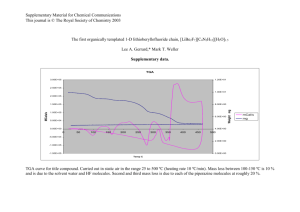
Figure 4.3
advertisement

MSc Chemistry Analytical Sciences Master thesis DEVELOPMENT OF BIOLOGICAL COMPOUND ARRAYS, EMBEDDED IN TISSUE MIMETIC MATERIALS FOR TESTING THE SENSITIVITY AND DETECTION LIMITS OF MASS SPECTROMETRY IMAGING TECHNIQUES by Skandalaki Eleni February 2014 Supervisor: Dhr. Dr. W.T. Kok Daily Supervisor: Prof.Dr.Ing. R. Heeren Research was carried out at the group of Bio Molecular Imaging Mass Spectrometry (BIMS) at AMOLF-FOM institute Eleni Skandalaki UvA-AMOLF Page 1 Abstract Mass spectrometry (MS) and the techniques based on it, are significant tools for researchers due to their high sensitivity, accuracy and capability of detecting and distinguishing compounds of high masses. Ergo, techniques depending on MS are suitable for applications in disciplines like chemistry, and biochemistry. One of the innovative techniques depending on MS that is widely employed in biochemistry is mass spectrometry imaging (MSI). With MSI, it is possible to localize, detect and visualize compounds of interest simultaneously; this is why it was considered an appropriate technique to use in this project. In particular, by using Maldi Assisted Laser Desorption Ionization (MALDI) mass spectrometry imaging it was attempted to create a quantification method for MALDI instruments, by developing test arrays of lipids (1, 2-Dipalmitoyl-sn-glycero-3-phosphocholine, Sphingomyelin), peptides (Substance P, Angiotensin II) and proteins (Bovine serum albumin, Trypsinogen) embedded in tissue mimetic materials such as gelatin and Carboxymethyl cellulose salt (CMC) solution. Even though the tissue mimetics appeared to mix with the compounds of interest, this did not occur in the test arrays developed and the compounds could not be distinguished by observing the images created after MSI analysis; therefore the creation of a quantification method was not achieved. Despite that fact, it was realized that in the event of using commercial hydrogels as tissue mimetics and different biological compounds for the development of the arrays, a qualification method could be created. Another part of this project included spatial resolution experiments with the biological compounds that are already mentioned. The results of these experiments showed that the MALDI- TOF MS instruments in the Bio Molecular Imaging Mass Spectrometry (BIMS) laboratory at AMOLFFOM institute have high sensitivity and high limit of detection. Eleni Skandalaki UvA-AMOLF Page 2 Table of Contents Abstract ................................................................................................................................................... 2 Introduction............................................................................................................................................. 5 Mass spectrometry and Mass Spectrometry Imaging......................................................................... 5 Matrix Assisted Laser Desorption Ionization (MALDI)......................................................................... 6 Principles of Time of Flight (TOF) MS .................................................................................................. 7 Principles of Secondary Mass Spectrometry (SIMS)............................................................................ 8 Compounds employed throughout the project .................................................................................. 8 Calibration lines ..................................................................................................................................... 12 Profiling of compounds of interest.................................................................................................... 13 DPPC calibration lines........................................................................................................................ 15 SM calibration lines ........................................................................................................................... 17 Peptide mixture calibration lines ...................................................................................................... 18 Protein mixture calibration lines ....................................................................................................... 19 An alternative normalization technique ........................................................................................... 20 Development of Test Arrays .................................................................................................................. 22 Choice of tissue mimetic materials ................................................................................................... 22 Choice and design of proper container ............................................................................................. 22 Sectioning .......................................................................................................................................... 23 Matrix application ............................................................................................................................. 24 MSI analysis in positive mode and Data Analysis .............................................................................. 25 MSI analysis in negative mode and Data analysis ............................................................................. 27 Principle Component Analysis (PCA) and Discriminant Analysis (DA)............................................... 28 Profiling of DPPC and SM arrays with Secondary Ionization Mass Spectrometry (SIMS) ................. 34 Spatial resolution experiments ............................................................................................................. 36 Sample preparation ........................................................................................................................... 36 Acquired Data from MSI analysis of DPPC ........................................................................................ 37 Acquired Data from MSI analysis of SM ............................................................................................ 38 Acquired Data from MSI analysis of Peptide Mixture -Substance p ................................................. 39 Acquired Data from MSI analysis for Angiotensin II.......................................................................... 40 Protein mixture- Sample preparation ............................................................................................... 42 Acquired Data from MSI analysis of BSA ........................................................................................... 42 Acquired Data from MSI analysis of Trypsinogen ............................................................................. 43 Results, discussion and suggestions ...................................................................................................... 45 Eleni Skandalaki UvA-AMOLF Page 3 Suggestions........................................................................................................................................ 46 References ............................................................................................................................................. 47 Appendix................................................................................................................................................ 