Here - ars-chemia.net

LABORATORY MANUAL FOR
CHEMISTRY 102
Prepared by
Department of Chemistry and Physics
Los Angeles Valley College
This Lab Book Belongs To:
Copyright © 2015 by the Department of Chemistry and Physics, Los Angeles Valley College. All rights reserved. No part of this publication may be reproduced or distributed in any form or by any means, electronic or otherwise, or stored in a database or retrieval system, without written permission of the copyright holder.
2
TABLE OF CONTENTS
Contents
BY SPECTROPHOTOMETRIC METHODS........................................................... 83
DETERMINATION OF PERCENT OXALATE BY OXIDATION-REDUCTION TITRATION ............................. 99
3
LABORATORY SAFETY RULES
5.
6.
3.
4.
7.
8.
Note: Failure to follow safety rules will result in expulsion from this course.
1.
2.
Wear approved safety goggles at all times in the laboratory.
It is not advisable to wear contact lenses during lab.
Do not wear loose clothing to lab. It is a fire hazard.
Tie back long hair. It too is a fire hazard.
Wear closed shoes to lab.
Never put anything into your mouth while in the lab.
Immediately wash off any chemicals spilled on your skin or clothes.
Keep the lab neat. Return reagent containers and equipment to proper locations.
for experimental work on the shelves provided.
Put any belongings not needed
9.
10.
Clean up all chemical spills or broken glass immediately.
Think about how much chemical you will need before you take it from a stock (reagent) bottle. Never return unused chemicals to stock bottles. Never dip into a reagent bottle with anything (spatula, dropper, pipet, etc.)!
11.
12.
13.
14.
Dispose of waste chemicals only as instructed.
Behave in a responsible manner.
You should be aware of the location and use of laboratory safety equipment.
15.
16.
Immediately report accidents and injuries to your professor.
Do not perform unauthorized experiments.
Thoroughly wash your hands any time you leave the lab.
17. No smoking on the Los Angeles Valley College campus.
I have carefully read all of the safety precautions summarized above and recognize that it is my responsibility to observe them throughout this course.
Chemistry 102
Printed Name
Date Section Number Signature
4
5
LABORATORY SAFETY RULES
Note: Failure to follow safety rules will result in expulsion from this course.
5.
6.
7.
8.
3.
4.
1.
2.
Wear approved safety goggles at all times in the laboratory.
It is not advisable to wear contact lenses during lab.
Do not wear loose clothing to lab. It is a fire hazard.
Tie back long hair. It too is a fire hazard.
Wear closed shoes to lab.
Never put anything into your mouth while in the lab.
Immediately wash off any chemicals spilled on your skin or clothes.
Keep the lab neat. Return reagent containers and equipment to proper locations.
Put any belongings not needed for experimental work on the shelves provided.
9.
10.
Clean up all chemical spills or broken glass immediately.
Think about how much chemical you will need before you take it from a stock (reagent) bottle. Never return unused chemicals to stock bottles. Never dip into a reagent bottle with anything (spatula, dropper, pipet, etc.)!
15.
16.
17.
11.
12.
13.
14.
Dispose of waste chemicals only as instructed.
Behave in a responsible manner.
You should be aware of the location and use of laboratory safety equipment.
Immediately report accidents and injuries to your professor.
Do not perform unauthorized experiments.
Thoroughly wash your hands any time you leave the lab.
No smoking on the Los Angeles Valley College campus.
Come to lab prepared!! Carefully read the experiment before coming to lab.
Quantity
2
1
1
1
1
1
1
1
3
1
1
1
1
2
2
2
2
2
4
10
10
1
2
1
1
1
1
1
1
1
Description
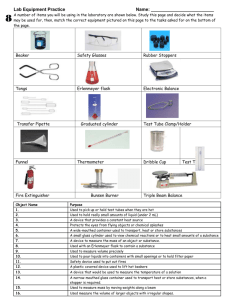
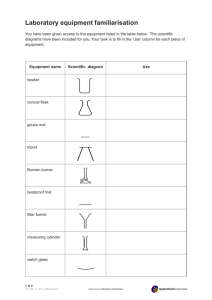
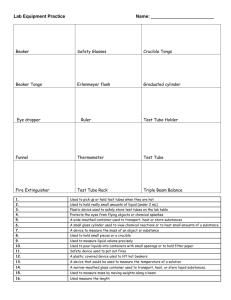
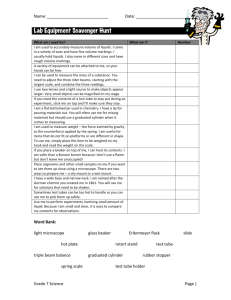
Beaker, 20 mL
Beaker, 50 mL
Beaker, 100 mL
Beaker, 150 mL
Beaker, 250 mL
Beaker, 400 mL
Beaker, 600 mL
Bottle, 500 mL, Screw Cap
Bulb, Pipet
Clamp, Buret
Cylinder, Graduated, 10 mL
Cylinder, Graduated, 50 mL
Flask, Erlenmeyer, 250 mL
Forceps
Funnel, Small, 45 mm
Holder, Test Tube
Microspatula
Pipet, Graduated 1.0 mL
Pipet, Graduated 5.0 mL or 10.0 mL
Pipet, Volumetric 10 mL
Pipet, Volumetric 25 mL
Rack, Test tube
Shell Vials
Test Tube, 10 mm x 75 mm
Test Tube, 13 mm x 100 mm
Test Tube Brush, 12 mm x 62 mm
Thermometer, -20°C to 110°C
Tongs
Wash Bottle, Polyethylene, 250 mL
Watch Glass, 75 mm
6
7
FACTORS AFFECTING THE RATE OF A REACTION
INTRODUCTION
There are several factors that affect the rate of a reaction. Some of these factors are:
• Mixing
• Concentration of a reactant
• Temperature
• The presence of a catalyst
• Surface area in a heterogeneous reaction
In this experiment we will examine these factors. This experiment is an introduction to the more commonly encountered factors that affect the rate of reactions. There are other factors that do influence the rate of a reaction such as light, molecular geometry and the type of solvent used; however, we do not have the time to explore all facets of all factors that affect reaction rate.
PROCEDURE
A. The effect of mixing.
1. Fill two small test tubes ¼ full with water.
2. Into each tube add a small crystal of solid potassium permanganate.
3. Let one tube sit undisturbed. Swirl the other tube to dissolve the potassium permanganate. Note the amount of time it takes the swirled sample to dissolve (form a solution).
4. Continue with the experiment (Parts B through E) and observe the undisturbed tube every few minutes. Note approximately how long it takes for the potassium permanganate to dissolve and diffuse throughout this tube.
To complete Parts B and C each group will need a timer, a total of 7.0 mL of 3%(m/m) hydrogen peroxide solution (H
2
O
2
) and a total of 15.0 mL of solution A (a mixture of starch (as an indicator), acetic acid, potassium iodide, and sodium thiosulfate). Do not waste reagents by taking more than you need for the experiment!
B. The effect of concentration of a reactant.
1. In your smallest beaker place 5.0 mL of solution A and add 5.0 mL of the hydrogen peroxide solution. Start your timer as soon as the solutions are mixed in the beaker.
2. Record the number of seconds that elapse until the solution turns blue/black.
3. Repeat steps 1 and 2 using 5.0 mL of solution A and 4.0 mL of deionized water which has been added to 1.0 mL of the hydrogen peroxide solution.
8
C. The effect of temperature.
1. Place a test tube containing 5.0 mL of solution A and another test tube containing 4.0 mL of deionized water combined with 1.0 mL of the 3% hydrogen peroxide solution into a warm water bath for about 5 minutes.
2. Measure the temperature of the water bath.
3. Mix the two solutions into a small beaker and start your timer.
4. Record the number of seconds required for the solution to turn blue/black.
5. Compare this number with the elapsed time from the second (diluted) mixture in part B.
D. The effect of a catalyst.
1. Fill a large (400 mL or larger) beaker about 2/3 full with water.
2. Fill a small test tube all the way with water. Place your finger over the opening and invert it into the beaker. Do not allow any gas to enter the tube as you remove your finger.
3. Obtain a gas collection apparatus. Place the gas evolution tube under the inverted test tube in the beaker.
4. Do you notice gas formation in the 3% H
2
O
2 reagent bottle?
5. Place 20 drops of 3 M copper(II) nitrate solution in the flask. Swirl the contents of the flask. Is any gas formed in the catalyst solution alone?
6. Quickly add about 20 mL of 3% hydrogen peroxide solution to the flask and quickly put the stoppered end of the tube into the flask. Continuously swirl the flask’s contents.
7. Record the number of seconds required for the tube to fill with the gas produced.
8. Empty the contents of the flask and beaker, clean them and set up the experiment for the next trial.
9. Place about 5 mL of 3% hydrogen peroxide solution in the flask. Remove the stopper just long enough to add 2 drops of 3 M iron(III) nitrate solution and quickly put the stoppered end of the tube into the flask. Continuously swirl the flask’s contents.
10. Record the number of seconds required for the tube to fill with the gas produced.
11. Set up the experiment again with 5 drops of 3 M copper(II) nitrate, 5 drops of 3 M iron(III) nitrate and 20 mL of 3% hydrogen peroxide solution in that order. Record the number of seconds required for the tube to fill with the gas produced.
9
E. The effect of surface area in a heterogeneous reaction.
1. Place a small iron nail that has been cleaned into a test tube. In a second test tube place a small ball of steel wool.
2. Into each test tube add 5 mL of 1 M Cu(NO
3
)
2
solution and place each tube into a warm water bath.
Occasionally stir the tubes and let them heat for at least 10 minutes.
3. Observe each tube closely and note any color change in the solutions. The intensity of the color change is an indication of the progress of the reaction. Which tube has a quicker color change?
Record the color intensity (lighter or darker) for each tube.
4. After 10 minutes, decant the solution from each tube and note the appearance of the nail and of the steel wool.
10
Report Name ___________________________
FACTORS AFFECTING THE RATE OF A REACTION Section ___________________________
A. The effect of mixing.
Time for color to diffuse throughout the tube stirred tube __________________________
__________________________ undisturbed tube
B. The effect of concentration of a reactant.
Time for color change in first trial _________________________
Time for color change in second trial
C. The effect of temperature.
Time for color change in heated trial
D. The effect of a catalyst.
Amount of gas formed in pure H
2
O
2
Amount of gas formed in just the catalyst solution
Time for tube to fill with gas when mixing
H
2
O
2
and Cu(NO
3
)
2
Time for tube to fill with gas when mixing
H
2
O
2
and Fe(NO
3
)
3
Time for tube to fill with gas when both catalysts are used with H
2
O
2
_________________________
________________________
_______________________
________________________
_______________________
_______________________
_______________________
11
Report
FACTORS AFFECTING THE RATE OF A REACTION
E. The effect of surface area
Color intensity of original copper solution
Appearance of the steel wool initially
Name __________________________
Appearance of the nail initially
_______________________
_______________________
After at least 10 minutes in the copper solution ______________________
Color intensity of solution with nail after 10 minutes _______________________
_______________________
After at least 10 minutes in the copper solution _______________________
Color Intensity of solution with steel wool after
_______________________ after 10 minutes
12
13
3.
2.
QUESTIONS FOR FACTORS EXP.
1.
NAME _____________________________
Looking at part A of this experiment, how would you expect the rate of the reaction to change if you were to stir a reaction mixture instead of just letting it sit?
Recall that the rate of the reaction is inversely proportional to the time measured. a. b.
Based on this, in part B, which tube had a higher rate of reaction?
Which tube had the higher concentrations of either or both reactants? c. How does concentration affect the rate of the reaction?
In part C of this experiment the temperature was raised above that used in part B. Comparing the rate of the reaction in the second part of part B and the rate of the reaction in part C, how does temperature affect the rate of the reaction?
14
5.
QUESTIONS FOR FACTORS EXP.
4. a.
NAME______________________________
In part D of this experiment how did the presence of the copper(II) nitrate affect the rate of the reaction? b. How did the presence of iron(III) nitrate affect the rate of the reaction? c. Which compound is a better catalyst? d. Does having both catalysts present increase the rate of the reaction more than either one alone?
If you have a choice of either to grind up a reactant or leave it in a large lump, which would you choose so that the reaction rate is increased? Refer to your results from part E.
15
CHEMICAL KINETICS
INTRODUCTION
It is not possible for one to predict a reaction rate or rate law from a balanced, overall equation.
Information about the reaction mechanism (pathway) must be known to make such predictions.
Through numerous laboratory studies, experimental rate laws have been found to obey the general expression: z
where [A], [B], [C], ... represent molarities of all chemical species that affect the rate, and x , y , z , ... are the experimentally determined exponents for each species. (The overall order of the reaction is equal to the sum of x + y + z +... .) The term k is known as the rate constant for the reaction.
Usually, when a reaction is initiated, the rate (known as the initial rate) is found to be at its maximum value. As the reaction progresses, reactants are consumed (lowering their concentrations) and the rate slows. One can avoid difficult concentration measurements by monitoring the initial rate. The concentrations at the time of the initial rate are simply the initial concentrations after taking dilutions into account. If the initial concentration of one reactant is varied while all others are held constant, then the resulting change in initial reaction rate yields the order with respect to that one reactant.
This is the initial rate method used to determine reaction order.
In this experiment you will be measuring the initial rate for the iodide ion and persulfate ion, S
2
O
8
2reaction:
,
2 I + S
2
O
8
(1)
To detect the extent to which reaction (1) has proceeded, an additional, simultaneous process must also occur:
2 I
2
+ 2 SO
4
2-
I
2
+ 2 S
2
O
3
2 2 I + S
4
O
6
2(2)
2, instantly reduces iodine molecules back to iodide ions. Only In reaction (2) the thiosulfate ion, S
2
O
3 when the thiosulfate ions have been completely consumed can the iodine formed in reaction (1) be available to react with the indicator to form the blue-black starch complex. Therefore, the rate for reaction (1) is equal to the change in thiosulfate concentration per time. What will be measured in this experiment is the time required to use all of the thiosulfate (the time required to change the thiosulfate concentration to zero). The elapsed time depends upon the rate of reaction (1) as well as the amount of thiosulfate added to the reaction mixture. (Thus, the amount of thiosulfate must be carefully controlled so that the only variables are the concentrations of iodide and persulfate ions.)
16
For reaction (1) the reaction rate is equal to k I − concentrations for the molarity terms). Thus, x
0
S O 2 −
2 8
y
0
(where the subscripts of zero indicate initial
Rate k I
0
S O 2 − y
0
Because of the experimental conditions employed, applying the initial rate method to the above expression yields
Rate
2
Rate
1
= k I − x
2 x
S O 2
2 8 k I −
S O 2
2 8
−
−
1
2 y y
" Exp 2"
" Exp 1"
Because the initial concentration of iodide ion in experiment 2 equals twice its initial concentration in experiment 1, after cancelling constant terms we obtain:
Rate
2
Rate
1
= 2.00
x
Solving for x is simplified by taking the logarithm of both sides. Thus, log
Rate
2
Rate
1
= log 2.00
x = x log 2.00
further reduces to: x = log
( )
1 log 2.00
Calculations similar to those presented above may be derived for y .
As we increase the temperature of a reaction, its rate typically increases. We find the temperature dependence of the rate of a reaction is a result of the temperature dependence of the rate constant.
In class, we examine two possible explanations of the temperature dependence of the rate constant.
Here, we will restrict ourselves to Arrhenius theory. Arrhenius conjectured that the rate constant would be a product of two factors; A, the pre-exponential factor, and an exponential factor that depends on the activation energy and the temperature: k Ae − Ea
Typically, the activation energy is in J mol -1 , R is in J mol -1
RT
K -1 , and the temperature is in K. This equation in itself cannot help us to determine the temperature dependence of k or the activation energy. To do this we need to use the two-point form of this equation. If we take the natural logarithm of both sides and subtract the equation at one temperature from the equation for another temperature we get: ln
k k
1
2
=
E a
2
T
1
17
Using this equation we can calculate the rate constant at different temperatures if we know the activation energy or calculate the activation energy if we know the rate constant at two different temperatures. In this experiment we will determine the rate constant at two different temperatures and then calculate the activation energy of the reaction.
PROCEDURE
1. Students may work in small groups (2 to 3 students/team). Each team needs a timer.
2. Each of the 4 experiments will be performed in duplicate. For each experiment, every team will need two clean flasks and two clean beakers – these can be wet, but should be well drained.
3. All reagent bottles have been fitted with Dispensette III bottletop dispensers. These are designed to deliver an exact volume when used properly. To dispense, turn the red cap counter- clockwise to remove, position the container you are using under the spout, pull the piston gently all the way up, then push gently all the way down dispensing into the container. Finally, replace the cap turning clockwise. Note: all of the Dispensette apparati are set to dispense 10.0 mL so for those experiments that require 20.0 mL you will need to dispense two times. Potassium chloride and sodium sulfate solutions are used to maintain a constant ionic strength while diluting reactants in this experiment. Read labels carefully.
4. Obtain reagents and perform one experiment at a time.
For experiment 1, prepare 2 flasks, each containing the volume of potassium iodide, potassium chloride, sodium thiosulfate, and starch
(indicator) solutions provided in the table below.
5. Also for experiment 1, prepare 2 beakers each with the volume of potassium persulfate shown in the table below.
Volumes of solution, mL
Ice bath
Experiment 4
0.200-
0.200-
M
M
0.00500-
Starch
KI
KCl
M Na
2
S
0.100M K
2
S
0.100M Na
2
2
O
SO
8
4
2
O
3
Experiment 1
Room Temperature
Experiment 2 Experiment 3
In a flask:
10.0
10.0
10.0
10 drops
20.0
0
10.0
10 drops
20.0
0
10.0
10 drops
20.0
0
20.0
0
In a beaker:
10.0
10.0
20.0
0
10.0
10 drops
20.0
0
18
6. Start the timer as the contents of one beaker are added to the contents of one flask. Mix the reagents by quickly pouring the contents of the flask into the beaker and then returning the solution to the flask. Allow the flask to sit undisturbed while observing its contents constantly.
Stop the timer when the blue-black color appears. (Constant observation of the flask is necessary because of the sudden appearance of the blue-black color.) Record the elapsed time in seconds.
Repeat step 6 using the second beaker and flask. Elapsed times for duplicate sets of experimental conditions should agree within about 2-3 seconds. (If not, do a third trial for that experiment.)
7. Clean your beakers and flasks and allow them to drain well.
8. Repeat steps 4, 5, 6, and 7 for experiment 2 and then experiment 3 with the same mixing procedure for each experiment. Note: In experiments 2, 3, and 4, no KCl is required in the flask, but sodium sulfate as well as potassium persulfate is needed for experiment 3. (Read steps 10 through 12 before doing experiment 4.)
9. Record the temperature of one of the reaction solutions. (Because all of the solutions have been sitting at room temperature, you can assume this is the temperature for all the solutions.)
10. For experiment 4, place the volumes of the solutions indicated into clean flasks and beakers. Fill four of your largest beakers with ice and set the reagent flasks and beakers onto the ice. Allow the flasks and beakers to remain on ice for at least 5 minutes.
11. Record the temperature of one of the solutions in the ice bath.
12. Repeat step 6. Return the solutions to the ice bath immediately after mixing.
When the reaction flask starts to show a color change, swirl the flask and determine the elapsed time.
13. Perform dilution calculations to determine the molarities at the instant of mixing for the iodide, persulfate, and thiosulfate ions used in each experiment. You can assume that the 10 drops of starch is 0.5 mL and that the total volume in each experiment is 50.5 mL.
14. Calculate the average elapsed time for each experiment (in seconds).
15. Calculate the Rate for each reaction. The Rate is equal to:
−
thiosulfate
f
−
thiosulfate
elapsed time
i
Remember that the final concentration of thiosulfate for each experiment is zero!
16. Using the average rates from experiments 1, 2, and 3 (but not 4), calculate the order of reaction
(1) with respect to the iodide ion and the persulfate ion. Record the orders using the number of significant figures appropriate for your data.
19
17. Round off the orders you have determined for iodide and persulfate to the nearest whole number and then calculate the overall order for the reaction (1). Use the Rate and the rounded orders to calculate the rate constant, k , for each of experiments 1, 2, and 3. Report your average value of k (include units).
18. Write the complete rate law for the reaction for the reaction that occurred at room temperature.
19. Determine the rate constant, k , (include units) for the reaction in the ice bath. Using the average rate constant from the room temperature experiments and the rate constant from the ice bath, determine the activation energy of the reaction.
20
REPORT
CHEMICAL KINETICS
Initial reactant concentrations:
Experiment
1
2
3
Temperature of solution, (°C)
NAME_______________________
SECTION _____________________
[Iodide] [Persulfate]
4
Temperature of solution, (°C)
Experiment
1
Elapsed time, (s)
Trial A Trial B
2
3
4 (ice bath)
For room temperature data:
Average time, (s)
Reaction Order
With correct sig figs
[Thiosulfate]
Rate, ( M /s)
Rounded to whole numbers
With respect to Iodide ion
With respect to Persulfate ion
Overall
Value for k from Experiment 1
Value for k from Experiment 2
Value for k from Experiment 3
Average value for k (including units)
Room Temperature Rate Law
SAMPLE CALCULATIONS (use separate sheets)
Include how you solved for both exponents in this experiment.
21
REPORT
CHEMICAL KINETICS (cont’d)
For ice bath data:
NAME________________________________
Value for k (including units)
SAMPLE CALCULATIONS (use separate sheets if needed)
Activation Energy, (kJ mol -1 )
SAMPLE CALCULATIONS (use separate sheets if needed)
22
23
QUESTIONS FOR CHEMICAL KINETICS EXP.
1. The reaction: 2 NO with respect to chlorine.
+ Cl
2
NAME _________________________________
2 NOCl has been studied and found to be second order with respect to nitrogen monoxide and first order a. What is the overall order for the reaction? b. How does the reaction rate change when the nitrogen monoxide concentration is halved and the chlorine concentration is doubled? Define terms (e.g., [NO]
1
for initial concentration in experiment 1, [NO]
2
for initial concentration in experiment 2, [NO]
2
= ½ [NO]
1
), set up the rate law ratios and show cancellations for
Exp 2
.
Exp 1
2. The initial rate of a reaction is found to increase by a factor of sixteen when the concentration of one reagent is doubled while all other reagent concentrations are held constant. What is the order of the reaction with respect to that one reagent? Define terms, set up the rate law ratios and show cancellations for
Exp 2
Exp 1
.
24
3. At 593K a particular decomposition’s rate constant had a value of 2.88×10 -4 and at 673K the same reaction’s rate constant was 1.94×10 -3 . It was noticed that when the reactant’s initial concentration was 0.1250 M (with a 593K reaction temperature), the initial reaction rate was identical to the initial rate when the decomposition was run at 673K with an initial reactant concentration of 0.04816 M . Recall that rate laws have the form rate = k [A] determine the order of the decomposition reaction. x and, showing work,
25
4. The following data was obtained for a reaction in which a chemical, X, decomposed.
Concentration of X (in Molarity)
5.00
Time (in seconds)
0
5.00 10 2 3.52
2.48
1.75
1.23
10.00 10 2
15.00 10 2
20.00 10 2
Chem 102 students are expected to prepare proper graphs (or lose points!) Appendix B of this document is a reprint of the Chem 101 lab manual’s graphing exercise which includes instructions for proper graph construction by hand and using Excel™. a. Prepare plots of concentration versus time using the provided data in a manner appropriate for zero, first and second order processes. You must include all three graphs with your report. b. Based on your graphs, is this reaction zero, first, or second order for X? c. Determine the slope for the straight line graph. Show how you arrived at the value for the slope of the line. Calculate the rate constant for this reaction from the slope. d. Write the complete rate law for the reaction including the value of k (with units).
26
27
LE CHÂTELIER'S PRINCIPLE
INTRODUCTION
When a chemical system at equilibrium is disturbed by a change in a component's concentration/pressure or by a temperature change, the system must shift to counteract the perturbation while simultaneously attempting to reestablish equilibrium. It is the description of this
"shifting" process that is referred to as Le Châtelier's Principle.
The equilibria to be studied in this experiment involve the formation of transition metal complex ions.
(You can refer to the text for more complete information.) Complex ions formed in this experiment are made from transition metal ions with Lewis bases (called ligands) attached to the central metal ion through coordinate covalent bonds.
In general, complex ion formation equilibria can be described by the following equation:
M x+ + y :LB M(:LB) y z+
Once a formation equilibrium is established, a change in temperature; in the concentration of the metal ion, (M); in the concentration of a ligand, (:LB), (which may or may not carry an overall charge); or in the concentration of the complex ion itself, would disturb the system.
Students will establish and perturb three different complex ion formation equilibria in this experiment, and will observe each system's response to these perturbations.
PROCEDURE
A. Fe 3+ , SCN-, and Fe(SCN) 2+
1. Work in groups of 2 or 3 students. Trays containing dropper bottles of the reagents will be provided.
2. Clean a 10 mL graduated cylinder, four test tubes (all of them must be the same size and hold at least 4 mL), and a 100 mL beaker. Use labeling tape to label the test tubes as 1, 2, 3, and 4.
