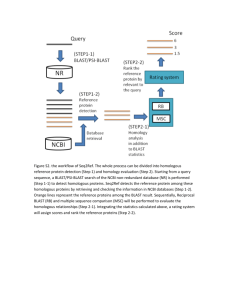
Program - blast - University of California, Santa Cruz
advertisement

BLAST XII MEETING HILTON TUCSON EAST TUCSON, ARIZONA JANUARY 20-25, 2013 Meeting Chairperson: Dr. Urs Jenal – University of Basel, Basel, Switzerland Meeting Vice-Chairperson: Dr. Karen Ottemann – University of California, Santa Cruz, CA Program Committee: Dr. John S. Parkinson (Chairperson) – University of Utah, Salt Lake City, UT Dr. Rasika Harshey – The University of Texas, Austin, TX Meeting Review Committee: Dr. Alan Wolfe (Chairperson) – Loyola University, Maywood, IL Dr. Christine Josenhans – Medical University Hannover, Hannover, Germany Dr. Kirsten Jung – Ludwig-Maximilians-Universitaet Muenchen, Munich, Germany Dr. Christopher Rao – University of Illinois at Urbana-Champaign, Urbana, IL Poster Awards Committee: Dr. John Kirby (Chairperson) – University of Iowa, Iowa City, IA Dr. Brian Crane – Cornell University, Ithaca, NY Dr. Sean Crosson – University of Chicago, Chicago, IL Dr. Kylie Watts – Loma Linda University, Loma Linda, CA Speaker Award Committee: Dr. Urs Jenal – University of Basel, Basel, Switzerland Dr. Karen Ottemann – University of California, Santa Cruz, CA Board of Directors – BLAST, Inc.: Dr. Robert Bourret (Chairperson) – University of North Carolina, Chapel Hill, NC Dr. Joe Falke – University of Colorado, Boulder, CO Dr. Michael Manson – Texas A&M University, College Station, TX Dr. John S. Parkinson – University of Utah, Salt Lake City, UT Dr. Phillip Matsumura (Chair Emeritus) – Molecular Biology Consortium, Chicago, IL Conference Coordinators: Ms. Tarra Bollinger – Molecular Biology Consortium, Chicago, IL Ms. Peggy O’Neill – Molecular Biology Consortium, Chicago, IL ii TABLE OF CONTENTS COMMITTEE INFORMATION ........................................................................................ii TABLE OF CONTENTS ................................................................................................. iii AWARDS INFORMATION.............................................................................................. iv MEETING SCHEDULE ...................................................................................................v SPEAKER PROGRAM ................................................................................................... vi POSTER LIST ................................................................................................................ix SPEAKER ABSTRACTS .................................................................................................1 POSTER ABSTRACTS ................................................................................................. 51 PARTICIPANT LIST .................................................................................................... 119 INDEX ......................................................................................................................... 135 iii AWARDS INFORMATION Robert M. Macnab Award for an Outstanding Poster Presentation by a Postdoctoral Scientist This award was established at BLAST VIII (2005) and is named in memory of the late Robert M. Macnab, Ph.D., who was an integral member of the Bacterial Locomotion and Signal Transduction Community. Dr. Macnab spent his 30 year career studying the assembly, structure and function of the bacterial flagellum. Bob actively participated in the BLAST meetings and served on the Program and Review Committees for BLAST IV. At the time of his death in 2003, Bob was a professor in the Department of Molecular Biophysics and Biochemistry at Yale University. Robert M. Macnab Memorial Travel Awards We are pleased to announce the establishment of the first travel awards for the BLAST meeting, remembering our colleague Dr. Robert M. Macnab on the 10th anniversary of his death. The intent of the awards is to help young scientists from outside the country that hosts BLAST to attend the meeting. The recipients chosen by the Board of Directors for BLAST XII are graduate students Ana Martinez del Campo from George Dreyfus' lab at the Universidad Nacional Autónoma de México and Ambroise Lambert from Mathieu Picardeau's lab at the Institut Pasteur - Paris. (The Macnab poster and travel awards are sponsored by generous donations from Mrs. May K. Macnab) Robert J. Kadner Award for an Outstanding Poster Presentation by a Graduate Student This award was established at BLAST IX (2007) and is named in memory of the late Robert J. Kadner, Ph.D., who was an integral member of the Bacterial Locomotion and Signal Transduction Community. Dr. Kadner spent his career studying microbial physiology of E. coli transport systems. Bob actively participated in the BLAST meetings and served as Chair of the Review Committee for BLAST V, Vice-Chair of BLAST VII and Meeting Chair of BLAST VIII. At the time of his death in 2005, Bob was the Norman J. Knorr Professor of Basic Sciences in the Department of Microbiology at the University of Virginia, School of Medicine. This award is sponsored by BLAST. Nucleic Acids Research Award for an Outstanding Poster Presentation by a Young Investigator This award was established at BLAST XI (2011) and is presented to a young investigator whose research is in on regulation of transcription. The award is sponsored by Nucleic Acids Research (NAR), an Oxford University Press Journal, which publishes the results of leading edge research into physical, chemical, biochemical and biological aspects of nucleic acids and proteins involved in nucleic acid metabolism and/or interactions (http://nar.oxfordjournals.org). BLAST Board of Directors' Award for an Outstanding Talk This award, the first for speakers, was established at BLAST XI (2011) by Phil Matsumura, Founding Chair of the BLAST Board of Directors. In contrast to the poster awards, which are open only to graduate students and/or postdoctoral scientists, all speakers are eligible for the BLAST Board of Directors Award. (This award is sponsored by a generous donation from Dr. Philip Matsumura) iii BLAST XII MEETING SCHEDULE TIME EVENT LOCATION Sunday, January 20, 2013 4:00 pm 4:00 pm – 7:00 pm 7:00 pm – 8:30 pm 9:00 pm – 11:00 pm Poster room available for poster setup Meeting Registration Dinner Welcome Reception Gala Rooms A & B Outer Vistas Private Dining Room Private Dining Room Monday, January 21, 2013 7:30 am 8:45 am 9:00 am 10:15 am 12:00 pm 2:00 pm 6:00 pm 7:30 pm 8:30 pm – 8:30 am – 9:00 am – 12:00 pm – 10:30 am – 1:30 pm – 4:00 pm – 7:30 pm – 10:00 pm – 8:45 pm Breakfast Welcome & Announcements Meeting Session – “Flagella 1” Coffee Break Lunch Poster Session – even numbered posters Dinner Meeting Session – “Two-Component Signaling” Coffee Break Private Dining Room Salon B & C Salon B & C Salon A Private Dining Room Gala Rooms A & B Private Dining Room Salon B & C Salon A Tuesday, January 22, 2013 7:30 am 8:45 am 10:15 am 12:00 pm 2:00 pm 6:00 pm 7:30 pm 8:30 pm – 8:30 am – 12:00 pm – 10:30 am – 1:30 pm – 4:00 pm – 7:30 pm – 10:00 pm – 8:45 pm Breakfast Meeting Session –“Chemoreceptor Arrays & Signaling” Coffee Break Lunch Poster Session – odd numbered posters Dinner Meeting Session – “Swarming & Gliding” Coffee Break Private Dining Room Salon B & C Salon A Private Dining Room Gala Rooms A & B Private Dining Room Salon B & C Salon A Wednesday, January 23, 2013 7:15 am 8:30 am 9:55 am 12:00 pm 12:15 pm – 8:15 am – 11:30 am – 10:10 am – 1:30 pm Breakfast Meeting Session – “Receptors” Coffee Break Tours Depart Lunch Private Dining Room Salon B & C Salon A Atrium Lobby Private Dining Room Thursday, January 24, 2013 7:30 am 8:45 am 10:15 am 12:00 pm 4:00 pm 5:30 pm 7:00 pm 7:30 pm 8:30 pm 9:40 pm 10:00 pm – – – – – – – – – – – 8:30 am 12:00 pm 10:30 am 1:30 pm 5:00 pm 7:00 pm 7:30 pm 9:40 pm 8:45 pm 10:00 pm 12:00 am Breakfast Meeting Session – “Flagella 2” Coffee Break Lunch Town Hall Meeting for students & postdocs Dinner Business Meeting for all attendees Meeting Session – “Regulation” Coffee Break Awards Presentation Reception Private Dining Room Salon B & C Salon A Private Dining Room Salon B & C Private Dining Room Salon B & C Salon B & C Salon A Salon B & C Private Dining Room Friday, January 25, 2013 7:00 am – 8:30 am Breakfast Private Dining Room v BLAST XII PROGRAM January 21, 2013 Flagella 1 Chair – Howard Berg Monday Morning (8:45 am – 12:00 pm) PRESENTER Morgan Beeby Lewis Evans Milana Fraiberg Hajime Fukuoka BREAK Lele, Pushkar ABSTRACT PAGE NO. TITLE Structural dissection of bacterial flagellar motors in live bacteria by electron cryo-tomography How does the flagellum grow at a constant rate? CheY acetylation in bacterial chemotaxis: How does it generate flagellar clockwise rotation? Direct imaging of CheY-binding to a functioning bacterial flagellar motor Mechanism for adaptive remodeling of the bacterial flagellar switch Flagella stator homologues power bacterial gliding motility by moving in Beiyan Nan helical trajectories Jonathan Partridge The FliL protein increases flagellar motor output in Salmonella Xiaowei Zhao 3-D visualization of bacterial flagellar assembly in Borrelia burgdorferi January 21, 2013 Lindsie Goss Daniela Keilberg BREAK Shonna McBride Tarek Msadek Hendrik Szurmant 4 5 6 7 8 9 Two-Component Signaling Chair – Steven Porter Monday Evening (7:30 pm – 10:00 pm) Rachel CreagerAllen 2 3 Autophosphorylation of the response regulator PhoB receiver domain is cooperative and saturable Ser/Thr phosphorylation regulates two-component systems involved in cell wall metabolism LinkedIn – finding connections: A response regulator links the Frz system to the MglA/MglB GTPase/GAP module to regulate polarity of motility systems in M. xanthus Resistance of Clostridium difficile to bacterial antimicrobial peptides through a disjoined two-component system that responds to multiple substrates The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response What sequence diversity teaches us about bacterial signaling vi 10 11 12 13 14 15 January 22, 2013 Chemoreceptor Arrays and Signaling Chair – Yuhai Tu Tuesday Morning (8:45 am – 12:00 pm) PRESENTER Ariane Briegel Christopher Jones Milena Lazova John S. Parkinson BREAK Kene Piasta Lynmarie Thompson Ady Vaknin Kylie Watts ABSTRACT PAGE NO. TITLE Structural changes of bacterial chemoreceptor arrays underlie different activation states Formation and segregation of the cytoplasmic chemosensory cluster in Rhodobacter sphaeroides Network-level properties of Salmonella typhimurium chemotaxis How HAMP domains and methylation bundles control stimulus response and sensory adaptation in chemoreceptor signaling Testing and refining the receptor-CheA regulatory domain interface in functional, membrane-bound arrays using disulfide mapping and TAMIDS Differences between signaling states of chemotaxis receptors revealed by hydrogen exchange mass spectrometry Prolonged stimuli alter the bacterial chemosensory clusters The sensing and signaling roles of multiple PAS-HEME domains in Aer-2 type chemoreceptors January 22, 2013 16 17 18 19 20 21 22 23 Swarming and Gliding Chair – Larry Shimkets Tuesday Evening (7:30 pm – 10:00 pm) Functions of Proteus mirabilis FliL in swarming and responding to surface viscosity The AAA+ protease LonA regulates SwrA levels and swarming motility in Sampriti Mukherjee Bacillus subtilis Daisuke Nakane Helical flow of surface protein required for Flavobacterium gliding motility BREAK Bacterial transitions: Optimization of biological and physical factors by Joshua Shrout Pseudomonas aeruginosa during swarming Structure of proteins involved in Mycoplasm mobile gliding revealed by Yuhei Tahara electron microscopy and high speed atomic force microscopy (AFM) Bacteriophytochromes 1 and LOV-HK work in a signaling network to Liang Wu regulate swarming motility of Pseudomonas syringae strain B728a Yi-Ying Lee vii 24 25 26 27 28 29 January 23, 2013 Wednesday Morning (8:30 am – 11:30 am) PRESENTER Tino Krell Caitlin Brennan Matthew Sears Andreas Möglich BREAK Michael Morse Christopher Rao Ben Webb Laurence Wilson Receptors Chair – Birgit Prüβ ABSTRACT PAGE NO. TITLE Multiple agonistic and antagonistic signals control the action of the complex sensor kinase TodS Determining chemoreceptor-ligand interactions in the light-organ symbiont Vibrio fischeri to understand niche specificity Chemotaxis to norepinephrine (Ne) requires the Tsr chemoreceptor and conversion enzymes induced by QseC Structure and function of a blue-light-regulated sensor histidine kinase Effects of molecular adsorption on the trajectories and accumulation of motile bacteria at the air/water interface Towards an integrated model of the chemotaxis pathway in Bacillus subtilis Methyl accepting chemotaxis protein U of Sinorhizobium meliloti binds the seed exudate component, proline Time-dependent chemotactic response in a large population January 24, 2013 Detecting a conformational change in the periplasmic region of the sodium-driven stator protein PomB by the disulfide crosslink Coordinated switching of bacterial flagellar motors: Evidence for direct Bo Hu motor-motor coupling? Helicobacter pylori flagellar motor: Structural insight into the mechanism Anna Roujeinikova of stator assembly and activation Shahid Khan Correlated mutation analysis of the flagellar motor protein FliG BREAK Ria Sircar Structure and activity of Thermotoga maritima FliY Borrelia burgdorferi flagella export apparatus and virulence: insight into Tao Lin type III secretion system Masayoshi Dynamic conformational changes of flagellar filament observed by highNishiyama pressure microscopy Novel Leptospira protein is essential factor in determining the flagellar Elsio Wunder sheath, translational motility, and virulence phenotype Shiwei Zhu January 24, 2013 Birgit Prüβ Ching Wooen Sze 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Regulation Chair – Tarek Msadek Thursday Evening (7:30 pm – 9:40 pm) Chakib Mouslim BREAK 31 Flagella 2 Chair – Kelly Hughes Thursday Morning (8:45 am – 12:00 pm) Susanne Gebhard Jürgen Michael Lassak 30 Interactions between a transporter and a sensor kinase mediate signal transduction in antimicrobial peptide detoxification modules A novel translational regulation mechanism mediated by elongation factor EF-P allows tight adjustment of protein copy numbers Transcriptional activation and repression of six flhDC promoters: An intricate promoter configuration for flhDC in Salmonella typhimurium Escherichia coli biofilms may favor a mixture of motile and non-motile bacteria Rrp1 regulates chitobiose utilization of the Lyme disease spirochete Borrelia burgdorferi viii 46 47 48 49 50 POSTERS - BLAST XII Poster # Lab Presenter Title Structure and function of chemoreceptors and the role of multiple CheW homologues in Rhodobacter sphaeroides Structure-function studies of the response regulator CheY6 from Rhodobacter sphaeroides Deciphering the role of the periplasmic domain of PilJ: Mechanisms of signal transduction Photosensory protein BphP1 initiates an unknown signal transduction pathway through phosphotransfer to Psyr_2449 in Pseudomonas syringae B728A A novel inducer of Roseobacter motility is also a disruptor of algal symbiosis Does the C-ring rotate? 1 Judith Armitage James Allen 2 Judith Armitage Sonia Bardy Gwyn Beattie Matthew Smith Robert Belas Howard Berg Robert Bourret Robert Belas 8 Robert Bourret Ruth Silversmith 9 Robert Bourret Sean Crosson Stephani Page Frederick Dahlquist Georges Dreyfus Georges Dreyfus Robert Levenson Marc Erhardt Joseph Falke Toshio Fukuda Rasika Harshey Marc Erhardt Penelope Higgs Michio Homma Michio Homma Penelope Higgs Michio Homma Seiji Kojima 3 4 5 6 7 10 11 12 13 14 15 16 17 18 19 20 21 Sonia Bardy Regina McGrane Basarab Hosu Bob Bourret Sean Crosson Ana Martinez-del Campo Manuel Osorio-Valeriano Joseph Falke Hirotaka Tajima Hyo Kyung Kim Mizuki Gohara Norihiro Takekawa A non-conserved active site residue influences response regulator reaction kinetics, phosphodonor specificity, and partner kinase interactions Nonconserved active site residues modulate CheY autophosphorylation kinetics and phosphodonor preference Experimental analysis of receiver domain autodephosphorylation kinetics guided by The Brucella abortus general stress response system regulates chronic mammalian infection, and exhibits non-canonical post-translational regulation Studies of flagellar motor protein FliG and its interactions with FliF and FliM Chemotaxis signaling pathway of the flagellar system 2 from Rhodobacter sphaeroides The C terminus of the flagellar muramidase SltF modulates the interaction with FlgJ in Rhodobacter spaheroides Profound effect of hook length on motility and virulence in Salmonella Structural and functional disulfide mapping of the CheACheW interface in the bacterial chemosensory array Analysis of the repellent-sensing mechanisms of Escherichia coli Identification four new GGDEF/EAL domain proteins that regulate motility in Salmonella , one of which localizes to the mid-cell and inhibits cell division Signalling systems controlling developmental cell fate segregation in Myxococcus xanthus Attempt to investigate the interaction between the rotor and the stator by using solution NMR Importance of charged residues of PomA and FliG for rotor-stator interaction in the sodium-driven flagellar motor of Vibrio alginolyticus Regulation of the flagellar number in Vibrio alginolyticus : Role of ATP binding motif of FlhG and the novel protein SflA Page # 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 POSTERS - BLAST XII Poster # Lab Presenter Title Kelly Hughes Kelly Hughes Masahiro Ito Kelly Hughes Effect of codon context on translation speed in vivo 73 Kelly Hughes Salmonella fliC mRNA translation 74 Masahiro Ito 75 Urs Jenal Barry Taylor Christine Josenhans Isabelle Hug Kirsten Jung Ikuro Kawagishi Daniel Kearns Keng Hwee Chiam John Kirby Stefan Behr Koji Nakayama Chunhao Li Keiko Sato Effect of a single motP mutation for motility at neutral pH of the Na+-driven flagellar motor of alkaliphilic Bacillus pseudofirmus OF4 Towards a mechanism for surface mechano-sensation in Caulobacter crescentus Surface accessibility study identifies domain interactions in the aerotaxis receptor Aer Role of energy sensor TlpD of Helicobacter pylor i in gerbil colonization and whole genome analyses after adaptation in the gerbil Interconnectivity between two LytTR-like twocomponent systems in Escherichia coli Localization control of chemotaxis-related signaling components of V. cholerae The role of FlgM in regulating flagellar assembly in Bacillus subtilis Role of the Pseudomonas aeruginosa flagellar motor in swimming motility and chemotaxis Binding affinity provides kinetic preference, specificity and signaling fidelity for two-component systems regulating development in Myxococcus xanthus Secretion and gliding of Bacterodetes phylum Jun Liu Jun Liu Michael Manson Jun Liu 38 Mark McBride Yongtao Zhu 39 Makoto Miyata Makoto Miyata Makoto Miyata MD Motaleb Dianne Newman Akihiro Tanaka 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 40 41 42 43 Darysbel Perez Wiebke Behrens Geetha Hiremath-Mendez Rebecca Calvo Chui Ching Wong John Kirby Chunhao Li Jun Liu Christopher Adase Hiroki Yamamoto Isil Tulum Syed Sultan Naomi Kreamer The response regulator Rrp1 regulates chitobiose utilization and virulence of the Lyme disease spirochete Borrelia burgdorferi Cryo-electron tomography of Escherichia coli minicells reveals molecular architecture of intact flagellar motor The torque generator of bacterial flagellar motor revealed in Borrelia burgdorferi The cytoplasmic aromatic anchor of TM2 regulates transmembrane signal transduction by the Tar chemoreceptor Comparative analysis of Flavobacterium johnsoniae and Cellulophaga algicola gliding motility and protein secretion Gliding of Mycoplasma mobile can be explained by directed binding Gliding machinery of Mycoplasma mobile observed by electron microscopy Gene manipulation of gliding bacterium, Mycoplasma mobile Is motility or possession of periplasmic flagella crucial for the pathogenesis of Lyme disease? Fe(II) sensing in Pseudomonas aeruginosa Page # 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 POSTERS - BLAST XII Poster # Lab 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 Presenter Title Takayuki Nishizaka Karen Ottemann Yoshiaki Kinosita John Parkinson John Parkinson John Parkinson Mathieu Picardeau Steven Porter Germán Piñas Simon Rainville Simon Rainville Christopher Rao Susan Rosenberg Edward Ruby Florian Schubot Guillaume Paradis Direct observation of unitary step of gliding machinery in Mycoplasma mobile The Helicobacter pylori TlpD chemoreceptor is critical for multiple stages of normal stomach colonization and mediates a response to iron and other reactive oxygen species-generating conditions The role of receptor-kinase interactions in array signaling Transmembrane signaling via synthetic control cables in the Tsr chemotaxis receptor Tsr-E502: An unorthodox chemoreceptor sensory adaptation site Characterization of a chemotaxis locus in Leptospira involved in motility and virulence Stimulatory interactions between hybrid histidine protein kinases in the virulence signalling network of Pseudomonas aeruginosa Taking control of the bacterial flagellar motor Karen Ottemann Smiljka Kitanovic Xuesheng Han Ambroise Lambert Steven Porter Ismael Duchesne The motility of bacteria in an anisotropic liquid environment Santosh Koirala Phenotypic variation and vistability within flagellar gene network in Salmonella enterica serovar Typhimurium Abu Amar Al Mamun Identity and function of a large gene network underlying mutagenic repair of DNA breaks under stress Marie-Stephanie Aschtgen Is bacterial twitching motility required to establish the squid/Vibrio symbiosis? Florian Schubot The Pseudomonas aeruginosa type three secretion system transcriptional activator ExsA interacts with the anti-activator protein ExsD in a temperature-dependent manner Liz David Milner A surface-regulated orphan ParA-like protein has an Sockett impact on gliding motility in Bdellovibrio bacteriovorus Liz Sarah Basford The role of polyphosphate related genes in the Sockett predatory bacteria Bdellovibrio bacteriovorus Lotte Beata Jakobczak Identification of a novel cell-envelope subcomplex Søgaard-Andersen involved in gliding motility in Myxococcus xanthus Lotte Carmen Friedrich Deciphering the assembly pathway of type IV pili in Søgaard-Andersen Myxococcus xanthus Lotte Kristin Wuichet Diversity of small Ras-like G-proteins in prokaryotes Søgaard-Andersen Claudia Claudia Studdert An engineered serine chemoreceptor with symmetric Studdert insertions mimics natural transducers of the 40H class Claudia Claudia Studdert Functional and structural analysis of homologous Studdert conserved residues CheW-R62 and CheA-R555 within the chemotaxis ternary complex Tom Francois Anquez Effect of chemoeffector on the stability of Shimizu chemoreceptors assembly studied by photo-activated localization microscopy Alan Alaa Abouelfetouh The flip of a coin: Acetylation or phosphorylation by Wolfe acetyl phosphate in biofilms Page # 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 POSTERS - BLAST XII Poster # Lab 66 67 Alan Wolfe Zhihong Xie Presenter Title Alan Wolfe Acetyl phosphate is a potent regulator of bacterial protein acetylation A novel chemoreceptor couples degradation of aromatic pollutants with chemotaxis Zhihong Xie Page # 117 118 SPEAKER ABSTRACTS 1 BLAST XII _____Mon. Morning Session STRUCTURAL DISSECTION OF BACTERIAL FLAGELLAR MOTORS IN LIVE BACTERIA BY ELECTRON CRYO-TOMOGRAPHY Morgan Beeby1, David R. Hendrixson2, Deborah Ribardo2, Patrizia Abrusci3, Steven Johnson3, Susan M. Lea3, Songye Chen4, and Grant J. Jensen4 1 Imperial College London, South Kensington Campus, London SW7 2AZ, UK 2 Department of Microbiology, University of Texas Southwestern Medical Center, Dallas, Texas 75390 3 Sir William Dunn School of Pathology, Oxford University, South Parks Road, Oxford, OX1 3RE, UK 4 Division of Biology, California Institute of Technology, Pasadena, CA, USA, and Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA, USA The bacterial flagellar motor spins the flagellar filament to propel the cell to favourable environments. While considerable insights into many facets of the motor's function and origins are promised by understanding the structure and assembly of the motor, such studies have historically been hampered by the intact motor's intimate association with the cell envelope and the lack of tools to image it there. The recent development of electron cryo-tomography remedies this problem by producing 3-D images of frozen, living bacteria at a resolution sufficient to resolve individual macromolecules. Because electron cryo-tomography can be used to image whole bacteria, and thus avoids the need for purification procedures, imaging the motor in situ is now routine and dependent only upon our ability to culture bacteria. We reasoned that a comparative study of a range of flagellar motors from phylogenetically diverse bacteria, including alpha-, gamma- and epsilon- bacteria, firmicutes and spirochaetes, would provide information on motor function, assembly, and evolution not apparent from studies focusing on a single model organism. Results revealed widespread elaboration upon the 'normal' Salmonella enterica or Escherichia coli motor that enabled us to combine our comparative electron cryo-tomography approach with genetic screens and bioinformatics to pinpoint the location of multiple constituent proteins in the 3-D structure of the motor. Some of these proteins are ubiquitous members of the flagellar export apparatus, while others are specific adaptations found only in a subset of the bacteria. In combination, these comparative results propose further testable hypotheses regarding universal flagellar motor function and suggest reasons why some bacteria have evolved elaborate structures around their motors that are not found in model organisms. Lab: Morgan Beeby ________________ 2 BLAST XII _____Mon. Morning Session HOW DOES THE FLAGELLUM GROW AT A CONSTANT RATE? Lewis D.B. Evans, Simon Poulter, Eugene M. Terentjev*, Colin Hughes & Gillian M. Fraser Department of Pathology, University of Cambridge, Tennis Court Road, Cambridge CB2 1QP, UK. *Cavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB3 OHE, UK. Bacteria swim by means of long flagella on the cell surface. Flagella are assembled from thousands of subunits that are translocated across the cell membrane by a dedicated export machinery at the base of each flagellum. Subunits then travel through a central channel in the flagellum to reach their assembly site at the tip. Remarkably, as the flagellum lengthens outside the cell the rate of flagellum growth does not appear to change, precluding the possibility that subunits diffuse passively through the external channel. Several studies have focused on the energy source for growth of flagella, but the mechanisms proposed appear incompatible with the observation that the rate of flagellum growth is independent of flagellum length. So the question remains, how do subunits move through the long inert channel in the external flagellum and, in particular, what is the energy source? We propose a simple physical mechanism that engenders a constant rate of subunit delivery to the flagellum tip independent of channel length. Lab: Colin Hughes ________________ 3 BLAST XII _____Mon. Morning Session CheY ACETYLATION IN BACTERIAL CHEMOTAXIS: HOW DOES IT GENERATE FLAGELLAR CLOCKWISE ROTATION? Milana Fraiberg1, Oshri Afanzar1, Yishai Levin2 and Michael Eisenbach1 1 Department of Biological Chemistry, The Weizmann Institute of Science, 76100 Rehovot, Israel. 2 Proteomics and Mass Spectrometry Teaching Unit, The Weizmann Institute of Science, 76100 Rehovot, Israel. N-acetylation of proteins (on lysine residues) is highly prevalent, from bacteria to mammals. One of the best-studied proteins that undergo this covalent modification is CheY, the excitatory response regulator of bacterial chemotaxis, whose acetylation is required for normal chemotaxis. In my talk I will focus on our studies aimed at resolving the enigma of how CheY is activated by acetylation to shift flagellar rotation from the default direction, counterclockwise, to clockwise, even though its binding to the switch of the flagellar motor does not increase. Lab: Michael Eisenbach ____________ 4 BLAST XII _____Mon. Morning Session DIRECT IMAGING OF CheY-BINDING TO A FUNCTIONING BACTERIAL FLAGELLAR MOTOR Hajime Fukuoka, Yuichi Inoue, Hiroto Takahashi, Akihiko Ishijima Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aobaku, Sendai, 980-8577, Japan Escherichia coli cell swims by rotating flagellar motors and migrates toward favorable environment by controlling the rotational direction of the flagellar motors. The rotational direction of flagellar motor is controlled by chemotaxis signaling system. When the chemoreceptors sense the chemotacitc signals, they modulate the auto-phosphorylation activity of a histidine protein kinase, CheA. The phosphoryl group on CheA is rapidly transferred to a response regulator, CheY. From the previous biochemically and genetically investigations, the binding of phosphorylated CheY (CheY-P) to the FliM, which is one of the components of flagellar motor, is believed to change the rotational direction of the motor from counterclockwise (CCW) to clockwise (CW). However, it is not directly observed the rotational switching induced by the binding of CheY-P. In this study, we tried to directly observe the rotational switching induced by CheY-binding in a single functioning flagellar motor. In order to observe the binding of CheY to the functioning flagellar motor, we constructed the expression system of GFP-fusion protein of CheY and observed the localization of CheY-GFP in a tethered cell under TIRF microscopy. In our microscopic system, the localization of CheY-GFP (fluorescence image) and the rotational motion of tethered cell (bright field image) were recorded simultaneously by using EMCCD and high-speed CCD cameras, respectively. By using our microscopic system, the fluorescent spot derived from CheY-GFP was observed at the rotational center of the tethered cell during CW rotation. The fluorescence intensity at the rotational center of the tethered cell coordinately changed with the rotational switching of the motor; the fluorescence intensity increased during CW rotation and decreased during CCW rotation. A cross correlation analysis between rotational switching and the change of the fluorescence intensity showed a major peak nearly 0s. The lag time of rotational switching after the binding or dissociation of CheYGFP, which was estimated from the peak-time of correlation, was within our sampling rate (50 ms/frame). The number of CheY-GFP molecules, which was estimated by the comparison with the fluorescence intensity of the motor constructed by the FliM-GFP, was smaller than that of FliM molecules in a motor. These results indicate that the conformational change of one of the FliM subunit by the binding of CheY-P should be spread to neighboring FliM subunits as proposed in previous theoretical models, and suggest that there is a strong cooperativity in the binding and dissociation of CheY-P to the flagellar motor in a rotational switching. The highly binding cooperativity would accelerate the conformational change of the basal body and contribute to the rapid response of motor to the intracellular signal. Lab: Akihiko Ishijima _______________ 5 BLAST XII _____Mon. Morning Session MECHANISM FOR ADAPTIVE REMODELING OF THE BACTERIAL FLAGELLAR SWITCH Pushkar P. Lele, Richard W. Branch, Vedhavalli S. J. Nathan and Howard C. Berg Department of Molecular and Cellular Biology, Harvard University, 16 Divinity Ave, Cambridge MA 02138 The bacterial flagellar motor has been shown to adapt to changes in the steady-state concentration of the chemotaxis signaling molecule, CheY-P, by changing the FliM content [Yuan J, Branch RW, Hosu BG, & Berg HC (2012) Adaptation at the output of the chemotaxis signalling pathway. Nature 484(7393):233-236]. We show here that the number of FliM molecules in the motor and the fraction of FliM molecules that exchange depend on the direction of flagellar rotation, not on CheY-P binding per se. Our results are consistent with a model in which the structural differences associated with the direction of rotation modulate the strength of FliM binding. When the motor spins CCW, FliM binding strengthens, the fraction of FliM molecules that exchanges decreases, and the ring content increases. The larger number of CheY-P binding sites enhances the motor’s sensitivity, i.e., the motor adapts. An interesting unresolved question is how additional copies of FliM might be accommodated. Lab: Howard Berg _________________ 6 BLAST XII _____Mon. Morning Session FLAGELLA STATOR HOMOLOGUES POWER BACTERIAL GLIDING MOTILITY BY MOVING IN HELICAL TRAJECTORIES Beiyan Nan1, Jigar N. Bandaria1,2, Amirpasha Moghtaderi1, Im-Hong Sun1, Ahmet Yildiz1,2* and David R. Zusman1* 1 Department of Molecular and Cell Biology 2 Department of Physics, University of California, Berkeley, CA 94720, USA. Many bacterial species use gliding motility in natural habitats since external flagella function poorly on hard surfaces. However, the mechanism(s) of gliding remain elusive since surface motility structures are not apparent. Here, we characterized the dynamics of the Myxococcus xanthus gliding motor protein AglR, a homologue of the Escherichia coli flagella stator protein MotA. We observed that AglR decorated a helical structure and the AglR helices rotated when cells were suspended in liquid or when cells moved on agar surfaces. We found that single molecules of AglR, unlike MotA/MotB, can move laterally within the membrane in helical trajectories. AglR slowed down transiently at gliding surfaces, accumulating in clusters. Our work shows that the untethered gliding motors of M. xanthus, by moving within the membrane, can transform helical motion into linear driving forces that push against the surface. Lab: David Zusman ________________ 7 BLAST XII _____Mon. Morning Session THE FliL PROTEIN INCREASES FLAGELLAR MOTOR OUTPUT IN SALMONELLA Jonathan D. Partridge* and Rasika M. Harshey Section of Molecular Genetics and Microbiology, University of Texas at Austin, Austin, TX 78712 The fliL gene is present in numerous flagellated bacteria, often co-expressed with genes encoding the switch complex or stator components. The precise function of FliL is not known, but its absence is associated with defects in both swimming and swarming motility. In Salmonella, absence of FliL leads to small defects in swimming but complete elimination of swarming, the latter being known to correlate with rod fracture1. Here we show a role for FliL beyond the structural. Using a variety of assays we show that FliL interacts strongly with itself, the MS ring protein (FliF) and also MotA and MotB proteins. Single motor experiments show that motor speed is slower in the absence of FliL. These and other experiments show that FliL increases PMF through the MotAB stators either by recruiting, by stabilizing, or by increasing proton flow through the stators. From this analysis, we place FliL outside and around the MS ring, in proximity to the stators and expand its previous remit beyond structural support at the rod. Overexpression of FliL in conjunction with the stator proteins MotA and MotB overcomes surface friction to allow swarming on harder agar surfaces. 1. U. Attmanspacher, B. Scharf and R. M. Harshey. 2008. Mol. Microbiol. 