6.06. Perturbational methods for dealing with correlation energy

MODELING MATTER AT
NANOSCALES
6. The theory of molecular orbitals for the description of nanosystems (part II)
6.06. Perturbational methods for dealing with correlation energy 1
Perturbed multielectronic
Hartree - Fock wave function
Multielectronic reference functions are antisymmetrized products or Slater determinants resulting from a Hartree – Fock calculation, a typical electronic configuration is that where each spin orbital represents a given existing electron.
I
( N !
)
1 y
1 y
2
( r
1 a
( r
1 a
)
)
.
y
1
( r
2
y
2
( r
2
)
)
.
...
...
...
y
1 y
2
( r
N
( r
N
)
)
.
y
N
( r
1 a
) y
N
( r
2
) ...
y
N
( r
N
)
( N !
)
1 det y i
( r n a
) y i
1
( r n
1
)
( N !
)
1 det y i
( t n
) where y i reference functions are spin orbitals describing the state of an electron of r n a n
≡ t n spatial and spin coordinates.
Perturbed multielectronic
Hartree - Fock wave function
The Hartree – Fock equation for eigenvalues and eigenvectors for a given I state taken as reference would be:
F
ˆ
I
E
I
I where the Fockian acts on the supposed unperturbed eigenfunction
I and gives an E
I eigenvalue corresponding to a certain I pure Hartree – Fock state (either ground or excited) as a reference.
Perturbed multielectronic
Hartree - Fock wave function
On the other hand, the “exact” quantum expression can be written as:
F
L
E
F
L L where the
F
L eigenfunction is referred to a certain system at the E
L
Hamiltonian.
energy. Ĥ
L state of the is the quantum time independent
Perturbed multielectronic
Hartree - Fock wave function
A certain operator can be defined to express the difference between the exact and the Fockian operators:
ˆ
Perturbed multielectronic
Hartree - Fock wave function
A certain operator can be defined to express the difference between the exact and the Fockian operators:
ˆ
Such difference would mean small values with respect to the total energy of the system. We treat it as the electron correlation energy, by definition.
Perturbed multielectronic
Hartree - Fock wave function
A certain operator can be defined to express the difference between the exact and the Fockian operators:
ˆ
Such difference would mean small values with respect to the total energy of the system. We treat it as the electron correlation energy, by definition.
It allows to treat correlation energy as a perturbation following the so called Rayleigh – Schrödinger (RS) theory.
Perturbed multielectronic
Hartree - Fock wave function
Then, in these conditions:
ˆ F
L
F
L
E
L
F
L
As the Hartree – Fock solution was:
ˆ
I
E
I
I
It can be considered that the Hartree – Fock system’s wave
I
E
I reasonably near to the corresponding
F
L exact wave function and E
L eigenvalue, respectively.
Perturbed multielectronic
Hartree - Fock wave function
The perturbations theory routine here consists in designing a procedure to systematically improve Fockian’s eigenfunctions and eigenvalues to approach the exact Hamiltonian’s eigenfunctions and eigenvalues.
Rayleigh – Schrödinger perturbation method
In the general case of Rayleigh – Schrödinger perturbation method a l variable ordering parameter is introduced as:
F
ˆ l
Rayleigh – Schrödinger perturbation method
In the general case of Rayleigh – Schrödinger perturbation method a l variable ordering parameter is introduced as:
F
ˆ l
It is related with the “intensity” of perturbations, taken as the value of electron correlation.
Rayleigh – Schrödinger perturbation method
In the general case of Rayleigh – Schrödinger perturbation method a l variable ordering parameter is introduced as:
F
ˆ l
It is related with the “intensity” of perturbations, taken as the value of electron correlation.
It can eventually become unitary depending on the way to build the perturbation operator.
Rayleigh – Schrödinger perturbation method
Developing a Taylor series on l to express the wave function and exact energy of the system in terms of the Hartree – Fock’ s solutions:
F
L
l
0
I
l
1
I
l
2
I
...
