of any natural Febinding ligands or to precipitate as insoluble ferric hydroxides. Chelating resins such as Chelex-100 and 8-Hydroxyquinoline (8-HQ) resins have also been used for the analysis of dissolved Fe (Bruland et al. 1994; Beauchemin and Berman 1989). For dissolved Fe measurements, filtered samples are first acidified to pH ~1.7-1.8 to solubilize any colloidal forms of iron and to dissociate any Fe-organic complexes. The samples are then adjusted to the appropriate pH (i.e., 5.8 for the Chelex-100 resin) just prior to passing them through the resin columns. A liability of these techniques is that the flow rates must be kept low (on the order of 1 mL min-1) to allow for the relatively slow kinetics of exchange with the active sites on the resin. For example, it takes approximately 8 hours to pass a 500 mL sample through a chelating resin column at these slow flow rates. The iron is then eluted from the chelating resin columns with acid and analyzed by GFAAS or ICPMS. De Baar et al. (1998) determined dissolved Fe by applying the flow injection analysis (FIA) method of Obata et al. (1993) to filtered samples that had been acidified with 1 mL of 15 M nitric acid per L to a pH of 1.8. They then mixed samples with ammonium acetate buffer to raise the pH to 4.2 before passing through an 8hydroxyquinoline column (immobilized on a hydrophilic vinyl polymer). These authors 14 commented that “The immobilized 8-hydroxyquinoline has very high affinity to iron and the resulting values are deemed to represent the complete dissolved fraction, although less than 100% extraction efficiency, due to competing very strong natural organic ligands, cannot be ruled out.” De Jong et al. (1998) reported a shipboard method in which they used a surface pumping system where the seawater was filtered in-line at a flow rate of ~0.5 L min-1 through a TeflonTM filter holder containing a 142 mm diameter 0.4 m poresize filter. Samples were left in the dark for one hour to allow any Fe(II) present to reoxidize in the absence of any photoreduction, and then strongly acidified to pH ~1.8 with nitric acid. The pH was readjusted to 3.5-4.0 with an ammonium acetate buffer just prior to concentrating the iron on an 8-HQ chelating resin column as part of the flow injection method of Obata et al. (1993). The storage of samples in the dark was performed to ensure that any Fe(II) initially present in the samples was oxidized to Fe(III) prior to acidification. This was to guarantee that total dissolved Fe was measured by this technique, particularly since they were loading the 8-HQ resin column at a pH of ~4.2 (see later discussion of resin performance). The authors presented an accuracy check of the method by regularly measuring NASS-4 reference seawater and carrying out an intercomparison study with the classic solvent extraction method. The accuracy obtained by this shipboard FIA method appeared to be excellent. 2.3.3 Direct Electrochemical Analysis of Dissolved Fe Van den Berg (1995) determined dissolved Fe by catalytic cathodic striping voltammetry (CSV) with a high concentration of the ligand 1-nitroso-2-naphthol (1N2N) added to sample aliquots that had been stored acidified at pH 2 with hydrochloric acid. Upon pH adjustment to pH 6.9, the added 1N2N forms a surface active complex with Fe(III) and adsorbs onto the hanging mercury drop electrode. The cathodic stripping peak is then proportional to the dissolved Fe concentration. Further UV-digestion of selected samples was carried out for 2 hours using a home-built apparatus containing a 500 W high-pressure mercury vapor lamp. Analyses carried out after UV-digestion did not show an increase in dissolved Fe suggesting that all iron had been released during the acidified storage. Wu and Luther (1995) used the same CSV method (van den Berg 1995). These authors acidified to pH < 2.0 with nitric acid followed by UV irradiation for 6 hours under a 1 kW UV lamp prior to analysis. They argued that dissolved Fe measured with this procedure should include all the organic and inorganic iron species. Data was not presented, however, on whether the UV oxidation was necessary in addition to the acidification step. Rue and Bruland (1997), using a CSV method with the ligand salicylaldoxime (SA), found that in surface waters from the equatorial Pacific and in low-iron samples 15 with various Fe-binding ligands present, that UV-oxidation without acidification was adequate to destroy the soluble organic ligands and allow for the determination of dissolved Fe. In contrast, Bruland et al. (in prep) have evidence that UV-oxidation alone does not appear to be adequate to release all the iron in high-iron, coastal shelf waters off central California. They observed that in high-iron shelf waters, the UV-oxidation treatment followed by CSV analysis yielded dissolved Fe concentrations only roughly 50% of what was determined on acidified (pH 1.7) samples analyzed by the dithiocarbamate solvent extraction/GFAAS method. There appeared to be a colloidal fraction in these high-Fe shelf waters that required acidification to pH 1.7 to release it. In the low-iron offshore waters, however, the extraction and electrochemical methods yielded similar values for dissolved Fe and there did not appear to be a significant photochemically inert colloidal fraction. 2.3.4 Operationally-defined, Labile Dissolved Fe Measurements Determined by On-line Methods All of the commonly used FIA methods that are proliferating in the literature currently all use an 8-hydroxyquinoline (8-HQ) resin column as the critically important preconcentration step. The development of this vinyl-polymer-agglomerate based 8-HQ resin by Landing et al. (1986) was an analytical breakthrough necessary for the subsequent development of the FIA methods. Landing and co-workers (Dierssen et al. 1999) have recently reported an improved one-step synthesis of this resin. Without this preconcentration step, the FIA systems are not sensitive enough to directly determine iron in open ocean surface seawater. The pH dependency of the recovery of Fe(III) and Fe(II) on the 8-HQ resin is an important consideration and is shown in Figure 4 (Obata et al. 1993). Only Fe(III) is recovered by the resin at pH values from 3 to 4.2, whereas at pH values of 5.2 to 6 both Fe(II) and Fe(III) are recovered. At ambient pH in surface seawater, the majority of the dissolved Fe exists chelated with strong Fe(III)-binding ligands. It is not known what the pH dependency of these Fe-binding ligands is with Fe(III) (or with any potential Fe(II) organic complexes). It is also not known what the time dependency of the pH adjustment is on releasing iron from these various forms. The contact time of the sample with the resin as it passes through the column is another important parameter since the dissociation kinetics of the various forms of iron can regulate what fraction is available to bind to the resin during the sample’s interaction time. As a result, the preconcentration of iron on the 8-HQ resin is an operationally defined measure of labile Fe(III) - if passed through the resin at pH 3 to 4.2, or the sum of labile Fe(II) and Fe(III) - if passed through the resin at pH 5.2 to 6. 