In vitro site specific mutagenesis
advertisement

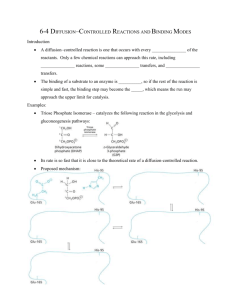
In vitro site specific mutagenesis Objectives: 1. To understand a range of purposes for conducting in vitro mutagenesis. 2. To understand a range of phenotypes that can be sought in mutants to clarify function of a protein. 3. To understand vector types most appropriate to different kinds of in vitro mutagenesis experiments. Background Site specific in vitro mutagenesis is the introduction of a specific preconceived mutation. This could be done to test a specific hypothesis about a DNA regulatory sequence, or a protein sequence, or an RNA sequence. Constraints on the vector. The mutated sequence will be subjected to an assay that may require it to be in some specific context within a vector. It is possible to conduct the mutagenesis in one vector, and then transfer the altered sequence to another vector more specifically designed for the assay. However, in vitro mutagenesis studies inevitably expand into a list of required mutations. It saves a lot of trouble to design the vector so that the mutagenesis is done directly in the final vector. Furthermore, it pays to anticipate alternative assays that may be conducted further ahead in the project and build those capabilities into the same vector. Promoter/Enhancer elements. Generally, the targeted element will need to be in the context of a functional promoter and set up do drive a reporter gene in an expression vector. If the vector will be assayed in a host other than E. coli, it will generally need both an E. coli ori and selectable marker, and an ori for the targeted host. This is because after mutagenesis, the clone will be grown up for sequence confirmation in E. coli, and then transferred into the appropriate host. If the assay is to be in vitro transcription, a convenient restriction site should be downstream to linearize the template. Other types of regulatory sites (splice sites, ori's, recombination sites, etc.) will similarly have to be within a context wherein they can be assayed. Promoter/enhancer regions are usually first characterized by a series of deletion mutants to localize a region for more detailed studies. Historically, one would take advantage of available restriction sites to make a nested set of deletions. PCR now makes it possible to form deletions starting at any arbitrary position. Then a finer survey of the implicated region would be done with some combination of the following: footprinting, motif searching, gel mobility shift assays, saturation mutagenesis, and linker scanning mutagenesis. Linker scanning mutagenesis is a systematic method of making a series of mutants where successive regions of about 10 bases are substituted by an arbitrary sequence (usually a restriction site). 1 Linker Scanning. After McKnight and Kingsbury, Science 217:316-324 (1982). Today, because of the reduced cost of making oligonucleotides, most people would make the 10 bp substitutions one at a time by the methods illustrated below. However, they often still call it linker scanning. Finally, given that a specific motif was implicated by the above methods, one might make single base changes at the conserved position of the motif, monitoring changes in gel mobility shift, a footprint, and expression in coordination. 2 Proteins that recognize a nucleic acid sequence. Proteins that recognize a nucleic acid sequence provide the possibility of mutating both the protein and the binding sequence. Successful prediction of a mutation in one that compensates a mutation in the other is generally considered as a powerful indication that one's model of the specific contacts that enforce specificity is accurate. Ribozymes One of the newest targets of mutagenesis are ribozymes, RNA molecules with catalytic activity. Ribozymes would generally be produced by in vitro transcription. If they are to be delivered in vivo, then a transcription terminator will probably be required. Since making ribozymes is an extensive design problem, some sort of evolutionary strategy is often used (several rounds of saturation mutagenesis interspersed with selection for improved function). Other RNAs rRNA, tRNA, snRNA, etc. can be the target of in vitro mutagenesis. Because these molecules are heavily modified, they will have to be produced in the appropriate host cells. Some provision will have to be made to separate them from the endogenous host product. For example, one might alter their length. Since these are usually essential genes, it will not be possible to make a null host strain, except by a colony sectoring approach. Signals that affect translation efficiency or half life of mRNAs may be identified by in vitro mutagenesis. Proteins Mutagenized proteins for biochemical characterization will generally have to be in an expression vector designed for mass production of the protein. The vector may also have features that aid the purification of the protein. The promoter should be regulatable. Mutagenized proteins designed for genetic testing will need to be in the appropriate expression vector for that target host cell. A regulatable promoter will aid proving that the phenotypic change is really due to expression of your protein. A regulatable promoter also helps prevent selection against your gene during construction and growth. The promoters in many expression vectors are leaky enough that selection against the insert is a problem even though the vector is grown under "non-expressing" conditions. Hence if the construction is complicated, many investigators would do all the steps in some other vector, including sequence verification. Then a restriction fragment carrying the gene would be moved to the expression vector in the last step. One will have to provide for separation of the mutagenized protein from the endogenous protein. Possible solutions are 1) use heterologous host, 2) delete the gene from the host, 3) use a thermostable subject protein, or 4) use an affinity tag purification system. 