50 Eleni Skandalaki UvA-AMOLF Page 4 Introduction Mass spectrometry and Mass Spectrometry Imaging “Mass spectrometry is the study of matter through the formation of gas-phase ions that are detected and characterized by their mass and charge” according to Murray, Boyd, Eberlin, Langley, Li & Naito (2013). Mass spectrometry and the techniques based on it, are significant research tools that are widely utilized in chemistry, biology, physics, and have numerous environmental, geological, biotechnological, pharmaceutical and clinical applications. Furthermore, mass spectrometry is extensively used in these fields, because of its high sensitivity, accuracy; resolution and wide mass range detection limit (Chughtai 2012). One of the innovations that have been developed in the field of mass spectrometry due to its technological and methodological benefits is mass spectrometry imaging (MSI). With MSI, the visualization of the spatial distribution of different compounds based on their molecular masses synchronously is possible (McDonnell & Heeren 2007, Caldwell & Caprioli 2005). For this reason, MSI has several applications in pharmaceutical and biomedical studies and in particular it has been used in many studies about tissue engineering with which this project is related. In this project, Matrix Assisted Laser Desorption (MALDI) mass spectrometry imaging was employed in the majority of experiments. Aim of project The objective of this project was to contribute in the on-going search of innovations in the mass spectrometry imaging, by creating test arrays of different biological compounds such as lipids, proteins and peptides embedded in tissue mimetic materials. The development of such arrays could be used for the creation of a quantification method that could indicate the sensitivity and the limit of detection of specific MSI techniques in different laboratories. Eleni Skandalaki UvA-AMOLF Page 5 Matrix Assisted Laser Desorption Ionization (MALDI) Matrix Assisted Laser Desorption Ionization (MALDI) is the most widely utilized MSI technique (Gode & Volmer 2013); it is a ‘’soft’’ ionization technique which has proved to be an important analytical tool (Hillenkamp & Karas 2007). A technique is defined as ‘’soft’’ when the energy that is supplied to the samples is lower compared to electron ionization, therefore the produced fragments are fewer and the obtained spectra are simple (Games 1978). MALDI was developed in the 1980s by F.Hillenkamp and M.Karas, and ever since it is a powerful source for producing intact gas-phase ions from numerous large, labile and nonvolatile compounds. Furthermore, as it is suggested from the name of MALDI the matrix that is applied on the sample is of tremendous significance because it determines the extent of the ionization and desorption of the analyte of interest. In particular, the MALDI process takes place in two parts. Firstly, the matrix solution is an aromatic acid dissolved in a mixture of organic solvents and it should have the capability of absorbing at a specific laser wavelength and remain stable under vacuum. The matrix is applied on the sample during the sample preparation. While it is applied, the solvents employed for the dilution of the matrix evaporate and it reacts with the sample leading to the formation of matrix crystals. The second step of MALDI, takes place under vacuum in the mass spectrometer source. During this stage, the sample and matrix co-crystals are “cut” by laser pulses that occur within a short window of time. Afterwards the co-crystals are heated quickly by the irradiation that the laser induces, to such an extent that localized sublimation occurs and intact analyte enters the matrix plume. Throughout this process, ionization reactions take place, and the mechanism that is presumed to be responsible for the ionization, is described according to Hoffmann & Stroobant (2007) with “proton transfer in the solid phase before desorption or gas-phase photon transfer in the expanding plume from photoionized matrix molecules”. Lastly, an electrostatic field induces the acceleration of the ions that are in the gas phase in the direction of the analyser. In the figure 1.1 there is a scheme depicting the principle of MALDI (Hoffmann& Stroobant 2007). Figure1.1 Principle of MALDI ionization, (Hoffmann& Stroobant 2007) Eleni Skandalaki UvA-AMOLF Page 6 In this project, the instruments used for the MSI analysis were the SYNAPTTM HDMS, Waters Corporation, Milford, MA and Ultraflex III, Brucker Daltonics. These instruments are MALDI tandem mass spectrometers, which are used for automated MS and MS/MS high throughput detection and identification of peptides and proteins. Principles of Time of Flight (TOF) MS After the sample-matrix- crystals are ionized, the ions head towards the mass analyser where they are separated according to their mass to charge ration (m/z) values. In this project, the mass analyser utilized and coupled with MALDI, was a time of flight (TOF) instrument with an orthogonal ion inlet. In particular, in a TOF analyser, the ions get separated based on the time they need to reach the detector after they get accelerated from an electric field and drift into a flight tube, which is a field free area. This procedure is showed in the Figure 1.2. Figure1.2 Ionization source and TOF mass analyzer (Hoffmann& Stroobant 2007). Additionally there are some mathematical formulas which contribute to the comprehension of how a TOF analyser works. Ek = 𝑚𝑢2 2 = 𝑞𝑉𝑠 = 𝑧𝑒𝑉𝑠 = 𝐸𝑒𝑙 (1) In formula (1) it is shown that the electric potential energy, Eel which is acquired by the ions while they are in the acceleration region of the source, is converted into kinetic energy, Ek which is identical for all the ions. Therefore, all ions move into the field free region called Eleni Skandalaki UvA-AMOLF Page 7 drift path, with the same initial Ek but different velocities (υ). As it is shown in formula (2), the velocities of the ions differentiate only if they have different masses. 2𝑧𝑒𝑉𝑠 1/2 ) 𝑚 u= ( (2) Moreover, the velocity of each ion is constant until it reaches the detector; but the time (t) an ion needs to move in the drift path until it reaches the detector is different. 𝑡= 𝐿 (3) 𝑢 As it is suggested from the name of the analyser, time formula (3) is the determining factor for the separation of the ions of a sample (Hoffmann & Stroobant 2007). MALDI-TOF MS is suitable for measuring compounds with molecular weight up to 200.000Da (Caprioli, Farmer & Gile 1997); ergo it was appropriate for the MS analysis of peptides, lipids and proteins that were used in this project. Principles of Secondary Mass Spectrometry (SIMS) SIMS is a mass spectrometry technique that has been used since the 1960s and it is especially employed for high spatial resolution MSI analysis. Its principle involves a primary ion beam, which hits the surface of a thin layered sample with 5-25kV energy and induces the production of secondary ions from the sample. Nowadays, primary ion guns, which are the cornerstone of a SIMS instrument utilize primary metal ions such as Au+, In+, Bi+, Xe+ and Ga+ . After the secondary ions of the sample are induced, they are accelerated due to a high voltage system, towards the mass analyser. One of the most regularly used mass analysers coupled with SIMS, is a TOF analyser (Chughtai 2012). The SIMS instrument in the BIMS laboratory is a Physical Electronics TRIFT II high resolution Secondary Ion Mass Spectrometry TOF-MS system and it was used only once for the profiling of a small area of a sample employed in this project, since it was too large to conduct a SIMS imaging experiment. Compounds employed throughout the project Sphingomyelin (SM) Figure 1.3. SM molecular structure,Sigma-Aldrich information sheet The sphingomyelin from chicken egg yolk used throughout this project was purchased from Sigma-Aldrich. Sphingomyelin is a sphingolipid, that is polar and it is widely used for Eleni Skandalaki UvA-AMOLF Page 8 pharmaceutical purposes, cosmetics and skin care (Karlsson, Michélsen & Odham 1998). Moreover, sphingomyelin is a compound of interest in several research studies because it is from the (significant) components of blood and nervous tissue and is contained in plasma membranes of higher animals. 1, 2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) Figure 1.4. DPPC molecular structure,Sigma-Aldrich information sheet The DPPC, which was also purchased from Sigma-Aldrich, is a phospholipid that consists of two palmitic acid moieties. DPPC is one of the various lipids that contain a choline group and it is found in abundance in eukaryotic cells (Attwood, Choi & Leonenko 2013). Angiotensin II (Asp-Arg-Val-Tyr-IIe-His-Pro-Phe) Figure 1.5. Angiotensin molecular structure,Sigma-aldrich information sheet The angiotensin II was a component of a peptide mixture prepared for the purpose of this project and was also purchased from Sigma-Aldrich. This peptide is synthesized when the angiotensin converting enzyme causes a cleavage in the C-terminal (-His-Leu) of angiotensin I. Angiotensin II, plays a significant role in controlling cardiovascular structure and hemodynamics. Last but not least, angiotensin II enhances the density of micro-vessels and triggers angiogenesis (Product Information sheet from Sigma- Aldrich). The peptide mixture used in this project was prepared according to a Brucker calibration standard, which is employed for the calibration of Matrix Assisted Laser Desorption Ionization -Time of Flight (MALDI-TOF) mass spectrometers (Instructions for use- Peptide calibration standard). Substance p Figure 1. 