16 The oxidation kinetics of Fe(II) to Fe(III) can be markedly slowed by acidifying the sample to a pH near 3. Thus, any Fe(II) present in the original sample freed up at pH 3 would tend to be stabilized at this pH, and would not be recovered by the 8-HQ column if the pH was not adjusted to a pH of 5.2 to 6,. It is also unclear what the effect of weak acidification to pH 3 - 3.5 (for variable lengths of time) would have on solubilizing any colloidal Fe phases that might be present in the dissolved fraction, an uncertainty of great concern especially in estuarine and coastal waters, where dissolved Fe concentrations may be higher and colloidal Fe comprises a greater fraction. Figure 4. Relationship between pH and percent recovery of iron in seawater on an 8hydroxyquinoline chelating resin column. The iron concentration equals 1.8 nM (from Obata et al. 1993). Open circles represent Fe(III) values and closed circles represent Fe(II). 2.4 ANALYSIS OF PARTICULATE FE Our lack of more detailed chemical knowledge of naturally occurring particulate Fe in various oceanic regimes is a barrier to progress in understanding the dynamics between 17 the dissolved and particulate phases of iron. A 25% acetic acid (HAc) leach has commonly been used to characterize the more “labile” fraction of particulate Fe. The 25% HAc phase is thought to include iron associated with organic phases, carbonates, or Mn/Fe oxides (Landing and Bruland 1987). This weak acid-leach has been followed by bomb digestion (Landing and Bruland 1987) utilizing aqua regia and hydrofluoric acid to release iron associated with refractory mineral phases. Other techniques to characterize the "labile" fraction of particulate Fe include the use of strong chelating agents such as 8hydroxyquinoline (Wells et al. 1991) or dilute acids to operationally define “reactive Fe”. Hudson and Morel (1990) used a reductive Ti-citrate EDTA rinse to distinguish between adsorbed versus incorporated Fe (extracellular versus intracellular) to distinguish biological uptake from surface or precipitated Fe in culture experiments with radiotracers. This approach has also been used in shipboard incubations of natural plankton assemblages with radiotracers (Hutchins and Bruland 1994; Wells et al. 1994). 2.5 ACIDIFICATION AND ANALYSIS OF UNFILTERED SAMPLES - AN OPERATIONALLY DEFINED MEASUREMENT OF "DISSOLVABLE" FE As mentioned earlier, particulate Fe is often the dominant form of iron in seawater. The weak acidification of unfiltered seawater samples can lead to highly variable amounts of this particulate Fe being solubilized. The amount solubilized from the particulate phases depends upon the type of particles (biogenic, amorphous oxyhydroxides, aluminosilicate clay minerals, etc.), upon the strength and type of acid used, the length of time the sample is acidified, the temperature, etc. This makes any measurement of iron in unfiltered, weakly acidified samples extremely operational. The analysis of weakly acidified (i.e., pH 3 to 3.5) unfiltered samples, however, is common among the shipboard flow injection analysis (FIA) methods that have been popularized recently (Elrod et al. 1991; Obata et al. 1993, 1995; Johnson et al. 1998). The advantage of such methods is the rapidity of sample analyses. A typical profile of 12 samples together with blanks and standards can be analyzed in triplicate in 4.5 hours (Elrod et al. 1991). The major disadvantage of these methods is the liability of the operational definition inherent for the method. For example, those that make the analyses at pH 3.2 are measuring some labile fraction of the dissolved Fe(III), plus some poorly understood labile fraction of the particulate Fe which has been solubilized under these weakly acidic conditions. It is truly an operational measurement, one that is difficult to compare with other measurements. An example of such a method is that of Takeda and Obata (1995). These researchers used an FIA method employing an 8-HQ preconcentration column followed by sensitive chemiluminescence detection to analyze unfiltered seawater samples that 18 were adjusted to pH 3.0 with formic acid/ammonium formate buffer prior to the preconcentration step. The result of this procedure is that it is a “labile Fe(III)” technique. The authors point out that their measured “dissolved” concentration also contains the chemically reactive fraction of particulate Fe at pH = 3.0. Therefore, the term "dissolved" is a misnomer in this case. As a result, some investigators are calling this operationally defined measurement "labile dissolvable Fe.” It includes some fraction of the dissolved Fe(III) and some fraction of the particulate Fe(III). Sohrin et al. (in press) used a FIA method with an 8-HQ preconcentration column (8-HQ immobilized on fluoride containing metal alkoxide glass ) followed by chemiluminescence detection to determine iron on unfiltered samples acidified to pH 3.2 with formic acid/ ammonium formate buffer. The authors define their technique as a measurement of “a labile species, which reacts with 8-hydroxyquinoline ... at pH 3.2.” The authors comment that this labile measurement “... may include dissolved and some colloidal or particulate species.” Johnson et al. (1998) determine what they refer to as “reactive” Fe by FIA using the luminol chemistry described by Obata et al. (1993) and Elrod et al (1991) and an 8-HQ preconcentration column. The unfiltered sample stream from a surface sampling system is merged with a stream of hydrochloric acid to obtain a final pH of 3.3 0.2. The acidified sample is then held in a delay loop for 1 minute before entering the FIA manifold. Johnson and his coworkers comment that this is “an operationally defined measurement that includes the dissolved monomeric iron and the fraction of colloidal and particulate iron that will dissolve in less than 1 minute at pH 3.3.” At pH 3.3, according to Obata et al. (1993), the resin would not isolate any Fe(II) that might be released. Powell et al. (1995) used a similar FIA method involving preconcentration onto an 8-HQ resin followed by chemiluminescent detection. Unfiltered seawater samples were acidified with 2 mL of 6 M HCl per L of seawater (12 mequiv/L H+ added - pH ~ 2). The authors stated that “The chemical nature of the iron found in these samples is somewhat uncertain since the samples were not filtered prior to acidification and storage (0.01 M HCl for 4 years).” Then, prior to analysis, the Fe(III) reducing agent ( HSO3 bisulfite) was added to the acidified samples and allowed to react for at least 1 hour. The sample was then buffered to pH 4.5 and passed through an 8-HQ column. This approach relied upon Fe(II) standard additions to the sulfite reduced samples for calibration. The pH dependency of Fe(II) recovery was presumably different on the 8-HQ resin used by Powell et al. (1995) compared to that of Obata et al. (1993). Measures, Yuan and Resing (1995) developed a novel and sensitive FIA catalytic spectrophotometric method combining the use of in-line preconcentration