Dimensionality reduction in the analysis of human
advertisement

Dimensionality Reduction in the
Analysis of Human Genetics Data
Petros Drineas
Rensselaer Polytechnic Institute
Computer Science Department
To access my web page:
drineas
Human genetic history
Much of the biological and evolutionary
history of our species is written in our
DNA sequences.
Population genetics can help translate
that historical message.
The genetic variation among humans is a
small portion of the human genome.
All humans are almost than 99.9% identical.
Our objective
Fact:
Dimensionality Reduction techniques (such as Principal Components Analysis
– PCA) separate different populations and result to “plots” that correlate
well with geography.
Our objective
Fact:
Dimensionality Reduction techniques (such as Principal Components Analysis
– PCA) separate different populations and result to “plots” that correlate
well with geography.
Our goal:
Based on this observation, we seek unsupervised, efficient algorithms for
the selection of a small set of genetic markers that can be used to
capture population structure, and
predict individual ancestry.
The math behind…
To this end, we employ matrix algorithms and matrix decompositions such as
the Singular Value Decomposition (SVD), and
the CX decomposition.
We provide novel, unsupervised algorithms for selecting ancestry informative
markers.
Overview
• Background
• The Singular Value Decomposition (SVD)
• The CX decomposition
• Removing redundant markers (Column Subset Selection Problem)
• Selecting Ancestry Informative Markers
A worldwide set of populations
Admixed European-American populations
The POPulation REference Sample (POPRES)
Single Nucleotide Polymorphisms (SNPs)
Single Nucleotide Polymorphisms: the most common type of genetic variation in the
genome across different individuals.
They are known locations at the human genome where two alternate nucleotide bases
(alleles) are observed (out of A, C, G, T).
SNPs
individuals
… AG CT GT GG CT CC CC CC CC AG AG AG AG AG AA CT AA GG GG CC GG AG CG AC CC AA CC AA GG TT AG CT CG CG CG AT CT CT AG CT AG GG GT GA AG …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AG AA CT AA GG GG CC GG AA GG AA CC AA CC AA GG TT AA TT GG GG GG TT TT CC GG TT GG GG TT GG AA …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AA AG CT AA GG GG CC AG AG CG AC CC AA CC AA GG TT AG CT CG CG CG AT CT CT AG CT AG GG GT GA AG …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AG AA CC GG AA CC CC AG GG CC AC CC AA CG AA GG TT AG CT CG CG CG AT CT CT AG CT AG GT GT GA AG …
… GG TT TT GG TT CC CC CC CC GG AA GG GG GG AA CT AA GG GG CT GG AA CC AC CG AA CC AA GG TT GG CC CG CG CG AT CT CT AG CT AG GG TT GG AA …
… GG TT TT GG TT CC CC CG CC AG AG AG AG AG AA CT AA GG GG CT GG AG CC CC CG AA CC AA GT TT AG CT CG CG CG AT CT CT AG CT AG GG TT GG AA …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AG AA TT AA GG GG CC AG AG CG AA CC AA CG AA GG TT AA TT GG GG GG TT TT CC GG TT GG GT TT GG AA …
There are ~10 million SNPs in the human genome, so this matrix could have ~10 million columns.
