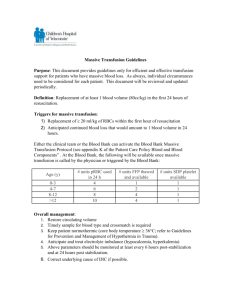
Quiz Yourself
advertisement

Quiz Yourself Hematology Please note that this was put together by a UNC MS2 for other UNC MS2s. If you find any mistakes or have any feedback, let us know – especially if this was remotely helpful in any way! Characteristics of Normal Adult Bone Marrow •Consists of approximately 50% fat cells; •Has more myeloid cells than erythroid cells (myeloid:erythroid ratio 2:1 to 7:1) •Megakaryocytes are 2-5 per high power field •Plasma cells <3% •Lymphocytes <20% Peripheral Blood Morphology 1. 2. Erythrocytes Granulocytes 1. 2. 3. 3. 4. 5. Neutrophils Eosinophils Basophils Platelets Monocytes Lymphocytes (B & T cells) Erythropoiesis – what is the order? Proerythroblast Basophilic erythroblast Orthochromatophilic erythroblast Reticulocyte Polychromatophilic erythroblast Mature RBC Granulopoiesis – what is the order? Myeloblast Metamyelocyte Promyelocyte Band Myelocyte* Neutrophil Identify the Type of Cell Basophil Eosinophil Monocyte Neutrophil Anemia: When do you give a transfusion right away? .Angina pectoris – coronary insufficiency .Shock .Surgery Iron kinetics as a function of time 1. Childhood – building both the hemoglobin and storage iron by the intake and conservation of iron- years 2. Adolescent and adult males – full complement of hemoglobin and storage iron 3. Adolescent and adult females – iron content is challenged by menses and childbirth Causes of iron deficiency 1. 2. 3. 4. inadequate intake Malabsorption diversion of iron during pregnancy blood loss RBC Characteristics in Iron Deficiency Anemia? Microcytic – small RBCs Hypochromatic – pale RBCs Ways to diagnose iron deficiency anemia? 1) Serum ferritin – will be decreased. 2) 3) Bone Marrow - staining Treatment – the simplest! Careful, it’ll also be low in diseased/ill patients (an acute phase reactant) Causes of Macrocytic Anemia B12 deficiency Folate deficiency Uses of B12 & Folate DNA synthesis B12 = co-factor Folate = transfer single carbon groups How do we get folate and B12? Folate In leafy green veggies, liver, yeast Destroyed by cooking Need 100-200 micrograms daily Vitamin B12 In animal products Unaffected by cooking Need 1-2 micrograms daily Folate Deficiency – 3 major causes Dietary Malabsorption Increased usage 3 ways to diagnose folate deficiency Morphology – macrocytic RBCs and hypersegmented neutrophils Serum folate Red cell folate What’s This? Megaloblastic Anemia Possible Causes? B12 or folate deficiency B12 Deficiency – 3 major causes Pernicious Anemia Pancreatic Insufficiency Malabsorption 3 ways to diagnose B12 deficiency Morphology Serum B12 Neurologic findings – Demyelination of spinal cord, cerebral cortex Treating B12 & Folate Deficiencies B12 IM B12 supplementation for life Folate Daily folate supplement (1mg/day) What do you see in the RBCs below? How would we quantitate this? Anisocytosis refers to red cells which vary widely in size. The RDW mathematically measures the range of red cell sizes. What do you see in the RBCs below? What diseases might they be associated with? Microcytosis refers to red cells that are small. You can use the lymphocyte nucleus as a visual reference, or you can use the MCV Associated with Iron deficiency Thalassemias Sideroblastic anemia What do you see in the top slide? Characterizes what diseases? Macrocytosis refers to large red cells. Associated with Elevated reticulocyte count B12/folate deficiency Liver disease Thyroid disease Chemotherapy Anti-retrovirals (AZT) What’s wrong with these RBCs? Measured how? A likely cause? Hypochromasia refers to red cells that have too little hemoglobin. The area of central pallor is more than 1/3 the total red cell diameter. This is measured by the MCH (mean cellular hemoglobin) Iron deficiency What do you see on this slide? Poikilocytosis refers to red cells that vary widely in shape. Remember that anisocytosis refers to red cells that vary widely in size. What do you see here? Diseases? Target cells look like bulls-eyes. Associated with Liver disease Thalassemias Hemoglobin C After splenectomy What do you see here? Diseases? Spherocytes have a loss of central pallor. Can be seen in Hereditary spherocytosis Autoimmune hemolysis If due to autoimmune hemolysis, the cells are smaller (i.e. microspherocytes) What do you see here? Diseases? Schistocytes are red cell fragments with sharp edges. They are a hallmark of Microangiopathic Hemolytic Anemia (MAHA) What do you see here? Sickle Cells are seen in sickle cell anemia. Notice that this slide has target cells as well as a sickled cell. What RBCs are here? How do you distinguish the two? Associated disease? Echinocytes, or burr cells, have small, regular projections. Seen in renal disease Acanthocytes, or spur cells, have larger, irregular projections, and are seen in liver disease. What do you see here? What causes it? Teardrop cells Seen in myelophthisic processes, or diseases of marrow infiltration. Deformed as it tries to squeeze out of the bone marrow And what have we here? What causes them? Howell-Jolly bodies are peripheral, small, round, purple inclusions within red cells that represent nuclear remnants. They are seen after splenectomy, or in cases of splenic hypofunction. What do you see here? Causes? Rouleaux are linear arrangements of red cells typically described as “piles of coins on a plate” They are typically seen in disorders with increased levels of immunoglobulin, such as Multiple Myeloma or Waldenstrom’s macroglobulinemia. Severe hypo-albuminemia can also lead to reouleux formation What do you see here? Red cell agglutination occurs when the red cells are coated with IgM. IgM is large enough to bridge two red cells and cause agglutination. Unlike rouleaux, the red cell clumps are not orderly and linear. General Clinical Features of Hemolytic Anemias Splenomegaly is generally present Patients have an increased incidence of pigmented gallstones. Dark urine (tea-colored or red), jaundice, scleral icterus Patients may have chronic ankle