3 Proteins without 3-D structures Many of the more sophisticated mutagenesis studies rely heavily on 3-D structure data. However, there are a variety of useful experiments that can be done without 3-D information: 1. Investigating modifications (phosphorylation, glycosylation, palmitylation, etc.) One can remove putative modification sites identified by sequence motifs to see if the resulting protein is less modified. Further one can ask if the unmodified protein produced in this way has altered function. 2. Investigating genetic disease: Now that defective genes of all descriptions are being isolated by positional cloning, one is often confronted with a defective gene with several differences from the "wild type". Mutagenesis may be conducted to isolate each of the differences to distinguish the actual defect from inconsequential polymorphisms. This will require an in vitro assay or a cell culture assay to act as a surrogate for the disease phenotype. One may wish to introduce other mutations into the target gene to see how specific the disease phenotype is with respect to different mutations. Site specific mutagenesis may also be used to introduce the same defect into a mouse by gene replacement. 3. Identify a compartmentalization signal. 4. Assign functions to particular protein domains. 5. Delete domains for functional assignment or to stabilize for crystallography. 6. Add a tag. 7. Alanine scanning: a strategy to produce a set of mutants containing a replacement to Ala at every position. Often this is reduced to substitution Ala at every charged residue (assuming that these have to be on the surface). Alanine scanning is used to assign functions to different domains, and to create tentative sets of active site residues. A variation called Cys scanning additionally allows one to covalently attach chemical moieties at the substituted positions. Trp scanning (PNAS 92(17):7946-50 (1995)), has been used to characterize proteins with multiple transmembrane helixes on the principle that Trp residues will be tolerated on surfaces exposed to the lipid environment, but not elsewhere. Alanine replacements in binding sites tend to give partial reduction in binding for reasons stated further below. Hence, one should employ this method with a quantitative assay for the target interaction. The loss of binding energy upon combining multiple Ala replacements at contact residues tends to be additive. For a good introduction see Cunningham and Wells 1993. J. Mol. Biol. 234: 554-563, followed by Lowman and Wells, 1993. J. Mol. Biol. 234:564-578. 4 8. Domain addition or swapping. For example, if it is postulated that a signaling protein is activated by regulating its access to the membrane, a constitutively active version might be made by adding a domain that always takes it to the membrane. 9. Traditionally, active site residues have been hypothesized by sequence conservation and then altered by in vitro mutagenesis in an attempt to define active site residues prior to obtaining 3D data. Much of this kind of investigation has been replaced by an emphasis to get 3D structural data first, and then work out the details of the active site by in vitro mutagenesis. Mutagenesis of proteins with 3D structures. Active sites Crystal structures generally lead to a hypothesis about the roles of active site residues. However, if a substrate analogue wasn't actually present in the crystal, the placement of substrate in the active site can be quite speculative. Even if the enzyme was crystallized with its substrate, the resolution of a crystal structure is generally insufficient to distinguish whether a proposed interaction (say a hydrogen bond) is positioned well enough to contribute positively to binding energy. Therefore, the contribution of all the proposed contacts to the activity of the enzyme still requires biochemical characterization. One is immediately faced with a decision of what to replace these residues with. Experience has shown that putting in a large side chain frequently kills the enzyme, leaving one to wonder if the substrate has been completely blocked out of the active site, or if the conformation of the enzyme has been distorted. Also, introduction of bulky side chains tend to promote aggregation either directly or by disrupting structure and exposing hydrophobic groups. So this kind of mutant is often uninformative. Substituting an Ala has been most consistently useful as a means of withdrawing a specific substrateenzyme contact without otherwise distorting the interaction. Gly tends to cause conformational distortion because too much of a hole has been left in the packing of the side chains. Other strategies are to pick chemically similar side chains to substitute, or to pick residues appearing at this position in homologous proteins (particularly useful if the homologous protein has altered function). One really should anticipate making a number of replacements at this position and finding that some of them fail to fold and