of iron onto a 19 column containing 8-HQ immobilized on vinyl polymer gel together with the spectrophotometric detection of the iron eluted from the column by its catalytic effect on the oxidation of N,N-dimethyl-p-phenylenediamine dihydrochloride (DPD) by hydrogen peroxide. The catalytic nature of the reaction enhances the sensitivity of the method since the amount of oxidized DPD is proportional to the amount of iron and the length of the reaction time. Unfiltered seawater samples are acidified with 1 mL of 6M HCl per liter of sample to a pH 3 for an unspecified period of time. For analysis, the seawater sample is buffered and brought to a pH of 5.2 with ammonium acetate buffer prior to passing through the preconcentration column. Thus, this method initially acidifies the sample to pH 2.5-3.0 and then readjusts the pH to 5.2 for the recovery of both labile Fe(III) and Fe(II) on the 8-hydroxyquinoline column. It was applied primarily to unfiltered samples, so it is an operationally defined measure of some labile fraction of particulate Fe together with some fraction of the dissolved Fe. Wu and Boyle (1998) developed a method for determining iron in unfiltered samples based upon a Mg(OH)2 coprecipitation followed by analysis by ICP-MS. Acidified samples were spiked with enriched 57Fe (a stable isotope of Fe) and allowed to equilibrate overnight. NH4OH was used to precipitate the Mg(OH)2 that coprecipitated approximately 90% of the Fe. The particulate Fe would have been centrifuged along with the Mg(OH)2 and then subjected to 0.6 N HCl acid treatment used to resolubilize the iron. Thus, this was a measure of dissolvable Fe, which included the dissolved Fe, and an acidlabile component of the particulate Fe. They utilized this isotope dilution method in a study of iron in the eastern North Atlantic near Bermuda. The usefulness or validity of these “dissolvable” or “reactive” iron measurements made on unfiltered samples is justified by the authors on the basis of a potential relationship between chemical lability and biological availability. There is little in the literature, however, to support this contention. We do not yet have a clear idea of what constitutes the "biologically available" fraction of iron. Research needs to be carried out to better characterize these operational definitions of "reactive" or "labile dissolvable" iron determined by these various methods. To this end, we need inter-laboratory comparisons at sea to better understand just what the different methods are measuring. 3. THE DETERMINATION OF THE CHEMICAL SPECIATION OF IRON IN THE DISSOLVED FRACTION 3.1 REDOX SPECIATION - FE(II) VS. FE(III) 20 A number of researchers have reportedly measured Fe(II) in surface seawater. The relative proportions of Fe(III) and Fe(II) dissolved in surface seawater is dependent on the relative rates of reduction and oxidation by various mechanisms (Waite, this volume; Moffett, this volume) and by the extent of stabilization of the oxidized and reduced iron by inorganic and organic complexation. Measurements of Fe(II) concentrations are difficult and subject to artifacts since most methods rely on the stabilization of Fe(II) by chelators and may measure “reducible” as well as reduced Fe (Hudson et al. 1992; Price and Morel 1998). Techniques to determine Fe(II) in seawater have generally used the Fe(II)-specific ligand Ferrozine that forms a Fe(FZ)23 species. The Fe(II)-ferrozine complex is preconcentrated onto a C-18 Sep-PakTM cartridge (Waters) (King et al. 1991; Yi et al. 1992). Yi et al. (1992) present limited data that suggests approximately 40% of the dissolved Fe in Narragansett Bay surface waters is Fe(II). These Fe(II) methods in general do not have the sensitivity or detection limits required to examine Fe(II) in lowiron open ocean regimes. The King et al. (1991) "FeLume" FIA-chemiluminescent method described by Powell et al. (1995) was able to measure Fe(II) directly down to about 0.2 nM without an 8-HQ preconcentration column. Using this approach, Landing et al. (1999) found high Fe(II) levels in surface Atlantic waters immediately after rain events, and undetectable Fe(II) levels when it wasn't raining. The technique did not appear to reduce Fe(III), which they tested by spiking samples with low levels (1-5 nM) of inorganic Fe(III) standards. Johnson et al. (1994) added 10 nM iron to low-iron equatorial Pacific water and were then able to examine the photochemical production of Fe(II) with a similar "FeLume" method in a diel study at this greatly enhanced concentration of iron. 3.2 INORGANIC SPECIATION - FE(III)' AND FE(II)' Computer modeling of the inorganic speciation of Fe(III) and Fe(II) has been carried out numerous times. Turner et al. (1981) provided one of the first comprehensive evaluations of inorganic trace metal speciation and included two pH values for seawater. Byrne et al. (1988) evaluated the influence of temperature and pH on inorganic trace metal speciation in seawater. Hudson et al. (1992) evaluated the inorganic speciation of Fe(III) with respect to studies with the well characterized chelator EDTA. Millero et al. (1987, 1995) has applied Pitzer equations to estimate the inorganic speciation of Fe(III) and Fe(II). The inorganic chemistry of Fe(III) is dominated by its hydrolysis species with the hydroxide ligand. Fe(III) has an overall inorganic side reaction coefficient, Fe(III) = [Fe(III)]/[Fe3+] estimated to be in the range of 1010 to slightly more than 1011 in surface 21 seawater at a pH of approximately 8. Hudson et al. (1992) estimated Fe (III ) to be 1 x 1010, a value which compared well with their EDTA data. Millero et al. (1995) arrived at a value of 3 x 1010, while Byrne et al. (1988) arrived at a value for Fe (III ) of 6 x 1011. Boye et al. (1998) used an ion-pairing model with metal stability constants from Turner et al. (1981) to calculate Fe (III ) to be 2 x 1011. Much of the discrepancy in these estimates revolves around uncertainty over the importance of the Fe(OH)03 species and estimates of its conditional stability constant and the solubility of Fe(III)' with respect to Fe(OH)3(s) (Millero 1998). There is still an unsettling degree of controversy over the inorganic speciation and solubility of Fe(III). On one hand, this is surprising considering the important role of Fe in the oceans, on the other it is not surprising considering the difficulty of making measurements of these Fe species at such low concentrations. The inorganic speciation of Fe(II) appears to be relatively straight forward. It is estimated that ~76% of Fe(II) is free hydrated Fe 2 , while 23% exists as the FeCO30 species (Millero et al. 1995). 