ulcers. Aplastic crises associated with Parvovirus B19, may occur Increased requirement for folate Post-splenectomy blood findings Howell-Jolly bodies - small round blue DNA remnants in periphery of RBCs Red cell abnormalities: target cells, acanthocytes, schistocytes, NRBCs Hemolytic Anemias Sites of Red Cell Destruction Extravascular Hemolysis Macrophages in spleen, liver, and marrow remove damaged or antibody-coated red cells Intravascular Hemolysis Red cells rupture within the vasculature, releasing free hemoglobin into the circulation (and the circulation does NOT like this!) Evidence for increased red cell production In the blood: Elevated reticulocyte count May be associated with high MCV Circulating NRBCs may be present In the bone marrow: erythroid hyperplasia reduced M/E (myeloid/erythroid) ratio In the bone: Deforming changes in the skull and long bones (“frontal bossing”) Evidence for Increased Red Cell Destruction Biochemical consequences of hemolysis in general Morphologic evidence of red cell damage Elevated LDH Elevated unconjugated bilirubin jaundice, scleral icterus Lower serum haptoglobin Hemoglobinemia Hemoglobinuria Hemosiderinuria Schistocytes Spherocytes Bite/blister cells Reduced red cell life-span Hemolytic Anemias Classification by Etiology Congenital Defects in membrane skeleton proteins Defects in enzymes involved in energy production Hemoglobin defects Acquired Immune-mediated Non-immune-mediated Most common defect leading to anemia? Frequency? Defect is in proteins of the membrane skeleton, usually spectrin or ankyrin Lipid microvesicles are pinched off in the spleen and other RE organs, causing decreased MCV and spherocytic change. Diagnosing? Autosomal dominant Pathophysiology? Affects 1/5000 Europeans Transmission? Hereditary spherocytosis Increased osmotic fragility Treatment? Supplemental folate Splenectomy (but carefully consider timing in children) • Functions of GP6D? • • Detoxification of metabolites of oxidative stress Elimination of methemoglobin • • NADPH Reduced glutathione • • Heinz bodies Causes the formation of bite/blister cells • • • • • Type B is more prevalent Type A is in 20% of healthy Africans In 10-14% of African American men Also prevalent in the Mediterranean X-linked • Important Products of GP6D? • Diagnostic methemoglobin precipitate? • Epidemiology of GP6D Deficiency? G6PD Deficiency Agents to avoid For SKAND… – – – – – – Fava beans Sulfa drugs Vitamin K Anti-malarials Naphtha compounds (mothballs) Dapsone What cell is below? Blister cell Sorry Skand – at least your girlfriend thought it was funny! How will you diagnose an autoimmune cause of hemolytic anemia? Coomb’s Test The Direct Coomb’s = DAT (Direct Antiglobulin Test) - tests for IgG or C3 DIRECTLY ON THE RED CELLS. You’re adding patient RBCs! The Indirect Coomb’s - tests for IgG or C3 in the serum which react with generic normal red cells. This is also known as the antibody screen in blood-banking. You’re adding patient serum! Warm-Antibody Hemolytic Anemias Clinical Features Splenomegaly, jaundice usually present. Depending on degree of anemia and rate of fall in hemoglobin, patients can have VERY symptomatic anemia Lab Dx reticulocytes, bili, LDH, positive Coomb’s test - both direct and indirect. SPHEROCYTES are seen on the peripheral smear. Warm-Antibody Hemolytic Anemias Treatment Immunosuppressive Treatment First line is corticosteroids (i.e. prednisone). If steroids fail to work, or if patient relapses after steroid taper, splenectomy may be necessary. Immunosuppressives such as cyclophosphamide (Cytoxan) or azathioprine (Immuran) may be required as third-line therapy. Folate repletion Transfusion – determining factors: Heart failure, shock? Inadequate reticulocyte count? Drug-Induced Immune Hemolysis Three general mechanisms Innocent bystander Hapten the Ab was directed at the drug, but it cross reacted w/ RBCs Drug must be present for hemolysis to occur Quinine, Quinidine, Isoniazide Drug binding to RBC Abs that react to this complex Penicillins, Cephalosporins True autoimmune You don’t need the drug in the body any more to get the hemolysis Alpha-methyldopa, L-DOPA, Procainamide Cold Agglutinin Disease IgM antibodies bind to I antigens of RBCs when cold (falls off when warm) Causes agglutination cyanosis & ischemia of extremities Direct Coomb’s test + for C3, but not IgM! Has both intravascular and extravascular hemolytic components Primary, or associated w/ Mycoplasma, Mononucleosis, or lymphoproliferative disease Treat by avoiding cold & folate repletion Corticosteroid and splenectomies uneffective (big difference from warm antibody-mediated hemolysis) Non-Immune Hemolytic Anemia Classification Mechanical trauma to red cells Microangiopathic Hemolytic Anemia Abnormalities in heart and large vessels March Hemoglobinuria Infections Drugs, Chemicals, and Venoms Chemical & Physical Agents Causing Hemolysis “BAr CoIns” Severe Burns Arsenic Copper Insect and spider bites Infections Causing Hemolysis Malaroa Babesia microti Clostridium welchii Bartonella bacilliformis Basic Structure of All Human Hemoglobin Each hemoglobin molecule is composed of: 4 iron-containing, tetrapyrrole heme rings 4 polypeptide globin chains 2 alpha chains 2 non-alpha chains Each globin chain has 141 amino acids All non- chains have 146 amino acids There is considerable structural homology among the non-alpha chains Normal Human Hemoglobins Gower Hemoglobin (Embryonic) Fetal Hemoglobin (HbF) 22 Major Adult Hemoglobin (HbA) 2ε2 22 Minor Adult Hemoglobin (HbA2) 22 Heme Synthesis Begins with condensation of glycine & succinyl CoA -amino levulinic acid (-ALA). The rate-limiting step in heme synthesis Requires intra-mitochondrial enzyme ALAsynthase -ALA travels to cytoplasm; converted to porphobilinogen (PBG), a monopyrrole. PBG converted from monopyrrole to biologically active form protoporphyrin IX, a tetrapyrrole. Iron inserted into tetrapyrrole ring n the mitochondria Heme synthesis stimulated by iron & repressed when iron is inadequate (e.g., iron deficiency) Location of the Globin Genes • Genes for the non- chains are located on Chromosome 11. This is referred to as the globin gene cluster • Chain genes are located on Chromosome 16 • There is duplication of the genes for: • • Globin Globin (G and A) * • • G and A differ from one another only at position 136 where they have glycine & alanine respectively Synthesis of the non- chains involves a coordinated switching that proceeds from embryonic (ε) to fetal () to adult () globin chains • Yolk sac (ε) liver/spleen () marrow () Structure of the Hemoglobin Molecule Each Hb is comprised of 4 subunits: 2 identical chains & 2 identical non- chains Each chain is arranged in the form of an -helix with 8 individual helical segments (labeled A - H) Each globin molecule has both hydrophobic & hydrophilic areas The iron-containing heme ring is buried within a very hydrophobic region of the globin that is called the “Heme Pocket” The hydrophobic nature of this region protects the iron residue from oxidation, thereby maintaining it in the active, reduced form Each iron atom in the center of the heme residue is held in place and kept in the active, reduced Fe++ state by two histidine residues Possible Consequences of a Hemoglobinopathy No detectable effect Instability of the hemoglobin molecule An increase or a decrease in oxygen affinity Inability to maintain the heme iron in its active, reduced state (methemoglobinemia) Decreased solubility of the hemoglobin molecule Unstable Hemoglobinopathies Most of the unstable hemoglobinopathies involve a mutation in the region of the heme pocket These mutations enable water to gain access to this very hydrophobic region of the molecule The end result is heme instability, denaturation, and release of heme from its binding site The demonstration of Heinz Bodies in these red cells is evidence of the presence of an unstable hemoglobin mutant Hemoglobinopathy Altering Oxygen Affinity Increased Oxygen Affinity Stabilization of the Oxy conformation increases the oxygen affinity of the hemoglobin molecule The presence of such an effect can be confirmed by demonstrating a left shift in the Oxygen Saturation Curve Individuals with an increase in oxygen affinity typically exhibit erythrocytosis Decreased Oxygen Affinity Stabilization of the Deoxy conformation produces a decrease in the the oxygen affinity of the hemoglobin molecule The presence of such an effect can be confirmed by demonstrating a right shift in the Oxygen Saturation Curve Individuals with a decrease in oxygen affinity are typically somewhat anemic Hemoglobin M Diseases The Hemoglobin M disorders are seen when a substitution has occurred at the locus of either the proximal or distal histidine Typically, this involves a his tyr substitution which then forms an iron-phenolate complex Hemoglobin with its iron in the oxidized Fe+++ state is incapable of binding oxygen This form of hemoglobin (called Methemoglobin) has a brownish appearance Patients with Hemoglobin M disease are typically cyanotic The Sickle Cell Diseases: Inheritance, Appearance of Symptoms, Diagnosis - The most common sickle cell disease (SCD) is called sickle cell anemia (HbSS) However, there are a number of other SCD genotypes - compound heterozygous states The sickle mutation is inherited in an autosomal co-dominant fashion Individuals with sickle cell trait (AS) have roughly equal amounts of HbA & HbS and are generally asymptomatic Compound heterozygotes (e.g., SC or S-Thalassemia) generally express a significant sickle cell disease We dx/ with electrophoresis: - Hb C has a positive; HbS is neutral, HB A is negative. Movement: HbA > HbS > HbC Sickle Cell Anemia Pathophysiology The presence of the abnormal (or sickle) hemoglobin (HbS) within the cells of the affected individuals The decreased solubility & the tendency of this abnormal hemoglobin to polymerize when it assumes the deoxy conformation In HbS, the negatively charged glutamic acid at 6 position is replaced by an uncharged valine residue In deoxy conformation, the valine at 6 position approaches the phenylalanine at 85 position on adjacent HbS molecule. Multiple critical contact points that enable the hemoglobin molecules to attach to one another The polymer begins as a small nucleus of hemoglobin molecules aligned polymer with a total of 7 antiparallel pairs (or 14 individual hemoglobin chains) SICKLE CELL DISEASE Clinical Features Painful Vaso-occlusive Crises Strokes Retinopathy Acute Chest Syndrome Pulmonary Hypertension Sickle Cell Nephropathy Biliary Tract Disease Leg Ulcers Avascular Necrosis of the Large Joints SICKLE CELL DISEASE Therapeutic Approaches Reactivate Fetal Hemoglobin Production using Hydroxyurea! Chemical inhibition of Hb S polymerization Increase in intracellular hydration Altering RBC/Endothelial cell interactions Bone marrow transplantation Gene therapy What is this an example of? Typical Diseases? Megaloblastic Anemia Red cells are macrocytic. Hypersegmented neutrophils can be seen. Vitamin B12 or folate deficiency What Disease? Sickle Cell Anemia Target Cell Sickled Cell Platelet Function in Hemostasis – what is it called? Primary Hemostasis! What are the functions? (1) Adhesion •exposure to collagen; binding to von Willebrand factor via GPIb receptor (4) Aggregation and Surface Coagulation (2) Accumulation and Shape Change (3) Granule Content Release •ADP released, integrin activation, fibrinogen binding Main Types of Coagulation Factors •Zymogens/active enzymes: Vitamin K-dependent -- factors II, VII, IX, X Vitamin K-independent-- XI, XII, and XIII •Cofactors: factor V, factor VIII, tissue factor, and von Willebrand factor •Non-protein cofactors: calcium and phospholipid surfaces •Fibrinogen: fibrinogen is converted to fibrin by thrombin Laboratory Assays to Monitor Coagulation Parameters •Prothrombin Time (PT) •Activated Partial Thromboplastin Time (APTT) •Thrombin Clot Time (TCT) Appropriate tube to use for specimen? Citrate solution as anticoagulant. Stops clotting by