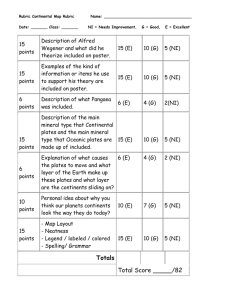
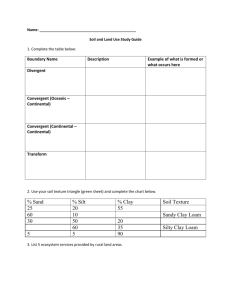
Laboratory 1
advertisement

Laboratory 1 Sampling and Field Measurement Introduction Analysis of the environment, whether chemical, physical or microbiological, begins with sampling the area of interest. It is important to understand how different factors involved in sampling influence the results of your analysis. One of these factors is where the sample is taken. Soil is particularly heterogeneous so the exact location and depth can contribute to variability of your results. Water is much more evenly mixed but its properties can change with depth and position in a body of water. Another important factor is the method of sampling. For instance, taking soil cores can lead to increased compaction and disturbance of aggregates among other things. Finally, storage and handling of samples after collection is very important. Removing soils or water from their original environment will result in changes in the samples’ properties. Changes in temperature and moisture etc. need to be controlled in order to minimize effects on the sample. This is particularly important for microbiological analysis. When you are designing an experiment it is important to try to anticipate how your analyses may be affected by the choices that you make about what gets sampled, how it is sampled and how the sample is handled after collection. The methods that you choose should be those that are likely to produce the most meaningful results. Another important consideration when sampling is to characterize the site that samples are taken from so that your results can be placed into an environmental context. This characterization can include location, depth, temperature, time and chemical properties among others. In this lab we will collect soil, sediment and water samples to be used in later experiments. We will also use a field portable lab kit to measure some of the physical and chemical properties (temperature, pH, turbidity, nitrate, ammonia and orthophosphate) of surface water at the sampling site. Materials Equipment - Hach portable lab (w/ turbidometer) - Soil corer - Water dipper - Trowel - Cooler - Rubber boots - Ruler Supplies - Sterile 1.0 liter bottles - Sterile sediment jars - Sterile funnel - Whatman No. 1 paper filters - Gloves - Paper towels - Waste bottles - markers Procedures Soil samples 1. 2. 3. 4. Use the soil corer to remove a soil core from a grassy area. Record the depth and location that the sample was taken from. Carefully remove any grass and large plant roots. Transfer the sample to a sterile jar, cap and put in the cooler. Sediment samples 1. 2. 3. 4. Locate an area near the shore of the pond with soft sediment. Use the soil corer or trowel to collect sediment from beneath the surface layer. Record the depth and location. Fill a sterile jar to the top, cap tightly and put in the cooler. Water samples 1. Locate the outlet of the pond. 2. Use the dipper and funnel to fill one sterile jar (for bacterial analysis) and put it in the cooler. 3. For the second bottle, put a piece of No. 1 filter paper in the funnel (this filtered water will be used for chemical tests). This jar only needs to be filled halfway. Water quality tests Temperature - Measure the temperature of the water at the point of sampling (top 6 inches) - Follow the instructions for the portable pH meter to measure the pH of each water sample. pH Turbidity 1. 2. 3. 4. 5. Shake the unfiltered water sample well to mix. Pour 5 ml of sample into the turbidimeter sample cell and cap. Wipe the outside of the cell with the oiling cloth. Apply a drop of silicone oil to the sample cell and wipe clean with the oiling cloth. Insert the sample cell into the instrument and cover the cell with the instrument cap. 6. Press and hold the [READ] key until the reading stabilizes. 7. Release the [READ] key and record the value. Nitrate [Method 8192, Cadmium reduction, Low Range (0 - 0.50 mg/L NO3-N)] 1. Press [PRGM] key. The display will read “PRGM?”. 2. Press [5] [5] [ENTER]. The display will read “mg/L, NO3-N” and the ZERO icon. 3. Fill a 25 ml graduated mixing cylinder to the 15 ml mark with sample. 4. Add the contents of one “NitraVer 6” packet to the cylinder and stopper. 5. Press [TIMER] [ENTER]. A 3-minute reaction period will begin. Shake the cylinder vigorously throughout this time period. 6. When the timer beeps, the display will read “2:00 TIMER 2”. Press [ENTER]. A 2-minute reaction period will begin. 7. When the timer beeps, pour 10 ml of the sample into a sample cell (be careful not to transfer any cadmium particles). 8. Add the contents of one “NitraVer 3” packet to the sample cell. Cap the cell and shake gently for 30 seconds. 9. The display will read “15:00 TIMER 3”. Press [ENTER]. A 15-minute reaction period will begin. 10. Fill another sample cell (the blank) with 10 ml of sample. 