pmic7841-sup-0007-SuppMat
advertisement

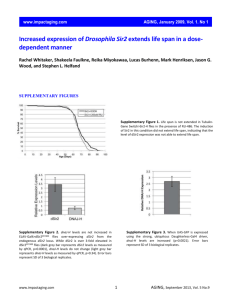
Supporting Information Supplementary Methods Sample preparation and analysis for LR-CID The phosphopeptides were enriched using titanium dioxide (TiO2) beads as described [1] with some modifications, including the use of batch-mode enrichment. Peptides from 500 g of protein were dissolved in 200 l loading buffer (1 M glycolic acid, 80% acetonitrile, and 2% TFA) and mixed with 1 mg of TiO2 beads (GL Sciences, Japan) in a tube and rotated gently for 15 min. After centrifugation, the supernatants were transferred to new Eppendorf tubes and incubated with TiO2 beads again, as described above. The beads were washed successively with glycolic acid loading buffer and wash buffer (80% acetonitrile, and 2% TFA). Then, the beads were loaded onto homemade StageTip-C18 columns in 200-μl yellow tips that were preconditioned with 80% acetonitrile and 2% TFA, washed once with the same buffer, and eluted with 100 μl elution buffer 1 (0.5% ammonium hydroxide solution pH 10.5) and 100 μl elution buffer 2 (30% acetonitrile). Ammonia and acetonitrile were evaporated using a vacuum centrifuge, and the peptides were acidified in 1% TFA and desalted using homemade StageTip-C18 Columns. Desalted phosphopeptide samples were dried in a vacuum centrifuge and stored at -80°C until LC-MS/MS analysis. The phosphopeptide samples from the traditional method were analyzed by LC-MS on an Easy-nLC II (Thermo Fisher Scientific, Odense, Denmark) that was coupled to a nanoelectrospray ion source (Thermo Fisher Scientific, Bremen, Germany) on an LTQ Velos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) as described (denoted LR-CID) [1]. Phosphopeptides were separated on a 2-column system, comprising a trap column (100 Å, 5 m particle, 75 m x 3 cm) and an analytical column (200 Å, 3 m particle, 75 m x 10 cm), with a 200-min gradient from 2% to 40% 1 acetonitrile at a flow rate of 300 nl/min and analyzed on a mass spectrometer. For low-resolution mass spectrometry on the LTQ Velos, a cycle of 1 full-scan MS survey spectra (m/z 300–1800) was acquired in the profile mode. CID peptide fragments were acquired at 35% normalized collision energy (NCE) for the LTQ Velos. All raw files were processed in Proteome Discoverer, version 1.4, using SequestHT against the IPI mouse database (version 3.87, 59,534 entries), containing forward and reverse protein sequences. MS/MS searches were performed with the following parameters: full enzyme digest using trypsin (After KR/-) with up to 2 missed cleavages; a precursor ion mass tolerance of 2.0 Da (average mass); CID fragment ion mass tolerance of 0.5 Da; carbamidomethylation as a fixed modification; and oxidation of methionine and phosphorylation of serine, threonine, and tyrosine as variable modifications. Percolator, with a semisupervised machine learning algorithm [2] in Proteome Discoverer, was used to merge the search results from the search engines (SequestHT) and subsequently filter PSMs at a false discovery rate (FDR) < 1% using q-values. In addition, PSMs that received a search engine rank of 1 were considered for further analysis. The location of phosphorylation sites from the CID was determined using the PhosphoRS 3.1 algorithm [3] in Proteome Discoverer. Phosphopeptides with a PhosphoRS probability ≥ 0.99 were considered to have confident phosphorylation site localization. 2 Supplementary Table 1. Summary of phosphoproteomic analysis by iMEED approach Summary of all identified phosphopeptides and phosphosites. Amount injected is 2 l or 4 l. Phosphorylation site localization was determined using the PhosphoRS 3.1 algorithm. Supplementary Table 2. List of all identified phosphopeptides using SequestHT_only All identified phosphopeptides are listed. Leading proteins in the corresponding protein groups are also shown. The list includes the IPI accession numbers of each identified protein group and the sequences of the phosphopeptides and nonphosphopeptides. Search results of the Proteome Discoverer 1.4-SequestHT engine and information on phosphorylation localization from the PhosphoRS 3.1 analysis are shown. Supplementary Table 3. List of all identified phosphopeptides by iMEED approach All identified phosphopeptides are listed. Leading proteins in the