Document
advertisement

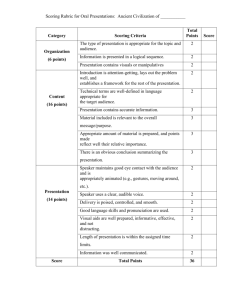
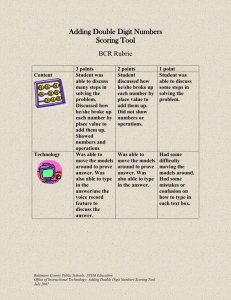
Predicting Protein-Ligand Binding Affinities: A Low Scoring Game? Dushyanthan Puvanendrampillai*, Philip M Marsden*, John BO Mitchell and Robert C Glen Unilever Centre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK *Corresponding authors – dp274@cam.ac.uk , pmm36@cam.ac.uk Introduction Methods Docking programs search for viable conformations of ligands to reside in target receptor sites and a scoring function ranks these docked conformations in terms of the quality of the fit between the ligand and receptor1,2. Preparation of the Test Set. •The test set used in this study consisted of 205 protein-ligand complexes with experimentally measured Kd values, which have been assembled from scoring function and/or docking evaluation studies. •Dissociation constants of the complexes ranged from -1.49 to -13.97 in log Kd units, spanning over 12 orders of magnitude and measured by a variety of experimental methods. •All ligand molecules bind non-covalently to their target proteins. The scoring function should ideally also be able to make predictions of binding affinity, allowing different candidate molecules to be ranked in terms of their predicted binding to a given target3. Analysis We have examined the square of the product moment correlation coefficient (r2) and Spearman’s rank correlation coefficient (Rs) between the binding free energies given by the five scoring functions with experimentally measured log Kd values for the 205 protein-ligand complexes in the dataset. This correlation between calculated binding energies and experimental values provides an indication of the performance of a scoring function, and of how well these values can predict the binding energies of proteinligand interactions. -16 -10 -8 -6 -4 0 -2 0 Dataset -2 -4 All -6 A -10 r2 0.59 0.32 GOLD Rs r2 0.31 0.11 Rs r2 0.50 0.20 Serine proteinases 35 0.82 0.74 0.75 0.51 0.61 B -14 Metalloproteinases 25 0.72 0.44 0.45 0.33 0.44 C -18 Experimental log Kd Figure 7. GOLD calculated log Kd vs. Experimental log Kd -16 -14 r2 0.43 0.20 Rs r2 0.45 0.18 0.47 0.83 0.69 0.13 0.00 0.14 0.42 0.20 0.43 0.17 -12 -10 -8 18 0.53 0.47 0.46 0.32 0.42 0.34 0.54 0.01 0.50 -14 -12 -10 -8 -6 -4 -2 -2 -6 -4 Sugar binding proteins 30 0.76 0.58 0.45 0.09 0.02 0.00 0.05 0.00 0.29 E Aspartic proteinases 38 0.08 0.01 - 0.52 0.13 0.02 0.00 0.08 0.01 -0.08 0.00 Figure 8. DOCK calculated log Kd vs. experimental log Kd 0 -2 0 -4 -6 -8 -10 -12 -14 -16 -18 -16 -14 -12 -10 -8 -6 -4 -6 -8 -10 -12 -14 -16 -18 Experimental log Kd Figure 9. ChemScore calculated log Kd vs. experimental log Kd 0 -2 -4 -6 -8 -10 -12 -14 -16 -18 2 r = 0.20 Experimental log Kd 0.09 0 -2 0 -4 0.28 D -2 r = 0.20 Experimental log Kd 0 -16 -14 -12 -10 -8 -6 -4 -2 -2 0 -4 -6 -8 -10 -12 -14 -16 r2 = 0.18 Experimental log Kd -18 Results Conclusions and Discussion For all 205 protein-ligand complexes – •The inescapable conclusion from these results is that the problem of accurately predicting the binding energies of a large and diverse set of protein-ligand complexes is a difficult one. •BLEEP gives the best agreement between its calculated binding free energy and experimental log Kd values with Rs=0.59. •None of the scoring functions tested here achieved r2 values above 0.32 when tested on the full 205 complex dataset. •GOLD, ChemScore and DOCK have a similar level of binding free energy agreement with Rs values of 0.50, 0.45 and 0.43 respectively. •This is a disappointing level of performance, although we should note in defence of the GOLD and DOCK functions that they were designed to identify the correct geometries of bound complexes and not intended to be applied to the problem of affinity prediction. •PMF gives an Rs value of 0.31. •The headline figures for the correlation coefficient given here seem less impressive than in previous work. Figure 3. Representation of the BLEEP scoring function for complex 1AAQ. Pairwise potentials are calculated between the ligand and protein atoms within a distance cut-off. Ligand atoms (red) and