11. When the timer beeps, place the blank into the cell holder and tightly cover with the instrument cap. 12. Press [ZERO]. The cursor will move to the right and the display will read “0.00 mg/L NO3-N” 13. Remove the blank and place the sample into the cell holder. Cover the sample cell with the instrument cap. 14. Press [READ]. The cursor will move to the right and the result will be displayed. Record this value. Ammonia [Method 8155, Salicylate (0 – 0.50 mg/L NH3-N)] 1. Press [PRGM] key. The display will read “PRGM?”. 2. Press [6] [4] [ENTER]. The display will read “mg/L, NH3-N” and the ZERO icon. 3. Fill a sample cell with 10 ml of deionized water (the blank). 4. Fill a second sample cell with 10 ml of the sample. 5. Add the contents of one “Ammonia Salicylate” packet to each sample cell. Cap the cells and shake gently until dissolved. 6. Press [TIMER] [ENTER]. A 3-minute reaction period will begin. 7. When the timer beeps, add the contents of one “Ammonia Cyanurate” packet to each sample cell. Cap the cells and shake gently to dissolve. 8. The display will read “15:00 TIMER 2”. Press [ENTER]. A 15-minute reaction period will begin. 9. When the timer beeps, place the blank into the cell holder and tightly cover with the instrument cap. 10. Press [ZERO]. The cursor will move to the right and the display will read “0.00 mg/L NH3-N” 11. Remove the blank and place the sample into the cell holder. Cover the sample cell with the instrument cap. 12. Press [READ]. The cursor will move to the right and the result will be displayed. Record this value. Orthophosphate [Method 8048, Ascorbic Acid (0 – 2.5 mg/L PO43-)] Press [PRGM] key. The display will read “PRGM?”. Press [7] [9] [ENTER]. The display will read “mg/L, PO4” and the ZERO icon. Fill a sample cell with 10 ml of the sample. Add the contents of one “PhosVer 3” packet to the sample cell. Cap the cell and shake for 15 seconds. 5. Press [TIMER] [ENTER]. A 2-minute reaction period will begin. Perform steps 6-8 during this period. 6. Fill another sample cell (the blank) with 10 ml of sample. 7. Place the blank into the cell holder and tightly cover with the instrument cap. Press [EXIT]. 8. Press [ZERO]. The cursor will move to the right and the display will read “0.00 mg/L PO4”. Remove the blank. 9. After the timer beeps, place the sample into the cell holder. Cover the sample cell with the instrument cap. 10. Press [READ]. The cursor will move to the right and the result will be displayed. Record this value. 1. 2. 3. 4. Nitrite [Method 8507, Diazotization, Low Range (0 - 0.350 mg/L NO2-N)] 1. Press [PRGM] key. The display will read “PRGM?”. 2. Press [6] [0] [ENTER]. The display will read “mg/L, NO2-N” and the ZERO icon. 3. Fill a sample cell with 10 ml of the sample. 4. Add the contents of one “NitraVer 3” packet to the sample cell. Cap the cell and shake gently until dissolved. 5. Press [TIMER] [ENTER]. A 15-minute reaction period will begin. 6. Fill another sample cell (the blank) with 10 ml of sample. 7. When the timer beeps, place the blank into the cell holder and tightly cover with the instrument cap. 8. Press [ZERO]. The cursor will move to the right and the display will read “0.000 mg/L NO2-N” 9. Remove the blank and place the sample into the cell holder. Cover the sample cell with the instrument cap. 10. Press [READ]. The cursor will move to the right and the result will be displayed. Record this value. LAB #1 (Due 9/08/09) 10 points Name: Date: Time: Sample site: SOIL Location: Depth: Description: SEDIMENT Location: Depth: Description: WATER Location: Depth: Temperature (C): pH: Turbidity (NTU): Orthophosphate: Nitrate (mg/L NO3-N): Nitrite (mg/L NO2-N): Ammonia (mg/L NH3-N): Laboratory 2 Media Preparation and Culture Handling Introduction In order to study environmental bacteria using traditional methods, it is necessary to isolate individual species and be able to cultivate them in the laboratory. Isolation is usually achieved by diluting a sample containing the organism until single cells are separated from each other. The dilution can be done by serial dilution in liquid media, by spreading the sample very thinly over the surface of an agar plate (streaking) or a combination of both. Once the cell is isolated from all others, it then needs to be able to grow and form a colony. Because the resulting colony is made up of only the offspring from one cell it is considered to be a pure culture. For some species, this is a relatively simple process. They grow readily on artificial media and have simple growth requirements. Other species require highly specific culture conditions and may only grow in the presence of other organisms. It is important to be aware that any culture media that is used will impose a selective pressure on the bacteria inoculated into it. An appropriate medium needs to be chosen for the organisms