Hematology/Oncology Board Review
advertisement

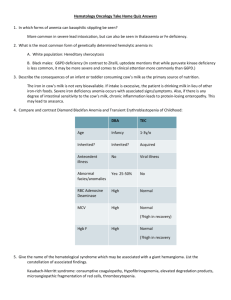
Survey Question Which source would you prefer to be assigned for next year’s board review sessions? A: Zitelli’s Atlas B: Peds In Review articles Tips: Normal Values MCV 70 to 90 Infants and Toddlers 80 to 100 Adults Hgb/Hct Ddx of hypochromic, microcytic anemia Iron deficiency Lead poisoning Thalassemia Chronic disease (may be normocytic, normochromic) Sideroblastic anemia Remember: Fe Def and Pb often coexist!! Tips If MCV< 70 with normal iron studies: Thalassemia and lead toxicity are most likely Iron deficiency Merely a lab finding, NOT a diagnosis Possibilities Poor nutrition (most common) Chronic hemorrhage Malabsorption 2 peaks 10-18 months Adolescent females Excessive cows milk intake Fe in cows milk is not bioavailable Leads to Fe deficiency anemia Clinical Large child Pallor Loose stools: +/- heme Protein losing enteropathy ○ Hypoalbuminemia ○ Edema Chronic Fe Deficiency Anemia Asymptomatic Irritability Tachycardia Fatigue Koilonychia or spooning of nails Glossitis Longstanding: Decreased IQ Start Fe supplementation at 4 mos old (Rice Cereal) Ddx: Fe Def, Thalassemia Peripheral smear: hypochromic microcytosis Thalassemia minor: RDW low Hgb ≥ 9 Lower MCV Fe Def RDW high Low RBC number Ddx: Fe Def, Thalassemia RDW Iron ThaLassemia hIgh Low Question 1 This finding may be seen in: A: Lead intoxication B: Thalassemia C: Iron Deficiency D: A, B, and C Hypochromic, microcytic anemia with basophilic stippling Ddx: Fe Def, Lead Toxicity Hypochromic, microcytic anemia Basophilic stippling Neither sensitive nor specific Ringed sideroblast Pb toxicity and sideroblastic anemia Free erythrocyte protoporphyrin (FEP) Elevated in both Fe Def and Lead Higher in Lead poisoning (Normal in thalassemia) Lead levels: Routine screening & high risk >15 μg/dL needs tretment Beta Thalassemia Minor (Trait) Defect in one of the beta globin alleles Asymptomatic Mild anemia Beta Thalassemia Major (Cooley’s) Mutation in both beta globin alleles Severe deficiency in amount of beta globin Profound microcytic anemia first year of life Frequent transfusions Beta Thalassemia Electrophoresis Low levels of Hgb A1 Increased levels of Hgb A2 Increased Hgb F Long term complications Cholelithiasis Hemosiderosis Alpha Thalassemia Defect in two alpha globin genes Mild microcytic anemia Fe Deficiency vs Chronic Illness Anemia Ferritin TIBC Chronic Illness High Low Iron Deficiency Low High • Chronic Illness: No need for Fe therapy!! • Adequate Fe stores • Not enough “carrier energy” (low TIBC) Physiologic Anemia of Infancy 6-10 weeks old Normocytic! MCV > 100 B12 (Cobalamin) deficiency Causes: Intrinsic factor deficiency (Pernicious anemia) Bacterial overgrowth Bowel resection Breastfed infants with vegetarian mothers Schilling test: Measures B12 absorption Tips Heme question with compromised gut: B12 deficiency ○ Glossitis, angular stomatitis Infant drinking goat’s milk Folic acid deficiency Patient taking phenytoin or methotrexate Folic acid deficiency Tips Serum B12 levels appear normal in 5% of patients with true deficiency If suspicion is high, measure Homocysteine Methylmalonic acid BOTH elevated in B12 def Only homocysteine elevated in folate def Administration of folate will correct anemia without addressing B12 deficiency: Irreversible neuro damage Tips Jaundice, dark urine, splenomegaly: Anemia involving hemolysis Question 2 What is the most common cause of genetically determined hemolytic anemia in the white population? A: Hereditary Spherocytosis B: G6PD Deficiency C: Pyruvate Kinase Deficiency D: Hereditary Elliptocytosis G6PD Deficiency Heinz Bodies Black (also Mediterranean) Most common X-linked disorder in black males (dark urine, jaundice, anemia) Heterozygous females may be affected Lyonization Causative agents Mothballs Antimalarials Nitrofurantoin Sulfa G6PD Deficiency Diagnosis: Fluorescent spot test Methemoglobin reduction test (methylene blue) !! Inaccurate following acute hemolysis !! Note: With G6PD deficiency, methylene blue is ineffective in treating methemoglobinemia. Pyruvate Kinase Deficiency AR Block in red cell’s energy pathway Depleted of lactate and ATP Buildup of DPG (shifts oxygen-binding curve to right) Dx: Assay of PK enzymatic activity Question 3 A defect in which RBC cytoskeletal membrane protein is responsible for Hereditary Spherocytosis? A: Kallerin B: Haematid C: Metalloprotein D: Spectrin E: Sphingomyelin Hereditary Spherocytois AD Defect in surface of RBC Loss of surface area Etiology: Spectrin deficiency Increased MCHC Cell is smaller with same amt Hgb Dx: Osmotic fragility test (Not pathognomonic) Tx: Folic acid Spelectomy is curative More prone to aplastic crisis Hereditary Elliptocytosis AD Similar RBC destruction as Spherocytosis Clinically insignificant unless splenic hypertrophy present (from another process) Tips G6PD much more common than Pyruvate Kinase deficiency However: PK deficiency more commonly symptomatic!! • Uptodate.com Sickle Cell Any severe, acute complications: Exchange transfusion is the answer! Symptoms of stroke: Exchange transfusion ○ Diagnostic tests are secondary Sickle Cell Howell-Jolly Bodies: Sickle Cell Heinz Bodies: G6PD deficiency Question 4 Which of the following is FALSE regarding Transient Erythroblastopenia of Childhood A: Most common in 1 to 4y/o children B: Responds well to steroids C: MCV is typically normal D: Normal amount of fetal hemoglobin DBA vs. TEC Diamond Blackfan: Arrest in maturation of RBCs Transient Erythroblastopenia of Childhood Suppression of erythroid production Similarities: DBA and TEC Profound isolated RBC anemia Low Hgb, low retic (Retic may be elevated in recovery phase of TEC) Present infancy/toddlerhood Gradual onset DBA may be anemic at birth Relatively asymptomatic (for degree of anemia)