lorenzos_background2
advertisement

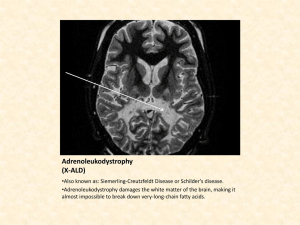
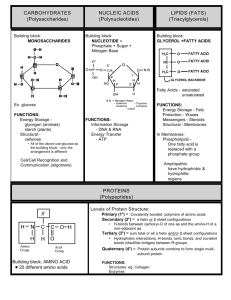
Day one Adrenoleukodystrophy • Called ALD • a rare, inherited metabolic disorder • the fatty covering (myelin sheath) on nerve fibers in the brain is lost, and the adrenal gland degenerates, leading to progressive neurological disability and death. • “Leuko” refers to white color of neuron sheath • “Dystrophy” refers to abnormal development • can become active at any point in a boy's life Symptoms • Pediatric onset occurs between the ages of 4 and 10 years. • The boys develop normally until the onset of symptoms that are often mistaken for attention deficit disorder. • There then appear to be signs of serious neurological involvement including impaired auditory discrimination, visual disturbances, impaired coordination, dementia and seizures. • These symptoms generally progress rapidly and lead to a vegetative state within two years and death anytime thereafter. Day two Testing The test for ALD and AMN is a blood test to determine the levels of very long chain fatty acids in the blood. Additionally, known carriers can submit their children to genetic testing to determine whether their sons have the gene for development, or if their daughters are in fact carriers as well. The figure above shows the location of the Adrenoleukodystrophy gene on the x chromosome. It was borrowed from http://www.ncbi.nlm.nih.gov/ disease/chr21-Y.html and modified in MS Paint v98 by the Microsoft Corporation. The above picture shows an affected boy. It was borrowed from http://www.mtsu.edu/~ jsanborn/dshp.htm There are many specific symptoms that lead to the total cognitive and behavioral impairment. The range of symptoms is: oAttention deficit disorder oImpaired vision oImpaired speech oImpaired motor function oCognitive and behavioral impairment oComa oSeizures oSwallowing difficulties Adrenomyeloneuropathy. (AMN) • When the gene becomes active in adulthood • Men that experience AMN also endure demyelination, however, it most commonly restricts itself to the long tracts of the spinal column causing increasing difficulty with walking, as well as bladder and bowel disturbances over a period of decades. • In approximately thirty-five percent of the men who develop AMN, demyelination of the brain does take place, with a much more debilitating effect. Up to 40% of the heterozygotes, or women who are carriers of these disorders, experience AMN like symptoms as their lives progress. Adrenal function is almost always normal. Adrenoleukodystrophy is an extremely rare disease, affecting only 1:75,000 live male births. The disease affects all races equally. There have been no known female sufferers of the childhood onset form of the disease, so the incidence in girls does not exist. It is estimated that 1:42,000 females in the United States carries the defective gene that causes Adrenolekodystrophy. People with ALD accumulate high levels of saturated, very long chain fatty acids in their brain and adrenal cortex because the fatty acids are not broken down by an enzyme in the normal manner. The ALD gene was discovered in 1993, it was a surprise that the corresponding protein was in fact a member of a family of transporter proteins, not an enzyme. It is still a mystery as to how the transporter affects the function the fatty acid enzyme and, for that matter, how high levels of very long chain fatty acids cause the loss of myelin on nerve fibers. Lorenzo’s Oil is a mixture of oleic and erucic acids, that helps to normalize the levels of very long chain fatty acids in boys with adrenoleukodystrophy. Having low levels of very long chain fatty acids in the body will help to slow and minimize the effects of the disease by preventing them from accumulating in the body. Unfortunately, use of the oil causes gross lowering of platelets in patients. Children with this disorder are deficient in docasahexaenoic acid, and clinical trials are being conducted with administration of this acid derived from algae. The picture shows the makeup of oleic and erucic acids and how they combine to make "Lorenzo's Oil." It was borrowed from http://carbon.cudenver.edu/~bstith/ loren.htm 1) Are they normally present in the body? YES! 2) What is their function? They are part of brain membranes, including myelin, the "insulation" around nerve fibers. 3) Where do they come from? - Dietary sources - Elongation of shorter fatty acids in the body 4) What could cause the increase levels of VLCFA in ALD/AMN? - Body makes too much - Body doesn't remove excess amounts 5) Which of these possibilities is correct? Studies with patient volunteers and in cells in the laboratory have shown that the process that normally breaks down or oxidizes VLCFA's is defective in ALD/AMN. Prognosis for the disease is very poor. The disease will disable and kill a child by age 14. Advances in ALD treatment have progressed rapidly over the last decade and clinical trials being conducted now may yield a cure in the future. Generally, if the disease is caught before the child is symptomatic, and aggressive treatment is used, the progression of the disease may be substantially slowed or halted. However, unless there is a family history of the disease, boys are generally not tested for it and diagnosed before they have symptoms. Currently, the Myelin Project is experimenting with ways to regenerate myelin in the brain and the central nervous system. Lorenzo’s Oil is a mixture of oleic and erucic acids, that helps to normalize the levels of very long chain fatty acids in boys with adrenoleukodystroph y. The Stop ALD Foundation was started immediately following the year 2000 ALD diagnosis of Oliver Abraham Lapin – at the time a sweet, caring, and extremely intelligent 8 year-old boy from Houston, Texas. Oliver was misdiagnosed for years and, by the time an accurate diagnosis was made, he was already severely affected with no treatment available to help him. That diagnosis did provide early warning for the 2 other affected boys in the family who could be “saved.” The expression “it takes a village” was taken to heart when Oliver’s extended family started The Stop ALD Foundation in early 2001. On Thursday, August 12, 2004, the day before Oliver’s 12th birthday, Oliver passed away from complications related to ALD. He was at home lovingly surrounded by his family. The Stop ALD Foundation wishes to thank the outpouring of support and donations that we have received in memory of Oliver. We will continue on our path and focus to make sure more children are diagnosed earlier, and that better, more effective, and safer treatments are available. We’ll so deeply miss you Oliver. We’ll never, ever forget you. Ideas for persuasive papers or essays: If a person knows they are a carrier for a fatal disease should they have children? Should they be allowed to manipulate genes (choose the sex of their child?) Should stem cell research be used? –from existing cell lines? From donors (dead/alive)? –from embryonic tissue from abortions? Should government funded research be limited to diseases which effect large numbers – or should we spend money on rare diseases? Is cloning an option as a treatment for disorders?