you wish to grow. In this lab we will be preparing a general nutrient media (both liquid and solid) and practicing aseptic culture handling technique and isolation. Materials Equipment - autoclave - balance - pH meter or pH strips - water bath (50C) - bunsen burners Chemicals and Reagents - agar - tryptic soy broth - 1N HCl - 1N NaOH - 95% Ethanol Supplies - pipettes (1ml and 10ml) - 16 X 150 culture tubes w/caps - graduated cylinders (1l) - Ehrlenmyer flasks (500 ml and 1l) - foam plugs - Petri plates - inoculating loops and spreaders - TSB plates and broth Cultures - Enterobacter aerogenes (plates) - Serratia marcescans (plates) - Mixed S. marcescans and E. aerogenes (broth and plates) Procedures Solid Media 1. Measure 250 ml of water in a graduated cylinder and pour it into a 500 ml Ehrlenmyer flask. 2. Mark the level of the water and empty the flask. 3. Weigh out media components according to the recipe. 4. Add the media components and 225 ml of distilled water to the flask and shake to dissolve (warm the flask in a water-bath if necessary). The agar will not dissolve. 5. Adjust the pH to ~7.2 with 1N HCl or NaOH if necessary. 6. Add distilled water to bring the level up to the 250 ml mark. 7. Plug the flask with a foam plug and cover loosely with aluminum foil. 8. Autoclave for 20 min. at 121C. 9. Place the media in a water bath and allow it to cool to ~50C. 10. Carefully pour the media into sterile petri plates being careful to avoid contamination. 11. Allow the plates to cool. Recipe: Tryptic Soy Media (0.25 liter) Tryptic soy broth Distilled water Agar 7.5 g 250 ml 3.75 g (1.5%) Liquid Media 1. Weigh out media components as for the solid media but leave out the agar. 2. Add the media components and 200 ml of distilled water to a pre-marked flask and shake to dissolve. 3. Adjust the pH to 7.2 with 1N HCl or NaOH if necessary. 4. Add distilled water to bring the level up to the 250 ml mark. 5. Using a glass pipette, dispense the media into 16 X 150 culture tubes (9 ml per tube) and cap. 6. Autoclave for 20 min. at 121C. Streak plates 1. Streak out three TSA plates (use the K, T and quadrant streaks) with colonies from the mixed E. aerogenes and S. marcescens plates. 2. Incubate at 25C for three days. Spread plates 1. Make a dilution series of the mixed broth into liquid TSB media. a. Aseptically transfer 1 ml of the culture into a tube containing 9 ml of TSB medium. b. Transfer 1 ml of the resulting tube into another tube with 9 ml of medium. c. Repeat this process until you have produced 5 tubes. (10-1 – 10-5). 2. Spread 0.1 ml of each dilution tube onto a TSA plate. 3. Incubate at 25C for three days. Dilution Series Streak Patterns LAB #2 (Due 9/15/09) 10 points Name: Date: Instructor’s approval Liquid Media Solid Media Streak Plates Isolated colonies? Spread Plates Isolated colonies? Questions: List three characteristics of the Tryptic Soy Agar plates and the way that we incubated them that impose a selective pressure on the bacteria that we plated on them. Why is it necessary to autoclave media before inoculating? Laboratory 3 Selective and Differential Media Introduction It is often helpful to take advantage of the selective nature of different types of bacterial media and incubation conditions in order to discourage the growth of organisms other than the ones you are interested in. For instance, by choosing to incubate plates under anaerobic conditions, you can easily exclude all of those bacteria that require oxygen. Some media are made even more selective by the addition of specific growth substrates or inhibitors. The dyes eosin and methylene blue in m-Endo agar discourage the growth of Gram positive bacteria. Cetrimide is an antiseptic compound that kills many bacteria but some species of Pseudomonas are resistant to it. In addition to being selective, bacterial growth media can also be differential. This means that components of the media will undergo a visible reaction in the presence of certain organisms but not others. This allows us to differentiate between species. Cetrimide agar contains high levels of potassium and magnesium that encourage the production of pigments in certain strains of Pseudomonas so that we are able to distinguish Ps. fluorescens from other colonies able to grow in the presence of cetrimide. In m-Endo agar those bacteria that can ferment lactose will show up as dark red because a change in pH causes the dyes to change color. If fermentation is very strong, a metallic sheen will develop. This is one of the most common ways to detect coliform bacteria. In this lab we will be inoculating different species of bacteria onto several types of selective and differential media and observing their reactions. We will also be plating our soil samples onto these same media. Materials Equipment - incubator (37C) - bunsen burners Supplies and Chemicals - inoculating loops and spreaders - pipettes (1ml) - TSA plates - Cetrimide agar plates - m-Endo plates - thioglycollate broth - sterile water (100ml) 95% Ethanol Cultures - Escherichia coli (plates) - Pseudomonas fluorescens (plates) - Enterobacter aerogenes (plates) - Serratia marcesens (plates) - Clostridium sporogenes (broth) - Bacillus megaterium (plates) - Soil samples Procedures Differential Plates 1. Divide two plates of each type (TSA, Cetrimide agar, and m-Endo) into three sections each. 2. Using strict aseptic technique, streak each of the sections with one of the bacterial cultures by picking a single colony from the plate (use the broth for Clostridium sporogenes). 