68: 328-341. Lab: Rasika Harshey _______________ 8 BLAST XII _____Mon. Morning Session 3-D VISUALIZATION OF BACTERIAL FLAGELLAR ASSEMBLY IN BORRELIA burgdorferi Xiaowei Zhao1*, Kai Zhang2*, Tristan Boquoi3, Bo Hu1, Chunhao Li2, Md A. Motaleb3, Kelly A. Miller4, Milinda E. James4, Nyles W. Charon4, Steven J. Norris1, Jun Liu1 1. Department of Pathology and Laboratory Medicine, Medical School, University of Texas Health Science Center at Houston, Houston, TX 77030 2. Department of Oral Biology, State University of New York at Buffalo, Buffalo, NY 14214 3. Department of Microbiology and Immunology, East Carolina University, Greenville, NC 27834 4. Department of Microbiology, West Virginia University, Morgantown, WV 26506 The bacterial flagellum is a remarkable self-assembled molecular machine, and is composed of the flagellar motor, the rod, the hook and the filament. The flagellar assembly in Escherichia and Salmonella, which has been a well-studied paradigm model, is a finely orchestrated biochemical process from highly regulated gene expression to protein assembly. The flagellum-specific type III secretion system (T3SS) is responsible for the translocation and the assembly of key proteins of the rod (FliE, FlgB, FlgC, FlgF and FlgG), the hook (FlgE) and the filament (FliC). Despite the recent progress in understanding overall flagellar structure and assembly pathway, direct visualization of the intermediates of this assembly process has not been achieved at the molecular level. In this study, cryo-electron tomography was utilized to determine three-dimensional (3-D) structures of flagellum from several key flagellar mutants (fliE, flgB, flgC, flhO (a homolog to E. coli flgF), flgG, flgE, flgI, and flaB (a homolog to E. coli fliC), which were all constructed in Borrelia burgdorferi, a Lyme disease spirochete. A complete set of 3-D intermediate structures of the T3SS mediated flagellar assembly indicated a clear progression of added densities. Evidently, the products of each rod gene form a small but distinct portion of the rod. This observation differs from the prior hypothesis that the lack of any of the rod proteins causes an overall failure of rod construction. Surprisingly, the export channel, which traverses the axial center of the MS ring, appears to be closed until the start of rod assembly. The modular composite structure of the rod may help to explain the intrinsic flexibility and robustness of the rod, which play a critical role in transmitting the motor rotation to the filament. Lab: Jun Liu ______________________ 9 BLAST XII _____ Mon. Evening Session AUTOPHOSPHORYLATION OF THE RESPONSE REGULATOR PHoB RECEIVER DOMAIN IS COOPERATIVE AND SATURABLE Rachel Creager-Allena, Ruth Silversmithb, Robert Bourretb Departments of aBiochemistry & Biophysics and bMicrobiology & Immunology, University of North Carolina, Chapel Hill, NC 27599-7290 Two component systems, comprised of histidine kinases and response regulators, are a common means of signal transduction in microorganisms. Response regulators can autophosphorylate using small-molecule phosphodonors. To date, comprehensive kinetic characterization of response regulator autophosphorylation is limited to CheY, which exhibits non-saturable autophosphorylation kinetics with respect to phosphodonor concentration. Non-saturable kinetics presumably reflect weak substrate binding. We characterized autophosphorylation of the receiver domain of the Escherichia coli response regulator PhoB (PhoB N ) by the small molecule phosphodonor phosphoramidate. The quench in intrinsic fluorescence of a tryptophan in the active site of PhoB N was used to monitor autophosphorylation time courses. In contrast to CheY, the rate of autophosphorylation of PhoB N was saturable and cooperative with respect to phosphoramidate concentration. Binding of a phosphoryl group analog was also cooperative. Saturability of autophosphorylation suggests that phosphodonors bind tighter to PhoB N than to CheY. Cooperativity indicates communication between PhoB N active sites and suggests that PhoB N dimerization might play a role in autophosphorylation kinetics. Consistent with such a possibility, disruption of the PhoB N dimerization interface by mutation1 led to autophosphorylation kinetics that were not cooperative and were much slower than wild type PhoB N . Furthermore, the rate of PhoB N autophosphorylation increased with protein concentration, as expected if dimerization enhances autophosphorylation. Mathematical modeling showed that the cooperativity of PhoB N autophosphorylation could be explained by formation of a heterodimer (one phosphorylated monomer and one unphosphorylated monomer of PhoB N ). Essential to the model was that the dimerization constants for the heterodimer and for the dimer consisting of two phosphorylated monomers of PhoB N were on the same order of magnitude (5 μM). Another essential feature of the model was that the autophosphorylation rate of the PhoB N monomer was at least 10-fold slower than the heterodimer. Phosphorylation-mediated dimerization allows many response regulators to bind to tandem sites on DNA and regulate transcription. Our data challenge the notion that response regulator dimers primarily form between two phosphorylated monomers, and raise the possibility that response regulator heterodimers containing one phosphoryl group may participate in gene regulation. 1. Mack, T.R., Gao, R., Stock, A.M. Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J Mol Biol, 2009. 389(2): p: 349-364. Lab: Robert Bourret _______________ 10 BLAST XII _____ Mon. Evening Session Ser/Thr PHOSPHORYLATION REGULATES TWO-COMPONENT SYSTEMS INVOLVED IN CELL WALL METABOLISM Lindsie Goss & Jonathan Dworkin Columbia University, 701 W 168th St., HHSC 1218, New York, NY 10032 Bacteria must be able to quickly respond and adapt to changing environments. The ability to sense and transduce these signals historically has been studied in the context of classical twocomponent systems (TCS), composed of histidine kinases and response regulators. Bacteria also contain eukaryotic-like Ser/Thr kinases (eSTKs) and eukaryotic-like Ser/Thr phosphatases (eSTPs) but the role of Ser/Thr phosphorylation has been unclear. My projects focus on the regulation of two different TCSs by Ser/Thr phosphorylation and suggest that these two types of signal transduction pathways convergently regulate physiological responses in bacteria involving the cell wall. Bacillus subtilis PrkC, a member of a well-conserved eukaryotic-like Ser/Thr kinase family, contains extracellular PASTA domains that bind cell wall peptidoglycan fragments. PrkC regulates expression of the autolysin YocH, which is also a primary target of the WalRK TCS, the only essential TCS in B. subtilis and a number of other Gram-positive bacteria. It is sensitive to cell wall perturbations but how it senses these effects is unclear. We find that PrkC phosphorylates WalR, the response regulator, both in vitro and in vivo, on T101. Allelic replacement strains that make a conservative serine substitution mimic phenotypes resulting from deletion of the threonine kinase, emphasizing the physiological relevance of this modification. This modification is conserved in S. aureus and we are examining whether the phenotypic consequences are also conserved. Finally, we are trying to understand the mechanistic implications of this modification for WalR. Some Enterococci contain a TCS (VanRS) that responds to vancomycin and induces the expression of genes that modify the cell wall such that the normally sensitive bacteria are now resistant to vancomycin. While it is clear that vancomycin induces VanRS activation, how it is detected remains mysterious. Vancomycin treatment results in the production of peptidoglycan fragments that are sensed by the enterococci PrkC homolog IreK. Thus, IreK could act as the activator of the VanRS system. We have both in vitro and in vivo data suggesting that threonine phosphorylation of VanS, the histidine kinase, is required for full induction of the VanRS operon. We find that application of staurosporine, an IreK inhibitor, blocks induction of the VanRS operon in response to vancomycin. Thus, the convergent action of eSTKs and TCS may have important clinical ramifications as well as implications for understanding the physiological regulation of TCS in bacteria. Lab: Jonathan Dworkin ____________ 11 BLAST XII _____ Mon. Evening Session LINKEDIN – FINDING CONNECTIONS: A RESPONSE REGULATOR LINKS THE Frz CHEMOSENSORY SYSTEM TO THE MglA/MglB GTPase/GAP MODULE TO REGULATE POLARITY OF MOTILITY SYSTEMS IN M. xanthus Keilberg, Daniela, Wuichet, Kristin, Drescher, Florian, Søgaard-Andersen, Lotte Max Planck Institute for Terrestrial Microbiology, Marburg, 35043 Germany M. xanthus cells possess two independent gliding machines: the adventurous (A) system and the social (S) system. A-motility allows movement of single cells, while S-motility is cell-cell contact dependent. Mutations which abolish both of these systems lead to non-motile cells, while mutations in only one of them allow bacterial cells to move by means of the intact system. While S-motility depends on extension and retraction of Type-4-pili powered by ATP, A-motility requires focal adhesion complexes connected to a MotAB-like motor powered by proton motive force. Furthermore, M. xanthus cells can reverse the direction of movement, which is accompanied by an inversion of the polarity of both motility systems. Reversals are induced by the Frz chemosensory system, acting upstream of a small GTPase, MglA and its cognate GTPase activating protein, MglB. RomR is a response regulator, which is required for motility and reversals and localizes in a bipolar asymmetric pattern with a large cluster at the lagging cell pole. We found that RomR can directly interact with MglA and MglB. Furthermore, RomR, MglA and MglB affect the localization of each other in all pair-wise directions suggesting that RomR stimulates motility by promoting correct localization of MglA and MglB in MglA/RomR and MglB/RomR complexes at opposite poles. Moreover, localization analyses suggest that the two RomR complexes mutually exclude each other from their respective poles. We further show that RomR interfaces with FrzZ, the output response regulator of the Frz chemosensory system, to regulate reversals. Thus, RomR serves at the interface to connect a classic bacterial signalling module (Frz) to a classic eukaryotic polarity module (MglA/MglB). This modular design is paralleled by the phylogenetic distribution of the proteins suggesting an evolutionary scheme in which RomR was incorporated into the MglA/MglB module to regulate cell polarity followed by the addition of the Frz system to dynamically regulate cell polarity. Lab: Lotte Søgaard-Andersen _______ 12 BLAST XII _____ Mon. Evening Session RESISTANCE OF CLOSTRIDIUM difficile TO BACTERIAL ANTIMICROBIAL PEPTIDES IS MEDIATED THROUGH A DISJOINED TWO-COMPONENT SYSTEM THAT RESPONDS TO MULTIPLE SUBSTRATES Jose M. Suárez, Adrianne N. Edwards and S. M. McBride* Department of Microbiology and Immunology, Emory University School of Medicine, Atlanta, GA U.S.A. Clostridium difficile is the major causative agent of antibiotic-associated diarrhea. To persist in the intestine the bacteria must cope with host innate defenses, including cationic antimicrobial peptides (CAMPs) made by the host and the indigenous microbial flora. We previously showed that C. difficile responds to CAMPs by inducing expression of genes that lead to CAMP resistance. The first such C. difficile gene cluster identified (cprABCK) encodes an ABC-type transporter and a sensor kinase typical of bacterial two-component systems (TCS). These genes are highly related to genes that encode immunity proteins in lantibiotic producer species. Using qRT-PCR and directed mutagenesis, we showed that the transporter and kinase genes are directly involved in resistance to CAMPs. However, no apparent response regulator is encoded in the vicinity of cprABCK. Here, we identify an orphan response regulator, CD3320, that encodes the response regulator that controls the cprABCK genes in response to antimicrobial peptides. We cloned the CD3320 gene and upstream region in a vector that replicates in C. difficile and demonstrated that cells adapted faster to CAMPs and expression of the cprABCK operon increased. By expressing these regulators and a Pcpr::lacZ reporter fusion in a heterologous host, Bacillus subtilis, we were able to show how CprK and CD3320 (renamed CprR) interact to activate expression of the cpr operon. In addition, we were able to identify putative residues of both the lantibiotics and of CprK that are involved in sensing and activation of this system. These results demonstrate how the Cpr ABC-transporter and regulators allow for substrate recognition that is broader than the typicallly narrow specificity of lantibiotic producer systems, resulting in resistance to multiple bacterial CAMPs through a single mechanism. The cpr lantibiotic immunity system represents a novel antimicrobial peptide resistance mechanism that has not been described in any other species. Lab: Shonna McBride ______________ 13 BLAST XII _____ Mon. Evening Session THE WalKR SYSTEM CONTROLS MAJOR STAPHYLOCOCCAL VIRULENCE GENES AND IS INVOLVED IN TRIGGERING THE HOST INFLAMMATORY RESPONSE Aurélia Delauné1, Sarah Dubrac1, Charlène Blanchet2, Olivier Poupel1, Ulrike Mäder3, Aurélia Hiron1, Aurélie Leduc4, Catherine Fitting2, Pierre Nicolas4, Jean-Marc Cavaillon2, Minou Adib-Conquy2 and Tarek Msadek1 1 Institut Pasteur, Biology of Gram-Positive Pathogens, Department of Microbiology, Paris, France 2 Institut Pasteur, Cytokines and Inflammation, Department of Infection and Epidemiology, Paris, France 3 Interfaculty Institute for Genetics and Functional Genomics, Department for Functional Genomics, Ernst Moritz Arndt University, Greifswald, Germany 4 Mathématique, Informatique et Génome, INRA, UR1077, Jouy-en-Josas, France The WalKR two-component system is essential for viability of Staphylococcus aureus, playing a central role in controlling cell wall metabolism. We produced a constitutively active form of the WalR response regulator in S. aureus through a phosphomimetic amino acid replacement (D55E). The strain displayed significantly increased biofilm formation and α-hemolytic activity. Using transcriptome analysis, we showed that transcription of 108 genes was increased in the presence of the walRc allele, including eleven involved in cell wall metabolism. Several of the upregulated genes encode major virulence proteins involved in host matrix interactions (efb, emp, fnbA, fnbB), cytotoxicity (hlgACB, hla, hlb), innate immune defense evasion (scn, chp, sbi) and signal transduction (saePQRS). For many of these, we showed that control by WalR occurs through increased activation of the SaeSR twocomponent system. The impact on pathogenesis of varying cell envelope dynamics through WalR activity was studied using a murine sepsis model, showing that strains producing constitutively active WalRc are strongly diminished in their virulence. We show that this is due to early and enhanced triggering of the host inflammatory response, associated with higher levels of released peptidoglycan fragments. Indeed, neutrophil recruitment and proinflammatory cytokine production (TNF-α, IL-6, IL-1β) were significantly increased when the walRc allele was expressed, leading to enhanced bacterial clearance. Taken together, our results indicate that WalKR play an important role in virulence and eliciting the host inflammatory response by controlling autolytic activity, suggesting that the precise finetuning of WalKR activity may be critical in the switch between S. aureus commensal and pathogenic lifestyles. References: 1. Delauné, A., S. Dubrac, C. Blanchet, O. Poupel, U. Mader, A. Hiron, A. Leduc, C. Fitting, P. Nicolas, J. M. Cavaillon, M. Adib-Conquy, and T. Msadek. 2012. The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infect. Immun. 80:3438– 3453. 2. Delauné, A., O. Poupel, A. Mallet, Y. M. Coic, T. Msadek, and S. Dubrac. 2011. Peptidoglycan crosslinking relaxation plays an important role in Staphylococcus aureus WalKR-dependent cell viability. PLoS One 6:e17054. Lab: Tarek Msadek ________________ 14 BLAST XII _____ Mon. Evening Session WHAT SEQUENCE DIVERSITY TEACHES US ABOUT BACTERIAL SIGNALING Hendrik Szurmant1, Alexander Schug2, Martin Weigt3 1 Department of Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, California, USA; 2 Steinbuch Centre for Computing, Karlsruhe Institute of Technology, Karlsruhe, Germany; 3 Laboratoire de Génomique des Microorganismes, Université Pierre et Marie Curie, Paris, France Two-component signal transduction mechanisms are woven in the fabric of cellular regulation in bacteria, plants and lower eukaryotic forms of life. These innate systems play a wide variety of roles from sensing the environment to controlling the cell cycle, cellular behavior and development. Twocomponent systems comprise a sensor protein, the histidine kinase and an output protein, the response regulator, which typical serves as a transcription factor. Since the completion of the first bacterial genome-sequencing project in 1995 more than 3000 bacterial genomes have been sequenced in part thanks to the rapid development of high throughput sequencing technology. Encoded by these genomes are more than 105 two-component system proteins, whose sequences have been sampled in evolutionary space to accomplish perfect functionality. Hidden within this sequence variation is a wealth of information that can inform about the molecular mechanisms underlying the signal transduction cascade, namely how the kinase modulates its activity in response to signals, how the kinase identifies its target response regulator from other response regulators of identical structure in the cell and how the kinase physically interacts with its target response regulator. This talk will discuss a computational method, called Direct Coupling Analysis that we developed in order to answer the questions of molecular activation and interaction specificity in two-component signaling. Going beyond the field of signal transduction, our technology proves promising in identifying protein interaction surfaces and protein complex structures formed by interacting proteins. Lab: Hendrik Szurmant 15 BLAST XII _____ Tue. Morning Session STRUCTURAL CHANGES OF BACTERIAL CHEMORECEPTOR ARRAYS UNDERLIE DIFFERENT ACTIVATION STATES Ariane Briegel1, Peter Ames2, James C. Gumbart3, Catherine Oikonomou1, John S. Parkinson2 and Grant J. Jensen3 1 California Institute of Technology, 1200 E. California Blvd., Pasadena, CA 91125 2 University of Utah, 257 South 1400 East Salt Lake City, UT 84112 3 Argonne National Laboratory, 9700 S. Cass Avenue, Argonne, IL 60439 Chemotactic bacteria utilize a highly sensitive and adaptable sensory system to swim towards attractants and away from repellents. A polar, highly organized sensory patch of transmembrane receptor proteins detects changes in nutrient concentrations. Attractants and repellents bind to the sensory domains of the receptors, thereby regulating the activity of the histidine kinase CheA, which phosphorylates a soluble messenger protein. This messenger protein in turn diffuses through the cytoplasm to the flagellar basal body, where it modulates the direction of flagellar rotation. EM maps generated by sub-volume averaging of wild-type chemoreceptor arrays in the adapted state, together with a new crystal structure, revealed the protein arrangement of the core chemotaxis proteins in the array. The trimers of receptor dimers lie at each vertex of a hexagon, arranged such that one dimer points towards the center of the ring. The receptors surround rings of alternating regulatory subdomains (P5) of CheA and the coupling protein CheW. The CheA kinase subdomains (P4) project toward the cytoplasm and away from the receptors, while the dimerization subdomains (P3) link neighboring rings together to form a stable, extended lattice. To get insight to the structural changes of the arrays during activation, we have now analyzed sub-volume averages from various E.coli mutants with chemoreceptor arrays locked in either constantly CheA-activating or inactivating states. Our results indicate that the chemoreceptors remain in a hexagonal arrangement with identical center-to-center spacing in all activation states. The EM maps do reveal a difference in the CheA packing, however: in the physiological CheA inactive state, both P1 and P2 subdomains of the CheA dimer are stably packed in a ‘rudder-like’ density beneath two adjacent receptor trimers. In the constantly CheA activating states, this rudder-like density is less prominent, suggesting more dynamic P1 and P2 subdomains. Lab: Grant Jensen ________________ 16 BLAST XII _____ Tue. Morning Session FORMATION AND SEGREGATION OF THE CYTOPLASMIC CHEMOSENSORY CLUSTER IN RHODOBACTER sphaeroides Christopher Jones, Nelly Dubarry, Mark Roberts and Judith Armitage Department of Biochemistry, University of Oxford, South Parks Road, Oxford, UK, OX1 3QU Rhodobacter sphaeroides, like many bacterial species, has a complex chemosensory network containing multiple homologues of each component of the paradigm chemosensory pathway of Escherichia coli. In R. sphaeroides this network is located to spatially distinct regions of the cell with proteins making complete pathways found at the poles with membrane spanning chemoreceptors and at around midcell with cytoplasmic chemoreceptors. New clusters develop during cell elongation so that upon cell division each daughter cell inherits one polar cluster and one cytoplasmic cluster. Recent work has shown that the segregation of the cytoplasmic cluster is dependent on a ParA ATPase homologue PpfA. Par systems are used by bacteria to segregate chromosomes and plasmids during cell division, and consist of ParA proteins which polymerise and ParB proteins which associate with DNA and activate ParA depolymerisation. An increasing number of ParA homologues have been shown to be involved in partitioning protein complexes. PpfA is encoded within the operon encoding the cytoplasmic chemosensory pathway of R. sphaeroides and interacts with one of the cytoplasmic chemoreceptors TlpT with the N-terminus of this protein acting as the ParB. Together these proteins use the chromosome to segregate the chemosensory clusters on division. PpfA localises over the chromosome, but forms foci around the cytoplasmic chemosensory cluster. Using two colour fluorescence time lapse microscopy cytoplasmic cluster components are shown to co-localise with PpfA foci throughout the cell cycle. Static images suggested that the cluster was positioned at midcell, and then once a new cluster was formed they were positioned at ¼ and ¾ positions but current results show that the chemosensory cluster and associated PpfA foci are highly mobile within the cell. Results will be presented on the pattern of chemosensory cluster positioning through the cell cycle and the possible mechanisms used for forming and segregating the chemosensory clusters. Lab: Judith Armitage ______________ 17 BLAST XII _____ Tue. Morning Session NETWORK-LEVEL PROPERTIES OF SALMONELLA TYPHIMURIUM CHEMOTAXIS Milena D. Lazova1, Bob Rosier1, Filippo Menolascina2, Roman Stocker2, Thomas S. Shimizu1* FOM Institute for Atomic and Molecular Physics (AMOLF), Amsterdam, The Netherlands. 2 Ralph M. Parsons Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA 1 Input-output relationships of the bacterial chemotactic response have been characterized in the model species Escherichia coli, using in vivo experiments (1-3) and theoretical modeling (4, 5). However, genomic studies have revealed that the E. coli system represents a streamlined example of bacterial chemotaxis: the inferred topologies of the chemotactic signaling networks demonstrate a great diversity across prokaryotes (6, 7). The dynamical features of chemotactic signaling, mediated by these varied topologies remains largely unknown. We examine here the chemotaxis system of the pathogen Salmonella typhimurium. It is a close relative to E. coli: chemotactic genes of S. typhimurium and E. coli are similar enough that deletions of chemotactic genes in one species are readily complemented by the orthologous genes from the other (8). However, differences in the chemoreceptor structure and chemoreceptor types (9, 10) suggest that the chemotactic signaling has diverged between the two species. We have used a quantitative physiology approach to characterize the chemotactic signaling dynamics of S. typhimurium in response to different chemoeffectors. Real-time fluorescence resonance energy transfer (FRET) measurements (11), combined with time-varying stimuli allowed us to characterize the chemotactic transfer functions. We have identified conserved features of E. coli and S. typhimurium chemotactic signaling, and highlighted differences in the receptor-kinase responses, as well as in the adaptation kinetics and frequency response. We probed how these physiological differences influence the performance of cells executing chemotaxis in controlled microenvironments, by observing the migration of bacterial populations in steady gradients created in microfluidic devices (12). We find striking differences between the transient drift velocity, as well as the steady state behavior of S. typhimurium and E. coli populations, which we explain in terms of the underlying control physiology. 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. Sourjik V & Berg HC (2002) Proc Natl Acad Sci U S A 99, 123-127. Shimizu TS, Tu Y, & Berg HC (2010) Mol Syst Biol 6, 382. Lazova MD, Ahmed T, Bellomo D, Stocker R, & Shimizu TS (2011) Proc Natl Acad Sci U S A 108, 13870-13875. Tu Y, Shimizu TS, & Berg HC (2008) Proc Natl Acad Sci U S A 105, 14855-14860. Jiang L, Ouyang Q, & Tu Y (2010) PLoS Comput Biol 6, e1000735. Szurmant H & Ordal GW (2004) Microbiol Mol Biol Rev 68, 301-319. Wuichet K & Zhulin (2010) IB Sci Signal 3, ra50. DeFranco AL, Parkinson JS, & Koshland DE, Jr. (1979) J Bacteriol 139, 107-114. Biemann HP & Koshland DE, Jr. (1994) Biochemistry 33, 629-634. Yamamoto K & Imae Y (1993) Proc Natl Acad Sci U S A 90, 217-221. Sourjik V, Vaknin A, Shimizu TS, & Berg HC (2007) Methods Enzymol 423, 365-391. Ahmed T, Shimizu TS, & Stocker R (2010) Nano Lett 10, 3379-3385. Lab: Tom Shimizu _________________ 18 BLAST XII _____ Tue. Morning Session HOW HAMP DOMAINS AND METHYLATION BUNDLES CONTROL STIMULUS RESPONSE AND SENSORY ADAPTATION IN CHEMORECEPTORS Runzhi Lai, Peter Ames and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112 In transmembrane chemoreceptors of the methyl-accepting chemotaxis protein (MCP) family, a HAMP domain at the cytoplasmic face of the membrane accepts stimulus signals from the periplasmic sensing domain and converts them to conformational changes that modulate activity of a CheA kinase at the hairpin tip of the MCP molecule. The dynamic bundle model proposes that chemoreceptor signaling involves opposed shifts in packing stability of the four-helix HAMP bundle and the adjacent methylation helix (MH) bundle, which contains the covalent modification sites for sensory adaptation. To test aspects of the dynamic bundle model, we measured the signaling behaviors of serine (Tsr) receptors that had structural alterations in the HAMP and/or MH bundles, using the FRET-based in vivo kinase assay pioneered by Sourjik & Berg (2002). The Tsr homodimer contains five modification sites in each subunit: E residues correspond to unmethylated sites; Q residues mimic methylated E sites. In a host strain lacking the CheR and CheB adaptation enzymes, we measured the serine sensitivities of Tsr variants that had all possible combinations of E and Q residues at sites 1-4, in combination with an E at site 5. We observed an exponential relationship between the K 1/2 for serine and the number of Q residues in the receptor, with little site specificity. This finding implies that each methylated (or Q) position makes an equivalent free energy contribution to transitions between kinase-on and kinase-off output states. Amino acid replacements in the HAMP domain can also alter Tsr response sensitivity by shifting the equilibrium between kinase-on and kinase-off output states. For example, Tsr-S255A is shifted toward the off state (K 1/2 = 2.7 µM, compared to 17 µM for wild-type in the QEQE modification state), whereas S255G (K 1/2 = 1.6 mM) is greatly shifted toward the on state. Yet, both mutant receptors mediate robust chemotactic responses in adaptation-proficient cells, suggesting that the adaptation system can offset S255 lesions by adjusting receptor modification state. Consistent with this interpretation, we found that increased Q:E ratios at the methylation sites shifted S255A to higher K 1/2 values (e.g., QQQE = 7 µM), whereas reduced Q:E ratios shifted S255G to lower K 1/2 values (e.g., EEEE = 33 µM). Despite their opposite shifts in response sensitivity, both S255A and S255G suppress the signaling defects of I229A and I229S lesions, which alone shift output to the kinase-on state. For example, in an adaptation-proficient background K 1/2 of I229S = 250 µM, whereas I229S+S255A = 11 µM and I229S+S255G = 27 µM). I229 and S255 reside in the same packing layer of the four-helix HAMP bundle. Structural interactions between these positions could restore response sensitivity by matching HAMP packing stability to the opposing structural effects of receptor methylation, thereby poising the receptor to respond to small attractant stimuli. This and other signaling implications of the structural interplay between the HAMP and MH bundles will be discussed in the talk. Lab: John S. Parkinson ____________ 19 BLAST XII _____ Tue. Morning Session TESTING AND REFINING THE RECEPTOR-CheA REGULATORY DOMAIN INTERFACE IN FUNCTIONAL, MEMBRANE-BOUND ARRAYS USING DISULFIDE MAPPING AND TAM-IDS Kene N. Piasta and Joseph J. Falke Department of Chemistry and Biochemistry, University of Colorado at Boulder, Jennie Smoly Caruthers Biotechnology Building, 596 UCB, Boulder, CO 80309 The bacterial chemotactic signaling array is composed of repeating core contacts between a receptor trimer-of-dimers, a homodimeric CheA kinase, and the CheW coupling protein. CheA incorporation into the array activates its kinase activity, while receptor-mediated attractant signals inhibit kinase activity. Structures of receptor fragments, CheA, CheW, and various combinations have been solved, but a molecular view of protein-protein contacts in the active, membrane-bound signaling array has proved elusive. The CheA regulatory domain, P5, shares an SH3-like domain fold with CheW, and NMR findings from the Dahlquist lab indicate P5 and CheW possess similar receptor binding sites. Two recent crystal structures solved in the Crane Lab reveal two distinct possibilities for the receptor-SH3-like domain interface. However, currently available information does not ascertain whether one of these two interfaces, or a different interface, most accurately describes the native contacts in chemotactic arrays. Here we construct two models of the receptor-P5 interface based on the two Crane lab structures, and then we test the predictions of each model in vitro utilizing a disulfide mapping approach in the active, membrane-bound signaling array. This approach can also reveal signaling events that move through the receptor-P5 interface as changes in disulfide bond formation rate. Finally, we utilize Tryptophan and Alanine Mutagenesis to Identify Docking Sites (TAM-IDS) to test predicted receptor-P5 contacts in vivo. Together these approaches are yielding an improved model of receptor-CheA-CheW contacts in the active, membrane-bound signaling array as well as providing new insights into signal transduction from receptor to CheA. Lab: Joseph Falke _________________ 20 BLAST XII _____ Tue. Morning Session DIFFERENCES BETWEEN SIGNALING STATES OF CHEMOTAXIS RECEPTORS REVEALED BY HYDROGEN EXCHANGE MASS SPECTROMETRY Seena S. Koshy, Stephen J. Eyles, Robert M. Weis, Lynmarie K. Thompson Department of Chemistry, University of Massachusetts, Amherst MA 01003 Bacterial chemotaxis receptors provide the sensory input needed to direct swimming towards favorable environments. Ligand binding to the receptor periplasmic domain modulates activation of a kinase CheA bound at the cytoplasmic tip of the receptor. Receptors operate in complexes with CheA and a coupling protein CheW that together form extended arrays in the membrane. Understanding the differences in conformation and dynamics between the kinase-activating and kinase-deactivating signaling states of the receptor will provide insight into the mechanism of transmembrane signaling. We have used hydrogen exchange mass spectrometry to probe the differences between the kinase-activating and kinase-deactivating signaling states of the receptor cytoplasmic domain assembled on membrane vesicles in functional complexes with CheA and CheW. Local exchange measurements reveal several differences between these states, including differences in the exchange pattern (EX1 vs. EX2) and in the fraction that are protected from exchange at long time (16 hours). Preliminary analysis shows that peptides corresponding to the adaptation region undergo EX1 exchange (long-lived unfolded state with complete exchange during each unfolding event) in the kinase-activating state and EX2 exchange (short-lived unfolded state with partial exchange) in the kinase-deactivating state. This suggests the adaptation region is destabilized in the kinase-activating state. Peptides corresponding to the signaling domain cytoplasmic tip of the receptor show greater protection from exchange in the kinase-activating state, which could be due to changes in interactions with CheA and CheW. This hydrogen exchange mass spectrometry approach is a promising means of detecting changes in structure and dynamics of a functional membrane-bound, multi-protein complex. This research supported by GM 47601, GM085288, and a Fellowship to Seena Koshy from the University of Massachusetts as part of the Chemistry-Biology Interface Training Program (NRSA T32 GM08515). Lab: Lynmarie Thompson __________ 21 BLAST XII _____ Tue. Morning Session PROLONGED STIMULI ALTER THE BACTERIAL CHEMOSENSORY CLUSTERS Vered Frank and Ady Vaknin The Racah Institute of Physics, The Hebrew University, Jerusalem 91904, Israel. While the principal structure of these receptor arrays is becoming clear, their dynamic nature and operation are not yet understood. By measuring the fluorescence-polarization of tagged receptorclusters in live Escherichia coli cells, we show that during a stimulus, the packing of the receptors in clusters slowly changes. Consistent with these physical changes, we find that the regulation of kinase activity by these clusters slowly evolves, altering the effective cooperativity of the response. Timelapse fluorescence imaging indicated that despite these changes, the clusters do not generally dissociate upon ligand binding. These data suggest that prolonged stimuli induce changes in receptor packing within chemosensory clusters, which, in turn, alters the coupling between the receptors, and thus leading to non-stationary signal transduction. Lab: Ady Vaknin __________________ 22 BLAST XII _____ Tue. Morning Session THE SENSING AND SIGNALING ROLES OF MULTIPLE PAS-HEME DOMAINS IN Aer-2 TYPE CHEMORECEPTORS Magi B. Ishak Gabra, Kahryl G. Bennett, Adwoa O. Wiafe, Andrew J. Hong, Lana S. Haddad, Mark S. Johnson and Kylie J. Watts Division of Microbiology and Molecular Genetics, Loma Linda University, Loma Linda, CA, 92350 Aer-2 is a soluble PAS-HAMP chemoreceptor that is found in a number of pathogens including Pseudomonas aeruginosa (PaAer-2) and Vibrio cholerae (VcAer-2). We previously showed that PaAer2 binds heme and can signal in response to various oxy-gases. The recently solved PaPAS structure revealed a uniquely placed histidine residue (H234 on E5η) that coordinates heme, and an unusual tryptophan residue (W283 on I8β) that may stabilize heme-oxygen binding. Unlike PaAer-2, which contains a single PAS domain, VcAer-2 contains two PAS domains. Here we show that both VcPAS domains bind heme and have spectra similar to PaPAS. The hemecoordinating His and oxygen-stabilizing Trp residues of PaPAS are both conserved in VcPAS-1 (H101, W151) and VcPAS-2 (H226, W276). We found that the VcAer-2 receptor signals only in response to oxygen and not in response to other oxy-gases. To identify which of the two VcPAS domains sense oxygen and to determine the importance of the His and Trp residues for Aer-2 function, we engineered amino acid substitutions into both truncated and full-length Aer-2 receptors for spectral and behavioral studies. The His substitutions, H234A in PaPAS, H101A in VcPAS-1, and H226A in VcPAS-2, each lowered the extent of heme bound to each PAS domain (24%, 15% and 2% of wild-type heme, respectively), indicating that the E5η histidine coordinates heme in Aer-2 type PAS domains. However, full-length receptors with heme-binding defects in PaPAS or VcPAS-2 retained partial function. In contrast, the heme-binding defect in VcPAS-1 caused a stronger response to oxygen, suggesting that VcPAS-1 is not required for oxygen sensing and may in part function to inhibit VcPAS-2 signaling. The Trp substitutions, W283L in PaPAS, and W276L in VcPAS-2, yielded aberrant PAS-heme peptides that by spectral analyses appeared to bind imidazole so tightly it was not removed by dithionite or by dialysis. The bound imidazole prevented the association of oxygen or carbon monoxide. Thus, the I8β tryptophan may be required for oxygen sensing by PaPAS and VcPAS-2. Notably, the Trp substitutions in PaPAS or VcPAS-2 created signal-on receptors, a state that typically requires gas binding. In contrast, the Trp substitution, W151L in VcPAS-1, significantly inhibited the ‘on state’ of VcAer-2, even though VcPAS-1 was still able to bind oxygen. This supports our hypothesis that VcPAS1 does not signal oxygen binding, and further suggests that although VcPAS-1 can bind oxygen, it does not stabilize its binding via the Trp-mediated mechanism that is used by PAS-heme ‘sensing’ domains. The results of this study have begun to reveal the similarities and differences between Aer-2 receptors from different species and the differences between PAS-heme domains from Aer-2 and those of other well-studied proteins such as FixL or DOS. Further studies are needed to elucidate the unique signaling mechanisms used by Aer-2 type receptors. Lab: Kylie Watts 23 BLAST XII _____ Tue. Evening Session FUNCTIONS OF PROTEUS MIRABILIS FliL IN SWARMING AND RESPONDING TO SURFACE VISCOSITY Yi-Ying Lee and Robert Belas Department of Marine Biotechnology, University of Maryland, Baltimore County, and Institute of Marine and Environmental Technology, 701 East Pratt Street, Baltimore, Maryland 21202 Proteus mirabilis is a dimorphic Gram-negative enterobacterium and well-known for its ability of move over agar surfaces by flagellar-dependent swarming motility. In liquid culture, P. mirabilis cells are uniformly 1.5-2.0 μm rods with 4-6 flagella, called swimmer cells. When P. mirabilis encounters a highly viscous environment or a solid surface, swimmer cells differentiate into elongated (10-80 μm), highly flagellated swarmer cells that lack of septa and contain multiple nucleoids. The bacteria detect a surface by monitoring the rotation of their flagellar motors. Conditions that prevent rotation of swimmer cell flagella trigger production of swarmer cells. This process involves a ubiquitous but functionally enigmatic flagellar basal body-associated protein called FliL. fliL is the first gene in an operon (fliLMNOPQR) that encodes proteins of the flagellar rotor switch complex and flagellar export apparatus. We used a fliL knock out mutant to gain further insight into the function of FliL. Loss of FliL results in cells that cannot swarm (Swr-), but do swim (Swm+), and in liquid produces cells that look like wild-type swarmer cells, “pseudoswarmer cells”, which are elongated, contain multiple, evenly spaced nucleoids, and lack septa. Unlike swarmer cells, pseudoswarmer cells are not hyperflagellated due to reduced expression of flaA (the gene encoding flagellin), despite an increased transcription of both flhD and fliA, two positive regulators of flagellar gene expression. We found that defects in fliL prevent viscosity-dependent sensing of a surface and viscosity-dependent induction of flaA transcription. Studies with fliL cells unexpectedly revealed a requirement of the fliL promoter, fliL coding region, and a portion of fliM DNA to complement the Swr- phenotype. The data support a dual role for FliL, as a critical link in sensing a surface and in the maintenance of flagellar rod integrity. Lab: Robert Belas _________________ 24 BLAST XII _____ Tue. Evening Session THE AAA+ PROTEASE LonA REGULATES SwrA LEVELS AND SWARMING MOTILITY IN BACILLUS subtilis Sampriti Mukherjee*, Joyce E. Patrick and Daniel B. Kearns Indiana University – Bloomington, IN, USA Bacillus subtilis exhibits robust swarming motility atop semi-solid media (1). Swarming motility is facilitated by secretion of a lipopeptide surfactant (surfactin) and powered by rotating flagella. Flagella are complicated, membrane-anchored, proteinaceous nanomachines that propel bacteria in their milieu. Each flagellum is built from over 30 proteins and has three architectural domains: basal body, hook and filament. Swarming motility is correlated with an apparent increase in flagellar number in swarm cells when compared to cells propagated in liquid media (swim cells). The mechanism and relevance of hyper-flagellation during swarming motility in B. subtilis is poorly understood. One way in which flagellar number could be increased is by an increase in the expression of the flagella and chemotaxis operon (fla/che) that encodes for the proteins that comprise the hook and basal body, and the alternate sigma factor σD required for the expression of the filament protein. The putative master regulatory protein SwrA increases transcription of the fla/che operon and is required for swarming in B. subtilis (2). Here we find that the increase in flagella number was correlated with an increase in the intracellular level of the SwrA. SwrA protein levels significantly increase in swarm cells when compared to swim cells and overexpression of SwrA results in hyperflagellation in swim cells. However, swrA gene transcription and translation do not change between swarm and swim cells implying post-translational regulation on SwrA. Swarming motility is regulated by Lon protease of the AAA+ protease family in diverse species such as Proteus mirabilis and Vibrio parahaemolyticus (3). We find that a lonA mutant exhibits increase in SwrA protein levels in swim cells. We therefore hypothesize that LonA potentially targets SwrA in swim cells but under conditions favorable to swarming LonA is inhibited and SwrA levels accumulate to allow hyper-flagellation and swarming motility. Thus although the master regulators of flagella biogenesis are distinct in different bacteria, the mechanism to regulate them appears to be similar. 1. Kearns, D.B., and Losick, R. (2003) Swarming motility in undomesticated Bacillus subtilis. Mol Microbiol 49:581-590. 2. Kearns, D.B., Chu, F., Rudner, R., and Losick, R. (2004) Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol Microbiol 52:357-369. 3. Patrick, J.E., and Kearns, D.B. (2012) Swarming motility and the control of master regulators of flagellar biosynthesis. Mol. Microbiol. 83:14-23. Lab: Daniel Kearns ________________ 25 BLAST XII _____ Tue. Evening Session HELICAL FLOW OF SURFACE PROTEIN REQUIRED FOR FLAVOBACTERIUM GLIDING MOTILITY Daisuke Nakane, Keiko Sato, Hirofumi Wada, Mark J. McBride and Koji Nakayama Department of Molecular Microbiology and Immunology, Graduate School of Biomedical Sciences, Nagasaki University, 852-8588 Nagasaki, Japan Cells of Flavobacterium johnsoniae and of many other members of the phylum Bacteroidetes exhibit rapid gliding motility over surfaces by a unique mechanism. These cells do not have flagella or pili, and instead rely on a novel motility apparatus comprised of Gld and Spr proteins. SprB, a 669 kDa cell-surface adhesion, is required for efficient gliding. SprB was visualized by electron microscopy as thin 150 nm long filaments extending from the cell surface. Fluorescence microscopy revealed movement of SprB proteins toward the poles of the cell at approximately 2 μm/s. The fluorescent signals appeared to migrate around the pole and continue at the same speed toward the opposite pole along an apparent left-handed helical closed loop. Movement of SprB, and of cells, was rapidly and reversibly blocked by the addition of CCCP, which dissipates the proton gradient across the cytoplasmic membrane. In a gliding cell, some of the SprB protein appeared to attach to the substratum. The cell body moved forward and rotated with respect to this point of attachment. Upon reaching the rear of the cell, the attached SprB was often released from the substratum, and apparently recirculated to the front of the cell along a helical path. The results suggest a model for Flavobacterium gliding, supported by mathematical analysis, in which adhesins such as SprB are propelled along a closed helical loop track, generating rotation and translation of the cell body. Lab: Koji Nakayama _______________ 26 BLAST XII _____ Tue. Evening Session BACTERIAL TRANSITIONS: OPTIMIZATION OF BIOLOGICAL AND PHYSICAL FACTORS BY Pseudomonas aeruginosa DURING SWARMING Morgen Anyan, Huijing Du, Oleg Kim, Zhiliang Xu, Mark Alber, and Joshua Shrout University of Notre Dame, 156 Fitzpatrick Hall, Notre Dame, IN 46556 USA Pseudomonas aeruginosa is one of several bacteria that swarms in groups. Often, but not always, P. aeruginosa forms branched tendril patterns during swarming; we find that cells propagate as high density waves that spread symmetrically as rings within swarms towards the extending tendrils. These marvelous tendril patterns only form when P. aeruginosa produces sufficient rhamnolipid at advancing swarm edge. During optimal swarm conditions we find that both cells and rhamnolipid propagate as high density waves that move symmetrically as rings within swarms towards the extending tendrils. Simulations conducted using a multi-scale model suggest that wave propagation and tendril formation depend upon competition between the changing viscosity of the bacterial liquid suspension and the liquid film boundary expansion caused by Marangoni forces. We have found that P. aeruginosa swarms spread due to oscillations between biological and physical factors. Essentially, we find that P. aeruginosa efficiently colonizes surfaces by controlling the physical forces responsible for expansion of thin liquid films and by propagating towards the tendril tips. We find that type IV pili interfere with optimal flagellar swarming; a hyperpiliated mutant (pilU) is swarm deficient. Alternatively, for bacteria devoid of type IV pili, these bacteria spread more easily than the wild type. A pilA mutant shows improved swarming and this phenotype can be complemented with PilA expressed in trans. We are studying this phenotype as part of our combined experimentalmodeling approach to study how the cells optimally align and coordinate during swarming. Lab: Joshua Shrout _______________ 27 BLAST XII _____ Tue. Evening Session STRUCTURE OF PROTEINS INVOLVED IN MYCOPLASMA mobile GLIDING REVEALED BY ELECTRON MICROSCOPY AND HIGH SPEED ATOMIC FORCE MICROSCOPY (AFM) Yuhei Tahara1, Noriyuki Kodera2, Toshio Ando2, Makoto Miyata1 1 Department of Biology, Graduate School of Science, Osaka City University, 3-3-138 Sugimoto Summiyoshiku, Osaka-shi, Japan 2 Department Physics, Kanazawa University, 920-1192 Kakuma-machi, Kanazawa, Japan Mycoplasma mobile, a fish pathogen, glides on solid surfaces with a unique mechanism. The gliding machinery is assembled on the protrusion mainly from three huge proteins, designated Gli521, Gli349, and Gli123, weighing 521k, 349k, 123kDa, respectively. In this study, we analyzed molecular shapes of these proteins essential for gliding, by negative-staining electron microscopy and high speed AFM. Gli521 has the role of “crank”, transmitting movements from motor to leg. Previously, rotary shadowing electron microscopy showed that the Gli521 molecule is shaped, “ ? ” ,120 nm long, and featured with a “hook” at the C terminus and an “oval” at the N terminus. In the present study, we have shown that the molecule is shaped, “ω”, and featured with a central hinge, based on better resolution of negative-staining electron microscopy. Analyses by high speed AFM have shown that the central hinge bends to only one direction. Gli349 is shaped, “♪ “, 100 nm long and function as “leg”. Analyses by high speed AFM have shown that the Gli349 molecule consists of a large and a small domains connected by a thin elastic filament. Gli123 has the role of “mount” essential for Gli349 and Gli521 subcellular localization. Analyses by electron microscopy have shown that the molecule of this protein is an ellipse of 12.5 nm and 15 nm axes, featured with a small protrusion and two dimples, reminiscent of “Japanese covered bowl”. These results give us better images for the machinery and the mechanism of M. mobile gliding. Lab: Makoto Miyata ________________ 28 BLAST XII _____ Tue. Evening Session BACTERIOPHYTOCHROME 1 AND LOV-HK WORK IN A SIGNALING NETWORK TO REGULATE SWARMING MOTILITY OF PSEUDOMONAS SYRINGAE STRAIN B728a Liang Wu, Regina S. McGrane and Gwyn A. Beattie Department of Plant Pathology and Microbiology, Iowa State University, Ames, IA, 50010 Photosensory proteins have long been studied in plants, algae, fungi and photosynthetic prokaryotes. Genome sequencing has indicated the presence of photosensory protein-encoding genes in non-photosynthetic bacteria, including the foliar plant pathogen Pseudomonas syringae. P. syringae has three genes that are predicted to encode two classes of photosensory proteins: the bacteriophytochromes, BphP1 and BphP2, and a LOV protein histidine kinase, LOV-HK. BphP1 is a red/far-red light receptor that uses biliverdin as a chromophore, whereas Lov-HK is a blue light receptor that uses a FMN (flavin mononucleotide)-binding LOV domain to sense light. BphP2 has eluded characterization because of its recalcitrance to purification. The biological roles of bacteriophytochromes and LOV proteins have been identified for a few non-photosynthetic bacterial species; these include light-mediated regulation of pigment production by a bacteriophytochrome in Deinococcus radiodurans, adhesion by LOV-HK in Caulobacter cresentus and Xanthomonas axonopodis, and cellular proliferation by LOV-HK in macrophages by Brucella abortus. The signal transduction pathways for these biological functions are unknown, as are the biological roles and signal transduction pathways for these proteins in P. syringae. Here we report that swarming motility of P. syringae strain B728a is regulated by both BphP1 and LOV-HK. Moreover, we discovered a novel regulatory network in which BphP1 functions to repress swarming motility in response to red/far-red light and blue light, and LOV-HK suppresses this unusual BphP1-mediated blue light repression. Lab: Gwyn Beattie _________________ 29 BLAST XII ____ Wed. Morning Session MULTIPLE AGONISTIC AND ANTAGONISTIC SIGNALS CONTROL THE ACTION OF THE COMPLEX SENSOR KINASE TodS Hortencia Silva-Jiménez, Jesús Lacal, Andreas Busch, M. Eugenia Guazzaroni, Juan Luis Ramos and Tino Krell Department of Environmental Protection, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, C/ Prof. Albareda, 1, 18008 Granada, Spain The TodS/TodT two component system (TCS) controls the expression of the toluene dioxygenase pathway in Pseudomonas putida for the metabolisation of the toxic compounds toluene, benzene and ethylbenzene into Krebs cycle intermediates. The TodS sensor kinase is characterized by a complex architecture and harbors two PAS domains, two transmitter modules and a receiver domain. Microcalorimetric studies of purified TodS have shown that a wide variety of aromatic signal molecules bind to the N-terminal PAS domain with high affinity (1,2). Interestingly, these molecules can be classified as agonists and antagonists (3). Only the binding of agonists (toluene for example) causes an upregulation of the autokinase activity of the N-terminal transmitter module and consequently gene expression in vivo. In contrast, antagonist binding has no effect on the autokinase activity and gene expression. We show that TodS employs an internal phosphorlay prior to the phosphorylation of the TodT of which its phosphorylated form stimulates gene expression (2). Using in vivo gene expression studies we show that the presence of antagonists (o-xylene for example) reduces the magnitude of agonist-mediated gene expression (2). Recent studies show that the activity of other TCSs is also based on the concerted action of agonists and antagonists (4). Crude oil can be understood as a mixture of agonists and antagonists and our data might provide an explanation for the reduced pathway expression observed in biodegradation experiments of complex mixtures such as petrol. In addition to aromatic compounds the TodS autokinase activity in vitro and consequently the in vivo expression is modulated specifically by the oxidative stress agent menadione (5). The amino acids necessary for the sensitivity to menadione have been identified using site-directed mutagenesis. Experiments with varying concentration of toluene and menadione show that the menadione-mediated reduction of TodS autokinase activity dominated over toluene-mediated stimulation. The physiological relevance of sensor kinases with a complex, multidomain architecture is still poorly understood. In this context, this work is one of the first demonstrations that multiple types of signal molecules regulate the action of complex sensor kinases. References: Lacal et al. (2006) Proc. Natl. Acad. Sci. USA 103, 8191-8196 Busch et al. (2009) J. Biol. Chem. 284, 10353-10360 Busch et al. (2007) Proc. Natl. Acad. Sci. USA 104, 13774-13779 Silva-Jiménez et al. (2012) Microb. Biotechnol. 5, 489-500. Silva-Jiménez et al., manuscript in preparation Lab: Tino Krell ____________________ 30 BLAST XII ____ Wed. Morning Session DETERMINING CHEMORECEPTOR-LIGAND INTERACTIONS IN THE LIGHT-ORGAN SYMBIONT VIBRIO fischeri TO UNDERSTAND NICHE SPECIFICITY Caitlin A. Brennan1,*, Run-zhi Lai2, Cindy R. DeLoney-Marino3, Mark J. Mandel4, John S. Parkinson2, and Edward G. Ruby1 *Presenting author 1 Department of Medical Microbiology and Immunology, University of Wisconsin, Madison, WI 2 Department of Biology, University of Utah, Salt Lake City, UT 3 Department of Biology, University of Southern Indiana, Evansville, IN 4 Department of Microbiology-Immunology, Northwestern University Feinberg School of Medicine, Chicago, IL The bioluminescent bacterium Vibrio fischeri enters into a monospecific association with the Hawaiian bobtail squid, Euprymna scolopes, that begins anew each generation. Symbionts from the surrounding seawater form mucus-bound aggregates on the light-organ surface, and then migrate to epithelium-lined crypts deep within its tissue, a distance of less than 100 micrometers. Despite this short distance, the initiation process is still exquisitely specific and complex. By using confocal microscopy to observe the behavior of a GFP-labeled cheA mutant, we have identified three stages at which chemotaxis likely mediates symbiotic initiation. To fully understand how V. fischeri senses and responds to the environment presented by the host, we sought to characterize the chemoreceptorligand pairings that underlie this behavior. Similar to other bacteria within the Vibrionaceae, the genome of V. fischeri encodes dozens of predicted methyl-accepting chemotaxis proteins (MCPs), none of which have been previously studied and cannot be well-characterized using bioinformatics. To identify the chemoattractants sensed by these MCPs, we used two complementary approaches: reverse genetics in V. fischeri, and heterologous expression of chimeric MCPs in Escherichia coli. We first generated 19 individual MCP mutants and screened for altered responses to six known V. fischeri chemoattractants using soft-agar assays, followed by capillary-assay analysis. Because functional redundancy by other V. fischeri MCPs could hide potential interactions in this approach, we also constructed chimeric proteins in which V. fischeri ligand-binding domains were fused to E. coli Tar, immediately upstream of the HAMP domain. These chimeras were expressed in a ΔMCP E. coli strain and analyzed by the FRET (Förster resonance energy transfer)-based in vivo kinase assay (adapted from Sourjik and Berg, 2002), in which pools of potential chemoattractants were used as stimuli. Using these two approaches, we have begun to clarify the chemotactic repertoire of V. fischeri, including the characterization of VfcA (Vibrio fischeri chemoreceptor A), which mediates chemotaxis towards multiple amino acids, and VfcB, which senses several sugars, including the chitinbreakdown products N-acetyl-glucosamine and chitobiose. Taken together, these approaches are providing insight into the role of chemotaxis in this symbiosis, as well as defining a system by which to identify chemoattractants in bacteria with large numbers of predicted MCPs. Lab: Edward Ruby _________________ 31 BLAST XII ____ Wed. Morning Session CHEMOTAXIS TO NOREPINEPHRINE (NE) REQUIRES THE Tsr CHEMORECEPTOR AND CONVERSION ENZYMES INDUCED BY QseC S. Pasupuleti1, M. R. Sears2, A. Jayaraman1, M. D. Manson2 Departments of 1Chemical Engineering and 2Biology Texas A&M University College Station, TX 77840 It has been reported that norepinephrine (NE) is an attractant sensed by E. coli and S. typhimurium. We have found that the actual attractant is the NE metabolite dihydroxymandelic acid (DHMA). NE is metabolized into DHMA in a two-step pathway involving the TynA tyramine oxidase and the FeaB aromatic aldehyde dehydrogenase. NE induces the synthesis of TynA and FeaB via the QseC sensor kinase. The peak response to DHMA is seen at concentrations of ~100 nM, a potentially physiological range in the gastrointestinal tract. Current data suggests that DHMA is sensed via Tsr and serves as an attractant at low concentrations and as an antagonist at higher concentrations. We present a model for how this behavior might arise through differential responses to DHMA mediated by the two serine-binding sites of the Tsr homodimer. Lab: Michael Manson _______________ 32 BLAST XII ____ Wed. Morning Session STRUCTURE AND FUNCTION OF A BLUE-LIGHT-REGULATED SENSOR HISTIDINE KINASE Ralph P. Diensthuber1, Martin Bommer2,3, Andreas Möglich1 Humboldt-Universität zu Berlin, Institut für Biologie, Biophysikalische Chemie. 2 Humboldt-Universität zu Berlin, Institut für Biologie, Strukturbiologie/Biochemie. 3 Helmholtz-Zentrum Berlin für Materialien und Energie, BESSY-II. 1 Two-component systems (TCS) which comprise sensor histidine kinases (SHK) and reponseregulator proteins represent the predominant strategy by which prokaryotes sense and respond to a changing environment. To date, no atomic structure of an intact SHK containing both sensor and effector (catalytic) modules has been elucidated. Here, we report the full-length crystal structure of the dimeric, blue-light-regulated SHK YF1 at 2.3 Å resolution in which two N-terminal light-oxygen-voltage (LOV) photosensors are connected by a coiled coil to the C-terminal effector modules. A second coaxial coiled coil derived from the N-termini of the LOV photosensors and inserted between them crucially modulates light regulation: single mutations within this coiled coil attenuate or even invert the signal response of the TCS. Structural motifs identified in YF1 recur in signal receptors, and the underlying signaling principles and mechanisms may be widely shared between soluble and transmembrane, prokaryotic and eukaryotic signal receptors of diverse biological activity. Lab: Andreas Möglich ______________ 33 BLAST XII ____ Wed. Morning Session EFFECTS OF MOLECULAR ADSORPTION ON THE TRAJECTORIES AND ACCUMULATION OF MOTILE BACTERIA AT THE AIR/WATER INTERFACE Michael Morse and Jay X. Tang Department of Physics, Brown University, Providence RI Motility allows bacteria to move through its environment in order to acquire nutrients, avoid danger, and perform other biological tasks. Cells can sometimes actively control their movement through processes such as chemotaxis. However, the environment itself plays a vital role determining cellular behavior due to the underlying interactions between the cell and its surroundings. For instance, micro-swimmers such as Escherichia coli, Salmonella enterica, and Vibrio cholerea are able to propel themselves through liquids by rotating their helical flagella. The cell's motility is dependent on many factors such as the viscosity of the medium and concentration of various chemicals. Any enhancements or obstacles to proper bacterial motility can affect cell lifetimes, reproduction rates, etc. We are particularly interested in bacterial swimming near an interface with other media (either a solid or a second fluid). Interfaces have interesting physical characteristics that affect cell motility. We are studying how different interfaces, including interfaces with different chemical compositions, affect bacterial motility and accumulation. In our experiments, we find that monotrichous bacterium Caulobacter crescentus travels in unique swimming trajectories near the air/water interface. Additionally, we find that the swimming behavior of a large population of swarmer cells is highly sensitive to adsorption of different classes of molecules. It is unclear whether this behavior has evolutionarily beneficial impacts on cell motility and growth or if near interface forces hinder certain biological activities. The wide variety of individual swimming and motility methods used by different bacterial species could be partially accounted for as coping methods for these interactions. We are also able to explain the large-scale behavior of a population of cells through examining the forces and torques acting on individual cells. The goal of our study is to quantitatively define these forces and their consequences on bacterial motility and accumulation near different surfaces. Lab: Jay Tang _____________________ 34 BLAST XII ____ Wed. Morning Session TOWARDS AN INTEGRATED MODEL OF THE CHEMOTAXIS PATHWAY IN BACILLUS subtilis Christopher Rao, George Ordal, Hanna Walukiewicz, George Glekas, Mathew Plutz, and Payman Tohidifar Departments of Biochemistry and Chemical & Biomolecular Engineering, University of Illinois at Urbana-Champaign Bacillus subtilis employs three adaptation mechanisms for chemotaxis: chemoreceptor methylation, the CheC/CheD/CheYp system, and the enigmatic CheV system. While there is some redundancy between these three systems, we now know that they do not function independently of one of another. Rather, they may function as integrated unit. E. coli, on the other hand, employs just one adaptation mechanism involving chemoreceptor methylation. Even then, this system functions differently in the two bacteria: methylation is selective and antagonistic in B. subtilis whereas it additive and synergistic in E. coli. During the past year, we have significantly advanced our understanding of the methylation and the CheC/CheD/CheYp systems. These results have enabled us to revise and extend our previous model of the B. subtilis chemotaxis pathway. In particular, our new model is now supported by extensive biochemical data. In this talk, I will discuss our recent progress modeling the chemotaxis pathway in B. subtilis. Using this model we have explored how the selective methylation system functions in B. subtilis. While the results are still preliminary, our models demonstrate that the receptor complexes must adapt at least three distinct conformations. A key advance was developing a robust in vitro receptor-coupled kinase assay. This assay enabled us to characterize the methylation sites on McpB, the paradigm chemoreceptor for B. subtilis chemotaxis. In addition, this assay has enabled us to characterize interactions between the methylation and CheC/CheD/CheYp adaptation systems. In particular, we have determined how CheD affects receptor-coupled kinase activity as a function of the methylation state. Collectively, these experimental data allowed us to extend our model to account for CheC and CheD. This new model offers fresh insights on the ultimate question: why does B. subtilis employ multiple adaptation systems when a single one would suffice? Lab: Christopher Rao ______________ 35 BLAST XII ____ Wed. Morning Session METHYL ACCEPTING CHEMOTAXIS PROTEIN U OF SINORHIZOBIUM meliloti BINDS THE SEED EXUDATE COMPONENT, PROLINE Benjamin A. Webb, Richard Helm, Sherry Hildreth, Angelica Thapa, Birgit E. Scharf. Virginia Tech, Blacksburg, Va. 24060 Sinorhizobium meliloti can exist as a free-living bacterium using chemotaxis to navigate through the soil and it can also live an endosymbiotic life with particular host-legume species. Host specificity has been shown to be determined mainly at the site of root hair infection, involving the exchange of host plant secreted flavonoids and rhizobial derived nodulation factors [1, 2]. In this study, we investigate chemotactic signals preceding the established line of communication and show that the seed exudate profiles of select host and non-host legumes differ significantly. We also demonstrate that S. meliloti shows strong positive chemotaxis toward the seed exudate of the agriculturally important host, Medicago sativa (alfalfa). S. meliloti has 8 chemoreceptors that contribute to chemotaxis including two cytosolic receptors [3]. Methyl accepting Chemotaxis Protein U (McpU) is one of the transmembrane receptors and plays a major role in the positive chemotaxis toward the seed exudate studied. McpU is maximally expressed during peak of cellular motility and localizes at the cell pole [4]. It is predicted to have a cytosolic domain for signal conversion, adaptation, CheA/CheW interaction, and a relatively large periplasmic region composed of 245 amino acids residues. Previous studies have shown it to be important for positive chemotaxis toward a wide range of nutrient attractants [3]. Here, we draw a correlation between the presence of proline in host seed exudates and the strong positive chemoattraction elicited by this amino acid. Amongst single mcp knock-out mutants, an mcpU deletion strain exhibits greatly diminished positive response toward seed exudateand proline. Complementation analyses confirmed that the response of S. meliloti to proline is mediated by McpU. Using isothermal titration calorimetry, we show that L-proline binds to the periplasmic domain of McpU with a binding constant of 148μM. These results show that McpU has a positive role in sensing the components of host seed exudate, suggesting that chemotaxis toward the germinating seed is a key step in the early establishment of the endosymbiosis. References 1. Goedhart J, Bono JJ, Gadella TW, Jr.: Rapid colorimetric quantification of lipochitooligosaccharides from Mesorhizobium loti and Sinorhizobium meliloti. Mol Plant Microbe Interact 2002, 15(9):859-865. 2. Phillips DA, Tsai SM: Flavonoids as plant signals to rhizosphere microbes. Mycorrhiza 1992, 1(2):55-58. 3. Meier VM, Muschler P, Scharf BE: Functional analysis of nine putative chemoreceptor proteins in Sinorhizobium meliloti. J Bacteriol 2007, 189(5):1816-1826. 4. Meier VM, Scharf BE: Cellular localization of predicted transmembrane and soluble chemoreceptors in Sinorhizobium meliloti. J Bacteriol 2009, 191(18):5724-5733. Lab: Birgit Scharf __________________ 36 BLAST XII ____ Wed. Morning Session TIME-DEPENDENT CHEMOTACTIC RESPONSE IN A LARGE POPULATION Laurence G. Wilson*, Rongjing Zhang The Rowland Institute at Harvard, 100 Edwin H Land Blvd., Cambridge, MA. 02142, USA We have previously reported techniques for quantifying motility, in terms of straight-line swimming speed (DDM) and motor speed (DFM) in free-swimming cells. These methods can be used with any standard microscope equipped with a fast camera (ideally capturing between 100-500 frames per second). Both techniques have recently benefitted from equipment modifications that have extended their applicability to faster-swimming organisms; we show data from a number of different species including E. coli, R. sphaeroides, S. typhimurium. In an extension to one of these techniques (DDM) we show how the spatial and temporal dependence of chemotactic behavior may be observed. Although much is now known about the biochemical signaling pathways that allow bacteria to respond to fluctuating chemostimulant concentration, the implications for the group behavior of cells are unknown. Previous microscopic studies have focused on single-cell behavior to build up a picture of how cells adapt to changing environmental conditions. We use our new methods to provide a comprehensive and highly automated characterization of collective bacterial behavior - around 5000 cells simultaneously - in the presence of a chemical stimulus. Lab: Laurence Wilson ______________ 37 BLAST XII ___ Thurs. Morning Session DETECTING A CONFORMATIONAL CHANGE IN THE PERIPLASMIC REGION OF THE SODIUMDRIVEN STATOR PROTEIN PomB BY THE DISULFIDE CROSSLINK Shiwei Zhu1, Masato Takao2, Na Li1, Michio Homma1, Katsumi Imada2 and Seiji Kojima1 1 Division of Biological Science, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan; 2Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan Bacterial flagellar motor consists of the rotor and stator, and their interaction that is coupled with the ion flux through the stator generates torque. The ion flux activity of the stator is activated only when it assembled around the rotor. Based on the crystal structure and functional analysis of the periplasmic region of the H+-driven stator protein MotB from Salmonella, a hypothesis of the stator activation is proposed: during the assembly step, a stator will undergo conformational change in the periplasmic region of the B subunit to activate the proton channel (Kojima et al, 2009). To test this model, we adopted disulfide crosslinking approach for the Na+-driven stator protein PomB from Vibrio, whose crystal structure of its periplasmic region (PomB C ) has recently been solved. If an intramolecular disulfide bridge is formed between predicted movable elements, it will suppress this conformational change, and consequently inhibit the function of the stator. Based on the structural information of PomB C , we introduced several pairs of cysteine replacements into cysteine-less PomB, one at the α1helix (residues, 154-169) and the other at the core domain (residues, 170~). As a result, we found that the pair of cysteine replacements M157C-I186C and that of I164C-L217C inhibited the function of the stator without affecting its stability. Addition of the reducing agent DTT in the medium rescued quickly the function of these stators. These results support the model that a large conformational change in the PomBC is required for stator activation. Kojima S, Imada K, Sakuma M, Sudo Y, Kojima C, Minamino T, Homma M, Namba K. Mol. Microbiol. (2009) 73, 710-718. Lab: Michio Homma ________________ 38 BLAST XII ___ Thurs. Morning Session COORDINATED SWITCHING OF BACTERIAL FLAGELLAR MOTORS: EVIDENCE FOR DIRECT MOTOR-MOTOR COUPLING? Bo Hu and Yuhai Tu IBM T.J. Watson Research Center, Yorktown Heights, NY, 10598, USA The swimming of Escherichia coli is propelled by its multiple flagellar motors. Each motor spins either clockwise (CW) or counterclockwise (CCW), under the control of an intracellular regulator, CheYP. A long standing question is whether these motors work independently or not. There can be two mechanisms (extrinsic and intrinsic) to coordinate the switching of bacterial motors. The extrinsic one arises from the fact that different motors in the same cell sense a common input (CheY-P) which fluctuates near the motors' response threshold. An alternative, intrinsic mechanism is direct motormotor coupling which makes synchronized switching energetically favorable. Here, we develop simple theoretical models for both mechanisms and uncover their different hallmarks. A quantitative comparison to the recent experiments suggests that the direct coupling mechanism may be responsible for the observed sharp correlation between motors in a single E. coli. We discuss possible origins of this coupling (e.g., hydrodynamic interaction) and present some feasible experimental suggestions. Lab: Yuhai Tu _____________________ 39 BLAST XII ___ Thurs. Morning Session H. pylori FLAGELLAR MOTOR: STRUCTURAL INSIGHT INTO THE MECHANISM OF STATOR ASSEMBLY AND ACTIVATION Daniel A. Andrews, Jenna O'Neill, Matthew C. Wilce and Anna Roujeinikova Department of Biochemistry and Molecular Biology and Department of Microbiology, Monash University, Clayton, Victoria 3800, Australia. Carcinogenic bacterium Helicobacter pylori uses the flagellar motor to drill into the mucus layer of the stomach and move towards the epithelial surface, where it colonizes [1]. Flagellar rotation is powered by the proton influx through the stator ring MotA/MotB [2,3]. The work presented at the meeting will inform the audience about the latest discoveries of novel factors that mediate stator/rotor interaction [4,5] and our latest findings that establish the relationship between the structure, dynamics and function of MotB. We have determined the first crystal structure of the protein domain that anchors the proton-motive-force-generating mechanism of the bacterial flagellar motor to the cell wall, and formulated a model of how the stator attaches to peptidoglycan (PG) [6,7]. Analysis of the accumulated structural, biochemical and mutagenesis data suggested a mechanism by which MotB’s cell wall binding activity is inhibited until the stator is incorporated into the motor [8]. Putting this in the perspective of the stator assembly and activation, we propose that in the pre-assembled stator units with the closed channel the linker is partially folded, stabilizing the dimer in the conformation that cannot insert into the PG mesh. This inhibition occurs via a dual mechanism: distance constraint and suboptimal geometry of two PG-binding sites. When the diffusing stator unit collides with the rotor, a drastic conformational change in MotB is induced, resulting in unwinding of the two beta strands at the edge of the beta sheet, extension of the linker, re-arrangement of the dimer geometry and insertion of the Pal-like conserved core domain into PG mesh. Implications for stator activation via rotational displacement of the transmembrane helices of MotB will be discussed. References: [1] Yoshiyama, H. & Nakazawa, T. (2000). Microbes Infect. 