6 Sunstance P molecular structure,Sigma-aldrich information sheet The substance p was the second component of the already mentioned peptide mixture and was purchased from Sigma-Aldrich too. Substance p is a peptide synthesised from 1 amino Eleni Skandalaki UvA-AMOLF Page 9 acid residues and it is abundant in the peripheral and central nervous system. In the former system, it induces vasodilation, smooth muscle-contracting and hypotension while in the latter it is believed to participate in the sensory nerve transmission (Product Information sheet from Sigma-Aldrich). Bovine serum albumin (BSA) Bovine serum albumin was purchased from Sigma-Aldrich as well, and was one of the proteins used in a protein mixture that was employed throughout this project. The proteins for the mixture were chosen based on the Brucker Protein standard II, which is a mixture of proteins useful for the calibrating and testing of MALDI-TOF mass spectrometers (Instructions for Use- Protein Standard II). Therefore, since BSA and trypsinogen were the only proteins included in the Brucker standard that were in the laboratory when this project was initiated, they were used until its completion. Trypsinogen As it is already mentioned, trypsinogen was also included in the protein mixture and was a purchased from Sigma too. Trypsinogen is a trypsin proenzyme that formulates in the bovine pancreas and contains a 229 amino acid chain that is cross-linked by six disulphide bridges (Product Information sheet from Sigma-Aldrich). Gelatin Gelatin is widely used as a thickener and stabilizer in foods, it is also a component of pharmaceuticals, paints, adhesives, films, artificial silk, matches and the first embedding material ever used, in 1802(Luft 1973, Product Information sheet from Sigma- Aldrich). It is a collagen derivative that is formed when the natural triple-helix structure of collagen, breaks into single-strand molecules. Additionally, it can readily form gels by altering its solution temperature and due to their biocompatibility, they have numerous applications in tissue engineering (Tabata & Ikada 1998, Product Information sheet from Sigma- Aldrich). In particular, gelatin has already been used in the formation of polymer scaffolds in tissue engineering (Yang, Leong, Zhaohui, Chua & Chee-Kai 2001). The gelatine utilized in this project was purchased from Sigma-Aldrich and the solution prepared for the needs of the project was a 10% solution. Carboxymethyl cellulose salt (CMC) Figure 1.7 CMC (monomer) molecular structure, product Information sheet from Sigma-Aldrich. Eleni Skandalaki UvA-AMOLF Page 10 CMC, which was purchased from Sigma-Aldrich, has several applications in tissue engineering. It is an ether derivate of cellulose, which is a natural chain-shaped polymer. Additionally, it has numerous applications in food industry as a thickener, in toothpastes, detergents, paints and so on. In chemistry and biochemistry it has proved to be useful because of its biocompatibility, solubility in water and low price (Jiang, Yubao, Wang, Xuejiang, Zhang, Wen, Jiqiu, Gong & Mei 2008, Product Information sheet from SigmaAldrich). For all the above reasons, CMC was chosen as a tissue mimetic material in this project and the CMC solution prepared was a 2% solution. Polyvinylpyrrolidone/ Carboxymethyl cellulose (PVP/ CMC) hydrogel According to Rosiak, Ulański , Pajewski , Yoshii & Makuuchi (2002), “hydrogels are two or multi-component systems consisting of a three-dimensional network of polymer chain and water that fills the space between macromolecules”. Natural and synthetic polymers have been used in biomedical applications such as drug delivery and tissue engineering. For the past decade in tissue engineering, hydrogels have been used as scaffold materials in order to engineer new tissues, which could result in revolutionary applications in medicine and could benefit many people who suffer from tissue or even organ failure. The hydrogel employed in this project, was synthesized in the laboratory and consisted from a polymer, polyvinylpyrrolidone (PVP) 0, 1% and CMC 1%, in two-to-eight ratio. PVP/CMC hydrogels have been used as wound dressing because of their good biocompatibility, moisture retention, ventilating capability and water absorption ability; consequently, such a hydrogel was considered a choice with great potential for this project (Rosiak, Ulański, Pajewski, Yoshii & Makuuchi 2002, Wang , Xu, Hu, Zhai , Peng , Nho , Li & Wei 2007). Eleni Skandalaki UvA-AMOLF Page 11 Calibration lines The plot of calibration lines for different concentrations of the DPPC, SM, peptide mixture and protein mixture with the tissue mimetic materials of interest, was an important first step in this project in order to get an idea of their ideal behaviour by using MALDI- TOF MS Imaging; and after taking the results into consideration the next step for the development of test arrays was decided. Sample preparation The calibration lines which will be shown in the successive paragraphs, are demonstrating the absolute intensities of the analytes’ protonated ions (M+H) + with regard to their concentrations. The concentrations, on which the calibration lines were based on, were chosen after some profiling tests at the SYNAPT and the Ultraflex III. Therefore, after conducting three series of spotting tests and by examining the spectra that were the outcome of this process, the concentrations that were chosen were 1,3,5,8,10μM for DPPC, 1, 2, 5, 7, 10μM for SM, 0.5, 1, 2, 3, 5μM for the peptide mixture and 0.5, 1, 2, 3, μΜ for the protein mixture. For the plot of the calibration lines, the experimental procedure contained the same steps for all the compounds of interest. Firstly, for the initial series of concentrations, H2O, purchased from Sigma-Aldrich and MeOH purchased from BiosolveChemicals were used to dilute the peptides, proteins and lipids respectively. Successively, PVP/CMC hydrogel and CMC were used because of their capability to mimic tissue behaviour in order to make new dilution series for the DPPC, SM, peptide and protein mixture and for the next step of the profiling process, the most suitable matrix should be chosen. Selection of matrix The choice of the matrix plays a significant role in the MALDI- TOF MS, as it is mentioned in the introduction chapter and its choice may determine the outcome of an experiment. The most commonly used matrixes are sinapinic acid (SA), 2, 5-dihrydoxy benzoic acid (DHB) and α-cyano-4-hydroxycinnamic acid (CHCA). SA is an appropriate choice for samples with molecular weights over 10.000Da while CHCA and DHB for masses under 10.000Da (Cohen & Chait 1996). Therefore either DHB or CHCA would be the more suitable choices for the peptides and lipids employed in this project. After applying both of them on the DPPC, SM, peptide, the CHCA, purchased from Fluka, Sigma-Aldrich, formed better co-crystals with them; and for the proteins SA, also purchased from Fluka, Sigma-Aldrich, was chosen. SA (MW: 224.21Da) Eleni Skandalaki UvA-AMOLF Page 12 CHCA (MW: 189.04 Da) Figure 2.1 Molecular structures, taken from Sigma-Aldrich product information sheets. Matrix solution preparation For the preparation of a matrix solution an organic solvent has to be chosen, such as acetonitrile (ACN) or methanol (CH3OH), then an acidic organic compound, which is the matrix itself, and trifluoacetic acid (TFA), is added. By adding TFA, there are an increased number of protons available for ionization (Chughtai 2012) CHCA preparation For the preparation of a 10mg/ml matrix solution, 100 mg of CHCA were weighed and diluted in a 10ml mixture of 70:30 H2O: ACN or CH3OH and 0.1% TFA. (TFA was purchased from Biosolve- Chemicals). SA preparation For the preparation of a 20mg/ml SA solution, 200mg of SA were weighed and diluted in a solution of 10ml 50:50 ACN: H2O and 0.1% TFA. Both matrix solutions were placed in ultrasonic bath for 10 minutes in order to ease the dilution. Profiling of compounds of interest The next step of the protocol followed included the profiling of the compounds. More specifically, a small amount (1μl) was prelevated from each concentration prepared and was mixed with the same amount of matrix in one-to-one ratio. Successively, 0.5μl of DPPC, SM and the peptides mixture- CHCA, was placed on different cells of a metal target plate and the spots were allowed to dry. Figure 2.2 Metal target plate Eleni Skandalaki UvA-AMOLF Page 13 When they dried, the plate was loaded in the SYNAPT and the software program Masslynx, Waters was used firstly for the calibration of the instrument, by profiling a polyethylene glycol (PEG) calibration standard mixed with matrix (CHCA), and then for the setting up of the experiment. After the calibration, the mode had to be set, which was positive in the