3. Incubate the plates upside down at 37C for 24 hrs. 4. Record whether or not growth occurred, and what reactions were evident on the plates. Soil plates 1. Prepare a soil slurry by mixing 1 gram of soil in 100 ml of sterile water and shake vigorously. 2. Aseptically remove 0.1 ml of the slurry and spread across the surface of each type of plate (TSA, Cetrimide agar and m-Endo) Selective Broth 1. Label three thioglycollate media tubes, one with the name of each organism (E. aerogenes, B. megaterium and Clostridium sporogenes). 2. Use an inoculating loop to transfer each culture from broth or plate into a tube of thioglycollate media. Make sure to inoculate all the way to the bottom of the tube. 3. Incubate at 37C for two days. 4. Record the position of growth for each tube. LAB #3 (Due 9/17/09) 10 points Name: Date: Differential Plates (record growth and reaction) TSA Cetrimide m-Endo Escherichia coli Psuedomonas fluorescens Enterobacter aerogenes Serratia marcesens Clostridium sporogenes Bacillus megaterium Soil Plates Observations TSA Cetrimide m-Endo Selective Broth E. aerogenes C. sporogenes Position of Growth Questions: Describe the difference between Selective and Differential media. B. megaterium Laboratory 4 Microscopy I (Protozoa) Introduction Although it is nearly impossible to specifically identify microorganisms by their appearance, microscopy is still an important tool in environmental microbiology. General classes of organisms (filaments, spirochetes, cocci etc.) can be distinguished by their appearance and certain features such as motility can be seen directly. We can often also tell how different organisms are associated with each other. By using differential stains, we can tell even more about the organisms (Gram reaction, spore formation, flagellation etc.). In order to get good images of microbes, though, we need to understand how to use the microscope correctly. In our microscopes, the light from a lamp is focused by the condenser and passes through the sample, then the image is magnified by the objective and ocular lenses. The quality of image that you see will depend on: 1) how well the incoming light is focused 2) the quality of sample preparation and 3) the quality and focus of the lenses. The factor that usually has the least attention paid to it is the focus of the incoming light. There are two types of illumination that are most often used for viewing bacteria. The first is called “bright-field” illumination where the incoming light is all focused on the same plane in the sample. This minimizes the amount of interference and maximizes the resolution. It is best used for viewing stained samples. For live mounts or other unstained samples, “phase-contrast” illumination is used. In this method, the incoming light is split in two and arrives at the sample in slightly different planes. This has the effect of giving contrast to the otherwise transparent microbes. In this lab we will be practicing the proper use of microscopes and using them to view several different types of protozoa. Materials Equipment - microscopes Supplies - prepared slides of various protozoa - slides and cover slips - Pasteur pipettes Cultures - pond water Procedures Adjusting the Microscope 1. Turn on the lamp and place a slide with an “X” drawn on with wax pencil on the stage. 2. Switch the objective lens to 20X and rotate the condenser turret to the “A” setting. 3. Adjust the lamp brightness. 4. Turn each diopter until the bottom of the ring lines up with the engraved base line. 5. Adjust the ocular lenses so that a single image is visible then carefully focus until the image is clear. 6. Adjust the aperture diaphragm to get the clearest image. 7. Focus the individual ocular lenses by using one eye at a time to look at the image and turning the diopter ring to focus. 8. Refocus with the main focus knob and repeat step 7. 9. Move the slide out of the field of view without refocusing. 10. Place a business card over the surface of the lamp so that one half of the field of view is dark. 11. Use the condenser focus knob to move the condenser up or down until the edge of the card is in sharp focus. 