3. Note the color of the reagents prior to mixing.
4. Add 20 mL of distilled water from a graduated cylinder to the 100 mL beaker. Next, add 10 drops of the iron(III) nitrate solution and 10 drops of the potassium thiocyanate solution to the beaker.
Stir the mixture thoroughly. The color in the beaker will be due to the formation of the complex
. Record your observations. ion, Fe(SCN) 2+
5. Using a 10 mL graduated cylinder, add 3 mL of the solution prepared in step 4 to each of the first three test tubes. Add 3.5 mL of the solution prepared in step 4 to the fourth test tube.
6. Add 10 drops of the 0.1M iron(III) nitrate solution to test tube 1. Stir the contents of this test tube.
7. Add 10 drops of the 0.1-
M
potassium thiocyanate solution to test tube 2. Stir the contents of this test tube.
28
B.
8. Add 10 drops of distilled water to test tube 3. Stir the contents of this test tube.
9. Compare the color of the solutions in test tubes 1, 2, and 3 with the color of the solution in tube
4. (For ease of comparisons, view each test tube's contents down its length against a white background.) Because the depth of solution and the final volume in all four test tubes are the same, the intensity of each solution's color is directly proportional to the complex ion's concentration. (The difference between tubes 3 and 4 may be difficult to see.)
10. Record your observations and determine which tube(s) contain(s) the highest concentration of the complex ion.
Ni 2+ , NH
3
and Ni(NH
3
)
6
2+
1. Clean a test tube.
2. Observe the 6M ammonium hydroxide, the 6M hydrochloric acid and the 0.1M nickel(II) nitrate.
Record the colors of the reagents.
3. Place 10 drops of 0.1M nickel(II) nitrate in the test tube.
4. Add 6M ammonium hydroxide (also known as aqueous ammonia) one drop at a time to the test tube from step 3 with stirring after each addition until there is a definite color change. Remember that aqueous ammonium hydroxide is primarily ammonia, with ammonium and hydroxide ions in equilibrium with the ammonia molecules. The ammonia molecules react with nickel(II) ions to form the colored complex ion, Ni(NH
6
2+ . Record your observations.
3
)
5. To the solution from step 4, add 6M hydrochloric acid (not 12M HCl) drop wise with stirring until the color changes once again. (The acid reacts with the basic molecules of ammonia to form ammonium ions. Ammonium ions have no lone pairs of electrons and therefore cannot act as
Lewis bases.) Record your observations.
C. Co 2+ , Cl and CoCl
4
2-
1. Place a small beaker containing tap water on the hot plate and heat to a gentle boil.
2. Place 5 drops of 0.1M cobalt(II) nitrate in a clean test tube. Record the color of this reagent.
Do not remove the concentrated hydrochloric acid from the fume hood! Immediately neutralize and clean up any spills!!
3. In a fume hood, add 8 drops of 12M hydrochloric acid (not 6M HCl) to the solution in the test tube from step 2. Stir the mixture and record the color. (This color is characteristic of the complex ion CoCl .)
4
2-
4. Add 5 drops of distilled water to the contents of the test tube from step 3. Stir to mix. Record the color. (There may or may not be a color change in this step.)
29
5. Place the test tube from step 4 in the hot water bath and wait a few minutes for a color change.
Record the color. What has been formed (as evidenced by the color change)?
6. Cool the test tube from step 5 in an ice-water bath until the color changes once more. Record the color. (Think about what has occurred that caused this color change.)
7. The next experiment requires clean, dry glassware. Always put your glassware away clean so that it will be dry by the next lab period. You will waste valuable lab time if you have to wash and dry glassware.
30
REPORT
LE CHATELIER’S PRINCIPLE
A. Record colors of:
Fe(NO
3
)
3
Compare the color of the solutions in: test tubes 1 and 4: test tubes 2 and 4: test tubes 3 and 4:
B. Record colors of:
Ni(NO
3
)
2
C. Record colors of:
Color after the addition of 6M HCl:
Co(NO
3
)
2
Color after addition of H
2
O:
Color after heating:
Color after cooling:
NAME ________________________________
SECTION _____________________
KSCN
NH
3
HCl
Fe(SCN)
Ni(NH
CoCl
3
4
2-
)
2+
6
2+
31
32
QUESTIONS FOR LE CHATELIER’S EXP.
2+ .
NAME _________________________________
1. a. Write a balanced net ionic equation for the equilibrium reaction in Part A, formation of
Fe(SCN) b. For each of the changes in Part A, give the immediate effect of each perturbation on the value for Q (increase, decrease, or no change). Do the color changes you observed agree with the shift predicted by the change in Q? Explain your answers. i. additional iron(III) is added ii. additional thiocyanate is added iii. additional water is added
a. Write a balanced net ionic equation for the equilibrium reaction in Part B, formation of
Ni(NH
3
)
6
2+
33
2. b. Select which component from the equilibrium mixture reacts with HCl and then write a net ionic equation for that reaction (NOT AN EQUILIBRIUM!). c. The addition of hydrochloric acid impacts one of the components in the equilibrium reaction shown in 2a. Determine the immediate effect on the value of Q due to the addition of HCl. d. Do the color changes you observed agree with the shift predicted by the change in Q?
Explain your answer.
NAME ________________________________
34
QUESTIONS FOR LE CHATELIER’S EXP.
3. a. Write a balanced net ionic equation for the (equilibrium) formation of the tetrachlorocobaltate(II) complex. b. Based on your observations of color changes in Part C, did heating the reaction mixture cause a shift in equilibrium? Which direction? Explain your answer based on the color changes you observed. c. Is the formation of tetrachlorocobaltate(II) complex ion exothermic or endothermic?
Explain your answer based on the shifts in equilibrium caused by heating and cooling the reaction mixture. d. What is the effect of an increase in temperature on the value for the equilibrium constant?
(Increase, decrease or no change)
35
QUESTIONS FOR LE CHATELIER’S EXP. NAME ________________________________
4. Consider the hypothetical equilibrium: A + B C + D a. Write the equilibrium expression for this reaction.
H<0 b. Suppose a change is made to the system. Fill in the following table—answer using one of the symbols given in each question. Note: NC means no change, and NS means no shift.
Changes
For each change given at the top of a column, answer the questions below
C is added A is added
D is removed
A catalyst is added
Temperature is decreased
What will be the immediate effect on Qc (↑, ↓, or NC)?
What will be the effect on Kc
(↑, ↓, or NC)?
In comparing the values from above how does the size of Qc compare to Kc (Q = K, Q < K, or
Q > K)?
Which way will the change cause the reaction shift to reestablish equilibrium, right
(→), left (←) or NS?
When the new A equilibrium has been established, B is the amount of each substance present C greater ( ), less ( ), or unchanged (NC) from what D it was before the change?
36
37
WEAK ACIDS AND BASES
INTRODUCTION
One method of measuring the acidity or basicity of a solution is to use a pH meter. A pH meter is a voltmeter that measures the potential of an electrical current flowing through a solution that is in contact with both a pH sensitive glass electrode (the measuring electrode) and a constant voltage
(reference) electrode. In many pH meters, these two electrodes are fused together into one
"combination" electrode. These electrodes feed their signals into a voltmeter that is calibrated so that the overall voltage is converted directly to pH units.
In this experiment, a pH meter will be used to study acid-base equilibria of a weak acid, acetic acid, and a weak base, ammonium hydroxide. Because pH = −
log
H O +
and
pOH = −
log
OH − =
14.00
− pH pH measurements can be used in the calculation of the equilibrium hydrogen ion and hydroxide ion concentrations in any aqueous solution. If the initial concentration of a weak acid is known and the hydrogen ion concentration at equilibrium is calculated from the pH, then the percent ionization
(dissociation) of the weak acid in solution can be determined. For example, let HA represent any monoprotic weak acid. Then
Initial
Change
Equilibrium
HA + H y
-x y-x
2
O A
0
+x x
+ H
3
O +
~0
+x x where y is the initial concentration of the acid. The percent ionization (dissociation) for the monoprotic acid is (x/y) times 100.
The effect of weak bases on pH is due to the ionization (hydrolysis) of water. If A- represents any weak base and y is the initial concentration of that base, then
Initial
Change
Equilibrium
A + H y
-x y-x
2
O HA + OH
0
+x x
-
~0
+x x and the percent of the weak base involved in ionization is (x/y) times 100.
38
1.
PROCEDURE
A. Calibration of the pH meter
The number of groups will be limited by the number of pH meters available. Follow your professor's instructions as to the number of students per group.
Instructions for standardizing the UB-5 pH meter. a. Immerse the electrode in a standard buffer solution. Stir gently. Allow the electrode to reach a stable value. b. If necessary, press and release the mode button until the display indicates pH mode. c. Clear existing buffers when performing a new standardization. Use the setup and enter buttons to clear existing buffers. d. Press standardize . The meter flashes the current buffer set and detects the flashing buffer.
When the signal is stable, or when you press enter, the buffer’s pH is stored. e. The meter displays the percent slope of the electrode as 100.0% on the first buffer. On entering a second or third buffer, the meter performs a diagnostic check on the electrode and displays the slope. f. To enter a second buffer, rinse the electrode with deionized water, gently dry it with a chemwipe and place the electrode in the second buffer solution. Stir and allow time for the electrode to stabilize, and press standardize again. The meter detects the buffer and when the signal is stable, or when you press enter, the buffer’s pH is stored. g. Next, the meter performs a diagnostic test of the electrode. The display indicates electrode’s condition. The meter displays the % slope obtained from the values read by the electrode. h. If Error displayed with the Slope symbol this indicates that your electrode is not working properly. The electrode response must be between 90 and 105% slope. Measurements causing Slope Error are not accepted, used or stored by the meter. Press enter to continue. i. To enter a third standard, clean the electrode as before and place the electrode in the third buffer solution, stir, allow it to stabilize, and press standardize . The results will be the same as in steps g and h. j. After entering each buffer, the Standardizing symbol goes off and the Measuring or Stable symbol appears on the display to indicate that the meter has returned to Measuring operation. k. Standardize your meter and electrode using at least two buffers with pH values above and below the expected pH of your samples.
39
B. The Effect of Dilution on the pH of a Weak Acid Solution
1. Clean a shell vial. When used in the experiment, the vial can be wet but should be well drained.
If you have not already done so, standardize your pH meter with the pH 4, 7 and 10 buffer solutions. Remember to rinse the pH meter’s probe well with distilled water between the measurements.
2. In a clean 10 mL graduated cylinder, obtain 4 to 5 mL of 1.0M acetic acid. Pour it into the clean shell vial. Measure and record the pH of the acetic acid. (The solution must cover the tip of the probe while the measurement is made.)
3. Pour exactly 1.0 mL of the acetic acid back into the 10 mL graduated cylinder. Discard the remaining acid. Add distilled water to the acid in the graduated cylinder until the total volume is exactly 10.0 mL (you have just made a 1 to 10 dilution). Mix well by carefully pouring the solution back and forth between the vial (the one from which you discarded the excess acid) and the graduated cylinder. Pour sufficient diluted acid into the shell vial to allow you to measure and record the pH of this diluted solution.
4. Save exactly 1.0 mL of diluted acid from step 3 in the cylinder and discard the remainder. Again add distilled water to the cylinder until the volume is 10.0 mL. You have now made a second 1 to 10 dilution. (What is the overall dilution?) Mix well and record the pH of this solution.
5. Again, save exactly 1.0 mL of the diluted acid from step 4 in the cylinder and discard the remainder. Again add distilled water to the cylinder until the volume is 10.0 mL. (What is the overall dilution now?) Mix well and record the pH of this solution.
C. The Effect of Dilution on the pH of a Weak Base
1. Clean a shell vial and repeat steps 2 through 5 for Part B above except use 4 to 5 mL of 1.0M ammonium hydroxide for the initial solution. ( Note: ammonium hydroxide is also known as aqueous ammonia and thus 1.0M NH
3
would also be an appropriate label for this solution.)
2. Rinse the probe well. If the probe had a cap and no storage solution is available, put a small amount of tap water into the cap before gently sliding it onto the probe. If your probe did not have a cap, leave the tip of the probe dipped into a beaker containing tap water. Save your standardization buffers for pH experiments that will be completed on other lab days.
D. Calculation of Equilibrium Constants
1. Calculate the initial molarity (before dissociation or hydrolysis) of the acid or base for each of the diluted solutions.
2. From the pH readings, calculate the hydronium ion concentration (in molarity) of each acidic solution and the hydroxide ion concentration for each basic solution. Use these data to calculate the percent dissociation for each acetic acid solution and percent hydrolysis for each ammonium hydroxide solution.
40
3. Calculate the K the K a a
for each of the acetic acid solutions, and the average K a
. Calculate the percent relative average deviation (see Appendix A at the end of the lab manual) for the four K a
’s. Use
value for acetic acid given in your textbook as the accepted value and calculate your percent error (see Appendix A at the end of the lab manual). Because you are calculating very small numbers and because this experiment was done at non-standard conditions, your experimental error may be quite large. Use analogous calculations to calculate percent relative average deviation and percent error for your K b
for ammonium hydroxide.
REPORT
WEAK ACIDS AND BASES
Acetic acid:
Solution from step
2
Molarity of
HC
2
H
3
O
2
1.0M
3
4
5 pH
Average K a
Percent Relative Average Deviation
Percent Error
Ammonium hydroxide (NH
3
):
Solution from step
2
Molarity of
NH
3
1.0M pH
3
4
5
Average K b
Percent Relative Average Deviation
Percent Error
SHOW CALCULATIONS ON SEPARATE PAGES:
NAME_________________________________
SECTION _______________________________
[H
3
O
[OH -
+ ],
],
M
M
% Dissociation
% Hydrolysis K b
K a
41
42
QUESTIONS FOR WEAK ACIDS & BASES EXP. NAME ______________________________
1. Examine the data for acetic acid and discuss the effects of dilution on the percent dissociation of this weak acid. a. What immediate effect did dilution have on Q? b. Did K change? Should it have changed? Why or why not? c. Which way did any changes cause the equilibrium to shift? Why? d. How did the shift affect the percent dissociation?
2. Should the effects of dilution on % dissociation for a weak acid be any different than % hydrolysis of a weak base undergoing dilution?
43
QUESTIONS FOR WEAK ACIDS & BASES EXP. NAME ______________________________
3. A 0.0100 M solution of a weak monoprotic acid is found to be 3.5% ionized. What is the pH of this acid solution? What is the K a
for this weak acid?
4. A weak base has a K b
of 4.6×10 -4 . Calculate the percent hydrolysis of the base and the pH of the solution if the initial concentration of the weak base is 2.5×10 -2 M .
44
45
DETERMINATION OF K
a
BY pH TITRATION
INTRODUCTION
From previous chemistry labwork students should already be familiar with acid-base titration techniques. Those experiments probably used a pH indicator (such as phenolphthalein) to determine the "endpoint" of the titration--the point at which a stoichiometrically equivalent amount of base had been added to the acid (or acid to base). In such a titration, the only data collected are the mass or volume of acid and base that have been added to the titration flask when the equivalence point is reached. However, to construct an acid-base pH titration curve, both pH and buret readings must be recorded after each addition of reagent from the buret. From the volume and molarity of the reagent added, the moles of reagent added can be calculated and then this is plotted against pH.
Acid-base titration curves for monoprotic acids have a characteristic shape. The titration curve shown below is typical of one obtained when a strong base is added to a weak acid. mol NaOH added
At the beginning of a titration, pH changes slowly as base is added. Acid is in excess and only a small percentage of the acid is neutralized after each addition of base. As more base is added, the ratio of the conjugate base formed to the remaining (unreacted) weak acid in the titration flask continues to increase. However, as the equivalence point is approached, very little acid remains and, as base continues to be added, there is a sudden excess of base. It is at this point in a titration that the pH changes very rapidly. After passing this rapid pH change region the pH becomes dependent only on the gradually increasing concentration of excess strong base and again changes slowly.
Acid-base pH titrations can provide information that titration to an indicator endpoint cannot. Both methods will identify the equivalence point, but the pH titration provides information which allows
46 the pK a
and K a
for the acid being titrated to be determined. One method of doing this is to plot pH as a function of the moles of base added. After the titration curve has been constructed, two straight lines can be drawn through the data that is almost horizontal (see the diagram on the previous page).
A vertical line that is parallel to the y-axis is drawn between the two "horizontal" lines. The midpoint of the vertical line (1/2 the distance between the horizontal lines) is the approximate equivalence point (moles of original Hydrogen ion equal to moles of Hydroxide ion added). Note: in a titration between a weak acid and strong base, at the equivalence point all the weak acid has been converted to its conjugate, weak base. An alternative method of determining the equivalence point is to construct, on the same graph, the first derivative curve. The first derivative shows how the pH changes for the amount of base added. The change in pH will be relatively constant at first and then start to increase as we approach the equivalence point. After the equivalence point the change in the pH will start to decrease and then become relatively constant again.
Now the pK a
and ultimately the K a
of the acid can be calculated. Remember, the equivalence point is the point at which the acid has been completely neutralized by the strong base. The weak acid has been converted completely to its conjugate base and water. To use the Henderson-Hasselbalch equation: a
+
log
conjugate base weak acid
you must determine from the graph at what point half of the acid was neutralized. It is only at this point that half the acid has been converted to its conjugate base and thus the concentration of the two are equal; when pH = pK a
.
Alternatively, we can think of the Henderson-Hasselbalch equation as: pH = pK
log
V
b
Where V b
is the volume of base added and V axis and log(V b
/(V e
-V b
. e a
+
V
e
−
V
b
is the equivalence point volume. If we plot pH on the y-
) on the x-axis for volumes from about 20% to 80% of the equivalence point we will get a straight line. The point on the pH scale where this line crosses 0 on the x-axis is the point at which the pH is equal to the pK a
The experimental value for K a
can then be determined from the equation: pK a
= −
log
So,
K a phthalate (KHP). KHP, KHC
8
H
4
O
4
K a
=
10
− pK a
In this experiment, an acid-base pH titration curve will be constructed for potassium hydrogen
, is a monoprotic acid having a structural formula of:
47
O
H
H
C
C
O
-
K
+
C C
C C
O H
H C
C
H
O
An experimental K a
for KHP will be determined in this experiment.
PROCEDURE
1. Each group should obtain a buret, a Vernier LabQuest, a pH probe and a Drop Counter.
2. Plug the pH Probe into the port on the top of the LabQuest labelled
“CH 1“ (on the top) and the Drop Counter in the port labelled “DIG
1” (on the side). The display should look like the image to the right.
3. Attach the Drop Counter and a buret clamp to a ringstand such that the Drop Counter is below the buret clamp.
4. Obtain about 100 mL of standardized (approximately 0.1-M) NaOH in a clean dry beaker. Record the exact molarity of the NaOH from the bottle.
5. Using the same techniques learned in previous titration experiments, clean the buret, rinse and flush it with 1 to 2 mL of the
NaOH solution, discard the rinsings and fill the buret with the NaOH solution.
6. You need to make sure that the Drop Counter can “see” each drop that passes through it. Place the buret filled with the NaOH solution in the buret clamp so that the tip of the buret is just above and approximately centered over the slot in the Drop Counter. Place a waste beaker under the
Drop Counter to collect the solution. Turn on the LabQuest. When it has started you should see that both probes are connected. Press the “Collect” button. Open the stopcock on the buret such that the NaOH solution comes out one drop at a time (about 1 drop every second or two). If it is aligned correctly you should see the volume increase incrementally on the screen.
If it is not, adjust the buret side-to-side until the LabQuest shows the volume changing. When everything is aligned correctly close the stop flow of the solution and press the “Collect” button to stop data collection.
7. Calculate the approximate mass of KHP (FM = 204.23) that would be required to neutralize about
25 mL of 0.1M NaOH.
8. Clean and label two 250 mL or 400 mL beakers (they can be wet). From your professor, obtain a small amount of KHP in a dry shell vial and take the KHP and titration beakers to the analytical balance room. Use the "weighing by difference" technique to place the approximate mass of
48
KHP determined in step 4 into each of the two beakers. Record the mass of KHP in each beaker
( ± 0.0001 g). Obtain approximately 50 mL of the supplied NaOH solution. Be sure to record its molarity.
9. Add approximately 50 mL of distilled water to each beaker and swirl until the KHP is dissolved.
10. Fill a small beaker with distilled water and stand the pH probe in the beaker. The probe should be free from the holder so that it can be moved easily between the beaker and titration beaker.
11. Add a magnetic stir bar to titration beaker 1 and place the beaker under the Drop Counter on top of the magnetic stir plate. Lower the tip of the pH probe through the hole in the Drop Counter into the solution of beaker 1. Turn on the magnetic stir plate and set the speed to the maximum setting.
12. Press “Collect” button on the LabQuest. Open the stopcock on the buret such that the solution flows out one drop at a time at a rate of no more than about 1 drop per second.
13. When pH reaches 11 to 12 and you have added about 2 to
3 mL of solution at that pH you can close the stopcock on the buret and stop the run by pressing the “Collect” button. At this time the contents of beaker 1 can be discarded.
14. Refill the buret with the NaOH solution.
15. Repeat Steps 11 through 13 for beaker 2. Before pressing the “Collect” button, click on the file cabinet icon to add another run to the data collection (Run 2).
16. Attach a USB flash drive to the USB port on the top of the LabQuest. It may take a few seconds for the device to recognize the USB drive. Click on “File” and then “Export.” Click on the USB icon and save the data as a text file onto the flash drive (both runs will be in the file). Give the file a meaningful name. Make sure that the data (including both runs) is on the drive and that all members of the group have a copy of the data. The data is saved as a tabdelimited ASCII file (.txt).
17. Using Excel, or another graphing program (i.e., Google Sheets, Origin, or Numbers on a Mac), open the file from your USB flash drive. It will recognize that it is a tab-delimited ASCII file. Just click on “Next” to choose the default option for everything. The spreadsheet should look like this:
18. In Excel, create a two new columns for your data. The first column should be labelled pH/ V, the second column is explained in Step 22. In this column starting with row containing the first
49 data point enter the formula “=(B9-B8)/(A9-A8)” (without the quotation marks). Here we used B8, B9, A8 and A9 because the data starts in row 8. Press ENTER. Put the cursor at the bottom right corner of the cell containing that formula (it should change into a bold cross) and click and drag it down to the last row of data for Run 1 to copy it down that column (there are likely about 1500 data points so be careful). Do this for both runs.
19. Construct two titration graphs, one for each run. Plot pH (vertical axis) as a function of the volume of NaOH added (horizontal axis) and
pH/ V (vertical axis) as a function of the volume of NaOH added on the same graph. You can do this by highlighting all three columns of data and selecting Insert/Chart/Scatter/Scatter with Smooth Lines . Be sure to properly title and label your graphs. Also be sure that each graph shows the correct precision. See Appendix B for a review on graphing. Do this for both runs.
20. You can more easily see where the peak of the pH/ V vs. volume graph is by plotting the
pH/ V data on a secondary axis. The point on the x-axis (volume) where this line has its maximum value is the equivalence point. Do this for both runs.
21. Determine the pK a
from each of your pH vs. volume graphs. Show on the graph how you determined the pK a
value.
22. The second column should be labelled log(V b
/(V e
-V b
)). Starting at a volume that is about 20% of the equivalence point enter the formula “=log(AXX/(yy.yy-AXX))” (again without the quotation marks). XX indicated the row number you are starting at and yy.yy is the equivalence point volume you determined in step 20. Copy this formula down to about 80% of the equivalence point. Do this for both runs.
23. Create a new graph (Scatter X-Y, Line) and plot pH on the y-axis and the new column of data on the x-axis. You should get a straight line. Where this line crosses the y-axis (x=0) is the point where the pH=pK a
. Do this for both runs.
24. Read the pK a
from each graph. Mark each graph to show how you got the pK a
.
25. Average the two pK a
values. Calculate the experimental K
26. Attach printouts of all 4 graphs to your lab report. a
for KHP from the average pK a
value.
50
REPORT
DETERMINATION OF K a
BY pH TITRATION
NAME_______________________________
SECTION _____________________________
Molarity of NaOH, M
Mass of KHP, g Trial 1 pK a
(trial 1) from pH vs. volume graph pK a
(trial 2) from pH vs. log(V b
/(V e
-V b
)) graph
Mass of KHP, g Trial 2 pK a
(trial 2) from pH vs. volume graph pK a
(trial 2) from pH vs. log(V b
/(V e
-V b
)) graph
Average pK
K a a
51
52
QUESTION FOR DETERMINATION OF K
A
… NAME _______________________________
1. Should the mass of KHP used for the pH titration change the experimental value for the K a
Explain your answer.
?
2. Calculate the experimental K for KHP. b
for the phthalate ion, C
8
H
4
O
4
2, from the average experimental K a
3. Using the experimental K b
in Question 2, calculate the pH of a 1.5M K
2
C
8
H
4
O
2
solution.
