acute inflammatory demyelinative polyneuropathy
advertisement

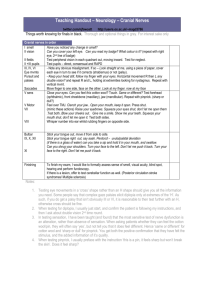
Polyneuropathies Mononeuropathies Motoneuron diseases Zsuzsanna Arányi Peripheral nerve Motor, sensory and autonomic fibers Fiber types according to diameter: • A fibers- 1-17 μm in diameter; myelinated motor and sensory fibers • B fibers- 1-3 μm in diameter; myelinated autonomic fibers • C fibers- 0.3-1.3 μm in diameter; non-myelinated autonomic and pain fibers Types of peripheral nerve damage Demyelination Slowed conduction: no symptoms Conduction block: weakness and sensory loss, but no atrophy Axonal damage (axonotmesis) Degeneration of axons distal to the lesion (denervation) Weakness, sensory loss, atrophy Neurotmesis Damage to axons and epineurium Weakness, sensory loss, atrophy No regeneration without nerve suture Nerve regeneration – reinnervation Remyelination 2-12 weeks Proximo-distal axon regeneration 1 mm/day Intact basal lamina/endoneurium is needed Collateral reinnervation (in case of partial nerve damage) Starts within 4-6 weeks Polyneuropathies Generalised disease of the peripheral nervous system (nerve roots and peripheral nerves) Usually the longest nerves are affected first Symptoms start on the toes, feet Usually the symptom of an underlying systemic disease Search for etiology! Classification of polyneuropathies Clinical presentation Time course Symmetric Asymmetric Acute Chronic Etiology Pathology Axonal Demyelinative Small-fiber Clinical forms of polyneuropathies Chronic, symmetric, distal and predominantly sensory polyneuropathies Mononeuropathy multiplex Purely motor or sensory polyneuropathies Autonomic polyneuropathies Acute polyneuropathies Typical symptoms of polyneuropathies Chronic course Symmetric, distal paraesthesia, pain and hypaesthesia in stocking – glove distribution; feet are affected first Allodynia Depressed or absent tendon reflexes Distally pronounced muscle weakness, with wasting, fasciculation Gait disorder Sensory ataxia Weakness Autonomic dysfunction (reduced sweating, tachycardia, urinary disturbances, gastroparesis etc.) Typical complaints of patients with polyneuropathies Tingling, pin-prick, numbness, burning or cold sensation, burning pain (especially during the night) ‘Ants crawling on my legs’ ‘As if I had tight boots on’ ‘As if I were walking on a duvet’ ‘As if I had stockings on when really not’ ‘As if my skin were thick on my soles’ Unstable gait, ‘dizziness’ Loss of dexterity of the hands: ‘I drop objects’ Causes of polyneuropathy Metabolic-endocrine disturbances: diabetes mellitus, uremia etc. Vitamin deficiencies: vitamin B1 -alcoholism, malabsorption, malnutrition, vitamin B12 Toxic causes: heavy metals, industrial solvents, drugs, alcohol Dysimmune polyneuropathies With manifestation only in the peripheral nervous system: acute inflammatory demyelinative polyneuropathy (Guillain-Barré syndrome), chronic inflammatory demyelinative polyneuropathy (CIDP), multifocal motor neuropathy (MMN) Systemic diseases: vasculitis (polyarteritis nodosa, SLE etc.), paraproteinaemias Paraneoplasia Infectious: lepra, Lyme-disease, HIV Hereditary: Charcot-Marie-Tooth disease etc. Other: critical illness polyneuropathy, small-fiber neuropathy Idiopathic Investigation of polyneuropathies ENG-EMG Blood tests: We, blood count, glucose, hepatic and renal function Vitamin B12 Thyroid function Se electrophoresis, autoanti-bodies, cryoglobulin Serological examinations (HIV, Lyme, HCV) Search for tumors CSF Toxicological investigations Sural nerve biopsy Genetic tests Treatment of polyneuropathies Treat the cause! Immune therapy plasmapheresis: Guillain-Barré syndrome, CIDP immunoglobulins: MMN, Guillain-Barré syndrome, CIDP corticosteroids: CIDP, systemic vasculitis Symptomatic treatment of paraesthesias and neuropathic pain antiepileptic medications (carbamazepine, gabapentin, pregabalin) tricyclic antidepressants (amitriptilin, clomipramin) SNRI antidepressants (duloxetin, venlafaxin) Vitamin B1: alcoholism, malabsorption, malnutrition Polyneuropathies associated with diabetes mellitus Distal symmetric sensory polyneuropathy Mononeuropathies- carpal tunnel syndrome, ulnar nerve lesion Cranial nerve lesionsoculomotor nerve palsy Autonomic neuropathysexual and urinary disturbance, gastroparesis and diarrhoea etc. Diabetic amyotrophy- painful, asymmetric, proximal weakness (plexopathy?) Radiculopathy- lumbar, thoraco-abdominal Diabetic chronic distal symmetric sensory polyneuropathy The most common form of diabetic neuropathy Prevalence among diabetic patients: 20-60% Present at the diagnosis of diabetes in 20% of patients May be the only manifestation of impaired glucose tolerance Severity is usually