binding calcium. Blood to additive ratio 9:1 Plasma, NOT Serum! Prothrombin Time (PT) XII “Extrinsic” Pathway XI IX VII VIII X “Common” Pathway V Prothrombin (II) Fibrinogen PT – what does it do? •Test plasma + tissue thromboplastin (source of tissue factor) + CaCl2 Fibrin clot •Tests extrinsic pathway: –Formation of tissue factor-factor VII complex to formation of fibrin. •Prolonged PT: –Deficiencies of factors II (prothrombin), VII, X, V, and fibrinogen. Activated Partial XII Thromboplastin Time (APTT) XI IX “Intrisic” Pathway VII VIII X “Common” Pathway V Prothrombin (II) Fibrinogen APTT – What does it do? •Test plasma + partial thromboplastin (lipid) + particulate activator + CaCl2 Fibrin clot •Tests intrinsic pathway: –Activation of factor XII to formation of fibrin •Prolonged APTT: –Deficiencies of factors XII, XI, IX, VIII, X, V, II, fibrinogen (and kallikrein and HMWK). Thrombin Clot Time (TCT) XII XI TCT – What does it do? •Test plasma + thrombin Fibrin clot IX VII VIII X V Prothrombin (II) Fibrinogen •Measures conversion of fibrinogen to polymerized fibrin •Sensitive to quantitative and qualitative fibrinogen deficiencies. Hemophilia A Factor VIII (VIII) ? Hemophilia B Factor X ? IXa (IX) VIIIa Factor Xa Fibrinogen Prothrombin Va Xa Thrombin Fibrin Thrombus Roles of Von Willebrand Factor von Willebrand Factor VIIIFactor (vWF) Primary Hemostasis Secondary Hemostasis von Willebrand Disease (vWD) How does it differ from classic hemophilia? (1) Suffer from mucocutaneous hemorrhage rather than hemarthroses like in hemophilia. (2) Autosomal inheritance trait, so men and women have similar prevalence, rather than X-linked like hemophilia. (3) Consistently had prolonged bleeding times unlike the normal bleeding times in hemophilia. Virchow’s Triad •Virchow (1845) thought that thrombosis was the result of abnormalities in: A) the vessel wall; B) blood flow, and C) the properties of blood. Thrombosis: 2 Types •Arterial: Injury to the endothelium; platelets adhere and a dense platelet aggregate is formed, and coagulation system activated. •”White thrombus” •Venous: Related to decrease blood flow (stasis); venous thrombosis is dominated by the coagulation system, the production of fibrin-rich thrombi. •”Red thrombus” Regulators of Blood Coagulation: 3 useful systems •Protein C Anticoagulant System: Thrombin-Thrombomodulin Protein C and S [Activated Protein C (APC) and Protein S] •Protease Inhibition by Antithrombin with Heparin: Antithrombin (ATIII) Heparin (drug)/Heparan Sulfate (naturally-occurring on vessel wall) •Fibrinolytic System: Tissue plasminogen activator (tPA) Plasminogen/Plasmin Plasminogen activator inhibitor-1 (PAI-1)/Antiplasmin How does the Protein C Anticoagulant System Work? Thrombin binds to thrombomodulin on vessel wall Thrombin, once bound to thrombomodulin, can no longer cleave fibrinogen into fibrin or activate factor V or platelets Thrombin-thrombomodulin complex activates Protein C (vit K dep) APC APC associates with Protein S APC + S cleaves/inactivates factors Va and VIIIa How Does the Antithrombin Anticoagulant System Work? Antithrombin = serine protease inhibitor (serpin) Circulates freely in the plasma Inhibits thrombin, IXa, Xa, XIa Activity increased by: Heparan sulfate (basement membrane) Heparin (drug) How does the Fibrinolysis/Clot Lysis Anticoagulant System Work? Plasminogen freely circulates in the plasma Endothelium secretes tissue-type plasminogen activator (tPA) tPA converst plasminogen plasmin Plasmin lyses clots Plasminogen activator inhibitor-1 (PAI-1) is secreted by endothelium PAI-1 downregulates tPA activity It’s another fine balancing act! Thrombosis Why heparin therapy? Inhibits further thrombus formation almost immediately. Why warfarin therapy? Depletes vitamin K-dependent factors to impair procoagulant function. Why tissue plasminogen activator therapy? Degrades thrombus to re-establish blood flow. Family history? Necessary to determine if familial or acquired clinical scenario. Common hereditary cause of venous thrombosis? factor V Leiden: a plasma protein “resistant’ to inactivation by the protein C system Some Acquired Causes of Venous Thrombosis Surgery and trauma Prolonged immobilization Older age Cancer Myeloproliferative disorders Previous thrombosis Pregnancy Use of contraceptives or hormone-replacement therapy Anti-phospholipid antibodies Anticoagulant Therapy: Medications •Heparin- Heparin binds to antithrombin, which converts it to a very potent and immediate inhibitor of thrombin, factor Xa and other proteases in the clotting cascade. IV •Warfarin (or Coumadin)- Oral anticoagulants produce their effect by interfering with the cyclic interconversion of vitamin K and its 2,3 epoxide (vitamin K epoxide). •Fibrinolytic enzymes- Induction of a fibrinolytic state by the infusion of plasminogen activators is used in massive pulmonary embolism and to restore the patency of acutely occluded arteries. Benefits of Low Molecular Weight Heparins (LMWH) •LMWHs have a higher affinity for antithrombinfactor Xa. •Longer plasma half-life. •Safe and effective for venous thromboembolism, and with unstable angina or acute thrombotic stroke. •Convenient, given subcutaneously without laboratory assay monitoring (allowing for patient and home care options). GENERAL Heparin Targets -Thrombin -IXa, Xa, XIa, XIIa -Measure efficacy with APTT Vitamin K Cycle and Effect of Warfarin •Vitamin K antagonists exert their anticoagulant effect by inhibiting vitamin K epoxide reductase and vitamin K reductase activities. •All vitamin K-dependent coagulant proteins are impaired: prothrombin, factor VII, factor IX, factor X, anticoagulant protein C and protein S •Oral anticoagulants cause hepatic production and secretion of partially and fully de--carboxylated and dysfunctional proteins. •Can reverse effects with emergency administration of Vitamin K •Monitored by PT; most closely reflects VII (shortest K-dep ½ life) •Must avoid use during pregnancy! •PO administration Treatment of Venous Thromboembolism •Treatment strategy differ between arterial from venous circulation. •Objective of treating/preventing venous thrombosis: -prevent extension of thrombus; -prevent thrombus from embolizing; -render fibrin more susceptible to fibrinolysis. (standard in threat of massive PE) -standard tx in acute venous thrombosis & PE heparin + oral vitamin K antagonists Arterial Thrombosis Main pathogenic mechanism for acute MI, unstable angina, sudden coronary death Tx: heparin, LMWH, warfarin, anti-platelet cmpds, fibrinolytics, ASPIRIN Clopidogrel and Ticlopidine inhibit platelet aggregation by blocking ADP receptor on platelet and inhibiting activation of GPIIb/IIIa. But Ticlopidine may cause TTP! Abciximab (ReoPro), Eptifibatide (Integrilin), Tirofiban (Aggrastat) bind GPIIa/IIIa receptor on platelets preventing fibrinogen binding What is this? What will it give rise to? A Megakaryocte that will shed off Platelets! Platelet Plug Formation Adhesion Aggregation Platelets stick to each other via fibrinogen bridges. GPIIb/IIIa to fibrinogen Activators: ADP, collagen, 5HT, Epi, TXA2, thrombin Providing a phospholipid scaffold for coagulation Secretion Platelets release granular contents and potentiate reactions, like generation clotting Spits out pro-clotting materials: ADP, Epi, factor V, offibrinogen Xa and thrombin! vWF, And another role of wall. platelets? Platelets stick to injured vessel GPIb/IX to vWF and collagen Thrombocytopenia three broad categories of causes Underproduction Peripheral Destruction Splenic sequestration If you saw this in a blood sample and were told the patient has too few platelets, what would you say? WRONG!!! It’s Pseudothrombocytopenia! Or in Dr. Ma’s words, you could say “damn, there’s more than one platelet on this field” YIKES! What’s this? Petechiae What’s the difference? ACK, and this? Purpura are formed when Purpura petichiae coalesce Thrombocytopenia – Underproduction Causes Marrow failure: myelodysplasia, aplastic anemia, vitamin deficiencies (B12/folate) Marrow infiltration: tumor, granulomatous diseases, fibrosis, leukemias, lymphomas Marrow toxins: drugs (esp. alcohol), radiation, infections Congenital: Wiskott-Aldrich Syndrome, Thrombocytopenia Absent Radius Syndrome (TAR), May-Hegglin DIC- Diagnosis Elevated PT - due to consumption of Factor VII, which has the shortest half-life (4 hrs) of all clotting factors. Low platelets Low/falling fibrinogen Elevated fibrin degradation products When advanced, the APTT can be prolonged as well, as the other clotting factor levels fall. (FDPs/FSPs) or D-Dimers Can see a few schistocytes on the peripheral smear in most cases. (MAHA) Low clotting factor levels DIC - Etiologies and Treatment Can be associated with: gram negative sepsis, severe burns, obstetrical disasters, certain leukemias or tumors, shock, insect or snake venoms TREAT THE UNDERLYING CAUSE!!! Supportive measures can include: transfusion of platelets clotting factors, fibrinogen +/- low dose heparin to halt thrombin generation. Low dose heparin can slow down the forest fire… TTP - Diagnostic Features (aka “The Pentad”) What’s normal in TTP that’s NOT in DIC? Microangiopathic Hemolytic Anemia (MAHA) – MUST BE PRESENT Elevated LDH, elevated bilirubin Schistocytes on the peripheral smear MUST BE PRESENT Low platelets - MUST BE PRESENT Fever Neurologic Manifestations - headache, sleepiness, confusion, stupor, stroke, coma, seizures Renal Manifestations hematuria, proteinuria, BUN/Creatinine PT, fibrinogen levels, FDPs/D-dimers TTP - etiology Associated with an antibody against or a deficiency of the protease (ADAMTS-13) that cleaves the very high molecular weight multimers of von Willebrand’s factor vWF accumulates abnormal platelet adhesion and activation Can be induced by drugs, including ticlopidine, quinine, cyclosporine, FK-506, mitomycin C Increased incidence with pregnancy or HIV TTP - Treatment Treatment relies on PLASMA EXCHANGE. Remove all inciting agents (ultra-high MW multimers of vWF) Restoring ADAMTS-13 Adjunct therapies, including glucocorticoids and anti-platelet agents can be used but are of uncertain benefit. Secondary measures if no response to plasma exchange include splenectomy, vincristine. AVOID PLATELET TRANSFUSIONS THEY “FUEL THE FIRE” Thrombocytopenia Drugs/Immune Mechanism Drugs can lead to immune-mediated thrombocytopenia by a variety of mechanisms. 1) directly stimulating anti-platelet antibody production 2) a hapten mechanism 3) “innocent bystander” phenomenon. Thrombocytopenia Drugs/Immune Mechanism “Qua – BASH” (don’t ask me, I’m sleepy) Quinine/quinidine Beta-lactam antibiotics Abciximab (ReoPro®) Sulfa drugs like Trimethoprimsulfamethoxazole Heparin ITP - Therapy Initial therapy relies on use of corticosteroids (e.g. prednisone). These can take 48-72 hrs to take effect. If platelet count is <10K or if patient is bleeding, need more rapid therapy--use IVIg If patient is Rh positive, can use Anti-D (WinRho®) in place of IVIg. (need a spleen) 2nd line – splenectomy 3rd line - immunosuppression Compare TTP, DIC, ITP TTP DIC ITP Low platelets Must have Usually Must have Schistocytes Must have Maybe Never PT/FDP/ Fibrinogen Normal Low n/A Transfuse platelets? NEVER Maybe Only if pt is bleeding Treatment Plasma exchange Tx under- Steroids, lying cause IVIG, AntiD Qualitative Platelet Disorders - Differential Congenital - Glanzmann’s thrombaesthenia - defect in IIb/IIIa Bernard-Soulier - defect in Ib/IX Acquired uremia Drugs - ASA, NSAIDs, antibiotics, ReoPro®, Herbs - ginkgo, garlic, Vitamin E Myeloproliferative diseases Diagnosis? Normal APTT/PT/TCT, prolonged bleed time Anti-Platelet Drugs Aspirin Thienopyridine Derivatives Inactivates COX-1, decreasing TXA2 (a platelet agonist) Prevent stroke, MI, CAD, peripheral arterial occlusion Ticlopidine, Clopidogrel Blocks ADP (platelet agonist) Ticlopidine may cause TTP GPIIb/IIIa inhibitors Abcizimab (ReoPro), Eptifibitide (Intergrillin), Tirofiban (Aggrastat) Blocks platelet aggregation by blocking fibrinogen receptor on platelets