53
QUESTION FOR DETERMINATION OF K a
… NAME _______________________________
4. Recall that an optimum buffer is one that contains equal (or close to equal) concentrations of a weak acid and its conjugate base. At approximately what pH reached during the titration would the solutions in titration flasks 1 and 2 meet the criterion for an "optimum" buffer? Explain your answer.
5. Using duplicate calculations (one for each graph), use the equivalence point on each graph to determine an experimental molar mass of KHP (remember it is monoprotic). Average your results. Now use the true molar mass of KHP (204.23 g mol -1 ) and calculate the percent error for this experiment (see appendix A of this lab manual). This is a measure of the accuracy of your work in this procedure. Show calculations.
54
55
BUFFERS AND pH
INTRODUCTION
An acid-base buffer is a solution that resists change in pH when small amounts of acid or base are added. This type of buffer contains two species, a weak acid and its conjugate base. The weak acid reacts with and partially removes from solution added base, and the weak acid’s conjugate base reacts with added acid. If hydrogen ion is removed from solution, the buffer’s weak acid dissociates to partially replace the hydrogen ion that was removed. If hydroxide is removed from the system, it is partially replaced through hydrolysis of water by the weak conjugate base. These processes are examples of Le Châtelier’s principle. The original buffer solution is at equilibrium. Added material temporarily disturbs this equilibrium, and the system shifts to restore equilibrium. Thus, concentrations of hydrogen ion and hydroxide ion are “buffered” and the pH of the solution remains relatively constant.
To be a pH buffer, both a weak acid or base and its conjugate base or acid must be initially present. In other words, both must be present before dissociation by the weak acid or hydrolysis by the weak base can be considered. An “optimum” buffer, which has equal capacity to neutralize either added acid or added base, is created when the concentrations of the conjugate acid/base pair in the buffer solution are equal. However, a solution does not have to contain equal amounts of the pair to be considered a buffer. The equation
K a
=
H + A −
HA can be rearranged into the Henderson-Hasselbalch equation: a
+ log
A −
HA
From the equation above, it can be seen that the ratio of the conjugate acid-base pair can be varied to create a buffer solution with a desired pH so long as that pH is close to the pK a
of the acid form of the weak pair. The buffer solution does not have to be made by combining the weak acid and its conjugate base directly. It also can be created by partial neutralization of a weak acid by a strong base, or by partial neutralization of a weak base using a strong acid. For example, if a weak acid (HA) is neutralized by a strong base, the net ionic equation for the reaction would be:
HA + OH A + H
2
O
If the hydroxide ion from the strong base were the limiting reactant, some weak acid, HA, would remain in solution after reaction was complete. The HA remaining in solution, along with its conjugate base, A , (formed in the partial neutralization) would create the buffer. An analogous approach would be to use an excess of weak base with a limited amount of strong acid.
56
In this experiment, various solutions will be prepared and studied. A pH meter will be used to determine the experimental equilibrium concentration of hydrogen ion in each solution. Using the
Henderson-Hasselbalch equation, the theoretical pH and hydrogen ion concentration can be determined from the K a
and the mole to mole ratio of the conjugate acid/base pair for each solution studied.
PROCEDURE
1. Due to the limited number of pH meters, students will work in groups. Follow your professor’s instructions regarding the number of students per group.
2. Each group will need to obtain pipet pump.
3. Prepare your meter’s electrode for use and standardize the pH meter (refer to the instructions provided in the “Weak Acids and Bases” experiment).
4. Obtain approximately 40 mL each of 0.20M acetic acid and 0.20M sodium acetate solutions in separate clean, dry 50 mL beakers. Obtain approximately 15 mL each of 0.10M hydrochloric acid and 0.10M sodium hydroxide solutions in separate clean, dry 20 mL beakers.
5. Pour enough of the 0.20M acetic acid solution into a clean, dry shell vial so that you can measure its pH. Record the pH.
6. Clean your pipet and use the solution remaining in the shell vial to rinse the pipet. Discard the solution used for rinsing.
Do not pipet by mouth; use a bulb or a pipet-pump!
7. Using the pipet, measure 25.0 mL of the acetic acid solution into a clean, dry 150 mL beaker. Save the acetic acid solution remaining in the 50 mL beaker.
8. Repeat steps 5 and 6 using the sodium acetate solution.
9. Using the freshly rinsed pipet, measure 25.0 mL of the sodium acetate solution and add it to the
150 mL beaker containing the acetic acid solution (Step 7) and mix well. This is the combined solution that will be referred to throughout this experiment. Save the sodium acetate solution remaining in the 50 mL beaker.
10. Pour enough of the combined solution into a clean, dry shell vial to measure its pH, and record.
Do not discard the remaining combined solution in the 150 mL beaker.
11. In all subsequent steps, you may use a clean shell vial that has been rinsed with deionized water and well-drained.
57
12. Use clean 10 mL graduated cylinders and follow the chart to carefully measure the volume of each reagent indicated into separate, well-drained shell vials.
Shell Vial
Number
Combined
Solution
0.20M acetic acid
0.20M sodium acetate
0.10M HCl
0.10M
NaOH
H
2
O
7
8
5
6
3
4
1
2
9
10
11
3.0 mL
4.0 mL
4.0 mL
3.0 mL
3.0 mL
4.0 mL
4.0 mL
2.0 mL
3.0 mL
4.0 mL
2.0 mL
3.0 mL
4.0 mL
3.0 mL
2.0 mL
3.0 mL
2.0 mL
3.0 mL
2.0 mL
3.0 mL
2.0 mL
3.0 mL
13. Cover each shell vial with Parafilm™. Mix the contents well by inversion, then measure and record the pH of each solution. Rinse the probe well with deionized water between every measurement.
14. Rinse the probe well. If the probe had a cap, put a small amount of storage solution or tap water into the cap before gently sliding it onto the probe. If your probe did not have a cap, leave the tip of the probe dipped into a beaker containing tap water.
15. Be sure to clean the pipet and rinse it with deionized water. Return the pipet pump if you borrowed one.
16. Calculate the initial molarity (after dilution but before any shift to achieve equilibrium) of the acetate and the acetic acid in the combined solution.
17. Calculate the initial moles (due to the combination of solutions or after any neutralization reaction but before any shift to achieve equilibrium) of the acetate ion and the acetic acid present in tubes
1 through 11. In some of the solutions these species come from more than one reagent. In others, acid-base neutralization calculations must be completed before the initial moles can be determined.
18. Determine the ratio of moles of acetate ion to moles of acetic acid for tubes 1 through 11. Express your ratios as 1:1, 1:3, 2:1, etc.
58
pH
NAME_________________________________
SECTION _______________________________
59
REPORT
BUFFERS AND pH
Solution
0.20 M
HC
2
H
3
O
2
0.20 M
NaC
2
H
3
O
2
Combined
Solution
Vial Number
1
2
3
6
7
4
5
Initial Molarities in Combined Solution acetate acetic acid
Initial millimoles acetate acetic acid acetate : acetic acid ratio
8
9
10
11
SAMPLE CALCULATIONS: (Use separate sheets if necessary)
60
QUESTIONS FOR BUFFERS AND pH EXP. NAME ____________________________
1.
In theory, which of the 14 solutions tested should have similar pH’s? Why? Use results from your calculations for step 18 of the Procedure to help explain your answer for each solution.
61
QUESTIONS FOR BUFFERS AND pH EXP. NAME ____________________________
2. The experimental pH values for the solutions should be in fairly good agreement with the theoretical pH values for each of the solutions tested. Why? What are some things that could cause the experimental pH to be different than the theoretical pH?
3. Which of the 14 solutions tested are buffers? Identify any of the solutions that would be considered “optimum” buffers (have the same number of moles of weak acid and conjugate weak base present).
62
63
ACID-BASE EQUILIBRIUM PROBLEMS
1. Calculate the pH of a solution that contains 0.15 M oxalic acid. Calculate the concentration of the oxalate ion in this solution.
2. Calculate the pH of a 0.0035 M solution of methylamine.
64
3. 65.3 mL of 0.156 M hydrochloric acid is added to 145.3 mL of 0.078 M aniline solution. What is the approximate pH of the resulting solution?
4. Out of the following, which is the best acid/base to use to prepare a buffer with a pH of 8.00? a. sodium cyanate b. sodium lactate c. hydrazine
What ratio of masses of the weak acid/base and its conjugate should you use to make the buffer of the required pH? Use the sodium salt of the conjugate base if you chose a weak acid or the chloride salt of the conjugate acid if you chose a weak base.
65
5. Calculate the pH at the equivalence point when 25.00 mL of 0.10 M iodic acid is titrated with
0.080 M barium hydroxide solution.
6. What is the pH of a solution obtained by adding 100.0 g of sodium benzoate to enough water to make 1.50 L of solution?
66
8. If K w
7. 25.00 mL of 0.15 M hydroxylamine is titrated with 0.20 M hydrochloric acid. When 12.56 mL of the acid have been added what should the approximate pH be?
at 40.0°C is 2.916×10 -14 , what is the pH of pure water at this temperature?
67
9. The pH of a 0.15 M solution of butanoic acid is 2.82. What is the K b
of the butanoate ion?
10. Ethanolammmonium ion has pK a
of 9.498. What is the pH of a 0.050 M solution of ethanolamine?
68
A SOLUBILITY INVESTIGATION
INTRODUCTION
Most metal ions are soluble when mixed with most anions. There are some exceptions as delineated in the solubility rules in your textbook. In this experiment we are going to examine some of these insoluble salts and the circumstances that affect their solubility.
One factor that can affect solubility is the pH of a solution. If the anion in the insoluble salt is the conjugate base of a weak acid, the salt will become more soluble as the pH decreases. For example, barium sulfate is an insoluble salt with an equilibrium reaction shown as
BaSO
4
Ba 2+ + SO
4
2 Eq. 1 and the solid will become more soluble as the pH decreases because sulfate ion will react with hydronium ions in an acid/base equilibrium
H
3
O + + SO
4
2 HSO
4
2 + H
2
O Eq. 2
As Equation 2 proceeds to the right it effectively removes sulfate ion from the first equilibrium (Eq. 1) causing the first reaction to shift to the right, (i.e., more barium sulfate dissolves), to re-establish equilibrium.
Increasing pH can also affect an equilibrium if the pH is raised in the correct manner. Silver chloride is an insoluble salt with an equilibrium reaction of
AgCl Ag + + Cl Eq. 3
If we increase the pH of the mixture shown in Equation 3 by adding aqueous ammonia, the ammonia forms a complex with the silver ion
Ag + + 2 NH
3
Ag(NH
3
)
2
+ Eq. 4 which removes silver ion from Eq. 3 causing more of the silver chloride to dissolve as equilibrium is reestablished.
Another example involves amphoteric hydroxides. Amphoteric hydroxides are compounds that can react with either acids or bases. Aluminum hydroxide is an amphoteric hydroxide. If we have aluminum ion in solution and we start to increase the pH by adding a strong base we initially produce an insoluble compound
Al 3+ + 3 OH Al(OH)
3
Eq. 5
Continued addition of hydroxide allows another equilibrium to occur in which a complex ion is formed between the aluminum ion and the hydroxide
Al(OH)
3
+ OH Al(OH)
4
Eq. 6
69 which results in the solid Al(OH)
3
dissolving. But, addition of a strong acid would also dissolve solid
Al(OH)
3
:
3 H
3
O + + Al(OH)
3
Al 3+ + 6 H
2
O Eq. 7
PROCEDURE
A. Effect of lowering the pH on the solubility of an insoluble salt.
1. Obtain 5.0 mL each of 1.0 M calcium chloride and 0.25 M sodium oxalate solutions
2. Pour both solutions into a 50 mL beaker (mixing well). What reaction has occurred?
3. Add approximately 10 mL of 6 M nitric acid and stir. What reaction has occurred? What do you observe? Dispose of the solution in the appropriate waste receptacle and thoroughly clean the beaker.
B. Effect of raising the pH on the solubility of an insoluble salt
1. Obtain 15.0 mL each of silver nitrate and sodium chloride solutions
2. Pour both solutions into a 100 mL beaker (mixing well). Allow the mixture to sit for 15 minutes and observe if any noticeable amount of silver chloride has precipitated.
3. Add approximately 25 mL of 6 M aqueous ammonia and stir. Allow the mixture to sit for 15 minutes and observe if any noticeable amount of silver chloride has dissolved.
4. Add approximately 25 mL of 6 M nitric acid and stir. Observe any changes that occur in the beaker.
Dispose of the solution from step 4 in the appropriate waste receptacle and thoroughly clean the beaker.
C. Effect of adding a strong acid base to an amphoteric hydroxide
1. Obtain approximately 20.0 mL of 1.0 M zinc nitrate solution and place it into a 150 mL beaker.
2. Add 6 M sodium hydroxide (with mixing), in a drop-wise fashion, until a reasonable amount of solid appears.
3. Divide the mixture from step 2 into approximately two equal portions. (This mixture contains the amphoteric hydroxide.)
4. To one of the two portions, continue to add 6 M sodium hydroxide (with mixing) until you see a distinct change in the mixture. Note how much sodium hydroxide solution was added. (Recall:
20 drops 1 mL)
5. To the other portion, add 6 M nitric acid (with mixing) until you see a distinct change in the mixture. Note how much nitric acid solution was added. Dispose of the solutions in the appropriate waste receptacle.
70
REPORT
A SOLUBILITY INVESTIGATION
NAME ______________________________
SECTION ___________________________
A. Effect of lowering the pH on the solubility of an insoluble salt.
Observations after mixing the two reagents
71
Observations after adding
HNO
3
to the mixture
B. Effect of raising the pH on the solubility of an insoluble salt
Observations after mixing the two reagents
Observations after adding
NH
3 to the mixture
Observations after adding
HNO
3
to the mixture
C. Effect of adding a strong acid or base to an amphoteric hydroxide
Observations after mixing the two reagents
To the first portion
Observation after adding mL NaOH
To the second portion
Observation after adding mL HNO
3
72
73
QUESTIONS FOR A SOLUBILITY INVESTIGATION NAME___________________________
1. Write the equilibrium reaction for the mixture in the beaker in Part A, step 2.
Write the net ionic equation for the reaction (which involves one of the species in the reaction that you’ve just written) that occurs when nitric acid is added to the beaker in Part A.
Examine the two reactions shown above for part A and explain, using Le Chatelier’s principle, why the changes occurred in the beaker after adding nitric acid.
2. Write the equilibrium reaction for the mixture in the beaker in Part B, step 2.
As in question 1, write the net ionic equation for the reaction (which involves one of the species in the reaction that you’ve just written) that occurs when aqueous ammonia is added to the beaker in Part B.
Again, according to Le Châtelier’s principle, why does the precipitate dissolve upon addition of ammonia?
As above, write the net ionic equation for the reaction that occurs when nitric acid is added to the beaker in part B.
As previously, explain why the precipitate reappears upon addition of nitric acid.
74
QUESTIONS FOR A SOLUBILITY INVESTIGATION NAME _________________________
3. Write the net ionic equation for the reaction that initially occurs when aqueous sodium hydroxide is added to the zinc nitrate solution. (Formation of the amphoteric hydroxide.)
Write the net ionic equation for the reaction that occurs when an excess of sodium hydroxide is added to the amphoteric hydroxide. (Step 4)
Write the net ionic equation for the reaction that occurs when nitric acid is added to the amphoteric hydroxide. (Step 5)
In the context of part C of this experiment, explain what an amphoteric hydroxide can do that:
• acetic acid can’t do
• aqueous ammonia can’t do
• sodium chloride can’t do
75
SOLUBILITY AND K
sp
DETERMINATION
INTRODUCTION
Calcium iodate is an ionic compound that is only slightly soluble in water. In aqueous solution, an equilibrium forms between the solid salt and its ions:
Ca(IO
3
+ 2 IO
3
)
2
Ca 2+
The solubility of calcium iodate can be determined by measuring the concentration of either the calcium ion or the iodate ion in a saturated solution. In this experiment the concentration of the iodate ion will be determined.
This analysis involves two reactions. First, the saturated solution of calcium iodate is acidified and reacted with excess potassium iodide, converting all the iodate ions into molecular iodine.
IO
3
+ 5 I + 6 H + → 3 I
2
+ 3 H
2
O
The molecular iodine formed is then titrated with standardized sodium thiosulfate.
I
2
+ 2 S
2
O
3
2 → 2 I + S
4
O
6
2
The titration uses as indicators, the brown color of the molecular iodine (the iodate and iodide ions are colorless) and the dark blue color of an iodine-starch complex, (seen in the chemical kinetics experiment).
PROCEDURE
1. Each group will need one buret and a pipet pump. In this experiment you will need a clean, dry shell vial, a clean, dry 10 mL graduated cylinder, three clean and dry filter funnels, and seven clean,
100 to 250 mL beakers (they don't need to be the same size). Four of the beakers must be dry, the other three can be wet. If your group does not have these available, clean them and put them in the oven now (remove any plastic parts from the graduated cylinders BEFORE putting them in the oven).
2. Label three beakers (the ones that can be wet) A-1, B-1, and C-1. Put about 50 mL of distilled water into each beaker. Bring a clean, dry shell vial to your instructor to obtain about 4.5 g of calcium iodate. Using the balances in the lab (NOT the analytical balances), weigh out approximately 1.0 g of calcium iodate and place it in beaker A-1. Again using the balances in the lab, weigh out approximately 1.5 g of calcium iodate and place it in beaker B-1. Weigh out approximately 2.0 g of calcium iodate and put it in beaker C-1.
3. Stir the contents of each beaker with a separate clean stir rod. Allow the solutions to sit for at least 20 minutes, stirring every few minutes. Calcium iodate is only slightly soluble and the saturated solution forms slowly. Use this time to prepare for titration (Steps 4-7). If you have put cylinders, beakers, and/or funnels into the oven, remove them now and allow them to cool.
76
4. Using one of the cooled, clean, and dry beakers, obtain about 100 mL of standardized sodium thiosulfate. Record the exact molarity of the sodium thiosulfate solution.
5. Using the clean, dry graduated cylinder that you prepared, measure out three separate samples of about 1 cm 3 (1 mL) each of solid KI. (A paper funnel might help you pour the KI into the cylinder without spills. Clean up any spilled KI!) Set these aside for use in step 14.
6. In each of three clean test tubes (they can be wet) put about 2.0 mL (40 drops) of 1% starch solution. You will need this indicator solution later in the titrations.
7. Clean the buret, rinse and fill it with the sodium thiosulfate solution.
8. Set up the three clean, dry funnels you have prepared using buret clamps. Place the three clean, dry beakers labeled A-2, B-2, and C-2 under these funnels. Put dry filter paper cones into each funnel.
9. After allowing the calcium iodate mixtures to come equilibrium (it takes at least 20 minutes) pour each solution through its own filter cone, catching each filtrate in its own dry beaker. Do not add water.
Any precipitate remaining in the beakers can be discarded. It is the solution filtering into the beakers that you will be titrating and you do not want to change its concentration by adding rinse water. After the solution has filtered through, discard the filter papers and precipitates.
10. Set up three clean 125 mL titration flasks (they can be wet) labeled A-3, B-3, and C-3.
11. Clean the 10.0 mL volumetric pipet. Shake as much water as possible from the pipet and then rinse the pipet twice (each time with 1 to 2 mL) with the filtered solution from beaker A-2.
Pipetting carefully (do not pipet by mouth; use a pipet pump or a bulb!), transfer exactly a 10.0 mL sample (aliquot) of the solution from beaker A-2 to flask A-3. Rinse the pipet twice with the filtered solution from beaker B-2 and transfer 10.0 mL of solution from beaker B-2 to flask B-3.
Repeat the procedure for beaker C-2/flask C-3.
12. Add about 20 mL of water to each flask. ( Think about why is it okay to add water now.)
13. Add about 8 drops of 6 M HCl to each flask.
14. Add about 1 cm 3 (1 mL) of solid KI to the flask that you are now ready to titrate. (As KI is added to the flask, it reacts with iodate to form brown I
2
.) Swirl each flask until the KI is dissolved.
15. Record the initial buret reading. Set flask A under the buret (a white piece of paper under the flask will help you see color changes).
16. Start adding sodium thiosulfate from the buret into the flask. Add about 1 mL at a time and swirl well after each addition. The sodium thiosulfate will react with the brown Iodine and will convert it to colorless iodide ions. When the color of the solution has faded to pale yellow , add one of the 2.0 mL aliquots of starch solution to the titration flask. The starch will react with the remaining iodine in the flask to produce a dark blue complex. (If the starch had been added at the beginning of the titration, the very large amount of iodine present would create numerous complex ions with the starch that would make it much more difficult to titrate.)
77
17. Continue to titrate slowly. The blue-black color will start to fade. The endpoint is when one drop of sodium thiosulfate causes the solution to become colorless. (Note: It is more difficult to titrate from a colored to a colorless solution than vice versa.)
18. At the endpoint, record the final buret reading and calculate the volume of sodium thiosulfate used.
19. Refill the buret and repeat steps 14 - 18 for flask B and then again for flask C.
20. Calculate the molarity of the iodate ion which was in the saturated solutions (in beakers A, B, and
C).
Note: You must use the mole to mole relationships from both of the chemical reactions provided in the Introduction to go from moles of thiosulfate to moles of iodate.
21. Determine the average molarity of the iodate ion in the saturated solutions.
22. Calculate the molar solubility, the solubility in g/100 mL, and the solubility product constant, K for calcium iodate. Include appropriate units. sp
,
78
REPORT
SOLUBILITY AND DETERMINATION OF K sp
Molarity of Na
2
S
2
O
3
solution, ( M )
Approx. mass of Ca(IO
3
)
2
used, (g)
Initial Na
2
S
2
O
3
buret reading, (mL)
Final Na
2
S
2
O
3
buret reading, (mL)
Volume Na
2
S
2
O
3
used, (mL)
Moles of IO
3
in aliquot, (mol)
Volume of aliquot, (L)
Molarity of IO
3
in saturated solution, ( M )
Average Molarity of IO
3
, ( M )
Molar Solubility of Ca(IO
3
)
2
Solubility in g Ca(IO
3
)
2
/100 mL
Ksp for Ca(IO
3
)
2
SAMPLE CALCULATIONS (use separate sheets)
NAME _______________________________
SECTION _____________________________
Beaker A
Flask A
0.0100 0.0100
Beaker B
Flask B
Beaker C
Flask C
0.0100
79
80
QUESTIONS FOR SOLUBILITY AND DETRMINATION OF K
SP
NAME_______________________
1. How do the molarities of the iodate ion in each of the saturated solutions compare? Should they be the same? Explain.
2. How would adding water in step 9 to wash the solid calcium iodate precipitate onto the filter paper change the K sp
value which was determined experimentally? Would the calculated value for the constant be higher, lower, or unchanged if extra water had been used in this step? Explain.
3. In step 12, extra water is added to the titration flask. This added water does not alter the value obtained for the K sp
. Explain why.
81
QUESTIONS FOR SOLUBILITY AND DETRMINATION OF K
SP
NAME_______________________
4. Should a precipitate form when 25.00 mL of 0.0100 M silver nitrate is added to 15.00 mL of 0.0500
M potassium acetate?
5. If a 6.00 M potassium chloride solution is added dropwise (no significant volume change) to a solution containing both 0.010 M silver nitrate and 0.010 M mercury(I) nitrate, which insoluble chloride starts to precipitate first?
What percent of the cation that precipitated first remains in the solution just as the other cation reaches its saturation point with the chloride?
82
6. What is the molar solubility of barium fluoride in a solution that contains 1.00 M acetic acid and
1.00 M sodium acetate? Hint: Combine three equilibria reactions to determine the K c
for:
BaF
2
+ 2 HC
2
H
3
O
2
Ba 2+ + 2 HF + 2 C
2
H
3
O
2
and then solve for the molar solubility using the approximation method. Be sure to validate!
83
DETERMINATION OF K
f
BY SPECTROPHOTOMETRIC METHODS
INTRODUCTION
This experiment will use a spectrophotometer to obtain the data needed to calculate an equilibrium constant, K f
, for the formation of a complex ion from iron(III) and thiocyanate ions. The net ionic equation for the reaction is :
Fe 3+ + SCN Fe(SCN) and the equilibrium constant is given by the expression
2+
K f
=
( ) 2 +
Fe 3 + SCN −
To determine the K f
value, one must be able to measure or calculate the equilibrium concentrations of the three ions that appear in the equilibrium expression. Because the reactants, iron(III) ion and thiocyanate ion, are colorless, and the complex ion product is red, spectrophotometry can be used to determine the equilibrium concentration of the complex ion. This data can then be used to calculate the equilibrium concentrations of iron(III) ion and thiocyanate ion, assuming the starting concentrations of those ions are known.
Spectrophotometry is based on the principle that the light absorbed by a solution is directly proportional to the concentration of a component of that solution. The relationship between absorbance and concentration is given by the equation:
A = ε bc where A represents absorbance, c represents concentration, and ε and b are constants.
A spectrophotometer operates by separating light into its component wavelengths and selectively measuring the intensity of a given wavelength of light before and after it passes through a solution.
