Document
advertisement

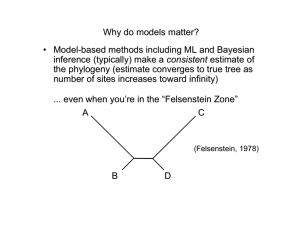
Tree Searching Methods • Exhaustive search (exact) • Branch-and-bound search (exact) • Heuristic search methods (approximate) – Stepwise addition – Branch swapping – Star decomposition Exhaustive Search 12 12 13 13 13 12 13 13 12 13 11 13 13 13 13 Searching for trees • Generation of all possible trees 1.Generate all 3 trees for first 4 taxa: Searching for trees 2. Generate all 15 trees for first 5 taxa: (likewise for each of the other two 4-taxon trees) Searching for trees 3. Full search tree: Searching for trees Branch and bound algorithm: The search tree is the same as for exhaustive search, with tree lengths for a hypothetical data set shown in boldface type. If a tree lying at a node of this search tree has a length that exceeds the current lower bound on the optimal tree length, this path of the search tree is terminated (indicated by a cross-bar), and the algorithm backtracks and takes the next available path. When a tip of the search tree is reached (i.e., when we arrive at a tree containing the full set of taxa), the tree is either optimal (and hence retained) or suboptimal (and rejected). When all paths leading from the initial 3-taxon tree have been explored, the algorithm terminates, and all most-parsimonious trees will have been identified. Asterisks indicate points at which the current lower bound is reduced. Circled numbers represent the order in which phylogenetic trees are visited in the search tree. Stepwise Addition (in a nutshell) 2 1 3 1 2 1 3 1 2 4 3 2 4 3 4 Searching for trees Stepwise addition A greedy stepwise-addition search applied to the example used for branch-and-bound. The best 4-taxon tree is determined by evaluating the lengths of the three trees obtained by joining taxon D to tree 1 containing only the first three taxa. Taxa E and F are then connected to the five and seven possible locations, respectively, on trees 4 and 9, with only the shortest trees found during each step being used for the next step. In this example, the 233-step tree obtained is not a global optimum. Circled numbers indicate the order in which phylogenetic trees are evaluated in the stepwise-addition search. Stepwise Addition Variants • As Is – add in order found in matrix • Closest – add unplaced taxa that requires smallest increase • Furthest – add unplaced taxa that requires largest increase • Simple – Farris’s (1970) “simple algorithm” uses a set of pairwise reference distances • Random – random permutation of taxa is used to select the order Branch swapping Nearest Neighbor Interchange (NNI) B C B E C A D D E A E D A C B Branch swapping Subtree Pruning and Regrafting (SPR) B A E B D A C D F C G D C E E A F F G B G a Branch swapping Tree Bisection and Reconnection (TBR) E D B A F G B A A C C E D D C B D F G E F G E E C D A F G F B G B A C Reconnection limits in TBR 3 2 r 4 x s 5 u t v z y w 1 3 2 6 4 u x x' 2 6 1 4 0 1 5 6 5 1 Reconnection distances: 3 w 3 2 4 v z 1 5 2 0 1 2 6 Reconnection limits in TBR 3 2 4 r (D) s 2 1 5 5 4 v y y' w 1 3 6 3 2 4 1 Reconnection distances: 1 1 6 5 1 0 0 1 6 In PAUP*, use “ReconLim” to set maximum reconnection distance Star-decomposition search Overview of maximum likelihood as used in phylogenetics • Overall goal: Find a tree topology (and associated parameter estimates) that maximizes the probability of obtaining the observed data, given a model of evolution Likelihood(hypothesis) Prob(data|hypothesis) Likelihood(tree,model) = k Prob(observed sequences|tree,model) [not Prob(tree|data,model)] Computing the likelihood of a single tree 1 j N (1) C…GGACA…C…GTTTA…C (2) C…AGACA…C…CTCTA…C (3) C…GGATA…A…GTTAA…C (4) C…GGATA…G…CCTAG…C (1) (3) C C A (5) (2) (4) (6) G Computing the