3.3 COMPLEXATION OR CHELATION WITH ORGANIC LIGANDS Voltammetric speciation methods have been developed recently to gain insight into the degree of complexation or chelation of Fe(III) with natural organic ligands in seawater. These methods involve a competitive ligand equilibration (CE) - followed by adsorptive cathodic stripping voltammetry (ACSV). Such CE-ACSV techniques are highly sensitive indirect methods that detect the concentration of an electrochemically-active, metal-added ligand (AL) complex. The measurement for iron is made after a competing equilibrium has been established between Fe3+ (or Fe(III)), a well-characterized added ligand, and any ambient, naturally-occurring, Fe(III)-binding organic ligand classes ( Li ). The Fe-AL complex formed during the competitive equilibration is subsequently adsorbed onto a hanging mercury drop electrode (HMDE) for an appropriate adsorption period, and the analytical signal is from the resultant reduction current as the Fe(III) in the adsorbed complex is reduced during the cathodic stripping step. Differential pulse (DP), square wave (SW), or rapid linear scan (LS) are common pulse forms used during the stripping step to enhance the analytical signal. Gledhill and van den Berg (1994) and van den Berg (1995) developed a CE-ACSV method using the added ligand 1-nitroso-2-naphthol (1N2N) to determine the complexation of Fe(III) with natural organic complexing ligands in seawater. Rue and Bruland (1995; 1997) developed a Fe(III) method involving salicylaldoxime (SA) as the added competing ligand. The “detection window” of the CE-ACSV method is framed by the analytical detection limit of the cathodic signal during the stripping step at one end and the ability to determine a small decrease of the peak height of the Fe(AL)n species by the competition 22 of the natural ligands classes ( Li) for Fe3+ at the other. In the CE-ACSV method using salicylaldoxime (Rue and Bruland 1995), the “analytical competition strength” ( Fe (SA) 2 ) of the surface-active complex formed by the added ligand with Fe3+ is a function of 2 Fecond [SA] . The analytical competition strength is determined by the comparative (SA) 2 , Fe 3 magnitudes of the side reaction coefficients between the ambient natural ligands and that of the added, well-characterized competitive ligand: cond FeL [FeLi ] / [Fe 3 ] KFeL [ Li] , Fe 3 i i and 2 Fe (SA) [Fe(SA)2 ] / [Fe 3 ] Fecond [SA] (SA) , Fe 3 2 2 The detection window of a CE-ACSV technique can be purposely varied by increasing or decreasing the concentration of the added competing ligand or by the selection of a different added ligand that forms weaker or stronger complexes with Fe 3 (or Fe(III)). The competitive equilibrium with the added ligand must not be too strong, or the added ligand will completely out compete [ Li] and result in all the Fe(III) existing as the electrochemically active and measured Fe(AL)n species, and not too weak, since the Fe(AL)n species is the electroactive or measured species in this approach. The added ligand must out compete the tendency for Fe(III) to form inorganic complexes with OH-; e.g., Fe (SA) 2 must be significantly greater than n 3 cond n Fe (III ) [Fe(OH)3 n ] / [Fe ] Fe(OH) [OH ] but less than or equal to FeL ). The ability to vary the analytical “detection window” or 3n n i "analytical competition strength" over many orders of magnitude is a unique property of CE methods (van den Berg 1984; Miller and Bruland, 1997). Either linearization or nonlinear calculations for fitting of the titration data are generally used to determine the total Fe-binding ligand concentrations and their respective conditional stability constants. Two linearization methods, based on discrete ligand binding models, have gained general use: one based on Scatchard’s work with small molecule binding by proteins (Scatchard 1949) described by Mantoura and Riley (1975) and the other from the Langmuir isotherm as described by Ruzic (1982). Other authors have suggested the use of non-linear curve fitting routines for direct fitting of the titration data (Fish et al. 1986: Gerringa et al. 1995). The linearization techniques of Scatchard or Ruzic for evaluating conditional stability constants and ligand concentrations have been criticized as being oversimplified (Buffle 1992). In particular, they are applicable only with caution to chemically heterogeneous ligands. Miller and Bruland (1997) evaluated these approaches on heterogeneous mixtures of ligands and 23 demonstrated that although they tended to represent complex mixtures of ligands as just two classes of ligands, the concentration of free or inorganic metal calculated was remarkably consistent and accurate. Titrations of natural samples with Fe(III), and resultant measurements of the Feadded ligand complex at each titration point, allow for the determination of the natural Fe(III)-binding ligand concentrations ([Li]) and their conditional stability constants cond cond ( KFeL 3 or KFeL , Fe (III ) ). These values allow for the original ambient, unperturbed i i , Fe Fe(III) speciation in the sample to be calculated. For a more detailed description of manipulations involved in this calculation see Rue and Bruland (1995). To demonstrate the effect of different analytical competition strengths in determining Fe-binding ligand concentrations and their conditional stability constants, we have provided an example of the use of the CE-ACSV approach in determining Febinding ligand concentrations and conditional stability constants. In Figure 5 we present model Figure 5. Model titration curves of a typical North Pacific surface sea water sample that cond contains the following: [L1] = 0.4 nM with a KFeL1 ,F e(III ) = 1013 M-1 and [L2] = 1.0 nM with 24 cond a KFeL2 , Fe ( III ) = 1011.5 M-1. A) [Fe(III)'] as a function of increasing [FeT] under ambient conditions (i.e., no added competing ligand). B) the concentration of electrochemically active species Fe(SA)2 as a function of increasing [FeT] using three different "analytical competition strengths" where (o) corresponds to [SA] = 15 M, () corresponds to [SA] = 30 M and () to [SA] = 90 M. titration curves of a typical North Pacific sample using three different concentrations of an added competing ligand to probe the natural Fe-binding ligands. The North Pacific surface sample is represented by two classes of Fe(III)-binding ligands; a stronger ligand cond class, [L1] = 0.4 nM with a KFeL1 ,F e(III ) = 1013 M-1, and a weaker ligand class, [L2] = 1.0 cond nM with KFeL2 , Fe ( III ) = 1011.5 M-1. Figure 5A shows an Fe(III) titration of this sample with no added competing ligand present. The natural Fe(III)-binding ligands are strong enough and their concentrations high enough to chelate the vast majority of the total dissolved Fe until the ligand classes are titrated completely ([FeT] > 1.4 nM). This figure also illustrates how the natural system would respond to Fe additions; [FeT] would remain effectively completely chelated until additions resulting in [FeT] being greater than 1.4 nM were made. Figure 5B shows iron titrations resulting from three different concentrations of SA; a concentration close to what was actually used to estimate the Fe speciation - 30 M salicylaldoxime (Rue and Bruland, 1995), 1/2 this concentration ([SA] = 15 M), and 3 times this concentration ([SA] = 90 M). This 6-fold variation in added ligand concentrations results in the analytical competition strength ( Fe (SA) 2 ) set up with the added ligand varying by a factor of 36. As the [SA] increases, the competing equilibrium is