E
L
l
0 E
I
l
1 E
I
l
2 E
I
...
Where and are the functions and energies of the n order perturbation, respectively, in the I state.
Rayleigh – Schrödinger perturbation method
Developing a Taylor series on l to express the wave function and exact energy of the system in terms of the Hartree – Fock’ s solutions:
F
L
l
0
I
l
1
I
l
2
I
...
E
L
l
0 E
I
l
1 E
I
l
2 E
I
...
Where and are the functions and energies of the n order perturbation, respectively, in the I state.
Observe that l
0 ≡ 1 for the 0 th order.
Rayleigh – Schrödinger perturbation method
The problem to solve is finding expressions to calculate the perturbation to the 0 th order Hartree – Fock’s wave function and energy:
I
I
E
I
E
I as they can be considered originated by electron correlation.
Rayleigh – Schrödinger perturbation method
The problem to solve is finding expressions to calculate the perturbation to the 0 th order Hartree – Fock’s wave function and energy:
I
I
E
I
E
I as they can be considered originated by electron correlation.
It can be achieved from the matrix elements of the perturbation operator among multielectronic reference functions, or 0 th order, in different I and J states that can be calculated according to:
E
L
E
I
E
I
l
1
E
I
l
2
E
I
...
E
I
f l
E
I
I
l
2 E
I
J
...
Term deduction and evaluation
Considering that reference multielectronic HF wave functions of any I state are orthonormalized, we can establish that the 0 th order wave function is normal with respect to a certain L state of the exact function to be represented:
I
F
L
I
I
l
I
I
l
2
I
I
...
1
Term deduction and evaluation
Considering that reference multielectronic HF wave functions of any I state are orthonormalized, we can establish that the 0 th order wave function is normal with respect to a certain L state of the exact function to be represented:
I
F
L
I
I
l
I
I
l
2
I
I
...
1
This forces all l values to vanish and means that the 0 ths order I reference state cannot be mixed, or is orthogonal, to perturbed wave functions of higher order of the same state:
I
I
0 for n
= 1, 2, 3,…
Higher order perturbed states cannot be mixed with the unperturbed.
Term deduction and evaluation
Departing from the exact L state already defined by
F
L
F
E
L
l
I
E
I
0
l l
E
I
I
1
l
2 l
2
E
I
I
2
L
...
...
: by substituting in:
ˆ F
L
l
F
L
E
L
F
L remains:
F
L
F
ˆ
E
I
l
l
E
I
I
l
2 l
I
E
I
...
l
2
I
I
...
l
I
l
2
I
...
Term deduction and evaluation
By collecting all terms with the same powers of l
, evaluated as unitary, equations corresponding to each perturbation order arise:
F
ˆ
F
ˆ
I
I
I
F
ˆ
I
E
I
I
I
I
E
E
I
I
I
I
E
E
I
1
I
I
1
I
ˆ
I
n
1
k n
0
E
I
I
n
k
l 0
0 th
order l 1
1 st
order
E
I
I
l 2
2 nd
order l n n th
order and their solution will allow finding all perturbational wave functions as
I
Term deduction and evaluation
If each of those equations are multiplied by the 0 th order
I
I
I
E
E
E
I
I
I
I
I
I
I
I
I
E
…
I
I
I
E
I
I
I
n 1
giving the energies for each perturbation order of each of the
I previously found functions.
Observe, for example, that the first order perturbed wave function is leading to second order perturbation energy.
Term deduction and evaluation
If we reorder the 1 st order energy equation above:
F
ˆ
I
I
E
I
I
E
I
I
matrix element:
E
I
F
ˆ
I
E
I
I
I
I
I
remains a case of an integro – differential equation in place of the eigenfunction and eigenvalue problem, and it requires the appropriate solution.