likelihood of a single tree Likelihood at site j = C Prob C A G C + Prob A C But use Felsenstein (1981) pruning algorithm A G A C … G C A + A + Prob C T T Computing the likelihood of a single tree L L1L2 N LN L j j1 ln L ln L1 ln L2 N ln LN ln L1 j1 Note: PAUP* reports -ln L, so lower -ln L implies higher likelihood Finding the maximum-likelihood tree (in principle) • Evaluate the likelihood of each possible tree for a given collection of taxa. • Choose the tree topology which maximizes the likelihood over all possible trees. Probability calculations require… • An explicit model of substitution that specifies change probabilities for a given branch length “Instantaneous rate matrix” A rAA CrAC G rAG T rAT A rCA CrCC G rCG T rCT Q A rGA CrGC G rGG T rGT A rTA CrTC G rTG T rTT Jukes-Cantor Kimura 2-parameter Hasegawa-Kishino-Yano (HKY) Felsenstein 1981, 1984 General time-reversible • An estimate of optimal branch lengths in units of expected amount of change ( = rate x time) P(v) e Q For example: Q Q Kimura (1980) “2-parameter” Jukes-Cantor (1969) A Q A A C G T G T C T C G Hasegawa-Kishino-Yano (1985) A rAA CrAC G rAG T rAT r r r r C CC G CG T CT Q A CA A rGA CrGC G rGG T rGT r r r r A TA C TC G TG T TT General-Time Reversible E.g., transition probabilities for HKY and F84: 1 j A 1e j e j j j j 1 e j e A Pij t j j 1 j j 1 e j (i j) (i j, transition) (i j, transversion) A Family of Reversible Substitution Models The Relevance of Branch Lengths C C C A A A A A A A A A C C A A A A C A A A A A When does maximum likelihood work better than parsimony? • When you’re in the “Felsenstein Zone” A C (Felsenstein, 1978) B D In the Felsenstein Zone A B 0.8 0.8 0.1 0.1 0.1 C Substitution rates: Base frequencies: D A C G T A 5 6 2 A=0.1 C=0.2 G=0.3 T=0.4 C 5 3 8 G 6 3 1 T 2 8 1 - In the Felsenstein Zone Proportion correct 1 0.8 parsimony ML-GTR 0.6 0.4 0.2 0 0 5000 Sequence Length 10000 The long-branch attraction (LBA) problem Pattern type 1 A I = Uninformative (constant) 4 A The true phylogeny of 1, 2, 3 and 4 (zero changes required on any tree) A 2 A 3 The long-branch attraction (LBA) problem Pattern type 1 A A I = Uninformative (constant) II = Uninformative 4 A G The true phylogeny of 1, 2, 3 and 4 (one change required on any tree) A 2 A 3 The long-branch attraction (LBA) problem Pattern type 1 A A C I = Uninformative (constant) II = Uninformative III = Uninformative 4 A G G The true phylogeny of 1, 2, 3 and 4 (two changes required on any tree) A 2 A 3 The long-branch attraction (LBA) problem Pattern type 1 A A C G I = Uninformative (constant) II = Uninformative III = Uninformative IV = Misinformative 4 A G G G The true phylogeny of 1, 2, 3 and 4 (two changes required on true tree) A 2 A 3 The long-branch attraction (LBA) problem … but this tree needs only one step G 1 A 2 A 3 G 4 Concerns about statistical properties and suitability of models (assumptions) Consistency If an estimator converges to the true value of a parameter as the amount of data increases toward infinity, the estimator is consistent. When do both methods fail? • When there is insufficient phylogenetic signal... 1 3 2 4 When does parsimony work “better” than maximum likelihood? • When you’re in the Inverse-Felsenstein (“Farris”) zone A C (Siddall, 1998) D B Siddall (1998) parameter space a b b a b 0.75 pa Both methods do poorly Parsimony has higher accuracy than likelihood 0 pb 0.75 Both methods do well Parsimony vs. likelihood in the Inverse-Felsenstein Zone 1 B B B B B B B B B B B 0.9 0.8 B J 15% 67.5% J 0.7 Accuracy 67.5% 0.6 J 0.5 J (expected differences/site) J J 0.4 J J J J J J 0.3 B Parsimony J ML/JC 0.2 0.1 0 20 100 1,000 Sequence length 10,000 100,000 Why does parsimony do so well in the Inverse-Felsenstein zone? C CA True