Signs of hemolytic anemia
advertisement

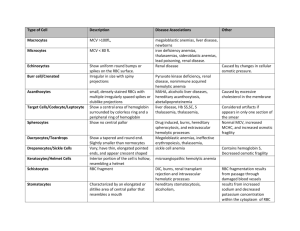
Course title: Hematology (1) Course code: MLHE-201 Supervisor: Prof. Dr Magda Sultan Outcome : The student will know : -The types of hemolytic anemias -The diagnosis of hemolytic anemias -The types of hereditary hemolytic anemias. -The diagnosis of hereditary hemolytic anemias -Types of acquired hemolytic anemias -The diagnosis of acquired hemolytic anemias -The laboratory tests needed for diagnosis Hemolytic anemia • Hemolytic anemia = decreased levels of red blood cells (anemia) because of their destruction (hemolysis) • A red blood cell survives 120 days • The spleen is the main organ which removes old RBCs from the blood. Causes of hemolytic anemias can be either: 1 - hereditary. 2 - acquired. Hereditary Hemolytic anemia Defects of hemoglobin Thalassaemia, Sickle cell anemia Defects of the red cell membrane Hereditary spherocytosis, Hereditary elliptocytosis Defective red cell metabolism (enzymes) G6PD deficiency. P K deficiency Acquired - Immune mediated : Autoimmune, isoimmune, drugs - Microangiopathic: DIC, HUS - Hypersplenism - Miscellaneous: drugs, toxin, infection, burn, chemical Signs of hemolytic anemia: Physical • • • • Symptoms of anemia Jaundice Pallor Splenomegaly / hepatosplenomegaly Laboratory features Anemia of increased destruction Normochromic, normocytic anemia Short RBC survival Reticulocytes increased Increased indirect bilirubin Increased LDH *Peripheral blood smear microscopy: fragments of the red blood cells and spherocytes Normoblasts can be present. Bone marrow smear microscopy: Erytrhroid hyperplasia Hereditary Hemolytic anemia Defects of hemoglobin Defects of the red cell membrane Thalassaemia, Sickle cell anemia Hereditary spherocytosis, Hereditary elliptocytosis Defective red cell metabolism G6PD deficiency. P K deficiency Sickle cell anaemia The abnormalities of the gene may result from substitution of single amino acid (Substitution of glutamic acid by valine ) The Hb is stable when oxygenated state and become unstable and polymerized on deoxygenated state Sickle cell anaemia Polymerization will lead to precipitation of Hb. The cell become deformed (sickle shape) and very sticky leading to vascular occlusion and small infarction to the affected areas. Short life span of cells leading to chronic anaemia, Sickle cell anaemia Diagnosis 1-Sickling test 2- Hemoglobin electrophoresis: Increased hemoglobin S (90% Hgb S, 10% Hgb F, small fraction of Hgb A2) HEMOGLOBIN NORMAL ADULT RBC CONSISTS OF 3 FORMS OF Hb: - HbA - 2 α and 2 β globin chains - HbA2 – 2 α and 2 δ globin chains - HbF - 2 α and 2 γ globin chains Thalassaemia Means decrease synthesis of one ofthe globin chain which form normal hemoglobin. (HbA - 2 α and 2 β globin chains HbA2 – 2 α and 2 δ globin chains HbF - 2 α and 2 γ globin chains ) . The defect may be in alpha chain ( thalassaemia), Beta chain ( thalassaemia) or Delta chain ( thalassaemia) Beta Thalassaemia Defective chain synthesis Excess chain Precipitation cell membrane damage Circulating Red cell Bone marrow Anaemia Hemolytic Erythropoietin increased Ineffective erythropoiesis blood transfusion Iron absorption Bone marrow expansion skeletal changes & hyper metabolism Iron overload Complication and death Beta-Thalassemia major laboratory features Severe anemia Blood film: microcytic hypochromic , target cells, basophylic stippling, reticulocytes increased and normoblasts . Marrow: marked erythroid hyperplasia, Shortened red cell survival Haemoglobin electrophoresis : Fetal hemoglobin > 90%, HbA absent, HbA2 low/normal/high HEREDITARY SPHEROCYTOSIS Defective or absent spectrin molecule Leads to loss of RBC membrane, leading to spherocytosis Decreased deformability of cell Increased osmotic fragility Extravascular hemolysis in spleen