Term deduction and evaluation
Searching for terms to develop perturbed wave functions we can develop the solution for the first order perturbation in terms of other states (or Slater determinants) being them also eigenfunctions of the Fockian:
I
J a
J
J
Term deduction and evaluation
Projecting any other 0
J th order J
I state onto this 1 st order perturbed wave function arises the following :
J
I
J
K a
K
K
d t
a
J as all HF solutions are orthonormal.
As perturbational wave functions are also orthonormal with respect to the 0 th order:
I
I
0 then the first order of the I th state can be developed in terms of other Hartree – Fock’s J states:
I
J
I
J
J
I
Term deduction and evaluation
In this condition, after multiplying all previous equations by ,
J projecting them onto all space, and knowing that all 0 th order functions are orthonormal:
E
I
J
I
J
F
ˆ
J
J
I
J
I
E
I
J
I
E
I
E
E
J
J
J
I
J
I
J
I
J
I
J
I
J
E
I
I
E
J
The projection of the 0 th order wave function of any J state onto that of the first order perturbation of any different I state depends on their of 0 th order wave functions perturbation matrix element as small was the difference between their expected energy values.
Term deduction and evaluation
As:
E
I
I
I then:
E
I
I
I
J
I
J
I
J
J
I
J
J
I
where substituting by:
J
I
J
E
I gives the second order expression as:
I
E
J
E
I
I
J
I
J
J
E
I
I
E
J
2
E
I
I
J
J
E
I
I
E
J
Term deduction and evaluation
Proceeding in a similar way, the third order perturbation energy expression is:
E
I
I
I
J
K
I
J
E
I
J
E
J
K
K
E
I
E
K
0
I
E
I
I
J
I
E
I
J
E
J
2
2
Higher order terms can also be obtained.
Møller – Plesset method
The total energy after perturbation corrections remains as:
E
I
E
I
E
I
E
I
E
I
being l
= 1.
The matter is to see how each perturbation term can be obtained from the output of a Hartree – Fock calculation and how important are each correction (perturbation order) on the E
I correlated total energy.
Møller – Plesset method
The Moller – Plesset (MP) method is the application of the previously detailed Rayleigh – Schrödinger (RS) perturbation theory to Hamiltonians as that of Hartee – Fock. Then:
H
ˆ
H
ˆ
F
ˆ
where the Fockian is considered as the 0 th order solution of the
Hamiltonian.
Møller – Plesset method
The energy operator for the whole multielectronic system can be expressed as a sum of monoelectronic terms, i.e. a non
interacting Fockian.
Then, for a 2 N electron system:
F
ˆ i
2 N
1 f
ˆ i
Møller – Plesset method
The energy operator for the whole multielectronic system can be expressed as a sum of monoelectronic terms, i.e. a non
interacting Fockian.
Then, for a 2 N electron system:
F
ˆ i
2 N
1 f
ˆ i
Slater determinants as obtained from spin orbitals of a HF solution are the 0 th order wave functions where monoelectronic operators apply to.
Møller – Plesset method
A Fockian on the basis of spin orbitals can thus be developed as:
F
ˆ i
N h
ˆ i j
N
1
1
J
ˆ ij
ij
i
N
1 h
ˆ i
i
N N
1 j
1
J
ˆ ij
K
ˆ ij
where double sums of two electron integrals through all molecular orbitals accounts twice the average HF interactions between every electron pair: i
N N
1 j
1
J
ˆ ij
ij
2 V
ˆ
F
ˆ i
N
1 h
ˆ i
2 V
ˆ
Møller – Plesset method
Being the exact Hamiltonian:
N i h
ˆ i
N N i j
i
1 r ij
N
h
ˆ i
V
ˆ i
Then, after cancelling the one electron term in both the Fockian and the exact Hamiltonian, the perturbation operator remains as:
F
ˆ
V
ˆ
2 V
ˆ exact operator of electron interactions (depending on the sum operator as deduced by the Hartree – Fock theory.