2, 55-60. [2] Kojima, S. & Blair, D.F. (2004) Biochemistry 42, 26-34. [3] Blair, D.F. & Berg, H.C. (1988) Science 242, 1678-1681. [4] Blair, K.M., Turner, L., Winkelman, J.T., Berg, H.C. & Kearns, D.B. (2008) Science 320, 1636-1638. [5] Boehm, A., Kaiser, M., Li, H., Spangler, C., Kasper, C.A. et al. (2010) Cell 141, 107-116. [6] Roujeinikova, A. (2008). Proc. Natl. Acad. Sci. USA, 105, 10348-10353. [7] Reboul, C. F., Andrews, D. A., Nahar, M. F., Buckle, A. M. & Roujeinikova, A. (2011). PLoS ONE, 6, e18981. [8] O'Neill, J., Xie, M., Hijnen, M. & Roujeinikova, A (2011). Acta Cryst. D67, 1009-1016. Lab: Anna Roujeinikova ____________ 40 BLAST XII ___ Thurs. Morning Session CORRELATED MUTATION ANALYSIS OF THE FLAGELLAR MOTOR PROTEIN FliG Shahid Khan%, Jens Kleinjung# and William R. Taylor# % Molecular Biology Consortium @ Lawrence Berkeley National Laboratory, Berkeley, CA 94720 USA # Division of Mathematical Biology, MRC National Unit for Medical Research, London MW7 1AA, UK We are applying correlated mutation analysis (CMA), an emerging technique for the analysis of protein-protein interactions, to the architecture of the bacterial flagellar basal body. The flagellar basal body is a multi-component, multi-subunit assembly embedded in the cytoplasmic membrane. It harbors machinery for flagellar assembly, energization and switching of flagellar rotation. The membrane protein FliF forms rings (MS-rings) that subsequently direct the assembly of the cytoplasmic C ring. Binding of the chemotaxis signal protein CheY to the C ring protein FliM switches rotation sense. There is a mismatch between the MS and C ring subunit stoichiometry. The mismatch is accommodated by the protein FliG that lies at the interface between the MS and C rings. FliG interacts with the force generating Mot membrane protein complexes, in addition to FliF and FliM. An atomic model of FliG and its binding partners within the MSC ring will be important for elucidation of the motility mechanism. CMA is one approach towards this goal. FliG is the most, well characterized protein of the flagellar basal body. FliG subunit stoichiometry has approximated from purified basal bodies. Membrane permeable cross-linkers, affinity chromatography and suppressor mutations have identified FliG interaction surfaces responsible for selfassociation and binding to FliF, FliM and MotA. FliG has been localized to basal bodies by immunoelectron microscopy. Native and mutant FliG X-ray crystal structures are available, including a complex of the FliG and FliM interaction domains. The structures have been fit to low resolution electron cryomicroscopy maps of native basal bodies and overproduced intact MSC rings. These data have mainly been obtained in the enteric bacteria Escherichia coli and Salmonella typhimurium; with Xray protein structures from the thermophiles Aquifex aeolicus and Thermatoga maritima. Flagellar motility is widespread in prokaryotes. A phylogenetic analysis is useful for revealing conserved design features. The FliG phylogenetic tree, based on over two thousand sequences, revealed several instances of horizontal gene transfer, particularly in spirochetes. Representative species covering phylogenetic spread were used to seed multiple sequence alignments. Where available, Pfam alignments were used. The alignments were used to generate model structures based on secondary structure prediction and correlation analysis for identification of residue contacts1. The top ranked models were validated by comparison with the structures of the two essential domains; the middle and carboxy-terminal domain. There are a number of reasons for false positives. These include artifacts arising from partial correlation as well as coupling due to function or self-association. Algorithm development has entailed elimination of partial correlations, utilization of filters based on the biochemical data and bootstrap analysis to assess variation. A model of FliG interaction surfaces and conformational transitions based on our CMA will be presented as well as ongoing developments of the method for its application to flagellar basal body architecture. ------------------------------------------------------------------------------------------------------------------1. Taylor, W. R. & Sadowski, M. I. (2011). Structural constraints on the covariance matrix derived from multiple aligned protein sequences. PLoS One 6, e28265. Lab: Shahid Khan 41 BLAST XII ___ Thurs. Morning Session CORRELATED STRUCTURE AND ACTIVITY OF THERMOTOGA maritima FLIY Ria Sircar, Anna R. Greenswag, Alexandrine M. Bilwes, Gabriela G. Bonet, and Brian R. Crane Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850 In bacterial chemotaxis, phosphorylated CheY levels dictate the direction of flagellar rotation. Switching between the counter-clockwise and clockwise directions dictates whether the bacteria will swim smoothly or tumble. For robust chemotaxis, the CheY-P signal must be terminated so that the cells can return back to the prestimulus level. In E.coli this function is accomplished by the phosphatase CheZ. However, in Thermotoga maritima, Bacillus subtilis and many other bacteria the CYX family of phosphatases, comprising of CheC, CheX and FliY, fulfills this role. Here we report the crystallographic structure of the last member of this family to have its structure determined: FliY. FliY, unlike CheC and CheX is an integral component of the flagellar rotor. FliY is a multidomain protein with a N-terminal CheY binding domain, a central CheC like domain and a C-terminal domain homologous to the conserved rotor protein FliN. The structure of the FliY middle domains is similar to CheC and the rotor protein FliM. Previous studies have identified two putative active sites on FliY to involve E35/N38 and E132/N135. We mutated these residues to assess their contribution to phosphatase activity in T. maritima FliY. We find that both the active sites are individually capable of dephosphorylating CheY but the second active site (E132/N135) is more reactive. Interactions of FliY with other rotor proteins were investigated and shown to have implications for rotor structure and function in FliY-containing bacteria. Lab: Brian Crane __________________ 42 BLAST XII ___ Thurs. Morning Session BORRELIA burgdorferi FLAGELLA EXPORT APPARATUS AND VIRULENCE: INSIGHT INTO TYPE III SECRETION SYSTEM Tao Lin, Lihui Gao, Xiaowei Zhao, Jun Liu, and Steven J. Norris UT Medical School at Houston, PO Box 20708, Room MSB 2.124, Houston, TX 77225-0708 The mechanisms of pathogenesis in Borrelia burgdorferi are largely unknown. The ability of this spirochete to migrate to distant sites in the tick and mammalian host is likely dependent on a robust chemotaxis response and motility. B. burgdorferi contains multiple copies of the chemotaxis genes cheA, cheB, cheR, cheW, cheX, and cheY. Multiple chemotaxis proteins may provide diversity in terms of function and/or structural location, or may be differentially expressed under varied physiological conditions. Proteins involved in motility are encoded by 38 genes including 36 flagellar & motor genes and 2 flagellin genes, whereas chemotaxis proteins are encoded by 20 genes including 15 chemotaxis genes and 5 chemotaxis receptor genes. To dissect the mechanism of Borrelia chemotaxis and motility and the relationship between the virulence and chemotaxis/motility, we examined Himar1 transposon mutants in 24 chemotaxis/motility genes including 10 motility genes, 10 chemotaxis genes and 4 chemotaxis receptor genes in the transformable and infectious B. burgdorferi strain 5A18NP1. The infectivity of these mutants was determined using a signature-tagged mutagenesis (STM) procedure in C3H/HeN mice and a newly developed, high throughput Luminex procedure. Among the mutants in 24 genes tested, mutations in flgI, flgJ, and cheW-3 exhibited reduced infectivity. Mutants in the 21 genes (cheW-1, fliG-1, fliZ, fliH, fliI, flgB, flaA, flbA, cheA-1, cheA-2, cheB-1, cheB-2, cheY-1, cheR-2, cheW-2, cheX, cheY-2, mcp-1, mcp-3, mcp-4, and mcp-5) showed no infectivity, indicating that these genes are required for full infectivity in B. burgdorferi. In examining morphology under dark-field microscopy, 18 mutants did not exhibit obvious morphologic defects. Seven mutants (fliH, fliI, flbA, flaA, cheA-2, cheB2, and cheR-2) exhibited elongated, string-like and/or rod-shaped morphology. In terms of motility, mutations in 13 genes did not induce obvious motility defects, whereas 7 mutants (flaA, flgI, fliG-1, fliW1, cheA-2, cheR-2, and mcp-5) showed a reduced motility, and string-like mutants (flbA, fliH, and fliI) were nearly non-motile, ‘trembling’ in a few sites of the cell. The string-shaped or rod-shaped mutants often bend at the cell center. The swimming ability of these mutants was evaluated by measuring their velocity in a highly viscous medium containing 1% methylcellulose. Seven mutants (fliH, fliI, flaA, flbA, flgI, cheX, and mcp-4) appeared to be incapable of translating motion, 8 mutants (fliG1, fliW-1, cheA-2, cheR-2, cheW-2, cheW-3, cheY-2, and mcp-5) exhibited reduced motility, and inactivation of 6 genes (flgJ, fliZ, cheA-1, cheB-1, cheB-2, mcp-1, and mcp-3) did not induce decreased motility in 1% methylcellulose medium. FliI and FliH, which are central to export apparatus function, appear to be required for mouse infection. fliH and fliI mutants also had reduced motility, growth and division defects (resulting in elongated organisms), and structural changes in the flagellar motor. Disruption of either fliH and fliI also resulted in altered cellular protein profiles; we are in the process of identifying the proteins that are underrepresented in the mutants. Inactivation of fliI and fliH genes resulted in the loss of the FliH/FliI complex as determined by Cryo-EM. These results represent the initial characterization of the flagellar export apparatus of B. burgdorferi, and continued studies will provide insight into export apparatus functions in this organism. Lab: Steven Norris ________________ 43 BLAST XII ___ Thurs. Morning Session DYNAMIC CONFORMATIONAL CHANGES OF FLAGELLAR FILAMENT OBSERVED BY HIGHPRESSURE MICROSCOPY Masayoshi Nishiyama1,2 and Yoshiyuki Sowa3 1 The Hakubi Center, Kyoto University, Kyoto 606-8302, Japan 2 WPI-iCeMS Complex2, Kyoto University, Kyoto 606-8501, Japan 3 Department of Frontier Bioscience, Hosei University, Tokyo 184-8584, Japan The bacterial flagellar motor is a molecular machine that rotates a flagellum in both directions. CCW rotation allows the left-handed helical filaments to form a bundle that propels the cell smoothly, whereas CW rotation of a filament leads to change the shape of filament in right-handed helix and break the bundle, and inhibits smooth swimming of the cell, called a tumble. The switching in the helical structure is thought to be caused by directional mechanical actions arising from abrupt change of exerted torque by the motor rotation. Here, we show that application of pressure can also change the helical structure of flagellar filaments. The flagellar filaments in E. coli cells were fluorescently labeled, and then the images were acquired by using a high-pressure microscope [1, 2] with some modifications. We measured the diameter and pitch of the individual filaments and then classified them into 11 possible waveforms which are predicted from structural data. At 0.1MPa (ambient pressure), all flagellar filaments formed left-handed helical structure (normal form). At 40 MPa, we found left-handed forms (normal and coiled forms) and right- (curly I (or II)). At 80 MPa, 80% flagellar filaments took curly I (or II) forms. After the pressure was released, most filaments returned to the initial left-handed structures. The application of pressure is thought to enhance the structural fluctuation and/or association of water molecules with the exposed regions of flagellin molecules, and results in switching the helical from left- to right-handed structure. [1] Nishiyama M. and Y. Sowa. 2012. Microscopic Analysis of Bacterial Motility at High Pressure. Biophys. J. 102:1872-1880. [2] Nishiyama M. and S. Kojima. 2012. Bacterial motility measured by a miniature chamber for highpressure microscopy. Int. J. Mol. Sci. 13:9225-9239. Lab: Masayoshi Nishiyama _______________ 44 BLAST XII ___ Thurs. Morning Session NOVEL LEPTOSPIRA PROTEIN IS ESSENTIAL FACTOR IN DETERMINING THE FLAGELLAR SHEATH, TRANSLATIONAL MOTILITY, AND VIRULENCE PHENOTYPE Elsio A. Wunder Jr.1,2, Cláudio P. Figueira2, Nadia Benaroudj3, Mitermayer G. Reis2, Nyles Charon4, Mathieu Picardeau3, and Albert I. Ko1,2 1 Yale School of Public Health and Medicine, New Haven, USA; 2Gonçalo Moniz Research Center, Oswaldo Cruz Foundation, Brazilian Ministry of Health, Salvador, Brazil; 3Unité de Biologie des Spirochètes, Institute Pasteur, Paris, France; 4West Virginia University, Department of Microbiology and Immunology, Morgantown, USA Background: Leptospires, the causative agent of leptospirosis, are unique among spirochetes and bacteria as a whole in that they have hooked-shaped ends and coiled periplasmic flagella, both of which are required for translational motility. However, the factors which produce these morphological phenotypes and the role of motility plays in leptospiral virulence have not been evaluated. Methods and Findings: We identified small and large colonies on agar when plating a Leptospira interrogans serovar Copenhageni isolate from a Brazilian patient with pulmonary hemorrhage syndrome. Leptospires from small colonies had straight terminal ends, and although their ends gyrated and produced spiral waves, they were unable to produce translational motility. Furthermore, purified periplasmic flagella from motility-deficient clones were straight in contrast to the coiled structure observed in those from motile clones and wild-type (wt) strains. LC-MS analysis of purified flagella and genomic DNA sequencing identified that the motility-deficient clones had a mutation that disrupted the gene encoding a novel 36kDa protein, which we called Flagella coil protein 1 (Fcp1). Fcp1 protein was distributed throughout the length of wt flagella. Cryo-electron tomography determined that the flagellar diameter in situ of motility-deficient clones were significantly smaller than that of motile clones (17.6±0.97 vs 22.8±1.99mm). We produced fcp1- mutants by performing allelic exchange in a wt strain and found that these mutants showed similar loss of terminal hook end morphology, coiled periplasmic flagellar structure, and translational motility, as did motility-deficient clone. We rescued these phenotypes by complementing motility-deficient clone and fcp1- mutants with the wt fcp1 gene. Finally disruption of fcp1 was associated with inability to penetrate epithelial tissue barriers and loss of virulence during experimental infection of hamsters while complementation of the gene restored these phenotypes. Conclusion: In summary, we identified a novel flagella structural protein unique to Leptospira and required for the formation of the flagellar sheath, and to maintain its coiled structure of periplasmic flagella and hook-shaped morphology, which allows the pathogen to perform translational motility. Moreover our studies provide the first evidence which use genetic knockout and complementation approaches to demonstrate that translational motility, a classic attribute of leptospires and pathogenic spirochetes, is essential for virulence. Support: NIAID 5 U01 AI088752, and 5 R01 AI052473, and FIC 7 D43 TW00919 Lab: Albert Ko ____________________ 45 BLAST XII ___ Thurs. Evening Session INTERACTIONS BETWEEN A TRANSPORTER AND A SENSOR KINASE MEDIATE SIGNAL TRANSDUCTION IN ANTIMICROBIAL PEPTIDE DETOXIFICATION MODULES Sebastian Dintner, Susanne Gebhard Ludwig-Maximilians-University Munich, Department Biology I, Microbiology, Grosshaderner Str. 2-4, 82152 Martinsried, Germany Resistance of many Firmicutes bacteria against antimicrobial peptides is mediated by so-called detoxification modules that are comprised of a two-component regulatory system (TCS) and an ATPbinding-cassette (ABC) transporter. Strikingly, the transporters and TCSs have an absolute and mutual requirement for each other in both sensing of and resistance against their respective antimicrobial compounds: expression of the transporter is regulated by the TCS, yet the sensor kinase is unable to detect a stimulus in the absence of an active transporter. These findings suggest a novel mode of signal transduction where the transporter constitutes the actual sensor of antimicrobial peptides. Database searches revealed the wide-spread occurrence of such modules among Firmicutes bacteria, and parallel phylogenetic analysis showed that transporters and TCSs have co-evolved. Based on these findings, we hypothesize the formation of a sensory complex between both components. In vivo and in vitro approaches studying the bacitracin-resistance module BceRS-BceAB of Bacillus subtilis support this hypothesis by indicating direct physical contacts between the sensor kinase, BceS, and the transport permease, BceB. Furthermore, random mutagenesis of BceB revealed that resistance and signaling are genetically separable traits of the transporter, showing that signal transduction is indeed mediated by the transporter directly and is not a secondary effect of substrate translocation. Taken together, our results show that Bce-type ABC-transporters and TCSs have co-evolved to form selfsufficient detoxification modules against antimicrobial peptides, and suggest a novel signaling mechanism involving formation of a sensory complex between transporter and sensor kinase. Lab: Susanne Gebhard _____________ 46 BLAST XII ___ Thurs. Evening Session A NOVEL TRANSLATIONAL REGULATION MECHANISM MEDIATED BY ELONGATION FACTOR EF-P ALLOWS TIGHT ADJUSTMENT OF PROTEIN COPY NUMBER Lassak, J.1,2,5, Ude, S.1,2,5, Starosta, A.L.1,3, Kraxenberger, T.1,2,4, Wilson, D.N.1,3, and Jung, K.1,2* 1 Center for integrated Protein Science Munich (CiPSM) 2 Department of Biology I, Microbiology, Ludwig-Maximilians-Universität München, Großhaderner Strasse 2-4, 82152 Martinsried, Germany 3 Gene Center and Department of Biochemistry, Ludwig-Maximilians-Universität München, FeodorLynen-Str. 25, 81377 Munich, Germany 4 Present address: AMSilk GmbH, Am Klopferspitz 19, 82152 Martinsried, Germany 5 These authors contributed equally to this work Elongation factor P (EF-P) binds to the ribosome and stimulates peptide-bond formation. It has a ubiquitous distribution, being conserved in all bacteria and orthologous to archaeal and eukaryotic initiation factor 5A (a/eIF-5A). E. coli EF-P is post-translationally β-lysinylated by the sequential action of YjeK, YjeA. Inactivation of the efp, yjeA or yjeK genes results in defects in bacterial growth, fitness and membrane integrity, as well as stress resistance and virulence. Despite the conservation and importance of EF-P, the biological function has remained enigmatic. We found that EF-P relieves ribosome stalling at polyproline stretches. We further demonstrate the relevance of EF-P in vivo to control the protein copy number and stress response of the polyproline-containing pH-receptor CadC. The E. coli proteome contains 95 proteins with a polyproline stretch, including the histidine kinases EnvZ and PhoR and the flagella master regulator FlhC. Our new findings now rationalize the observed defects in motility and attenuated virulence in efp mutants. Bacterial, archaeal and eukaryotic cells contain 100-1000’s of polyproline-containing proteins of diverse function and we suggest that EF-P plays a central role for the tight copy number adjustment of various pathways across all kingdoms of life. Lab: Kirsten Jung _________________ 47 BLAST XII ___ Thurs. Evening Session TRANSCRIPTIONAL ACTIVATION AND REPRESSION OF SIX flhDC PROMOTERS: AN INTRICATE PROMOTER CONFIGURATION FOR flhDC IN SALMONELLA typhimurium Chakib Mouslim and Kelly Hughes University of Utah In Salmonella typhimurium, the transcriptional factor FlhDC is involved in coordinating the expression of flagellar genes and pathogencity island 1 (Spi1). Transcription of flhDC is controlled negatively and positively by multiple regulatory factors including, RcsB, LrhA, SlyA, DskA, EcnR, HNS, cAMP/CRP, CsrA, PefI-SrgD, HilD, RtsB and QseB. In Salmonella, the flhDC promoter region contains 6 transcriptional start sites that drive the transcription of the flhDC operon. The detailed mechanism of this regulation and the mechanism by which these transcriptional factors integrate signals to regulate the expression of flhDC are largely unknown. We characterized individual promoters within the flhDC promoter region. We investigated the effect of the six individual promoters and their role in flhDC transcription in addition to the effect of the different transcriptional regulators on each promoter. These promoters can respond differently to growth conditions that activate specific regulators leading to the flexibility in the adjustment of flhDC expression. Depending on their arrangement these multiple promoters drive transcription on their own or interact with each other. These interactions particularly coupled with the effect of selective transcriptional regulators generate regulatory circuits between expression of flagellar genes and Spi1. Lab: Kelly Hughes _________________ 48 BLAST XII ___ Thurs. Evening Session ESCHERICHIA COLI BIOFILMS MAY FAVOR A MIXTURE OF MOTILE AND NON-MOTILE BACTERIA Shelley M. Horne, Joseph Sayler, Ty Lynnes, Priyankar Samanta, & Birgit M. Prüβ* Department of Veterinary and Microbiological Sciences, North Dakota State University, Fargo ND To shed light into the ongoing controversy on motility constituting an advantage or disadvantage for Escherichia coli in a biofilm, we performed two long-term experiments. First, we recovered colonies from 7 and 14 days old biofilms, formed by the partially motile and good biofilm forming E. coli K-12 strain AJW678 and the highly motile, poor biofilm former MC1000. Up to 5% of the colonies that were recovered from either parental strain had altered motility. In the case of AJW678, the motility phenotype changed to hyper motile. For MC1000, motility changed to entirely or partially non-motile. Many of the MC1000 derived non-motile colonies contained insertions of IS1 in their flhD promoter or the open reading frame of FlhC. Some of these colonies produced slightly more biofilm biomass than their MC1000 parent. To further investigate the moderate inverse correlation between motility and biofilm biomass, we performed a competitive experiment, where MC1000 and its isogenic flhD::kn mutant were inoculated at a 1:1 ratio. Biofilms maintained a mixture of both bacterial populations over the first two weeks post inoculation. Towards the end of week 4, the flhD mutant took over the population. In a spatial experiment, flhD was expressed higher at the top layer of the biofilm than at its bottom. Altogether, we will come to the conclusion that E. coli biofilms may need both, parent bacteria and flhD mutants for optimal functionality, at least during the first two weeks of their formation. Motility may constitute an advantage in certain areas of the biofilm. Lab: Birgit. Prüβ __________________ 49 BLAST XII ___ Thurs. Evening Session Rrp1 REGULATES CHITOBIOSE UTILIZATION OF THE LYME DISEASE SPIROCHETE BORRELIA burgdorferi Ching Wooen Sze1, Alexis Smith3, Young Hee Choi2, Xiuli Yang3, Utpal Pal3, Aiming Yu2, and Chunhao Li1* Department of Oral Biology, The State University of New York at Buffalo, Buffalo, New York1, Department of Pharmaceutical Sciences, The State University of New York at Buffalo, Buffalo, New York2, Department of Veterinary Medicine, University of Maryland and Virginia-Maryland Regional College of Veterinary Medicine, College Park, Maryland3 Life cycle alternating between arthropod and mammals forces the Lyme disease spirochete, Borrelia burgdorferi, to adapt to different host milieus by utilizing diverse carbohydrates. B. burgdorferi lacks the de novo biosynthesis pathway of N-acetylglucosamine (GlcNAc) but encodes a set of chitobiose metabolic genes that allows conversion of chitobiose to GlcNAc. The chbC gene, a key chitobiose transporter, is dually regulated by RpoD and RpoS. Rrp1 is a response regulator that synthesizes cyclic diguanylate (c-di-GMP) in B. burgdorferi. In this report, we found that the rrp1 mutant had growth defects and formed membrane blebs when GlcNAc was replaced by chitobiose in growth medium. The expression level of chbC was significantly repressed in the mutant and constitutive expression of chbC successfully reversed the phenotype. Immunoblotting and transcriptional studies revealed that Rrp1 is required for the activation of BosR and its impact on chbC is likely mediated by the BosR-RpoS signaling pathway. Although the rrp1 mutant was unable to be transmitted via tick bite, exogenous supplementation of GlcNAc into unfed ticks partially rescued the transmission. Based on these results, we propose that Rrp1 governs the chitobiose metabolism of B. burgdorferi via a BosRRpoS-ChbC regulatory network essential for the transmission of the spirochete. Lab: Chunhao Li __________________ 50 POSTER ABSTRACTS 51 BLAST XII ________________ Poster #1 STRUCTURE AND FUNCTION OF CHEMORECEPTORS AND THE ROLE OF MULTIPLE CheW HOMOLOGUES IN RHODOBACTER SPHAEROIDES James R. Allen and Judith P. Armitage Department of Biochemistry, South Parks Road, Oxford, OX1 3QU Rhodobacter sphaeroides is a model organism for investigating complex chemosensory pathways now identified in many bacterial species. The proteins that make up the complex chemotactic system of R. sphaeroides are encoded over 3 distinct major operons as well as additional other smaller loci. 13 chemoreceptors are encoded in the genome; 9 localise to a membranous cluster much like that seen in Escherichia coli (known as Methyl-accepting Chemotaxis Proteins or MCPs) while 4 have no transmembrane regions and localise to a cytoplasmic chemosensory cluster (these are designated Transducer-like proteins or Tlps). Previous analysis of the roles of individual MCP homologues as has proved difficult. A new approach is needed if the complexity of signalling through multiple receptors is to be analysed in full. R. sphaeroides also contains 4 homologues of the scaffold protein CheW and 4 homologues of the scaffold and phosphotransfer protein CheA. CheW 2 , CheW 3 and CheA 2 have been shown to be required to localise the MCP receptors into a tight, cooperative signalling array while CheW 1 and CheA 1 are not involved. An additional CheW, CheW4, localises to the cytoplasmic cluster, but not to the membrane cluster, even when over expressed. All R. sphaeroides CheWs are able to complement E. coli deletions. Why are 3 CheW homologues required in R. sphaeroides? Structural analysis of the MCPs suggests that most fall into the 34H category (designating the number of heptad repeats and thus length of cytoplasmic domain) with MCPA being 36H and MCPM being 38H. Previous deletion studies of the R. sphaeroides MCPs also showed that once a certain subset had been deleted, no tight receptor cluster could be observed with CheWs still present. This not only prevented individual MCPs from being tested for their activity to various substrates but also suggests complex interactions with to the scaffold. In order to analyse the binding of the MCPs to individual components of the scaffold and, in turn, analyse their substrate specificity, a new approach is needed. The E. coli receptor deletion mutant UU2612 (courtesy of S Parkinson) contains all of the E. coli scaffold proteins but has been deleted for all known E. coli MCPs. Plasmid-based expression of individual MCPs has shown that E. coli scaffold proteins are insufficient for full localisation of R. sphaeroides MCPs tagged with RFP. Co-expression with R. sphaeroides CheW proteins shows that MCPB and MCPJ can localise into a tight cluster using CheW3, indicative of that seen in the wild type system. CheW2 is unable to localise these MCPs. This shows that UU2612 containing the R. sphaeroides CheW scaffold is a viable approach to assess CheW specificity and cluster formation. We will describe recent data on the structure and function of the R. sphaeroides chemoreceptors and the roles of the CheWs. Lab: Judith Armitage ______________ 52 BLAST XII STRUCTURE-FUNCTION STUDIES RHODOBACTER SPHAEROIDES ________________ Poster #2 OF THE RESPONSE REGULATOR CheY 6 FROM Matthew W Smith, Nick Delalez, Christina Redfield, Judy Armitage University of Oxford Oxford Centre for Integrative Systems Biology New Biochemistry Building South Parks Road Oxford OX3 1QU Rhodobacter sphaeroides has a complex chemosensory network which comprises multiple homologous genes and involves metabolic sensing (also known as energy taxis). R. sphaeroides has two different flagellar motors but only one is expressed under laboratory conditions (Fla1). Unlike most bacteria, the expressed motor displays a stop-and-go phenotype. R. sphaeroides genome also encodes six different CheY proteins but only three (CheY 3 , CheY 4 and CheY 6 ) have been shown to interact with the Fla1 motor. Deletions of CheY 3 , CheY 4 and CheY 6 result in the flagellar motor being unable to stop (smooth swimming). CheY 6 has been shown to be essential for chemotaxis whereas CheY 3 and CheY 4 have some functional redundancies. Although CheY 6 can stop the motor alone, R. sphaeroides requires the presence of either CheY 3 or CheY 4 to be chemotactic. To date, not much is known about how these three CheY proteins interact to stop the flagellar motor. Structural studies of CheY 6 using NMR highlighted a flexible loop region (residues G109-K118) that is not present in the other CheYs. It was hypothesised that this flexible loop might confer to CheY 6 a different mechanism compared to other CheY proteins. Deletion mutants were made to investigate the role of this loop in CheY 6 function. Phenotypical studies were performed using swim plates and tethering assays. The mutants showed smooth swimming but wild type phenotype was restored upon expression of wild type CheY 6 from plasmid, indicating that CheY 6 ∆loop is unable to stop the motor. However, results also revealed that CheY 6 ∆loop is able to compete with wild type CheY 6 for phosphorylation by the kinase protein CheA 3 . Current studies using phosphotransfer assays and NMR aim at further characterising the mechanism of CheY 6 function in the Fla1 chemotaxis pathway. The results of these studies will be presented and the importance of the findings discussed. Lab: Judith Armitage ___________ 53 BLAST XII ________________ Poster #3 DECIPHERING THE ROLE OF THE PERIPLASMIC DOMAIN OF PilJ: MECHANISMS OF SIGNAL TRANSDUCTION Vishwakanth Y. Potharla, Geoff T. Riddell, and Sonia L. Bardy Department of Biological Sciences, University of Wisconsin-Milwaukee, Milwaukee WI 53211 The Chp chemosensory cluster is one of four chemosensory signal transduction systems in Pseudomonas aeruginosa. While this signal transduction pathway has long been recognized to regulate twitching motility (via type IV pili) (1), it has recently been discovered to regulate intracellular levels of cAMP, by modulating activity of the adenylate cyclase CyaB (2). Intracellular levels of cAMP correlate with levels of pilin on the cell surface, but not pilin function. Encoded within the chp gene cluster is one MCP, two CheW-like proteins, one CheA-CheY hybrid, a methyltransferase and methylesterase and two response regulator proteins. Mutational analysis of the sole MCP (PilJ) was used to investigate the role of PilJ in signal transduction through the Chp chemosensory system. Deletion of the putative periplasmic domain of PilJ resulted in a reduction of intracellular cAMP levels. This reduction was equivalent to the reduction of cAMP seen in a ΔpilJ strain, which is expected to have a non-functional Chp system. In contrast, deletion of the periplasmic domain reduced, but did not inhibit twitching motility, although strain dependent differences were observed for this phenotype. Furthermore, deletion of the periplasmic domain did not prevent directional twitching in response to the chemoattractant (18:1) phosphatidylethanolamine (PE). Replacement of PilJ periplasmic and transmembrane domains with those from Tsr resulted in only a minor reduction in twitching motility and surface piliation. Our results suggest that neither the periplasmic domain nor the transmembrane domains of PilJ are essential for twitching motility, or directional twitching towards PE, but that the periplasmic domain/protein conformation has a significant effect on cAMP levels. (1) Darzins, A. 1994. Characterization of a Pseudomonas aeruginosa gene cluster involved in pilus biosynthesis and twitching motility: sequence similarity to the chemotaxis proteins of enterics and the gliding bacterium Myxococcus xanthus. Mol. Microbiol. 