following cases and the detector had to be at 1850V. Then a cell on the target plate was chosen and in the end the laser was fired at 250eV all around the cell, for the same number of laser shots (about 50 shots) for all the compounds and concentrations. From this process, a chromatogram and a spectrum were produced, for each cell of the target plate. Furthermore in each spectrum, it is possible to find the absolute intensity of the m/z of interest, by choosing the appropriate setting in Masslynx. The molecular masses for the DPPC, SM, Angiotensin II, Substance p, Trypsinogen and Albumin bovine are listed in a table below. The peaks taken into consideration for the plotting of the calibration lines that follow, were the intact protonated ions of the analytes, (M+H)+ where M represents the average molecular weight and the m/z of the ions that are detected and measured by the TOF analyser of the SYNAPT and the Ultraflex III. Compounds Sphingomyelin (SM) 1,2-Dipalmitoyl-sn-glycero-3phosphocholine (DPPC) Substance p Angiotensin II Trypsinogen Bovine serum albumin (BSA) (M+H)+ 703.5 734.3 1046.54 1347.73 23982 66463 Figure 2.2 Intact protonated molecules Eleni Skandalaki UvA-AMOLF Page 14 DPPC calibration lines In the Figure 2.3, there is an example of a combined spectrum based on which one of the calibration lines of DPPC was plotted. Figure 2.3 Combined spectrum from profiling of 1, 3, 5, 8, 10μM of DPPC-CMC 6.00E+02 Absolute Intensity 5.00E+02 4.00E+02 3.00E+02 DPPC DPPC -CMC 2.00E+02 DCCP-HYDROGEL 1.00E+02 0.00E+00 1 3 5 8 10 Concentration ( μΜ) Figure 2.4 Calibration lines for DPPC Eleni Skandalaki UvA-AMOLF Page 15 In Figure 2.4, the calibration lines for DPPC are not completely linear and it was assumed that normalization was needed in order to overcome this problem. 2.50E-01 Absolute Intensity 2.00E-01 1.50E-01 DPPC 1.00E-01 DPPC -CMC DCCP-PVP:CMC 5.00E-02 0.00E+00 1 -5.00E-02 3 5 8 10 Concentration ( μΜ) Figure 2.5 Normalized calibration lines for DPPC The Figure 2.5 occurred after normalizing the values of the absolute intensities by dividing them by the Total Ion Count (TIC) from every profiling of each concentration used for the construction of the calibration lines. Even though normalization took place, the calibration lines did not become linear thus it was assumed that either there was a human error in the sample preparation, or the normalization that was conducted was not the appropriate one. Eleni Skandalaki UvA-AMOLF Page 16 SM calibration lines 1.40E+03 Absolute Intensity 1.20E+03 1.00E+03 8.00E+02 SM 6.00E+02 SM-CMC SM-Hydogel 4.00E+02 2.00E+02 0.00E+00 1 2 5 7 10 Concentration (μΜ) Figure2.6 Calibration lines for SM In Figure 2.6 it is shown that the calibration lines are not linear, thus the next step was to normalize the results, similarly to the case of DPPC. Based on the normalization applied, the following plot was constructed. 5.00E-01 Absolute Intensity 4.00E-01 3.00E-01 SM 2.00E-01 SM-CMC SM-PVP:CMC 1.00E-01 0.00E+00 1 -1.00E-01 2 5 7 10 Concentration (μΜ) Figure 2.7 Normalized calibration lines for SM Once again the normalized plot did not follow a linear trend; as a consequence it was assumed again that either the normalization was not the appropriate one or that there was an error in the sample preparation. Eleni Skandalaki UvA-AMOLF Page 17 Peptide mixture calibration lines 1.20E+05 Absolute Intensity 1.00E+05 8.00E+04 Angiot 2 Substance p 6.00E+04 Angiot 2-CMC 4.00E+04 Subs p- CMC Angiot 2-hydrogel 2.00E+04 Subst p-hydrogel 0.00E+00 0.5 -2.00E+04 1 2 3 Concentration (μΜ) Figure 2.8 Calibration lines for peptide mix. In Figure 2.8 it is shown that the trend of the calibration lines for the peptide mixture is close to linear but normalization with TIC was also conducted in order to check if the calibration lines could be further optimized. 2.00E+01 Absolute intensitiy 1.50E+01 Angiot 2 1.00E+01 Subst p Angiot 2-CMC Subs p-CMC 5.00E+00 Angiot 2-CMC:PVP Sub p -CMC:PVP 0.00E+00 0.5 -5.00E+00 1 2 3 Concentration (μΜ) Figure 2.9 Normalized Calibration lines for peptide mix. Eleni Skandalaki UvA-AMOLF Page 18 Figure 2.9 indicates that the normalization based on the TIC optimised the initial results as the normalized calibration lines to a small degree as they seem to follow a similar trend with the non-normalized ones. Protein mixture calibration lines For the plot of the protein mixture calibration lines, the Ultraflex III was used instead of the SYNAPT due to the fact that its detection range is too low for such large proteins as Bovine Serum Albumin and Trypsinogen. It has not been possible to obtain a calibration line for the proteins in the PVP/CMC hydrogel because of the low signal-to noise (S/n) ratio. For this reason it was not feasible to detect any clear peak signals and to assign them to the proteins. 8.00E+04 7.00E+04 Absolute Intensity 6.00E+04 5.00E+04 BSA 4.00E+04 Tripsinogen 3.00E+04 Trypsinogen-CMC 2.00E+04 BSA-CMC 1.00E+04 Prot mix- Hydrogel 0.00E+00 -1.00E+04 0.5 1 2 3 -2.00E+04 Concentration (μΜ) Figure 2.10 Calibration lines for protein mix. In Figure 2.10 the calibration lines for the protein mixture appear to be linear except for the one that corresponds to trypsinogen, for this reason they were not normalized. Based on the non-normalized calibration lines, CMC seems to be a better embedding medium than the PVP/CMC hydrogel for SM and DPPC, while the PVP/CMC appears to be a better choice for the peptide mix because it causes less suppression of the absolute signal of the protonated ions of interest. As far as it regards the protein mixture, both CMC and the hydrogel supress the signal of the proteins involved, consequently they were excluded from the main part of the project, where the rest of the compounds of interest were used in order to develop arrays while they were “diluted” and embedded in materials that mimic tissue behaviour. Eleni Skandalaki UvA-AMOLF Page 19 Because of the fact that the normalization of the calibration lines with TIC was overlooked and was performed after the main part of the project, the normalized calibration lines were not taken into account for the actual choice of the tissue mimetic material that was used until the end of the project. An alternative normalization technique Another normalization technique that was tried out was by using Cresyl Violet (CV) as an Internal Standard (IS). It was only employed for the compounds for SM, DPPC and the peptide mixture that were mixed with the CMC, after it was chosen as the appropriate tissue mimetic material for this project. Cresyl violet preparation For the preparation of a 10mg/ml CV solution, 100mg of CV power were weighed and diluted in 10ml CH3OH. Normalization of DPPC-CMC with CV The sample preparation and the profiling of the solutions of interest for the construction of the calibration lines similar to the one explained in par graph “sample preparation””, except for the fact that the same amount (10μl) of CV solution was added to the each concentration of DPPC with CMC. 5.00E+00 Absolute Intensity 4.00E+00 3.00E+00 2.00E+00 DPPC-CMC-CV 1.00E+00 0.00E+00 1 -1.00E+00 3 5 8 10 Concentration (μΜ) Figure 2.11 DPPC-CMC with CV Eleni Skandalaki UvA-AMOLF Page 20 Normalization of SM-CMC with CV 1.80E+01 1.60E+01 Absolute Intensity 1.40E+01 1.20E+01 1.00E+01 8.00E+00 SM-CMC-CV 6.00E+00 4.00E+00 2.00E+00 0.00E+00 -2.00E+00 1 2 5 7 10 Concentration (μΜ) Figure 2.12 SM-CMC with CV Normalization of Peptide mixture-CMC with CV 1.20E+02 Absolute Intensity 1.00E+02 8.00E+01 6.00E+01 4.00E+01 Substance P-CMC-CV 2.00E+01 Angiotensin II-CMC-CV 0.00E+00 -2.00E+01 -4.00E+01 0.5 1 2 3 Concentration (μΜ) Figure 2.13 Peptide mixture-CMC with CV Apart from the calibration line that refers to DPPC which is close to linear, from the rest of the normalized calibration lines it is indicated that adding CV was not a good normalization method which could explained due to the fact that the CV solution was prepared with CH3OH which diluted even further the concentration series, especially for DPPC and SM. Eleni Skandalaki UvA-AMOLF Page 21 Development of Test Arrays For the development of the test arrays the protocol that was developed, included the following steps. Firstly, the tissue mimetic materials that were chosen were frozen with different shaped stamps in them, in a suitable container. Successively, the stamps were removed and the cavities formed were filled with the analytes “diluted” in the tissue mimetic materials and the block created was frozen again. The newly formed solid block was dissected and after matrix application on the sections, they were loaded on the MALDI-TOF MS instrument in