Bioinformatics 2-Chen 2011
advertisement

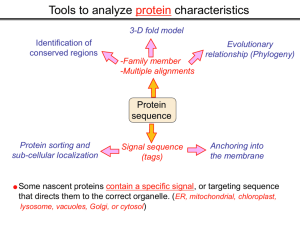
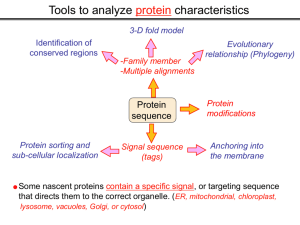
Tools to analyze protein characteristics 3-D fold model Identification of conserved regions -Family member -Multiple alignments Evolutionary relationship (Phylogeny) Protein sequence Protein sorting and sub-cellular localization Some Signal sequence (tags) Anchoring into the membrane nascent proteins contain a specific signal, or targeting sequence that directs them to the correct organelle. (ER, mitochondrial, chloroplast, lysosome, vacuoles, Golgi, or cytosol) Questions Can we train the computers: To detect signal sequences and predict protein destination? To identify conserved domains (or a pattern) in proteins? To predict the membrane-anchoring type of a protein? (Transmembrane domain, GPI anchor…) To predict the 3D structure of a protein? Learning algorithms are good for solving problems in pattern recognition because they can be trained on a sample data set. Classes of learning algorithms: -Artificial neural networks (ANNs) -Hidden Markov Models (HMM) Artificial neural networks (ANN) Machine learning algorithms that mimic the brain. Real brains, however, are orders of magnitude more complex than any ANN. ANN is composed of a large number of highly interconnected processing elements (neurons) working simultaneously to solve specific problems. like people, learn by example. ANNs cannot be programmed to perform a specific task. ANNs, The first artificial neuron was developed in 1943 by the neurophysiologist Warren McCulloch and the logician Walter Pits. Hidden Markov Models (HMM) Used to answer questions like: What is the probability of obtaining a particular outcome? What is the best model from many combinations? HMM is a probabilistic process over a set of states, in which the states are “hidden”. It is only the outcome that visible to the observer. Hence, the name Hidden Markov Model. HMM has many uses in genomics: Gene prediction (GENSCAN) SignalP Finding periodic patterns The ExPASy (Expert Protein Analysis System) Expasy server (http://au.expasy.org) is dedicated to the analysis of protein sequences and structures. Sequence analysis tools include: DNA -> Protein [Translate] Pattern and profile searches Post-translational modification and topology prediction Primary structure analysis Structure prediction (2D and 3D) Alignment PredictProtein: A service for sequence analysis, and structure prediction http://www.predictprotein.org/newwebsite/submit.html TMpred: TMHMM: Predicts transmembrane helices in proteins (CBS; Denmark) http://www.ch.embnet.org/software/TMPRED_form.html http://www.cbs.dtu.dk/services/TMHMM-2.0/ big-PI : Predicts GPI-anchor site:http://mendel.imp.univie.ac.at/sat/gpi/gpi_server.html DGPI: Predicts GPI-anchor site: http://129.194.185.165/dgpi/index_en.html SignalP: Predicts signal peptide: http://www.cbs.dtu.dk/services/SignalP/ PSORT: Predicts sub-cellular localization: TargetP: Predicts sub-cellular localization: http://www.cbs.dtu.dk/services/TargetP/ NetNGlyc: Predicts N-glycosylation sites:http://www.cbs.dtu.dk/services/NetNGlyc/ PTS1: Predicts peroxisomal targeting sequences http://www.psort.org/ http://mendel.imp.univie.ac.at/mendeljsp/sat/pts1/PTS1predictor.jsp MITOPROT: Predicts of mitochondrial targeting sequences http://ihg.gsf.de/ihg/mitoprot.html Hydrophobicity: http://www.vivo.colostate.edu/molkit/hydropathy/index.html Multiple alignment Used to do phylogenetic analysis: Same protein from different species Evolutionary relationship: history Used to find conserved regions Local multiple alignment reveals conserved regions Conserved regions usually are key functional regions These regions are prime targets for drug developments Protein domains are often conserved across many species Algorithm for search of conserved regions: Block maker: http://blocks.fhcrc.org/blocks/make_blocks.html Multiple alignment tools Free programs: Phylip and PAUP: http://evolution.genetics.washington.edu/phylip.html Phyml: http://atgc.lirmm.fr/phyml/ The most used websites : http://align.genome.jp/ http://prodes.toulouse.inra.fr/multalin/multalin.html http://www.ch.embnet.org/index.html (T-COFFEE and ClustalW) ClustalW: Standard popular software It aligns 2 and keep on adding a new sequence to the alignment Problem: It is simply a heuristics. Motif discovery: use your own motif to search databases: PatternFind: http://myhits.isb-sib.ch/cgi-bin/pattern_search http://meme.nbcr.net/meme4_6_0/intro.html Phylogenetic analysis Phylogenetic Describe Major trees evolutionary relationships between sequences modes that drive the evolution: Point mutations modify existing sequences Duplications (re-use existing sequence) Rearrangement Two most common methods Maximum parsimony Maximum likelihood The most useful software: http://www.megasoftware.net/mega4/m_con_select.html Parsimony vs Maximum likelihood Parsimony is the most popular method in which the simplest answer is always the preferred one. It involves statistical evaluation of the number of mutations need to explain the observed data. The best tree is the one that requires the fewest number of evolutionary changes. In contrast, maximum likelihood does not necessarily satisfy any optimality criterion. It attempts to answer the question: What parameters of evolutionary events was likely to produce the current data set? This is computationally difficult to do. This is the slowest of all methods. Likelihood generally performs better than parsimony Definitions Homologous:Have Orthologous: Paralogous: a common ancestor. Homology cannot be measured. The same gene in different species . It is the result of speciation (common ancestral) Related genes (already diverged) in the same species. It is the result of genomic rearrangements or duplication Determining protein structure Direct measurement of structure X-ray crystallography NMR spectroscopy Site-directed mutagenesis Computer modeling Prediction of structure Comparative protein-structure modeling Comparative protein-structure modeling Goal:Construct 3-D model of a protein of unknown structure (target), based on similarity of sequence to proteins of known structure (templates) Procedure: Template selection Template–target alignment Model building Model evaluation Blue: predicted model by PROSPECT Red: NMR structure The Protein 3-D Database The Protein DataBase (PDB) contains 3-D structural data for proteins Founded in 1971 with a dozen structures As of June 2004, there were 25,760 structures in the database. All structures are reviewed for accuracy and data uniformity. 80% come from X-ray crystallography 16% come from NMR 2% come from theoretical modeling Structural data from the PDB can be freely accessed at http://www.rcsb.org/pdb/ High-throughput methods Most used websites for 3-D structure prediction Protein Homology/analogY Recognition Engine (Phyre) at http://www.sbg.bio.ic.ac.uk/phyre/html/index.html PredictProtein at http://www.predictprotein.org/newwebsite/submit.html UCLA Fold Recognition at http://www.doe-mbi.ucla.edu/Services/FOLD/ Commercial bioinformatics softwares CLC Genomics Workbench Genomics: 454, Illumina Genome Analyzer and SOLiD sequencing data; De novo assembly of genomes of any size; Advanced visualization, scrolling, and zooming tools; SNP detection using advanced quality filtering; Transcriptomics: RNA-seq including paired data and transcript-level expression; Small RNA analysis; Expression profiling by tags; Epigenetics: Chromatin immunoprecipitation sequencing (ChIP-seq) analysis; Peak finding and peak refinement; Graph and table of background distribution; false discovery rate; Peak table and annotations; VectorNTI: Sequence analysis and illustration; restriction mapping; recombinant molecule design and cloning; in silico gel electrophoresis; synthetic biology workflows AlignX: BioAnnotator: ContigExpress: GenomBench The bioinformatics not covered in this class Comparative genomics and Genome browser: http://genome.lbl.gov/vista/index.shtml http://www.sanger.ac.uk/resources/software/artemis/ Genome annotation: http://linux1.softberry.com/berry.phtml http:// rast.nmpdr.org/ Metagenomics: http://metagenomics.anl.gov/ System biology tools.