becoming stronger and therefore shifting the equilibrium away from FeLi and more towards Fe(AL)2. It can be seen from the titration data in Figure 5B and the results calculated from the Scatchard linearizations in Figure 6A, that the competition set up at the lowest concentrations of SA ([SA] = 15 M) yields a good estimate of the total ligand concentration of 1.4 nM, but is unable to distinguish the presence of the two classes of cond ligands. In this case a [LT] = 1.4 nM was calculated with a KFeL ,Fe (III ) = 1011.7 M-1, a cond value slightly higher than the actual value of KFeL2 , Fe ( III ) . In contrast, the highest concentration of added ligand ([SA] = 90 M) is able to distinguish the L1 ligand class, but slightly underestimates the concentration of the weaker L2 ligand class. In this case cond the Scatchard plot (Figure 6B) yielded [L1] = 0.43 nM with a KFeL1 ,F e(III ) = 1012.9 M-1 and cond [L2] = 0.90 nM with a KFeL2 , Fe ( III ) = 1011.8 M-1. Thus, the choice of analytical competition strength employed can bias the estimates of both ligand concentrations and conditional stability constants. With too weak an 25 analytical competition strength, only the sum of the ligands will be "seen" and the conditional stability constant approaches that of the weaker ligand class (L2) and underestimates the effect of the stronger ligand class, L1. With too strong an analytical competition strength, the presence of the weaker ligands can be missed and only the concentration of the strongest ligand classes can be estimated. In this case, however, better estimates can be made of the conditional stability constants of the stronger ligand classes. Thus, it is imperative to recognize the limitations of these approaches and, when necessary, alter the analytical competition strength of the approach appropriately (by Figure 6. Scatchard linearization plots from model titration curves of a typical North Pacific Surface sea water sample (same conditions as Figure 5) at 2 different "analytical competition strengths." A) [SA] = 15 M and B) [SA] = 90 M. either varying the concentration of added competing ligand or the choice of a new ligand which is either weaker or stronger depending upon the requirement - see Bruland et al. 1999 for an analogous and more detailed description of an intercomparison study on copper speciation involving multiple voltammetric approaches). A fundamental problem underlying efforts to characterize trace metal organic complexation phenomena in seawater is the heterogeneity of the natural environment. Both the character and concentrations of metal binding ligands in different seawater samples, and the extent to which ligand coordination sites are naturally occupied by a variety of metals are variable. In iron speciation studies, differences in estimates of classes of discrete Fe-binding ligand concentrations and their conditional stability 26 constants could be due to systematic errors in the analytical or curve fitting techniques used. Differences may also be due to the presence of a mixture of natural chelators that form complexes with a range of stabilities that may or may not be distinguishable using the particular analytical competition strength employed. Due to the potential large spread of stability constants of the natural ligands, the characteristics (i.e., conditional stability constants and concentrations) of the classes of Fe-binding ligands detected can depend on the “detection window” or analytical competition strength of the analytical technique used (Bruland et al. Submitted). This, in turn, may influence estimates of the ambient [Fe(III)]. Ideally, an investigator would use a variety of analytical competition strengths to probe the natural iron speciation and characterize the Fe-binding ligand classes. Potential problems that also might effect the use of CE-ACSV methods include: 1) possible competitive adsorption of natural and added ligands (or their metal complexes) at the HMDE surface, 2) the possibility that slow kinetic dissociation of the strong natural complexes of Fe might not allow equilibration with the added ligand, and 3) possible formation of mixed ligand complexes between the added ligand, the natural ligand and the metal ion. To a large extent, these potential problems have been considered or addressed. Open ocean seawater, with its relatively low dissolved organic matter (DOM) concentration, is a much simpler solution to analyze than high DOM fresh waters. As a result, these artifacts appear to be minimized in the analysis of open ocean samples. Rue and Bruland (1995) compared the sensitivity of seawater samples from the North Pacific with that of UV oxidized seawater and found the sensitivities to be the same, inferring that there was not a problem with competitive adsorption of the natural ligands. Rue and Bruland (1995), van den Berg (1995), and Wu and Luther (1995) have all evaluated the kinetics of ligand exchange and have accounted for these rates and employed appropriate equilibration times in their analytical techniques. The problem of possible formation of mixed ligand complexes, however, is more difficult to evaluate. Rue and Bruland (1995) did check the accuracy of their CE-ACSV method by determining concentrations and conditional stability constants of the well-characterized, commercially available siderophore, Desferal. They found good agreement between their analytical results and those expected. That is not to say, however, that the potential for adsorption of mixed ligand complexes, especially at high ligand concentrations, does not exist. Regardless of these potential problems, a clear picture is emerging that the chemical speciation of dissolved Fe is dominated by complexation or chelation with Fe(III)-binding organic ligands of biological origin. Depending upon estimates of the concentrations and stability constants of these natural Fe-binding ligands, estimates of the inorganic side reaction of Fe(III), and estimates of the photochemical reactivity of iron, various 27 investigators suggest that between 90 and 99.9% of the dissolved iron in the open oceans exists as organically bound FeLi species. A recent example (Rue and Bruland 1997) of an iron speciation study in a low-iron HNLC area was performed as part of the Iron-Ex II mesoscale iron addition experiment (Coale et al. 1996). Rue and Bruland (1997) observed two classes of Fe(III)-binding cond organic ligands: a strong ligand class (L1) with a conditional stability constant KFeL 1 ,Fe (III ) 12 -1 = 5 x 10 M and a mean concentration of 310 pM, and a weaker class (L2) with a cond conditional stability constant KFeL = 6 x 1011 M-1 and a mean concentration of 190 2 , Fe (III ) pM. The total Fe(III)-binding organic ligand concentrations were ~25 times higher than total dissolved Fe concentrations of only 20 pM (0.02 nM). Thermodynamic equilibrium calculations suggest that 99.9% of the ambient dissolved Fe(III) would be complexed with these organic ligands and results from ultrafiltration experiments indicated that they exist as low-molecular weight Fe(III) chelates. Rue and Bruland (1997) and Sunda (this