Hereditary spherocytosis (HS) Laboratory features - hemolytic anemia - blood smear spherocytes - increased osmotic fragility time G6PD DEFICIENCY Function of G6PD Infections Drugs 2 H2O GSSG H2O2 2 GSH NADPH NADP 6-PG G6P G6PD Hgb Sulf-Hgb Heinz bodies Hemolysis Glucose 6-Phosphate Dehydrogenase Functions Regenerates NADPH, allowing regeneration of glutathione Protects against oxidative stress Lack of G6PD leads to hemolysis during oxidative stress Infection Medications Fava beans Oxidative stress leads to Heinz body formation, extravascular hemolysis G6PD DEFICIENCY DIAGNOSIS : QUANTITATIVE ASSAY DETECTING LOW ENZYME TREATMENT – SUPPORTIVE AND PREVENTATIVE Acquired hemolytic anaemia Due to Antibodies directed against RBC membrane = autoimmune hemolytic anemia destruction of RBC in an enlarged spleen Introduction Increased RBC Destruction – Short RBC life span <120 days. Normocytic normochromic, reticulocytosis. Anemia, Jaundice, marrow hyperplasia Splenomegaly, increased bilirubin Types of acquired HA AutoImmune Haemolytic Anemias (+ve Direct CoombꞋs) Alloimmune haemolytic anemias Drug-induced immune haemolytic anemias Assesment of HA Clinical features: - pallor - jaundice - splenomegaly Laboratory features: 1. Laboratory features - normocytic, normochromic anemia - reticulocytosis - antiglobulin Coombs’ test is positive 2. Blood smear - anisopoikilocytosis, spherocytes - normoblasts - schistocytes 3. Bone marrow smear - erythroid hyperplasia DIRECT ANTIGLOBULIN TEST (DAT)Coomb′s test Procedure of DAT 1. 2. 3. 4. 5. 6. Take 2-3 drops of blood to be tested in a clean labeled tube. Wash the red cells 3-4 times in a large volume of saline to remove free globulin molecules. Remove all supernatant after each wash. Completely decant the final supernatant wash. Add 2 drops of polyspecific AHG serum in 1 drop of sensitized washed red cells or in 1 drop of 3-5 % suspension of sensitized cells immediately. Mix, Centrifuge at 1000 rpm for 1 minutes immediately. Gently shake the tube to dislodge the cell button and see for agglutination, use optical aid if needed. Record the result. Add 1 drop of IgG coated red cells to a negative test. Mix, centrifuge at 1000 rpm for 1 min. Immediately look for agglutination. If a negative result (no agglutination) is obtained the test result is invalid and whole test should be repeated. If agglutination is obtained, the result is valid. Indirect antiglobulin (coomb′s ) test Procedure: 1. 2. 3. 4. 5. 6. Place 2-3 drops of the test serum in a tube. Serum should be fresh for detecting complement components and complement binding antibodies, otherwise, fresh AB serum should be added to it. Add 1 drop of 3-5% suspension of washed O Rh (D) positive red cells to the serum in the tube. Mix and incubate at 37°C for 30-40 minutes. Centrifuge at 1000 rpm for 1 minutes. Examine for hemolysis and/or agglutination. Use optical aid if necessary. Agglutination at this stage indicates the presence of saline (complete) antibodies. If no agglutination is seen, wash cells 3-4 times in large volume of saline. Decant supernatant in each wash as completely as possible. Procedure: 7. 8. 9. 10. 11. Add 2 drops of AHG serum to the cells. Mix and centrifuge at 1000 rpm for 1 minutes immediately. Gently shake the tube to dislodge the button and examine for agglutination, using optical aid. Record the result. Add 1 drop of IgG coated red cells to any test that is negative. Mix and centrifuge at 1000 rpm for 1 minutes. Look for agglutination. If there is no agglutination, the test result is invalid and the whole test is repeated. If agglutination is obtained the result is valid. Auto control should be kept with IAT. Training questions : What are the tests of hemolysis ? How to diagnose Autoimmune hemolytic anaemia ? Reference book : Essential Hematology . Dacie .