12. Move the slide back into place and check the focus. Prepared Slides 1. Use the microscope to locate and identify protozoa from prepared slides. 2. Draw each of the four types of protozoa. Environmental Samples 1. Locate different kinds of protozoa in the environmental samples. 2. Draw at least two of the protozoa. LAB #4 (Due 9/17/09) 10 points Name: Date: Cilliate Dinoflagellate Amoeba Cryptosporidium or Giardia Environmental sample I Environmental sample II Identity:___________________________ Identity:___________________________ Laboratory 5 Microscopy II (Bacteria and Staining) Introduction Bacteria are very small and generally colorless. This makes them very difficult to view under the microscope. In order to effectively view most live bacteria it is necessary to use a phase-contrast microscope with an oil immersion lens to reduce distortion. It is often much easier to see details of a bacterium’s morphology when the cells are fixed and stained. Stains can be simple, where the dye used stains all of the cells equally, or they can be differential. Differential stains are designed to stain certain cells or parts of cells differently than others. The Gram stain is a differential stain that distinguishes between cells with a thick cell wall from those with a thin wall and an outer membrane. The crystal violet / iodine complex is retained within the thick peptidoglycan layer of Gram-positive cells but is easily washed out of the thin walls of Gram-negative cells by alcohol. By using these and other differential stains it is possible to visualize cell structures that are not otherwise visible. Materials Supplies - inoculating loops - slides and coverslips - staining racks and trays - Pasteur pipettes and bulbs Cultures - Escherichia coli (plates and broth) - Ps. fluorescens (broth) - Serratia marcescans (broth) - S. cerevisia (brewers yeast) (broth) - Bacillus megaterium (plates and broth) Reagents - crystal violet - Gram’s iodine - safranin - decolorizing solution Equipment - phase-contrast microscopes - bunsen burner Procedures Wet Mounts (Yeast and 2 Bacteria) 1. Using aseptic technique, transfer an inoculating loop full of broth culture onto a microscope slide. 2. When viewing a sample from an agar plate, first put a small drop of water on the slide then touch the colony with your inoculating loop and mix it into the water. The water should be only slightly cloudy. 3. Carefully place a cover slip over the sample. 4. View under the microscope. Simple Stain (2 Bacteria, separate) 1. 2. 3. 4. 5. 6. 7. Prepare smears in the same way as for wet mounts but do not add a cover slip. Allow the smear to dry completely. Fix the cells by passing the slide through a bunsen burner flame. Flood the smear with crystal violet stain and let it sit for 60 seconds. Rinse the slide with water, being careful to not wash the cells off. Blot the slide dry. View under the microscope. Gram Stain (E. coli and Bacillus, separate and mixed) 1. 2. 3. 4. 5. 6. Prepare and fix cells in the same way as for the simple slide. Flood the smear with crystal violet stain and let it sit for 60 seconds. Rinse the slide with water, being careful to not wash the cells off. Flood the slide with iodine and let it sit for 60 seconds. Rinse with water. Decolorize by dripping the decolorizing solution across the smear until no more color runs out (~30 sec.). 7. Immediately rinse with water. 8. Flood the slide with safranin and let it sit for 60 seconds. 9. Rinse with water and blot the slide dry. 10. View under the microscope. LAB #5 (Due 9/22/-09) 10 points Name: Date: Instructor’s approval Wet Mounts Simple Stain Gram Stain Questions: What is the purpose of passing the microscope slides through a flame before staining? What is the purpose of the decolorizing step in the Gram’s stain? What will happen to bacterial cells if the slide they are on is heat fixed before it is completely dry? Laboratory Setup Soil Microcosms Materials Equipment - Balance - Scissors Supplies - Plastic flowerpots - Dry, sieved soil - Yeast extract - Fertilizer (15-30-15) - Markers - Filter paper - Microscope slides Activities - Set up four soil microcosms with different fertilizer treatments - Bury microscope slides in the soil for Rossi-Cholodny slides Treatment Organic Inorganic Combined Unamended Fertilizer 0.0 g 0.2 g 0.2 g 0.0 g Yeast Extract 1.6 g 0.0 g 1.6 g 0.0 g Procedure 1. 2. 3. 4. 5. 6. Cut out a circle of filter paper to fit the bottom of the flower pot. Use a glass beaker to measure out ~300ml of soil. Weigh out the appropriate amount of fertilizer and mix it into the soil. Put the filter paper circle into the bottom of the flowerpot Pour in half of the soil and fertilizer mix. Insert four glass slides into the soil so that the top of the slide is even with the top of the flower pot. 7. Pour in the remaining soil until it is ~1/2 inch from the top. 8. Water the soil until it is all moist.