Lecture_6 (2014)
advertisement
")
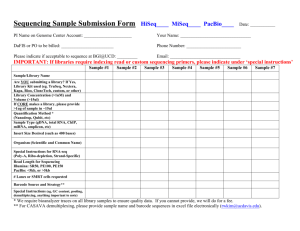
High Throughput Sequencing Agenda • • • • Introduction to sequencing Applications Bioinformatics analysis pipelines What should you ask yourself before planning the experiment Introduction to sequencing What is sequencing? Finding the sequence of a DNA/ RNA molecule What can we sequence? http://cancergenome.nih.gov/newsevents/multimedialibrary/images/CancerBiology Sanger sequencing • Up to 1,000 bases molecule • One molecule at a time • Widely used from 1970-2000 • First human genome draft was based on Sanger sequencing • Still in use for single molecules http://www.genomebc.ca/education/articles/sequencing/ High Throughput Sequencing Next Generation Sequencing (NGS) / Massively parallel sequencing • Sequencing millions of molecules in parallel • Do not need prior knowledge of what you’re sequencing Platform Read length 454 sequencing Up to 1,000 bp SOLiD 50-75 bp HiSeq 100-150 bp No. of reads per run ~1 M (1 million) ~1 G (1 billion) ~0.5 G We will discuss Illumina’s platform only Sequencing Workflow 1. 2. 3. 4. 5. Extract tissue cells Extract DNA/RNA from cells Sample preparation for sequencing Sequencing Bioinformatics analysis Why is it important to understand the “wet lab” part? Sample Prep Random shearing of the DNA Size selection Amplification Adding adaptors and barcodes Sequencing Sequencing process Sequencing process Leave only sequences from one direction Sequencing process Sequencing process Sequencing process Applications DNA sequencing • Resequencing – sequencing the genome of an organism with a known genome • Exome sequencing / Targeted sequencing – sequencing only selected regions from the genome • De-novo sequencing– sequencing the genome of an organism with a unknown genome RNA-Seq Sequencing of mRNA extracted form the cell to get an estimate of expression levels of genes. Counting vs. Reading RNA-Seq vs. DNA-sequencing ChIP-Seq Sequencing the regions in the genome to which a protein binds to. Basic concepts Insert – the DNA fragment that is used for sequencing. Read – the part of the insert that is sequenced. Single Read (SR) – a sequencing procedure by which the insert is sequenced from one end only. Paired End (PE) – a sequencing procedure by which the insert is sequenced from both ends. Bioinformatics Analysis Pipelines Demultiplexing Unknown inserts Lane Demultiplexing Mapping Sample Reference Genome Example of mapping parameters: • Number of mismatches per read • Scores for mismatch or gaps Mapping parameters affect the rest of the analysis Demultiplexing Mapping Removing duplicates and non-unique mappings Reference Genome ? Reference Genome 𝒂𝒗𝒆𝒓𝒂𝒈𝒆 𝒄𝒐𝒗𝒆𝒓𝒂𝒈𝒆 = 𝑟𝑒𝑎𝑑 𝑙𝑒𝑛𝑔𝑡ℎ ⋅ 𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑟𝑒𝑎𝑑𝑠 ⋅ % 𝑢𝑛𝑖𝑞𝑢𝑒𝑙𝑦 𝑚𝑎𝑝𝑝𝑒𝑑 𝑟𝑒𝑎𝑑𝑠 𝑔𝑒𝑛𝑜𝑚𝑒 𝑠𝑖𝑧𝑒 Resequencing/ Exome Pipeline Demultiplexing Mapping Removing duplicates and non-unique mappings Coverage profile and variant calling G …ACTTCGTCGAAAGG… Reference Genome Demultiplexing Frequency >= 20% Mapping Removing duplicates and non-unique mappings Coverage profile and variant calling …ACTTCGTCGAAAGG… Variant filtering Reference Genome Coverage >= 5 …ACTTCGTCGAAAGG… Reference Genome Demultiplexing Mapping Removing duplicates and non-unique mappings G Variant calling A Variant filtering Genes and known variants …ACTTCGTCGAAATG… Gene X …GTCCCGTGATACTCCGT… rs230985 Reference Genome Resequencing results Example for further analysis Demultiplexing Mapping Removing duplicates and non-unique mappings Coverage profile and variant calling Variant filtering Genes and known variants Finding suspicious variants Recessive disease: 1. Variant not in known databases 2. Homozygous variant shared by all affected individuals 3. Same variant appears in healthy parents at heterozygous state 4. Healthy brothers can be heterozygous to the same variant Dominant disease: 1. Variant not in known databases 2. Heterozygous variant shared by all affected individuals 3. The variant doesn’t appear in healthy individuals Quality control steps in the pipeline Demultiplexing QC Mapping QC Removing duplicates and non-unique mappings Coverage profile and variant calling QC QC Variant filtering QC Genes and known variants Finding suspicious variants How is de-novo assembly different from resequencing analysis ? RNA-Seq Pipeline Demultiplexing Mapping Removing duplicates and non-unique mappings FPKM Normalization: 𝑟𝑎𝑤 𝑐𝑜𝑢𝑛𝑡 𝑔𝑒𝑛𝑒 𝑙𝑒𝑛𝑔𝑡ℎ ⋅ 𝑠𝑎𝑚𝑝𝑙𝑒 ′ 𝑠 𝑚𝑎𝑝𝑝𝑒𝑑 𝑟𝑒𝑎𝑑𝑠 𝑖𝑛 𝑚𝑖𝑙𝑙𝑖𝑜𝑛𝑠 Gene expression levels Unannotated genes Demultiplexing Mapping Removing duplicates and non-unique mappings Gene expression levels Differential gene expression Differential expression parameters: • Threshold - Minimum number of reads for pair testing • Normalization • Replicates Differential expression parameters affect the results RNA-Seq results Coverage Coverage – resequencing Number of bases that cover each base in the genome in average. 𝒂𝒗𝒆𝒓𝒂𝒈𝒆 𝒄𝒐𝒗𝒆𝒓𝒂𝒈𝒆 = 𝑟𝑒𝑎𝑑 𝑙𝑒𝑛𝑔𝑡ℎ ⋅ 𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑟𝑒𝑎𝑑𝑠 ⋅ % 𝑢𝑛𝑖𝑞𝑢𝑒𝑙𝑦 𝑚𝑎𝑝𝑝𝑒𝑑 𝑟𝑒𝑎𝑑𝑠 𝑔𝑒𝑛𝑜𝑚𝑒 𝑠𝑖𝑧𝑒 “Coverage” – RNA-Seq • Depends on the expression profile of each sample. • Highly expressed genes will be detected with less “coverage” than lowly expressed genes. What should you ask yourself before sequencing when planning the experiment • Reference genome: – – – – What is my reference genome? Does it have updated annotations? What annotations are known? Are my samples closely related to the reference genome? • Do I expect to have contaminations in my sample? • Do I have validations from other technologies? (RT-PCR, SNPchip…) • Do I have controls and replicates? • RNA-Seq: am I interested in alternative splicing? • Resequencing: What kind of mutations do I expect to find?