volume) have made estimates of potential photoreduction rates of the FeLi complexes, and coupled these with estimates of the re-oxidation rates of Fe(II) and the re-chelation rates of Fe(III). Such an active photoreduction cycle could potentially increase Fe(III) concentrations in surface waters during daytime with full-sunlight conditions by over two orders-of-magnitude, from less than 0.1% of the dissolved Fe to on the order of 10% or even 20% of the dissolved Fe. Rue and Bruland (1997) estimate that there could be as much as 2 to 5 pM of Fe(III) and 1 pM of Fe(II) under full sunlight conditions, but less than 0.01 pM of either species during nighttime conditions. This demonstrates the analytical challenges researchers face in speciation studies with iron, in trying “...to demonstrate and quantify the existence of fractions of chemical constituents as picomolar concentrations of perhaps ephemeral species” (Morel and Hering 1993). In studies of iron speciation in HNLC areas, our thinking should perhaps be changed from picomolar to femptomolar concentrations. It should be pointed out that these equilibration techniques outlined above do not allow us to look at Fe(II) vs. Fe(III) speciation. During the dark storage time prior to analysis or during the equilibration with the competing ligand, all of the Fe(II) would likely be oxidized to Fe(III) and become complexed with the excess ligand(s) or the added competing ligand. An approach to measure Fe(II) speciation in-situ with organic ligands similar to the Fe(III) CE-ACSV techniques needs to be developed. 4. NEW DIRECTIONS 28 Our ability to learn more about the dynamic cycling of iron has been, and most likely will continue to be, limited by the availability of highly sensitive analytical techniques that are well-characterized with respect to what forms or chemical species of iron they determine. To continue making progress in our understanding of the biogeochemical cycling of iron in seawater, we must remain vigilant about collection, filtering, and subsequent sample handling. We need to continue to develop clean, fast, simple and very well defined (species specific) analytical methods that we can use at sea so that scientists will be able to carry out near real time determinations that will allow for rapid evaluation of field results in order to alter the research plan and make efficient use of ship time in process studies. Two different analytical approaches for the rapid shipboard determination of total dissolved Fe that show substantial promise are 1) flow injection analysis with either chemiluminescence (Obata et al. 1993; Elrod et al. 1991; Johnson et al. 1997) or catalytically enhanced spectrophotometric detection (Measures et al. 1995) and 2) automated flow-through ACSV approaches (Colombo et al. 1997). Both of these approaches, however, need more attention paid to the basic steps of filtration and acidification. Methodological questions to consider are: 1) What is the best in-line method to effectively filter samples prior to analyses? 2) What conditions of preacidification and/or UV-oxidation treatment are necessary to release colloidal-Fe and organically bound iron for quantitative determination? 3) For the flow injection analysis (FIA) methods, more attention needs to be placed on characterizing the chelating resin columns used to pre-concentrate the iron. If the trend in research initiatives moves towards autonomous ocean observatories, expect to see the development of in-situ iron speciation analyzers. Evidence is mounting that the availability of iron influences the productivity, species composition and trophic structure of planktonic communities in the oceans. Addressing the processes responsible for the role of iron forces us to look more closely at its marine chemistry. Analytical techniques to probe the chemical species or forms of iron in the oceans have been developed and applied. This has provided us with the observation that the majority of dissolved iron exists as Fe(III)-chelates with relatively specific and strong Fe(III)-binding ligands. Very little information exists, however, as to the molecular structure of these Fe-binding ligands or the mechanisms involved in their production or function. The need for future research elucidating the molecular structure and character of the strong Fe-binding ligands observed in seawater has been recognized and has begun. In addition, research into the biological availability of the various 29 chelated forms of iron and mechanisms various microorganisms utilize to access these species will be a research area of importance in the next decade. 30 5. REFERENCES Barbeau, K., Moffett, J.W., Caron, D.A Croot, P.L. and Erdner, D.L. 1996. Role of protozoan grazing in relieving iron limitation of phytoplankton. Nature 380: 61-64. Beauchemin, D. and Berman, S. S. 1989. Determination of trace metals in reference standards by inductively coupled plasma mass spectrometry with on-line preconcentration. Anal. Chem. 61:1857-1862. Boye, M., Sarthou, G. and van den Berg, C.M.G. 1998. Organic speciation of iron and zinc in the northeastern Atlantic Ocean. Deep-Sea Res. in press. Boyle, E.A., Edmond, J.M. and Sholkovitz, E.R. 1976. The mechanism of iron removal in estuaries. Geochim. Cosmochim. Acta, 41: 1313-1324. Brand, L. E. 1991. Minimum iron requirements of marine phytoplankton and the implications for the biogeochemical control of new production. Limnol. Oceanogr. 36: 1756-1771. Bruland, K. W.,Franks, R. P. , Knauer, G. A. and Martin, J. H. 1979. Sampling and analytical methods for the determination of copper, cadmium and nickel in seawater. Anal. Chim.Acta 105: 233-245. Bruland, K.W., Coale, K.H. and Mart, L. 1985. Analysis of seawater for dissolved cadmium, copper and lead: an intercomparison of voltammetric and atomic absorption methods. Mar. Chem. 17: 285-300. Bruland, K.W., Donat, J.R. and Hutchins, D.A. 1991. Interactive influences of bioactive trace meetals on biological production in oceanic waters. Limnol. & Oceanogr. 36: 15551577. Bruland, K.W., Orians, K.J. and Cowen, J.P. 1994. Reactive trace metals in the stratified central North Pacific. Geochemica et Cosmochemica Acta. 58: 3171-3182. Bruland, K. W., Rue, E.L. , Donat, J. R., Skrabal,S. and Moffett, J. W. ( submitted). An intercomparison of voltammetric approaches to determine the chemical speciation of dissolved copper in a coastal seawater sample. Anal. Chim. Acta. Bruland,K.W., Rue, E.L. and Smith, G. (in prep.) Iron and zinc in coastal upwelling regimes off central California: implications for diatom blooms. Buffle, J., Pernet, D. and Newman, M. The use of Filtration and Ultrafiltration for size fractionation of aquatic particles, colloids, and macromolecules. In “Characterization of 31 Environmental Particles”, J. Buffle and H. P. van Leeuwen, Eds. IUPAC Environmental Analytical Chemistry Series, Vol. 1, Chelsea, MI. Lewis Publishers, 1992, Chapter 5. Byrne, R., Kump, L.R. and Cantrell, K.J. 1988. The influence of pH and temperature on trace metal speciation in seawater. Mar. Chem. 25: 163-181. Chisholm, S. 1992. Phytoplankton Size. In Primary Productivity and Biogeochemical Cyles in the Sea. Falkowski, P. G. and Woodhead, A. D. (Eds.). Plenum Press, New York, 213. Chisholm, S, Olson, R. J., Zettler, E. R., Goericke, R., Waterbury, J. and Welschmeyer, N. 1988. A novel;, free-living prochlorophyte abundant in the oceanic euphotic zone. Nature, 334:340. Danielsson, L.