The absorbance (A) is then calculated (by the spectrophotometer) using the relationship
A = −
log
I
I
0 where I
0
is the intensity of the light entering the solution and I is the intensity of the light that has passed through the solution. It is customary to "zero" the spectrophotometer using the solvent that will be used for the test solution. This "zeroing" process accounts for light that is absorbed by the solvent or is scattered by the cuvet (a special test tube made of optically uniform glass).
Before determining the concentration of a particular solute in a solution, a “standard curve” for the solute must be prepared. The standard curve (which is actually a straight line) is prepared by measuring the absorbances of solutions having known concentrations of the solute. The absorbances of the known solutions are plotted as a function of their concentrations. The unknown's concentration is then obtained from that solution's absorbance and the “standard curve.”
84
PROCEDURE
A. Preparation of a Standard Curve
1. Each group will need one 1.00 mL pipet and three 5.00 or 10.00 mL graduated pipets, a pipet pump and a cuvet. Also obtain some Parafilm™.
2. In separate clean, dry labeled beakers or shell vials, obtain about 25 mL of 0.000075 M Fe(NO
3
)
3
, about 40 mL of 1 M KSCN, and about 45 mL of 0.1 M HNO
3
. Be sure to use the correct concentrations. Different concentrations are used in part B.
Do not take more reagent than you need.
If you do not have clean, dry beakers or shell vials (you should always put your glassware away clean) you will need to rinse the clean, wet beaker and/or vials with 2 to 3 mL of the reagent that you are obtaining. Discard the rinse solution.
3. Set up 12 large test tubes (they should each hold at least 8 mL). They should be clean and dry. If you need to wash them, rinse them with 1 to 2 mL of 0.1 M HNO3 and discard rinse.
4. Label the 1 mL pipet for Fe(NO
3
)
)
3
3 that are 1.0 mL or
(to use for volumes greater than 1.0
3
. Use this pipet for the volumes of Fe(NO
3
) smaller. Label the three 5 or 10 mL pipets, one for Fe(NO
3 mL) , one for KSCN, and one for HNO
3 it will be used.
. Rinse each pipet with 0.5 to 1.0 mL of the reagent for which
5. Using the appropriate pipet for each reagent, add the amount of each reagent to each tube that is shown on the chart below. Pipet carefully using the bulb!
)
3
(mL) Tube No. 0.000075 M Fe(NO
3
1
2
7
8
9
10
5
6
3
4
4.00
3.50
3.00
2.50
11
12
2.00
1.50
1.00
0.80
0.60
0.40
0.30
0.20
6. Cover each tube with parafilm and mix well.
1 M KSCN (mL) 0.1 M HNO
3
3.00
3.00
3.00
3.00
3.00
3.00
3.00
3.00
3.00
3.00
3.00
3.00
1.00
1.50
2.00
2.50
3.00
3.50
4.00
4.20
4.40
4.60
4.70
4.80
(mL)
85
7. The spectrophotometer must be “zeroed.” This means that the light absorbed by the solvent
(aqueous Nitric acid in this experiment) and light scattered by the cuvet must be blanked out so that it does not register on the display. The procedure below must be followed each time you
“zero” the spectrophotometer.
GENESYS 20 INSTRUCTIONS a. Set the wavelength to 450 nm. b. Rinse a cuvet, first with distilled water, and then with 0.5 to 1 mL of 0.1-M HNO
3
. Discard the rinse and fill the cuvet about three-fourths full with 0.1-M HNO
3 with a chem wipe.
. Wipe any fingerprints off the cuvet
Cuvets are made of special optically uniform plastic that needs to be protected against scratches. Use only chem wipes to clean them and do not allow chemicals to stand in them. Rinse them well with distilled water immediately after using them. c. Place the cuvet containing the HNO
3
into the cuvet holder. Position the cuvet so the light passes through the clear walls . Close the lid. d. Press the A/T/C button to select absorbance (A) mode. Press the 0 Abs/100%T button. After a few seconds, 0.000 should be displayed. e. The spectrophotometer has now been “zeroed.” Discard the HNO
3
in the cuvet, rinse the cuvet with 1 to 2 mL of the well-mixed solution in tube 1 and discard the rinse. Fill the cuvet about three-fourths full with the solution from tube 1, wipe the cuvet with a chem wipe and place it in the cuvet holder with the label on the cuvet facing forward and close the lid. Read and record the absorbance reading from the display. f. Repeat this procedure for each of the other solutions in tubes 2 through 12. (You do not have to re-zero the machine; just continue with the next solution, rinsing the cuvet as before.) g. Clean all your glassware and return the cuvet and pipet pump. h. Calculate the molarity of complex ion, Fe(SCN) 2+ , that was present at equilibrium for tubes 1 through 12. Note that the concentration of KSCN was very large compared to the concentration of iron(III) nitrate. This resulted in the reaction for the formation of the complex ion being driven essentially to completion and you can assume that all the iron(III) ion was converted to the complex ion. However, to calculate the concentrations you must take dilution into account. (Note: the final volume for all 12 tubes was 8.00 mL.) i. Construct a graph (using the graph paper provided after Appendix D) plotting absorbance (vertical axis) as a function of concentration (horizontal axis). Be sure to properly label and title your graph.
Draw a best fit straight line through the data. (See Appendix C for a review of graphing.) Include
0.00, 0.00 as a data point.
86
B. Determination of K f
for the Fe(SCN) 2+ complex ion.
1. Each group will need a cuvet, a 1.00 mL, a 5.00 mL and a 10.00 mL graduated pipet and a pipet pump. If you must wash beakers or test tubes, follow the same rinse procedure as for Part A.
2. In separate clean, dry beakers obtain about 15 mL 0.0025 M Fe(NO
KSCN, and about 60 mL of 0.1 M HNO
3
3
)
3
, about 20 mL of 0.0025 M
. Be sure to use the correct concentrations. Different concentrations were used in part A.
Do not take more reagent than you need.
3. Label the 1 mL pipet for Fe(NO
3
)
3
, the 5 mL pipet for KSCN and the 10 mL pipet for HNO
3
. Prior to using, rinse each pipet with between 0.5 and 1.0 mL the reagent to be used in that pipet.
4. Set up 10 large clean dry test tubes. Pipetting carefully, using the pipets you have just prepared, transfer the following amounts of reagent to each tube.
0.0025 M Fe(NO
3
)
3
(mL) 0.0025 M KSCN (mL)
0.50
0.50
0.50
0.50
0.50
0.50
1.00
Tube No.
1
2
3
6
7
4
5
1.50
2.00
2.50
0.50
1.00
1.50
2.00
2.50
0.1 M HNO
3
(mL)
7.00
6.50
6.00
5.50
5.00
6.50
6.00
8
9
10
1.00
1.00
1.00
1.00
1.00
5.50
5.00
4.50
5. Cover each tube with Parafilm™ and mix well.
6. Following the procedure from Part A, set your spectrophotometer on 450 nm, zero the machine using 0.1 M Nitric acid solution and determine the absorbances of the solutions in the tubes 1 through 10.
7. Clean and return borrowed items.
8. Using your standard curve prepared in Part A, determine the equilibrium concentration of the complex ion in tubes 1 through 10.
9. Calculate the initial concentration of Fe 3+ and SCN in each tube. You must account for dilution.
10. Calculate the equilibrium concentrations of Fe 3+ and SCN .
11. For each tube, calculate the experimental equilibrium constant, K f
, for the formation of the complex ion. Calculate the average K f
and the percent relative average deviation (see Appendix
A).
REPORT
DETERMINATION OF K f
…
A. Standard Curve
Tube No.
1
2
3
4
7
8
5
6
9
10
11
12 b. K f
Determination
Tube No.
1
2
8
9
6
7
3
4
5
10
Tube No.
1
2
5
6
3
4
7
8
9
10
Initial [Fe 3+ ]
Absorbance
Absorbance
NAME ____________________________
SECTION __________________________
Initial [SCN ]
Eq [Fe(SCN) 2+
Eq [Fe(SCN)
Eq [Fe
] (from calculations)
3+ ]
2+ ] (from graph)
Eq [SCN ]
87
REPORT
DETERMINATION OF K f
…
Tube No.
1
2
3
4
5
6
7
8
NAME _____________________________
9
10
Average K f
Percent Relative Average Deviation
SAMPLE CALCULATIONS (use separate sheets if necessary)
K f
88
89
QUESTIONS FOR DETERMINATION OF K f
… NAME _____________________________
1. In your opinion, what most affects the precision for this experiment?
2. In your opinion, what most affects the accuracy for this experiment?
3. Would you expect all the K
Explain your answer. f
values determined in this experiment to be the same (or nearly so)?
90
QUESTIONS FOR DETERMINATION OF K f
…
4. The reaction:
NAME _____________________________
Co 2+ + 6 NH
3
Co(NH
3
)
6
2+ has an equilibrium constant of 5.0 x 10 4 . Solutions were mixed so that the initial concentration of the cobaltous ion was 0.100 M and the ammonia was 1.00 M . What are the equilibrium concentrations of all three species in the reaction?
91
K
sp
,
G
,
H
, AND
S
OF POTASSIUM NITRATE DISSOLVING IN WATER
INTRODUCTION
Solubility Equilibrium
When potassium nitrate (KNO
3 ions (NO
3
solid KNO
). Once sufficient quantities of K
3
) dissolves in water, it dissociates into potassium ion (K +
+ and NO
3
-
) and nitrate
are in solution, however, the ions recombine into
. Eventually, for every pair of ions that forms, another pair recombines. As a result, the concentrations of the ions remain constant; we say the reaction is at equilibrium. This solubility equilibrium of KNO
3
is shown in Equation 1,
KNO
3
K + + NO
3
undissolved solid is in equilibrium with its dissolved ions, a saturated solution.
(Eq. 1) where the opposing arrows indicate that the reaction is reversible. We call this system, where
We can describe the saturated solution with its fixed concentrations of ions with an equilibrium constant expression. Equation 2 defines the equilibrium constant, K sp
, for KNO
3
dissolved in water.
K sp
K NO
3
− (Eq. 2)
The sp stands for solubility product and the square brackets around the ions symbolize molar concentration ( M or mol/L). The equation serves as a reminder that the equilibrium constant not only is concerned with solubility but also is expressed as a product of the ions’ molarities. The value for K can be large, greater than 1, for the very soluble KNO temperature, its K sp
Thermodynamics
3
, or small, less than 10
is likewise a function of the temperature.
-10 sp
, for an insoluble compound such as silver chloride. In addition, because the solubility of a compound changes with the
We use thermodynamics to understand how and why KNO
3
dissolves in water. The enthalpy change,
H , for KNO
3
dissolving in water provides the difference in energy between solid KNO dissolved ions. If H is positive, heat must be added for KNO
3
3
and its
to dissolve. On the other hand, if H is negative, dissolving KNO
3
ion water gives off heat. The entropy change, S , for KNO
3
dissolving in water indicates the higher number of possible energy states being occupies by the dissolved ions with respect to the lower number of energy states occupied by the solid KNO
3
KNO
3
. We expect Δ S for solid
dissolving in water to be positive because the two ions on the product side of Equation 1 can occupy more possible energy states than the KNO
3 energy change, Δ G , for KNO
3
If Δ G is negative, solid KNO
3 crystal lattice can as a reactant. Finally, the free
dissolving in water indicates whether this process occurs spontaneously.
spontaneously dissolves in water.
We relate the equilibrium constant to the standard free energy change by Equation 3,
∆ = − ln sp
(Eq. 3)
92 where R is the ideal gas constant, 8.314 J K -1 mol -1 , T is the temperature in Kelvin, and ln K sp
is the natural logarithm of the equilibrium constant. Like K sp
, the free energy change for a reaction also changes with temperature.
We also relate the standard free energy change to standard enthalpy and standard entropy changes by the Gibbs–Helmholtz equation, Equation 4.
(Eq. 4) ∆ G = ∆ H − ∆
Substituting Equation 3 into Equation 4 yields Equation 5.
− ln sp
H T S
(Eq. 5)
Using algebra, we rearrange the equation into the form for a straight line, y = mx + b
ln
K sp
= −
∆ H
R
1
+
∆
R
S
(Eq. 6) so that a plot of ln K sp
on the y-axis, versus 1/T on the x-axis, is linear with a slope, m, of
– Δ H°/R and a y-intercept, b, of Δ S°/R. One assumption in this derivation is that Δ H° and Δ S° are constant, independent of the temperature.
PROCEDURE
1. Prepare a hot water bath by placing a 400-mL beaker half-filled with tap water on a hot plate.
2. On a balance, weigh about 20 g of KNO
3
on a tared piece of weighing paper. Record the exact mass
(to ±0.0001 g) of KNO
3
on your report sheet. Transfer the KNO
3
to a clean 25×200-mm test tube.
3. Using a graduated cylinder, add 15 mL of distilled or deionized water to the test tube containing the KNO
3
. Clamp the test tube in the beaker. Heat the test tube in the assembled hotwater bath. Stir the mixture with a thermometer until all of the KNO
3
dissolves.
4. Determine the volume of the KNO
3
solution by filling another 25 × 200-mm test tube with tap water until the volumes in both test tubes are the same. Measure the volume in the test tube filled with tap water by pouring this water into a graduated cylinder. Record this volume on your report sheet.
5. Remove the test tube with the KNO
3
solution from the hot-water bath and allow it to cool while slowly and carefully stirring the solution with your thermometer.
6. Record the temperature when crystals first appear. This is the temperature at which the solution is just saturated with potassium nitrate (the very small amount of solid is assumed to be in equilibrium with the ions in solution).
7. Add 5 mL of distilled water to the test tube containing the KNO
3
solution. Warm and stir the mixture in the hot-water bath until the solid has completely redissolved. Using the same method
93 as in Step 4, determine and record on your report sheet the new solution volume.
8. Remove the test tube containing the KNO
3
solution from the hot-water bath. Allow it to cool slowly. Record on your report sheet the temperature at which crystals first appear.
9. Repeat Steps 7 and 8 for a total of 6 determinations. Record all volume and temperature measurements on your report sheet.
10. Pour the contents of your test tube containing KNO
3
into the container labeled “Discarded KNO
3
Solution”.
11. Use the mass of the KNO
3
to calculate the number of moles of KNO
3
present.
12. Use the number of moles of KNO
3
and the volumes you determined at each temperature to calculate the molar concentration of KNO
3
in the solution at each temperature. Because, with only a very small amount of solid present, nearly all the KNO
3
is still in solution, its molar concentration equals the molar concentrations of K + temperature.
and of NO
13. Use Equation 2 to calculate the equilibrium constant, K
3
in the saturated solution sp
, for dissolving KNO
3
in water at each
14. Convert the temperatures in degrees Celsius (°C) to Kelvin (K).
15. Determine the natural logarithm of K sp
(ln K sp
) at each temperature.
16. Use Equation 3 to calculate Δ G° at each temperature.
17. Calculate the reciprocal of each Kelvin temperature, 1/T (K -1 ).
18. Using the graph paper provided at the end of this lab manual or a computer spreadsheet or graphing program, construct a graph with the y-axis as ln K sp
and the x-axis as 1/T (K
19. Determine the slope of the resulting straight line on this graph by choosing two widely separated points on the line that are not data points.
-1 ).
20. Calculate Δ H° for the reaction. Remember that the slope of the straight line in the ln K plot equals – Δ H°/R, according to Equation 6. sp
versus 1/T
21. Calculate Δ S° at each temperature using Equation 4. Determine the average Δ S.
22. Calculate S° from the y-intercept from your graph ( average S° from step 21. b = ∆ S
R
) and compare this value to the
94
95
REPORT
K sp
, G , H , and S of KNO
3
Mass of KNO
3
used, (g)
Moles of KNO
3
used, (mol)
NAME ___________________________
SECTION _________________________
Volume of
Solution, (mL)
Temperature at which crystals appear, (°C)
Solution 1
Solution 2
Solution 3
Solution 4
Solution 5
Solution 6
Solution 1
Solution 2
Solution 3
Molar solubility of
KNO
3
K sp ln K
Solution 4
Solution 5
Solution 6
Show Calculations (use separate sheets if necessary) sp
Temperature at which crystals appear, (K)
G°
(J mol -1 )
H
(J mol
-1 )
1
T
, (K -1 )
S°
(J mol -1 K -1 )
96
QUESTIONS FOR K sp
, G , H , and S of KNO
1. (a) Is the process of KNO
3 explain.
3
NAME____________________________
dissolving in water spontaneous at all temperatures studied? Briefly
(b) Is the reaction in (a) one that gives off heat or requires heat? Briefly explain.
(c) Is your value of Δ S° consistent with the expected change in disorder for the reaction in Equation
1? Briefly explain.
2. A few compounds exist whose solubility decreases as the temperature increases. How would the values for Δ G°, Δ H°, and Δ S° for these reactions be different from those values observed for the solubility of KNO
3
? Briefly explain.
97
QUESTIONS FOR K sp
, G , H , and S of KNO
3
NAME____________________________
3. (a) Why must the temperature be measured when only a small amount of solid has been formed?
(b) What could not be calculated if the temperature was measured after a large quantity of crystals precipitated?
(c) If you calculated G using temperatures when a large amount of solid had been formed, disregarding the error of doing so, how would the result impact G ’s value? Would it be higher or lower? Explain why.
98
99
DETERMINATION OF PERCENT OXALATE BY OXIDATION-REDUCTION
TITRATION
INTRODUCTION
The fundamental event in an oxidation-reduction reaction is electron transfer. Balancing an oxidation-reduction equation requires that the quantity of electrons lost by the reducing agent be equivalent to those gained by the oxidizing agent. Determination of the equivalence point in an oxidation-reduction reaction can be accomplished by using titration techniques.
In this experiment each student will work alone and: a. prepare an approximately 0.02 M potassium permanganate solution. b. standardize the permanganate solution against pure, solid sodium oxalate. c. determine the percent by mass of sodium oxalate in an impure sample.
Heat is required to catalyze (speed up) the permanganate-oxalate ion reaction. Manganese(II) sulfate will also be present to catalyze the reaction. A catalyst speeds up a reaction but is not consumed in the reaction.
Notice that when permanganate reacts with oxalate in the presence of an acid, two of the products are manganese(II) ion and carbon dioxide. If acid is not present in sufficient quantity, the permanganate ion will instead react according to:
4 MnO
4
-
2
+ 3 O
2
+ 4 OH + 2 H
2
O → 4 MnO -
Evidence that this reaction has occurred and contaminated the titration is the appearance of a muddy brown color due to manganese(IV) oxide. This reaction must be avoided ! Should a brown color persist in a flask during a titration, then that trial must be discarded. A momentary brownish discoloration, which completely disappears, is nothing to worry about. (however, you should add the permanganate solution more slowly during subsequent titrations to avoid the chance of permanent contamination by manganese(IV) oxide.)
100
PROCEDURE
A. Preparation of the Potassium Permanganate solution
1. Thoroughly clean one of your large screw cap bottles. Also clean its cap. (If the bottle or cap has been stained brown see your professor for cleaning instructions.)
2. Calculate the mass of solid potassium permanganate required in the preparation of approximately
300 mL of a 0.02 M solution.
3. Potassium permanganate is very corrosive to metal and can destroy analytical balances. Use the beam balances in the lab room to weigh out the approximate mass of potassium permanganate needed and place it in the clean bottle. Add ten drops of 0.001-M sulfuric acid solution and add approximately 100 mL of deionized water.
4. Cap the bottle and mix well. After all solid potassium permanganate has dissolved; fill the bottle to the 300 mL mark with deionized water. Mix well.
5. Put your name and/or locker number on the bottle.
Note: The permanganate solution must "rest" for several days before it can be standardized. It must be shaken before each use to insure uniformity. Also, because a potassium permanganate solution will degrade if exposed to light for extended periods of time, you should store the solution in your dark lab drawer as much as possible.
B. Standardization of the Potassium Permanganate solution
1. Obtain and clean the buret assigned to your locker number (see signs posted in the lab).
2. Rinse the buret three times with 2 to 3 mL of your potassium permanganate solution. If you wish to use a beaker or funnel to help fill your buret you must also clean them and then rinse them with your potassium permanganate solution prior to their use.
3. Fill the buret with the permanganate solution, drain the solution through the buret tip to eliminate air bubbles, and note the initial buret reading. Due to the intense color of the permanganate solution it will be easier to read the volume at the top of the meniscus.
4. Obtain a shell vial with pure sodium oxalate standard (FM=134.00 g mol keep it capped when it is not in use.
-1 ). Label this vial and
5. Clean a 250 mL Erlenmeyer flask (it does not have to be dry).
6. Calculate the mass of pure sodium oxalate that will require about 25 mL of approximately
0.02 M potassium permanganate solution for complete reaction with the sodium oxalate, you must have the balanced chemical equation for this step.
7. Take the vial of pure sodium oxalate, your clean flask, and the data sheet to the analytical balance room and measure sodium oxalate into the flask. Use the approximate mass (+/- 0.05 g) calculated in step 6 as a guide. Place the vial of sodium oxalate on the analytical balance pan and
101 zero the balance while the vial remains on the pan. Carefully avoiding spills, transfer some of the sodium oxalate into the flask and then place the vial back on the balance pan (without re-zeroing).
The negative mass displayed is the mass of sodium oxalate dispensed into the flask. Record the exact mass of sodium oxalate dispensed into the flask to the nearest 0.0001 g.
8. Add about 25 mL of distilled water to the flask.
9. Add about 25 mL of the 3 M sulfuric acid solution (that contains some manganese(II) sulfate catalyst) to the flask. The oxidation-reduction reaction is catalyzed by manganese(II) sulfate but the catalyst in no way affects experimental results.
10. Set up a water bath using about 200 mL of tap water in your largest beaker on a hot plate. Heat the water to a gentle boil and then place the Erlenmeyer flask containing the acidified oxalate solution in this bath for 1-2 minutes (until the flask feels quite warm).
11. Titrate the warm, acidic oxalate solution in the flask with the permanganate solution, swirling
12. Record the final buret reading and calculate the total volume of permanganate used for the titration. The contents of the flask can now be discarded. Save your oxalate for additional trials. constantly. When the flask starts to cool, return it to the hot water bath for a minute or two and then continue the titration. The end point is the persistence of the (diluted) permanganate color
(a very light purple). If you are not extremely careful as you approach the end point you will add too much permanganate solution, the end point color will be too dark, and you will have to discard that trial.
Note: If your titration volume was at least 10.00 mL (4 sig. figs.) this titration can be included in your calculations. However, a larger titration volume (closer to 25 mL) will give better precision.
On the other hand, an unnecessarily large titration volume (more than 25 mL) is time consuming.
The volume of potassium permanganate solution required is directly proportional to the mass of oxalate titrated. If the volume for your first your titration was not between 20 and 25 mL, adjust the mass used for the rest of your trials.
13. Clean three Erlenmeyer flasks (they can be wet) and label them #1, #2, and #3.
14.
Take your oxalate, the flasks, and your report sheet to the balance room and weight out a sample of oxalate into each flask using the method in Step 7. Record the mass of oxalate transferred to each flask.
Note : You should refill the buret for each titration. The potassium permanganate solution remaining in your buret at the end of each lab session should be saved in a clean dry beaker and used for rinsing the buret at the next lab session. Never put any unused solution back into your stock bottle . You risk contaminating your solution.
15.
Titrate each flask following the procedures in Steps 8-12.
16.
From the volume of the potassium permanganate solution used in each titration and the mass of sodium oxalate in each flask, calculate the molarity of the potassium permanganate solution ( at least three values are necessary for a good average).
102
17.
Using at least three molarity values calculate your percent relative average deviation (see
Appendix A at the end of this lab manual). (Note: Percent relative average deviation is a measure of precision and at least 3 trials are required for the calculation to be meaningful.). If your average deviation is less than 2%, it means that the data you have collected show good precision and you have performed enough trials. If it is greater than 2%, then additional trials are needed.
18.
Determine and record the average molarity for your potassium permanganate solution. This solution will be used to analyze your unknown. Take good care of it!
C. Determination of percent sodium oxalate in an impure sample
1. Obtain a shell vial containing an impure sodium oxalate unknown. Record your unknown number and label your unknown vial. Keep this unknown in your locker until you have received your graded lab report.
Note: The shell vial of impure sodium oxalate (unknown) contains enough sample for at least six trials. No additional unknown will be provided! Should you spill your unknown, a different unknown will be obtained and you will start the unknown’s analysis from the beginning.
2. Check out and clean the buret assigned you your locker number. Rinse and fill the buret with your permanganate solution, as before.
3. Do one titration with the unknown using two to three times as much mass as was used for pure sodium oxalate (record the exact mass of unknown used). Use the same titration procedure as was used in steps 7-12 for Part B.
4. You will need to do at least two more trials. If the total volume of potassium permanganate solution used in your first titration was less than 20 mL use a little more unknown for your subsequent titrations. If your titration volume was greater than 25 mL use a little less unknown.
(The mass of impure sodium oxalate and the volume of potassium permanganate solution used in the titration are directly proportional .)
5. Using the average molarity of your potassium permanganate from part B in your calculations, determine the percent by mass of sodium oxalate in your impure sample for each trial.