order to acquire spectra that were lastly converted into images. These steps are described in detail in the following paragraphs. Choice of tissue mimetic materials At first, it was thought that one tissue mimetic material would be used as an embedding media both for the formation of a solid frozen block and as a ‘’solvent’’ of the compounds that would be placed in the different shaped stamps of the block. Nevertheless, the choice of an embedding material was difficult, even though it was indicated based on the calibration lines that the CMC (2%) led to a lesser suppression of the compounds of interest signal, compared to CMC/PVP; it was important to test the actual behaviour of CMC/PVP too. As a result, CMC (2%) and CMC/PVP and gelatine (10%), were tested to check if they would freeze homogeneously, if they would be able to get dissected and stay intact in the process and also if they diffuse in such a degree that the signal of the lipids and peptides of interest would be suppressed. Moreover, after testing PVP/CMC, gelatine and CMC as embedding mediums in all combinations possible, for the formation of the block and the “dilution” of the compounds, gelatine proved to be the most suitable for the block formation and CMC for the dilutions. PVP/CMC has not been found useful because it does not freeze homogeneously thus the sectioning of its solid block, was impossible and also in the event of a relatively intact section, when it was desiccated, the hydrogel diffused and covered the compounds. To conclude, after several tests CMC (2%) proved suitable as a tissue mimetic material with which the lipids and peptides were mixed, and gelatine (10%) resulted in being the best material for the formation of a solid block that contained the compounds in the CMC. Choice and design of proper container The design and construction of a proper container for the freezing of a tissue mimetic material that could be able to hold stamps of different shapes, was a real challenge. At first it was considered a good idea to 3D- print a box; yet the plastic that is used in this kind of printers was not firm and it broke very easily after it froze. Another problem emerged when removing the frozen block from of the box and it resulted in the breaking of the plastic into small fibber-like pieces. Consequently, the plastic box was rejected and the next idea involved utilizing a metal one. The metal box was constructed in the workshop of AMOLF and the stamps on the cap of the box are also metallic and they have three different shapes; squares, circles and triangles. The stamps are connected to the cap by screws so they could Eleni Skandalaki UvA-AMOLF Page 22 be readily removed. In the end, this metal box worked perfectly for the formation of a block of embedding material; however the block that came out of the box was too large for the Cryostat, a device used for the dissection of the block. Thus, the metal cap with the shaped stamps was put into the embedding material in a plastic ice cube tray which resulted in being the most efficient and simple container. Figure 3.1 Metal stamps of different shapes Sectioning The next step of the developed protocol included the formation of a solid block of gelatine and its dissection. In particular, gelatine (10%) was poured into an ice cube tray with the metal stamps inside and the tray was placed in the freezer until a solid block was created. Then, the stamps were taken out and different shaped cavities were formed. Specifically, the circle- cavities were filled with different concentrations of SM-CMC, the triangle- cavities were filled with various concentrations of DPPC-CMC and in the squared- cavities the peptide mixture-CMC was deposited. Successively, the ice cube tray was placed again in the freezer and the result was a solid block of gelatine with the compounds of interest also frozen in the shapes of the stamps. Eleni Skandalaki UvA-AMOLF Page 23 Figure 3.2 Gelatin block with developed arrays Figure 3.3 Microtome Cryostat, Microm HM535 Moreover, after removing the gelatine block out of the ice cube tray, it was placed in a instrument called Microtome Cryostat, Microm HM535. With Microtome, the intact sections that were obtained were 10μm thick and the sample stage temperature was around -25OC. Figure 3.4 Gelatin section on a glass slide. Matrix application The application of CHCA had to be performed, after the section was placed in the dessicator for some time. For the homogeneous application of the matrix, a sprayer device, ImagePrep (Bruker, Bremen, Germany) was employed at first, but because of the fact that application Eleni Skandalaki UvA-AMOLF Page 24 was not always homogeneous and because it is a time consuming device, another device SunCollect (SunChrom, Friedrichsdorf, Germany) was utilized. With SunCollect, an aerosol of matrix is applied on the sample and a homogeneous layer is formed within 10 to 30min. In this device, the air pressure is applied is 2 bar, the distance between the sample and the spray nozzle is manually set at 25cm, the layers of matrix are also chosen by the operator and are between 10 and 20 and the flow of the matrix solution, starts from 5uL/min and builds up to 30uL/min. The matrix is deposited on the section and then the organic solvent of the CHCA solution in this case, extracts the molecules of the section and rapidly evaporates. After the evaporation, the formation of matrix crystals occurs from the organic acid of the solution. MSI analysis in positive mode and Data Analysis When the coating of the section with CHCA was homogeneous, afterwards it was loaded into the mass spectrometer, SYNAPT in order to perform the MSI analysis. During the imaging experiment, firstly the sample was scanned and an area of interest was selected. Then the experiment was set up in MALDI-TOF linear mode via the Masslynx Software. In addition, after choosing the area of interest of each sample, setting the resolution at 250x250μm and the laser frequency at 250-300Hz, the experiment is ready to start. Data Analysis- Biomap After each analysis, the raw data acquired which include numerous spectra, were converted into imaging data that are displayed with the Biomap Software (Novartis, Basel, Switzerland, www.maldi-msi.org). In Biomap, differences at specific values in an area of interest can be observed readily and the changing in certain pixels can be further analysed when combined with the observing of the related spectrum. Below in Figure 3.5 there are some images at the m/z of (M+H)+ of each compound of interest, in particular for SM: 703.5Da, for DPPC: 734.5Da, for Angiotensin: 1046.6Da and Substance p: 1347.50Da. The Figures 3.5 and 3.6 show two different sections and their difference lands on the sample preparation; in the Figure 3.5 the CHCA application was performed with the ImagePrep device while in the section depicted in Figure 3.6, CHCA was applied with the Suncollect device. Eleni Skandalaki UvA-AMOLF Page 25 SM (M+H) + m/z: 703.5 DPPC m/z (M+H) + 734.4 Angiotensin II (M+H) + m/z: 1046.6 Substance p (M+H) + m/z: 1347.5 Figure 3.5 Section 1 - CHCA applied with ImagePrep If the sample preparation and the MSI analysis had succeed ,in the m/z values that correspond to the (M+ H) +of each compound of interest, only one array should illuminate. In particular, at 703.5Da, only the array of the circles should be illuminated and because in each circle there was a different concentration of the SM-CMC mixture the intensity of the emitted light should be different. Likewise, at 734.5Da only the triangle array with the DPPCCMC should illuminate and at 1046.5 and 1347.6Da for angiotensin II and substance p –CMC the square-array should illuminate similarly. Eleni Skandalaki UvA-AMOLF Page 26 SM (M+H) + m/z: 703.5 DPPC m/z (M+H+Na) + 756.4 Angiotensin II (M+H) + m/z: 1046.6 Substance p (M+H) + m/z: 1347.5 Figure 3.6 Section 2 – CHCA applied with Suncollect In Figure 3.6, where a second section (section 2) is depicted, it can be observed that CHCA is more homogenously layered compared to the first section, however at the (M+ H) + m/z values of the compounds of interest, there is no difference at the light intensity of the arrays. The next step was to check other m/z values that correspond to (M+ H+ Na) +, (M+H+NH4) +, (M+ H+ K) +, (M+ H+ ACN+ Na) +of each compound. Unfortunately, this step did not bear fruits for all the compounds but only for the (M+H+ Na) + of DPPC-CMC as it shown at the Figure 3.6. MSI analysis in negative mode and Data analysis It was also decided to try a change in the protocol followed, and tune the SYNAPT in negative mode. Furthermore, in