behave in a tractable manner. Strategies that involve changing more than one residue at a time have an increased risk that the final product will not fold or suffer from intractable aggregation problems.. Active site residues can be conceptualized as performing two different kinds of roles: substrate binding, and catalysis. Mutation of catalytic residues (those that specifically stabilize the transition state) generally kill the activity of the enzyme. One certainly has to do this experiment, because if the enzyme still worked fine the experiment would reject the proposed mechanism. However, the negative result (dead enzyme) is a weak result. The mutation may have killed the enzyme by altering its conformation in some unexpected way, and the residue may in fact have nothing to do with catalysis. The overall structural integrity of the mutant enzyme could be supported by the following observations: unaltered proteolysis, substrate(s) still binds, other aspects of the function still carried out (eg. partial reactions not including the mutated step), physical properties unaltered (eg. fluorescence, circular dichroism, quaternary 5 structure). For small proteins (<45 kD), HSQC could both confirm minimal conformational distortion and that the substrate still binds. Here's an example of mutations at catalytic residues involved in the conversion of ATP + tyrosine to tyr-AMP by tyr-tRNA synthetase. In this case there are two catalytic residues, neither one of which is completely essential. 6 Enzyme wild type His-45 --> Gly Thr-40 --> Ala His-45 --> Gly & Thr-40 --> Ala k(rate limiting) 38 s-1 0.16 0.0055 0.00012 KD tyr 12 uM 10 8.0 4.5 KD ATP 4.7 mM 1.2 3.8 1.1 When ATP reacts with tyrosine, the alpha phosphate must go from its normal tetrahedral configuration through a five bonded intermediate in the transition state. This causes the other two phosphates to be physically displaced in the active site. Thr-40 and His-45 are thought to bind the gamma phosphate in the transition state, thus stabilizing it. From Leatherbarrow et al. (1985) PNAS 82, 7840-7844. 7 The effects of altering a substrate contact. Mutations to binding residues normally have subtle effects on the Km, KD, or substrate specificity. The assumption is that these residues bind the substrate the same whether or not it is in the transition state. The substrate is usually bound by many such contacts, so disrupting one of them is insufficient to kill activity. The binding energy contributed by the various contacts is often approximately additive. Again looking at tyrosyl-tRNA synthetase, Thr 51 was proposed as a hydrogen bonding contact for the ribose of the ATP. Tyrosyl-AMP in the active site of tyrosyl-tRNA synthetase from Fersht et al., Bioch 24, 5858 The numbers tabulated below are relative Gibbs Free energies of binding determined from a combination of equilibrium dialysis and kinetic measurements. A number of different measurements could be compared in this way. KM values are popular to compare, because the measured KM value is independent of enzyme concentration. This provides relief from the concern that the mutant protein is unstable and really exists as an equilibrium between some amount that is active and some that is not. In a simple Michaelis-Menton treatment, the KM is the same as the equilibrium dissociation constant for binding of the substrate. However, many enzymes do not exhibit simple Michaelis-Menton behavior, and treating them this way may cause you to attribute a mutant's effect to substrate binding when it is really affects other aspects of enzyme structure or function. A thorough kinetic treatment (as was done here) yields the true substrate dissociation constant, and requires actually determining how much of the enzyme 8 is active, and having a detailed description of the enzyme mechanism. A final alternative is to directly measure the substrate dissociation constant under conditions where the substrate is not turned over. This also requires knowing how much of the enzyme is active (at substrate binding), but has the advantage that it can be applied to mutants that are catalytically dead (at some other step besides substrate binding). Kinetic effects of various changes to Thr-51 Enzyme wild type (Thr-51) Cys-51 Ala-51 Ser-51 Gly-51 kcat(s-1) KM (mM ATP) 8.35 12.4 8.75 1.88 6.0 1.08 0.35 0.54 1.16 1.25 delta G -0.90 -0.44 +0.92 +0.28 From Fersht and Wilkinson (1985) Bioch. 24, 5858-5861. The tabulated delta G values are computed from kcat and KM for ATP and represent the binding energy in the transition state. Here we see an advantage of making the comparisons as delta G values. These values are additive, and can be broken down to individual contributions of different atoms in the side chains. If you just look at thr -> gly, you would see that you lost .28 kcal/mol in binding energy, and you might (mistakenly) attribute that to the loss of the hydrogen bond to the substrate. However, you really removed a hydrogen bond, and potential interactions with 2 carbon groups (beta and gamma). The change from thr -> ser shows that you lose -0.92 kcal/mol by withdrawing the contact with the gamma methyl group. The change from ser -> ala shows that you actually gain 1.36 kcal/mol from withdrawing the hydrogen bond, and the change from ala -> glycine shows that you lose another 0.72 kcal/mol from withdrawing the beta methyl group. So thr 51 really contributes favorable hydrophobic contacts plus an unfavorable hydrogen bond. group gamma methyl gamma SH gamma OH beta methyl contribution to binding energy (kcal/mol) -0.92 -0.46 +1.36 -0.72 A negative number denotes improved binding. Note that these numbers are in the range you would expect for individual hydrogen bonds and Van der Waals contacts. The hydroxyl forms an unfavorable H-bond because the distance is 0.5 angstroms too long. The unfavorable bonding energy means that this thr residue forms a 1.36 Kcal better bond to water than the substrate, therefore it disfavors substrate binding. Sulfhydrals tend to form longer H-bonds, therefore the cys residue makes a better contact from this position. You could not get this information by gazing at the crystal structure, because X-ray structures are not accurate to 0.5 angstrom resolution. 