6. Using the percent by mass calculated for each trial, determine the percent relative average deviation. If it is greater than 2%, do additional trials.
7. Report the average percent by mass of sodium oxalate for your unknown.
103
REPORT
RE-DOX TITRATIONS EXP.
A. Preparation of KMnO
4
solution
NAME _______________________________
SECTION ____________________________
Show calculations used to estimate the mass of potassium permanganate needed to prepare the solution.
B. Standardization
Show the calculation used to estimate the mass of sodium oxalate needed to do the first titration, include the balanced chemical equation:
Mass of pure Na dispensed, (g)
2
C
2
O
4
Trial 1 Trial 2 Trial 3 Trial 4
Final buret reading, (mL)
Initial buret reading,
(mL)
Volume of solution dispensed, (mL)
Molarity of solution, ( M )
Average molarity, ( M )
Percent Relative
Average Deviation, (%)
SAMPLE CALCULATIONS (use additional sheets if necessary)
REPORT FOR RE-DOX EXP. (cont.) NAME _______________________
C. Determination of percent sodium oxalate in an impure sample.
UNKNOWN #
Mass of unknown dispensed, (g)
Final buret reading, (mL)
Initial buret reading, (mL)
Volume of solution dispensed, (mL)
Percent Na
2
C
2
O
4 by Mass, (%)
Trial 1 Trial 2
Average Percent,
(%)
Percent Relative
Average
Deviation, (%)
SAMPLE CALCULATIONS (use additional sheets if needed)
Trial 3 Trial 4
104
105
QUESTIONS FOR RE-DOX EXP. NAME _________________________
1. Dichromate and ferrous ions react in acidic solution to form chromic and ferric ions, respectively.
If 1.285 grams of iron(II) bisulfate dissolved in sulfuric acid solution requires 35.78 mL of sodium dichromate solution for complete titration, what is the molarity of the sodium dichromate solution? Show the balanced net ionic and complete chemical equations for this reaction.
2. The sodium dichromate solution from problem 1 was used to titrate a solution made by dissolving
3.500 g of a pure ferrous salt in sulfuric acid. The titration required 47.22 mL of the sodium dichromate solution. Calculate the percent by mass of iron in the pure salt. The net ionic equation here is the same as in problem 1. You will not be able to write a balanced molecular equation for this because the anion in the ferrous salt was not specified in this problem.
106
107
ELECTROCHEMISTRY
INTRODUCTION
Electrochemistry involves the transfer of electrons from a reducing agent to an oxidizing agent. For the electrons involved in the transfer to be used in a productive fashion (e.g. electroplating flatware, starting a car, etc.), an electrochemical cell is usually set up. An electrochemical cell is a device that converts the energy of a chemical reaction into electrical energy. In such a cell, the reaction proceeds by the transfer of electrons, producing an electric current. A reaction involving the transfer of electrons is called an oxidation-reduction reaction. If the oxidized and reduced species are separated from each other in different containers but are allowed to maintain contact through a salt bridge or porous cup, the electron transfer can be made to occur through a wire which is in contact with the oxidized and reduced species. The flow of electrons through the wire, called the current, can be used to produce electrical work. The common dry cell, for example, is an electrochemical cell. When the terminals of the dry cell are connected to a motor, electrons flow from the cell through the motor, producing work.
An electrochemical cell can only function when there is a complete electric circuit. In a cell in which there are two half-cells, a salt bridge must be used to maintain electrical neutrality.
An electrolytic cell uses current from an outside source (a battery or other power supply) to cause a reaction to run in the direction that is “non-spontaneous.” In this laboratory exercise, the electrolysis of aqueous potassium iodide will be studied.
Students will record observations and information, and then perform calculations pertaining to the electrolytic cell. (Examples of common electrolytic processes are recharging “dead” batteries and anodizing metals such as aluminum.)
If a current spontaneously flows when an electrochemical cell's circuit is complete, then the cell is referred to as a voltaic or galvanic cell. (Examples of these cells are cell phone and laptop batteries.)
This experiment will include the study of voltaic cells formed from half-cells involving pairs of the following half-reactions:
Cu 2+ + 2 e Cu
Fe 2+
Fe 3+
+ 2 e -
+ e -
Fe
Fe 2+
Zn 2+ + 2 e Zn
If a solid metal is a component in a half-reaction then that metal will be used as the electrode for that half-cell. A graphite rod will serve as the electrode in a half-cell which involves no solid metal.
The voltaic cells in this experiment will not be “standard” cells. In standard cells all molarities are 1-
M, all partial pressures of reactant and product gases are 1 atmosphere. In addition, the “ideal” standard cell would be constructed with perfect electrical connections and zero resistance electrical leads and utilizes circuits that draw no current. The imperfect voltages obtained from the nonstandard cells in this exercise will be compared to standard potentials for that type of cell.
The next exercise in this experiment will be to construct a concentration cell. This cell will measure the potential generated by a difference in copper(II) concentrations in copper/copper(II) half cells.
Lastly, you will construct an electrochemical cell to determine the solubility product constant of
108 copper(II) carbonate. This can be accomplished by measuring the potential of a cell which has a saturated solution of copper(II) carbonate in one of the half cells. This potential, compared to the cell potential of a standard cell allows us to determine the copper(II) ion concentration and the K sp
.
PROCEDURE
Due to equipment limitations, students will work in groups during this experiment.
Each group will need a voltmeter, a porous cup, a 100-mL graduated cylinder, and 2 copper electrodes.
Voltaic Cells Day 1 – A.
1. Often, the metal electrodes are stored in oil, which must be removed before use. If so, pour a small amount of acetone on to a paper towel and wipe the metal electrodes well. Rinse the electrodes with tap water and then distilled water.
DO NOT COMPLETE A CELL'S CIRCUIT UNTIL YOU ARE READY TO MEASURE ITS VOLTAGE.
2. Obtain a volt meter and insert the red plug into the “V ” connector on the meter and the black plug into the black “COM” connector. Press the button in the center of the dial and turn the voltmeter’s dial to “V.” You should hear a beep from the meter. Set the meter display to read three places after the decimal. The meter will run through an internal self- check and will be ready for use when the display reads approximately 0.000 VDC. (The meter should read zero if you clip the leads together.)
3. Take the volt meter to the various cell set-ups and measure the voltage of each cell.
4. For example, go to the Cu|Cu 2+ Fe|Fe 2+ cell. Attach one of the lead's alligator clips to the top of the copper electrode. Attach the other lead's alligator clip to the iron electrode. If the meter indicates a negative voltage then it has been hooked up backwards. (This meter is designed to yield positive voltages when its black lead is connected to the anode.) Swap the positions of the alligator clips. Record the voltage for this cell. As soon as the voltage is read, remove one of the alligator clips to break the circuit and stop the current flow.
5. Determine and record the half reactions for the cell. Write the cathode reaction as a reduction and the anode reaction as an oxidation. Write the overall chemical reaction.
6. Using the half-reaction potentials in your text, calculate the standard voltage potential for this cell.
7. Write the shorthand cell notation for each cell that you tested. Remember that these cells were not standard cells.
8. Repeat steps 4 through 7, for each of the half-cell combinations listed on the report sheet.
B. Concentration Cell
1. Measure 1.0 mL of 0.10 M copper(II) nitrate in a clean 10 mL graduated cylinder. Transfer this
109 solution to a clean 100 mL graduated cylinder and add distilled water to bring the volume to the
100 mL mark. (Use some of the water to rinse out the smaller cylinder. Add the risings to the larger cylinder, and then finish the dilution by adding water directly to the larger cylinder.) Pour this solution into a clean 250-mL beaker. Stir to mix well.
2. Use some of the diluted solution to rinse the 10 mL graduated cylinder before measuring 1.0 mL of the diluted copper(II) nitrate solution. Transfer the 1.0 mL of diluted solution to the well rinsed
100 mL graduated cylinder and make a second dilution by adding distilled water to bring the total volume to 100 mL as before. Mix well.
3. Transfer ~50 mL of solution from the second dilution into a clean 150 mL beaker.
4. Place a clean copper electrode in the diluted solution to form a half-cell.
5. Place ~30 mL of 0.10 M copper(II) nitrate solution into a porous cup and carefully place the porous cup into the 150 mL beaker from step 3. Place a copper electrode into the porous cup.
6. Record the temperature of the solution in the beaker.
7. Set the voltmeter to 300 mV. Connect the clips to the electrodes and measure the concentration cell's voltage. The display will be in mV.
8. Add 10 drops of 0.10 M copper(II) nitrate to the more dilute solution in the cell apparatus. Stir the mixture (you can use the electrode to stir the solution).
9. Measure the concentration cell's new voltage in mV.
10. Rinse the electrodes with distilled water. Dry the electrodes with paper towels.
11. Calculate the concentration of the copper(II) nitrate in the diluted solution taking both dilutions into account (you have to calculate each dilution separately). Use the data to do the calculations and answer the questions in the lab report.
12. Return all borrowed equipment.
110
For Day 2, each group will need a voltmeter, a porous cup, a timer, a set of electrodes with transformer, an electrode holder, and the following electrodes: 1 copper, 1 zinc.
Day 2 – C. Electrolysis of Aqueous Potassium Iodide
1. Clean the 100-mL beaker and place 50.0 mL of distilled water into it.
2. Weigh out 1.00 g of potassium iodide and put it into the water in the beaker. Stir until the potassium iodide is completely dissolved.
3. Take the aqueous KI solution to the pH meter that has been set up for use by the class. Measure and record the initial pH of the solution.
Be careful of the platinum electrodes because they can be easily damaged! Do not twist or bend them.
4. Insert the ends of the platinum electrodes into the glass tubes of the electrode holder and place the entire assembly in the beaker containing the aqueous KI solution (see diagram). (If necessary, the cork electrode holder can rest on top of the beaker.) The wires should exit through the spout of the beaker and the electrodes should rest on the bottom of the beaker. Adjust the glass tubes of the electrode holder up or down so that the glass holds just the very tip of each electrode. The purpose of the holder is to make sure the electrodes do not touch each other during the electrolysis. Do not plug in the transformer until you have the electrodes in the proper orientation!
5. Start the timer as you plug in the transformer. Observe and record what is happening at each electrode, initially and several times during the electrolysis.
6. Allow the electrolysis to proceed for 20-25 minutes. (You can start Part D of the experiment during this time.) Do not move the electrodes until you have unplugged the transformer!
Stop the timer as you unplug the transformer. Record the exact amount of time elapsed.
7. Remove the electrode assembly. Measure and record the pH of the solution after electrolysis.
8. Dip the electrodes into the sodium thiosulfate cleaning solution provided in a container in the hood. Gently swirl the electrodes in the solution for about 30 seconds to remove any iodine adhering to the electrodes.
9. Carefully rinse the electrodes 2-3 times with distilled water and gently blot them with a paper towel to dry them. Rinse and dry the glass tubes in the electrode holder.
10. The data that was collected in this experiment will used in calculations in the Report and Questions section of the lab.
111
D. Determination of the solubility product constant of copper(II) carbonate.
1. Obtain a volt meter and insert the red plug into the “V ” connector on the meter and the black plug into the black “COM” connector. Push the button in the center of the dial and turn the voltmeter’s dial to “V.” The meter will beep. Set the meter display to read three places after the decimal. The meter will run through an internal self-check and will be ready for use when the display reads approximately 0.000 VDC. (The meter should read zero if you clip the leads together.)
2. Place ~50 mL of 1.0 M sodium carbonate and a clean copper strip into a 150-mL beaker. Add 5 drops of 1.0 M copper(II) nitrate solution to form a precipitate (stir the solution). Record the temperature of the solution in the beaker.
3. Place ~30 mL of 1.0 M zinc nitrate solution into the porous cup. Place a clean zinc electrode into the porous cup.
4. Carefully place the porous cup into the 150 mL beaker from step 2.
5. Connect the volt meter to the metal strips and record the voltage. Switch the connections if you get a negative voltage.
6. Return all borrowed equipment.
REPORT
ELECTROCHEMISTRY
A. Voltaic Cells
Cu/Cu 2+ ; Fe/Fe 2+
Cathode half-reaction
Anode half-reaction
Overall chemical reaction
Experimental Cell Notation
Cathode half-reaction
Anode half-reaction
Cu/Cu 2+ ; Fe 2+ /Fe 3+
Overall chemical reaction
Experimental Cell Notation
Cu/Cu 2+ ; Zn/Zn 2+
Cathode half-reaction
Anode half-reaction
Overall chemical reaction
Experimental Cell Notation
NAME___________________________
SECTION _________________________
Experimental
Cell Voltage
E red
− E red
E cell
Experimental
Cell Voltage
E red
− E red
E cell
Experimental
Cell Voltage
E red
− E red
E cell
112
REPORT
ELECTROCHEMISTRY (cont.)
Cathode half-reaction
Anode half-reaction
Fe/Fe 2+ ; Fe 2+ /Fe 3+
Overall chemical reaction
Experimental Cell Notation
Fe/Fe 2+ ; Zn/Zn 2+
Cathode half-reaction
Anode half-reaction
Overall chemical reaction
Experimental Cell Notation
Cathode half-reaction
Anode half-reaction
Fe 3+ /Fe 2+ ; Zn/Zn 2+
Overall chemical reaction
Experimental Cell Notation
NAME ___________________________
Experimental
Cell Voltage
E red
− E red
E cell
Experimental
Cell Voltage
E red
− E red
E cell
Experimental
Cell Voltage
E red
− E red
E cell
113
REPORT
ELECTROCHEMISTRY (cont.)
B. Concentration Cell
Temperature of copper(II) nitrate solution
Molarity of the diluted copper(II) nitrate solution
NAME ___________________________
114
Initial Voltage
Voltage after addition of 10 drops of 0.10 M copper(II) nitrate
C. Electrolysis of Aqueous Potassium Iodide
Volume of solution
Initial pH
Initial [OH ]
Elapsed time of electrolysis
Final pH
Final [OH ]
Change in [OH ]
Observations:
Before electrolysis
During electrolysis
After electrolysis
REPORT ELECTROCHEMISTRY EXP. (cont.) NAME _____________________________
D. Determination of the solubility product constant of copper(II) carbonate.
Standard Cell Potential of a copper/zinc cell, V
Cell Potential of experimental cell, V
Number of moles of electrons transferred in the chemical equation, n
Concentration of copper(II) ions in the saturated copper(II) carbonate half-cell, M
Temperature of solution in beaker, C
Experimental K sp
SAMPLE CALCULATIONS (Use additional paper as needed)
115
116
QUESTIONS FOR ELECTROCHEMISTRY EXP. NAME __________________________
1. Use the standard reduction potential tables to answer the following questions, (show the voltages in justifying your answers):
(a) What will happen to an iron nail in cupric nitrate solution?
(b) What will happen when a piece of copper metal is added to a solution of zinc nitrate?
2. Use the Nernst equation to calculate the expected voltage of the concentration cell before the addition of the 10 extra drops of 0.10 M copper(II) nitrate solution. Use your experimental temperature.
How did the addition of 10 drops of 0.10 M cupric nitrate affect the concentration cell's voltage?
Why?
117
QUESTIONS FOR ELECTROCHEMISTRY EXP. NAME___________________________
3. How do your experimental voltages obtained for the voltaic cells compare with the standard potentials for those cells? Give some reasons to explain why the experimental voltages are probably different than the standard voltages.
4. (a) Assuming that there was no overvoltage, what half reaction occurred at the anode of the electrolytic cell?
(b) At the cathode?
(c) Write a balanced net ionic equation for the overall reaction that occurred in the cell.
(d) Calculate the standard potential for the reaction.
5. Calculate the average current that flowed through the electrolytic cell (using the change in hydroxide ion concentration, the volume of the solution, and the total elapsed time of the electrolysis).
118
QUESTIONS FOR ELECTROCHEMISTRY EXP. NAME___________________________
6. An experimental cell is set up such that one half cell contains a solid silver electrode dipping into a saturated solution of silver oxalate. The oxalate ion comes from added sodium oxalate so that equilibrium concentration of oxalate ions is 0.50 M . This half-cell is connected to a standard hydrogen electrode (SHE) half-cell. The measured potential for this cell is 0.476 V at 25°C. What is the experimental value of the solubility product constant for silver oxalate?
119
ELECTROLYTIC DETERMINATION OF THE MOLAR MASS OF LEAD
INTRODUCTION
If an electric current is allowed to pass through a solution containing ionic species, the ions experience a force that causes the positive ions to move in one direction while the negative ions move in the opposite direction. This movement of ions allows the current to pass through the solution. In order to maintain the current, oxidation-reduction chemical reactions must occur in the solution.
The amount of electric current that passes through the solution and the amount of chemical reaction that occurs are related by Faraday's Laws. The transfer of Avogadro's number of electrons corresponds to one faraday of charge. One faraday is equal to 96,485.3399±0.0024 coulombs of electrical charge.
The number of electrons transferred, and hence the number of faradays, can be found by multiplying the current by the time during which the current flowed.
In this experiment, two lead strips are placed in a lead(ll) nitrate solution and a wire is attached to each of these strips. The wires are then connected to the Constant Current System. The Constant
Current System is also connected to the Vernier™ LabQuest to monitor current flowing in the cell.
Electrons flow between the current source and the lead strips. One of the strips receives electrons while the other strip loses electrons. In order to have electron flow, electrons must be used up at the strip that gains electrons and must be released at the strip that loses electrons.
The positively charged ions will move to the electron-rich strip and accept electrons; thus, a reduction occurs at this strip which can be represented by the half-reaction:
Pb 2+ + 2 e Pb
The strip where reduction occurs is called the cathode. Negatively charged ions migrate to the other strip and electrons are released; thus, an oxidation must occur which can be represented by the half-reaction:
Pb Pb 2+ + 2 e -
The strip where oxidation takes place is called the anode.
The mass of lead that dissolves at the anode must be the same as the mass of lead deposited at the cathode.
120
PROCEDURE
1. Obtain two lead strips from the laboratory instructor. Sandpaper each strip to remove surface oxides and dirt. Use a file to mark each strip. One scratch mark will designate the strip to be used as the anode and two scratch marks will identify the strip to be used as the cathode. Weigh each strip to the nearest ±0.0001 g. Record the mass of the anode and cathode strips on the Data
Sheet.
2.
Assemble the apparatus by adding about 100 mL of 0.50 M lead(ll) nitrate solution to a 250-mL beaker containing the two lead strips from step 1.
3.
Gently turn the dial of the Constant Current System™ counterclockwise to confirm that it is in the minimum current position.
4.
Place the lead strips into the solution in the beaker. Be sure to keep them as far apart as possible.
You may find it easier if you bend the strips over the edge as shown in the figure on the previous page.
5.
Connect the lead strip you marked as the anode (two scratches) to the positive (red) clip of the
Constant Current System. Connect the other Lead strip (the cathode, one scratch mark) to the negative (black) clip.
6.
Plug the Constant Current System™ into a powered electrical outlet. Connect the sensor cable to the LabQuest™ and choose New from the File menu.
7.
Start data collection by pressing the start button or the green arrow on the screen and now adjust the current to the 0.1–0.2 A range. Data collection will run for 30 minutes.
8.
When data collection is complete, disconnect the DC power source and carefully remove the lead strips from the solution. Gently rinse with distilled water. Allow the strips to dry in air at room temperature. Weigh each strip to the nearest ±0.0001 gram, adding the mass of any solid that may have fallen off to the cathode and record on the Data Sheet.
9. Perform a second determination following the same procedure as before. Use the same equipment as before. Swap the cathode and the anode from the first trial. The initial masses of the cathode and anode lead strips will be the final masses of the anode and cathode from step 8.
REPORT
ELECTROLYTIC DETERMINATION …
NAME ____________________________
SECTION __________________________
Before Electrolysis
Mass of Anode, g
Mass of Cathode, g
Initial Current, A
Current @ 5 min, A
Current @ 10 min, A
Current @ 15 min, A
Current @ 20 min, A
Current @ 25 min, A
Current @ 30 min, A
Trial 1
After Electrolysis
Mass of Anode, g
Mass of Cathode, g
Mass of lead transferred, g
Time Elapsed, s
Average Current, A
Moles of electrons transferred, mol
Moles of lead transferred, mol
Molar mass of lead, g mol -1
Trial 2
After Electrolysis
Mass of Anode, g
Before Electrolysis
Mass of Anode, g
Mass of Cathode, g
Initial Current, A
Current @ 5 min, A
Mass of Cathode, g
Mass of lead transferred, g
Time Elapsed, s
Current @ 10 min, A Average Current, A
Current @ 15 min, A
Current @ 20 min, A
Current @ 25 min, A
Current @ 30 min, A
Moles of electrons transferred, mol
Moles of lead transferred, mol
Molar mass of lead, g mol -1
Average molar mass of trials 1 & 2,
SHOW CALCULATIONS (attach separate sheets if necessary)
121
122
QUESTIONS FOR ELECTROLYTIC DETERMINATION … NAME______________________________
1. A electrolytic cell is set up as was done in this experiment but with a different metal. An average current of 135.4 mA is delivered for 15 minutes and 23 seconds. The cathode gains 0.0728 g in mass. If there are two moles of electrons transferred per mole of the metal, what is the molar mass of the metal?
2. What mass of sodium metal can be obtained from the electrolysis of molten sodium chloride if a current of 10.0 amps is allowed to pass though the cell for 45 minutes?
3. How many hours are required to obtain 150.0 g of chromium metal from a solution of chromium(III) nitrate if a current of 2.50 amps is passing through the cell?
123
DETERMINATION OF THE HALF-LIFE OF POTASSIUM-40
INTRODUCTION
All radioactive isotopic decay follows first order kinetics. We explored kinetics in the first week of this class and found that first order kinetics obeys the following equation:
−
∆ N
=
∆ t kN
0 where k is the rate constant with units of reciprocal time and N
0
is the initial number of nuclei. We can get a measurement of the rate (the left hand side of the equation above) by using a Geiger-Müller detector. The detector we are using is the Digital Radiation Monitor made by Vernier. Once we have a rate measurement, we can calculate the number of radioactive nuclei in the sample from the mass.
The rate and the number of nuclei then give us the rate constant. The half-life, t the following equation:
1/2
, can be found from t
1
2
= ln2 k
In this experiment we will make adjustments to the count to take into account the beta counting efficiency of the detector (not all beta particles emitted get measured by the detector) and to adjust to a (hypothetically) infinitely thin disk of KCl (the KCl will start to absorb some of the beta particles as the sample gets thicker).
PROCEDURE
1. Obtain a Digital Radiation Monitor (DRM). Turn it on using the bottom switch on the front of the device. In order to avoid annoying your instructor and classmates do not put the switch in the
“Audio” position.
2. The first task is to obtain a background count. The background count is more accurate the longer it is measured. We are going to do a 30 minute count for the background. To set the timer on the
DRM for 30 minutes we need to put the top switch into the “Total/Timer” position. The display should show a time measure and the word “SET” in the upper right. Use the “+” and “-” buttons on the top left of the DRM to set it to 30 minutes (display will show “0:30”).
3. Press the “Set” button (between the “+” and “-” buttons). The DRM will start totaling the counts it measures. At the end of the counting period the DRM will beep three times. Record the number in the display as the Background Count on your Report Sheet. Divide this number by 30 and record it as the “Background Counts Per Minute (CPM)” on your Report Sheet. After the background count has been completed, reset the timer on the DRM by placing the switch in the CPM/CPS position and then moving it back to the “Timer/Total” position. Use the “+” and “-” buttons to adjust the timer to 10 minutes (0:10 in the display) which is the time used for the remainder of the experiment.
4. Measure approximately 0.7 gram (±0.0001 g) of potassium chloride on a piece of tared weighing paper. Record the mass in your Report Sheet. Place the potassium chloride into a clean dry shell vial. Tightly cover the shell vial with a piece of Parafilm™. This is your sample holder. We will
124 place the shell vial upside-down in a buret clamp to hold it in place over the counter window of the DRM (located on the top right side of the device). We will also clamp the DRM in place with a buret clamp to make sure that it doesn’t move out of position (See picture below).
5. Press the “Set” button to start the timer and the count. When the
DRM beeps, record the total count in your Report Sheet. Divide the count by 10 to get the CPM and record this number as the
CPM in your data sheet.
6. Measure approximately 0.3 gram (±0.0001 g) of potassium chloride and add it to the potassium chloride already in the shell vial. Record the amount measured in your data sheet. Cover the shell vial tightly with another piece of Parafilm™. Reset the timer as before. Set everything up as before and press the “Set” button to start the count again for 10 minutes. Record the total and the
CPM as before.
7. Measure approximately 1.0 gram (±0.0001 g) of potassium chloride and add it to the potassium chloride already in the shell vial. Record the amount measured in your data sheet. Cover the shell vial tightly with another piece of Parafilm™. Reset the timer as before. Set everything up as before and press the “Set” button to start the count again for 10 minutes. Record the total and the CPM as before.