order to conduct a MALDI-TOF MS analysis in negative mode, the matrix choice should be different thus 9-Aminoacridine (9AA) was applied in a third section and the rest of the protocol remained the same to the one followed in the positive mode. After acquiring the raw data of the analysis were converted into Biomap data and led to the below images. Eleni Skandalaki UvA-AMOLF Page 27 SM (M-H) - m/z: 702.5 DPPC m/z (M-H) - 733.4 Angiotensin II (M-H) - m/z: 1045.6 Substance p (M-H) - m/z: 1346.5 Figure 3.7 Section 3. Biomap images- negative mode After examining the Biomap images at the m/z values corresponding to the (M-H) - , (M+ Na H) - , (M-TFA-H)-of each compound, it was shown that neither in the negative mode the developed arrays were able to deliver the desired results and show different light intensities at different m/z values. Principle Component Analysis (PCA) and Discriminant Analysis (DA) Principle Component Analysis was also conducted as a complimentary procedure to the data acquired from the MSI analysis. PCA is the foundation of multivariate data analysis and is an important statistical tool for researchers. (Wold, Esbensen & Geladi 1987). According to H. Abdi and L.J Williams (2010) “PCA is a multivariate technique that analyses a data table in which observations are described by several inter-correlated quantitative dependent variables”. With PCA, it is possible to acquire significant information that concerns the table of data, to convert them into orthogonal variables which are called principal components and present the correlation of the variables as points in histograms. Discriminant analysis (DA) is also used in statistics in order to classify samples of interests (Poulsen & French 2004). DA in combination with PCA can offer researchers useful information that could verify Eleni Skandalaki UvA-AMOLF Page 28 or disprove initial indications of experiments. In this project, PCA and DA were combined, but the information acquired was limited. PCA and DA analysis for section2- positive mode In order to start the PCA analysis, a conversion of the Biomap file of the section 2 into a MATLAB file has to be performed. Subsequently, the functions ‘’Peakpicking’’ and ‘’Regions of interest’’ (ROIs) in MATLAB were employed and each shape was considered a ROI. In particular, in the 1, 2, 3, 4 there was SM-CMC; where in 1, the highest concentration (10μM) of the mixture was contained. Likewise in 5, 6, 7 ROIs there was DPPC-CMC and in 8, 9, 10, 11 ROIs there was the peptide mixture. The combination of the ROIs of PCA and Discriminant Analysis (DA) let to Figure 3.8. 1 2 4 3 5 8 9 6 7 10 11 Figure 3.8 Image of ROIs When using the DA function, there seemed to be a classification that showed a correlation of the peptide mixture and the CHCA and a correlation between the DPPC, SM and the CMC. These correlations were deduced from the Figure 3.8 and its conversion into Figure 3.9. Eleni Skandalaki UvA-AMOLF Page 29 Peptides Lipids CHCA CMC Figure 3.9 CMC m/ z CHCA , Gelatin Figure 3.10 Based on the reference spectra of the gelatine and CMC with CHCA, it is known that most of the m/z values at the negative part of the histogram correspond to the gelatine and the CHCA and the majority of the values at the positive part occurred due to the CMC. Additionally, due to the PCA and DA, some histograms presented some indications of the existence of the SM-CMC solution in the array of circles where it was supposed to be in the Eleni Skandalaki UvA-AMOLF Page 30 second section. + (M+H+Na) m/ z Figure 3.11 Normalized Histogram In particular, as it is highlighted by the arrow in Figure 3.11, the peak at the m/z: 725.2Da could have occurred due to (M+ H+ Na) +. While in the non-normalized histogram, the signalto-noise ratio (S/n) was low, as a result no conclusions could be drawn except for when it is converted to another also non-normalized histogram-Figure 3.13, where it seems that the ROIs 1, 2, 3, 4 are separated; presumably based on the different concentrations of the SMCMC solutions in the developed arrays. In particular, the lowest concentration which was 2μM and the highest which was 10μM appeared to separate completely, while the concentrations 5,7μM seemed to overlap. Eleni Skandalaki UvA-AMOLF Page 31 m/z Figure 3.12 Non- normalized histogram- SM Figure 3.13 Non-normalized histogram2- SM PCA and DA for third section- negative mode Even though there were no certain conclusions from the PCA analysis for the second section which was previously described, due to the fact that there were some indications about the SM-CMC developed array, the same PCA and DA analysis took place for the third section for which the MSI analysis was in negative mode. Eleni Skandalaki UvA-AMOLF Page 32 m/ z Gelatin Figure 3.14 Non-normalized histogram- SM m/z :(M-H)- = 702.4 m/ z Figure 3.15 Normalized histogram- SM Eleni Skandalaki UvA-AMOLF Page 33 From the PCA and DA conducted for the SM-CMC array, in negative mode, the only indication of existence of SM was a small peak at the m/z: 702.4Da that could be assigned to the (M-H)-, but since there were no further indications, no other histograms are to be shown. Therefore, the peak at 702.4Da could be coincidental and belong to gelatine and not be an SM peak. Profiling of DPPC and SM arrays with Secondary Ionization Mass Spectrometry (SIMS) As it is already mentioned in the introduction chapter, SIMS was not repeatedly employed in this project. This was the case because of the fact that the samples (sections) were too large for this technique; despite that fact SIMS was used once in order to conduct a profiling of the arrays developed for DPPC and SM, but not for the peptides and the protein mixtures as the detection range of SIMS is not suitable for such compounds. For the profiling, the only condition was that the sample should be placed on a conductive Indium Tin Oxide (ITO) coated slide and then the slide could be placed into the TRIFT II high resolution Secondary Ion Mass Spectrometry TOF-MS DPPC array Figure 3.16 Eleni Skandalaki UvA-AMOLF Page 34 In the Figure 3.16 there is a combined spectrum and each spectrum corresponds to a different DPPC-CMC concentration of the developed arrays, and the peak observed at m/z: 184Da could be assigned to the phosphorylcholine head group. The fact that this peak is so small and the fact its size is almost the same to every spectrum doesn’t seem to shown any correlation with the different concentrations. If the DPPC was actually in this area, the peak would be much higher. SM array Figure 3.17 The SM has the same phosphorylcholine head group as DPPC, therefore the peak at m/z 184 could also be an indication of the existence of SM in the developed array. Despite the observed peak at m/z 184, no certain conclusions could be drawn either for the DPPC or for the SM, since no other adduct ions of them were observed. Last but not least, due to a high resolution camera of SIMS, it became clear that even though the sections from the gelatin block with the arrays of compounds seemed to be consistent, the arrays were not homogeneous. This also led to the realization that methanol used for the initial dilution of SM and DPPC, was responsible for the non-homogeneous freezing of the arrays. Eleni Skandalaki UvA-AMOLF Page 35 Spatial resolution experiments One of the targets of MSI is the visualization of spatial organization of biological compounds (Jungmann & Heeren 2012) such as the lipids, peptides and proteins employed in this project. As an extension of that, the last part of this project included a series of experiments regarding the spatial resolution imaging, based on which, conclusions about the sensitivity of MSI techniques such as MALDI TOF-MS and the instruments employed, can be drawn. Sample preparation The sample preparation included similar steps for the lipids and peptides used throughout this project. First of all, the highest concentrations of the solutions for each compound of interest were chosen, hence 10μΜ solution of DPPC and SM, and 5μΜ solution of peptide mixture was used. Each compound was mixed in one-to-one ratio with a suitable matrix, which was CHCA for the lipids and peptides and SA for the protein mixture, and they were sprayed with Suncollect (SunChrom, Friedrichsdorf, Germany) or ImagePrep (Bruker, Bremen, Germany) (whichever instrument was working) through a plastic mold. With this process, each solution was sprayed on the glass and the shapes of the cavities of the plastic mold were formed on the slide. The plastic mold was constructed by a 3D printer and it had four arrays of different shapes; circles, triangles, squares and rectangles of different sizes. . Figure 4.1 3D- printed plastic mold The last step of the sample preparation was to load the glass slides with the lipids and the peptide mixture into the SYNAPT and set up the experiments in order to conduct the MSI analysis. The settings of each spatial resolution experiment, included laser frequency of 250300Hz, and the resolution was set at 200μm x 200μm. Eleni Skandalaki UvA-AMOLF Page 36 Acquired Data from MSI analysis of DPPC The acquired data from each experiment were converted into Biomap files and then they were processed further with a Biomap function with which Regions of Interest (ROIs) were selected in order to determine the exact areas with the DPPC-CHCA in mm2. Successively, a setting in Biomap called Statistics calculated the mean intensity of the ROIs, at m/z value of 734.5Da which is the (M+H) + of DPPC, as it is already mentioned. The results of this process let to the following images and plots. Figure 4.2 DPPC-CHCA Biomap image . 