Our data as a matrix
… AG CT GT GG CT CC CC CC CC AG AG AG AG AG AA CT AA GG GG CC GG AG CG AC CC AA CC AA GG TT AG CT CG CG CG AT CT CT AG CT AG GG GT GA AG …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AG AA CT AA GG GG CC GG AA GG AA CC AA CC AA GG TT AA TT GG GG GG TT TT CC GG TT GG GG TT GG AA …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AA AG CT AA GG GG CC AG AG CG AC CC AA CC AA GG TT AG CT CG CG CG AT CT CT AG CT AG GG GT GA AG …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AG AA CC GG AA CC CC AG GG CC AC CC AA CG AA GG TT AG CT CG CG CG AT CT CT AG CT AG GT GT GA AG …
… GG TT TT GG TT CC CC CC CC GG AA GG GG GG AA CT AA GG GG CT GG AA CC AC CG AA CC AA GG TT GG CC CG CG CG AT CT CT AG CT AG GG TT GG AA …
… GG TT TT GG TT CC CC CG CC AG AG AG AG AG AA CT AA GG GG CT GG AG CC CC CG AA CC AA GT TT AG CT CG CG CG AT CT CT AG CT AG GG TT GG AA …
… GG TT TT GG TT CC CC CC CC GG AA AG AG AG AA TT AA GG GG CC AG AG CG AA CC AA CG AA GG TT AA TT GG GG GG TT TT CC GG TT GG GT TT GG AA …
SNPs
Individuals
Individuals
SNPs
0 0 0 1 0 -1 1 1 1 0 0 0 0 0 1 0 1 -1 -1 1 -1 0 0 0 1 1 1 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 0 1 0 1 -1 -1 1 -1 1 -1 1 1 1 1 1 -1 -1 1 -1 -1 -1 -1 -1 -1 1 -1 -1 -1 1 -1 -1 1
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 1 0 0 1 -1 -1 1 0 0 0 0 1 1 1 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 0 1 1 -1 1 1 1 0 -1 1 0 1 1 0 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
-1 -1 -1 1 -1 -1 1 1 1 -1 1 -1 -1 -1 1 0 1 -1 -1 0 -1 1 1 0 0 1 1 1 -1 -1 -1 1 0 0 0 0 0 0 0 0 0 1 -1 -1 1
-1 -1 -1 1 -1 -1 1 0 1 0 0 0 0 0 1 0 1 -1 -1 0 -1 0 1 -1 0 1 1 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 1 -1 -1 1
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 0 1 -1 1 -1 -1 1 0 0 0 1 1 1 0 1 -1 -1 1 -1 -1 -1 -1 -1 -1 1 -1 -1 -1 0 -1 -1 1
example: ΑΑ = 1
ΑG = 0
GG = -1
Forging population variation & structure
Genetic diversity and population (sub)structure is caused by…
• Mutation
Mutations are changes to the base pair sequence of the DNA.
• Natural selection
Genotypes that correspond to favorable traits and are heritable become more common in
successive generations of a population of reproducing organisms.
Mutations increase genetic diversity.
Under natural selection, beneficial mutations increase in frequency, and vice versa.
Forging population variation & structure
• Genetic drift
Sampling effects on evolution:
Example: say that the RAF of a SNP in a small population is p.
The offspring generation would (in expectation) have a RAF of p as well for the same SNP.
In reality, it will have a RAF of p’ (a drifted frequency) …
• Gene flow
Transfer of alleles between populations (immigration)
• Non-random mating
Reduces interaction between (sub)populations.
• Other demographic events
Dimensionality reduction
Individuals
SNPs
0 0 0 1 0 -1 1 1 1 0 0 0 0 0 1 0 1 -1 -1 1 -1 0 0 0 1 1 1 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 0 1 0 1 -1 -1 1 -1 1 -1 1 1 1 1 1 -1 -1 1 -1 -1 -1 -1 -1 -1 1 -1 -1 -1 1 -1 -1 1
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 1 0 0 1 -1 -1 1 0 0 0 0 1 1 1 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 0 1 1 -1 1 1 1 0 -1 1 0 1 1 0 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
-1 -1 -1 1 -1 -1 1 1 1 -1 1 -1 -1 -1 1 0 1 -1 -1 0 -1 1 1 0 0 1 1 1 -1 -1 -1 1 0 0 0 0 0 0 0 0 0 1 -1 -1 1
-1 -1 -1 1 -1 -1 1 0 1 0 0 0 0 0 1 0 1 -1 -1 0 -1 0 1 -1 0 1 1 1 -1 -1 0 0 0 0 0 0 0 0 0 0 0 1 -1 -1 1
-1 -1 -1 1 -1 -1 1 1 1 -1 1 0 0 0 1 -1 1 -1 -1 1 0 0 0 1 1 1 0 1 -1 -1 1 -1 -1 -1 -1 -1 -1 1 -1 -1 -1 0 -1 -1 1
Mutation/natural selection/population history, etc. result to significant structure in the data.
