Training
advertisement

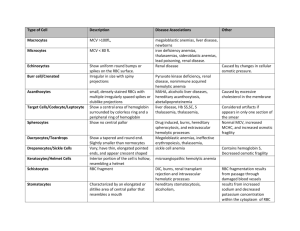
Anemias and Red Cell Disorders Jeffrey T. Reisert, DO University of New England Physician Assistant Program 25 FEB 2010 Contact Information Jeffrey T. Reisert, DO Tenney Mountain Internal Medicine, PLLC 16 Hospital Rd. Plymouth, NH 03264 603-536-6355 603-536-6356 (fax) Jeffrey.T.Reisert@Hitchcock.org Case You are referred a case from one of your healthy patients. He was told the “Red Cross didn’t want my blood.” You rarely see this healthy fellow You have no additional information Case work up Does he have anemia Does he have AIDS or something? What do you need to – What tests are needed – What is considered a reasonable work-up with his story Objectives Participants will be able to analyze the CBC (carry over from prior coursework on CBC) Develop an understanding of anemias and polycythemias Given a hematologic case presentation, students will define a common disorder and its treatment. Agenda Analyzing the Complete Blood Count (CBC)-A Quick Review Abnormalities of Hemoglobin synthesis Microcytic Anemias Normocytic Anemias Macrocytic Anemias Polycythemias Other disorders The CBC Complete Blood Count You have had this, right? First focus, the red blood cell Red Blood Cell Carries oxygen and nutrition to cells Survive about 120 days Contains hemoglobin molecule Requires erythropoietin for production (kidneys) Red Blood Cell synthesis Pluripotential precursor (bone marrow)---> Proerythroblasts--->Erythroblast Normoblast--->Normocytes (Nucleated RBC) --->Reticulocyte --->Erythrocyte (RBC) Reticulocyte Immature RBC Usually less than 1.5% of total RBC’s Seen in larger quantities after bleed, about 72 hours later (Stress reticulocytosis) Stains for reticulin – New methylene blue stain, others – Must be requested specifically by lab (not reported on normal CBC) Larger than mature RBC – Will increase average cell size (MCV) – If plentiful will increase RDW Erythrocyte (RBC) Normal, mature, red blood cell Biconcave disk – Like a bagel (or donut…..Your choice!) No nucleus Hemoglobin molecule Normal Hb (Hb A) 2 alpha chains, 2 beta chains Hb F= 2 alpha, 2 gamma (Fetal Hb) Hb A2= 2 alpha, 2 delta (Thalassemia) Hb S= Single AA substitution on beta chain (Beta s (βs) Sickle disease if in both beta globins) Hb C=Beta c genotype (βc) Hb Barts=γ 4. Seen in children Hb H=β4 in adults Example of geneticsHemoglobin C disease If Hb C trait no symptoms If two Hb C traits, mild anemia If combined with Hb S, sickle variants with sickling under stress Hemoglobin electrophoresis Adults 97% Hb A About 3% Hb A2 Handouts CBC (See overhead, too) Red Cell Count-Number Hemoglobin-Weight Hematocrit-Percentage of red cells MCV-Volume MCH-Hemoglobin MCHC-Hemoglobin concentration Rule of 3’s Hematocrit is generally 3x the Hemoglobin Eyeball for accuracy Is HCT better or Hb? CBC continued RDW-Distribution width Variability of size Reticulocyte count – Increased in Hemolysis, Acute blood loss, Treatment Other tests-I Iron – Low in iron deficiency and chronic disease – Test reduces Fe+3 and adds reagent that forms color to measure TIBC-iron binding – High in iron deficiency Body’s attempt to increase iron carrying capacity – Low in chronic disease, Hemochromatosis, sideroblastic Other tests-II Ferritin – Reflective of iron stores – Phase reactant protein (Goes up in acute illness) % Transferrin sat. (Fe x TIBC x 100%). Elevated in: – Hemochromatosis – Sideroblastic anemia Other Acute Phase ReactantsFYI Go up in face of acute illness – – – – – – C reactive protein Alpha 1 antitrypsin Haptoglobin Fibrinogen Ceruloplasmin C-3 