9 Note that Cys actually makes a better contact with the substrate than the wild type residue, thr. If you look at the velocity versus [ATP] curves for these two enzymes, you see that each has a range of [ATP] where the velocity is greater than the other. This serves to show that tighter binding is not necessarily better. Figure modified from Fersht and Wilkinson Biochemistry 24: 585-5861 (1985) 10 Mutations that alter structure. Mutations that alter structure often have non additive effects on activity when coupled with other mutations. In the example of Thr 51 given above, you would expect a proline at residue 51 to contribute no binding energy. Unexpectedly, pro 51 actually improves binding by -1.9 kcal. This is an example of a conformational effect. Pro 51 improves substrate binding by disrupting an alpha helix and making a better contact for His 48. This is shown by observing non additivity in double mutant Thr -> Pro 51, His -> Gly 48 (panel d below). Panels b and c show that there is not much of a conformational effect of pro 51 on binding by Cys 35, or between mutations at residue 48 and 35. From Carter et al. (1984) Cell 38, 835-840. 11 Characterization of conformation. In the context of active site investigation given above, a conformational effect usually arises as an unanticipated side effect of a site directed mutation. Our current understanding of protein structure is generally insufficient to build a preconceived conformational change into a protein. However, after the fact, a conformational mutant can yield considerable information about structure [Biochem. 33(15):458593], stability [Biochem. 32(39):10371-7, 1993], function [J. Biol. Chem. 269(15):11578-83], or folding [Biochem. 32(49):13566-74 (1993)]. Those who intentionally want to seek out conformational mutants might try inserting prolines, or conducting saturation mutagenesis and screening for temperature sensitivity. Characterization of quaternary structure. Refs: Jones et al., (1985) Bioch. 25, 5852-5857. Ward et al. (1986) JBC 261, 9576-9578. The interface between enzyme subunits can be disrupted by placing charged residues in it. The following is an example where complementary changes are made at an interface to convert a homodimer into a heterodimer. Tyrosyl-tRNA synthetase is a dimeric enzyme that shows half-of-the-site reactivity. That is, although there are two active sites, they interact with negative cooperativity such that only one is active at a time. The experiment is to set up a situation where one can maneuver a mutation into only one of the two subunits and thereby measure the effect on the other. In this case to physically demonstrate the heterodimer, one of the subunits will be a version carrying a deletion to alter its size. By engineering a negatively charged residue into the subunit interface, it was possible to cause dissociation under native conditions by varying the pH. By engineering a positively charged residue into the subunit interface of another variant, it was possible to form heterodimers wherein the two residues formed a salt bridge. This verified that the pH effect was due to subunit dissociation and not due to a generalized unfolding of the enzyme. Further, other mutations could be introduced 12 specifically into one of the subunits of the heterodimer. Mutagenesis to add a label to a protein. Physical-chemical studies of proteins often make use of fluorescence of endogenous trp residues to report on the conformation of the protein. With site-specific mutagenesis, one might arrange for the placement of trp residues in ideal positions for the anticipated study. This may involve both removing endogenous trp residues and adding new ones. (JBC 269(11):7919-25, 1994). Other kinds of reporter groups may be added to the protein. One relatively flexible strategy is to add a Cys at a predetermined position, and to chemically modify the Cys to attach some other moiety. 13 Study Questions 1. It is proposed that the following network of hydrogen bonds is involved in stabilizing substrate binding in a particular enzyme. How could you test this by in vitro mutagenesis? substrate--HO Ser118 main chain NH--OH Thr46 2. What would be a good method if you wanted to map monoclonal antibody epitopes on a protein? 3. A protein has 5 trp residues. You try to change 4 to Ala so that the 5th can be used as a probe of conformational change. This results in a protein that aggregates intractably. How might you approach solving this problem? 4. A particular 200 bp promoter fragment is sufficient to confer a heat shock response on a reporter gene. Can you think of a reason why a complete set of linker scanning mutants might fail to identify the heat shock response element? Last revised 3/13/2005 - Steve Hardies 14