This structure can be extracted via dimensionality reduction techniques.
In most cases, unsupervised techniques suffice!
In most cases, linear dimensionality reduction techniques (PCA) suffice!
Why study population structure?
Mapping causative genes for common complex disorders
(e.g. diabetes, heart conditions, obesity, etc.)
History of human populations
Genealogy
Forensics
Conservation genetics
Population stratification
Definition:
Correlation between subpopulations in the case/control samples and the phenotype
under investigation.
Effects:
Confounds the study, typically leading to false positive correlations.
Solution:
The problem can be addressed either by careful sample collection or by statistical
post-processing of the results (Price et al (2006) Nat Genet).
Population stratification (cont’d)
Population 1
AA
AC
Cases
Population 2
CC
Example of the confounding
effects of population
stratification in an
association study.
Marchini et al (2004) Nat Genet
Controls
Recall our objective…
Develop unsupervised, efficient algorithms for the selection of a small set of SNPs
that can be used to
capture population structure, and
predict individual ancestry.
Why? cost efficiency.
Let’s discuss (briefly) prior work …
Inferring population structure
Africa
Europe
Middle East Central Asia
Oceania
East Asia
America
377 STRPs, Rosenberg et al (2004) Science
Examples of available algorithms/software packages:
• STRUCTURE
Pritchard et al (2000) Genetics
• FRAPPE
Li et al (2008) Science
Selecting ancestry informative markers
Existing methods (Fst, Informativeness, δ)
Rosenberg et al (2003) Am J Hum Genet
Allele frequency based.
Require prior knowledge of individual ancestry (supervised).
Such knowledge may not be available.
(e.g., populations of complex ancestry, large multi-centered studies of anonymous samples, etc.)
Unsupervised “feature selection” techniques are often preferable because
they tend to not overfit the data.
The Singular Value Decomposition (SVD)
Matrix rows: points (vectors) in a Euclidean space,
e.g., given 2 objects (x & d), each described with
respect to two features, we get a 2-by-2 matrix.
feature 2
Let A be a matrix with m rows (one for each subject)
and n columns (one for each SNP).
Object d
(d,x)
Two objects are “close” if the angle between their
corresponding vectors is small.
Object x
feature 1
SVD, intuition
Let the blue circles represent m
data points in a 2-D Euclidean space.
5
2nd (right)
singular vector
Then, the SVD of the m-by-2 matrix
of the data will return …
4
1st (right) singular vector:
direction of maximal variance,
3
2nd (right) singular vector:
1st (right)
singular vector
2
4.0
4.5
5.0
5.5
6.0
direction of maximal variance, after
removing the projection of the data
along the first singular vector.
Singular values
5
2
2nd (right)
singular vector
1: measures how much of the data variance
is explained by the first singular vector.
4
2: measures how much of the data variance
is explained by the second singular vector.
3
1
1st (right)
singular vector
2
4.0
4.5
5.0
5.5
6.0
SVD: formal definition
0
0
: rank of A
U (V): orthogonal matrix containing the left (right) singular vectors of A.
S: diagonal matrix containing the singular values of A.
Let 1 ¸ 2 ¸ … ¸ be the entries of S.
Exact computation of the SVD takes O(min{mn2 , m2n}) time.
The top k left/right singular vectors/values can be computed faster using
Lanczos/Arnoldi methods.
Rank-k approximations via the SVD
A
=
U
S
VT
features
objects
noise
=
significant
sig.
significant
noise
noise
Rank-k approximations (Ak)
Uk (Vk): orthogonal matrix containing the top k left (right) singular vectors of A.
Sk: diagonal matrix containing the top k singular values of A.
Principal Components Analysis (PCA) essentially amounts
to the computation of the Singular Value Decomposition
(SVD) of a covariance matrix.