11:137-53. (2) Fulcher, N., P. Holliday, E. Klem, M. Cann, and M. Wolfgang. 2010. The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol. Microbiol. 76:889-904. Lab: Sonia Bardy ________________________ 54 BLAST XII ________________ Poster #4 PHOTOSENSORY PROTEIN BphP1 INITIATES AN UNKNOWN SIGNAL TRANSDUCTION PATHWAY THROUGH PHOSPHOTRANSFER TO Psyr_2449 IN PSEUDOMONAS SYRINGAE B728A Regina S. McGrane, Liang Wu, and Gwyn A. Beattie Department of Plant Pathology and Microbiology, Iowa State University, Ames, IA 50010 Many organisms perceive and respond to light and thus require photosensory proteins. Photosensory proteins can be grouped into six families based on the specific wavelength that induces a conformational change in the chromophore. Until recently, the study of light-induced signaling pathways has been limited to plants, animals, fungi and cyanobacteria. However, with the availability of sequenced bacterial genomes, photoreceptors have been identified in a wide range of heterotrophic bacteria, but their functional roles remain unknown. The plant pathogen Pseudomonas syringae is among these heterotrophs; it encodes two bacteriophytochromes (BphPs) and a LOV (light-oxygenvoltage)-histidine kinase protein (LOV-HK). Bacteriophytochromes are the most abundant photosensory proteins found in bacteria and are responsive to red/far-red light through an association with bilin chromophores. They convert between two stable conformations, a red light-absorbing Pr form and a far-red light-absorbing Pfr form. P. syringae expresses bphP1 in an operon with bphO, which encodes a heme oxygenase that breaks down heme into the chromophore biliverdin. It expresses bphP2 in an operon with bphR, which encodes a response regulator, BphR. Both BphP1 and BphP2 are histidine kinases that are likely to be involved in two-component systems. Little is known of BphP2 because of its resistance to purification. Previous studies demonstrated that BphP1 from P. syringae absorbs red light (698nm) to convert to its Pfr form, which is the active kinase, and also that biliverdin binding is required for kinase activity. In addition to providing the chromophore, BphO has been shown to directly interact with BphP1 and to likely assist in folding. Although biochemical analysis of BphP1 is available, the target(s) of its kinase activity have not been identified, and the potential biological impacts of the BphP1 signaling pathway remain unknown. Identification of a BphP1-mediated signaling pathway is especially difficult because it is an orphaned histidine kinase with no associated response regulator. To further elucidate this pathway we utilized a tool to predict kinase/regulator interactions, http://www.swissregulon.unibas.ch/cgi-bin/TCS.pl, which uses Bayesian network methods applied to multiple sequence alignment. We tested the predicted response regulators using yeast-twohybrid analyses and phosphotransfer assays and identified Psyr_2449 as a target for BphP1-mediated phosphotransfer. Lab: Gwyn Beattie ______________ 55 BLAST XII ________________ Poster #5 A NOVEL INDUCER OF ROSEOBACTER MOTILITY IS ALSO A DISRUPTOR OF ALGAL SYMBIOSIS Robert Belas and Preeti Sule Dept Marine Biotechnology, University of Maryland Baltimore County, IMET, 701 East Pratt St., Baltimore, MD 2102 Silicibacter sp. TM1040, a member of the Roseobacter clade, forms a symbiosis with unicellular phytoplankton, which is inextricably linked to the biphasic “swim or stick" lifestyle of the bacteria. Mutations in flaC bias the population towards the motile phase. Renewed examination of the FlaCstrain (HG1016) uncovered that it is composed of two different cells: a pigmented type, PS01, and a non- pigmented cell, PS02, each of which has an identical mutation in flaC. While monocultures of PS01 and PS02 had few motile cells (0.6% and 6% respectively), co-culturing the two strains resulted in a 10-fold increase in the number of motile cells. Cell-free supernatants from co-culture or wild-type cells were fully capable of restoring motility to PS01 and PS02, which was due to increased fliC3 (flagellin) transcription, FliC3 protein levels per cell, and flagella synthesis. The motility-inducing compound has an estimated mass of 226 Da, as determined by mass spectrometry, and is referred to as Roseobacter Motility Inducer (RMI). Mutations affecting genes involved in phenyl acetic acid synthesis significantly reduced RMI, while defects in tropodithietic acid (TDA) synthesis had marginal or no effect on RMI. RMI biosynthesis is induced by p-coumaric acid, a product of algal lignin degradation. When added to algal cultures, RMI caused loss of motility, cell enlargement, and vacuolization in the algal cells. RMI is a new member of the roseobacticide family of troponoid compounds whose activities affect roseobacters, by shifting their population toward motility, as well as their phytoplankton hosts, through an algicidal effect. Lab: Robert Belas ______________ 56 BLAST XII ________________ Poster #6 DOES THE C-RING ROTATE? Basarab G. Hosu, Veda Nathan, Howard C. Berg Department of Molecular and Cellular Biology, Harvard University, 16 Divinity Ave, Cambridge, MA 02138 E. coli is propelled by a set of rotary motors. Each motor drives a helical filament, which rotates in the external aqueous environment. Although the rotation of the extracellular components of the flagellum (hook and filament) is well documented, the actual mechanism of rotation of the intracellular components remains to be clarified. We investigate the rotation of the C-ring, believed to be part of the rotor, by polarized microscopy. We expect to learn whether the C-ring rotates with respect to the cell body, at what rate and whether a vernier mechanism is involved in driving the filament. Lab: Howard Berg ______________ 57 BLAST XII ________________ Poster #7 A NON-CONSERVED ACTIVE SITE RESIDUE INFLUENCES RESPONSE REGULATOR REACTION KINETICS, PHOSPHODONOR SPECIFICITY, AND PARTNER KINASE INTERACTIONS Robert M. Immormino and Robert B. Bourret Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, NC 27599-7290 USA Two-component regulatory systems, minimally composed of a sensor kinase and a partner response regulator protein, are common mediators of signal transduction in microorganisms. All response regulators share a receiver domain with conserved active site residues that catalyze phosphorylation and dephosphorylation reactions. To understand the factors that endow different response regulators with their distinctive characteristics, our laboratory investigates the consequences of altering non-conserved portions of receiver domains, particularly the impact on reaction kinetics and specificity. In this study, we explored the effects of various amino acids at non-conserved position T+1 (one residue C-terminal of the conserved Thr/Ser) in the receiver domain active site. Position T+1 is most commonly occupied by the small residues Ala or Gly, sometimes by the slightly larger Ser or Thr, and occasionally by the much larger Val or Met. Karl Volz proposed 20 years ago (Volz, 1993) that the prevalence of small amino acids at position T+1 serves to allow access to the site of phosphorylation. Distinctive differences in amino acid preference at position T+1 between different response regulators suggest the T+1 residue has functional consequences. We chose Escherichia coli CheY, CheB, and NarL as representative of the Ala/Gly, Ser/Thr, and Val classes respectively and then used wildtype and mutant response regulators carrying various substitutions at position T+1 to assess (1) selfcatalyzed dephosphorylation; (2) self-catalyzed phosphorylation with phosphoramidate, acetyl phosphate, or monophosphoimidazole; and/or (3) phosphotransfer from the CheA or NarX sensor kinases. To help clarify the molecular mechanisms underlying the observed effects on reaction kinetics and phosphodonor specificity, we determined the X-ray crystal structures of four CheY mutants containing substitutions at position T+1 and constructed molecular models of phosphodonors in the active site. Our results help explain the distinctive distributions of amino acids at position T+1 in the CheB, CheY, and NarL response regulator families and may contribute to prediction of response regulator self-catalyzed reaction rates based on amino acid sequence. Reference Volz, K. (1993) Structural conservation in the CheY superfamily. Biochemistry 32, 11741-11753. Lab: Robert Bourret ____________ 58 BLAST XII ________________ Poster #8 NONCONSERVED ACTIVE SITE RESIDUES MODULATE CheY AUTOPHOSPHORYLATION KINETICS AND PHOSPHODONOR PREFERENCE Stephanie A. Thomas, Robert M. Immormino, Robert B. Bourret, and Ruth E. Silversmith Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, NC 27599-7290 In two-component signal transduction, response regulator proteins contain the catalytic machinery for their own covalent phosphorylation and can catalyze phosphotransfer from a partner sensor kinase or autophosphorylate using various small molecule phosphodonors. Although response regulator autophosphorylation is physiologically relevant and a powerful experimental tool, the kinetic determinants of the autophosphorylation reaction and how those determinants might vary for different response regulators and phosphodonors are largely unknown. We characterized the autophosphorylation kinetics of 21 variants of the model response regulator Escherichia coli CheY that contained substitutions primarily at nonconserved active site positions D+2 (CheY residue 59) and T+2 (CheY residue 89), two residues C-terminal to conserved D57 and T87, respectively. Overall, the CheY variants exhibited a >105-fold range of rate constants (k phos /K S ) for reaction with phosphoramidate, acetyl phosphate, or monophosphoimidazole, with the great majority of rates enhanced over wild type CheY. Although phosphodonor preference varied substantially, nearly all the CheY variants reacted faster with phosphoramidate than acetyl phosphate. Correlations between charge of the D+2/T+2 side chains and rate indicated electrostatic interactions are a kinetic determinant. Moreover, the effect of ionic strength varied with phosphodonor but not active site composition, suggesting at least two types of electrostatic interactions influence autophosphorylation kinetics. Nonpolar surface area of the D+2/T+2 side chains also correlated with autophosphorylation rate, especially for reaction with phosphoramidate and monophosphoimidazole. Computer docking strongly suggested that highly accelerated monophosphoimidazole autophosphorylation rates for CheY variants with a tyrosine at position T+2 likely reflect structural mimicry of phosphotransfer from the sensor kinase histidyl phosphate. Lab: Robert Bourret ____________ 59 BLAST XII ________________ Poster #9 EXPERIMENTAL ANALYSIS OF RECEIVER DOMAIN AUTODEPHOSPHORYLATION KINETICS GUIDED BY BIOINFORMATICS Stephani Page1, Robert Immormino2, Robert Bourret2 Departments of Biochemistry & Biophysics1 University of North Carolina Chapel Hill, NC 27599 and Microbiology & Immunology2 Receiver domains of response regulators contain the site of phosphorylation, which mediates responses by switching output function on and off. Receiver domains have highly conserved active site residues that catalyze phosphorylation and dephosphorylation. Despite the conserved active site and similarities in structure and sequence amongst receiver domains, self-catalyzed dephosphorylation of receiver domains reportedly occurs over a nearly million-fold range of reaction rates. Receiver domain autodephosphorylation rates appear to be tuned to the timescales of the different biological processes in which response regulators participate. For example, receiver domains involved in chemotaxis typically autodephosphorylate faster than those involved in transcriptional regulation. We want to understand what factors influence receiver domain autodephosphorylation rates and the mechanisms by which those factors exert influence. Sequence, structural, and kinetic analyses have drawn our focus to variable residues at the active site of receiver domains, in particular, residues at positions D+2 and T+2, two positions C-terminal to a conserved Asp and Thr/Ser, respectively, that mediate dephosphorylation chemistry. Specifically, residues at D+2 and T+2 (i) have the strongest coevolutionary relationship amongst residues found at any two positions in receiver domains, (ii) are positioned appropriately to influence catalysis, and (iii) affect autodephosphorylation rates when changed. For a more comprehensive assessment of the influence of D+2 and T+2 on receiver domain autodephosphorylation, we used bioinformatics to select D+2/T+2 pairs representing a large fraction of naturally occurring receiver domain sequences. Our collection of 48 mutants contains combinations of amino acids at D+2 and T+2 that are found in 60% of wild type receiver domain sequences in a nonredundant database of nearly 14,000 response regulators. Using E. coli response regulators CheY and PhoB as scaffolds, we assessed the effects of changing residues at positions D+2 and T+2 on autodephosphorylation rates. Autodephosphorylation rates of 42 CheY mutants spanned a 120-fold range. Six mutants of PhoB had autodephosphorylation rates spanning a 60-fold range. The range of autodephosphorylation rates observed was the result of both enhancing and diminishing substitutions. By changing just two variable active site residues, we have been able to “tune” CheY and PhoB to a range of different autodephosphorylation timescales, thus accounting for two out of six orders of magnitude in the reported range of receiver domain autodephosphorylation rates. We are currently exploring the mechanistic basis by which positions D+2 and T+2 influence receiver domain autodephosphorylation rates. Lab: Robert Bourret ____________ 60 BLAST XII _______________ Poster #10 THE BRUCELLA ABORTUS GENERAL STRESS RESPONSE SYSTEM REGULATES CHRONIC MAMMALIAN INFECTION, AND EXHIBITS NON-CANONICAL POST-TRANSLATIONAL REGULATION Hye-Sook Kim1,2 , Clayton C. Caswell3, Robert Foreman2,4, R. Martin Roop II3, and Sean Crosson1,2,4,* 1 Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, IL 60637, USA. 2 Howard Taylor Ricketts Laboratory, Argonne National Laboratory, Argonne, IL, 60439, USA. 3 Department of Microbiology and Immunology, East Carolina University School of Medicine, Greenville, NC 27834, USA. 4 The Committee on Microbiology, The University of Chicago, Chicago, IL 60637, USA. Brucella spp. are adept at establishing a chronic infection of mammalian host cells, though the mechanisms by which Brucella maintain long-term host residence remain poorly defined. We demonstrate that core components of the α-proteobacterial general stress response (GSR) system, PhyR and σE1, are top-level regulators of Brucella abortus virulence that are specifically required for maintenance of chronic animal infection. ∆phyR and ∆rpoE1 null mutants exhibit decreased survival in the face of oxidative and acid stress in vitro, but are not defective in infection of primary murine macrophages or initial colonization of BALB/c mice. However, ∆phyR and ∆rpoE1 mutants are cleared from a murine host beginning one month post-infection. Thus, the B. abortus GSR system affects a transcriptional program that is dispensable for initial host colonization, but is required to maintain longterm infection. We experimentally define the B. abortus GSR transcriptional regulon and show that it contains genes previously linked to virulence including genes required for oxidative and acid stress survival, and genes that affect immunomodulatory components of the cell envelope. These data support a model in which the GSR system regulates stress survival, and affects how B. abortus interfaces with the host immune system. We further present a molecular-level analysis of purified GSR proteins that defines unique features of regulation in B. abortus. In particular, we demonstrate that the cellular level of PhyR is controlled post-translationally by regulated proteolysis. PhyR proteolysis provides a mechanism to attenuate spurious PhyR protein interactions that would inappropriately activate the GSR transcriptional program. We conclude that the B. abortus GSR system regulates transcription of genes required for acute stress survival and long-term interaction between the bacterium and its mammalian host, and that PhyR proteolysis is a novel regulatory feature in B. abortus that ensures proper control of the GSR. Lab: Sean Crosson _____________ 61 BLAST XII _______________ Poster #11 STUDIES OF FLAGELLAR MOTOR PROTEIN FliG AND ITS INTERACTIONS WITH FliF AND FliM Robert Levenson, Armand Vartanian, Susan Gelman, Hongjun Zhou, Frederick W. Dahlquist Department of Chemistry and Biochemistry, University of California at Santa Barbara, Santa Barbara, CA, 93106 The cytoplasmic ring (C-ring) of the flagellar motor is composed of three proteins: FliG, FliM, and FliN, each present in different copy numbers. These proteins perform the function of transmitting torque from the stators to the basal body, as well as regulating the rotational direction of the flagellum. FliG is an entirely alpha-helical protein composed of three domains, FliG N , FliG M , and FliG C . There remain significant questions regarding the organization of the FliG domains within the C-ring. To gain insight into these questions, we have been studying these proteins using a variety of biophysical methods. The N-terminal domain of FliG, FliG N , has been known to interact with the membrane protein FliF in one of the first interactions during the assembly of the flagellar motor. We have previously studied the strong interaction of T. maritima FliG N with an NMR-unlabeled synthetic peptide corresponding to a 38 amino acid C-terminal portion of FliF (FliF C ). We have now extended the study of this complex by producing a fusion protein that coexpresses the two proteins in one polypeptide, and we present preliminary structural NMR data of this fusion protein. We also present NMR data from the FliF C -bound full-length FliG protein. This data, together with the recently-published crystal structure of the FliG MC -FliM complex from our lab as well as other results, combine to support a structural model of the C-ring that agrees very well with available cryo-EM data. We have also used NMR spectroscopy to study the interplay between FliG MC , FliM, and CheY. We present experiments using methyl-labelled FliG MC that indicate that FliG C and CheY competitively bind to FliM, extending previous in vitro results that used individual FliG M or FliG C domains to the full FliG MC protein. Lab: Frederick Dahlquist ________ 62 BLAST XII _______________ Poster #12 CHEMOTAXIS SIGNALING PATHWAY OF THE FLAGELLAR SYSTEM 2 FROM RHODOBACTER SPHAEROIDES Ana Martínez del Campo1, Teresa Ballado1, Javier de la Mora1, Laura Camarena2 and Georges Dreyfus1. 1 Departamento de Genética Molecular, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México. Circuito Exterior S/N Ciudad Universitaria, 04510 México D.F. 2 Departamento de Biología Molecular y Biotecnología, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México. Rhodobacter sphaeroides posses several reiterated chemotactic genes: 4 cheW, 2 cheB, 3 cheR, 4 cheA and 6 cheY, which are encoded in three loci cheOp1, cheOp2 and cheOp3. Moreover, R. sphaeroides genome contains two flagellar gene clusters. The genes of the first cluster are constitutively expressed under laboratory conditions and are required for the synthesis of the wellcharacterized single subpolar flagellum (Fla1); which allows the swimming of the WS8 wild-type strain. The genes of the second flagellar cluster are not expressed in the wild-type strain, however strains that express it have been isolated. When expressed, the Fla2 genes of R. sphaeroides produce polar flagella that are required for swimming. When the cell is swimming with the Fla1 flagellum only cheOp2 and cheOp3 are essential. In contrast CheY1, CheY2 and CheY5, which are encoded in cheOp1, are required for the chemotactic control of the Fla2 flagella. Here we report that when R. sphaeroides swims with Fla2 flagella, cheOp1 controls its chemotactic behavior. Additionally, the molecular dissection of cheOp1 allowed us to identify a novel component of chemotaxis signaling pathway of the Fla2 flagella from R. sphaeroides; which we are currently investigating. This project is supported by grants IN-206811 from DGAPA and 106081 from CONACyT. A.M.-D.C. is a recipient of a Robert M. Macnab Memorial Grant to attend BLAST XII. Lab: Georges Dreyfus __________ 63 BLAST XII _______________ Poster #13 THE C TERMINUS OF THE FLAGELLAR MURAMIDASE SltF MODULATES THE INTERACTION WITH FlgJ IN RHODOBACTER SPHAEROIDES Manuel Osorio-Valeriano1, Javier de la Mora1, Teresa Ballado1, Laura Camarena2 y Georges Dreyfus1*. Instituto de Fisiología Celular1, Instituto de Investigaciones Biomédicas2, Universidad Nacional Autónoma de México. Ap. Postal 70-243, 04510 México, D.F., México. Tel. 56225618. Macromolecular structures such as the bacterial flagellum must traverse the cell wall, for this reason, the assembly of some of these structures requires the activity of peptidoglycan degrading enzymes. In Salmonella enterica, FlgJ has been characterized as a bifunctional protein, acting as a scaffold for rod assembly and also acting as a muramidase degrading the PG layer to facilitate rod penetration. The FlgJ homologue in R. sphaeroides lacks the muramidase domain. In a previous study we have characterized a flagellum specific muramidase SltF, whic interacts with the scaffold protein FlgJ. We proposed that this interaction occurs in the periplasm during rod formation and helps localize SltF at the flagellar assembly site. In this study, we demostrate that the export of SltF to the periplasm is dependent on the SecA pathway. Additionally we carried out a deletion analysis of the C-terminal portion of SltF, the resulting truncated mutants were unable to restore wild-type swimming in a Δsltf genetic background. These mutants retained their catalytic activity, however they lost the SltF-SltF interaction and also show a higher affinity binding FlgJ, when compared to the wild-type protein. We propose that this region modulates the interaction with the scaffold protein FlgJ during the flagellum biogenesis. This study was supported by grants from CONACyT (106081) and DGAPA/UNAM (IN206811-3). Lab: Georges Dreyfus __________ 64 BLAST XII _______________ Poster #14 PROFOUND EFFECT OF HOOK LENGTH ON MOTILITY AND VIRULENCE IN SALMONELLA Christian Hotz1, Hanna M. Singer1, Carole Bourquin2 and Marc Erhardt1 1 Département de Médecine, Université de Fribourg, 1700 2 Helmholtz Centre for Infection Research, 38124 Braunschweig, Germany Fribourg, Switzerland Introduction and Aims Bacteria swim through liquid environments by rotating a rigid, helical organelle, the flagellum. The flagellum enables bacteria to swim towards nutrients and away from harmful substances. In addition, flagellar motility contributes to bacterial pathogenesis by promoting bacteria-host interactions, biofilm formation, adherence and invasion of epithelial cells. The structure of the bacterial flagellum consists of a long external filament connected to a membrane-embedded basal-body at the cell surface by a short curved structure, the hook. The hook is a flexible universal joint and transmits the motor torque to the helical propeller over a wide range of its orientation for swimming and tumbling. In Salmonella enterica the hook structure extends 55 nm beyond the cell surface. The length of the hook is controlled via intermittent secretion of a molecular ruler molecule. To address the question why length of the hook structure is tightly controlled, we analyzed various hook length variation mutants for their effects in motility and invasion assays. Material and Methods Various deletion and insertions mutants of FliK, the molecular ruler protein responsible for hook length control, were analyzed in a battery of swimming and swarming motility assays, as well as for their capability to infect macrophages. Deletions and insertions in FliK give rise to shorter and longer hook structures, respectively. Key Results We demonstrate that the variation of flagellar hook length has a profound effect on motility and virulence in Salmonella. Deletion and insertion variants of the FliK molecular ruler were secreted and resulted in induced the substrate specificity switch as shown by assembly of flagellar filaments. Long hook mutants displayed a defect in swarming and swimming motility, as well as a profound defect in their capability to invade both macrophages and epithelial cells in a Gentamicin protection assay. Conclusion Our results suggest that the flagellar hook length of 55 nm in Salmonella enterica has evolved to match the requirements for optimal motility. The ability to efficiently move in a directed manner confers distinct advantages. Potential benefits of efficient motility in Salmonella include the ability to search for nutrients, to avoid toxic substances and facilitate contact with eukaryotic host cells for invasion. The costs of motility are significant, including the metabolic burden of flagellar biosynthesis and the energetic expenses for flagellar rotation. Thus, it is not surprising that the flagellar structure is subject to strict control and has evolved to provide the optimal requirements for efficient motility. Lab: Marc Erhardt ______________ 65 BLAST XII _______________ Poster #15 STRUCTURAL AND FUNCTIONAL DISULFIDE MAPPING OF THE CheA-CheW INTERFACE IN THE BACTERIAL CHEMOSENSORY ARRAY Andrew M. Natale and Joseph J. Falke Molecular Biophysics Program, and Department of Chemistry and Biochemistry, University of Colorado at Boulder, UCB 596, Boulder, CO 80309-0596 The bacterial chemosensory array, which senses gradients of attractant and repellent ligands and accordingly modulates cellular swimming behavior, is composed of a highly ordered, ultrastable protein lattice. The repeating core unit within the lattice consists of a membrane-spanning trimer of receptor dimers, a dimeric histidine kinase CheA bound at the receptor tips, and two scaffolding CheW monomers. Signals are transduced from the periplasmic ligand binding receptor domains to the cytoplasmic receptor tips where they control the activity of the kinase. An increasing body of recent evidence has begun to paint a picture of the large scale architecture of the lattice, while other studies have illuminated more of its specific protein-protein contacts. While the picture of the array is becoming clearer, questions remain about the identities of the packing interfaces between core components and about the nature of signal transduction through these interfaces. To address some of these questions, the present study examines the CheA kinase – CheW scaffolding protein interface using a strategy of disulfide mapping. While the structure of isolated CheA regulatory domain in complex with CheW has been solved, we aim to probe the interface structure in active, membrane bound arrays reconstituted in vitro from the three full length core proteins. Using combinations of single cysteine CheA and CheW mutants we are able to form interfacial disufides and map contact points. We can use similar experimental techniques to identify signaling induced changes in packing or dynamics at the interface, as well as to test whether this interface plays a role in mediating the significant cooperativity observed in the array. Our latest findings will be presented at the meeting. Lab: Joseph Falke ______________ 66 BLAST XII _______________ Poster #16 ANALYSIS OF THE REPELLENT-SENSING MECHANISMS OF ESCHERICHIA COLI Hirotaka Tajima1, Takaya Inui2, Eri Iijima3, Ken Konemura3, Hisako Matsuzawa3, Toshio Fukuda1, Michio Homma4, Ikuro Kawagishi2,3 1: Graduate School of Engineering, Nagoya Univesity, 2: Graduate School of Engineering and 3: Faculty of Bioscience and Applied Chemistry, Hosei University, Koganei 184-8584, and 4: Graduate. School of Science, Nagoya University Chikusa-ku, Nagoya 464-8603, Japan E. coli recognizes the environmental factors such as chemical compounds, temperature, and osmotic pressure, and migrates toward or from the factors by altering the direction of flagellar rotation. Above all, behavior of to the chemical compounds, including some amino acids, sugars, metal ions, termed as chemotaxis, is the most famous and best characterized. All components of chemotaxis have been identified and studied about them. Four chemoreceptors of E. coli (Tar, Tsr, Trg and Tap) can sense variety of factors such as amino acids, sugars, metal ions, indole, pH and temperature. Chemotactic signals are transferred by phosphor-relay of histidine kinase CheA and response regulator CheY. Chemoreceptors produce signals by the regulation of CheA, however, the mechanism is poorly understood. Especially, the sensing mechanisms of repellents, such as, Ni2+ and indole, are less understood than that of attractants. Then we analyzed the repellent-sensing mechanism of chemoreceptors. kcal/mol µcal/sec. time (min.) time (min.) time (min.) Tar of E. coli mediates 0 50 100 0 50 100 0 50 100 repellent responses to the divalent 0 0 0 cations, Ni2+ and Co2+. NikA was -0.03 -0.025 presumed to serves as a primary -0.06 -0.3 receptor. However, Englert et al. (2010) -0.05 -0.2 0 reported that E. coli requires neither the 0 -0.3 Ni2+-binding protein (NikA) nor the -0.4 -2 2+ -0.6 transmembrane components of the Ni -0.6 transporter (NikB and NikC) for a Ni2+ -4 -0.9 0 3 6 0 3 6 0 3 6 molar ratio molar ratio molar ratio response. Then we performed ITC Tsr / NiSO4 / NiSO Tarof/ CoSO measurements of Ni2+ binding to the FigureTar 1: ITC measurements of binding Ni2+ to the fragment 4 4 Tar periplasmic periplasmic fragment of Tar. The addition of Ni2+ to the Tar fragment caused heat release, indicating the direct binding of Ni2+ (Figure 1). Furthermore, we examined the properties of chimeric proteins Tar and Tsr to determine the region of Tar responsible for Ni2+ sensing. Indole is known as an autoinducer, and increases the drug resistance and inhibits the biofilm formation of E. coli. It is known that repellent response to indole is mediated by Tsr. However, we found that cells expressing Tar as a sole chemoreceptor attracted to high concentration of indole (2.5 mM) (Figure 2). The analysis using chimeric receptors and pin-head Tar indicated that periplasmic domain of Tar is dispensable for indole sensing. Here, we will show the current progress of analysis of the repellent-sensing mechanisms. Figure 2: tethered cell assay with indole Lab: Toshio Fukuda ____________ 67 BLAST XII _______________ Poster #17 IDENTIFICATION FOUR NEW GGDEF/EAL DOMAIN PROTEINS THAT REGULATE MOTILITY IN SALMONELLA, ONE OF WHICH LOCALIZES TO THE MID-CELL AND INHIBITS CELL DIVISION Hyo Kyung Kim, Vincent Nieto and Rasika M. Harshey Section of Molecular Genetics and Microbiology, University of Texas at Austin, Austin, TX 78712 Cyclic-di-GMP (c-di-GMP) levels control the transition between motility and sessility. This molecule is produced by diguanylate cyclases (DGCs) having a catalytic GGDEF domain and broken down by phosphodiesterases (PDEs) containing an EAL domain. Our model organism Salmonella enteric has 5 GGDEF domain proteins, 8 EAL domain proteins and 7 proteins having both GGDEF and EAL domains. Thus far, absence of only the specific phosphodiesterase YhjH impairs motility in this bacterium. Using the yhjH mutant as the starting strain, we have identified 4 additional GGDEF/EAL domain proteins which affect motility. Most of these proteins have an N-terminal receiver domain, coupling environmental signals to c-di-GMP levels in the cell. To test whether these proteins provide a spatially compartmentalized c-di-GMP pool in the cell, we initially localized them by using fluorescent fusion proteins. 