Complement Bone Marrow Biopsy Perhaps most invasive hematologic test Uses core needle to obtain aspirate and core biopsy Slides prepped at bedside – Microscopic – Iron assessment Genetic tests – I.e.: Philadelphia chromosome See handouts for techniques Pause here Any questions so far? Diseases next….. Basic Definitions Anemia-Low Red cell count Polycythemia-High Red cell count Disorders of Hb synthesis Sideroblastic anemias – – – – A general term for failed synthesis Also can be idiopathic (Idiopathic sideroblastic anemia) Results in a delay……..Process is held up/stuck As a result, abnormal red cell Sickle cell anemia Thalassemia These change the O2 affinity of the molecule or make molecule unstable Sideroblastic anemias Two categories conceptually – Sideroblastic anemia-Idiopathic/Primary – Sideroblastic anemias-A broad term incorporating several different anemia types with key feature Etiology: Problem with globin synthesis/maturation – Thalassemia, Sickle cell, Idiopathic Also caused by alcohol, lead, INH (a treatment for TB), chemotherapy Increased ring sideroblasts in bone marrow Sideroblastic anemias-II Often dimorphic (two different populations of cells) May be micro-, normo-, or macrocytic Iron Nl or Inc., % transferrin Sat high Abnormal amount of red cell precursors – Immature cells that stain with Prussian blue stain Sickle cell disease African Americans – Trait 8%, 1/625 disease Single amino acid substitution (valine for glutamic acid at 6th AA in beta chain due to substitution of thiamine for adenine in DNA) Hemoglobin Beta s (βs ) Sickle disease if in both beta globins affected Sickle cell disease-Symptoms Sickling occurs at low oxygen tension Chronic anemia, abnormal smear Painful crises due to blood flow obstruction Typically die of infection SC variant (Hb S + Hb C), Hb S/Beta Thal Sickle cell-Treatment O2 IV fluids Transfusion Analgesia during crisis Asplenic-Need Strep. pneumoniae vaccine (Pneumovax®), meningococcal (N meningiditis) vaccine, H. influenza vaccine- “Encapsulated organisms.” Hydroxyurea chronically – Increases Hb F (Increases gamma chains) – Resultant decrease crises and mortality Thalassemias Mediterranean's, N. Africans – Thalassa (Greek for the sea) Most common monogenic disease in humans – Up to 3-10% of population in some regions Develop ineffective erythropoeisis – Expanded bone marrow – Splenomegaly – Extramedullary erythropoesis Typically Microcytic, hypochromic anemias Typically normal iron, TIBC, ferritin Typically very microcytic, not very anemic Thalassemia types Minimal, Minor, Intermedia, or Major Alpha Beta Alpha Thalassemia Alpha globin is deficient Relative excess of beta chain Result is decreased Hb A (70-95%) – If none at all, fatal (Hydrops fetalis) May require splenectomy Alpha thalassemias cont. Deletion of 1 alpha chain in about 30% of African descent-Asymptomatic Deletion of 2 called “alpha trait” Deletion of all but one, Hb H in adults or Hb Barts in children Deletion of all 4 alpha incompatible with life Beta Thalassemia 200 different mutations can cause Beta (Can’t make beta globin) – – – – Increased Hb A2 (> 4-5%) and Hb F Excess of α-globin chains Β0 thal-no beta globin B+ thal-some beta globin Beta thalassemia types Major – – – – No beta globin (B0 thal or alpha 4) Diagnosed first year of life Cooley’s anemia--->Hemolysis Transfusion dependent Intermedia – Presents later – Typically both parents are heterozygotes Minor – Common – 1 mutant Beta gene Minima – Silent carrier – 1 mutant Beta gene Thalassemia treatments Screening Blood transfusion with iron chelation – Iron overload is biggest problem – Deferoxamine (Desferal®)-iron chelator IM or IV – New one coming (NEJM 2006) Splenectomy Give folic acid if deficient Hydroxyurea – Increases HbF Bone marrow transplant Gene therapy G6PD deficiency Most common red cell enzyme deficiency Sex linked/X linked 400 different variations 12% African Americans, 20-30% Greeks Increased risk of hemolysis under stress Many drugs including sulfa drugs, nitrofurantoin and others, fava beans Diagnosis made by measurement of the RBC enzyme Treatment: Avoid these medications, stress How to think about anemias Two ways I suggest: – By size – By marrow activity Anemia classification by size Uses MCV off of the CBC Small cell sized anemias-Microcytic Normal cell size anemias-Normocytic Large cell size anemias-Macrocytic We will discuss these at length One downfall is that some anemias can be two – I.e.: anemia of chronic disease may be normo or microcytic – Thus, anemia is more of a spectrum of diseases, but still helpful to use this categorization (I think) Anemia by marrow activity Hypoproliferative anemias – – – – Sluggish marrow Uses the reticulocyte as gauge Low retic count, low bone marrow activity Example: Aplastic anemia Normoproliferative anemias – Reticulocyte count is usually normal – Example: Sickle cell anemia I don’t use this one – I don’t find as helpful – Some do use the term, however Anemias by size “The diseases” Here we go…….. Microcytic anemias Iron deficiency Anemia of chronic disease Sideroblastic anemias Lead-Children Thalassemia-As discussed previously Iron deficiency MC cause of anemia in world (tapeworm) MC in USA (menses) Low iron (<50), increased TIBC, Low Ferritin Hypochromic GI vs. Gyn loss – Must look for cause Iron deficiency-treatment 325mg FeSO4 TID – Contains 50mg elemental iron – 3-4 days to respond Also available as gluconate, IM, IV We use sodium ferric gluconate complex (Ferrlecit®) 125mg IV weekly for 2-8 weeks. Others too. Anemia of Chronic disease/inflammation Low iron (50-100), Low TIBC, Nl or Inc Ferritin Often normocytic early Treatment: Treat cause IDA-vs-ACD FE TIBC Ferritin Iron deficiency Low High Low Anemia of Chronic Disease Low Low High Normocytic Anemias Acute blood loss Pregnancy – Dilutional – Iron deficiency (though often microcytic) Hemolysis (Include G6PD defic, PNH) Hypersplenism Spherocytosis, Myelophthesis, Aplasia Normocytic Anemias-Part II Renal failure – Low erythropoietin – Rarely is macrocytic Myelophthesis (BM space occupying lesion) Mixed (i.e.: Fe and Folate defic.) Others Hemolytic anemia Elevated LDH, serum bilirubin and urinary bilinogen COOMBS test for circulating antibody to red cell Haptoglobin low (used up) Causes multiple Hemolytic anemia-Etiology Congenital – Spherocytosis, elliptocytosis, G6PD, Sickle cell – A sort of chronic hemolysis – Would these be hypo or normoproliferative? Acquired – Drugs, Infection (HIV, Ebstein Barr virus (EBV), chlamydia), – Immune, Splenomegaly, DIC, – Paroxysmal Nocturnal Hemoglobinuria, Trauma (heart valve) Hemolytic anemia-Treatment Supportive Immune suppression Intravenous immunoglobulin (IVIG) Folic acid (Folate) – Helps as cofactor for BM production – 1g per day po Hypersplenism Collection of different etiologies – Infiltrative – Malignancies – Cirrhosis Result in pancytopenias due to destruction and engulfment of cells Not same a big spleen Macrocytic Anemias B12/Folate deficiency Liver disease Alcoholism Aplastic anemia (lowers B12). IE: chemotherapy Hypothyroidism Myelophthistic diseases-Replacement of marrow with fibrosis, infiltration (sarcoidosis), infection (TB) Myelodysplastic diseases-Preleukemic state