0 th order Møller – Plesset energy
(MP0)
The 0 th order equation is obtained after applying such Fockian to the ground state Hartree – Fock determinant wave function:
I
( 0 )
E
I
( 0 )
I
( 0 )
After applying the previously taken non – interacting Fockian:
F
ˆ
2 N f
ˆ i i
1 to spin orbitals in the determinant, it can be proven that if e i is the eigenvalue of the i’ s HF molecular orbital of those N all in the I state of the system, then:
E
I
( 0 )
2 i
N
0
I e i
0 th order Møller – Plesset energy
(MP0)
The 0 th order equation is obtained after applying such Fockian to the ground state Hartree – Fock determinant wave function:
I
( 0 )
E
I
( 0 )
I
( 0 )
After applying the previously taken non – interacting Fockian:
F
ˆ
2 N f
ˆ i i
1 to spin orbitals in the determinant, it can be proven that if e i is the eigenvalue of the i’ s HF molecular orbital of those N all in the I state of the system, then:
E
I
( 0 )
2 i
N
0
I e i
It must be observed that the total 0 th order energy by the non – interacting Fockian is less exact than the total HF energy because missing the explicit two – electron terms.
1 st order Møller – Plesset energy
(MP1)
We know that for the I ’s Hartree – Fock state:
E
I
V
I
I
I
V
I
2 V
2
I
V
ˆ
I
If we consider that the “exact” electron interaction energy could be similar to the corresponding expectation Hartree – Fock’ s value, it becomes as the first order perturbation energy:
E
I
V
1 st order Møller – Plesset energy
(MP1)
Therefore:
E
I
( 0 )
E
I
( 1 )
2 i
N
0
I e i
2 i
N
0
E
HF
I e i
V
I
V
ˆ
I
Observe that the matrix element corrects double counting of electron repulsions after taking HF orbital eigenvalues at the 0 th order.
1 st order Møller – Plesset energy
(MP1)
Therefore:
E
I
( 0 )
E
I
( 1 )
2 i
N
0
I e i
2 i
N
0
E
HF
I e i
V
I
V
ˆ
I
Observe that the matrix element corrects double counting of electron repulsions after taking HF orbital eigenvalues at the 0 th order.
The 1 st order Møller – Plesset perturbative method does not improve Hartree – Fock’s energy of a given I state.
2 nd order Møller – Plesset energy
(MP2)
The 2 nd order perturbation contribution to the total energy is taken from the previous Rayleigh – Schrödinger development that implies mixing of 0 th order states (interactions of the given I with other J states):
2
E
I
I
J
J
E
I
I
E
J
I
J
J
V
ˆ
I
E
I
E
J
2
2 nd order Møller – Plesset energy
(MP2)
Taking I as the ground state of a certain Hartree – Fock solution, then:
E
0
J
0
J
V
ˆ
0
E
0
E
J
2 being J’s some other electronic configurations or “excited” states of the same 0 th Hartree – Fock solution for the ground state.
2 nd order Møller – Plesset energy
(MP2)
A J state could be either a configuration where occupation of orbital i is transferred to the formerly unoccupied orbital a
(singly excited) or those where occupation of orbital i is transferred to a together with that of orbital j to orbital b
(doubly excited).
As the Brillouin theorem (shown when variational methods were explained) discards interactions between singly excited configurations and the ground state, in the case of 2 nd order perturbations:
0
J
ij ab are allowed for excited terms as well as:
0
J
i a are disallowed.
2 nd order Møller – Plesset energy
(MP2)
Therefore the second order term substracts the following energy to the Hartree – Fock’s ground state in order to account the correlation energy at the second order:
E
0
i occ virt
j a
b
0
V
ˆ
E
0 ab ij
ij ab
E ab ij
V
ˆ
0
2 nd order Møller – Plesset energy
(MP2)
Therefore the second order term substracts the following energy to the Hartree – Fock’s ground state in order to account the correlation energy at the second order:
E
0
i occ virt
j a
b
0
V
ˆ
E
0 ab ij
ij ab
E ab ij
V
ˆ
0
After transforming it by directly taking into account spin orbital matrix elements, the MP2 correction is:
E
0
i occ vir
j a
b
y i y j y e i a y
e b j
y i y e a
e j b y b y a
2
Higher order Møller – Plesset energies (MPn)
Higher order MP terms are being deduced in a similar way. Most programs implement up to MP5 (to the 5 th order).