4 of the 5 GGDEF/EAL proteins showed polar localization, suggesting that this location was either non-specific or that they were associating with the chemoreceptor complex at the poles. Interestingly, 1 GGDEF protein showed a striking spiral pattern all along the length of the cell that was most obvious at the mid-cell position. This pattern is remarkably similar to that of FtsZ tubulin spirals, thought to be early intermediates in formation of the division ring. Accordingly, mutant cells of this protein were shorter than wild-type cells, and increased protein expression (native protein or GFP fusion protein) produced longer cells. The localization of a DGC at division sites suggests a role for cdi-GMP signaling in cell division. Lab: Rasika Harshey _______________ 68 BLAST XII _______________ Poster #18 SIGNALLING SYSTEMS CONTROLLING DEVELOPMENTAL CELL FATE SEGREGATION IN MYXOCOCCUS xanthus Andreas Schramm, Vidhi Grover, and Penelope I. Higgs Max Planck Institute for Terrestrial Microbiology, Dept. of Ecophysiology, 35043, Marburg, Germany Myxococcus xanthus is a model organism for regulation of complex behavior. Under nutrient limiting conditions, these bacteria enter a multicellular developmental program wherein cells follow different fates: aggregation into mounds (fruiting bodies) followed by differentiation into environmentally resistant spores; differentiation into a persister-like state termed peripheral rods; programmed cell death; or cell clusters, a recently identified population whose role is not well understood. At least four distinct histidine-aspartate phosphorelay (aka “two-component”) signaling systems are necessary for appropriate cell fate segregation. Our results suggest that each of these signaling systems independently regulates accumulation of an important transcriptional regulator, MrpC. Consistently, we have demonstrated that MrpC accumulates heterogeneously in the developing cell population; rapid MrpC accumulation is observed in cells induced to migrate into fruiting bodies, while little accumulation is observed in cell clusters or in cells destined to become peripheral rods. Our data indicate that one of these signaling systems, Esp, regulates proteolytic turnover of MrpC. The Esp signaling system consists of two hybrid histidine kinases, EspA and EspC. Phosphorylation of the receiver domains from both EspA and EspC is necessary to stimulate MrpC turnover, and this occurs via a novel inter- and intra-hybrid kinase signaling mechanism which will be presented. Interestingly, the EspA:EspC ratio varies between developmental subpopulations suggesting that the relative per cell signaling efficiency is controlled not just by signals perceived by EspA and EspC , but also by the respective levels of the signaling components themselves. A model for the role of Esp and the remaining signaling systems which control production of MrpC and cell fate segregation will be presented. Lab: Penelope Higgs ___________ 69 BLAST XII _______________ Poster #19 ATTEMPT TO INVESTIGATE THE INTERACTION BETWEEN THE ROTOR AND THE STATOR BY USING SOLUTION NMR Mizuki Gohara1, Rei Abe-Yoshizumi1, Shiori Kobayashi1, Yohei Miyanoiri2, Yoshikazu Hattori3, Chojiro Kojima3, Masatsune Kainosho2,4, Michio Homma1 1 Division of Biological Science, Graduate School of Science, Nagoya University, Chikusa-ku, Nagoya, Aichi, Japan 464-8602; 2Structural Biology Research Center, Graduate School of Science, Nagoya University, Chikusa-ku, Nagoya, Aichi, Japan 464-8602; 3Institute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka, Japan 565-0871; 4Center for Priority Areas, Tokyo Metropolitan University, 1-1 Minamiosawa, Hachioji, Toyko, Japan 192-0397; In the bacterial flagellar motor, torque is generated by the interactions between rotor and stator. In the rotor, a component FliG, especially at its C-terminal domain, is mainly participated in torque generation. In the H+-driven motor of Escherichia coli, mutational analyses suggested that the interaction is mediated with charged residues between the FliG C-terminal domain and the MotA cytoplasmic loop region. However, in the Na+-driven motor of Vibrio, the effects of mutations in the charged residues on motility were very weak. So the important residues for torque generation in the Na+-driven motor may be different from the H+-driven motor. We want to observe the interaction between FliG and PomA (MotA ortholog) in the Na+-driven motor and decided to use the solution NMR. In the previous meeting, we reported that N-terminally truncated FliG variants G214-FliG (FliGC) gave much better NMR spectra by 1H-15N TROSY-HSQC. Here, we would like to present the results of the measurement of the 3D-NMR(1H-13C-15N) and the on going assignments of the signals to amino acids. Furthermore, we purified intact PomA, the PomA/B complex, and the PomA fragments and currently we are trying to measure NMR spectra of FliGC in the presence and absence of the purified proteins to investigate residues involved in the rotor-stator interaction. Lab: Michio Homma ____________ 70 BLAST XII _______________ Poster #20 IMPORTANCE OF CHARGED RESIDUES OF POMA AND FLIG FOR ROTOR-STATOR INTERACTION IN THE SODIUM-DRIVEN FLAGELLAR MOTOR OF VIBRIO ALGINOLYTICUS Norihiro Takekawa, Seiji Kojima and Michio Homma Division of Biological Science, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan Bacterial flagellar motor is a reversible rotary motor located at the base of the flagellum. The flagellar motor is consisted of two parts, rotor and stator. Essential components for torque generation in the rotor is located at the C-ring composed of FliG, FliM, and FliN, and the stator is composed of the membrane proteins MotA and MotB in H+-driven motor and PomA and PomB in Na+-driven motor that form ion channel structure. In the H+-driven motor of E. coli, it was suggested that the interactions between conserved charged residues of FliG and MotA were important for the torque generation of the motor. However, the presence of such interactions had been unclear in the Na+-driven motor of V. alginolyticus. In this study, we systematically made mutants whose charged residues of FliG and PomA were replaced to uncharged or charge-reversed ones, and examined motility of those Vibrio mutants. We found that some fliG/pomA double mutations gave the synergetic effects or suppression on the motor rotation. Our results suggested that interactions between some charged residues of FliG and those of PomA were important for force generation of Na+-driven flagellar motor in V. alginolyticus. In the H+-driven motor, smaller number of electrostatic interactions between charged residues at the rotor-stator interface are good enough for motor function, whereas in the Na+-driven motor of Vibrio requires more charged residues at the rotor-stator interface. Such larger number of interactions may cause robustness against the mutations at the rotor-stator interface in the Vibrio motor, and its remarkably high-speed rotation. Lab: Michio Homma ____________ 71 BLAST XII _______________ Poster #21 VIBRIO ALGINOLYTICUS REGULATION OF THE FLAGELLAR NUMBER IN: ROLE OF ATP BINDING MOTIF OF FlhG AND THE NOVEL PROTEIN SflA Seiji Kojima, Takehiko Nishigaki, Hiroki Ono and Michio Homma Division of Biological Science, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan Bacterial flagella are expressed in a wide variety of locations and numbers. Marine bacterium Vibrio alginolyticus has a single polar flagellum whose number is regulated positively by putative GTPase FlhF and negatively by putative ATPase FlhG. FlhF is also involved in its polar positioning in flagellar formation. Our mutational and biochemical analyses showed that FlhG interacts with FlhF to prevent FlhF from localization at cell pole. Here we investigated the role of ATP binding motif of FlhG. Since FlhG is the homolog of the cell division inhibitor MinD, we mutated four residues in the ATP binding motif of FlhG, whose corresponding mutations in MinD disrupted its ATPase activity. Two of them (K31A and K36Q) could not complement the flhG defective mutant on motility. Investigation of subcellular localization and binding to FlhF of these FlhG mutants are now ongoing. We also isolated a suppressor mutant from the ∆flhFG strain that only occasionally confers peritrichous flagella in a small number of cells. The responsible mutation was mapped to the non-flagella-related gene. This novel gene, named sflA, is specific for Vibrio and is speculated to encode a transmembrane protein with a DnaJ domain. Expression of the C-terminal cytosolic region of SflA containing DnaJ domain is sufficient to suppress the flagellar generation. SflA is detected in the membrane fraction by immunoblot using an antibody raised against the N-terminal region of SflA. Further analyses using the C-terminal GFP-fusion variant are now ongoing and SflA function will be discussed. Lab: Michio Homma ____________ 72 BLAST XII _______________ Poster #22 EFFECT OF CODON CONTEXT ON TRANSLATION SPEED IN VIVO Kelly T. Hughes, Chakib Mouslim and Valentina Rosu Department of Biology, University of Utah, Salt Lake City, UT 84112 Codon context plays a significant role in determining the speed at which the ribosome translates through codon pairs. We have isolated a translation-slow “silent” mutant in the flgM gene of Salmonella at amino acid codon 8. This results in inhibition of flgM translation and a restart of flgM translation at a methionine codon near the 3’end of the 97 amino acid coding sequence. We developed a regulatory system that provides a simple, yet elegant mechanism to determine the relative, in vivo speed of the ribosome as it translates through codons and codon pairs in a manner that is independent of protein and mRNA stability. The system utilizes the leader sequence of the Salmonella histidine biosynthetic operon. Surprisingly, the rate at which the ribosome translates through single codon pairs in the his leader RNA shows significant variation even for different codons used by a single tRNA species. Our system allows us to rank codon pairs according to in vivo translational speed, and readily identifies the fastest and slowest among the 3,721 coding pairs. Choice of slow codon pairs might be important to allow proper protein folding, or differential expression levels in the products of polycistronic transcripts. Of all possible amino acid-coding codon pairs, approximately 50, including all 16 Pro-Pro pairs result in ribosome stalling in the His leader peptide equivalent to what is observed with a stop codon. Implications for gene expression and evolution are discussed. Lab: Kelly Hughes ______________ 73 BLAST XII _______________ Poster #23 SALMONELLA fliC mRNA TRANSLATION Fabienne F.V. Chevance, Soazig Le Guyon and Kelly T. Hughes Department of Biology, University of Utah, Salt Lake City, UT 84112 Production of the filament subunit protein FliC is regulated by multiple mechanisms. The fliC gene is transcribed from a σ28-dependent flagellar class 3 promoter, which is subject to FlgM inhibitory activity coupled to hook-basal body formation. Production of FliC is also regulated at the posttranscriptional. The FljA protein binds sequences in the 5’-untranslated region of fliC mRNA to inhibit translation. A mutant duplicated for FljA binding domain in the fliC 5’UTR is actually dependent on FljA for fliC mRNA translation. We have uncovered a second translational control mechanism in the 5’UTR of fliC mRNA that is independent of FljA. A predicted stem-loop region near the 5’-end of the transcript, SL2, was discovered as an inhibitory element for fliC mRNA translation. A single base change that is predicted to destabilize SL2 formation results in the inhibition of fliC mRNA translation. An “opened” SL2 region is believed to act to inhibit fliC mRNA translation because complete deletion of the SL2 sequence results in an increase in FliC production over wild-type. Furthermore, an 12 base sequence coding for amino acids 12 through 15 of FliC (12-15 coding) is complementary in sequence to SL2. Mutations that are predicted to increase 12-15 coding sequence interaction with SL2 sequence result in decreased FliC production. Mutations that are predicted to decrease 12-15 coding sequence interaction with SL2 sequence result in increased FliC production. A transposon mutagenesis experiment was developed to identify genes whose products are required for fliC mRNA translation, but not for fliC transcription. This was done using either wild-type fliC sequence, mutants that have enhanced SL2 interaction with the fliC amino acid 12-15 coding region (translation-down) and mutants that have decreased interaction between SL2 and the fliC amino acid 12-15 coding region (translation-up). Insertions mutants isolated included those affecting multi-drug efflux pumps, tolA and acrB, and a number of genes affecting outer membrane/cell wall assembly, surA, rfaC, rfaQ, rfbA and rfbH, in addition to known genes required for general mRNA translation rne and prfA. The role of outermembrane/cell wall integrity in fliC mRNA translation will be discussed. Lab: Kelly Hughes ______________ 74 BLAST XII _______________ Poster #24 EFFECT OF A SINGLE motP MUTATION FOR MOTILITY AT NEUTRAL pH OF THE Na+-DRIVEN FLAGELLAR MOTOR OF BACILLUS PSEUDOFIRMUS OF4 Yuka Takahashi, Yukina Noguchi, Masahiro Ito Graduate School of Life Sciences, Toyo University Many bacteria swim by rotating helical flagellar filaments that act as screw propellers. There is a motor that consists of a rotor and some stators at the base of the flagellar. Stators such as the MotAB complex work as an ion channel. The flagellar motor is driven by the electrochemical potential of a coupling ion, H+ or Na+. Direct interactions between the rotor protein FliG and the stator protein MotA are thought to generate the rotational torque. Escherichia coli and Salmonella typhimurium have a MotAB complex as a stator and the flagellum is driven by a proton-motive force. On the other hand, alkaliphilic Bacillus and marine bacteria, such as Vibrio species, have a MotPS or PomAB complex as a stator and the flagellum is driven by a Na+-motive force. Alkaliphilic Bacillus pseudofirmus OF4 has a MotPS complex as a stator and the flagellum is driven by a sodium-motive force. Previous studies showed that B. pseudofirmus OF4 swimming is dependent on the Na+ concentration at pH 8-10, but showed poor motility at neutral pH even in the presence of a high Na+ concentration. It was hypothesized that there could be competitive inhibition by H+ of the Na+ translocation by the stator-force generator MotPS (1). On the other hand, B. subtilis has a MotPS complex similar to B. pseudofirmus OF4. However, it was reported that B. subtilis swimming is dependent on the Na+ concentration at neutral pH (2). Here, we made a mutant strain of motPS from B. pseudofirmus OF4 that was expressed in the stator deletion strain of B. subtilis (ΔmotABΔmotPS), and we investigated whether the mutant strain can complement motility on a soft agar plate. In addition we observed whether the mutant strain exhibited motility at neutral pH. In this study, we identified that an amino acid residue in MotP is involved in competitive inhibition by H+ in the Na+-driven flagellar motor of B. pseudofirmus OF4. 1) Fujinami, S. et al. (2007) Arch. Microbiol. 187: 239-247 2) Ito, M.et al. (2005) J. Mol. Biol., 352: 396-408 Lab: Masahiro Ito ______________ 75 BLAST XII TOWARDS A MECHANISM CRESCENTUS _______________ Poster #25 FOR SURFACE MECHANO-SENSATION IN CAULOBACTER Isabelle Hug1, Siddharth Deshpande2, Thomas Pfohl2, Urs Jenal1 1 Biozentrum, 2Department of Chemistry, University of Basel, Switzerland The bimodal reproductive program of C. crescentus upon division generates two distinct daughter cells, a sessile stalked and a flagellated swarmer cell. While the newborn stalked cell reinitiates growth, replication and division immediately, the swarmer progeny remains in G1 for a defined time before differentiating into a sessile stalked cell. During this process it looses its polar flagellum and pili and assembles an adhesive organelle, the holdfast, at the same pole. The progression through the cell cycle is controlled by the interplay between cyclic di-GMP signaling and phosphorylation networks, following the dogma that high intracellular cyclic di-GMP levels favor settlement and surface attachment, whereas low levels correlate with planktonic behavior. Recently it was shown that swarmer cells can produce holdfast and attach immediately when encountering surface (Li G. et al., Mol Microbiol, 2012). This suggested that the cell cycle program that directs the motilesessile transition can be overridden and accelerated when cells mechanically sense solid surface. To decipher the molecular mechanisms responsible for mechano-sensation and to explore a potential role of cyclic di-GMP regulation we used microfluidic devices to observe single dividing C. crescentus cells and their offspring when challenged with surface. Using this assay we could confirm rapid surfacemediated attachment of newborn swarmer cells and the strict dependence of this process on pili, a rotating flagellar motor and an intact holdfast machinery. Furthermore, surface induced attachment was also dependent on the diguanylate cyclase DgcB, indicating a role for cyclic di-GMP signaling in this process. Interestingly, the rapid surface-mediated transition to sessility was not accompanied by cell cycle progression into S-phase, arguing that two c-di-GMP dependent processes in C. crescentus, surface adherence and S-phase entry, are not interconnected. These findings suggest that C. crescentus can effectively and rapidly uncouple the tight interplay between cell cycle progression and pole differentiation and that mechano-sensing processes override the cell cycle program when cells encounter surface. Lab: Urs Jenal _________________ 76 BLAST XII _______________ Poster #26 SURFACE ACCESSIBILITY STUDY IDENTIFIES DOMAIN INTERACTIONS IN THE AEROTAXIS RECEPTOR Aer Darysbel Perez, Kylie J. Watts, Mark S. Johnson, and Barry L. Taylor Division of Microbiology and Molecular Genetics, Loma Linda University, Loma Linda, CA, 92350 The Escherichia coli aerotaxis receptor, Aer, monitors cellular redox changes via FAD, which is non-covalently bound to a PAS sensing domain. Changes in the redox state of FAD propagate from the PAS domain to downstream HAMP and proximal signaling domains. Our recent disulfide crosslinking studies demonstrate that the PAS β-scaffold can collide with the HAMP domain, and suggest that the PAS β-scaffold is a site of PAS-HAMP interaction. If so, one would predict that surface residues in the PAS β-scaffold would be partially hidden by the HAMP domain and would be less accessible to a reactive hydrophilic probe. To map PAS surface regions that are shielded in Aer, we created cysteine replacements for 57 PAS residues predicted to be surface accessible in an Aer-PAS homology model. Two additional substitutions predicted to face the PAS interior served as inaccessible controls. The accessibility of the substitutions was tested in vivo using the probe methoxypolyethylene glycol-maleimide 5000 (PEGmal). All mutants were tested for function as a proxy for native folding; only Aer-W94C abolished aerotaxis. Residues were classified as having low (0-30% PEGylation), intermediate (31-50% PEGylation), or high (≥51% PEGylation) accessibility according to the extent that the residue bound PEG-mal in vivo. Residues in the N-terminus of the PAS domain (the N-cap) were highly accessible (70-113% PEGylated), suggesting that the N-cap is flexible and/or does not stably interact with other domains in Aer. In contrast, a large area in the PAS β-scaffold (1,497 Å2) was inaccessible (0-26% PEGylated, similar to inaccessible controls), and overlapped with a cluster of previously identified PAS signal-on lesions, and with the proposed PAS-HAMP interaction site. Of the remaining residues tested, 9 had low, 13 had intermediate, and 13 had high accessibility. Notably, nine of the residues with intermediate accessibility bordered the hidden β-scaffold region and may delineate the boundary of the PAS-HAMP contact surface. To highlight potential PAS-PAS interaction sites, we tested whether the PAS cysteine replacements could form disulfides in vivo in response to the oxidant copper phenanthroline. We found that several of these cysteine replacements could form disulfides. Some of them occurred at scattered positions around the PAS domain, but most of the PAS-PAS disulfides were in the flexible, Nterminal region, and were likely between (not within) dimers. Importantly, none of the cysteine replacements in the inaccessible part of the PAS β-scaffold formed PAS-PAS disulfides. To further map the PAS-HAMP interacting region, we chose 10 cysteine replacements in the hidden region of the PAS domain and monitored disulfide formation between these and HAMP-Q248C. Two of these cysteine replacements formed disulfides (V100C-Q248C and M112C-Q248C) (11-16% crosslinking) and were located in the inaccessible region on the PAS β-scaffold. The remaining PASCys mutants surrounded this region but failed to crosslink, giving a clearer understanding of the PASHAMP interaction site. Together these data provide a putative contour map of PAS contact surfaces, and provide novel insights into the PAS-HAMP signaling mechanisms used by Aer-type chemoreceptors. Lab: Barry Taylor ______________ 77 BLAST XII _______________ Poster #27 ROLE OF ENERGY SENSOR TlpD OF HELICOBACTER PYLORI IN GERBIL COLONIZATION AND WHOLE GENOME ANALYSES AFTER ADAPTATION IN THE GERBIL Wiebke Behrens1, Tobias Schweinitzer1, Sarah Klose1, Friederike Kops1, Birgit Brenneke1, Martina Dorsch2, André Bleich2, Sebastian Suerbaum1, Peter Loewen3, Jonathan McMurry4, Christine Josenhans1 1 Department for Medical Microbiology and Hospital Epidemiology, Medical School Hannover, Carl-Neuberg-Straße 1, 30625 Hannover, Germany 2 Central Animal Laboratory, Medical School Hannover, Hannover, Germany 3 Department of Microbiology, University of Manitoba, Winnipeg, Canada 4 Department of Chemistry and Biochemistry, Kennesaw State University, Kennesaw, USA Helicobacter pylori maintains colonization in its human host using a limited set of taxis sensors. One sensor which appears dominant under environmental conditions of low bacterial energy yield is TlpD, a proposed energy taxis sensor of H. pylori (Schweinitzer et al., 2008). Previous in vivo colonization data of a H. pylori TlpD mutant in a mouse model (Williams et al., 2007) had indicated that TlpD may not be essential in vivo. In addition, the mechanism of TlpD function remained unclear. One important open question is the impact of H. pylori TlpD in vivo which was explored here using a gerbil infection model, which closely mimicks the gastric physiology of humans. This approach included whole genome analyses and comparisons of gerbil-adapted H. pylori strains. A gerbil-adapted H. pylori strain, HP87 P7, was able to perform TlpD-dependent energy behavior, while its isogenic mutant in tlpD had lost it. A complemented strain regained the ability. Gerbil infections demonstrated that TlpD was essential for the initial colonization in the antrum as well as for persistence. A long-term effect of tlpD-deficiency in reducing H. pylori colonization was observed both in the antrum and corpus region of the gerbil stomach. The complete genome sequence of the gerbiladapted strain HP87 P7 was elucidated and compared with HP87 pre-gerbil, with a HP87 P7 tlpD clone pre-gerbil and with a reisolate pool of HP87 P7 tlpD which persisted during the gerbil infection. Genetic differences of the strains after gerbil adaptation were observed but no major losses or gains of genes or gene functions. The tlpD mutant gerbil reisolate also had acquired several SNPs in comparison to the gerbil-adapted wild type strain and the HP87 P7 tlpD input strain. The occurrence of potential adaptations to the animal host or mutations compensating for the loss of TlpD in the gerbil model will be discussed. Schweinitzer T, Mizote T, Ishikawa N, Dudnik A, Inatsu S, Schreiber S, Suerbaum S, Aizawa S, Josenhans C, 2008. J. Bacteriol. 190:3244-55. Williams SM, Chen YT, Andermann TM, Carter JE, McGee DJ, Ottemann KM, 2007. Infect Immun. 75:3747-57. Lab: Christine Josenhans _______ 78 BLAST XII INTERCONNECTIVITY ESCHERICHIA COLI _______________ Poster #28 BETWEEN TWO LytTR-LIKE TWO-COMPONENT SYSTEMS IN Stefan Behr, Luitpold Fried, and Kirsten Jung Center for Integrated Protein Science Munich (CiPSM) at the Ludwig-Maximilians-University Munich, Microbiology, Großhaderner Str. 2-4, 82152 Martinsried, Germany Bacteria use two-component systems (TCSs) to encounter fluctuating environmental conditions. A membrane-bound histidine kinase (HK) senses a stimulus and transduces it into a cellular signal via phosphorylation. The transfer of this phosphoryl group to a response regulator (RR) with DNA-binding properties mediates the inert reaction, generally an alteration in gene expression (1). Based on the limited number of TCSs in Escherichia coli (30/32 HK/RR) it is necessary to coordinate cellular adaptions in order to respond to a multitude of environmental signals. To this end many so called auxiliary proteins have been described recently (2). These proteins can be involved in sensing, scaffolding or connecting TCSs and evolved to an emerging field of bacterial signal transduction. Both LytS/LytR-like TCSs of E. coli, the YehU/YehT TCS (3) and YpdA/YpdB TCS are studied in our laboratory. Based on bioinformatical data these two TCSs share an amino acid identity of more than 30%. They are found in many γ-proteobacteria (4). The characterization of the YehU/YehT and the YpdA/YpdB systems revealed reversed transcriptional effects on target genes. Using the bacterial adenylate cyclase-based two-hybrid system YehS was uncovered as hub connecting the two TCSs via protein-protein interactions. Surface plasmon resonance spectroscopy with purified YehS and the RRs confirmed the interactions and suggest an interconnectivity between YehU/YehT and YpdA/YpdB. References: 1) Stock et al. (2000): Two-component signal transduction. Annu Rev Biochem 69:183-215 2) Jung et al. (2011): Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 2:118-24 3) Kraxenberger et al. (2012): First insights into the unexplored two-component system YehU/YehT in Escherichia coli. J. Bacteriol. 194(16):4272-84 4) Szklarczyk et al. (2011): The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39:561-568 Lab: Kirsten Jung ______________ 79 BLAST XII _______________ Poster #29 LOCALIZATION CONTROL OF CHEMOTAXIS-RELATED SIGNALING COMPONENTS OF V. CHOLERAE Geetha Hiremath1, Hiroki Okabe2, So-ichiro Nishiyama2, Ikuro Kawagishi3 1 Research Center for Micro-Nano Technology, and 2Dept. of Frontier Bioscience, Hosei University, Japan. The genome sequence of V. cholerae has revealed that this bacterium possesses three sets of proteins with similarity to conventional chemotaxis-signaling proteins (Che proteins). Each set constitutes a distinct system, and only System II is directly involved in chemotaxis. To investigate the roles of Systems I and III and also to understand how parallel (closely related) signaling systems can perform distinct functions in V. cholerae, subcellular localization of their components were observed using GFP fusions. Whereas System II components localized constitutively to a cell pole, the histidine kinases (CheA1and CheA3) and adaptor proteins (Putative CheW, CheW2 and CheW3) of Systems I and III showed clustering at a cell pole and lateral regions of the membrane in cells cultured with standing (microaerobic) but not in cells cultured with shaking (aerobic), implying that Systems I and III functions under oxygen-limiting conditions. Polar localization of the histidine kinases of Systems I and III was induced by the addition of NaN 3 , but not that of chloramphenicol, to shaking cultures. Thus, it is neither mechanical stimuli, oxgen limitation nor growth retardation per se, but changes in energy metabolism that triggers the localization of these components. We further examined the effect of NaN 3 under various conditions. Increasing concentrations of NaN 3 at 30˚C enhanced localization of CheA1 and CheA3 to a cell pole and lateral membrane regions. Changes in temperature from 30˚C to 37˚C showed that CheA1 localized to the pole in the presence of lower concentrations of NaN 3 , and CheA3 localized to the pole in the absence of NaN 3 . CheA2, which always localized to a cell pole at 30˚C even in the presence of NaN 3 , altered its localization to cell pole and lateral membrane regions at 37˚C with 0.2% NaN 3 . Immunoblotting analyses demonstrated that these changes in localization of the CheA proteins are not due to those in their expression levels. These results suggest that components of Systems I and III of V. cholera may be assembled into active signaling complexes under energylimitating conditions. Lab: Ikuro Kawagishi ___________ 80 BLAST XII _______________ Poster #30 THE ROLE OF FlgM IN REGULATING FLAGELLAR ASSEMBLY IN BACILLUS SUBTILIS R. A. Calvo and D. B. Kearns Indiana University, Bloomington, IN, USA Flagella are composed of over thirty different proteins, and transit both the membrane and peptidoglycan to provide motility to diverse bacterial species. The flagellum is composed of three parts: a membrane-embedded anchor called the basal body, a universal joint called the hook, and an extracellular helical propeller called the filament. Coordinated assembly of the flagellum requires bacteria to couple gene expression with specific stages in assembly. Early flagellar genes are transcribed by the housekeeping sigma factor σA. The early genes encode components of the hookbasal body (HBB) and a type-three secretion apparatus. The HBB serves as channel through which extracellular components of the flagellum are secreted by the type-three secretion apparatus. Late flagellar genes, such as the filament protein, are transcribed by the alternative sigma factor σD. σD activity is inhibited by the anti-sigma factor FlgM prior to HBB completion. Once HBB assembly is complete, FlgM is antagonized and inhibition of σD is relieved. Relief of σD inhibition allows transcription of the filament gene and leads to the assembly of a functional flagellum. FlgM plays a crucial role in flagellar morphogenesis in that it couples gene expression to assembly state. In Salmonella enterica, FlgM is antagonized by secretion through the HBB. This regulatory paradigm has only been observed in the γ-proteobacteria, and the mechanism by which the HBB antagonizes FlgM in B. subtilis is not known. By precipitating proteins from culture supernatant, we found that FlgM is secreted through the HBB in B. subtilis. Furthermore, we determined that FlgM is degraded extracellularly by a specific subset of the seven proteases secreted by B. subtilis. Although FlgM is secreted, secretion may not be sufficient to regulate the anti-σD activity of FlgM. Mutations in the HBB abolish FlgM secretion and σD activity is inhibited. However, certain HBB mutants that fail to secrete FlgM have active σD. Currently, we are screening for regulation blind alleles of FlgM in an effort to understand the role of FlgM in regulating σD activity in response to the assembly state of the flagellum. Lab: Daniel Kearns _____________ 81 BLAST XII _______________ Poster #31 ROLE OF THE PSEUDOMONAS AERUGINOSA FLAGELLAR MOTOR IN SWIMMING MOTILITY AND CHEMOTAXIS Chui Ching Wong, Chen Qian and Keng-Hwee Chiam Mechanobiology Institute, National University of Singapore, Singapore Flagellar-driven swimming motility is well-established in some bacterial model organisms, and it is best described in the case of Escherichia coli. However, increasing genetic and structural data show that diversity in flagellar motors exists across the bacterial kingdom, where new paradigms of swimming motility may be discovered. In this report, we describe the flagellar motor function of monotrichous P. aeruginosa, and show that unlike E. coli, it is a motor that rotates in both CCW and CW directions giving rise to a ‘run-and-reverse’ trajectory. Additionally, the flagellar motor exhibits two-speeds in CCW and CW direction. Using a microfluidic-based assay, we show that in the presence of a chemoattractant (serine), the cells alter their run-length, switching frequency and motor speeds in order to move toward favourable environments. Therefore, in chemotaxis, apart from varying the switch frequency, the P. aeruginosa flagellar motor has an additional mechanism that allows it to favour the higher rotation speed state. These findings are validated in a computational model of P. aeruginosa swimming and chemotaxis. Lab: Chiam Keng Hwee _________ 82 BLAST XII _______________ Poster #32 BINDING AFFINITY PROVIDES KINETIC PREFERENCE, SPECIFICITY AND SIGNALING FIDELITY FOR TWO-COMPONENT SYSTEMS REGULATING DEVELOPMENT IN MYXOCOCCUS XANTHUS Jonathan W. Willett1, Nitija Tiwari2, Katherine Hummels1, Ernesto Fuentes2, John R. Kirby1 1 University of Iowa, Department of Microbiology 2 University of Iowa, Department of Biochemistry Two-component signal transduction systems, composed of histidine kinases and their cognate response regulators provide bacteria with the ability to sense and respond to a wide variety of signals. Upon activation the histidine kinase, can both phosphosphorylate and subsequently dephosphorylate the cognate response regulate. These reactions are highly specific and take place via a transiently formed histidine kinase and response regulator complex. A highly tractable model system for studying signaling specificity, fidelity and HK-RR interactions can be found in the organism Myxococcus xanthus. Here we report a systems-level analysis on a highly homologous family of two-component systems. The genome of M. xanthus encodes for over 125 two-component systems and as such has one of the largest number of signal transduction proteins in any bacterium. Bioinformatic analyses led us to conclude that M. xanthus possesses 27 NtrC homologs, which display high degrees of sequence similarity. 