synapomorphy A A C C CA A CA Apparent synapomorphies actually due to misinterpreted homoplasy A C C CA GA G C C A A Parsimony vs. likelihood in the Felsenstein Zone 1 J 0.9 J J J 0.8 J 0.7 Accuracy J J 67.5% 67.5% J 0.6 15% 0.5 J (expected differences/site) J 0.4 0.3 J J B Parsimony J ML/JC 0.2 B 0.1 B B 0 20 100 B B B B 1,000 Sequence length B B 10,000 B B B 100,000 From the Farris Zone to the Felsenstein Zone A B A A C C C D D D B A B B A C C D D B External branches = 0.5 or 0.05 substitutions/site, Jukes-Cantor model of nucleotide substitution G 1.0 H J Simulation results: H G J H G J H G J H G J H G J Accuracy 0.8 0.6 Parsimony 0.4 0.2 0 0.05 J 100 sites G 1,000 sites H 10,000 sites 0.04 0.03 0.02 Farris zon e 1.0 H H J 0.01 J H G 0 J G H 0.01 H H H G H Accuracy J J J 0.4 0.2 0 0.05 H G 0.03 G H 0.04 G H 0.05 Felsenstein zone H H G H G H G J G J J G J J G G 0.6 G H 0.02 Length of internal branch ( d) G 0.8 J J J 100 sites G 1,000 sites H 10,000 sites 0.04 Farris zon e 0.03 0.02 J Likelihood J G J H G J G HJ ML/JC 0.01 0 0.01 0.02 Length of internal branch ( d) 0.03 0.04 0.05 Felsenstein zone Maximum likelihood models are oversimplifications of reality. If I assume the wrong model, won’t my results be meaningless? • Not necessarily (maximum likelihood is pretty robust) Model used for simulation... A B 0.8 0.8 0.1 0.1 0.1 C Substitution rates: Base frequencies: D A C G T A 5 6 2 A=0.1 C=0.2 G=0.3 T=0.4 C 5 3 8 G 6 3 1 T 2 8 1 - Performance of ML when its model is violated (one example) Among site rate heterogeneity equal rates? Lemur Homo Pan Goril Pongo Hylo Maca AAGCTTCATAG AAGCTTCACCG AAGCTTCACCG AAGCTTCACCG AAGCTTCACCG AAGCTTTACAG AAGCTTTTCCG TTGCATCATCCA TTGCATCATCCA TTACGCCATCCA TTACGCCATCCA TTACGCCATCCT TTACATTATCCG TTACATTATCCG …TTACATCATCCA …TTACATCCTCAT …TTACATCCTCAT …CCCACGGACTTA …GCAACCACCCTC …TGCAACCGTCCT …CGCAACCATCCT • Proportion of invariable sites – Some sites don’t change do to strong functional or structural constraint (Hasegawa et al., 1985) • Site-specific rates – Different relative rates assumed for pre-assigned subsets of sites • Gamma-distributed rates – Rate variation assumed to follow a gamma distribution with shape parameter . .... Performance of ML when its model is violated (another example) Modeling among-site rate variation with a gamma distribution... 0.08 =200 Frequency 0.06 =0.5 =2 0.04 =50 0.02 0 0 1 2 Rate …can also estimate a proportion of “invariable” sites (pinv) Performance of ML when its model is violated (another example) = 0.5, pinv=0.5 Tree = 1.0, pinv=0.5 1 1 1 0 .9 0 .9 0 .9 0 .8 0 .8 0 .7 0 .4 0 .3 0 .2 0 .1 100 1000 1 0 0 00 GTRg 0 .4 HK Yg 0 .2 GTRi HKYi GTRer HKYer 0 .1 p arsimo n y 0 .1 0 .3 1 0 0 0 00 HYYig 0 .5 0 .4 0 0 100 GTRi 0 .3 1000 1 0 0 00 100 1 0 0 0 00 1 0 .9 0 .9 0 .9 0 .8 0 .8 GTRig 0 .7 GTRig 0 .7 0 .6 HKYig 0 .6 0 .6 0 .5 GTRg HKYg GTRi HKYi GTRer 0 .5 HKYig GTRg HKYg GTRi HKYi GTRer HKYer 0 .3 p arsimo n y 0 .1 0 .4 0 .3 0 .2 HKYer 0 .1 p arsimo n y 100 1000 1 0 0 00 0 .1 1 0 0 0 00 100 1 0 0 00 1 0 0 0 00 100 0 .9 0 .9 0 .9 0 .8 0 .8 GTRig 0 .7 GTRig 0 .7 0 .6 HKYig 0 .6 HKYig GTRg 0 .6 0 .5 0 .2 0 .3 HKYer p arsimo n y 0 .1 0 100 1 0 0 00 1 0 0 0 00 0 .2 1000 1 0 0 00 1 0 0 0 00 0 .3 HKYer p arsimo n y 0 .1 100 HKYer p arsimo n y 0 .4 0 1000 GTRg HKYg GTRi HKYi GTRer 0 .5 HKYg GTRi HKYi GTRer 0 .4 HKYi GTRer GTRig HKYig 0 .8 0 .7 0 .3 1 0 0 0 00 0 1000 1 0 .4 1 0 0 00 0 .2 1 0 .5 1000 0 .4 1 GTRg HKYg GTRi parsim ony 0 .5 0 0 HK Yer 0 .8 0 .7 0 .2 GRTer 0 1 0 .3 HK Yi 0 .2 1 0 .4 GTRig 0 .6 GTRg HKTg 0 .5 GTRer HKYer Parsimo n y 0 .7 GTRig HKYig 0 .6 GTRg HKYg GTRi HKYi 0 .5 0 .8 0 .7 GTRig HKYig 0 .6 Proportion Correct a = 1.0, pinv=0.2 1000 1 0 0 00 Sequence Leng t h 1 0 0 0 00 0 .2 0 .1 0 100 1000 1 0 0 00 1 0 