corresponding protein groups are also shown. The list includes the IPI accession number of each identified protein group and the sequences of the phosphopeptide and nonphosphopeptide. Search results of the Proteome Discoverer 1.4-SequestHT and MS Amanda engines and information on phosphorylation localization from the PhosphoRS 3.1 analysis are shown. Supplementary Table 4. Overlap between technical and biological replicates. The results for the overlap analysis between technical and biological replicates are summarized in each fraction. The overlap analysis involved 2 sets, A and B. Percentage (%) of overlap (A) represents the percentage of overlap for set “A” in the 2-set comparison. Percentage (%) of overlap (B) 3 represents the percentage of overlap for set “B” in the 2-set comparison. The sharp sign (#) represents the number of identified phosphopeptides. Supplementary Table 5. MS1 intensity of identified phosphopeptides. Supplementary Table 6. Correlation of phosphopeptide intensity between biological replicates Pearson correlation of intensity between biological replicates, demonstrating high reproducibility in phosphopeptide intensity for biological replicates 1-3. Supplementary Table 7. Distribution of pI and GRAVY values pI values of phosphopeptides were calculated using the pI calculator tool (https://trac.nbic.nl/picalculator/) and ExPASY pKa values of amino acids. The pKa values of pS/T and pY were set to program default values. Percentages of identified phosphopeptides by pI value in the E1, E2, and E3 fractions are listed. Cumulative percentages of phosphopeptides from low to high pI are also shown. GRAVY values were calculated using the GRAVY calculator (http://www.gravycalculator.de/). Percentages of identified phosphopeptides by GRAVY value in the E1, E2, and E3 fractions are listed. Cumulative percentages of phosphopeptide from low to high GRAVY value are also shown. Supplementary Table 8. Summary of comparison between this study and other previous studies 4 Supplementary Figure 1. Detailed flowchart of database search using Proteome Discoverer 1.4 5 Supplementary Figure 2. Distribution of PhosphoRS peptide scores Distribution of PhosphoRS binomial peptide scores of the identified peptides in the control (red), E1 (green), E2 (purple), and E3 (blue) fractions. 6 Supplementary Figure 3. Reproducibility of proteomic analysis Reproducibility between 4 technical and 3 biological replicates is expressed as Pearson correlation value (R). bio1_E1_tech1 represents technical replicate 1 of biological replicate 1 in fraction E1, etc. Red boxes represent the comparison between technical replicates. 7 Supplementary Figure 4. Evaluation of phosphopeptide identification by multistep enrichment Multistep enrichment improves the identification of phosphopeptides. The pie chart shows the number of fractions in which any given phosphopeptide was identified. Sixty-six percent of phosphopeptides appeared in only 1 fraction. 8 Supplementary Figure 5. Comparison of multi-phosphopeptide identification from each fraction Numbers of identified phosphopeptides in each fraction with phosphoRS score ≥ 99% are shown in the bar graph. Phosphopeptides with mono- and multiple phosphate groups are shown in blue and red, respectively. Percentages of identified multiply phosphorylated peptides of all identified phosphopeptides in each fraction are shown with a dashed line. 9 Supplementary Figure 6. Comparison with the PhosphositePlus database Comparison of all phosphosites of human, rat, and mouse orthologs extracted from the Phosphosite Plus database (http://www.phosphosite.org)—1262 (17.6%) phosphosites in this study are unknown. 10 Supplementary References [1] Han, D., Moon, S., Kim, Y., Ho, W. K., et al., Comprehensive phosphoproteome analysis of INS-1 pancreatic beta-cells using various digestion strategies coupled with liquid chromatography-tandem mass spectrometry. Journal of proteome research 2012, 11, 2206-2223. [2] Spivak, M., Weston, J., Bottou, L., Kall, L., Noble, W. S., Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. Journal of proteome research 2009, 8, 3737-3745. [3] Taus, T., Kocher, T., Pichler, P., Paschke, C., et al., Universal and confident phosphorylation site localization using phosphoRS. Journal of proteome research 2011, 10, 5354-5362. 11