21 of these NtrC homologs are encoded next to predicted cognate kinases. All of these TCS have homology to the prototypical E. coli TCS system NtrB and NtrC. Using in vitro phosphotransfer and phosphatase profiling methods we show that each tested HK has kinetic preference for its predicted cognate regulator. Using isothermal titration calorimetry we demonstrate that cognate HK-RR pairs display simmilar dissociation constants (K d ) of approximately 1 μM while non-cognate pairs have no measureable binding affinity in this assay. Lastly, we generated a chimeric HK to demonstrate that previously known residues conferring phosphotransfer specificity also impart binding preference. These data suggest that phosphotransfer and phosphatase activities are predicated by discrete and measurable protein-protein interactions. Lab: John Kirby ________________ 83 BLAST XII _______________ Poster #33 SECRETION AND GLIDING OF BACTERODETES PHYLUM Keiko Sato, Daisuke Nakane, Mark J. McBride and Koji Nakayama Department of Molecular Microbiology and Immunology, Graduate Sciences, Nagasaki University, 852-8588 Nagasaki, Japan School of Biomedical Flavobacterium johnsoniae is a Gram-negative bacterium, which moves rapidly over surfaces at a speed of 1-3 um/s in gliding motility. In F. johnsoniae, the ahesion complexes containing SprB adhesin and two proteins that are involved in the bacteriodete protein translocation systems (referred to as the Por secretion system (PorSS)) are essential for gliding motility. The disruption of the PorSS proteins resulted in defects in translocation and assembly of SprB to cell surfaces and therefore in motility in F. johnsoniae. Many members of the phylum Bacteroidetes, including gliding bacteria such as F. johnsoniae and Cytophaga hutchinsonii, and nomotile bacteria such as Porphyromonas gingivalis, encode the genes of related to PorSS. Not only F. johnsoniae but also P. gingivalis secrete many proteins ossessed the C-terminal domains (CTDs), which have been suggested to form the CTD protein family including pathogen cysteine proteinase and hemagglutinin by PorSS. Lab: Koji Nakayama ____________ 84 BLAST XII _______________ Poster #34 THE RESPONSE REGULATOR RRP1 REGULATES CHITOBIOSE UTILIZATION AND VIRULENCE OF THE LYME DISEASE SPIROCHETE BORRELIA BURGDORFERI Ching Wooen Sze1, Alexis Smith3, Young Hee Choi2, Xiuli Yang3, Utpal Pal3, Aiming Yu2, and Chunhao Li1* Department of Oral Biology, The State University of New York at Buffalo, Buffalo, New York1, Department of Pharmaceutical Sciences, The State University of New York at Buffalo, Buffalo, New York2, Department of Veterinary Medicine, University of Maryland and Virginia-Maryland Regional College of Veterinary Medicine, College Park, Maryland3 Life cycle alternating between arthropod and mammals forces the Lyme disease spirochete, Borrelia burgdorferi, to adapt to different host milieus by utilizing diverse carbohydrates. B. burgdorferi lacks the de novo biosynthesis pathway of N-acetylglucosamine (GlcNAc) but encodes a set of chitobiose metabolic genes that allows conversion of chitobiose to GlcNAc. The chbC gene, a key chitobiose transporter, is dually regulated by RpoD and RpoS. Rrp1 is a response regulator that synthesizes cyclic diguanylate (c-di-GMP) in B. burgdorferi. In this report, we found that the rrp1 mutant had growth defects and formed membrane blebs when GlcNAc was replaced by chitobiose in growth medium. The expression level of chbC was significantly repressed in the mutant and constitutive expression of chbC successfully reversed the phenotype. Immunoblotting and transcriptional studies revealed that Rrp1 is required for the activation of BosR and its impact on chbC is likely mediated by the BosR-RpoS signaling pathway. Although the rrp1 mutant was unable to be transmitted via tick bite, exogenous supplementation of GlcNAc into unfed ticks partially rescued the transmission. Based on these results, we propose that Rrp1 governs the chitobiose metabolism of B. burgdorferi via a BosRRpoS-ChbC regulatory network essential for the transmission of the spirochete. Lab: Chunhao Li _______________ 85 BLAST XII _______________ Poster #35 CRYO-ELECTRON TOMOGRAPHY OF ESCHERICHIA COLI MINICELLS REVEALS MOLECULAR ARCHITECTURE OF INTACT FLAGELLAR MOTOR Bo Hu1, Xiaowei Zhao1, William Margolin2 and Jun Liu1 1. Department of Pathology and Laboratory Medicine, University of Texas Medical School at Houston, Houston, TX 77030, USA 2. Department of Microbiology & Molecular Genetics, University of Texas Medical School at Houston, Houston, TX 77030, USA Cryo-electron tomography (cryo-ET) has emerged as a powerful technology to study intact flagellar motor structure in living cells. However, the power of cryo-ET is significantly compromised in Escherichia coli because of their larger diameters, resulting in limited structural information. To circumvent this limitation, we constructed a mreB mutant of E. coli that produces cells with smaller diameters. Additional inactivation of clpX and min and overexpression of flhD/flhC genes resulted in highly motile and tiny minicells, which are about 0.3 micrometers in diameter and also contain one to three flagella. Cryo-ET and image analysis of several thousands of the wild-type cells yielded a more detailed three-dimensional (3-D) structure of the intact E. coli flagellar motor. Our comparative analysis of the flagellar motor structures from the wild-type cells, non-motile motA/B deletion mutants and purified flagellar motors provides new insights into the dynamic of the major components of the motor: the stator, C ring and L/P rings. Our results suggest that the flagellar motor is not a rigid, but a highly dynamic and flexible structure, which could undergo large elastic deformation in cells. Lab: Jun Liu ___________________ 86 BLAST XII _______________ Poster #36 THE TORQUE GENERATOR OF BACTERIAL FLAGELLAR MOTOR REVEALED IN BORRELIA BURGDORFERI Xiaowei Zhao1, Kihwan Moon2, Tristan Boquoi2, Steven, J. Norris1, Md A. Motaleb2, Jun Liu1 Department of Pathology and Laboratory Medicine, University of Texas Medical School at Houston, Houston, TX 77030 2 Department of Microbiology and Immunology, Brody School of Medicine, East Carolina University, Greenville, NC 27834 1 The bacterial flagellum is the core organelle of motility in bacteria, and is often important for the virulence of many bacterial pathogens including Lyme disease spirochete Borrelia burgdorferi. Powered by the membrane ion-motive force, the flagellar motor converts electrochemical energy into torque through an interaction between the rotor and its surrounding stator. The stator is the torque-generating unit composed of the membrane protein complex MotA/MotB. The mechanism of how stator interacts with the rotor remains elusive due to the lack of structural information of these components. Here we present the three-dimensional (3-D) structures of B. burgdorferi periplasmic flagellar motor in wild-type (WT), motA, and motB mutants by utilizing high-throughput cryo-electron tomography (cryo-ET) and sub-volume analysis. Comparative analysis of those motor structures revealed the 3-D in situ structure of the torque generating unit and its interaction with the rotor. Sixteen stator units are assembled peripherally around the rotor. The cytoplasmic domain of the stator is located adjacent to the C-terminal domain of the rotor protein FliG. This stator-rotor interaction induces an unexpected conformational change of FliG. We also mapped the association of stator-rotor in a nonmotile motBD24N mutant and a less motile motBD24E mutant, in which the proton translocation is blocked or limited, respectively. The results show that the incorporation of stators into the motor is closely associated with the proton flux. Collectively, our results provide novel structural insights into the rotor-stator interaction, which is fundamentally important for understanding the mechanism of flagellar rotation and bacterial motility. Lab: Jun Liu ___________________ 87 BLAST XII _______________ Poster #37 THE CYTOPLASMIC AROMATIC ANCHOR OF TM2 REGULATES TRANSMEMBRANE SIGNAL TRANSDUCTION BY THE Tar CHEMORECEPTOR Christopher Adase, Roger Draheim, Raj Desai and Michael D. Manson Texas A&M University, BSBE Rm 303, MS3258, College Station, TX 77843 The chemoreceptor Tar of Escherichia coli (Tar Ec ) interacts with two of its principal attractant ligands very differently. L-aspartate binds directly to the receptor; whereas maltose interacts with the receptor indirectly though the maltose-binding protein. The repellent nickel also appears to interact directly with the periplasmic domain, although the exact binind site is currently not known. Thus, Tar must communicate transmembrane signals in response to at least three modes of ligand binding. Our specific interest is in how particular combinations of aromatic residues at the cytoplasmic (C-terminal) end of transmembrane helix 2 (TM2) contribute to baseline signal output and to ligand sensitivity. Each of the four MCPs of E.coli has a unique aromatic anchor composition, and Tar Ec has been subjected to evolutionary selection for the ability to respond to divergent ligands. This process is occurs for each transmembrane sensor leading to the variety of aromatic anchor residue combinations and number of aromatic anchors found in various transmembrane sensors of two component systems. We report here studies in which both the position and residue composition of the aromatic anchor of Tar Ec have been altered. Our results indicate which parameters of the aromatic anchor are most critical to its function. They also demonstrate the importance of Gly-211, which immediately follows Trp-209/Tyr-210 of the aromatic anchor of wild-type Tar Ec anchor, in the response to ligands. Together with previously published information on the TM2-HAMP connector cable of Tsr (Kitanovic et al., 2011), our data can be used to generate a model for tunable signal transduction by transmembrane receptors. Lab: Michael Manson ___________ 88 BLAST XII _______________ Poster #38 COMPARATIVE ANALYSIS OF FLAVOBACTERIUM JOHNSONIAE ALGICOLA GLIDING MOTILITY AND PROTEIN SECRETION AND CELLULOPHAGA Yongtao Zhu and Mark J. McBride Department of Biological Sciences, University of WI-Milwaukee Many members of the phylum Bacteroidetes glide rapidly over surfaces. Bacteroidete gliding has been well studied in Flavobacterium johnsoniae. Gliding is powered by proton motive force and involves the rapid movement of adhesins such as SprB along the cell surface. Nineteen gld and spr genes are required for efficient gliding. The distantly related bacteroidete Cellulophaga algicola has orthologs for most of these but lacks gldA, gldF, and gldG. F. johnsoniae GldA, GldF and GldG are similar in sequence to ATP-binding-cassette (ABC) transporter components. The absence of these proteins in C. algicola suggests that they do not have a central or irreplaceable role in gliding. To address the possibility of the involvement of another transporter in gliding we developed techniques for HimarEm1 transposon mutagenesis, site-directed mutagenesis, and complementation analyses for C. algicola. Sixty-one HimarEm1-induced motility mutants that formed nonspreading colonies were isolated and characterized. Mutants containing transposon insertions in gldB, gldI, gldJ, gldK, gldL, gldM, gldN, sprA, sprB, sprC, sprE, sprF, and sprT, which correspond to F. johnsoniae gliding motility genes, were identified. No ABC transporter genes were identified and no novel genes required for C. algicola gliding were found. The results suggest that an ABC transporter is not required for C. algicola gliding motility. They also reinforce the importance of the other Gld and Spr proteins for gliding. gldD and gldH are required for F. johnsoniae gliding and orthologs are found in C. algicola but no insertions were identified in these genes. gldD and gldH are small, which probably explains the absence of transposon insertions. We disrupted gldD and gldH by insertion mutagenesis and demonstrated that each mutant had severe motility defects. Surprisingly, unlike F. johnsoniae gldD mutants the C. algicola gldD mutants exhibited some motility. The results suggest that GldD may be less critical than some of the other components of the motility machinery. Some of the motility proteins (GldK, L, M, N, SprA, E, T) are components of a novel Bacteroidetes-specific protein secretion system, the Por secretion system, also referred to as the Type IX secretion system (T9SS). The F. johnsoniae T9SS is needed for secretion of SprB to the cell surface and for secretion of an extracellular chitinase. The C. algicola T9SS is probably involved in secretion of SprB, but it also appears to be required for secretion of one or more proteases. Mutations in gldK, L, M, N, sprA and sprT resulted in inability to digest casein. Surprisingly, sprE mutant cells digested casein as well as the wild type. This suggests that SprE may have a less central role in protein secretion than the other components. These studies of C. algicola complement those performed on F. johnsoniae and help to identify the most critical components of the gliding motility and T9SSs of members of the phylum Bacteroidetes. Lab: Mark McBride _____________ 89 BLAST XII _______________ Poster #39 GLIDING OF MYCOPLASMA MOBILE CAN BE EXPLAINED BY DIRECTED BINDING Akihiro Tanaka, Daisuke Nakane, Takayuki Nishizaka and Makoto Miyata 3-3-138 Sugimoto Sumiyoshi-ku Osaka, Japan Mycoplasma mobile, a fish pathogen, forms a membrane protrusion, namely the gliding machinery, and glides on solid surface smoothly up to 4.5 μm/s and 27 pN in the direction of protrusion. The “directed smooth gliding” has been suggested to occur through repeated catch-pull-release of sialylated oligosaccharide existing on animal cells by hundreds of 50 nm flexible “legs” sticking out from the gliding machinery. However, the directed displacement is difficult to explain, because the legs responsible for gliding is very flexible under electron microscopy and fast scan AFM (atomic force microscopy). In the present study, we measured the force to detach the cell laterally from glass surface by using laser trap. A single cell binding to sialylated oligosaccharide fixed on glass was pulled forward and backward by manipulating the plastic bead attached to front or tail of the cell. The forces required to detach the cell were 22.5 ± 13.9 pN and 48.4 ± 28.0 pN for forward and backward, respectively. These results suggest that M. mobile binds to solid surface in a direction dependent manner, and this feature may cause the directed gliding. Lab: Makoto Miyata _____________ 90 BLAST XII _______________ Poster #40 GLIDING MACHINERY OF MYCOPLASMA MOBILE OBSERVED BY ELECTRON MICROSCOPY Hiroki Yamamoto, Makoto Miyata 3-3-138 Sugimoto Sumiyoshi-ku Osaka Japan Mycoplasma, a fish pathogen, forms a membrane protrusion at a cell pole and glides in the direction of protrusion, by a unique mechanism. The structural information on the gliding machinery is critical to elucidate this unique mechanism. The gliding machinery is composed of mainly three huge proteins, Gli521, Gli349, and Gli123, weighing 521, 349, and 123 kDa, respectively. To date, the “leg” structure on the surface of the gliding machinery and the molecular shapes of isolated component proteins have been clarified, by freeze-fracture and rotary-shadowing electron microscopy (EM), respectively. However, the whole structure of the machinery or the assignment of component proteins in the machinery remains unclear. We examined negative staining and rotary-shadowing EM under reduced backgrounds, and then achieved the better image of gliding machinery featured with two types of fibers. To assign filamentous proteins, Gli349 and Gli521, involved in the gliding mechanism, onto two types of fibers, we examined the subcellular localization and amounts of gliding proteins by immunofluorescence microscopy, for the wild type and mutant strains, and compared these results with their EM images. The results suggested that Gli349 sticks out from the membrane and Gli521 lines along the membrane. Lab: Makoto Miyata _____________ 91 BLAST XII _______________ Poster #41 GENE MANIPULATION OF GLIDING BACTERIUM, MYCOPLASMA MOBILE Isil Tulum, Atsuko Uenoyama, and Makoto Miyata Graduate School of Science, Osaka City University Mycoplasma mobile, a fish pathogen, is the fastest-gliding mycoplasma. It glides on solid surfaces at an average speed of 2.0 to 4.5 μm/s, exerting a force up to 27 pN. M. mobile exhibits gliding motility in the direction of the protrusion, which is essential for mycoplasma infection. This motility is caused by a unique mechanism composed of at least three huge surface proteins and an ATPase, and probably more than ten internal proteins. To elucidate this mechanism, genetic manipulation should be a powerful tool. However, this organism has not been transformed so far by standard procedures used for other Mycoplasma species. In this study, we examined and improved each step of transformation, and found and solved the possible problems. (i) The electroporation conditions have been adjusted to fit to the M. mobile cells which are much tougher than the other species. (ii) The recovery time after electroporation was elongated from 2 to 12 h. (iii) The growth medium was modified to get clearer colony shapes. Then, we achieved the transformation of M. mobile with efficiencies ranging 10-5 to 10-7 per competent cells by using 10 μg of oriC plasmids. Transformation was confirmed by colony PCR, and labeling of cells by EYFP. In order to improve the transformation efficiencies, we have developed vectors containing different marker genes under the control of different M. mobile specific promoters. The results suggested the best combination for transformation. Lab: Makoto Miyata _____________ 92 BLAST XII IS MOTILITY OR POSSESSION OF PATHOGENESIS OF LYME DISEASE? _______________ Poster #42 PERIPLASMIC FLAGELLA CRUCIAL FOR THE S. Z. Sultan1, A. Manne1, X. Zhao2, Y. Chien2, Jun Liu2, P. Sekar3, R. M. Wooten3 and M. A. Motaleb1. 1 Dept. of Microbiology and Immunology, East Carolina University; 2Dept. of Pathology and Lab Medicine, University of Texas Medical School at Houston; 3Dept. of Medical Microbiology and Immunology, University of Toledo College of Medicine, Toledo, OH. Motility and chemotaxis are known to be important for host tissue colonization and disease production by many bacteria, including the spirochetes. Rotation of flagellar motors is required for propulsion of bacteria. However, flagellin molecules are recognized by Toll-like receptor 5, which activates host inflammatory responses. Additionally, bacterial flagella are potent immunogens and elicit adaptive immune response, and the immunogenicity of flagellin has been the basis for several vaccine strategies. However, the role of Borrelia burgdorferi motility in the pathogenesis of Lyme disease has not been rigorously studied. The genome of B. burgdorferi contains more than fifty genes encoding the motility and chemotaxis proteins. We have recently inactivated the flaB gene, which encodes the major periplasmic flagellar filament. The flaB mutants were rod shaped and non-motile, whereas the wild-type cells are motile and flat-wave. The flaB mutants were unable to infect experimental mice by needle or ticks, suggesting that either motility and/or the presence of the periplasmic flagella is crucial for infection. To verify if the mere presence of structural periplasmic flagella without their motor function can establish an infection, we created a motB-specific mutant. B. burgdorferi ΔmotB retained their periplasmic flagella, but were paralyzed. ΔmotB display a flat-wave morphology similar to wild-type cells, however, they possess a rod-shaped section in the middle of the cell body. Using cryo-electron tomography, we revealed that ΔmotB periplasmic flagella form a ribbon in the flat-wave region, while the ribbon is not well-organized in the rod-shaped area. Mouse and tick-mouse infection studies using ΔmotB determined that motility, and not just the possession of periplasmic flagella, is important for the pathogenesis of Lyme disease. Further study of ΔmotB and ΔflaB may discriminate the effect of these immune agonists during the disease process. Lab: Md Motaleb _______________ 93 BLAST XII _______________ Poster #43 Fe(II) SENSING IN PSEUDOMONAS AERUGINOSA Naomi Kreamer, Maureen Coleman, James Boedicker, Dianne Newman California Institute of Technology 1200 E. California Blvd. Pasadena, CA 91125 Pseudomonas aeruginosa is a ubiquitous Gram-negative bacterium best known as the predominant opportunistic pathogen infecting the lungs of cystic fibrosis patients. In this context, it is thought to form biofilms, within which locally reducing and acidic conditions can develop that favor the stability of ferrous iron [Fe(II)]. Because iron is a signal that stimulates biofilm formation within a particular concentration range, we performed a microarray study to determine whether P. aeruginosa exhibits a specific transcriptional response to extracellular Fe(II). We identified a novel two-component sensor histidine kinase (BqsS) – response regulator system (BqsR) that responds specifically to Fe(II). BqsS distinguishes between Fe(II) and Fe(III) and other dipositive ions, responding solely to Fe(II). The periplasmic domain of the sensor contains a motif similar to a known Fe-binding motif: RExxE. Sitedirected mutants in this putative Fe(II)-binding motif abolish the Fe(II) shock transcriptional response. Using bioinformatics, we identified a putative DNA sequence that BqsR binds based on identifying conserved patterns in regions upstream of the genes most highly upregulated in an Fe(II) shock microarray experiment. We are in the process of testing this prediction biochemically using gel-shift assays with purified BqsR. In parallel, we are attempting an in vivo assay to verify the specificity of this motif by employing a LacZ-reporter construct to measure transcription from engineered promoter sites. Lab: Dianne Newman ___________ 94 BLAST XII _______________ Poster #44 DIRECT OBSERVATION OF UNITARY STEP OF GLIDING MACHINERY IN MYCOPLASMA MOBILE Yoshiaki Kinosita1, Daisuke Nakane2, Tomoko Masaike1, Kana Mizutani1, Makoto Miyata2, Takayuki Nishizaka1 1 Dept. Phys., Gakushuin Univ.,2Dept. Biol., Osaka City Univ. Mycoplasma mobile (M.mobile) is a fish pathogenic flask-shaped bacterium of about 0.8 μm long and glides on the substrate at speed of 2.5-4.0 μm/sec. The mechanism of the gliding motility has been unique because the gene of M. mobile contains no homologs related to known bacterial motility or conventional motor proteins such as kinesin and myosin. With a series of recent contributions, it was revealed that M. mobile has four novel proteins, all of which are involved in the gliding motion, Gli123, Gli349, Gli521 and P42 (1). To establish the molecular basis of the gliding mechanism, we here develop a method to detect the unitary step of the gliding machinery comprised of these proteins under the changing ATP concentrations in the medium. Three strategies were adopted: 1) The cell membrane was stained with a fluorescent dye, Cy3, so that we could observe the motility of single bacterium under fluorescent microscope at high temporal resolution (2 msec). 2) The addition of Triton X-100 permeabilized the cells, called them gliding ghosts (2). M. mobile gliding is supplied by ATP hydrolysis and so we could control its gliding speed by changing ATP concentrations in the medium 3) To reduce the number of legs contributing to the motility, free sialylated oligosaccharide, binding target for the gliding motility, were added. Through these improvements we successfully observed repetitive stepwise motions with the size of 70 nm. The speed during each stepping coincides with the maximum speed, suggesting that the continuous gliding motion comprises assemblies of the unitary step we detected. The 70 nm step size is the largest one ever reported amongst all motor proteins and possibly contributes to the characteristic fast gliding of M. mobile. (1) Miyata, M. Annu Rev Microbiol 64, 519-37 (2010). (2) Uenoyama A, Miyata M. Proc. Natl. Acad. Sci. USA 102:12754–58 (2005). Lab: Takayuki Nishizaka ________ 95 BLAST XII _______________ Poster #45 THE HELICOBACTER PYLORI TlpD CHEMORECEPTOR IS CRITICAL FOR MULTIPLE STAGES OF NORMAL STOMACH COLONIZATION AND MEDIATES A RESPONSE TO IRON AND OTHER REACTIVE OXYGEN SPECIES-GENERATING CONDITIONS Susan M. Williams, Tessa M. Andermann, Lisa Sanders, Sarina Porcella, Kieran Collins, and Karen M. Ottemann Department of Microbiology and Environmental Toxicology, UC Santa Cruz, Santa Cruz, CA 96054, USA Helicobacter pylori is an epsilon proteobacterium that uses chemotaxis to infect the human stomach and promote inflammation in tissues. The H. pylori chemotaxis system contains four chemoreceptors, called TlpA, TlpB, TlpC and TlpD. TlpD is a soluble chemoreceptor that has been implicated in an energy taxis response (J Bacteriol, 2008 190: 3244-55). We have explored TlpD’s role in stomach colonization and conditions that it responds to. Toward this inquiry, we generated a tlpD mutant in strains SS1 and mG27. tlpD mutants have a substantial initial colonization defect, and also show defects after two weeks of colonization, suggesting tlpD is needed for both initial and sustained colonization. The tlpD-associated defect could be complemented by expression of tlpD from a heterologous locus, confirming that TlpD itself is required for mouse colonization. To determine what TlpD responds to, we employed a variety of chemotaxis assays. Initially, we observed that tlpD mutants fail to form a bacterial band in response to iron chloride in a modified agarose plug bridge assay. The wild type band formed at a slight distance from the plug that could represent either an attractant or repellent response. We thus analyzed bacterial temporal swimming behavior, and found that iron caused a tlpD-dependent repellent response in a strain mG27 isolate, but an attractant response in strain SS1. We furthermore used the temporal swimming assay to determine that H. pylori uses TlpD to detect hydrogen peroxide as a repellent. These studies indicate that wild-type sensing of iron and hydrogen peroxide require TlpD, suggesting that TlpD is part of H. pylori’s ability to sense intracellular stress, particularly oxidative stress. Lab: Karen Ottemann ___________ 96 BLAST XII _______________ Poster #46 THE ROLE OF RECEPTOR-KINASE INTERACTIONS IN ARRAY SIGNALING Germán Piñas and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112 Recent models of chemoreceptor array organization (Briegel et. al, 2012; Liu et al., 2012) predict a contact between the tip of the receptor dimer and the P5 domain of the CheA dimer that might be important to signaling. The CheA kinase has five domains with different signaling roles: P1, the phosphorylation site domain; P2, a phosphotransfer target acquisition domain; P3, the dimerization domain; P4, the ATP-binding domain; and P5, a domain that resembles the CheW protein required for coupling CheA to chemoreceptor control. P5 appears to mediate binding interactions with CheW and with receptors, but only the P5-CheW interaction is known to be critical for receptor coupling control. To explore the signaling role of the predicted P5-receptor interaction, we constructed a series of amino acid replacements in E. coli CheA at P5 residues proposed, by biochemical and structural studies from various groups, to lie at the receptor-binding surface: L528, V531, S534, I581, L599. The mutant CheA proteins were encoded on a regulatable plasmid that also expressed CheW, to maintain normal stoichiometry of these interacting components. The mutant plasmids were transferred to various host strains to test their behaviors in soft agar chemotaxis assays and with in vivo FRET kinase assays. We found that most amino acid replacements at V531 and S534 had little effect on chemotaxis in plate assays, whereas roughly one-half of the amino acid replacements at L528, I581, and L599 impaired chemotactic ability on plates. The most deleterious amino acids at all three positions were ones with charged sidechains (D, E, K, R) or with very small (G) or very large (W) sidechains. These findings indicated that putative P5-receptor contact residues were important for operation of the signaling array. These P5 mutants evinced a variety of signaling defects, often rather subtle ones, in FRET kinase assays with wild-type Tsr partners in the QEQE modification state. Replacements at L528 typically increased serine sensitivity; those at I581 mainly reduced cooperativity; and those at L599 usually exhibited changes in both parameters. To ask whether these P5 lesions might have additive effects on signaling proficiency, we constructed and characterized a series of triple mutants with G, S, or W replacements at all three residues (L528/I581/L599). The S and W triple mutants still formed ternary complexes in vivo and showed chemotactic ability on soft agar plates, but had threshold and cooperativity shifts in FRET assays. The triple G mutant could not support ternary complex formation or chemotaxis, and showed no serine responses in FRET tests. These results show that a P5-receptor interaction is important for chemotaxis, but that the P5 residues that comprise the interaction surface are not individually critical for array signaling. The shifts in serine response threshold and cooperativity observed in the CheA-P5 mutants imply that the multiple connections between CheA, CheW and receptors in the array probably shift, break, and reform during a serine-induced signaling cycle. We are currently studying the effect of these P5 mutations on sensory adaptation. Lab: John Parkinson ___________ 97 BLAST XII TRANSMEMBRANE SIGNALING IN THE SERINE CHEMORECEPTOR TSR _______________ Poster #47 VIA SYNTHETIC CONTROL CABLES Smiljka Kitanovic and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112 Ligand binding in the periplasmic domain of the E. coli serine receptor promotes inward piston displacement of one TM2 helix in the Tsr homodimer. Piston motions in turn influence the structure or stability of the cytoplasmic HAMP domain to control receptor output signals to the flagellar motors. A five-residue control cable connects TM2 to the AS1 helix of the HAMP bundle and transmits conformational signals between them. To assess the role of control cable secondary structure in Tsr signaling, we constructed 32 synthetic control cable mutants that had all possible combinations of alanines (A) and glycines (G) at the five wild-type control cable residues (G213, I214, K215, A216, S217). We assumed that alanines would favor helical secondary structure of the control cable, whereas glycines would destabilize control cable helicity. We characterized the synthetic control cable mutants for serine chemotaxis on soft agar plates, for pre- and post-stimulus flagellar rotation pattern, for ability to undergo adaptational modifications, and for serine responses in FRET-based in vivo kinase control assays. The Tsr synthetic control cable mutants fell into several distinct groups, based on their serine response thresholds. In hosts lacking the CheR and CheB adaptation enzymes, some Tsr mutants, e.g., AAAAA, failed to respond to any level of serine. In contrast, the GGGGG mutant exhibited the most sensitive serine response. Surprisingly, in hosts containing the CheR and CheB adaptation enzymes, every mutant receptor except the one with an all-glycine control cable (GGGGG) supported chemotaxis at some serine concentration in soft agar assays. In vivo FRET kinase assays revealed that the AAAAA and other control cable mutants with high serine thresholds in adaptation-deficient hosts had greatly reduced serine thresholds and underwent sensory adaptation in adaptation-proficient hosts. In contrast, the GGGGG mutant had low serine response thresholds, but failed to undergo sensory adaptation. The adaptation defect of the GGGGG mutant most likely accounts for its inability to mediate serine chemotaxis in soft agar assays. These results suggest a trade-off between two receptor functions: stimulus response and adaptational modification. The AAAAA control cable is defective in responding to a serine stimulus, but its output is still subject to sensory adaptation control. The GGGGG control cable, on the other hand, mediates very sensitive responses to serine, but is not subject to sensory adaptation control. These behaviors imply that attractant stimuli may initiate HAMP signaling by relaxing control cable helicity. The GGGGG control cable is less helical than the AAAAA control cable and, therefore, more easily driven to the attractant-stimulated signaling state. Additionally, a glycine in the first position of the control cable appears to play a prominent role in lowering the serine response threshold, possibly functioning as a structural swivel. Adaptational modifications, however, may depend on a more stable helical control cable. Lab: John Parkinson ___________ 98 BLAST XII _______________ Poster #48 TSR-E502: AN UNORTHODOX CHEMORECEPTOR SENSORY ADAPTATION SITE Xue-sheng Han and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112, USA Methyl-accepting chemotaxis proteins (MCPs) mediate diverse chemotactic responses in motile microbes. These chemoreceptors sense temporal changes in chemoeffector concentrations by comparing current ligand occupancies with a memory of the recent chemical past, recorded in the form of reversible methyl-group modifications of glutamate residues in the receptor’s cytoplasmic signaling domain. MCP methylation state is regulated by the stimulus-modulated interplay of two MCP-specific enzymes, CheR, a methyltransferase, and CheB, a methylesterase. The best-studied MCPs, Tar (aspartate/maltose sensor) and Tsr (serine sensor) of E. coli, have four canonical methylation sites in each subunit, located near the dimer interface. Methylation at these sites should enhance inter-subunit packing of the four-helix methylation (MH) bundle. Tsr has a fifth methylation site, E502, located on a different helical face, that would most likely modulate intra-subunit packing interactions in the MH bundle. Does E502 play a different signaling role than the four conventional modification sites in Tsr? Is E502 essential for Tsr function? Is it sufficient for Tsr function? We took three approaches to answer these questions. We first constructed and characterized Tsr receptors with all possible amino acid replacements at E502. Nearly all of the mutant receptors mediated robust serine chemotaxis on tryptone soft agar plates, indicating that E502 is not critical to Tsr function. Only two mutant receptors, E502P and E502I, lacked function in soft agar assays. Flagellar rotation tests and in vivo FRET kinase assays demonstrated that these mutant receptors had aberrant signal outputs. Tsr-E502P was shifted toward the attractant-induced kinase-off state; Tsr-E502I was shifted toward a kinase-on state. Both mutant receptors mediated responses to serine stimuli, but with higher (E502P) or lower (E502I) detection sensitivity than wild-type Tsr. To determine whether E502 underwentboth methylation and demethylation reactions, we used combinations of D and N replacements to approximate the wild-type E and Q residues found at the other four Tsr methylation sites. D produced signaling effects similar toE, but proved to be an ineffective substrate for CheR-dependent methylation; N produced signaling effects similar to Q, but was refractory to CheB-dependent deamidation. Tsr variants with only the E502 site intact underwent methylation and demethylation, but could not support chemotaxis in soft agar assays, demonstrating that E502alone is not sufficient for Tsr signaling and sensory adaptation. To compare the signaling effects of modifications at E502 with those at the canonical methylation sites, we analyzed, with in vivo FRET kinase assays, the serine thresholds and response cooperativities of receptors with different mutationally adjusted modification states. All five sites influenced Tsr signaling in the same manner, implying that overall MH bundle packing stabilitydetermines receptor signaling characteristics, rather than a particular intra- or inter-subunit packing conformation. In sum, our study of Tsr-E502supports predictions of the dynamic bundle model of HAMP signaling, which proposes that stimulus responses and sensory adaptation occur through shifts in the relativepacking stabilities of the oppositionally coupled HAMP and MH bundles. The especially strong effects of E502 modifications on signaling behavior probably reflect the facts that this site lies closest to the adjoin HAMP domain and that it uniquely influences intra-subunit helix packing in the receptor’s methylation bundle. Lab: John Parkinson ___________ 99 BLAST XII _______________ Poster #49 CHARACTERIZATION OF A CHEMOTAXIS LOCUS IN LEPTOSPIRA INVOLVED IN MOTILITY AND VIRULENCE Ambroise Lambert1,2, Jérôme Ng Wong3, Mathieu Picardeau1 1 Institut Pasteur, Unité de Biologie des Spirochètes, Paris, France 2 Université Paris Diderot, Sorbonne Paris Cité, Cellule Pasteur, Paris, France 3 Institut Pasteur, Unité de Physique des Systèmes Biologiques Leptospira belong to the spirochete family of bacteria existing as both saprophytes and pathogens. Pathogenic species are the causative agents of leptospirosis, which is currently recognized by the WHO as an emerging zoonotic disease. Leptospires express flagella that are localized in the periplasm and attached at each polar end of the cell. Leptospira coordinate their flagellar motor together asymetrically to allow bacterial motility. Although chemotaxis and motility might have an important role in the infection process (Lambert et al., 2012), the regulation of the flagellar-based motility in relation to chemotaxis remains unexplored in these atypical bacteria. We generated a library of random transposon mutants in the pathogen L. interrogans which included insertions in genes located in the same chromosomal locus. This operon is constituted by genes encoding homologues of chemotaxis proteins CheA, CheW, CheD, CheB, CheY and MCP. Swarm plate assays suggested that the disrupted chemotaxis genes were involved in chemotaxis. Microscopy of Leptospira in liquid medium demonstrated loss of motility of one of the mutants and this mutant was attenuated in the gerbil model of infection. Analysis via videomicroscopy combined with determination of bacterial trajectories has allowed us to show significant differences in the motility of the mutants compared to the wild-type. This is the first study demonstrating that a chemotaxis regulatory system is required for both motility and virulence of Leptospira. Lambert A, Takahashi N, Charon N, Picardeau M. Chemotactic behavior of pathogenic and nonpathogenic Leptospira species. Appl Environ Microbiol. 2012 Sep 21. Lambert A, Picardeau M, Haake DA, Sermswan RW, Srikram A, Adler B, Murray GA. FlaA proteins in Leptospira interrogans are essential for motility and virulence but are not required for formation of the flagellum sheath. Infect Immun. 2012 Jun;80(6):2019-25. Epub 2012 Mar 26. Lab: Mathieu Picardeau _________ 100 BLAST XII _______________ Poster #50 STIMULATORY INTERACTIONS BETWEEN HYBRID HISTIDINE PROTEIN KINASES IN THE VIRULENCE SIGNALLING NETWORK OF PSEUDOMONAS AERUGINOSA Vanessa I. Francis and Steven L. Porter Biosciences, College of Life and Environmental Sciences, Geoffrey Pope Building, Stocker Road, University of Exeter, Exeter, Devon, EX4 4QD, United Kingdom The GacS signalling network of P. aeruginosa plays a key role in regulating the transition between acute and chronic modes of infection. This network comprises multiple histidine protein kinases (HPKs) that have the remarkable ability to directly affect one another’s signalling through physical interactions. The HPK, RetS has previously been reported to inhibit the signalling of the HPK, GacS (1). In this study, we have identified stimulatory interactions between several of the HPKs comprising this network. In particular, we find that the phosphorylation of the HPKs, RetS and PA1611, can be stimulated by other HPKs in the GacS network. PA1611-P levels are increased by over 15-fold of that seen from autophosphorylation alone, while RetS, which is unable to autophosphorylate, becomes highly phosphorylated upon coincubation with the activating HPKs. We discuss the potential implications of this HPK to HPK signalling for signal integration and downstream signalling. 