SVD is the algorithmic tool behind MultiDimensional
Scaling (MDS) and Factor Analysis.
feature 2
PCA and SVD
Object d
(d,x)
Object x
feature 1
The data…
European
Americans
South Altaians
-
Spanish
Chinese
African
Americans
Japanese
Puerto Rico
Mende
Nahua
Mbuti
Mala
Burunge
Quechua
Africa
Europe
E Asia
America
274 individuals from 12 populations genotyped on ~10,000 SNPs
Shriver et al (2005) Human Genomics
A
America
Africa
Asia
Europe
B
Paschou et al (2007) PLoS Genetics
A
America
Africa
Asia
Europe
B
Not altogether satisfactory: the singular vectors are linear combinations of all
SNPs, and – of course – can not be assayed!
Can we find actual SNPs that capture the information in the singular vectors?
(E.g., spanning the same subspace …)
Paschou et al (2007) PLoS Genetics
SVD decomposes a matrix as…
The SVD has strong
optimality properties.
Top k left singular vectors
X = UkTA = Sk VkT
The columns of Uk are linear combinations of up to all columns of A.
CX decomposition
Carefully
chosen X
Goal: make (some norm) of A-CX small.
c columns of A
Why?
If A is an subject-SNP matrix, then selecting representative columns is
equivalent to selecting representative SNPs to capture the same structure
as the top eigenSNPs.
We want c as small as possible!
CX decomposition
Carefully
chosen X
Goal: make (some norm) of A-CX small.
c columns of A
Theory: for any matrix A, we can find C such that
is almost equal to the norm of A-Ak with c ≈ k.
CX decomposition
c columns of A
Easy to prove that optimal X = C+A. (C+ is the Moore-Penrose pseudoinverse of C.)
Thus, the challenging part is to find good columns (SNPs) of A to include in C.
From a mathematical perspective, this is a hard combinatorial problem.
A theorem
Drineas et al (2008) SIAM J Mat Anal Appl
Given an m-by-n matrix A, there exists an algorithm that picks, in expectation,
at most O( k log k / 2 ) columns of A
runs in O(mn2) time, and with probability at least 1-10-20
The CX algorithm
Input:
m-by-n matrix A, target rank k, number of columns c
Output:
C, the matrix consisting of the selected columns
CX algorithm
• Compute probabilities pj summing to 1
• For each j = 1,2,…,n, pick the j-th column of A with probability min{1,cpj}
• Let C be the matrix consisting of the chosen columns
(C has – in expectation – at most c columns)
Subspace sampling (Frobenius norm)
Vk: orthogonal matrix containing the top
k right singular vectors of A.
S k: diagonal matrix containing the top k
singular values of A.
Remark: The rows of VkT are orthonormal vectors, but its columns (VkT)(i) are not.
Subspace sampling (Frobenius norm)
Vk: orthogonal matrix containing the top
k right singular vectors of A.
S k: diagonal matrix containing the top k
singular values of A.
Remark: The rows of VkT are orthonormal vectors, but its columns (VkT)(i) are not.
Subspace sampling in O(mn2) time
Leverage scores
(many references in the
statistics community)
Normalization s.t. the
pj sum up to 1
Deterministic variant of CX
Input:
m-by-n matrix A,
integer k, and
c (number of SNPs to pick)
Output:
the selected SNPs
CX algorithm
• Compute the scores pj
• Pick the columns (SNPs) corresponding to the top c scores.
Paschou et al (2007) PLoS Genetics
Mahoney and Drineas (2009) PNAS
Deterministic variant of CX (cont’d)
Input:
m-by-n matrix A,
integer k, and
Paschou et al (2007) PLoS Genetics
Mahoney and Drineas (2009) PNAS
c (number of SNPs to pick)
Output:
the selected SNPs: we will call them PCA Informative Markers or PCAIMs
CX algorithm
• Compute the scores pj
• Pick the columns (SNPs) corresponding to the top c scores.
In order to estimate k for SNP data, we developed a permutation-based test to determine
whether a certain principal component is significant or not.