0 0 00 “MODERATE”–Felsenstein zone = 1.0, p inv =0.5 1 0.9 0.8 JCer 0.7 JC+G 0.6 JC+I JC+I+G 0.5 GTRer GTR+G 0.4 GTR+I 0.3 GTR+I+G parsimony 0.2 0.1 0 100 1000 10000 100000 “MODERATE”–InverseFelsenstein zone 1 0.9 0.8 JCer 0.7 JC+G 0.6 JC+I JC+I+G 0.5 GTRer GTR+G 0.4 GTR+I 0.3 GTR+I+G parsimony 0.2 0.1 0 100 1000 10000 100000 Bayesian Inference in Phylogenetics • Uses Bayes formula: Pr(|D) = Pr(D|) Pr() Pr(D) Pr(D|) Pr() L() Pr() ( =tree topology, branch-lengths, and substitution-model parameters) • Calculation involves integrating over all tree topologies and model-parameter values, subject to assumed prior distribution on parameters Bayesian Inference in Phylogenetics • To approximate this posterior density (complicated multidimensional integral) we use Markov chain Monte Carlo (MCMC) – Simulated Markov chain in which transition probabilities are assigned such that the stationary distribution of the chain is the posterior density of interest – E.g., Metropolis-Hastings algorithm: Accept a proposed move from one state to another state * with probability min(r,1) where r = Pr(*|D) Pr(| *) Pr(|D) Pr(*| ) – Sample chain at regular intervals to approximate posterior distribution • MrBayes (by John Huelsenbeck and Fredrik Ronquist) is most popular Bayesian inference program A brief intro to Markov chain Monte Carlo (MCMC) B “burn in” C B Likelihood C A D B A C B D B D A B D AB|CD D C C B B A C D A A B C B A D A C ... D A B A A C AB|CD C C D AB|CD D BC|AD BC|AD BC|AD BC|AD D AC|BD Iterations 1. Initialize the chain, e.g., by picking a random state X0 (topology,branch lengths, substitution-model parameters) from the assumed prior distribution 2. For each time t, sample a new candidate state Y from some proposal distribution q(.|Xt) (e.g., change branch lengths or topology plus branch lengths) PrY | Dq(X |Y) (Y) Pr(D |Y) q(X |Y) (X,Y) min 1, min Calculate acceptance probability 1, Pr X | D q(Y | X) (X) Pr(D | X) q(X |Y) 3. If Y is accepted, let Xt+1 = Y; otherwise let Xt+1 = Xt If the chain is run “long enough”, the stationary distribution of states in the chain will represent a good approximation to the target distribution (in this case, the Bayesian posterior) Model-based distances • Can also calculate pairwise distances based on these models • These distances estimate the number of substitutions per site that have accumulated since the two sequences shared a common ancestor, allowing for superimposed substitutions (“multiple hits”) • E.g.: – Jukes-Cantor distance – Kimura 2-parameter distance – General maximum-likelihood distances available for other models Distance-based optimality criteria “Additive trees” 1 2 3 4 d12 a d13 d23 d14 d24 d34 1 2 3 4 p12 = a+b p13 = a+c+d p14 = a+c+e p23 = b+c+d p24 = b+c+e p34 = d+e 3 1 b 2 c d e 4 pij = dij for all i and j if the tree topology is correct and distances are additive Distances in gene ral will not be add itive, so choose opt imal tree acco rding to one of the following c riteria (object ive funct ions ): "Goodness - of - fit" : minimize wij i<j pij dij r Typicall y, r = 2 (l east-squares) and wij = 1/dij2 ("FitchMargoliash" metho d) #branches "Minimum - evolution" : minimize k =1 # branches vk or k =1 vk Distance-based optimality criteria Minimum evolution and least-squares Homo sapiens Pan 0.045 0.050 Minumum evolution (ME) 0.015 0.050 0.044 0.286 0.085 LS branch lengths Lemur catta 0.28611 0.04436 0.01511 0.04463 0.05044 0.05038 0.08485 0.57588 Least-Squares Gorilla Pongo dij pij SS 0.39646 0.39838 0.09506 0.37222 0.11172 0.11431 0.37096 0.18107 0.19399 0.18820 0.39021 0.39602 0.09507 0.38084 0.11011 0.11592 0.37096 0.18894 0.19475 0.17958 0.000039 0.000006 0.000000 0.000074 0.000003 0.000003 0.000000 0.000062 0.000001 0.000074 0.000261