protein atoms (green) within 8Å of the ligand are shown in ball and stick representation. Rs -16 r2 = 0.32 2 function for complex 1AAQ. Based on van der Waals interactions, hydrogen bonds (yellow dashed lines), and ligand internal torsional energy. External van der Waals shown by the two complementary surfaces of the ligand (green) and the protein (brown). ChemScore ChemScore calculated log Kd r 2 = 0.11 Carbonic anhydrase ii -18 Figure 2. Representation of the GOLD scoring DOCK -12 GOLD calculated log Kd The development of such scoring functions to accurately predict experimental binding free energies for protein-ligand complexes is currently a major challenge in structure-based drug design. We expect that improvements in the scoring functions will be crucial in addressing this problem. Rs PMF 0 -16 5,6 We used the knowledge-based methods and BLEEP and empirical scoring functions of GOLD7, DOCK8 and ChemScore9, as implemented by Sybyl® 6.910. BLEEP Figure 6. BLEEP calculated log Kd vs. Experimental log Kd -8 We have compared the results of five different scoring functions to determine the binding energy of a protein-ligand complex with known three-dimensional structure on a diverse dataset of 205 protein-ligand complexes, and also on various subsets of mutually similar complexes. PMF4 No. of complexes 205 DOCK calculated log Kd isostere inhibitor (PDB ID 1AAQ). Protein secondary structure illustrated in cartoon representation coloured by secondary structure type. A Connolly molecular surface shown of the cavity coloured by electrostatic potential. Ligand shown in ball and stick representation. -12 PMF calculated log Kd Figure 1. Crystal structure of HIV-1 protease complexed with an hydroxyethylene -14 Table 1. Correlations Between Experimental and Calculated log Kd Values Given by Five Scoring Functions. BLEEP calculated log Kd Figure 5. PMF calculated log Kd vs. experimental log Kd •All five scoring functions give modest r2 •This is partly due to the choice of dataset. Six outliers were excluded from that previous analysis of BLEEP6, which raised the r2 value from 0.40 to 0.55. values, the highest being the 0.32 given by BLEEP. References Figure 4. Representation of the DOCK scoring function for complex 1AAQ. Based on shape complementarity. Molecular surface of the ligand (yellow) and the complementary surface of the protein (green). 6. Mitchell,J.B.O., Laskowski,R.A., Alex,A., Forster,M.J. and Thornton,J.M. (1999) BLEEP - potential of mean force describing protein-ligand interactions: II. Calculation of binding energies and comparison with experimental data. J. Comp. Chem., 20, 1177-1185. 7. Jones,G., Willett,P., Glen,R.C., Leach,A.R. and Taylor,R. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol., 267, 727-748. 8. Kuntz, I.D., Blaney, J.M., Oatley, S.J., Landridge, R. and Ferrin, T.E. (1982) A geometric approach to macromolecule-ligand interactions. J. Mol. Biol., 161, 269-288. Muegge,I. and Martin,Y.C. (1999) A general and fast scoring function for protein- ligand interactions: a simplified potential approach. J. Med. Chem., 42, 791-804. 9. Eldridge,M.D., Murray,C.W., Auton,T.R., Paolini,G.V. and Mee,R.P. (1997) Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des., 11, 425-445. Mitchell,J.B.O., Laskowski,R.A., Alex,A. and Thornton,J.M. (1999) BLEEP - potential of mean force describing protein-ligand interactions: I. Generating potential. J. Comp. Chem., 20, 1165-1176. 10. SYBYL 6.9, Tripos Inc., 1699 South Hanley Road, St. Louis, Missouri, 63144, USA 1. Stahl,M. and Rarey,M. (2001) Detailed analysis of scoring functions for virtual screening. J.Med.Chem., 44, 1035-1042. 2. Peréz,C.and Ortiz,A.R. (2001) Evaluation of docking functions for protein-ligand docking. J.Med.Chem., 44, 3768-3785. 3. Böhm,H.J. (1994) The development of a simple empirical scoring function to estimate the binding constants for a protein-ligand complex of known three-dimensional structure. J. Comput.-Aided Mol. Des., 8, 243-256. 4. 5. Acknowledgements We thank Unilever, the EPSRC and the Newton Trust for their funding, and Tripos Inc. for the use of Sybyl®.