8. Measure approximately 1.0 gram (±0.0001 g) of potassium chloride and add it to the potassium chloride already in the shell vial. Record the amount measured in your data sheet. Cover the shell vial tightly with another piece of Parafilm™. Reset the timer as before. Set everything up as before and press the “Set” button to start the count again for 10 minutes. Record the total and the CPM as before.
9. Return the potassium chloride to the container. Turn off the DRM and return it to the same place from which you got it.
10. For each of the samples, calculate the Net CPM by subtracting the background CPM from the measured CPM. Then calculate CPM/g KCl and ln(CPM/g KCl) for each sample and record these values on your Report Sheet.
11. Construct a graph of ln(CPM/g KCl) vs g KCl and extrapolate back to zero grams KCl. The intercept corresponds to an infinitely thin disk of KCl. This process eliminates the effect of the selfabsorption of beta particles by the potassium chloride. Calculate the extrapolated CPM/g KCl from the intercept and record this on your Report Sheet.
12. Next we need to make an adjustment for the efficiency of the detector. Prior measurements of samples with known activities have determined the efficiency of the detector to be about 1.5%.
We can then calculate the Adjusted CPM/g KCl from
Adjusted CPM/g KCl =
Extrapolated CPM/g KCl efficiency
=
Extrapolated CPM/g KCl
0.015
125
Record the Adjusted CPM/g KCl on your report sheet.
13. The Adjusted CPM/g KCl is our experimental activity. We need to calculate the number of potassium-40 nuclei in a 1.000 g sample (because we adjusted everything to be per gram). To accomplish this you will need the isotopic abundance of potassium-40 which is 0.0118%. Record the number of K-40 nuclei/g KCl in your Report Sheet.
14. Calculate the rate constant using
Adjusted CPM/g KCl k = number of K-40 nuclei/g KCl
Record this value on your Report Sheet.
15. Calculate the half-life, t
1/2
, by taking ln2
2
= k
This value will have units of minutes. Convert your answer to years. Record this on your Report
Sheet. t
1
126
Net
CPM
Net CPM g KCl
127
REPORT
DETERMINATION OF THE HALF-LIFE OF K-40
30 min background count
Initial mass, g
Added mass, g
Total mass, g
Background
CPM
Counts in
10 min
NAME___________________________
SECTION_________________________
CPM
Extrapolated ln(CPM/g)
Extrapolated CPM/g
Adjusted CPM/g
Number of K-40 nuclei in 1.00 g
Rate constant (1/min)
Half-life (min)
Half-life (yr)
SAMPLE CALCULATIONS: (Use separate sheets if necessary) ln
Net CPM g KCl
128
QUESTIONS FOR HALF-LIFE OF K-40 EXP.
1. Calculate the activity of a 15.0 g sample of natural potassium. Express your answer both in Curies
(Ci) and in Becquerel (Bq).
NAME ______________________________
1 Bq = 1 nuclei s -1 1 Ci = 3.700×10 10 nuclei s -1
2. Technetium-99m is a metastable form of technetium-99 (isotopic mass = 98.9062547 amu) and has a half-life of 6.0058 hours. How many grams of Tc-99m are required to have an activity of
1.00 µ Ci?
3. Cesium-137 is a radioactive isotope. 10.0 g of pure Cs-137 (isotopic mass = 136.9070835) has an activity of 871.8 Ci. What is the half-life of Cs-137 in years?
129
COORDINATION COMPOUNDS AND COMPLEX IONS
Nomenclature System
The textbook provides an account of how to write names of complex ions and coordination compounds. However, the method in the textbook is not entirely correct according to the
International Union of Pure and Applied Chemistry (IUPAC). IUPAC is the governing body that sets the rules for naming compounds and approving the names of newly created elements. For inorganic compounds, compounds that are not comprised mainly of carbon and hydrogen, the document is
“NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005” also known as the Red
Book. For organic chemistry, the relevant document is “A Guide to IUPAC Nomenclature of Organic
Compounds Recommendations 1993,” also known as the Blue Book. The rules outlined here are from the Red Book.
The following general rules are used when naming coordination compounds:
(i)
(ii) ligand names are listed before the name of the central atom, no spaces are left between parts of the name that refer to the same coordination entity,
(iii) ligand names are listed in alphabetical order (multiplicative prefixes indicating quantities are not considered in determining that order),
(iv) the use of abbreviations in names is discouraged.
The general process for naming a complex ion or coordination compound is:
• identify the central atom
• identify the ligands
• name the ligands
• specify the mode of coordination (which atom provides the lone pair of electrons that forms the coordinate covalent bond to the central atom, if known)
• order the ligands – this will be alphabetical without regards to any multiplicative modifiers
The central atom (in this class) will be a transition metal. This can become more complicated in higher level chemistry classes.
The ligands can be small molecules or monatomic ions or polyatomic ions or more complicated organic molecules.
In order to indicate the number of a particular ligand we use the prefixes that are used in the nomenclature of covalently bonded binary compounds.
2
3
4
2
3
4 di- tri- tetra-
If the ligand is a complex ligand (for example a larger molecule, usually an organic compound) we use the following prefixes with parentheses around the ligand name: bis- tris- tetrakis-
For example, we would write diammine for (NH
3
)
2
but we would write bis(methylamine) for (CH
3
NH
2
)
2 to avoid confusion with dimethylamine ((CH
3
)
2
NH). Use of bis-, tris-, etc. does not require the ligand
130 name to contain a prefix as stated in the textbook.
Consecutive vowels are retained in the names (tetraammine or diaqua, for example).
The names of ligands which are anions are altered to end it “o.” –ide becomes –ido (not –o as in the textbook), –ate becomes –ato, and –ite becomes –ito.
The names of neutral and cationic ligands (which are rare) are used in an unaltered form:
(NH
H
2
SO
O
CO
2
2
)
2
CO
Exceptions to this are: urea aqua carbonyl sulfonyl
NH
2
CH
2
CH
2
NH
2
ethylenediamine
CH
3
CONH
2
N
2
H
4
NH
3
NO acetamide (not acetamido) hydrazine ammine nitrosyl
Enclosing marks (parentheses) are required for ligand names in order to avoid ambiguity. The ligands in the list above generally do not require enclosing marks unless there is an ambiguity.
Which atom connects to the central atom is denoted with a κ (Greek letter kappa) followed by the atom bonded to the central atom in italics. This is not required for single atom ligands or ligands where it is obvious which atoms is forming the bond (i.e., water or ammonia). If there is more than one atom in a ligand that bonds to the central atom in the ligand, then we use κ the bonds in alphabetical order (κ 2
2 followed by the atoms forming
N,O , use a ‘ if the atoms are the same kind, i.e., - κ 2 N,N’ ). The difference in bonding of the cyanide ion, the nitrite ion, the cyanate ion and thiocyanate ion is indicated in this way. They are NOT nitro and nitrito for NO
2
cyanide (CN ) it’s either cyanido (the rules specify that the – κ
, they are nitritoκ N or nitritoκ O . For is not required when CO or CN bonds via the carbon atom, via the oxygen atom in carboxylate (like acetate), or via the nitrogen atom in NO) or cyanidoκ N . For cyanate (OCN ) it’s either cyanatoκ N or cyanatoκ O . For thiocyanate (SCN ) it’s either thiocyanatoκ N or thiocyanatoκ S . The textbook is incorrect about this. There is no iso- prefix according to IUPAC. However, sometimes the κ may be omitted such as cyanatoO or nitritoN .
The name of the central atom is modified with the Latin form of the element’s name and an –ate ending when the complex ion is an anion. If the complex is neutral or a cation, the IUPAC name of the central atom is unaltered.
Element Name
Iron copper gold
Tin silver
Lead
Latin Form ferrum cuprum aurum stannum argentum plumbum
Anionic Complex Ion Form ferrate cuprate aurate stannate argentate plumbate
A couple of exceptions to this are mercury which will be mercurate instead of hydrargyrate (from the
Greek hydrargyrum –“ water + silver” ) and tungsten which will be tungstate instead of wolframate
(from the German wolfram –“ wolf’s rahm ” or “ wolf’s cream ”).
The last part of the name is either the oxidation state of the central atom, which is represented in
Roman numerals in parentheses, or the overall charge of the complex, which is represented with
131
Arabic numerals with the sign of the charge after the number in parentheses.
Examples:
[CoCl(NH
3
)
5
[Fe(CH
[AuBr
6
[Fe(NH
3
COO)
] 5-
] 2+
3
2
CH
2
CH
(H
2
2
O)
NH
2
3
)
3 pentaamminechloridocobalt(III) or pentaamminechloridocobalt(2+)
] triaquatris(acetato)iron(III) or triaquatris(acetato)iron(0) hexabromidoaurate(I) or hexabromidoaurate(5-)
] 3+ tris(ethylenediamineκ 2 N , N’ )iron(III) or tris(ethylenediamineatoms in the ligand.
κ 2 N , N’ )iron(3+)
This is indicating that the bonds are formed by both of the nitrogen
Writing Formulas of Complex Ions
The central atom (metal ion) is listed first.
Polyatomic ligands are enclosed in parentheses. Usually, the element that is forming the bond to the central atoms is listed first in the ligand formula (even for water, for example (OH
The ligand symbols are then listed alphabetically without regard to charge.
2
) instead of (H
2
O)).
The entire complex is then enclosed in square brackets regardless if it is charged.
If there is a charge and no counter-ion is listed, the charge is a following superscript with the number preceding the sign of the charge.
Abbreviations of complicated organic ligands may be used in formulas even though the same is discouraged in writing names.
Examples: tetraamminechloridonitritoκ N -cobalt(III) amminebromidochloridonitritoκ O -platinate(1-) bis(ethylenediamineκ
(ONO)
[CoCl(NH
2 N,N’ )diiodidonitritoκ O -chromate(III) [CrI
3
)
[PtBrCl(NH or [PtBrCl(NH
3
2
(NH
2
4
3
CH
(NO
)(NO
2
)(ONO)] -
CH
2
2
2
)] +
)]
NH
-
2
)
2
(NO
2
)] or or [Cr(en)
2
I
2
(NO
2
)] or (ONO)
(because e comes before i) or [Cr(C
2
H
8
N
2
)
2
I
2
(NO
2
)] or (ONO)
132
EQUILIBRIUM BETWEEN TWO COMPLEX IONS OF Co
2+
IN SOLUTION
INTRODUCTION
Co 2+ in solution can be surrounded by either four or six species in tetrahedral or octahedral geometries, respectively. Such structures, called complex ions, are stable because the central, positively charged Co 2+ attracts the negatively charged, or electron-rich, portions of the coordinating species. The number of species surrounding the Co 2+ depends on the charges and the sizes of the ligands. The complex’s structure determines its resulting color: tetrahedral Co 2+ complex ions are deep blue, while octahedral ones are light pink.
When we dissolve cobalt(II) chloride (CoCl
2
) in water (H
2
O), the Co 2+ retains one chloride ion and attracts the electronegative, electron rich, oxygen end of water molecules. The resulting complex ion
2+ ion, one chloride ion and five water molecules in an octahedral configuration with consists of one Co a light pink color.
The size of the ligands is one of the factors that determine the structure of a Co
1 shows the geometries and colors of the complex ions formed when CoCl
2
2+ complex ion. Table
is dissolved in a variety of solvents. Alcohols are structurally similar to water in that they all have –OH groups with the other hydrogen replaced by an organic group. We can see in Table 1 that the geometry of all Co 2+ complex ions is either tetrahedral or octahedral and not anything else.
Table 1 Co 2+
Solvent
coordination complex ion color and structure for CoCl
Water methanol ethanol
1-propanol
2-propanol
[CoCl(CH
3
Density
(g cm
When we dissolve CoCl
2
-3
1.000
0.7914
0.7893
0.7796
0.7851
)
Solvent molecule: group attach to –OH
(CH
H-
CH
3
)
2
3
-
CH
3
-CH
2
-
CH
3
-CH
2
-CH
2
-
-CH-
in a mixture of methanol (CH
3
)
2
2
dissolved in various alcohols solution color light pink light pink dark blue dark blue dark blue
(CH
3
OH)
3
] +
Co 2+
+ CH
coordination
geometry octahedral octahedral tetrahedral tetrahedral tetrahedral
OH) and 2-propanol (CH
3
3
CH(OH)CH ones (with 2-propanol). An equilibrium will be established between the two complex ions.
CHOHCH
3
3
) the solution will contain two types of Co 2+ complex ions: octahedral ones (with methanol) and tetrahedral
CH(OH)CH
3
)
3
] + + 3 CH
This equation can be simplified as
3
OH [CoCl(CH
3
CH(OH)CH
3
[CoClP
3
] + + 3 M [CoClP
2
M
3
] + + P where P and M represent 2-propanol and methanol molecules, respectively. Because this equilibrium is between tetrahedral and octahedral complex ions, we can also represent it as
133
[Co
(tet)
] + 3 M [Co
(oct)
] + P where the subscripts indicate the geometries of the complex ions. We can then use this last equation to write an equilibrium constant expression for this system
K eq
=
Co
Co
P
M 3
This equilibrium constant, like all equilibrium constants, depends only on the temperature of the system.
The color of the system is determined by the relative proportions of the two complex ions. If we add methanol to the equilibrium mixture, the equilibrium position will shift in accordance with Le
Châtelier’s principle. Thus, the dark blue solution of Co 2+ in pure 2-propanol becomes a lighter blue upon addition of methanol, because of production of the octahedral complex ion at the expense of the tetrahedral complex ion.
As previously stated, the equilibrium constant for a system will only change if the system temperature changes. Octahedral complex ions of Co 2+ are reported to be favored over tetrahedral complex ions by
31 kJ/mol. Given this information, we can conclude that the reaction to form an octahedral Co 2+ complex ion from a tetrahedral Co 2+ complex ion is exothermic, with H equal to -31 kJ/mol.
Color results from the absorption or transmittance by matter of certain wavelengths of light within the visible spectrum. Tetrahedral Co 2+ complex ions have a strong absorption of light in the yellow-tored wavelengths that results in a dark blue solution color. Octahedral Co 2+ complex ions have a weak absorption of light in the blue-to-green wavelengths that results in a pale pink solution color.
Initially, we use molecular models to demonstrate the relationship between the geometry and color of a Co 2+ coordination complex ion. You will construct models of different Co 2+ coordination complex ions in order to illustrate how the geometry of a complex ion depends on the sizes of the coordinating molecules. Then you will identify the color associated with each of the geometries.
In this experiment you will take advantage of the intense absorbance of the tetrahedrally coordinated
Co 2+ in solution. The wavelength of maximum absorbance for solutions of CoCl
2
dissolved in 2- propanol is 657 nm, so we use 657 nm as the analytical wavelength for these solutions. The absorbance at 657 nm for a CoCl
2
–2-propanol solution is proportional to the concentration of tetrahedrally coordinated Co 2+ present. The general mathematical relationship between absorbance
(A) and concentration (c) of the absorbing species is known as Beer’s law. One form of Beer’s law is
A = ε bc where ε is the proportionality constant, relating absorbance to concentration for solutions measured in cuvets of a constant size, b. For the absorbance at 657 nm for solutions of tetrahedrally coordinated
Co 2+ formed by dissolving CoCl
2
in 2-propanol, we can write Beer’s law as
A = ε
134
We can determine the value of ε as follows. First, we measure the absorbances for various CoCl propanol solutions with known concentrations of tetrahedrally coordinated Co absorbance as a function of the corresponding Co
2+
2
–2-
. We plot each
2+ concentration, and then draw the best straight line through the data points and the origin (because a solution with zero concentration has zero absorbance). The slope of this straight line is the value of ε. Consequently, we can determine [Co
–2-propanol solution from its absorbance at 657 nm and this value of ε.
(tet)
] for any CoCl
2
You will follow the above method to determine the value of ε for solutions of CoCl
2
in 2-propanol. To do so, you will prepare several solutions, each containing a known concentration of CoCl
2
in 2- propanol. All of the Co 2+ in these solutions is in a tetrahedral geometry, and the intensity of the blue solution color is proportional to [Co
(tet)
]. You will use a spectrophotometer to measure the absorbances of the solutions at 657 nm. Based on these data, we determine the value of ε.
You will then determine the equilibrium constant for the conversion of tetrahedrally coordinated Co 2+ to octahedrally coordinated Co 2+ in solutions of CoCl
2
in 2-propanol and methanol. To do so, you will prepare solutions of CoCl
2
in various mixtures of 2-propanol and methanol, measure the absorbance of each solution at 657 nm, and relate the absorbance to the corresponding concentration of tetrahedrally coordinated Co of tetrahedrally coordinated Co 2+
PROCEDURE
2+ . The equilibrium constant for this system defines the relative stability versus that of octahedrally coordinated Co 2+ .
1. Rinse a clean, dry 10-mL graduated cylinder with about 1 mL of 2-propanol. Rinse again with another 1 mL of 2-propanol. Also rinse a clean, dry 13×100-mm test tube twice, using about 1 mL of 2-propanol each time. Pour all rinses into your “Discarded Solutions’’ container. Use a spatula to place about 0.01 g of cobalt chloride hexahydrate, CoCl
2
· 6H to dissolve the solid. Record the color of the resulting solution.
2
O, in the test tube. Using the graduated cylinder, add 5 mL of 2-propanol to the test tube. Use a clean glass stirring rod and stir
2. Rinse the 10-mL graduated cylinder twice, using about 1 mL of methanol each time. Rinse a second clean, dry 13×100-mm test tube twice, using about 1 mL of methanol each time. Pour all rinses into your ‘‘Discarded Solutions’’ container. Use a spatula to place about 0.01 g of CoCl
2
·6H
2
O in the test tube. Using the graduated cylinder, add 5 mL of methanol to the test tube. Use a second clean, dry glass stirring rod to dissolve the solid. Record the color of the resulting solution.
3. Pour the contents of the two test tubes into your “Discarded Solutions” container.
135
4. Use your molecular model kit to construct models of the following Co 2+ coordination complex ions:
(a) [CoClP
(b) [CoClP
5
3
]
]
+ : one Cl and three 2-propanols tetrahedrally arranged around a central Co
+ : one Cl and five 2-propanols octahedrally arranged around a central Co 2+
2+
Remember that the oxygen atom in the 2-propanol is what links the alcohol to the central Co 2+ .
Compare the two structures. Identify and record which structure is too crowded for all of the 2propanols to easily fit around the Co 2+ , and therefore will be unstable.
5. Using your molecular model kit, construct models of the following Co 2+ coordination complex ions:
(a) [CoClM
3
] + : one Cl and three methanols tetrahedrally arranged around a central Co 2+
(b) [CoClM
5
] + : one Cl and five methanols octahedrally arranged around a central Co 2+
Compare the two structures. Identify and record which structure has too much open space between the methanols to be stable.
6. Turn on the spectrophotometer, and adjust the wavelength control to 657 nm. Allow the spectrophotometer to stabilize while you do Steps 7–14.
7. Attach a clean, dry 50-mL buret to a support stand using a buret clamp. Rinse the buret with three
5-mL portions of 2-propanol. Collect the rinses in the “Discarded Solutions” container.
8. Fill the buret to the 0.00-mL mark with 2-propanol. Use tape to label the base of the support stand “P”.
9. Weigh between 0.16 and 0.20 g of CoCl
2
· 6H
2
O. Transfer the CoCl
Erlenmeyer flask. Record your exact mass of CoCl
2
· 6H
2
O.
2
· 6H
2
O to a clean, dry 250-mL
10. Dispense exactly 50.00 mL of 2-propanol from buret P into the Erlenmeyer flask. Swirl the flask to dissolve the solid. Make sure that the entire solid has dissolved before going on to the next step. Use tape to label the flask “stock CoCl
2
solution”.
11. Attach another clean, dry 50-mL buret to the same support stand (on the other side of the buret clamp). Rinse this buret with three 5-mL portions of your CoCl
2
stock solution. Pour the rinses into your “Discarded Solutions” container. Pour all of the remaining CoCl
2
stock solution into the buret.
The stock solution will only fill the buret to about the 25-mL mark. Use tape to label the base of the support stand “Co”.
12. Refill buret P with 2-propanol to the 0.00-mL mark.
13. Label four clean, dry 3-oz plastic cups “1”, “2”, “3”, and “4”. The cups must be free of all traces of water.
14. Prepare various mixtures of your CoCl
2
stock solution and 2-propanol in the cups by dispensing the following volumes from buret Co and buret P. Record the exact volumes dispensed from each
136 buret. Swirl the mixtures. Each cup now contains a dilution of the CoCl
2 propanol. Record the colors of the four solutions.
stock solution in 2-
Cup Volume from Buret Co, mL Volume from Buret P, mL
1
2
3
4
0.50
1.00
1.50
2.00
15. Press the A/T/C button until the display shows “A.”
9.50
9.00
8.50
8.00
16. Rinse a clean spectrophotometer cuvet twice, using about 1 mL of 2-propanol from buret P each time. Pour the rinses into your “Discarded Solutions” container. Fill the cuvet with 2- propanol from buret P, wipe the outside of the cuvet with lint-free tissue, and place the cuvet in the spectrophotometer’s sample holder. Always position the grooved sides of the cuvet in the spectrophotometer in the same orientation, facing the sides, for this and all subsequent analyses.
Close the sample holder cover.
17. Press the 100%T/0A button to zero the spectrometer. Remove the cuvet from the sample holder, and pour the 2-propanol from the cuvet into your “Discarded Solutions” container.
18. Rinse the cuvet twice, using about 1 mL of the solution from cup 1 each time. Pour the rinses into the “Discarded Solutions” container. Fill the cuvet with the solution from cup 1. Wipe the outside of the cuvet with lint-free tissue, and place the cuvet in the spectrophotometer’s sample holder.
Close the sample holder cover. Record the absorbance of this solution. Remove the cuvet, and pour its contents into your “Discarded Solutions” container.
19. Repeat Step 18 using the solutions in cups 2, 3, and 4. Remember to rinse the cuvet twice, using
1-mL portions of the solution each time before filling the cuvet and determining the solution’s absorbance. Record the absorbance of each solution.
20. Attach a third clean, dry 50-mL buret to a second support stand, using another buret clamp. Rinse the buret with three 5-mL portions of methanol. Pour the rinses into your “Discarded Solutions” container. Fill the buret to the 35-mL mark with methanol. Use tape to label the base of the support stand “M”.
21. Refill buret P to the 0.00-mL mark with 2-propanol.
22. Label six clean, dry, 3-oz plastic cups as “A”, “B”, “C”, “D”, “E”, and “F”. The cups must be free of all traces of water.
137
23. Prepare six solutions as prescribed in the following table, using the liquids in your three burets.
Swirl the cups to ensure complete solution mixing.
Cup
Volume from Buret P, mL
8.00
7.50
A
B
C
D
E
F
1.00
1.00
1.00
1.00
1.00
1.00
1.00
1.50
2.00
2.50
7.00
6.50
6.00
7.00
Record the exact volumes dispensed from each buret. Note that the contents of cup F are the same as those of cup C.
3.00
2.00
24. Prepare an ice-water bath by adding about 25 mL of water to about 100 mL of ice in a 250-mL beaker. Position cup F in the ice-water bath in a way that will prevent it from tipping over.
25. Repeat Steps 15–17 to recalibrate the spectrophotometer at 0 %T and 100 %T.
26. Measure the temperatures of the solutions in cups A–E by inserting a thermometer in each and allowing 1 min for equilibration. Record the temperatures.
27. Rinse the cuvet with two 1-mL portions of the solution in cup A. Pour the rinses into your
“Discarded Solutions” container. Fill the cuvet with the solution in cup A, place the cuvet in the spectrophotometer’s sample holder, and measure and record the solution’s absorbance. Also record the solution color. Remove the cuvet, and pour its contents into your “Discarded Solutions’’ container.
28. Repeat Step 27 for the solutions in cups B–E.
29. Remove cup F from the ice-water bath, insert a thermometer into the solution, and record the solution temperature. Repeat Step 27 for the solution in cup F.
30. Remove the cuvet from the spectrophotometer. Pour the contents of the cuvet, the three burets, and the ten cups into your “Discarded Solutions” container. Discard the material in this container as directed by your laboratory instructor. Wash all glassware with detergent. Discard the plastic cups and tissue as directed by your laboratory instructor. Turn off the spectrophotometer.
138
D
E
F
A
B
C
REPORT
EQUILIBRIUM BETWEEN TWO COMPLEX IONS …
Co 2+ in 2-propanol
Co 2+ in methanol
NAME _________________________
SECTION _______________________
Unstable Coordination Complex ions of Co 2+
Too much crowding in 2-propanol structures
Color
Too much room in methanol structures
Mass of CoCl
2
6H
2
O in 50.00 mL 2-propanol, g
Cup
Volume of stock
CoCl2 solution, mL
Volume of 2- propanol, mL
1
2
Solution color Absorbance
3
4
Cup
Volume of
CoCl
2
, mL
Volume of M, mL
Volume of P, mL Solution color Abs
139
T, °C
REPORT (Cont’d) NAME ________________________________
EQUILIBRIUM BETWEEN TWO COMPLEX IONS OF Co 2+ IN SOLUTION
Solvent Molecular formula of complex Geometry of complex Color
2-propanol methanol
Co 2+ concentration of stock CoCl
2
solution, M
Cup [Co(tet)], M
1
2
Abs
3
4
140
Slope = ε, M -1
C
D
E
Cup [Co
(total)
]
A
B
F
Average K eq
in cups A-E
Abs [Co
(tet)
] [Co
(oct)
] [P] [M]
Average temperature in cups A-E, °C
K eq
T
141
QUESTIONS FOR EQUILIBRIUM BETWEEN TWO… NAME ___________________________
1. The equation in the introduction is one way to represent the equilibrium between Co 2+ ions in mixtures of 2-propanol and methanol.
complex
[CoClP
3
] + + 3 M [CoClP
2
M
3
] + + P
(a) For the five solutions of CoCl
2 temperature (cups A–E):
, 2-propanol, and methanol at approximately the same
(i) Did you find that K eq
is indeed constant for these solutions? Briefly explain, stating your criteria for constancy.