(A similar test was presented in Patterson et al (2006) PLoS Genetics)
Worldwide data
European
Americans
South Altaians
-
Spanish
Chinese
African
Americans
Japanese
Puerto Rico
Mende
Nahua
Mbuti
Mala
Burunge
Quechua
Africa
Europe
E Asia
America
274 individuals, 12 populations, ~10,000 SNPs using the Affymetrix array
Shriver et al (2005) Human Genomics
Selecting PCA-correlated SNPs for individual assignment to four continents
(Africa, Europe, Asia, America)
Afr
Eur
Asi
Africa
Ame
Europe
Asia
America
PCA-scores
* top 30 PCA-correlated SNPs
SNPs by chromosomal order
Paschou et al (2007) PLoS Genetics
Correlation coefficient between true and predicted membership of an
individual to a particular geographic continent.
(Use a subset of SNPs, cluster the individuals using k-means.)
Paschou et al (2007) PLoS Genetics
Cross-validation on HapMap data
Paschou et al (2007) PLoS Genetics
A
B
•
9 indigenous populations from four different continents (Africa, Europe, Asia, Americas)
•
All SNPs and 10 principal components: perfect clustering!
•
50 PCAIMs SNPs, almost perfect clustering.
Africa
Oceania
East Asia
South Central
Asia
America
Middle East
Europe
The Human Genome Diversity Panel
Appox. 1000 individuals – 650K Illumina Array
Li et al (2008) Science
Highest scoring genes in HGDP dataset
Gene
Function (RefSeq)
EDAR*
Ectodermal development, hair follicle formation.
PTK6
Intracellular signal transducer in epithelial tissues. Sensitization of cells to
epidermal growth factor.
GALNT13
Initiates O-linked glycosylation of mucins.
SPATA20*
Associated with spermatogenesis.
MCHR1
Plasma membrane protein which binds melanin-concentrating hormone. Probably
involved in the neuronal regulation of food consumption.
FOXP1*
Forkhead box transcription factors play important roles in the regulation of
tissue- and cell type-specific gene transcription during both development and
adulthood.
PSCD3*
Involved in the control of Golgi structure and function.
CNTNAP2*
Member of the neurexin family which functions in the vertebrate nervous
system as cell adhesion molecules and receptors.
OCA2*
Skin/Hair/Eye pigmentation.
EGFR*
This protein is a receptor for members of the epidermal growth factor family.
Associated with the melanin pathway.
* Barreiro et al (2008) Nat Genet, Sabeti et al (2007) Nature, The International HapMap Consortium (2007) Nature
A problem with the CX decomposition
Input:
m-by-n matrix A, integer k, and c (number of SNPs to pick)
Output:
the selected PCA Informative Markers or PCAIMs
CX algorithm
• Compute the scores pj
• Pick the columns (SNPs) corresponding to the top c scores.
Problem:
Highly correlated SNPs (a.k.a., SNPs that are in LD) get similar – high – scores,
and thus the deterministic variant would select redundant SNPs.
How do we remove this redundancy?
Column Subset Selection Problem (CSSP)
Definition:
Given an m-by-c matrix A, find k columns of A forming an m-by-k matrix C that are
maximally uncorrelated (a.k.a., select maximally uncorrelated SNPs).
Column Subset Selection Problem (CSSP)
Definition:
Given an m-by-c matrix A, find k columns of A forming an m-by-k matrix C that are
maximally uncorrelated (a.k.a., select maximally uncorrelated SNPs).
Metric of correlation:
A common formulation is to select a set of SNPs that span a parallel-piped of maximal
volume.
This formulation is NP-hard (i.e., intractable even for small values of m, c, and k).
Column Subset Selection Problem (CSSP)
Definition:
Given an m-by-c matrix A, find k columns of A forming an m-by-k matrix C that are
maximally uncorrelated (a.k.a., select maximally uncorrelated SNPs).
Metric of correlation:
A common formulation is to select a set of SNPs that span a parallel-piped of maximal
volume.
This formulation is NP-hard (i.e., intractable even for small values of m, c, and k).
Significant prior work:
The CSSP has been studied in the Numerical Linear Algebra community, and many
provably accurate approximation algorithms exist.
Prior work on the CSSP: 1965 – 2000
Boutsidis, Mahoney, and Drineas (2009) SODA, under review in Num Math
The greedy QR algorithm
We use a standard greedy approach (the Rank-Revealing QR factorization).