proportional to the duration and severity of hyperglycemia Prevalence increases with age and duration of diabetes Small fibers (pain, temperature, light touch) are preferentially affected → painful diabetic neuropathy in about 20-35% Autonomic dysfunction Trophic alterations → diabetic foot Small fiber neuropathy- skin biopsy Normal Small fiber neuropathy Epidermal nerve fibers (arrow): anti PGP 9.5 antibodies Fibrous tissue and basal lamina: anti collagen IV antibodies Symptoms of sensory diabetic neuropathy I. Length-dependent: first symptoms on the toes and feet Later stocking-gloves distribution Usually doesn’t go above the knees and elbows If symptoms appear on the hands first → carpal tunnel syndrome Areflexia Trophic changes Symptoms of sensory diabetic neuropathy II. Positive sensory symptoms: burning pain (pronounced during the night) hyperesthesia, allodynia paresthesia Negative sensory symptoms: hypesthesia (loss of sensation) Diabetic foot Related to diabetic sensory neuropathy and peripheral artery disease Diabetic foot ulcers precede 85% of nontraumatic lower limb amputations Life-time prevalence of foot ulcers is 15% in diabetic patients Guillain-Barré syndrome Acute immunmodulated poly-radiculo-neuro-pathy Pathology: perivascular lymphocyte-macrophage infiltration in the peripheral nervous system leading to macrophage mediated segmental demyelination Incidence: 1.5-2.0/100 000/year In most cases preceded by an infection (upper respiratory tract infection, diarrhoea) Infectious agents associated with Guillain-Barré syndrome: CMV, EBV, HIV, Campylobacter jejuni, Mycoplasma pneumoniae The infectious agent is usually unidentified Pathomechanism of GBS Guillain-Barré syndrome- symptoms Acute, symmetric ascending flaccid paralysis Variable severity Respiratory insufficiency Bilateral facial palsy Ascending numbness to a lesser degree Radicular pain Areflexia Autonomic symptoms- tachycardia, cardiovascular instability Guillain-Barré syndrome- time course Symptoms evolve over 1-2 weeks Plateau is reached within 2-3 weeks Spontaneous recovery within a few months Good prognosis Prognosis is determined mainly by complications of being bed-bound (infection, thrombosis etc.) Guillain-Barré syndrome- diagnosis Normal neurography Segmental demyelination Conduction block Temporal dispersion Guillain-Barré syndrome- diagnosis and treatment Diagnosis Clinical symptoms Electroneurography- confirms segmental demyelination Cerebrospinal fluid examination: elevated protein content with normal cell count (starting from the 2nd week) Treatment Plasmapheresis, immunoglobulin (IVIG) Supportive treatment! Chronic inflammatory demyelinative polyneuropathy (CIDP) Autoimmune disease Prevalence: 1-2/100 000 Course: chronic monophasic (15%) chronic relapsing-remitting (34%) step-wise progressive (34%) continuously progressive (15%) Symptoms: proximal and distal motor and sensory symptoms, cranial symptoms (not a length-dependent neuropathy) Rarely associated with central nervous system demyelination (3%) Diagnosis of CIDP ENG/EMG: segmental (non-uniform) demyelination CSF: protein >45 mg/dl, cell count <10 Histology (biopsy): not obligatory, may be normal chronic demyelination-remyelination may lead to Schwanncell proliferation (‘onion bulb’ formation) infiltration of inflammatory cells MRI: hypertrophy of peripheral nerves and nerve roots, contrast enhancement CIDP- nerve biopsy ‘onion bulbs’ CIDP- MRI Hypertrophied trigeminal nerves CIDP treatment IVIG Corticosteroids 2 g/kg bw in 2-5 days, monthly for 3 months maintanance treatment methylprednisolon 1 mg/kg bw, later gradual reduction Plasmapheresis Mononeuropathies- causes Trauma Compression cutting, laceration and stretching of the nerve often iatrogenic Tunnel syndromes Ischemia Localisation of focal nerve lesions • A partial proximal nerve lesion may selectively affect only one nerve fascicle → clinically the lesion appears more distal • The longer axons are more sensitive to compression → distal symptoms are more pronounced Median nerve Distal median nerve damage: carpal tunnel syndrome Incidence: 200-500/100 000/year, 3 times more common in women Symptoms: Painful paraesthesia of the hand during the night, pain in the whole arm First the dominant hand is affected, but bilateral involvement in most cases Advanced symptoms: sensory loss on digits 1-3, thenar atrophy and weakness Causes: idiopathic, overuse, change of tunnel anatomy (fracture, arthrosis, oedema etc.), diabetes Treatment: Splinting of the hand during the night Surgery Proximal median nerve damage 1. 1. Weakness of all median nerve muscles ‘oath hand’ 2. 