Higher order Møller – Plesset energies (MPn)
Higher order MP terms are being deduced in a similar way. Most programs implement up to MP5 (to the 5 th order).
MP3 only uses double excitations as well as MP2.
Higher order Møller – Plesset energies (MPn)
Higher order MP terms are being deduced in a similar way. Most programs implement up to MP5 (to the 5 th order).
MP3 only uses double excitations as well as MP2.
MP4 and MP5 involve singly (S), double (D), triple (T) and quadruple (Q) excited determinants.
Examples
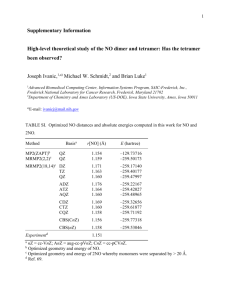
Calculated and experimental data of methyl amine*
4-31G 4-31G(d,p)
MP2|
4-31G(d,p)
MP3|
4-31G(d,p)
MP4|
4-31G(d,p) r
(CN)
<
HNH
<
CNH
1.450
113.2
116.3
1.450
107.1
110.9
1.460
105.7
109.4
1.461
105.8
109.4
1.466
105.4
109.0 t
E tot
-95.071663 -95.1303976 -95.4589619 -95.4828086 -95.4901785
E corr
0 0
-0.329023
(0.34%)
-0.352805
(0.37%)
-0.360498
(0.38%)
E corr
0 0 0 -0.023782 -0.007693
1 1.5 2.1 5.0 257.7
*Distances in Å, angles in degrees , energy in Hartree’s and time in seconds exp 4
1.472
106.0
111.5
Some remarks
Perturbative methods have no clear limits to correct Hartree –
Fock’s energies and it could even occur that a truncation (i.e. applying MP2 without MP3) could lead to energies even smaller than the exact, although MP2 alone is generally a very good level for common calculations.
Some remarks
Perturbative methods have no clear limits to correct Hartree –
Fock’s energies and it could even occur that a truncation (i.e. applying MP2 without MP3) could lead to energies even smaller than the exact, although MP2 alone is generally a very good level for common calculations.
The main shortcoming is that a low quality Hartree – Fock multilectronic wave function (i.e. a very small atomic basis set) gives larger perturbations and the method’s consistency becomes less trustworthy in such cases.
Some remarks
It has been considered that results could become erratic when degenerate or quasi degenerate electronic states are involved.
Some remarks
It has been considered that results could become erratic when degenerate or quasi degenerate electronic states are involved.
For open shell molecules, MPn-theory can directly be applied only to unrestricted Hartree–Fock reference functions.
However, the resulting energies often suffer from severe spin contamination, leading to very wrong results. A much better alternative is to use one of the MP2-like methods based on restricted open-shell Hartree–Fock references.
Some remarks
Møller – Plesset (MPn) perturbative methods are the most used
ab initio tool to improve Hartree –Fock’s wave functions, energies and resulting optimized geometries.
Some remarks
Møller – Plesset (MPn) perturbative methods are the most used
ab initio tool to improve Hartree –Fock’s wave functions, energies and resulting optimized geometries.
This methods are size – consistent. Therefore the resulting energies can be used to compare different structures and molecular compositions, even for thermochemical evaluations.
Some remarks
Møller – Plesset (MPn) perturbative methods are the most used
ab initio tool to improve Hartree –Fock’s wave functions, energies and resulting optimized geometries.
This methods are size – consistent. Therefore the resulting energies can be used to compare different structures and molecular compositions, even for thermochemical evaluations.
MP2 calculations are estimated to account up to 80 – 90 % of the correlation energy and then cover most chemical problems at a relatively modest cost, being fully a priori originated results.