The algorithm performs k iterations:
In the first iteration, the top PCAIM is picked;
In the second iteration, a PCAIM is picked that is as uncorrelated to with the
previously selected PCAIM as possible;
In the third iteration the chosen PCAIM has to be as uncorrelated as possible
with the first two previously selected PCAIMs;
And so on…
Efficient implementations are available, and run in minutes for typical values of m, c, and k.
Paschou et al (2008) PLoS Genetics
A European American sample
Datasets
CHORI dataset
980 European Americans, ~300,000 SNPs (Illumina chip)
Simon et al (2006) Am J Cardiol & Albert et al (2001) JAMA
Coriell dataset
541 European Americans, ~300,000 SNPs (Illumina chip)
Fung et al (2006) LANCET
HapMap
90 Yoruba (YRI), 90 CEPH (CEU), 90 Han Chinese & Japanese (CHB-JPT)
Paschou et al (2008) PLoS Genetics
Europeans
Africans
Asians
PCA plot of European-American populations and HapMap 2 populations.
(The top three eigenSNPs are presented.)
Paschou et al (2008) PLoS Genetics
POPRES - The Population Reference Sample
~6,000 individuals of European, African-American, East Asian, South
Asian, and Mexican origin
Genotyped with Affy 500K array set
Nelson et al (2008) Am J Hum Genet
3,192 Europeans
Correlation between genetic structure and geographic origin
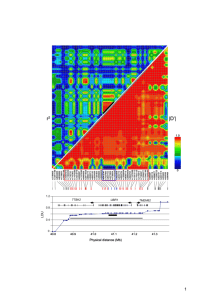
Lao et al (2008) Current Biology
Novembre et al (2008) Nature
We analyzed 1,387 individuals from Novembre et al.
Randomly included only 200 UK and 125 Swiss French (even out sample sizes)
excluded
Europeans not sampled in Europe
Putative relatives
Outliers in preliminary PCA
Conclusions
Using linear algebraic techniques (e.g., matrix decompositions) we selected markers
that capture population structure.
Our technique requires no prior assumptions and builds upon the power of SVD and
PCA to identify population structure in various settings, including admixed populations.
Prior theoretical work and mathematical understanding of the underlying problem
was fundamental in designing our algorithm!
Future research
Unsupervised dimensionality reduction techniques are NOT successful in separating
cases from controls in GWAS studies.
Why?
Because the disease signal is too “weak”.
Potential remedies?
Supervised techniques (Fischer Discriminant Analysis or LDA, etc.).
Sparse approximations for regression problems.
Goal?
Design a global test that may help uncover effects of gene-gene interactions in
disease risk.
Acknowledgements
Collaborators
Students
P. Paschou, Democritus University, Greece
E. Ziv, UCSF
E. Burchard, UCSF
K. K. Kidd, Yale University
M. Shriver, Penn State
R. Krauss, Oakland Research Institute
Asif Javed, RPI (now at IBM)
Jamey Lewis, RPI
Funding: NSF (Drineas), Tourette Syndrome Association (Paschou), NIH (Ziv)
P. Paschou, M. W. Mahoney, A. Javed, J. Kidd, A. Pakstis, S. Gu, K. Kidd, and P. Drineas. (2007) Intra- and interpopulation genotype reconstruction from tagging SNPs, Genome Research, 17(1), pp. 96-107.
P. Paschou, E. Ziv, E. Burchard, S. Choudhry, W. Rodriguez-Cintron, M. W. Mahoney, and P. Drineas. (2007) PCAcorrelated SNPs for structure identification in worldwide human populations, PLoS Genetics, 3(9), pp. 1672-1686.
P. Paschou, P. Drineas, J. Lewis, C. Nievergelt, D. Nickerson, J. Smith, P. Ridker, D. Chasman, R. Krauss, and E.
Ziv. (2008) Tracing sub-structure in the European American population with PCA-informative markers, PLoS
Genetics, 4(7), pp. 1-13.
M. W. Mahoney and P. Drineas. (2009) CUR matrix decompositions for improved data analysis, Proceedings of
the National Academy of Sciences, 106(3), pp. 697-702.