Renal disease (Usually normocytic) Pseudomacrocytosis Folic acid (folate) deficiency More common Seen perhaps most often in alcoholism Elevated LDH Vitamin B12 deficiency Pernicious anemia (MC) – Lack of intrinsic factor or ileum Malabsorption – Alcoholism – Diphyllobothrium latum (fish tapeworm) – Dietary deficiency rare due to fortification of flour with Elevated LDH Treatment – Oral supplement if able to absorb – IM B12 if not (i.e.: PA) typically monthly – Nasal spray (expensive) daily Liver disease Several reasons for anemias Include problems with synthesis, destruction, loss Alcohol affect on blood Macrocytosis Anemia, leukopenia, thrombocytopenia Decreased synthesis in marrow Increased destruction – Toxic effect – Hypersplenism Increased loss – Hemorrhage (IE: varices) Pancytopenias/Bicytopenias Know this differential! B12/Folate deficiency, Hypersplenism Aplastic anemia, Alcohol, Lupus Viral (HIV, others) Myelophthesis – Space occupying lesion of marrow Myelodysplasia – Unstable, unregulated growth of marrow Paroxysmal nocturnal hemoglobinuria PNH-(next slide) Myelodysplasia Aged bone marrow – Old people – Pre-malignant state Clue is abnormal counts – Macrocytosis common Treatment difficult – Erythropoietin? – Transfusion dependence? See handouts for types Paroxysmal Nocturnal Hemoglobinuria (PNH) Defect in production of Glycosylphosphatidyl-inositol (anchor protein) Hemolytic anemia – Increased reticulocyte count Pancytopenia Intermittent hemoglobinuria Increased risk of venous thrombosis PNH Continued Diagnosis – Genetic test – Ham’s test (lysis at pH 6.2) – Sugar (sucrose) water test-lysis Treatment – – – – – – – Supportive Iron if needed Folic acid (hemolysis) Possibly steroids (debated) Anticoagulation/Warfarin Androgens? Gene therapy? Elevated iron Not an anemia, but due to excess of metal, iron. Never the less, cover here. Hemochromatosis – 1% of Caucasians – TIBC low, % saturation high Aplastic anemia Myelodysplasia Myelophthesis Iron deposition Heart (Cardiomyopathy) Liver (Function affected, or even cirrhosis) Pituitary Pancreas (Diabetes can develop) Thyroid Parathyroid Adrenals Polycythemias Rubra Vera (Spurious or primary polycythemia) – Increased RBC mass, normal O2, splenomegaly – Treatment: Phlebotomy Secondary – COPD – Stress-Gaisböck's syndrome (tobacco) – Treatment: Treat disease Treatment of Anemias and Coagulopathies Transfusion Phlebotomy Others – – – – Fix cause Iron B12 Etc. Transfusion Therapy Whole blood Packed red blood cells Platelets – Derived from several donors – Single phoresis donation Require type and cross match Can request type and screen if may or may not need transfusion Transfusions cont. Fresh frozen plasma – Long storage – Coagulopathies with factor deficiencies – Volume expansion Cryoprecipitate – Contain extra Factor VIII and vWF Transfusion Reactions Immediate type immune mediated hemolysis Delayed type Febrile non-hemolytic Graft vs. host disease (bone marrow transplant) Fluid overload Infection (bacterial, viral) Phlebotomy To obtain blood products – Whole blood – Pheresis Therapeutic – Iron overload – Polycythemias Case study What do you want to know? Letter from Red Cross? Serologies to r/o infection (Hepatitis, HIV)? Repeat CBC-Most definitely Iron levels? Other tests? ?GI work up (endoscopy?) PA’s and PCP’s should all be able to do a work-up for anemia…..doesn’t need a hematologist Look for common things Where to get more information Harrison’s or Cecil’s textbooks of Internal Medicine Any Hematology text (i.e...: Hoffman et al) The Beta-Thalassemias (NEJM Vol 341, No. 2, July 8, 1999, pgs 99-109)