Donor screening criteria: Allogeneic (volunteer) Hgb >12.5 BP, pulse: healthy Uniform Donor screening questionnaire Infectious Disease Screening of donor Hepatitis B Hepatitis C HIV I/II HTLV I/II Syphilis Autologous (for self): Less stringent criteria Parts Collected Out of a Whole Blood Collection pRBC Platelet rich plasma (platelet concentrate) Plasma (FFP) pRBC Storage RBCs suspended in anticoagulant (citrate based) and, Additive Solution - AS Provides nutrients to support RBC metabolism 42 days = Shelf life Volume= 250 to 300 mL 65% RBCs, 35% plasma and AS contains WBC’s and some platelets may be frozen w/ glycerol (cryoprotectant) for 10 yrs pRBC Transfusion 1 unit = 1 g/dL Hb; 3% hematocrit Decide w/ clinical judgement NOT lab values Transfuse slowly, so you can catch adverse rxn RBCs should be infused alone or with 0.9% NaCl through a 170µm clot-screen filter NEVER mixed with : Calcium containing solutions Dextrose May cause clumping or clots Hypotonic,may cause hemolysis or clumping Medications Hypertonic solutions AVOID infusing with Lactated Ringers Fresh Frozen Plasma: Storage, Contents, Tx? Frozen w/in 8hrs of collection Stored -20º C for up to 1 year Once thawed, can be kept at 1-6º C for 24 hrs Contents: 1 unit/mL of all clotting factors including labile Factors V and VIII ~400 mg fibrinogen Citrate as anticoagulant No platelets Treatment of multiple coagulation factor deficiencies Massive transfusion Trauma Liver disease DIC Unidentified deficiency Platelets: Storage, Dosing, Treatment, Matching Pooled platelet concentrates (PC’s) from several whole blood donations or apheresis Suspended in citrated plasma Stored @ 20-24º C for 5 days only highly susceptible to shortages!!! One therapeutic dose platelet count 30-50k PLT surface Trace amts RBC’s Rh type important ABO antigens but not Rh Platelet specific Ags HLA- A and HLA-B Rh- female gets Rh- PLT Tx: thrombocytopenia, qualitative defects Monitor efficacy of transfusion via PLT count w/in 1hr of transfusion conserve resources… Cryoprecipitate: Contents, How to Get it, Tx, Dosing? Cold insoluble white precipitate Forms when FFP is thawed at 1-6º C Removed from FFP by centrifugation, then refrozen at –20º C CONTAINS: 80 to 150 IU Factor VIII:C (antihemophilic factor) 150 mg fibrinogen Von Willebrand Factor Tx: Deficiency of fibrinogen, Factor VIII Improve platelet function in uremia Dose calculation based on Patient’s weight and hematocrit : plasma volume Desired increase in Factor level ABO Blood Group: Population Frequency O A B AB 45% 41% 10% 4% What is ABO? specific terminal sugar residues on a large glycolipid backbone on the RBC membrane The ABO genes Codominant inheritence Encode for a glycosyl transferase enzyme Adds the specific terminal sugar to the glycolipid backbone Convey immunogenicity O = fucose A = N-acetyl galactosamine B = galactose ABO Discrepancy when the front and back types do not match Front = antigen on cells Back = antibody in serum Must resolve prior to transfusion Common Causes: Cold agglutinin Weak or absent antibodies in elderly or infants Interfering substance: protein, dextran Weak subgroup of A or B RECIPENT BLOOD TYPE RED CELLS PLASMA B Whose Whose O O, A, B, AB RBCs Plasma A, O A, AB Can they Can they B, O B, AB Take? Take? AB AB, A, B, O O A AB ABO Antibodies ABO Antibodies anti A , anti B Predominantly IgM >> IgG > IgA IgM reacts at room temperature Can bind complement intravascular hemolysis Naturally occurring Anti A and B form due to similar antigens in nature (bacteria, pollen, etc) Transfusion exposure or pregnancy NOT required IgM pentamer can agglutinate RBCs Immediate transfusion reactions possible Rh Blood Group: the 2nd most impt Rh System : family of 51 antigens Integral membrane proteins, well formed during fetal development Rh Antigens of routine importance: D, C, c, E, e Rh null are individuals lack all Rh proteins Clinically significant in: Transfusion practice Transfusion reactions Hemolytic disease of newborn D Antigen (Rh Type) D+ 85% prevalence D- 15% Highly immunogenic = Rh+ = Rh- Clinically significant with RBC transfusion & platelet transfusion Females of child bearing potential need Rh- blood Immune sensitization required to develop Rh system antibodies Transfusion or pregnancy IgG , react at 37° C - must incubate in the lab to demonstrate them Typically cause extravascular hemolysis, if present Some may activate complement and cause intravascular hemolysis Comparison b/tw ABO and Rh Blood Groups ABO IgM >> IgG > IgA Test at room temp Causes intravascular hemolysis Naturally occurring Bacteria, pollen Rh IgG Test at body temp Causes extravascular hemolysis Sensitization required Pregnancy, transfusion RBC Blood Groups and Antibodies Other protein blood groups Integral membrane proteins are well formed at birth Antibodies predominantly IgG Delayed Transfusion Reactions Kell K Duffy Fy a , Fy b Kidd Jk a, Jk b Extravascular hemolytic reactions Intravascular hemolysis more likely with Kidd Jka, Jkb due to complement binding Hemolytic disease of newborn Front & Back Types FRONT TYPE –what’s on the cells? BACK TYPE – what’s in the serum? Mix 2 drops of patient cells with 2 drops of reagent antibodies to A, B and D antigens in different test tubes, Agglutination indicates presence of antigen Mix 2 drops patient serum with both A and B reagent cells. Agglutination indicates presence of antibody Front and back types should match Antibody Screen Determines if patient has antibodies to the other major blood groups Requires Combining pt serum with 3 different RBCs with known blood group phenotype Incubate at 37 C to detect IgG antibodies Addition of Coombs serum Anti-human IgG : enables in vitro agglutination if IgG present due to monomeric structure of IgG If screen is +, antibody specificity is determined by a more extensive panel of testing RBCs Types of Crossmatch Immediate Spin Crossmatch Full Crossmatch Rapid, room temp mixing of patient serum with donor RBCs to confirm ABO compatibility For patients with