300.00 Mean Intensity 250.00 200.00 150.00 Mean Intensity 100.00 Normalized Intensity 50.00 1.000 1.360 1.480 1.760 1.840 1.920 2.000 2.400 1.840 2.960 2.760 3.240 3.320 3.920 4.200 4.680 4.760 0.00 Area (mm2) Figure 4.3 Eleni Skandalaki UvA-AMOLF Page 37 In Figure 4.3, the red line refers to the mean intensity in relation to the regions of interest in mm2 and although it was expected for the trend of the line to be straight or constant, this was not the case. This could be explained because of the non-homogenous spraying of the DPPC-CHCA, or because of the manual drawing of the regions of interest. Subsequently, the data were normalized by dividing the mean intensity of each square with the area that corresponds to it. The normalization process is indicated with the blue line in Figure 4.3 and it is possible to observe the better linearity of the line. Acquired Data from MSI analysis of SM The data were acquired similarly to DPPC and processed correspondingly with Biomap software at the m/z: (M+H) +is 703.5Da for SM and the following image and plot were the results of the MSI analysis. Figure 4.3 SM-CHCA Biomap Image Eleni Skandalaki UvA-AMOLF Page 38 Mean Intensity 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 Mean Intensity Normalized Intensity 1.8 2.08 2.12 1.6 1.2 1.28 1.32 1.2 0.88 0.84 1.32 Area ( mm2) Figure 4.4 In Figure 4.4, the red line refers to the mean intensity in relation to the ROIs in mm2 and even though it was expected the trend of the line to be straight or constant again, this is not the case. This could occur due to similar errors to the DPPC in the spraying step or again due to the manual drawing of the ROIs. Successively normalization was performed similarly to the normalization of DPPC-CHCA. The normalization is depicted with the blue line in Figure 4.4 but there was not any significant improvement in the linearity of the trend. Acquired Data from MSI analysis of Peptide Mixture -Substance p After the MSI analysis, the data processing was similar to the one for DPPC and SM. Moreover, the only difference was the m/z ratio that corresponds to the protonated ions of Substance p which is (M+H) +: 1347.5Da. Figure 4.4 Substance P- CHCA Biomap image Eleni Skandalaki UvA-AMOLF Page 39 35 Mean Intensity 30 25 20 15 Mean Intensity Normalized Intensity 10 5 1.28 3.24 3.52 2.84 4.8 3.96 4.04 4.68 5.32 5.64 5.16 5.32 6.4 6.48 8.16 8.44 8.4 0 Area (mm2) Figure 4.5 In Figure 4.5, the blue line refers to the mean intensity in relation to the ROIs in mm2 and despite the fact that we would expect the trend of the line to be straight or constant, this did not occur. This could be explained similarly to the already mentioned DPPC and SM-CHCA due to errors in spraying of peptide mixture-CHCA onto the glass slide, because of the fact that the mixture kept clogging the spraying head of the ImagePrep, or due to the manual drawing of the ROIs. Furthermore, the following step was to perform normalization of the results similarly to the normalization of DPPC-CHCA. The normalization is depicted with the red line in Figure 4.5 and an improvement of the line linearity is observed. Acquired Data from MSI analysis for Angiotensin II Successively, when acquiring the data from the MSI analysis, the following step was similar to the step followed for Substance p. Moreover, the m/z chosen in Biomap corresponds to (M+H) +: 1046.5Da and due to the afore- mentioned process, the following image and plot were constructed. Eleni Skandalaki UvA-AMOLF Page 40 Figure 4.6 Substance p-CHCA Biomap image 50 45 Mean Intensity 40 35 30 25 20 Mean Intensity 15 Normalized Intensity 10 5 1.44 3.12 3.04 4.12 4.64 4.2 5.16 4.8 5.32 6.12 6.68 6.56 7.08 7.36 8.16 8.36 8.72 0 Area (mm2) Figure 4.7 In the Figure 4.7, the red line indicates the mean intensity in relation to the ROIs and the trend of the lines are not again what was expected. The reasons which could be responsible for that are already mentioned in the other peptide used in the peptide mixture. In order to check if the results could be optimized, they were normalized and in this case, the normalization was better than for any other compound so far, as it is shown with the blue line of the plot, which is very close to linear. Eleni Skandalaki UvA-AMOLF Page 41 Protein mixture- Sample preparation As it is already mentioned, the sample preparation of the protein mixture was different to the other compounds to some extent. First of all, the protein mixture was not mixed with the matrix and sprayed with ImagePrep because this mixture would clog the spray head of the device; thus the protein mixture was firstly sprayed through the plastic mold and then the matrix, SA was sprayed over it, onto a conductive ITO slide. Furthermore, due to the fact that the deposit of the SA was not enough for an MSI analysis and due to lack of time, an extra small amount (1μΜ) of SA was deposited on each sprayed on area of the ITO slide with protein mixture; this is why some of the shapes on the image below were irregular. The MSI analysis was conducted with the Ultraflex III, Brucker as it is more appropriate for such large compounds as the proteins used, but the same resolution with the rest of the compounds was set at 200μm x200μm and a linear positive mode was also set. Acquired Data from MSI analysis of BSA After acquiring the data from the MSI analysis of the protein mixture, the following steps included converting the data into Biomap data, processing them further in Biomap by drawing the ROIs and calculating the mean intensity of BSA with SA in the ROIs at m/z (M+H)+ : 66.294 Da. Figure 4.8 BSA Biomap image Eleni Skandalaki UvA-AMOLF Page 42 5 4.5 4 Mean Intensity 3.5 3 2.5 Mean Intensity 2 Normalized Intensity 1.5 1 0.5 0 Area (mm2) Figure 4.9 In Figure 4.9 the blue line refers to the mean intensity in relation to the ROIs. Due to the unorthodox sample preparation and the manual drawing of the ROIs, it was expected that the blue line would not be linear or constant; hence the results should be normalized. Nevertheless, even with the normalization, the trend of the line did not become linear but it only got smoothened in some areas. Acquired Data from MSI analysis of Trypsinogen The process of acquiring and processing further the data from the MSI analysis, was performed similarly to BSA, with the only difference lying in the m/z that corresponds to the (M+H) + which is 22.223 Da. Based on this process, the following image and plot were constructed. Eleni Skandalaki UvA-AMOLF Page 43 Figure 4.10 Trypsinogen Biomap image 6 Mean Intensity 5 4 3 Mean Intensity 2 Normalized Intensity 1 0 Area (mm2) Figure 4.11 In the Figure 4.11, the blue line shows to the mean intensity in respect to the ROIs. Because of the unorthodox sample preparation and the manual drawing of the ROIs, it was expected that the blue line would not be linear or constant; consequently, the results should be normalized. Despite the normalization conducted, the trend of the line did not become linear as it can be observed with the red calibration line. Eleni Skandalaki UvA-AMOLF Page 44 Results, discussion and suggestions There were several conclusions drawn from this research project. First of all, the initial idea to employ a non-commercial hydrogel prepared from PVP/CMC as a tissue mimetic material, proved to be a non- efficient choice for this project as it suppresses the absolute intensity of the intact, protonated and other adduct ions of DCCP, SM and peptide mixture. Moreover it completely suppresses the signals of the protein mixture. PVP/CMC was also considered unsuitable for this project, due to its difficulty