-G., Magnusson, B. and Westerlund, S. 1978. An improved metal extraction procedure for the determination of trace metals in seawater by atomic absorption spectrometry with electrothermal atomization. Anal. Chim. Acta 98: 47-57. Davison, W. and DeVitre, R. Iron particles in freshwater. In In “Characterization of Environmental Particles”, J. Buffle and H. P. van Leeuwen, Eds. IUPAC Environmental Analytical Chemistry Series, Vol. 1, Chelsea, MI. Lewis Publishers, 1992, Chapter 8. de Baar, H.J.W., de Jong, J.T.M., Nolting, R.F. , van Leeuwe, M.A. , Timmermans, K.R. Bathmann, U. , van der Loeff, M.R. and Sildam, J. 1998. Low dissolved Fe and the absence of diatom blooms in remote Pacific waters of the Southern Ocean. Mar. Chem. in press. de Jong, J.T.M., den Das, J. , Bathmann, U. , Stoll, M.H.C. , Kattner, G. , Nolting, R.F. and de Baar, H.J.W. 1998. Dissolved iron at sub-nanomolar levels in the Southern Ocean as determined by ship-board analysis. Anal. Chim. Acta, in press. Dierssen et al. 1999 ****** Elrod, V. A., Johnson, K. S. and Coale, K. H. 1991. Determination of subnanomolar levels of iron(III) and total dissolved iron in seawater by flow injection analysis with chemiluminescence detection. Anal. Chem. 63: 893-898. Falkowski, P.G. 1997. Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean. Nature 387: 272-274. Falkowski, P.G., Barber, R.T. and Smetacek, V. 1998. Biogeochemical controls and feedbacks on ocean primary production. Science 281: 200-206. 32 Farias, P.A.M., Liohara, A. and Ferreira, S.L.C. 1992. Adsorptive preconcentration for voltammetric measurements of trace level of iron(III) with 2-(2-thiazolylazo)-4methylphenol. Anal. Lett. 25: 1929-1939. Fish, W., Dzombak, D.A. and Morel, F.M.M. 1986. Environ. Sci. Technol.20: 676. Gerringa, L.J.A., Herman, P.M.J. and Poortvliet, T.C.W. 1995. Comparison of the linear van den Berg/Ruzic transformation and a non-linear fit of the Langmuir isotherm applied to Cu speciation data in the estuarine environment. Mar. Chem. 48: 131-142. Gledhill,M. and van den Berg, C.M.G. 1995. Measurement of the redox speciation of iron in seawater by catalytic cathodic stripping voltammetry. Mar. Chem. 50: 51-62. Gordon, R.M., Martin, J.H. and Knauer, G.A. 1982. Iron in north-east Pacific waters. Nature 299: 142-143. Howard, A. G. and Statham, P. J. Inorganic Trace Analysis: Philosophy and Practice. John Wiley, and Sons, Ltd. N.Y. 1993. Hudson, R. J. M., and Morel, F. M. M. 1990. Iron transport in marine phytoplankton: Kinetics of cellular and medium coordination reactions. Limnol. Oceanogr. 35: 10021020. Hudson, R.J.M., Covault, D.T. and Morel, F.M.M. 1992. Investigation of iron coordination reactions in seawater using 59Fe radiometry and ion-pairing solvent extraction of amphiphilic iron complexes. Mar. Chem. 38: 209-235. Hudson, R.J.M. 1998. Which aqueous species control the rates of trace metal uptake by aquatic biota? Observations and predictions of non-equilibrium effects. The Science of the Total Environment. in press. Hutchins, D.A. 1995. Iron and the marine phytoplankton community. In Chapman, D. and F. Round (Eds.) Progress in Phycological Research, Vol. 11. Biopress Ltd. pp 1-49. Hutchins, D.A., DiTullio, G.R.and Bruland, K.W. 1993. Iron regenerated production: evidence for biological iron recycling in two marine environments. Limnol. & Oceanogr. 38: 1242-1255. Hutchins, D. A. and Bruland, K. W. 1994. Grazer-mediated regeneration and assimilation of Fe, Zn, and Mn from planktonic prey. Mar. Ecol. Prog. Ser. 110: 259269. Hutchins, D.A., DiTullio, G.R., Zhang, Y. and Bruland, K.W. 1998. An iron limitation mosaic in the California upwelling regime. Limnol. & Oceanogr. 43: 1037-1054. 33 Johnson, K.S., Gordon, M.R. and Coale, K.H. 1997. What controls dissolved iron concentrations in the world ocean? mar. Chem. 57: 137-161. Johnson, K.S., Coale, K.H., Elrod, V.A. and Tindale, N.W. 1994. Iron photochemistry in seawater from the equatorial Pacific. Mar. Chem. 46: 319-334. King, J.N. and Fritz, J.S. 1985. Concentration of metal ions by complexation with sodium bis(2-hydroxyethly)dithiocarbamate and sorption on XAD-4 resin. Anal. Chem., 75: 1016-1020. King, D.W., Lin, J. and Kester, D.R. 1991. Spectrophotometric determination of iron(II) in seawater at nanomolar concentrations. Anal. Chim. Acta 247: 125-132. Kozelka, P. B. and Bruland, K. W. 1998. Chemical speciation of dissolved Cu, Zn, Cd, Pb in Narragansett Bay, Rhode Island. Mar. Chem. 60: 267-282. Kuma, K., Katsumoto, A., Nishioka, J. and Matsunaga, K. 1998. Size fractionated iron concentrations and Fe(III) hydroxide solubilities in various coastal waters. Est. Coast. Shelf Sci. 47: 275-283. Landing, W.M. and Lewis, B. L. 1991. Collection, processing, and analysis of marine particulate and colloidal material for transistion metals. In: D. C. Hurd and D. W. Spencer (Editors), Marine Particles: Analysis and Charterization. Am. Geophys. Union Geophs. Monogr., 63: 263-272. Landing, W.M. and Yeats, P.A. 1991. The analysis and characterization of marine particles: sample manipulation and speciation. In: D. C. Hurd and D. W. Spencer (Editors), Marine Particles: Analysis and Charterization. Am. Geophys. Union Geophs. Monogr., 63: 23-32. Landing, W. M., Haraldsson, C. and Paxeus, N. 1986. Vinyl polymer agglomerate based transition metal cation ion-exchange resin containing the 8-hydroxyquinoline funtional group. Anal. Chem. 58: 3031-3035. Landing, W.M. and Bruland, K.W. 1987. The contrasting biogeochemistry of iron and manganese in the Pacific Ocean. Geochim. Cosmochim. Acta, 51: 29-43. Maldonado, M.T. and Price, N.M. 1998. Iron and nitrate reductase activities in membranes of Thalassiosira oceanica. Limnol. & Oceanogr. in press. Mantoura, R.R.C., and Riley, J.P. 1975. The use of gel filtration in the study of metal binding by humic acids and related compounds. Anal. Chim. Acta 78: 193-200. Maranger, R., Bird, D. F. and Price, N. M. 1998. Iron aquisition by photosynthetic marine phytoplankton from injected bacteria. Nature 396: 248-251. 34 Martin, J.H and Gordon, R.M. 1988.Northeast Pacific iron distributions in relation to phytoplankton productivity. Deep-Sea Res. 35:177-196. Martin, J.H., Fitzwater, S.E., Gordon, R.M., Hunter, C.N. and Tanner, S.J. 1993. Iron, primary production and carbon-nitrogen flux studies during the JGOFS North Atlantic Bloom Experiment. Deep-Sea Res. 40: 115-134. Martin, J. H., K. H. Coale,K. S. Johnson, S. E. Fitzwater, R. M. Gordon, S. J. Tanner, C. N. Hunter, V. A. Elrod, J. L. Nowicki, T. L. Coley, R. T. Barber, S. Lindley, A. J. Watson, K. Van Scoy, C. S. Law, M. I. Liddicoat, R. Ling, T. Stanton, J. Stockel, C. Collins, A. Anderson, R. Bidigare, M. Ondrusek, M. Latasa, F. J. Millero, K. Lee, W. Yao, J. Z. Zhang, G. Friederich, C. Sakamoto, F. Chavez, K. Buck, Z. Kolber, R. Greene, P. Falkowski, S. W. Chisholm, F. Hoge, R. Swift, J. Yungel, S. Turner, P. Nightingale, A. Hatton, P. Liss, and N. W. Tindale. 