antibodies Requires incubation and Coombs serum to confirm the patient’s IgG will not react with donor RBCs Electronic Crossmatch Alternative for Immediate spin crossmatch for patients without antibodies Special Circumstances Emergency Release When delaying transfusion poses risk of death Insufficient time to perform type screen and crossmatch Requires MD signature Conditional Release Blood may be crossmatch compatible however, blood bank testing is incomplete or cannot completely resolve antibody testing (ex: warm auto antibody) Requires MD signature Component Modifications Leukocyte reduction Filtration with specialized leukocyte removing filters 3 log leukocyte reduction Prevent CMV transmission Prevent alloimmunization to leukocyte antigens for those w/ chronic transfusion Prevent recurrent febrile non-hemolytic transfusion reactions Washing Removal of plasma by washing RBC or platelets with saline For prevention of severe allergic reactions Anaphylaxis IgA deficiency Time consuming , labor intensive, delays transfusion, decreases transfusion increment slightly Does not substitute for leukocyte reduction Irradiating Prevent graft versus host disease GVHD (Transfusion is a transplant) Indicated in severe immunodeficiency settings BMT Hematopoietic malignancies undergoing chemotherapy Premature infants Severe combined immunodeficiency Blood products from relatives must also be irradiated due to HLA antigens Adverse Effects of Transfusion Acute Immune Transfusion Reactions < 24 hours Allergic Hemolytic Febrile, non-hemolytic Anaphylactic Transfusion related acute lung injury (TRALI) Delayed Immune Transfusion Reactions > 24 hours Hemolytic GVHD Platelet refractoriness Post transfusion Purpura (development of anti-platelet antibodies) Acute Non-Immune Transfusion Reactions Circulatory Overload (Volume excess) Septic shock from bacterial contamination of blood product Delayed Non-Immune Transfusion Reactions Iron Overload Infectious Disease transmission Suspected Transfusion Reaction Hemolytic reaction symptoms are not specific and include: Fever Chills Hypotension Oozing from IV site Back pain Hemoglobinuia – red urine If any of these occur STOP transfusion, provide appropriate supportive care, notify blood bank Send repeat samples for blood bank evaluation DO NOT restart the unit Exceptions: mild urticaria that responds to antihistamine White Cells What are the cell types? Granulocytes Neutrophils Band forms Eosinophils Basophils Lymphocytes Monocyte/Macrophages ID the cell! Band Cell Eosinophil Basophil Monocyte Lymphocyte Neutrophil What is this? What does it do? Neutrophils! PMNs (polymorphonuclear neutrophils) polys segs (short for segmented neutrophils) Most common white cell Pale pink granular cytoplasm with condensed, segmented nucleus 7 hr ½ life Functions include chemotaxis, phagocytosis, killing of phagocytosed bacteria What is this? What does it do? Eosinophil Granulocytes with large, refractile, orange-pink granules. Nucleus is typically bilobed. Functions include all PMN functions, Chemotaxis Phagocytosis Killing of phagocytosed bacteria serving as effector cells for antibodydependent damage to parasites, regulation of immediate-type hypersensitivity reactions inactivation of histamine and leukotrienes released by basophils and mast cells What is this? What does it do? Basophils Large, dark blue granules which overlie the nucleus. The most uncommon of all granulocytes Functions include mediation of immediate-type hypersensitivity modulation of inflammatory responses by releasing heparin and proteases Precursor of tissue mast cells What is this? What does it do? Lymphocyte Lymphocytes have an oval nucleus, with a thin rim of blue cytoplasm. There may be a few very fine purplish-red granules. The nuclear border is smooth. Functions in immune regulation and production of hematopoietic growth factors. Functions in immune regulation and production of hematopoietic growth factors. What is this? What does it do? Monocyte Largest white cell normally found in the periphery Has a folded nucleus with uneven countour Slate grey cytoplasm--there may be vacuoles Functions Include: chemotaxis, phagocytosis, killing of some microbes, antigen presentation, release of IL-1 and TNF, which stimulates bone marrow stromal cells to produce growth factors, including: GM-CSF, G-CSF, M-CSF, and IL-6. Precursors of tissue macrophages Causes of Elevated Neutrophil Count Physiologic – exercise, pregnancy, lactation, neonates Acute infections Acute inflammation – surgery, burns, infarcts, crush injuries, acute gout, rheumatoid arthritis Acute hemorrhage Non hematologic malignancies Myeloproliferative disorders, esp CML Drugs: corticosteroids, G-CSF, lithium Misc: seizures, electric shock, post-splenectomy, Leukocyte Adhesion Deficiency Causes of Neutropenia Physiologic - in African-Americans Drugs – anti-psychotics, anti-epileptics, anti-thyroid, and some antibiotics (gold, sulfa) Chemotherapy Infections: viral, overwhelming bacterial sepsis, TB, fungal Immune - lupus, rheumatoid arthritis (Felty syndrome) Familial Hypothyroidism, hypopituitarism What is Agranulocytosis? Major Sxs? This is the complete or near-complete absence of neutrophils in the peripheral blood, with a normal platelet count and hgb Almost always drug-induced Clozaril (and other newer antipsychotics) Propythiouracil (antithyroid) Anti-convulsants Sulfa and chloramphenicol antibiotics Causes severe necrotizing ulcers in the mouth and throat What is basophilia a major symptom of? chronic myeloproliferative diseases Causes of Eosinophilia Differential Diagnosis for elevated eosinophil count “NAACP”: Neoplasm, Allergy/asthma, Addison’s disease, Collagen vascular disease Parasites Causes of Lymphocytosis Viral infections Bacterial infections - whooping cough (pertussis), TB, syphilis, brucellosis Chronic Lymphocytic Leukemia (CLL) Lymphomas and Waldenstrom’s macroglobulinemia Causes of Lymphocytopenia Immunodeficiencies, including HIV/AIDS Immunosuppresive drugs, including