to be sectioned. Taking the above into account and after many sets of trial experiments, CMC and gelatin proved to be better tissue mimetic materials. Even though they met our expectations, the results about the developed arrays from the MSI were not satisfactory, due to the fact that the developed arrays were not distinguished just by observing an image created at specific m/z values. Nearly at the end of the project, it was realized that the developed arrays did not behave as expected, because of the fact that the DPPC and SM were initially dissolved in methanol and then for the creation of the concentration series- arrays they were dissolved in CMC. In particular, the high intensity of water in CMC prevented its mixing with methanol, thus the lipids-CMC solutions had two phases and even though they seemed homogeneous after vortex, when the solutions were placed into the cavities of the gelatine to form a frozen solid bock, the freezing was not homogeneous either. In addition, the fact that the freezing point of MeOH is around -100oC, made its freezing impossible. Figure 5.1 Two phases in DPPC and SM with CMC solutions. All the above led to the conclusion that this project could not be employed as a quantification method for MSI techniques and instruments involved in it. As far as it regards the spatial resolution experiments, from the MSI analysis and the plots constructed for DPPC, SM, peptide and protein mixture mixed with CHCA and SA respectively, it became clear that the Suncollect is a better device than ImagePrep for the sample preparation in such experiments. It was also indicated that the correlation between the intensity of the solutions at the (M+H) + values of each compound of interest and the ROIs was not completely linear but it could probably be, if the human error was reduced and if the Suncollect was employed for all the experiments. Last but not least because of the Eleni Skandalaki UvA-AMOLF Page 45 spatial resolution experiments, it was indicated that the MALDI-TOF instruments used in the BIMS laboratory are of high sensitivity and their limit of detection is also high. Suggestions In the event of a repetition of this project a suggestion would be to employ a commercial hydrogel as a tissue mimetic material. Moreover for the plot of the calibration lines of the biological compounds of interest it would be more accurate to use more adduct ions and not just their protonated ions. As far as it concerns a normalization method for the calibration lines, a good idea would be to employ deuterium (2H) as an internal standard. On the other hand, if the biological compounds chosen for a future project are diluted in a solvent that actually mixes with the tissue mimetic material, then maybe the normalization methods would not be necessary. Eleni Skandalaki UvA-AMOLF Page 46 References Abdi & Williams 2010 Hervé Abdi & Lynne J. Williams, 'Principal component analysis', Wiley Interdisciplinary Reviews: Computational Statistics 2010-4, p. 433-459. Attwood, Choi & Leonenko 2013 Simon J. Attwood, Youngjik Choi & Zoya Leonenko, 'Preparation of DOPC and DPPC Supported Planar Lipid Bilayers for Atomic Force Microscopy and Atomic Force Spectroscopy', International Journal of Molecular Sciences 2013-2, p. 3514-3539. Caldwell & Caprioli 2005 Robert L. Caldwell & Richard M. Caprioli, 'Tissue profiling by mass spectrometry A review of methodology and applications', Molecular & Cellular Proteomics 2005-4, p. 394-401. Caprioli, Farmer & Gile 1997 Richard M. Caprioli, Terry B. Farmer & Jocelyn Gile, 'Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS', Analytical Chemistry 1997-23, p. 4751-4760. Chughtai 2012 K. Chughtai, 'Multimodal Imaging of Hypoxia in Breast Cancer', 2012. Cohen & Chait 1996 Steven L. Cohen & Brian T. Chait, 'Influence of matrix solution conditions on the MALDI-MS analysis of peptides and proteins', Analytical Chemistry 1996-1, p. 31-37. Games 1978 David E. Games, 'Soft ionization mass spectral methods for lipid analysis', Chemistry and Physics of Lipids 1978-4, p. 389-402. Gode & Volmer 2013 David Gode & Dietrich A. Volmer, 'Lipid imaging by mass spectrometry–a review', Analyst 2013-5, p. 1289-1315 Hillenkamp & Karas 2007 Franz Hillenkamp & Michael Karas, 'The MALDI process and method', Maldi Ms 2007. Hsu & Turk 2000 Fong-Fu Hsu & John Turk, 'Structural determination of sphingomyelin by tandem mass spectrometry with electrospray ionization', Journal of the American Society for Mass Spectrometry 2000-5, p. 437-449. Hoffmann & Stroobant 2007 Edmond Hoffman &Vincent Stroobant, ‘Mass Spectrometry: Principles and applications’, Third edition, Wiley; 2007 Eleni Skandalaki UvA-AMOLF Page 47 Jiang, Yubao, Wang, Xuejiang, Zhang, Wen, Jiqiu, Gong & Mei 2008 Liuyun Jiang, Li, Yubao, Wang, Xuejiang, Zhang, Li Wen, Jiqiu, Gong, Mei., 'Preparation and properties of nano-hydroxyapatite/chitosan/carboxymethyl cellulose composite scaffold', Carbohydrate Polymers 2008-3, p. 680-684. Jurinke, Oeth & van den Boom 2004 Christian Jurinke, Paul Oeth & Dirk van den Boom, 'MALDI-TOF mass spectrometry', Molecular Biotechnology 2004-2, p. 147-163. Jungmann & Heeren 2012 Julia H. Jungmann & Ron Heeren, 'Emerging technologies in mass spectrometry imaging', Journal of Proteomics 2012-16, p. 5077-5092. Karlsson, Michélsen & Odham 1998 Anders Å. Karlsson, Peter Michélsen & Göran Odham, 'Molecular species of sphingomyelin: determination by high‐performance liquid chromatography/mass spectrometry with electrospray and high‐performance liquid chromatography/tandem mass spectrometry with atmospheric pressure chemical ionization', Journal of Mass Spectrometry 1998-12, p. 11921198. Luft 1973 John H. Luft, 'Embedding media—old and new', in: Advanced techniques in biological electron microscopy, Springer 1973, p. 1-34. McDonnell & Heeren 2007 Liam A. McDonnell & Ron Heeren, 'Imaging mass spectrometry', Mass Spectrometry Reviews 2007-4, p. 606-643. Murray, Boyd, Eberlin, Langley, Li & Naito 2013 Kermit K. Murray, Robert K. Boyd, Marcos N. Eberlin, G. John Langley, Liang Li and Yasuhide Naito, 'Definitions of terms relating to mass spectrometry (IUPAC Recommendations 2013)', Pure and Applied Chemistry 2013. Poulsen & French 2004 John Poulsen & Aaron French, 'Discriminant function analysis (DA)', San Francisco State University, Available at: http://online.Sfsu.Edu/efc/classes/biol710/discrim/discrim.Pdf#Search¼ 2004. Rosiak, Ulański , Pajewski , Yoshii & Makuuchi 2002 JM Rosiak, P. Ulański , L.A. Pajewski, F. Yoshii , K. Makuuchi, 'Radiation formation of hydrogels for biomedical applications', Radiation Physics and Chemistry, 1995-2 p. 161-168. Tabata & Ikada 1998 Yasuhiko Tabata & Yoshito Ikada, 'Protein release from gelatin matrices', Advanced Drug Delivery Reviews 1998-3, p. 287-301. Eleni Skandalaki UvA-AMOLF Page 48 Wang , Xu, Hu, Zhai , Peng , Nho , Li & Wei 2007 Min Wang, Ling Xu, Hui Hu, Maolin Zhai, Jing Peng, Youngchang Nho, Jiuqiang Li & Genshuan Wei, 'Radiation synthesis of PVP/CMC hydrogels as wound dressing', Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2007-1, p. 385-389. Wold, Esbensen & Geladi 1987 Svante Wold, Kim Esbensen & Paul Geladi, 'Principal component analysis', Chemometrics Intelligent Laboratory Systems 1987-1, p. 37-52. Yang, Leong, Zhaohui, Chua & Chee-Kai 2001 Shoufeng Yang, Kah-Fai Leong, Zhaoui Du & Chee-Kai Chua, 'The design of scaffolds for use in tissue engineering. Part I. Traditional factors', Tissue Engineering 2001-6, p. 679-689. Eleni Skandalaki UvA-AMOLF Page 49 Appendix The combined spectra base on which the calibration lines where constructed DPPC Figure 6.1 Combined spectrum of DPPC Figure 6.2 Combined spectrum of DPPC- PVP/CMC Eleni Skandalaki UvA-AMOLF Page 50 SM Figure 6.3 Combined spectrum of SM Figure 6.4 Combined spectrum of SM-CMC Eleni Skandalaki UvA-AMOLF Page 51 Figure 6.5 Combined spectrum of SM-PVP/CMC Peptide mixture Figure 6.6 Combined spectrum of Peptide mixture Eleni Skandalaki UvA-AMOLF Page 52 Figure 6.7 Combined spectrum of Peptide mixture-CMC Figure 6.8 Combined spectrum of Peptide mixture- PVP/CMC Eleni Skandalaki UvA-AMOLF Page 53 Intens. [a.u.] Protein Mixture x104 6 4 2 0 30000 40000 50000 60000 70000 80000 90000 100000 m /z Intens. [a.u.] Figure 6.10 Combined spectrum of Protein Mixture 2000 1500 1000 500 30000 40000 50000 60000 70000 80000 90000 100000 m /z Figure 6.11 Combined spectrum of Protein mixture- CMC Eleni Skandalaki UvA-AMOLF Page 54 Intens. [a.u.] 175 150 125 100 75 50 20000 25000 30000 35000 40000 45000 50000 55000 60000 65000 70000 m /z Figure 6.12 Combined spectrum of Protein mixture- PVP/CMC Eleni Skandalaki UvA-AMOLF Page 55