(ii) Describe the effect on [Co
(tet)
] of increasing the proportion of methanol in the solutions. Is this effect consistent with Le Châtelier’s principle? Briefly explain.
(b) For the two solutions with the same composition (cups C and F):
(i) Describe the effect of increasing temperature on [Co
(tet)
].
(ii) Describe the effect of increasing temperature on K eq
.
(iii) Determine the sign of H for the conversion of tetrahedrally coordinated Co 2+ to the octahedral form. Briefly explain.
142
QUESTIONS FOR EQUILIBRIUM BETWEEN TWO… NAME ___________________________
(iv) The van’t Hoff equation uses the temperature dependence of the equilibrium constant to calculate the value of the enthalpy change, H , for a reaction. One form of this equation is, ln
K
K
1
2
=
∆ H
R
T
1
2
−
T
1
1
where R is the ideal gas constant (8.314 J/mol · K) and T is the temperature, in Kelvin. Use this equation and your experimental data to calculate H for the conversion of tetrahedrally coordinated Co 2+ to the octahedral form. Compare your answer to the reported value for for this conversion in the introduction.
H
2. An alternative equilibrium that could be used to explain the conversion of tetrahedrally coordinated Co 2+ in 2-propanol to the octahedral form upon addition of methanol is represented by the following equation:
[CoClP
3
] + + 5M [CoClM
5
] + + 3P
In this equilibrium, five methanol molecules replace the three 2-propanol molecules in the complex ion.
(a) Write an expression for K eq
for this equilibrium.
(b) Use your experimental data and the expression for K eq
from (a) to calculate the values of K eq for the solutions in cups A, C, and E.
(c) Based on your answers to (b), confirm or reject this alternative equilibrium. Briefly explain.
143
SYNTHESIS AND ANALYSIS OF A NICKEL COMPLEX
x
Introduction
Two important tasks many chemists perform are the synthesis and analysis of compounds. Synthesis involves not only preparing a compound, but also maximizing the yield of pure product. After isolating the product, the chemist must analyze it to ascertain its chemical composition (formula). Both tasks require good lab technique and close attention to what might seem minor procedural details.
Therefore, a technically skilled chemist with a good understanding of the purposes of each step in both the synthesis and analysis procedures will get the most accurate results. This experiment involves the preparation of a coordination compound containing nickel(lI) ion (Ni 2+ ), ammonia (NH
3
), and chloride ion (Cl ) and the determination of its empirical formula. Until the formula has been determined, it will be represented as [NiCI x
(NH
3
) n
]Cl
2-x
, with n representing a small whole number and x is an integer from 0 to 2.
Synthesizing [NiCI x
(NH
3
) n
]Cl
2-x
You will synthesize [NiCI
2
(NH
3
) n
] by reacting nickel(II) chloride hexahydrate (NiCI
2
6H
2
O) and NH
3
. This reaction is shown below
Ni 2+ (green) + 2 CI + n NH
3
[NiCI x
(NH
3
) n
]Cl
2-x
(bluish purple) (Eq. 1)
A complication arises because NH
3
in aqueous solution is involved in the equilibrium reaction shown as follows:
NH
3
+ H
2
O NH
4
+ + OH (Eq.2)
Although the equilibrium constant for Equation 2 is small (1.75 x 10 -5 ) some of the Ni 2+ ion will react
, as shown below: with hydroxide ion (OH ) to form nickel(lI) hydroxide, Ni(OH)
2
Ni 2+ (green) + 2 OH Ni(OH)
2
(green) (Eq. 3)
To the extent that the reaction in Equation 3 occurs, the product formed in Equation 1 will be impure, and the synthesis reaction yield will therefore decrease. Water is a convenient solvent for the synthesis reaction because the reactants are water soluble. However, because [NiCI
Unfortunately, the water solubility of NH
3 x
(NH
3
) n
]Cl
2-x
is also somewhat soluble in water, you must keep the volume of water used in the synthesis to an absolute minimum. Nickel(lI) chloride hexahydrate is more soluble in hot water than in cold water, so heating the reactants will enable you to dissolve more of this compound in a smaller volume of water.
is greatly decreased with increasing temperature. In this case, at temperatures approaching 100°C, NH
3
volatilizes before it can react with the NiCI maintaining the reaction temperature at 60°C, you will maximize the [NiCI x
(NH
3
) n
]Cl
2-x
yield. Once the reaction is complete, you will cool the reaction mixture to 0°C in an ice-water bath. Because
]Cl
2-x product’s solubility, because [NiCI x
(NH
3
) n
]Cl
2-x
crystals from the cold ethanolic solution and wash them with cold concentrated NH should help to convert any Ni(OH)
2
Ni(OH)
2
(green) + n NH
3
+ 2 Cl
is insoluble in ethanol. You will filter the [NiCI x
(NH
3
in the sample to [NiCI
[NiCI x
(NH
3
) n
]Cl
2
(NH
3
) n
], as shown in Equation 4.
2-x
(bluish purple) + 2 OH (Eq.4)
2
. By
[NiCI x
(NH
3
) n
is less soluble in cold water than in hot water, this step decreases the solubility of the product in Equation 1. You will add cold ethanol to the cold reaction mixture to further reduce the
) n
]Cl
2-
3
. This treatment
144
Finally, you will dry and weigh the crystals to determine the actual yield of your synthesis.
Analyzing [NiCI
3
) n
]Cl x
(NH
2-x
Determining the Molar Mass of the Compound, Mass % Ni 2+
The [Ni(NH
3
)
Ion n
] 2+ ion absorbs light in the visible region of the spectrum. You will take advantage of this property in order to determine the molar mass of the [NiCI
2
(NH
3
) n
]. Solutions containing [Ni(NH
3
) n
] 2+ ion are colored. The observed color is produced by those visible wavelengths that are not being absorbed. You can determine which wavelengths are absorbed by using a spectrophotometer to measure the absorbance of the solution throughout the visible region of the spectrum. The wavelength at which the species absorbs the most light is called the analytical wavelength ( λ max
) for that species. Absorbance is directly proportional to the concentration of the absorbing species in solution. This relationship, known as Beer's law, is represented by Equation 8. A is absorbance, ε is molar absorptivity, b is the length of the light path through the solution, and c is the molar concentration of the absorbing species.
A = ε bc (Eq. 8)
Molar absorptivity is a proportionality constant relating absorbance and molar concentration of the absorbing species at the wavelength being measured. The value of ε varies with wavelength, reaching a maximum at the analytical wavelength.
You will prepare a standard [Ni(NH hexahydrate (NiSO
4
6H
2
3
) n
] 2+ ion solution by dissolving a known mass of nickel(II) sulfate
O) in water and adding excess concentrated NH
3 mixture with water to a known volume. The Ni 2+ use your standard [Ni(NH
3
) n
] 2+
. Then you will dilute the
ion in the sample converts to [Ni(NH
3
) n
] 2+ ion. You will
ion solution to establish the analytical wavelength ( λ max
) for [Ni(NH
3
) n
] 2+ ion, the wavelength where the complex has the maximum absorbance. Then, you will compare the absorbance of your standard [Ni(NH a known mass of the [NiCI
[Ni(NH
3
) n
] 2+
3 x
(NH
)
3 n
)
] n
2+ ion solution with that of a solution you will prepare from
]Cl
2-z determine the concentration of the Ni(NH
you synthesized. Because the absorbing species, the
ion, is identical in both solutions, ε is the same for both solutions. This will allow you to n
2+
3
) and therefore the molar mass of the synthesized complex. This will allow you to determine the empirical formula.
PROCEDURE
DAY ONE – SYNTHESIZING [NiCI x
(NH
3
) n
]Cl
2-x
Prepare a warm-water bath. Half fill a 600-mL or larger beaker with tap water and place the beaker on a hot plate. Monitor the water temperature with a thermometer. Suspend the thermometer into the beaker making sure that the thermometer extends into the water but does not touch the side or bottom of the beaker. Heat the beaker and its contents until the water temperature reaches 50°C.
Adjust the hot plate setting so that the water temperature remains between 50 and 60°C.
145
On a tared piece of weighing paper, weigh 8.0 g of NiCI
2
6H
2
O. Record the mass of the solid on the
Data Sheet for day 1. Transfer the solid to a clean 125-mL Erlenmeyer flask. Add 10 mL of distilled or deionized water to the solid in the flask. Place the flask in the 60°C water bath and clamp the flask in position. Stir the mixture in the flask with a clean glass stirring rod until it has all dissolved.
Loosen the clamp on the ring stand, and while holding the end of the clamp, remove the flask from the bath. Attach the clamp to another ring stand, and let the flask and contents cool in air for 1-2 min.
6H
2
O Slowly, with stirring, in the fume hood, add 25 mL of concentrated NH
3
solution to the NiCI
2 solution in the flask.
Cover the top of the flask with a wet paper towel and suspend the flask in the warm-water bath by clamping it to the ring stand. Make sure the water temperature is between 50°C and 60°C. Leave the flask in the bath for 15 min. During this time, periodically swirl the mixture.
Prepare an ice-water bath in another 600-mL beaker by adding 150 mL of water and several pieces of ice to the beaker. Transfer 20 mL of concentrated NH
3
solution into a labeled, 18 x 150-mm test tube.
Stopper the test tube with a No. 2 solid rubber stopper. Place the test tube in the ice-water bath.
Obtain 60 mL of 95% ethanol in a labeled 100-mL beaker. Place the beaker and its contents in the ice- water bath.
Assemble a second ice-water bath in a third 600-mL beaker. After the reaction in the warm-water bath has proceeded for 15 min, unclamp the flask from the ring stand. Carefully clamp the reaction flask on another ring stand so that the flask is suspended in the second ice-water bath. Remove the damp paper towel covering the mouth of the flask. While holding the flask, loosen the clamp and swirl the reaction mixture for 5 min while it is cooling. Add 10 mL of ice-cold 95% ethanol to the flask and stir.
Remove the reaction flask from the ice-water bath. Wipe any water off the bottom of the flask using a paper towel.
Place a Büchner funnel with an adapter in a 250 mL filter flask. Place a circle of filter paper in the funnel. Wet the paper with a small amount of distilled water and turn on the vacuum, making sure the paper is firmly seated. Slowly pour the liquid-solid mixture from the flask into the Büchner funnel, as follows. Pour the mixture smoothly at such a rate that it passes through the filter quickly, but does not cause a build-up of liquid in the funnel.
Rinse any remaining solid down the inside wall of the reaction flask using 5 mL of ice-cold, concentrated NH
3
solution from your test tube. Swirl the solid and rinse solution mixture, and quickly pour the mixture into the Büchner funnel.
In the same manner, rinse any remaining solid from the flask using two additional 5-mL portions of the cold, concentrated NH
3
solution.
Dry the solid by drawing air through the solid for 3-5 min. Break up the solid with a spatula, being careful not to tear the filter paper. Pour 15 mL of cold 95% ethanol over the solid. Repeat the ethanol washing two more times, using 15 mL of cold 95% ethanol each time. Make sure to carefully break up the solid before each ethanol wash.
Pour 15 mL of acetone over the solid in the funnel. Break up the solid with a spatula to expose all its surfaces to the acetone. Draw air through the solid for 10-15 min. Turn off the vacuum.
146
Determine the mass of a labeled beaker and record this mass on the Data Sheet for day 1. After the solid is completely dry, add it to the beaker. Determine the mass of the beaker and solid and record this mass on your Data Sheet. Cover your beaker with a watch glass and store it in your locker.
Transfer the solution in the filter flask into the container provided that is labeled "Discarded
NiCl
2
/Ethanol/Acetone Solution Mixture." Rinse the filter flask and Büchner funnel once with 20 mL of tap water. Transfer the rinse to the same discard container.
DAY TWO – DETERMINING THE MOLAR MASS OF [NiCI x
(NH
3
) n
]Cl
2-x
BY SPECTROPHOTOMETRY
On the frosted or white circle on a clean, dry 100-mL beaker, write "std" to indicate the NiSO
4
6H
2
O standard sample solution. Write "unk" on a second clean, dry 100-mL beaker, which you will use for your [NiCI x
(NH
3
) n
]Cl
2-x
sample solution with unknown %Ni 2+ ion. Clean two 50-mL volumetric flasks and stoppers. Label one flask "std" and the other flask "unk."
Using an analytical balance, weigh on tared piece of weighing paper approximately 0.30 g NiSO
4
6H
2
O.
Transfer the sample to the "std" beaker. Record the mass of NiSO approximately 0.35 g sample of your [NiCI x
(NH
3
) n
]Cl
2-x
4
6H
2
O to the nearest 0.1 mg on the
Data Sheet for day 2. Using an analytical balance, weigh on a tared piece of weighing paper
. Transfer the sample to the "unk" beaker.
to the nearest 0.1 mg on the Data Sheet for day 2 (near the Record the mass of [NiCI x
(NH
3
) n
]Cl
2-x bottom of the page).
Using a graduated cylinder, add 20 mL of distilled water to both samples. Note and record the color and appearance of each solution on the Data Sheet for day 2. Using separate glass stirring rods, stir each mixture until most of the solid has dissolved. Leave the rods in the beakers to avoid losing any solution adhering to the rods.
Measure out 10 mL of concentrated NH dry. Add the NH
3
3
solution in a 10-mL graduated cylinder, which need not be
solution to the solution in the "std" beaker. Then measure another 10 mL of concentrated NH
3
solution, and add it to the solution in the "unk" beaker. Stir each mixture until no solid remains. Note and record the color and appearance of each solution on The Data Sheet for day
2.
Using a short-stem funnel, transfer the "std" solution into the "std" 50-mL volumetric flask. Rinse the beaker and rod with a minimum amount of distilled water from a wash bottle, and pour the rinses into the "std" volumetric flask. Rinse the beaker two more times, using distilled water, but do not allow the volume of solution in the volumetric flask to exceed 50 mL.
Add distilled water to the solution in the flask until the solution level coincides with the junction of the neck and body of the flask. Stopper the flask. Firmly holding the stopper in place, invert the flask
10 times to thoroughly mix the solution. Then fill the flask exactly to the etched mark, 50.00 mL, by adding distilled water from a disposable pipet or medicine dropper. Stopper the flask. Thoroughly mix the solution by inverting the flask at least 25 times, while holding the stopper firmly in place.
Follow the same procedure to transfer your "unk" solution to the "unk" 50-mL volumetric flask. Dilute the "unk" solution to the 50.00 mL mark, stopper the flask, and thoroughly mix.
147
Obtain two spectrophotometer cuvets, and place them in a dry beaker or test tube rack. Clean the cuvets and rinse them distilled water. Fill one cuvet about three-quarters full with distilled water. Set the wavelength on the spectrophotometer to 620 nm. Place the cuvet in the holder in the spectrophotometer. Press the 100%T/0A button to zero the spectrophotometer. Press the A/T/C button until the display shows an “A”.
Rinse the other cuvet three times, using a 1-2 mL portion of your "std" solution each time. Dispose of the rinses into the container provided that is labeled "Discarded NiCI
2
/NiSO
4
/NH
3
Solutions." Fill this second cuvet about three-quarters full with "std" solution. Place the cuvet in the holder in the spectrophotometer and read the absorbance. Record this in your Data Sheet.
Adjust the wavelength to 600 nm. Following the above procedure, first with the reference (H
2
O-filled) cuvet and then the sample cuvet obtain and record the absorbance for the standard solution at 600 nm. Repeat the procedure at wavelength intervals of 20 nm down to 540 nm. From among your five absorbance readings, determine the approximate λ
Data Sheet for day 2. To more precisely establish λ max max
for the [Ni(NH
for the [Ni(NH
3
3
) n
] 2+ ion and record it on The
] 2+ ion, measure the absorbance
Select the λ max
for the [Ni(NH
3
) n
] 2+
) n of the standard solution at wavelengths 10 nm less and 10 nm greater than the wavelength you estimated as λ max
. Record these additional absorbance measurements on The Data Sheet for day 2.
ion and record it on the Data Sheet for day 2.
Empty the sample cuvette into the "Discarded NiCI
2
/NiSO
4
/NH
3
Solutions" container. Rinse the cuvette with distilled water, and then rinse it three times with your "unk" solution, using 1 mL of solution each time. Transfer all rinses to the discard container. Fill the cuvette about three-quarters full with "unk" solution. Set the spectrophotometer at the λ max
you determined earlier. Check the 0% T setting with the cuvette compartment empty and its cover closed. Check the 0 Abs setting using the water-filled reference cuvette. Determine the absorbance of the unknown solution at λ max
. Record this absorbance on The Data Sheet for day 2. Transfer the solutions in your cuvettes to the discard container. Rinse and wash the cuvettes and add any rinses and washings to the discard container.
Transfer the solutions in your volumetric flasks into the appropriate discard container. Rinse the volumetric flasks twice with 10 mL of tap water each time and twice with 10 mL of distilled water each time. Transfer the rinses into the appropriate discard container. Allow the flasks to drain. Empty the cuvettes appropriately and allow them to dry.
Calculate the molar mass of the synthesized compound using the mass of the unknown,
[NiCI x
(NH
3
) n
]Cl
2-x
, dissolved in the 50.0 mL solution and the solution’s molarity obtained from its absorbance.
Recall that there must be 1 mole of Ni 2+ ions and 2 moles of Cl ions per mole of the synthesized compound and that any remaining mass in the compound must be due to the ammonia ligands.
Calculate the number of moles of ammonia equivalent to the “remaining mass” and round to the nearest whole number to arrive at the empirical formula of the synthesized compound.
148
REPORT
SYNTHESIS & ANALYSIS OF A Ni 2+
NAME _________________________________
COMPOUND SECTION _______________________________
DAY ONE – SYNTHESIZING [NiCI x
(NH
3
) n
]Cl
2-x
Mass of NiCl
2
6H
2
O, g
149
Molar mass of NiCl
2
6H
2
O, g mol -1
Moles of NiCl
2
6H
2
O used in the synthesis, mol
237.69
Mass of capped shell vial and synthesized [NiCI x
(NH
3
) n
]Cl
2-x
, g
Mass of capped shell vial, g
Mass of synthesized [NiCI x
(NH
3
) n
]Cl
2-x
, g
150
DAY TWO – SPECTROPHOTOMETRIC DETERMINATION OF MASS PERCENT OF Ni 2+ ION IN
[NiCI x
(NH
3
) n
]Cl
2-x
Initial color and appearance of NiSO
4
solution
Initial color and appearance of [NiCI x
(NH
3
) n
]Cl
2-x
Solution
Color after adding NH
3
to NiSO
4
solution
Color after adding NH
3
to [NiCI x
(NH
3
) n
]Cl
2-x solution
Absorbance for [Ni(NH
3
) n
] 2+ ion in solution using NiSO
4
6H
2
O
Mass of NiSO
4
6H
2
O used, g
Molar mass of NiSO
4
6H
2
O, g mol -1
Concentration of standard [Ni(NH
3
) n
] 2+ , M
Wavelength, nm
620
610
600
590
580
570
560
550
Approximate λ max
of the [Ni(NH
3
) n
] 2+ ion, nm
Approximate λ max
- 5 nm, nm
Approximate λ max
+ 5 nm, nm
Chosen λ max
of the [Ni(NH
3
) n
] 2+ ion, nm
Abs of the standard [Ni(NH
3
) n
] 2+ solution at λ max
Absorbance of the unknown [Ni(NH
3
) n
] 2+ ion solution
Chosen Wavelength, nm
Abs of the [Ni(NH
3
) n
] 2+ ion in the synthsized solution at the chosen wavelength
Mass of [NiCI x
(NH
3
) n
]Cl
2-x used, g
Concentration of [NiCI x
(NH
3
) n
]Cl
2-x
synthesized, M (from calculation)
APPROXIMATE molar mass of [NiCI x
(NH
3
) n
]Cl
2-x
, g mol -1
262.85
Abs
REPORT NAME _________________________________
DETERMINATION OF THE EMPIRICAL FORMULA AND PERCENT YIELD OF [NiCI
2
(NH
3
) n
]
151
Empirical formula for [NiCI
2
(NH
3
) n
]
*Mass of [NiCI
2
(NH
3
) n
] synthesized, g
Molar mass of [NiCI
2
(NH
3
) n
], g mol from empirical formula)
-1 (calculated
Moles of [NiCI
2
(NH
3
) n
] synthesized, mol
*Moles of NiCl
2
6H
2
O used in the synthesis, mol
Percent yield of [NiCI
2
(NH
3
) n
], %
* these are the values from Day 1.
152
QUESTIONS FOR SYNTHESIS & ANALYSIS …
1. Based on your determined empirical formula for [NiCI
2 of the nickel(II) ion?
NAME _________________________________
(NH
3
) n
], what is the coordination number
2. Based on the electron configuration of the nickel(II) ion and the coordination number stated above, what is the hybridization used by the Ni 2+ ion (i.e.,sp 3 , dsp 2 , d 2 sp 3 or sp 3 d 2 )? Why? Explain your answer based on the electron configuration coordination number and what you know about the different types of hybridization.
3. Do you expect the [NiCI
2
(NH
3
) n
] complex to be paramagnetic or diamagnetic? Use the answers to questions 1 and 2 to support your answer.
153
MOLECULAR MODELS OF TRANSITION METAL COMPLEXES
INTRODUCTION
The chemical and physical properties of a substance are influenced by the distribution of outer shell
(valence) electrons and the three-dimensional arrangement of its nuclei. A variety of experimental methods are employed to map out the relative positions of the nuclei in a molecule or an ion. We will be using molecular models to determine some of the properties of transition metal complexes.
All transition metal complexes in this lab have a transition metal ion as the central atom. You will determine the distribution of electrons and bonded atoms about a central atom. In so doing, you will be able to determine the probable hybridization of the central atom, electron pair and molecular geometries, and polarity of the species in question. We can also determine if the species is optically active by looking at the presence or lack of a superimposable mirror image of the compound or ion.
PROCEDURE
All of the models we will be constructing will use the gray 14 sided polyhedron (with a hole in each face) representing the transition metal as the central atom. Only the square sides will be used. In order to simplify things we will be omitting the hydrogen atoms on ethylenediamine. Carbon atoms are the black polyhedra with 4 holes, nitrogen atoms are the blue polyhedra with 4 holes, oxygen atoms are the red polyhedra with 4 holes and chlorine atoms are the green polyhedra with 4 holes.
A. Square Planar Complexes
H
H
N
1. Construct four ammonia molecules:
“extra” bond position on the nitrogen.
H
These will attach to the metal atom through the
2. Construct a model of cis -diamminedichloridoplatinum(II).
3. Construct a model of trans -diamminedichloridoplatinum (II).
B. Tetrahedral Complexes.
1. Construct a water molecule. This will attach to the metal atom through one of the “extra” bond positions on the oxygen.
2. Construct a model of ammineaquabromidochloridoiron(II).
3. Using the model from step 1 as guide construct the mirror image of the model.
4. Rotate one of the models to try to make it match exactly (superimpose) with the other model.
5. Replace the aqua ligand with another ammine ligand in both of the models.
154
6. Rotate one of the models to try to make it match exactly (superimpose) with the other model.
C. Octahedral complexes.
1. Construct 6 ethylenediamine molecules: (we are not showing the hydrogen atoms here). These will attach to the metal atom through the nitrogen atom’s lone pairs.
2. Construct the model for the tris(ethylenediamine)cobalt(II) ion.
3. Using the model from step 2 as a guide, construct the mirror image of the model in step 2.
4. Rotate the model from step 3 to try to make it match up exactly (superimpose) with the model from step 2.
5. Replace one of the ethylenediamines in each model with 2 chloride ions. The two chloride ions should be in adjacent positions around the central metal atom ( cis - conformation).
6. Rotate one of the modified models to try to make it match up exactly (superimpose) with the other one.
7. Replace one more of the ethylenediamines with 2 ammonia molecules in each of the models. a. While the chlorides are in the cis- position, examine the structures to see if the models match up exactly (superimpose).
8. In each model assembled in step 7 swap the positions of two of the appropriate ligands so that the chloride ions are on opposite sides of the metal atom ( trans- conformation).