1994. Testing the iron hypothesis in ecosystems of the equatorial Pacific Ocean. Nature 371:123-129. Mayer, L. M. 1982. Aggregation of colloidal iron during estuarine mixing: Kinetics mechanism, and seasonality. Geochim. Cosmochim. Acta. 46:2527-2535. Measures, C.I., Yuan, J. and Resing, J.A. 1995. Determination of iron in seawater by flow injection analysis using in-line preconcentration and spectrophotometric detection. Mar. Chem. 50: 1-2. Miller, L.A. and Bruland, K.W. 1997. Competitive equilibration techniques for determining transition metal speciation in natural waters: evaluation using model data. Anal. Chim. Acta 343: 161-181. Millero, F. J., Sotolongo, S. and Izaguirre, M. 1987. The oxidation kinetics of Fe(II) in seawater. Geochim. Cosmochim. Acta 51: 793-801. Millero, F.J., Yao, W. and Aicher, J. 1995. The speciation of Fe(II) and Fe(III) in natural waters. Mar. Chem. 50: 21-40. Millero, F.J. 1998. Solubility of Fe(III) in seawater. Earth and Mar. Sci. Lett. 154: 323329. Morel, F.M.M. and Hering, J. 1993. Principles and Applications of Aquatic Chemistry. New York: Wiley, 588 p. Nolting, R.F., Gerringa, L.J.A., Swagerman, M.J.W., Timmermans, K.R. and de Baar, H.J.W. 1998. Fe(III) speciation in the high nutrient, low chlorophyll Pacific region of the Southern Ocean. Mar. Chem. 62: 335-352. 35 Obata, H., Karatani, H. and Nakayama, E. 1993. Automated determination of iron in seawater by chelating resin concentration and chemiluminescence detection. Anal. Chem. 65: 1524-1528. Obata, H., Karatani, H., Matsui, M. and Nakayama, E. 1997. Fundamental studies for chemical speciation of iron in seawater with an improved analytical method. Mar. Chem. 56: 97-106. Powell, R.T., King, W. and Landing, W.M. 1995. Iron distribution in surface waters of the south Atlantic. Mar. Chem. 50: 13-20. Powell, R.T., Landing, W.M., and Bauer, J.E. 1996. Colloidal trace metals, organic carbon and nitrogen in a southeastern U.S. estuary. Mar. Chem. 55: 165-176. Price, N. Ahner, M.,B.A.and Morel, F.M.M. 1994. The Equatorial Pacific Ocean: grazercontrolled phytoplankton populations in an iron-limited ecosystem. Limnol. & Oceanogr. 39: 520-534. Price, N. M. and Morel, F.M.M. 1998. Biological cycling of iron in the sea. In:Iron Transport and Storage in Microorganisms, Plants, and animals. Vol. 35 of Metal Ions in Biological systems A. Sigel and H. Sigel, eds. M. Dekker, Inc. New York. Price, N. M. and Maldonado, M. T. 1995. Iron acquisition by marine diatoms consuming different nitrogen sources. Abstract in: Iron speciation and its biological availability in seawater: a workshop. Institute of Marine Sciences, U.C. Santa Cruz. Raven, J. 1997. Phagotrophy in phototrophs. Limnol. Oceanogr. 42: 198-205. Rue, E. L., Bruland, and K. W. 1995. Complexation of iron(III) by natural organic ligands in the Central North Pacific as determined by a new competitive ligand equilibration / adsorptive cathodic stripping voltammetric method. Mar. Chem. 50: 117-138. Rue, E. L., and Bruland, K. W. 1997. The role of organic complexation on ambient iron chemistry in the equatorial Pacific Ocean and the response of a mesoscale iron addition experiment. Limnol. Oceanogr. 42: 901-910. Ruzic, I. 1982. Theoretical aspects of the direct titration of natural waters and its information yield for trace metal speciation. Anal. Chim. Acta 140: 99-113. Scatchard, G. 1949. "The Attractions of Proteins for Small Molecules and Ions." Ann. New York Acad. Sci. 51:660-672. Sholkovitz, E.R. and Copeland, D. 1981. The coagulation, solubility and absorption properties of Fe, Mn, Cu, Ni, Cd,Co and humic acids in a river water. Geochim. Cosmochim. Acta, 45: 181-189. 36 Skoog, A. and Benner, R. 1997. Aldoses in various size fractions of marine organic matter: Implications for carbon cycling. Limnol. Oceanogr. 42: 1803-1813. Sohrin, Y., Iwamoto, S., Matsui, M. Obata, H. Nakayama, E. Handa, N., Suzuki, K. and Ishii, M. 1998. in press. The distribution of Fe in the Australian sector of the Southern Ocean. Stumm, W. and Morgan, J. 1996. Aquatic Chemistry. New York: Wiley. Sunda, W. G., and Huntsman, S. A. 1995. Iron uptake and growth limitation in oceanic and coastal phytoplankton. Mar. Chem. 50: 189-206 Sunda, W. G., and Huntsman, S. A. 1997. Interrelated influence of iron, light and cell size on marine phytoplankton growth. Nature 390: 389-392. Takeda,S.A. and Obata, H. 1995. Response of equatorial Pacific phytoplankton to subnanomolar Fe enrichment. Mar. Chem. 50: 219-228. Turner, D,R, Whitfield, M. and Dickson, A.G. 1981. The equilibrium speciation of dissolved components in freshwater and seawater at 25o C and 1 atm pressure. Geochim. Cosmochim. Acta 45: 855-881. van den Berg, C.M.G. 1984. Determination of the complexing capacity and conditional stability constants of cpmplexes of copper (II) with natural organic ligands in seawater by cathodic stripping voltammetry of copper-catechol complex ions. Mar. Chem. 15: 1-18. van den Berg, C.M.G. 1995. Evidence for organic complexation of iron in seawater. Mar. Chem. 50: 139-157. van Geen, A. and Boyle, E. 1990. Automated preconcentration of trace metals from seawater and freshwater. Anal. Chem., 62: 1705-1709. Wells, M. L. and Mayer, L. M. 1991. The photoconversion of colloidal iron oxyhydroxides in seawater. Deep-Sea Res. 38: 1379-1395. Wells, M.L., Mayer, L.M. and Guillard, R.R.L. 1991. A chemical method for estimating the availability of iron to phytoplankton in seawater. Mar. Chem. 33: 23-40. Wells, M.L., Price, N.M. and Bruland, K.W.. 1994. Iron limitation and the cyanobacterium Synechococcus in equatorial Pacific waters. Limnol. & Oceanogr. 39: 1481-1486. Wells, M.L., Price, N.M. and Bruland, K.W. 1995. Iron chemistry in seawater and its relationship to phytoplankton: A workshop report. Mar. Chem. 48: 157-182. 37 Wells, M.L., Smith, G.J. and Bruland, K.W. submitted. The distribution of colloidal and particulate bioactive metals in Narragansett Bay, RI. Mar. Chem. Wells, M.L. and Bruland, K.W. 1998. An improved method for rapid preconcentration and determination of bioactive trace metals in seawater using solid phase extraction and high resolution indictively coupled plasma mass spectroscopy. Mar. Chem. in press. Wu, J. and Luther III, G.W. 1995. Complexation of Fe(III) by natural organic ligands in the Northwest Atlantic Ocean by a competitive ligand equilibration method and a kinetic approach. Mar. Chem. 50: 159-178. Wu, J. and Luther III, G.W. 1996. Spatial and temporal distribution of iron in the surface water of the northwestern Atlantic Ocean. Geochim. Cosmochim. Acta 60: 2729-2742. Wu, J. and Boyle, E. 1998. Determination of iron in seawater by high-resolution isotope dilution inductively coupled mass spectrometry after Mg(OH)2 coprecipitation. Anal. Chim. Acta. 367:183-191. Yi, Z., Zhuang, G. Brown, P.R. and Duce, R.A.. 1992. High-performance liquid chromatographic method for the determination of ultratrace amounts of iron(II) in aerosols, rainwater and seawater. Anal. Chem. 64: 2826-2830. 38