2. Weakness of flexion of the distal phalanx of digit 1-2 no sensory loss Ulnar nerve Ulnar nerve lesion at the elbow- two types Extension • Retroepicondylar lesion (more common) • Compression, elbow fracture, arthrosis, diabetes • Real cubital tunnel syndrome Flexion Ulnar nerve lesion Numbness of digit 4-5 and ulnar edge of the hand Atrophy and weakness of hypothenar, interosseus muscles and adductor pollicis muscle Tinel-sign at the elbow Claw hand Radial nerve Radial nerve lesion on the upper arm ‘Saturday night palsy’: nerve compression during sleep common in alcoholics Symptoms: weakness of wrist and finger extension (wrist drop); triceps is normal; loss of sensation on the dorsal-radial aspect of the hand Radial nerve lesion on the forearm • Weakness of finger extension (‘finger drop’), extension of the wrist is only sightly weak, oftens starts on digit 4-5 → may be confused with ulnar nerve lesion • No sensory loss • Causes: supinator tunnel syndrome due to overuse Common peroneal nerve Peroneal nerve damage at the fibular head Foot drop, steppage gate Supination (inversion) and plantarflexion is normal Sensory loss on the lateral aspect of the leg and dorsal aspect of the foot Causes: compression During sleep, in coma During surgery Cast Crossed legs Squatting (strawberry pickers) Peroneal tunnel syndrome? Motoneuron diseases Progressive loss/degeneration of motoneurons Weakness Atrophy No sensory or autonomic symptoms Two major types: Amyotrophic lateralsclerosis (ALS): both upper and lower motoneurons are affected Spinal muscular atrophies / lower motoneuron syndromes ALS First described by Jean Martin Charcot in 1874 Incidence: 2 / 100 000 / year Prevalence: 6 / 100 000 ‘Lou Gehrig’s disease’ ALS- Clinical forms Sporadic ALS Lower motoneuron onset Familial ALS (5-10%) SOD1 mutations No SOD1 mutations Autosome recessive Classic ALS Autosome dominant Progressive muscular atrophy (PMA) Classic ALS Progressive muscular atrophy (PMA) Primary lateralsclerosis Progressive bulbar paralysis Progressive pseudobulbar palsy Bulbar onset SOD1 mutation Chronic juvenile ALS Upper motoneuron onset X-linked Frontotemporal dementia + ALS (ubiquitin positive) Progressive bulbar paralysis Primary lateralsclerosis ALS- symptoms and course Mixed signs of upper and lower motor neuron lesion Atrophy, fasciculation, cramps Spasticity, increased reflexes, Babinski Relentlessly and quickly progressive Average survival: 2-5 years Cause of death: respiratory insufficiency ALS- Clinical syndromes at onset Asymmetric small hand muscle atrophy and weakness (segmental distribution)- 60-85% Diff. dg.: radiculopathy, ulnar nerve lesion Proximal arm muscle atrophy and weakness (‘flail’ arm) Diff. dg.: radiculopathy Bulbar onset- 15-40% Dysarthria and dysphagia Diff. dg.: myasthenia gravis, pseudobulbar paresis Spastic paraparesis Diff. dg: spinal disease ALS symptoms ALS- treatment No cure Only drug approved for ALS: riluzol (inhibits the presynaptic release of glutamate), survival on riluzol increases by 3-6 months Supportive treatment: Riluzol trials Muscle relaxants Antidepressants, anxiolytic drugs PEG in case of severe dysphagia Assistive devices Ventilation??? (moral issue) Infantile and juvenile spinal muscular atrophies (SMA I-III) 1 / 6-20 000 live births Autosome recessive In 95% of patients the mutation is found in the SMN (survival motoneuron) gene (chr. 5) Infantile and juvenile spinal muscular atrophies (SMA I-III) SMA I: Werdnig-Hoffmann disease. Symptoms are present at birth‘floppy baby’. Death within 1-2 years. SMA II.: Intermediate form SMA III: Kugelberg-Welander disease Symptoms start at age 12-15 years: proximal, symmetric weakness in the legs Progression is variable Differential diagnosis: muscle dystrophies Dg.: EMG (chronic neurogenic findings), genetic testing Adult onset spinal muscular atrophies / lower motoneuron diseases SMA IV: 'adult onset' proximal spinal muscular atrophy Onset: 20-40 years of age Inheritance: 70% AR, 30% AD Gene is unknown Symptoms: very slowly progressive limb girdle weakness and atrophy. May be asymmetric, the quadriceps muscle is very often affected. No bulbar involvement. Differential diagnosis: muscle dystrophies, ALS Adult onset spinal muscular atrophies / lower motoneuron diseases dSMA V: 'adult onset' distal spinal muscular atrophy Onset: 20-40 years of age Inheritance : AD Gene is unknown Symptoms : slowly progressive distal weakness and atrophy Differential diagnosis: polyneuropathies Adult onset spinal muscular atrophies / lower motoneuron diseases Benign focal amyotrophy Usually sporadic More common in men Starts in young adulthood, slow progression over a few years, then stagnation Symptoms: small hand atrophy on one side Differential diagnosis: ALS, ulnar nerve lesion