9. Rotate one of the modified models to try to make it match up exactly (superimpose) with the other one.
155
REPORT
MOLECULAR MODELS OF TRANSITION…
NAME ________________________________
SECTION ______________________________
A. Refer to the models that you constructed for this portion of the lab.
Which conformation ( cis - or trans -) should be polar? _____________________________
Which conformation should be soluble in water? _____________________________
B. Refer to the models that you constructed for this portion of the lab.
Did the mirror images of the models of ammineaquabromidochloridoiron(II) match up exactly
(superimpose) with one another?
Did the mirror images of the models of diamminebromidochloridoiron(II) match up exactly
(superimpose) with one another?
Which of these compounds exists as stereoisomers?
C. Refer to the models that you constructed for this portion of the lab.
Did the mirror images of the models of the tris(ethylenediamine)cobalt(II) ion match up exactly (superimpose) with one another?
Does this complex ion exist as enantiomers?
Did the mirror images of the models of the cis-dichloridobis(ethylenediamine)cobalt(II) match up exactly (superimpose) with one another?
Does this compound exist as enantiomers?
Did the mirror images of the models of either cis or transdiamminedichlorido
(ethylenediamine)cobalt(II) match up exactly (superimpose) with one another?
Which of these compounds exist as enantiomers if any?
156
CHECK OUT INSTRUCTIONS
1.
Clean!
•
West Bench – Benchtop, West Fume Hoods, Balance Room
•
Middle Bench – Benchtop, Rear Balance Area, Rear Fume Hoods
•
East Bench – Benchtop, East Fume Hoods, Rear Sink Area
2.
Return Locks!
•
Obtain a white tag (if you no longer have the original)
•
Write your combination on the tag
•
Lock it to your lock
•
Place the lock on the middle bench in the front
3.
Equipment Check
•
Ensure your drawer has a complete set of equipment
•
Remove extra items
•
CLEAN
AND RETURN SHELL VIALS
•
Obtain missing items
•
When
EVERYONE
is ready the stockroom will verify that your drawer is complete
THANK YOU & ENJOY!
¦|
157
Quantity
2
1
1
1
1
1
1
1
3
1
1
1
1
2
2
2
2
2
4
10
10
1
2
1
1
1
1
1
1
1
Description
Beaker, 20 mL
Beaker, 50 mL
Beaker, 100 mL
Beaker, 150 mL
Beaker, 250 mL
Beaker, 400 mL
Beaker, 600 mL
Bottle, 500 mL, Screw Cap
Bulb, Pipet
Clamp, Buret
Cylinder, Graduated, 10 mL
Cylinder, Graduated, 50 mL
Flask, Erlenmeyer, 250 mL
Forceps
Funnel, Small, 45 mm
Holder, Test Tube
Microspatula
Pipet, Graduated 1.0 mL
Pipet, Graduated 5.0 mL or 10.0 mL
Pipet, Volumetric 10 mL
Pipet, Volumetric 25 mL
Rack, Test tube
Shell Vials
Test Tube, 10 mm x 75 mm
Test Tube, 13 mm x 100 mm
Test Tube Brush, 12 mm x 62 mm
Thermometer, -20°C to 110°C
Tongs
Wash Bottle, Polyethylene, 250 mL
Watch Glass, 75 mm
158
159
APPENDIX A
Calculations Involving Precision and Accuracy
Precision
Precision is a measure of how well multiple (repeated) measurements agree with each other. It is an indication of consistency. One method of evaluating the precision of a set of data is to determine the percent relative average deviation. The procedure for this calculation is as follows:
1. Determine the average value for at least three experimental trials.
2. Subtract each individual value from the average value to get the deviation for each trial.
3. Add together the absolute values of the deviations and divide by the number of trials and the average value to get the relative average deviation.
4. Multiply by 100 to get percent relative average deviation.
Percent Relative Average Deviation = average deviation average value
× 100 = i n
∑
= 1 x i
− x nx
× 100
Accuracy
Accuracy is a measure of how close an experimental value (usually an average value) is to the accepted value (also called the "true" value). One method of evaluating the accuracy of an experimental result is to determine the percent error as follows:
Percent Error = experimental value - accepted value accepted value
× 100
Do not use absolute values when calculating accuracy. The sign simply indicates that the experimental value is higher than the accepted value when the percent error is a positive number, lower if negative.
160
APPENDIX B
GRAPHS
INTRODUCTION
Relationships between experimental quantities are often represented in the form of graphs. Straight line graphs are easier to construct and to interpret than curved ones. Data that initially result in a curve when graphed are sometimes mathematically rearranged to result in a straight line relationship.
This can often be accomplished by taking the logarithms of the values for one or both of the quantities that were being plotted and then graphing these new log values. When data that has been graphed forms a straight line plot, the mathematical relationship between the quantities can be determined from the equation for a line:
= +
PROCEDURE
A. Construction of a graph
A number of rules must be followed when constructing graphs. Your score for this exercise will depend upon how well you follow these rules.
1.
Select a good quality graph paper that is easy to use with the metric scale. Graph paper that has divisions marked in blocks with different shades of lines is easier to use (less counting) than paper that has uniform shading. Choose paper that is divided into five by five or ten by ten small squares within a larger grid. Avoid paper in which the large squares are divided into four by four or eight by eight blocks (this type of graph paper is for drafting classes that use English system units).
2.
It is customary to plot the quantity that is varied (the independent variable) on the x (horizontal) axis and the quantity that is measured (the dependent variable) on the y (vertical) axis. In mathematical terms, the quantity on the y-axis is a function of the quantity on the x-axis.
3.
Use a scale for each axis that will spread the data points to be plotted over the full page (or over the space assigned). Do not crowd the data into one corner. However, your scale should result in convenient units (such as 10, 20, 30, etc. or 2, 4, 6, 8, etc.) for each major division on the graph.
A compromise may be necessary.
4.
Use a constant scale (the same number of divisions/unit) along each axis. However, because different quantities are plotted on each axis you would not necessarily expect the scale on the x and y axes to be the same.
5.
It is only necessary to mark (and label) the intervals at 4 to 6 places along an axis (more than that gets cluttered). For example, if you had mass readings ranging from 7 to 68 g, you might mark and label the axis at 0, 20, 40, 60, and 80 g. Do NOT mark your axes at the data points. The coordinates for the data plotted on the graph should be presented in a table on an unused section of the graph paper (away from the data points) or on a separate piece of paper.
6.
The precision in the labels for the axes intervals should reflect the precision in the data being plotted. For example, if masses were determined to one place after the decimal (such as 9.1 g,
15.4 g, etc.) the intervals on the graph should be labeled 0.0, 20.0, 40.0 and so on.
161
Note: The precision for measurements plotted on the y-axis may differ from those for the x axis.
7.
If you do not have any data close to a zero value, you need not place “zero” in the lower left-hand corner of the graph. The graph origin can begin at any convenient value (provided it is labeled).
However, if the graph is to be assessed to determine a “straight-line” relationship between data, and you wish to read the y-intercept directly from the graph, then you must use intervals and plot the data so that the y-intercept is NOT off the graph.
8.
Label each axis with the appropriate label.
9.
Title each graph. The title should reflect what quantities are being plotted. The title might simply be an equation that has been provided or it might be the description of experimental quantities.
10.
After the data have been plotted, draw either a straight line or a smooth curve that best represents the data points. Do NOT connect the dots with individual straight lines. When data being plotted has been experimentally obtained, you should not expect the line to pass directly through every data point due to experimental errors. Construct a “best-fit” plot in which the points that do not fall on the line are randomly scattered. The sum of the distances between the line and the points above it should be the same as the sum of the distances between the line and the points below it. In addition, the line should be drawn so that these distances are minimized.
B. Determination of a Mathematical Relationship from a Straight Line Graph.
The straight line relationship between quantities x and y can be represented by: where y (the quantity plotted on the vertical axis) is a function of x (the quantity plotted on the horizontal axis). The “m” is the slope of the line and “b” is called the y-intercept. Linear regression analysis and substitution can be used to obtain the exact value for the slope and y-intercept, but in this exercise these values will be estimated by reading them directly from the graph.
1.
Graph the data and draw a “best-fit” straight line (see Part A of the Procedure).
2.
Determine the slope of the line. Choose two points on the line (not necessarily data points) that can be read accurately. To maximize precision, these two points should be fairly far apart. Read the coordinate values for each point. Point number one is the data point having an x value closest to the origin and the values for point one will be (x
1
, y l
). The other point will have values of (x
2
, y
2
). The slope of the line is: m = y x
2
2
−
− y x
1
1
162
3.
The sign of the slope can be negative (indicating an inverse relationship between the quantities x and y). Note that the number of significant figures for the slope will be artificially reduced if the points on the line selected for slope determination are too close together. Be sure to include units
(unit for y/unit for x) with the value for the slope.
4.
To determine the y-intercept value from the graph, extrapolate (extend) the line until it reaches the y-axis (x = 0) and read the value for y at that point (include units).
5.
Write the mathematical relationship for the quantities that have been graphed. Into the equation:
= +
substitute (each with its appropriate unit): for y – the quantity (what is being graphed) on the y axis for m – the value (number) for the slope for x – the quantity (what is being graphed) on the x axis for b – the value (number) for the y-intercept
For example: distance(m) = time(s) 5.26 m/s + 6.35 m
163
Constructing Scientific Graphs in Excel™
Excel™ makes constructing scientific graphs easy. You can plot points and find the line of best fit
(linear or otherwise) and the equation for that line. You can also import scientific data (such as that from the Vernier™ LabQuest) and visually represent the data. Excel’s “Scatter Plot” chart function allows you to do both.
Input the data
One version of the scatter chart is used when you only have a few data points (5 to 15 or so). You will enter the independent variable (x-axis) in the first column, “A,” and the dependent variable (yaxis) in the second column, “B.” You can also add descriptions of the data in the first row if you like.
For example:
Once the data has been entered you can use the cursor to select all of the data, including the header row. Then you will click on the “Insert” tab.
On the “Insert” tab you want to choose the icon under the “Charts” section that indicates a scatter chart “Insert Scatter (X, Y) or Bubble Chart” and click on it.
After clicking the icon you will have a choice of what kind of scatter plot you want to make. Click on the first one which just shows the points.
164
With this data set you will have something that looks like this.
y data
Double-click here to change the title.
120
100
80
60
40
20
0
0 2 4 6 8 10 12
Presently, this is not that useful. We need to have the graph properly formatted with the axes labelled, the correct precision, and a descriptive title. We should also have more grid lines. In Excel
2013 clicking on the Scatter Chart icon will then bring up a two new toolbars at the top. One for the
Design and one for the Format of the chart. We are mainly interested in the Design toolbar. The first part of the toolbar is labelled “Add Chart Element.” In Excel 2010, three new toolbars are created, “Design,” “Layout,” and “Format.” The Chart Elements are in the “Layout” toolbar in Excel
2010. With this we can add our axes labels and the minor gridlines and format them as we need.
Click on “Add Chart Element” then click on “Axis Title” then on “Primary Horizontal.” In the formula bar just under the toolbar you can then put in the title for that axis. For instance, time in minutes
(“Time (min)”) and press ENTER. We can do the same for the y-axis by clicking through to “Primary
Vertical” and put in the label in the box. For instance, the distance in kilometers (“Distance (km)”) and press ENTER. If you double click on the chart title, where it currently says “y data,” you can
165 change that to a descriptive title such as “Plot of distance vs. time for a road trip.” The graph should now look like this:
Plot of distance vs. time for a road trip
120
100
80
60
40
20
0
0 2 4 6
Time (min)
8 10 12
Going back to the “Add Chart Element” box we can add minor gridlines by clicking on “Gridlines” then either “Primary Minor Horizontal” or “Primary Minor Vertical.” You will want to change both of them but you can only change on at a time. Adding these with the default values gives this:
Plot of distance vs. time for a road trip
120
100
80
60
40
20
0
0 2 4 6
Time (min)
8 10 12
The values on the axes can also be modified to indicate values with appropriate precision by doubleclicking on the numbers on the axis. On the “Format” panel that opens on the right click on the icon with 3 vertical bars and the last option is “Number.” Clicking this and changing the option from
“General” to “Number” allows you to specify the number of decimal places the values on that axis have.
166
We can also make the data points smaller (the default is too big).
On the right side of the program is the formatting options. Clicking on the drop down arrow where it says “AXIS OPTIONS” and then clicking on the “Series ‘y data’” option (the last one in the list), allows you to change the size, type and color of the data point markers. In the new panel that comes up click on the icon that looks like a paint can and then click on “MARKER” then on “MARKER
OPTIONS.” Change it to “Built In” and reduce the size. The smallest you can make it is “2” which works well.
At this point we also want to add a trend line which will be the best-fit line for the data.
We can do this with the “Add
Chart Element” option under
“Trendline.” I would suggest using the “More Trendline
Options.” Here on the right side under the “Format…,” click on the icon that looks like three vertical bars. You can then select which kind of trend line you want and click on the option to display the equation on the chart.
You can also choose to set the intercept to 0.0 (or any value that you know it should be). You can also format the line under the icon that looks like a paint can being poured out. Set the trend line to be a solid line and the thickness to be thin (0.5 pt). Our finished graph then looks like:
167
120
Plot of distance vs. time for a road trip
100
80
60
40
20
0
0 y = 8.2517x
2 4 6
Time (min)
8 10 12
Clicking on a blank area of the chart and pressing “Ctrl-P” will allow you to print the chart. You will usually want to print only one graph on a page.
Smooth Line Scatter Chart
Inputting the data
In this case we will usually input data from another source so we won’t we typing it in by hand. The process for creating the chart will be essentially the same as above, we’ll select “Insert Scatter (X, Y) or Bubble
Chart” and choose the option “Scatter with
Smooth Lines.” Here we have spectrum data created with the Vernier™ SpectroVis
Module from three different discharge lamps. There are 645 rows of data!
Obviously, there’s far too much data to enter (or graph) by hand but Excel will handle it nicely. We need to select the data we want to plot. Here, we will just plot the first spectrum out of the three. To select the first spectrum we can just click on the column “A” label and drag over to the column “B” label. This gives us:
168
As before, we then click on the
“Insert” tab and click on the
“Insert Scatter (X, Y) or Bubble
Chart” option. Then well click on the option that shows a smooth line. If we wanted to plot all three spectra on the same plot we would then hold down the
Ctrl key and click on the other two intensity columns.
This will create a chart that looks like this:
Intensity
1.2
1
0.8
0.6
0.4
0.2
0
0 200 400 600 800 1000
Again, we need to do some formatting here to make it useful as a scientific graph. First we need to set the x-axis correctly because we do not need to show the area from 0 to almost 400 nm and from about 900 nm to 1000 nm. To set the x-axis scale correctly we then click in the “Format” area on the right on the “Horizontal (Value) Axis.” Click on the icon that looks like three vertical bars and on
169
“Axis Options.” The first section here is “Bounds.” Change the Minimum to 380 and the maximum to 900.
Next we need to add the axis labels. This is done exactly as we did before. Click on “Add Chart
Elements” then “Axis Titles” then “Primary Horizontal.” Type “Wavelength (nm)” in the box and press ENTER. Do the same for “Primary Vertical” and type “Intensity” in the box. This has no units so we don’t have to add anything else. We should also change the graph title to something more descriptive such as “Plot of Intensity vs. Wavelength for the Hydrogen lamp.” We can also add in the minor gridlines to make the graph easier to read by clicking “Add Chart Elements” then
“Gridlines” then “Primary Minor Vertical.” Then in the “Format” section on the right click on
“Vertical (Value) Axis” and click on the icon that looks like 3 bars. Click on “Axis Options” and change the value for the minor units to 0.02 (1/10 th of the major unit). Then do the same thing for the Horizontal axis. We can change the precision of the labels on the axes in the same way we did before with the “Number” option at the bottom of the “Format Axis” panel (3 decimals for intensity and 1 for the wavelength).
Finally, again the default value for the line is too thick. Click on “Series ‘Intensity’” and then on the paint bucket icon and change the line thickness to 0.5 pt. We then have a graph that looks like:
1.200
Plot of intensity of light vs. wavelength for the hydrogen lamp
1.000
0.800
0.600
0.400
0.200
0.000
380.0
480.0
580.0
680.0
Wavelength (nm)
780.0
At this point, you can click on a blank area of the graph and press Ctrl-P you can print it.
880.0
170
APPENDIX C
BALANCING REDOX REACTIONS USING THE HALF-REACTION METHOD
There are many methods that can be used when balancing chemical reactions that involve oxidationreduction. The following steps are used in a “half-reaction” method:
1. The initial “skeleton” reaction to be balanced for an oxidation-reduction reaction occurring in aqueous solution often does not include the H
2
(in acid), or OH O, H + (in base) that will be added later as the reaction is balanced. Sometimes spectator ions are not included either.
Write the skeleton reaction and assign oxidation numbers to each element.
2. Split the reaction into two half-reactions, one containing the oxidation and one containing the reduction. (Note: In some reactions, more than one element is oxidized or more than one is reduced. Sometimes the mole to mole relationships between these elements can be determined from the formulas of the chemicals involved in the reaction. However, in some cases, experimental data is needed to help determine the correctly balanced equation.)
3. For each half-reaction, balance of all the elements present except oxygen and hydrogen.
4. Balance oxygen by adding H
2
O to the side of each half-reaction needing oxygen.
5. The method for balancing hydrogen in each half-reaction depends on whether the reaction is taking place in acidic or basic solution. a. in acid, add H + to the side of the reaction needing more hydrogen. b. in base, count the number of hydrogen atoms that are needed. Add one H
2
O for every hydrogen atom needed to the side with insufficient hydrogen and simultaneously add the
ions to the opposite side same number of OH -
Note: # of H needed = # of H
2
O added to the side with insufficient H = # of OH added to opposite side
6. Balance overall charge by adding electrons (e ) to the more positive side of the half-reaction.
7. Multiply each half-reaction by the factors needed to make the electrons in each half-reaction equal.
8. Add the half-reactions (combining any like terms) and cancel species that appear on both sides of the equation (electrons must cancel).
9. If needed, divide by the largest common factor to reduce the coefficients to the lowest whole number ratio.
10. CHECK to make certain that the number of atoms of each element and overall charge are balanced.
171
This method of balancing redox reactions will now be applied to a problem. The numbers shown to the left of each step in the process correspond to the numbers for the steps in the instructions given on the previous page.
Balance the following redox reaction:
N
2
H
4
+ Pu
2
O
3
→ N
2
O + Pu(OH)
2
(in base)
N
2
H
4
+ Pu
2
O
3
→ N
2
O + Pu(OH)
2
(in OH and H
2
O)
-2 +1 +3 -2 +1 -2 +2 -2 +1 -2 +1 +1 -2
3.
2
1.
N
N
2
2
H
H
4
4
→ N
→ N
2
2
O
Nitrogen is balanced
O
H
2
O + N
2
H
4
→ N
2
O
5. (in base)
(Pu needs to be balanced)
Pu
2
O
3
→ 2 Pu(OH)
2
4. one oxygen needed on the reactant side, one oxygen needed on the reactant side,
add one H
2
O to the reactant side add one H
2
O to the reactant side
H
2
Pu
O + Pu
2
2
O
O
3
3
→
→
Pu(OH)
2
2 Pu(OH)
2
6 H needed on the product side, add 6 H
2
O to 2H needed on the reactant side, add 2 H
2 product side and 6 OH to the reactant side reactant side and 2 OH -
6OH + H
4
→ N
2
O + 6 H
to the product side
2
O + N
2
H
2
O 2 H
(H
2
| 3 H
2
O + H
2
2
O + Pu
O + Pu
2
O
3
→
2
O
3
→ 2 Pu(OH)
2 Pu(OH)
2
2
+ 2 OH
O on the reactant side could be combined)
+ 2 OH -
-
O to the
Note: It does not matter that there is H
2
O on both sides of the nitrogen equation at this point. They will be canceled later. Hydrogen and Oxygen are balanced in each half reaction.
6. add 6 e to the product side
→ N 2 e + 3 H
2
O + Pu
2
O add 2 e
3
to the reactant side
→ 2 Pu(OH)
2
+ 2 OH 6 OH
8.
+ H
2
6 e
O + N
-
2
H
+ 6 OH
4
-
2
O + 6 H
2
O + 6e -
7. Multiply equation above by 1
6 OH
+ 10 H
(cancel 6 e
-
-
2
+ H
2
O + N
, 6 OH
O + N
-
2
H
4
2
6 e + 9 H
2
O + 3 Pu
2
H
4
O
3
+ 3 Pu
→
2
O
N
3
Multiply the equation above by 3
2
→
O + 6 H
→ 6 Pu(OH)
N
2
2
2
O + 6e -
+ 6 OH -
(add equations and combine like terms)
O + 6 H
2
O + 6 Pu(OH)
4 H
2
O + N
2
H
4
+ 3 Pu
2
O
3
→ N
2
O + 6 Pu(OH)
2
2
, and 6 H
2
O from each side of the reaction)
+ 6 OH + 6e -
9. Because the coefficients are in the lowest whole number ratio, the equation is complete.
10. Check to make sure the number of atoms and overall charge are balanced in the completed equation.
Apply the method outlined for the half-reaction method to balance the following redox reactions.
1. NO
3
+ Zn → Zn 2+ + N
2
(in acid)
2. O
2
+ I → I
2
(in base)
Hint: one of the half-reactions has nothing on the product side.
172
3. CrO
4. HNO
2
+ ClO → Cl + CrO
4
2 (in base)
3
+ Bi
2
S
3
→ Bi(NO
3
)
3
+ NO + S (in acid)
Hint: remember that most metal sulfides are insoluble.
5. S
2
O
3
2-
6. Cr
2
O
7
2-
+ I
2
→ S
4
O
6
+ Sn
7. SCN + H
2
O
2
2+ →
→
2-
Sn
NH
+ I
4+
4
+
(in base)
+ Cr 3+
+ HCO
(in acid)
3
+ HSO
Hint: same as in number 2.
4
(in acid)
173
DERIVING CHEMICAL EQUATIONS FROM BALANCED NET IONIC EQUATIONS
For oxidation-reduction reactions, often it is easier to balance the net ionic form of the equation first and then to derive the chemical equation from the net ionic equation. The following is a method for this procedure:
1. Write the skeleton equation from the information given for the reactants and products.
2. Assign oxidation numbers to every element (including the elements of any acids and bases).
3. The elements which are spectator ions do not change oxidation numbers. However, sometimes an ion can be involved as both a spectator ion and in oxidation-reduction.
4. Balance the net ionic equation following the rules given in the previous section. Remember to add in the spectator ions on the side needing them when you balance the atoms other than oxygen and hydrogen.
5. If needed divide to reduce the coefficients to the lowest whole-number ratio. At this point you will need to add in the counter ion for the acid or base used. Add one counter ion for each H +
(for sulfuric acid you will add HSO
4
) or OH in the equation to each side. The result of this step is the ionic equation. Check to make sure that the net charge on each side of the reaction is zero.
6. Combine anions and cations to create the balanced chemical equation. No uncombined ions should remain. Check to make sure the number of atoms is still balanced and that the coefficients are in the lowest whole-number ratio.
Potassium permanganate reacts with chromium(III) chloride to produce manganese(IV) oxide and the chromate ion in potassium hydroxide.
KMnO
4
+ CrCl
3
MnO
2
+ CrO
+1 +7 -2 +3 -1 +4 -2 +6 -2
4
2-
3 e + 4 H
2
O + KMnO
4
MnO
2
+ K + + 2 H
2
O + 4 OH -
8OH + 4 H
2
O + CrCl
3
CrO
4
2 + 3 Cl + 8 H
2
O + 3 e —
4 OH + KMnO
4
+ CrCl
3
MnO
2
+ CrO
4
2 + 3 Cl + K + + 2 H
2
O
+ 4 K + + 4 K +
4 KOH + KMnO
4
+ CrCl
3
MnO
2
+ K
2
CrO
4
+ 3 KCl + 2 H
2
O
Balance atom other than H or O.
Balance O by adding water.
Balance H by adding H + .
Balance charge by adding e .
Add in counter ion to acid or base.
(in H
2
SO
4
)
(in KOH)
(in NaOH)
(in HI)
(in KOH)
(in HCl)
(in KOH)
(in NaOH)
174
MORE PRACTICE REDOX PROBLEMS
1. NaCl + MnO
2
→ Mn 2+ + Cl
2
2. K
4
Fe(CN)
6
+ CeCl
4
→ Ce(OH)
3
+ Fe(OH)
3
+ CO
3
3. NaNO
2
+ Al → NH
3
+ AlO
2
4. NaIO
3
+ NaI → NaI
3
5. Fe + HCl → HFeCl
4
6. Fe(OH)
2
+ H
2
+ H
2
O
2
→ Fe(OH)
3
7. Na
2
S
2
O
8
+ CrCl
3
8. KCN + KMnO
4
→ Cr
2
O
7
2 + SO
4
2
→ CNO + MnO
9. CrI
3
+ Cl
2
→ CrO
4
2 + IO
4
+ Cl
2
2 + NO
10. Potassium permanganate and nitrous acid react in sulfuric acid. Two of the products of this reaction are manganese(II) bisulfate and nitric acid.