corticosteroids Lymphomas Granulomatous diseases, including sarcoid, TB Alcoholism, malnutrition, zinc deficiency Causes of Monocytosis Bacterial infex: TB, syphilis, subacute bacterial endocarditis, typhoid, brucellosis Protozoal infex: malaria Rickettsial infex: RMSF, typhus Myelodysplastic syndromes (just one of them) Leukemias Inflammatory bowel disease Normal Neutrophil Function Adherence Chemotaxis Moving along a concentration gradient to higher [ ]s Recognition/Phagocytosis Rolling mediated by selectins Adhesion mediated by beta-2 integrins Via complement and IgG Once it eats, it’s got a phagosome Degranulation Granules are released INTO the phagosome NADPH oxidase: O2 O2Superoxide dismutase: O2- H2O2 MPO: H2O2 HOCl Oxidative Metabolism and Bacterial Killing Defects in Neutrophil Function Acquired Defects Corticosteroid Use Alcoholism Leukemias Myelodysplasia Myeloproliferative disorders Congenital Defects Leukocyte Adhesion Deficiency Chronic Granulomatous Disease Myeloperoxidase Deficiency Chediak Higashi Syndrome Erythrocytosis – What is it? An increase in the number of circulating RBCs per volume of blood. Reflected as an elevated hemoglobin and hematocrit. Relative Erythrocytosis (Gaisbock’s syndrome) Depressed plasma volume, RBC mass is normal Common in middle age men w/ HTN, smoking hx Secondary Erythrocytosis >60% in man, >57% in woman = true erythrocytosis Red cell mass study if elevated but not quite these levels Erythropoietin production increased by kidney/liver Tissue hypoxia, tumors, genetic disorders, drugs for athletics will all increase Epo Primary Erythrocytosis The bone marrow is going crazy without outside input Why is erythrocytosis bad? Hyperviscosity Syndrome Your blood gets too sludgy Symptoms include: Headaches Visual changes Tinnitus Dizziness Paresthesias Decreased mental acuity Erythrocytosis due to appropriate increases in epo Life at high altitude High affinity hemoglobins Cardiopulmonary disease Obesity-Hypoventilation syndrome Obstructive sleep apnea High carboxyhemoglobin levels What are the Myeloproliferative Diseases? Definition? Associated sxs? Includes: Myeloproliferative disorders are Stem Cell Disorders leading to autonomous production of hematopoietic cells from ALL THREE LINEAGES (red cells, white cells, platelets). All of these disorders are clonal (except for a subset of ET cases) Associated sxs: Polycythemia vera Essential Thrombocythemia Myelofibrosis Chronic Myelogenous Leukemia Basophilia Splenomegaly Potential to develop into AML Polycythemia vera Most of cells in circulation are derived from a single, neoplastic stem cell Does not need Epo to produce more cells… Diagnosis based on low/absent levels of Epo Natural History – 4 phases: Latent phase - asymptomatic Proliferative phase -pts may have sxs of: Hypermetabolism Hyperviscosity Thrombosis Spent phase - red cell mass, anemia, leukopenia, secondary myelofibrosis, increasing HSM. 20% of pts Secondary AML aka when the body says “screw it, I’m not differentiating anymore” 1-2% of pts treated with phlebotomy alone Symptoms of Polycythemia Vera Those common to ALL erythrocytosis Pruritis after bathing Hypermetabolic sxs Erythromelalgia Thrombosis Hemorrhage PE findings Headache Decreased mental acuity Weakness Facial plethora Splenomegaly Hepatomegaly Retinal vein distension Lab findings BASOPHILIA Low EPO levels Increased Hbg/HCT, WBCs, platelets, uric acid, B12, leukocyte alkaline phosphatase score P vera - Treatment Phlebotomy – Draw 500 cc blood 1-2x/wk to target Hct 45%; maintain BP w/ saline Generally, the best initial treatment for P vera – rapid onset Downsides: Increased risk of thrombosis No effect on progression to spent phase May be insufficient to control disease Myelosuppressive agents Hydroxyurea can be used in conjunction with phlebotomy May increase the risk of leukemic transformation from 1-2% to 4-5% 32P – kills some of the proliferating cells! increase the risk of leukemic transformation from 1-2% to 11% Single injection may control hemoglobin and platelet count for a year or more. Alkylating agents such as busulfan Interferon alpha Benefits No myelosuppression No increase in progression to AML No increase in thrombosis risk Drawbacks Must be given by injection up to daily Side effects may be intolerable in many pts: flu-like symptoms, fatigue, fever, myalgias, malaise So what disease are you thinking? Arrow indicates a giant platelet, larger than the red cells or lymphocyte Essential Thrombocythemia Essential Thrombocythemia Increased megakaryocyte production of platelets Must exclude secondary causes of thrombocytosis and other myeloproliferative disorders Major complications: Thrombosis: 20-30% of all patients; Budd-Chiari Microvascular thrombi/digital ischemia Pruritis & erythromelalgia Acquired von Willebrand’s disease Will see clusters of abnormal megakaryocytes on smear Platelet morphology will be big and odd-shaped Treatment: Anagrelide – platelet lineage specific Hydroxyurea Interferon alpha Myelofibrosis Clonal stem cell disorder affecting megakaryocytes predominantly All myeloproliferative disorders can result in a spent phase which can be difficult to distinguish from primary MF Myeloid metaplasia refers to earlier proliferative phase where extramedullary hematopoiesis predominates. WILL become AML median survival is 5yrs Splenomegaly and hepatomegaly Aspirate is a “dry tap” Peripheral blood smear: leukoerythroblastic Teardrop RBCs Nucleated RBCs Early granulocytes/precursors Myelodysplastic Syndromes Definition: disordered maturation in 1 or more cell lines producing cytopenias: anemia, leucopenia, thrombocytopenia or combos More common in the elderly Based on cytogenic abnormalities Peripheral cell abnormalities Macrocytic RBCs Large platelets Hypogranular or bilobed nuclei neutrophils Megaloblastic erythropoeisis Ringed sideroblasts Abnormal nucleus of RBC precursors (dyserythropoiesis) Small megakaryocytes with abnormally hypolobate nuclei Blast cells should account for <30% of marrow cells