1. Goodman, A. L., M. Merighi, M. Hyodo, I. Ventre, A. Filloux, and S. Lory. 2009. Direct interaction between sensor kinase proteins mediates acute and chronic disease phenotypes in a bacterial pathogen. Genes Dev. 23:249-259. Lab: Steven Porter _____________ 101 BLAST XII _______________ Poster #51 TAKING CONTROL OF THE BACTERIAL FLAGELLAR MOTOR Guillaume Paradis, Ismaël Duchesne and Simon Rainville Department of Physics, Engineering Physics and Optics and Centre of Optics, Photonics and Lasers, Laval University, Québec, Québec, CANADA The bacterial flagellar motor is a fairly complex machine embedded in the multiple layers of the bacterial membrane. The focus of our laboratory for the past years has been the development of a unique in vitro assay to study the bacterial flagellar motor. Our setup consists of a filamentous Escherichia coli bacterium partly introduced inside a micropipette. Femtosecond laser pulses (60 fs and ~ 15 nJ/pulse) are then tightly-focused on the part of the bacterium that is located inside the micropipette. This vaporizes a small portion of the membrane, leaving an essentially permanent hole in the wall of the bacterium. Using a patch-clamp amplifier, we then apply an external voltage between the inside and the outside of the micropipette. That voltage then directly contributes to the proton-motive force (pmf) that powers the flagellar motor. As we change the applied potential, variations in the motor's rotation speed are observed. In addition to granting us full access to the inside of the cell, this in vitro assay gives us direct control over both components of the proton-motive force. Indeed, we can apply an electric potential across the membrane, control the pH both inside and outside of the cell and expose the motor to various concentrations of proteins. That system therefore opens a world of new possibilities. Interestingly, we recently used this novel system to expose a ∆FliL mutant of Salmonella enterica (from R. Harshey) to very high pmf in an attempt to explain why these mutants lose their filaments when swarming. Our system allowed us to expose these cells to pmf up to -320mV and invalidate the hypothesis that the high pmf caused the filament to fall off. The rotation speed is measured using 2 different techniques: high-speed video microscopy of fluorescently labeled filaments and gold nanoparticules using an avalanche photodiode. Image sequences from a fast EMCCD camera are analyzed with custom MatLab code while signal from the photodiode is analyzed with LabView code. Using the strong link between gold and the thiol group genetically added to the end of the fliC protein we can label the filaments with gold nanoparticules. This technique gives the opportunity to have a high ratio of filament labeled. It should also be possible to shine a new light on the switching mechanism as each event can be recorded and analyzed in an easier way using those probes. Finally, the fabrication of the micropipettes was recently automated. Using image analysis in real time, the system allows a much more precise measurement of the constriction in the micropipette and hence a more flexible and robust fabrication process. Lab: Simon Rainville ____________ 102 BLAST XII _______________ Poster #52 THE MOTILITY OF BACTERIA IN AN ANISOTROPIC LIQUID ENVIRONMENT Ismaël Duchesne1, Anil Kumar2, Guillaume Paradis1, Tigran Galstian1 and Simon Rainville1 1 Department of Physics, Engineering Physics and Optics and Centre of Optics, Photonics and Lasers, Laval University, Québec, Québec, Canada 2 Department of Chemical Engineering, Indian Institute of Technology Delhi, Hauz Khas, New Delhi, India While the influence on bacterial motility of many genetic and biochemical factors has been extensively studied, there have been limited studies of the impact of physical parameters. Indeed, despite the fact that natural environments are often asymmetric (such as stretched supramolecular structures), the majority of behavioural experiments with bacteria have been done in isotropic liquid solutions. In the present work, we show that the behaviour of living microorganisms is dramatically different in media that are asymmetric. The example of E. coli bacteria swimming in a bulk uniaxial liquid environment is used to demonstrate this phenomenon. The results of our study shine light on the behaviour of bacteria in conditions that they may encounter in real environments and open new avenues for the control of their movements. Lab: Simon Rainville ____________ 103 BLAST XII _______________ Poster #53 PHENOTYPIC VARIATION AND BISTABILITY WITHIN FLAGELLAR GENE NETWORK IN SALMONELLA ENTERICA SEROVAR TYPHIMURIUM Santosh Koirala, Phillip Aldridge, Christopher V. Rao University of Illinois at Urbana Champaign, Newcastle University Availability of nutrients in the cellular environment plays a key role in switching between motile or sessile phenotypes. However, the response to nutritional cues could be very different even in closely related species. For example, E. coli up regulates flagellar synthesis whereas Salmonella enterica serovar Typhimurium downregulates flagellar synthesis under low-nutrient condition. YdiV is responsible for the repression of flagellar synthesis in Salmonella enterica serovar Typhimurium which acts as an anti-FlhD 4 C 2 factor. Moreover, FliZ-dependent activation of P class2 promoters is more pronounced in low nutrient condition and is achieved by repression of the ydiV gene by FliZ. YdiV expression is enhanced in poor media and greatly reduced in rich media. Thus, FliZ and YdiV, in poor media, form an overall positive feedback loop which results in a bistable motility phenotype. This study aims to characterize the extent of bistability within the flagellar network in Salmonella enterica serovar Typhimurium. Our results suggest that two distinct phenotypes, motile and sessile, coexist in an isogenic cell population under limited nutrient condition and the heterogeneity is enabled by FliZ-YdiV feedback loop. Lab: Christopher Rao ___________ 104 BLAST XII _______________ Poster #54 IDENTITY AND FUNCTION OF A LARGE GENE NETWORK UNDERLYING MUTAGENIC REPAIR OF DNA BREAKS UNDER STRESS Abu Amar M Al Mamun1, Mary-Jane Lombardo1*, Chandan Shee1, Andreas M Lisewski1, Caleb Gonzalez1‡, Dongxu Lin1†, Ralf B Nehring1, Claude Saint-Ruf2, Janet L Gibson, Ryan L Frisch, Olivier Lichtarge1,3, P. J. Hastings1, Susan M Rosenberg1,,3,4,5 1 Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030-3411 USA; 2INSERM, France; 3Department of Biochemistry and Molecular Biology, 4Department of Molecular Virology and Microbiology, and 5The Dan L Duncan Cancer Center, Baylor College of Medicine, Houston, TX 77030. Mechanisms of DNA repair and mutagenesis are defined based on relatively few proteins acting on DNA, yet the identities and functions of all proteins required are unknown. Here, we identify the network that underlies mutagenic repair of DNA breaks in stressed Escherichia coli and define functions for much of it. Using a comprehensive screen, we identified 77 new, and a total network of ≥93 genes that function in mutation. Most operate upstream of activation of three required stress responses (RpoS, RpoE and SOS), key network hubs, apparently sensing stress. An autophagy-like starvation-signaling pathway is identified. The results identify how a network integrates mutagenic repair into the biology of the cell, reveal specific pathways of environmental sensing, demonstrate the centrality of stress responses and imply their attractiveness as potential drug targets for blocking evolution of pathogens. Lab: Susan Rosenberg __________ 105 BLAST XII IS BACTERIAL TWITCHING SYMBIOSIS? _______________ Poster #55 MOTILITY REQUIRED TO ESTABLISH THE SQUID/VIBRIO Aschtgen, MS., Brennan, C.A., Schaefer, A.L.*, Ruby, E.G. Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, Madison, WI 53705, * Department of Microbiology, University of Washington, Seattle, WA 98195 The symbiotic infection of the light organ of the squid Euprymna scolopes by the luminous bacterium Vibrio fischeri has been studied experimentally to define the bacterial basis for the biochemical and molecular events that characterize the colonization of animal epithelial tissue. The light organ consists of a specific superficial ciliated epithelium and a series of deeply invaginated epitheliumlined crypt spaces, the site where symbiont colonization can occur. Within the first 2-3 hours after inoculation, V. fischeri cells form external aggregates, migrate to and through surface pores leading into ducts, and finally arrive in the crypt spaces. It has been previously shown that flagellar swimming motility is not required for the migration from the aggregate to the pores, suggesting that another kind of motility could be involved. For pathogenic associations, twitching motility is an essential virulence factor allowing bacteria to contact specific cells and disseminate within a host. In that context, we aim to (i) determine whether twitching motility is also essential for establishing a symbiotic association, and (ii) characterize the underlying molecular mechanism. In a preliminary study, we made a pilT/U mutant to describe the importance of this locus in the ability of V. fischeri to move by twitching on 1% agar plates. We next showed that twitching motility is an important determinant for host colonization. We also determined that the loss of twitching motility results in a decrease in biofilm formation in V. fischeri. This last observation and the fact that the colonization efficiency for a twitching motility mutant is dose and time dependent indicate, for the first time, the importance of this particular form of motility in the first steps of colonization in this model symbiosis. Lab: Edward Ruby ______________ 106 BLAST XII _______________ Poster #56 THE PSEUDOMONAS AERUGINOSA TYPE THREE SECRETION SYSTEM TRANSCRIPTIONAL ACTIVATOR ExsA INTERACTS WITH THE ANTI-ACTIVATOR PROTEIN ExsD IN A TEMPERATUREDEPENDENT MANNER Robert C. Bernhards, Anne E. Marsden, Shannon K. Schubot Virginia Polytechnic Institute & State University Esher, Timothy L. Yahr and Florian D. The opportunistic pathogen Pseudomonas aeruginosa ranks among leading causes of nosocomial infections. The type III secretion system (T3SS) aids acute P. aeruginosa infections by injecting potent cytotoxins into host cells to suppress the host's innate immune response. Expression of all T3SS-related genes is strictly dependent upon the transcription factor ExsA. Consequently, ExsA and the biological processes that regulate ExsA function are of great biomedical interest. The presented work focuses on the ExsA-ExsC-ExsD-ExsE signaling cascade that ties host cell contact to the up-regulation of T3SS gene expression. Prior to induction, the anti-activator protein ExsD binds to ExsA and blocks ExsA-dependent transcription by interfering with ExsA dimerization and promoter interactions. Upon host cell contact, ExsD is sequestered by the T3SS chaperone ExsC resulting in the release of ExsA and an up-regulation of the T3SS. Previous studies have shown that the ExsD-ExsA interactions are not freely reversible. Because independently folded ExsD and ExsA were not found to interact, it has been hypothesized that folding intermediates of the two proteins form the complex. Here we demonstrate for the first time that ExsD alone is sufficient to inhibit ExsA-dependent transcription and that no other cellular factors are required. More significantly, we show that independently folded ExsD and ExsA are capable of an interaction, but only at 37°C and not at 30°C. Guided by the crystal structure of ExsD, we designed a monomeric variant of the protein and demonstrate that ExsD trimerization prevents ExsD from inhibiting ExsA-dependent transcription at 30°C. We propose that this unique mechanism plays an important role in T3SS regulation. Lab: Florian Schubot ___________ 107 BLAST XII _______________ Poster #57 A SURFACE-REGULATED ORPHAN ParA-LIKE PROTEIN HAS AN IMPACT ON GLIDING MOTILITY IN BDELLOVIBRIO BACTERIOVORUS David S. Milner, Emma Saxon and Liz Sockett The Centre for Genetics and Genomics, University of Nottingham, NG7 2UH, United Kingdom Bdellovibrio bacteriovorus is a small, liquid- and surface- motile, predatory bacterium which invades other Gram-negative bacteria to replicate. Inside the periplasm of the host bacterium, Bdellovibrio do not replicate by binary fission, but by synchronous septation of a multiploid filament. Bdellovibrio has two genes encoding orphan ParA proteins which have no partner ParB, in addition to the usual parAB gene pair coexpressed in an operon. Whilst ParA ATPases (usually with ParB partners) are involved in chromosome segregation in some other bacteria; orphan ParAs have a wide variety of functions, including aiding localisation of the Z ring and positioning of large chemotaxis clusters. It was hypothesised that one of the orphan ParA proteins would assist in division site selection, due to the complex division strategy of Bdellovibrio. Instead we discovered that one of these Bdellovibrio orphan ParA proteins (ParA2) is regulated in a surface-responsive manner. We show that although transcription of parA2 was unaltered upon surface exposure, fluorescent tagging of the protein revealed localisation is affected when Bdellovibrio cells are applied to a solid surface. The incidence of ParA2 foci alters, and the sites where these foci reside correlates with the surface-related gliding motility of that cell. Directed gene deletion demonstrated that although not essential for predation, loss of the parA2 gene alters surface gliding motility behaviour by Bdellovibrio. Lab: Liz Sockett _______________ 108 BLAST XII _______________ Poster #58 THE ROLE OF POLYPHOSPHATE RELATED GENES IN THE PREDATORY BACTERIA BDELLOVIBRIO BACTERIOVORUS Sarah M Basford, Andrew Gilbert, Liz Sockett. Centre for Genetics and Genomics, School of Biology, Medical School, QMC, University of Nottingham, Nottingham, NG7 2UH UK. Bdellovibrio bacteriovorus is a small predatory Gram-negative bacterium which invades other Gram-negative bacteria. Once inside prey, the Bdellovibrio grow and divide before causing lysis. This lifestyle means that Bdellovibrio has potential as a “living antibiotic” as it attacks and kills pathogens. They small numbers of the cells also have the ability to grow host independently in rich media. Survival in the time between prey invasions and the active processes of invasion itself are energetically costly. This is especially important as Bdellovibrio don’t “feed” extensively outside prey to produce the ATP required. Like many other bacteria, Bdellovibrio are reported to have phosphate-rich granules, hundreds of nanometers in diameter. They also have genes encoding enzymes with homology to exopolyphosphatases (Ppx), which could break down polyphosphate or the secondary messenger pppGpp. Many roles have been assigned to polyphosphate in other bacterial species, including uses as an intermediate-energy or phosphate store for metabolism, and (p)ppGpp is important in the stringent response during amino acid starvation. This project is investigating how the exopolyphosphatase homologues are used in the unique Bdellovibrio system. Directed mutagenesis was used to test the role of polyphosphate degradation during Bdellovibrio development and survival, in order to assess the ideal storage conditions to preserve Bdellovibrio actively for medical application. One ppx gene is involved in development as its deletion causes cells to be shorter and wider than wild type cells and slows their development inside prey, whereas deletion of a different ppx gene homolog does not cause such a phenotype. The Ppx gene product of this second homolog is involved in allowing Bdellovibrio to grow host independently, as its removal decreases the number of cells which are able to convert from host dependent growth to host independent growth. Lab: Liz Sockett _______________ 109 BLAST XII _______________ Poster #59 IDENTIFICATION OF A NOVEL CELL-ENVELOPE SUBCOMPLEX INVOLVED IN GLIDING MOTILITY IN MYXOCOCCUS XANTHUS Beata Jakobczak, Daniela Keilberg, Lotte Søgaard-Andersen Max Planck Institute for Terrestrial Microbiology, Marburg, 35043 Germany Myxococcus xanthus is a rod-shaped, Gram-negative bacterium that has two different motility systems: the A- and S-motility system. A-motility, also referred to as gliding motility, allows movement of single cells, while S-motility is cell-cell contact-dependent and is similar to twitching motility. If genes of one of the motility systems are deleted, cells remain motile by the means of the remaining system. Mutations which abolish both systems lead to non-motile cells. While S-motility depends on extension and retraction of type-IV-pili, the A-motility is powered by the pH gradient across the cytoplasmic membrane through the AglQRS and/or the AglXV complex. The exact mechanism of Amotility is not known; however, numerous proteins essential for A-motility have been described, four of them (AglQ, AglZ, mxan_4868/GltF and AgmU/GltD) have been shown to localize to focal adhesion complexes (FACs). FACs are distributed along the cell body and are stationary with respect to the substratum in moving cells. We identified two genes, agmO and mxan_2541, by a transposon mutagenesis screen to be required for A-motility. Further experiments gave evidence that the two genes between agmO and mxan_2541 (mxan_2539, mxan_2540) are also required for A-motility. agmO encodes a protein with a lipoprotein signal peptide. Furthermore, AgmO belongs to the group of contact dependent gliding motility proteins and is likely localized to the outer membrane. In contrast, mxan_2539, mxan_2540 and mxan_2541 encode proteins with a signal peptide I. Moreover mxan_2539 and mxan_2540 are predicted to contain a domain similar to the the outer membrane domain of OmpA, while mxan_2541 is predicted to contain TPR domain. Bioinformatic analyses showed conservation of these genes in Myxococcales genomes suggesting that the four proteins interact. Interestingly, the three genes mxan_2539, mxan_2540 and mxan_2541 were often found in the genetic context of homologs of known A-motility genes, such as agmU and aglT. Our data demonstrate that these four proteins localize to the cell envelope as predicted from the bioinformatic analyses and are dependent on each other for stability. Furthermore, interaction studies strongly indicate that these four A-motility proteins form a complex in the cell envelope. We are currently determining the localization of the four proteins as well as their putative interaction with known FAC components using bioinformatic, biochemical and localization studies. Lab: Lotte Søgaard-Andersen ____ 110 BLAST XII _______________ Poster #60 DECIPHERING THE ASSEMBLY PATHWAY OF TYPE IV PILI IN MYXOCOCCUS XANTHUS Carmen Friedrich, Iryna Bulyha & Lotte Søgaard-Andersen Max Planck Institute for Terrestrial Microbiology, Marburg, 35043 Germany Myxococcus xanthus moves by gliding. For this purpose, M. xanthus uses two independent motility engines. The Social motility system is powered by type IV pili (T4P). T4P are built at the leading cell pole and generate movement by undergoing cycles of extension, attachment to the surface of other cells and retraction, which provides the force to pull a cell forward. When cells reverse, T4P switch poles. T4P are composed of thousands of copies of the type IV pilin. The pilin subunits are incorporated into the base of the growing pilus from a reservoir of pilins in the inner membrane. Similarly, when pili retract, pilin subunits are removed from the base of the pilus and incorporated back into the inner membrane. The channel in the outer membrane through which the pilus is extruded is built by the secretin PilQ. The energy required for T4P extension and retraction is provided by two cytoplasmic ATPases PilB and PilT, respectively. Furthermore, the inner membrane platform protein PilC is required for T4P biogenesis. While some of the T4P-proteins, i.e. PilC, PilM and PilQ, localize to both poles and remain stationary during a reversal, PilB and PilT localize mainly to one pole and switch poles upon a reversal. These data suggest that some T4P-proteins build a preassembled complex at both poles and are complemented by the dynamic proteins PilB or PilT to build a functional pilus extension and retraction complex. The proteins PilM, PilN, PilO and PilP are also important for T4P-dependent motility, but their function remains unclear. Approximately 10 pil-genes are conserved and essential for T4aP assembly and function. We have as a working hypothesis that these proteins interact to form a trans-envelope macromolecular complex supporting T4P assembly and dynamics. However, it is currently unknown how the T4Pproteins interact to make a functional T4P machinery. To analyze in which order and dependence these proteins work together in the T4P assembly machinery, we determined the stability of every T4P protein in the absence of each individual T4P protein. Similarly, we determined the localization of every T4P protein and how correct localization of each T4P protein depends on other T4P proteins. Our protein stability and localization data suggest that PilM/N/O/P/Q interact and that PilM/N/O/P depend on the outer membrane secretin PilQ and the outer membrane lipoprotein Tgl for correct bipolar localization. To verify the interconnection between single components of the protein complex, we are currently analyzing direct interactions between PilM, PilN, PilO PilP and PilQ via co-purification experiments or site-directed mutagenesis. Further localization experiments revealed that the inner membrane platform protein PilC depends on PilN, PilQ and Tgl for correct localization. In contrast, the two cytoplasmic ATPases PilB and PilT, which switch poles during a reversal, localize independently from the presence of PilM/N/O/P/Q/Tgl. This suggests that the assembly of the T4P machinery does not follow a hierarchical pathway, but involves at least two independent pathways. Lab: Lotte Søgaard-Andersen ____ 111 BLAST XII _______________ Poster #61 DIVERSITY OF SMALL Ras-LIKE G-PROTEINS IN PROKARYOTES Kristin Wuichet, Stuart Huntley, and Lotte Søgaard-Andersen Max Plank Institute for Terrestrial Microbiology, Marburg, 35043 Germany Members of the Ras superfamily of small G-proteins are ubiquitous in eukaryotes and function as nucleotide-dependent molecular switches that cycle between an active GTP-bound form and an inactive GDP-bound form. The inherent GTPase activity of these proteins is low and requires the assistance of a GTPase activating protein (GAP). Additionally, they have a high affinity for both GTP and GDP and utilize guanine nucleotide exchange factors (GEFs) to exchange GDP for GTP. Ras-like G-proteins were initially identified in and considered to be exclusive to eukaryotes. Experimental studies demonstrated that Ras-like G-proteins have important functions in e.g. signal transduction, cell polarity and motility. Experimental studies and sequence characterization revealed MglA, the master motility regulator of Myxococcus xanthus, to be a Ras-like small G-protein and its partner protein, MglB, which is a member of the superfamily of Roadblock proteins, functions as a GAP. Currently, no GEF has been identified as part of this system. Interestingly, the MglA/MglB module functions to regulate cell polarity and motility. Here, we performed a computational analysis of 1611 prokaryotic genomes to tally the distribution of small Ras-like G-proteins in prokaryotes. Using sequence similarity and genome context methods, we expanded on previous studies, identifying and computationally characterizing 630 prokaryotic Ras-like G-proteins in 277 genomes from diverse lineages including Acidobacteria, Actinobacteria, Aquificales, Bacteriodetes, Chlorobi, Chloroflexi, Cyanobacteria, Deferribacteres, Deinococcus/Thermus, Dictyoglomi, Fibrobacteres, Gemmatimonadetes, Proteobacteria, Verrucomicrobia as well as archaeal species. MglA-like sequences make up the largest family of prokaryotic Ras-like G-proteins and co-evolve with MglB-like sequences. MglA-like proteins can be divided into five subfamilies. Our previous structural analysis of a close homolog of the canonical MglA from M. xanthus revealed a novel catalytic mechanism. Interestingly, sequence diversity among signature motifs of the other four subfamilies suggests additional G-protein catalytic mechanisms that have yet to be revealed. While MglA-like proteins only maintain conservation of four of the five Ras signature motifs that were established in eukaryotic studies, a few families of prokaryotic Ras-like Gproteins maintain conservation of all five Ras signature motifs. Genome context analyses hint at coevolving proteins that may function as GAPs or GEFs for these family members; however, none of these G-proteins or neighboring sequences have been characterized experimentally. The abundance and wide distribution of small Ras-like G-proteins suggest that they may have critical functions in prokaryotic physiology most of which have yet to be uncovered. Our analysis provides valuable predictions that can be tested experimentally. Lab: Lotte Søgaard-Andersen ____ 112 BLAST XII _______________ Poster #62 AN ENGINEERED SERINE CHEMORECEPTOR WITH SYMMETRIC INSERTIONS MIMICS NATURAL TRANSDUCERS OF THE 40H CLASS Herrera Seitz, M.K, Massazza, D.A. and Studdert, C.A Instituto de Investigaciones Biológicas, Universidad Nacional de Mar del Plata, Buenos Aires, Argentina Bacterial chemoreceptors usually detect extracellular signals through a periplasmic sensing domain and transmit them to a highly conserved intracellular domain. The signal then reaches the flagellar motors to control swimming behavior. The cytoplasmic signaling domain consists of a long alpha-helical hairpin that forms, in the dimer, a coiled-coil four-helix bundle. The huge variety of chemoreceptors identified from genomic analysis in Bacteria and Archaea can be classified into a small number of classes according to the length of their cytoplasmic signaling domain. Differences in length are due to the presence of pairs of insertions or deletions of seven-residue stretches (heptads), located symmetrically with respect to the hairpin turn. The size and location of the indels highlight the importance of the coiled-coil structure and suggest the existence of specific interactions between the two arms of the hairpin, that are needed to preserve proper signal transmission. To understand the structural requirements of signal transmission that led to this peculiar evolution pattern, in our lab we are engaged in the construction and characterization of derivatives of the serine chemoreceptor of E. coli, Tsr (36H-class because it possesses 36 heptads in its cytoplasmic domain), to mimic natural transducers of different length classes. In this work, we built a 40H-class derivative of Tsr through the introduction of symmetric 14-residue insertions, whose sequence and position were chosen based on the alignment of Tsr with PctApp, a 40H-class receptor from Pseudomonas putida. The 40H-class derivative, TsrH18, was able to activate the CheA kinase at a high level. However, it was insensitive to serine and unable to mediate serine taxis in soft agar plates. Random mutagenesis applied to the construct allowed the isolation of two functional derivatives (TsrH18*) that carry single point mutations, indicating that subtle changes restored signaling abilities to the enlarged hairpin. TsrH18* derivatives were able to control CheA activity in response to serine. They also localized to the poles of the cell, as assessed with the fluorescent reporters YFP-CheZ or YFP-CheR. In contrast, the original TsrH18 construct failed to mediate YFP-CheZ localization, suggesting its inability to form properly assembled ternary complexes. Unlike the derivative belonging to the 34H class that he had characterized in previous work, this 40H-class derivative does not interfere with the chemotactic function of native Tar (36H class), and it does not form mixed trimers of dimers with Tar, probably due to the larger length difference. The availability of functional derivatives that mimic receptors of different classes represent useful tools to study interactions between different receptors in the same cell and the requirements for co-operation within the same chemoreceptor cluster. Lab: Claudia Studdert ___________ 113 BLAST XII _______________ Poster #63 FUNCTIONAL AND STRUCTURAL ANALYSIS OF HOMOLOGOUS CONSERVED RESIDUES CheWR62 AND CheA-R555 WITHIN THE CHEMOTAXIS TERNARY COMPLEX Pedetta A.1, Parkinson J.S.2, Studdert C.A.1 1 Instituto de Investigaciones Biológicas, Universidad Nacional de Mar del Plata, Mar del Plata, Argentina 2 Biology Department, University of Utah, Salt Lake City, USA The small protein CheW, structurally homologous to the P5 regulatory domain of CheA, couples the receptors with the kinase, playing an essential role in CheA control. Various studies in recent years indicate that the role of CheW may be more complex than that of a simple bridge between the two proteins. We focused our study on the conserved residues R62 in CheW and R555 in CheA. These arginine residues lie in equivalent positions in CheW and the homologous P5 domain in CheA, and replacements in either of them cause severe impairment of chemotactic behavior. However, the mutant proteins mediate the formation of ternary complexes with normal kinase activity when studied in vitro, using chemoreceptor-containing membranes. To assess the effect of these replacements on the signaling ability of the complex in living cells we performed a FRET-based assay. The assay, based on CheY-CheZ interaction as a read-out of kinase activity, allowed us to compare the sensitivity and cooperativity of the response in cells expressing the wild-type versions of CheA and CheW with those of cells expressing the mutant variants. Whereas in CheW-R62 mutants the sensitivity and cooperativity were slightly reduced, in CheA-R555 mutants both parameters were more significantly affected. In order to contribute to the understanding of the arrangement of CheW within the signaling complex, CheW R62C was co-expressed with Tsr variants carrying single cysteine replacements, and cells were subjected to oxidizing conditions through diamide treatment. The co-expression of CheW R62C with Tsr V398C (but not Tsr carrying other cysteine replacements) at physiological levels led to the formation of a disulfide-bonded product between the two proteins, indicating that the crosslinking reflects a specific interaction. Moreover, the presence of Tsr V398C corrects the chemotaxis defect of cells expressing CheW R62C, indicating that this protein pair is part of a fully functional complex. The extent of crosslinking is somehow increased in cells that lack the CheA kinase. This fact could be explained if there exists some competition between CheW and CheA (P5 domain) for interaction with Tsr. However, we could not detect any disulfide bond formation between CheA R555C and Tsr V398C. Our results suggest that small changes in the conformation of the ternary complexes may lead to reduced sensitivity and/or cooperativity of the system, with consequent alterations in signal amplification and in the chemotactic response. Lab: Claudia Studdert ___________ 114 BLAST XII _______________ Poster #64 EFFECT OF A CHEMOEFFECTOR ON THE STABILITY OF CHEMORECEPTORS CLUSTERING STUDIED BY PHOTOACTIVATED LOCALIZATION MICROSCOPY F. Anquez and T.S. Shimizu FOM Institute AMOLF, Amsterdam Bacterial chemoreceptors cluster into partially ordered arrays which provides cooperative signal amplification and their sensitivity can be tuned via covalent modification, allowing adaptation. A number of studies have addressed the in vivo stability of the large polar clusters upon addition of chemoattractants with conflicting results : while some studies [1-3] yielded data indicating that polar clusters are disrupted upon attractant stimulation, others found that similar stimuli do not appear to significantly affect the appearance of clusters [4,5]. Notably, all of these studies have employed diffraction-limited fluorescence microscopy, the resolution of which is limited to ~250 nm. Here, we employ photoactivation localization microscopy (PALM) which can achieve resolution down to 10nm [6,7], to probe the distribution of Tar-receptor cluster sizes (a) in the absence of chemoeffector stimuli, (b) shortly after exposure to the attractant methylaspartate (MeAsp), and (c) after adaptation to sustained MeAsp stimulation. Preliminary results suggest that exposure to this chemoeffector can disrupt polar clusters shortly after MeAsp addition, but the cluster size distribution is restored to its pre-stimulus state upon prolongated stimulation. [1] Lamanna et al., Mol. Microbiol. 57 (3) pp. 774-785 (2005) [2] Borrock et al., ACS Chem. Biol. 3 (2) pp. 101-109 (2008) [3] Wu et al., J. Biol. Chem. 286 pp. 2587-2595 (2011) [4] Homma et al., PNAS 101 (10) pp. 3462-3467(2004) [5] Liberman et al. J. Bacteriol. 186 (19) pp. 6643-6646 (2004) [6] Betzig et al. Science 313 (5793) pp.1642-1645 (2006) [7] Greenfield et al. Plos Biol.7 (6) pp. e1000137 (2009) Lab: Tom Shimizu ______________ 115 BLAST XII _______________ Poster #65 THE FLIP OF A COIN: ACETYLATION OR PHOSPHORYLATION BY ACETYL PHOSPHATE IN BIOFILMS Abouelfetouh, Alaa A. Y.1,2, Zemaitaitis, Bozena1 and Wolfe, Alan J.1 1 Department of Microbiology and Immunology, Loyola University Chicago, Stritch School of Medicine, 2160 S. First Ave. Bldg. 105, Maywood, IL 60153. 2 Department of Pharmaceutical Microbiology, Alexandria University, Faculty of Pharmacy, 1 Khartoum Sq., Alexandria, Egypt 21521. The high-energy intermediate acetyl phosphate (acetyl-P) plays a pivotal role in metabolism. It accumulates during growth in the presence of large amounts of carbon. As the flux of carbon through glycolysis puts pressure on the limited supply of coenzyme A, the acetyl group from acetyl-CoA is transferred to inorganic phosphate (by the enzyme phosphotransacetylase, Pta). Because acetyl-P retains the high-energy status of acetyl-CoA, it can donate its phosphoryl group to ADP to generate ATP (by the enzyme acetate kinase, AckA). Acetyl-P can also donate its phosphoryl group to certain two-component response regulators (e.g. RcsB and CpxR). We recently discovered a third function: acetyl-P donates its acetyl group to a large set of proteins that function in diverse cellular processes, including many critical to the development of biofilms. We previously reported that acetyl-P influences biofilm development by E. coli K-12 (Wolfe et al., 2003) and that this effect acts in part through the response regulator RcsB (Fredericks et al., 2006). Since acetyl-P can donate its phosphoryl group to response regulators and its acetyl groups to diverse proteins (including some response regulators), it is clear that acetyl-P could influence biofilm development by many mechanisms. This abstract describes early studies designed to dissect those mechanisms. One strategy is to identify genes whose deletion either enhances or suppresses phenotypes of an ackA mutant, including reduced motility, increased mucoidy, defective biofilm development and increased protein acetylation. We therefore have generated and screened double mutants of the type ackA yfg. We have begun by constructing double mutants with genes that encode proteins involved in acetyl transfer, e.g. the protein deacetylase CobB, one known protein acetyltransferase (YfiQ/Pat) and several predicted GCN5-like acetyltransferases. At present, we can conclude that acetyl-P and CobB affect protein acetylation by independent pathways: the double mutant is more hyper-acetylated and more defective for biofilm development than either single mutant. On the basis of the behaviors of the acetyltransferase mutants, this inverse relationship may be general. A complex relationship is emerging between the deacetylase CobB and the response regulator RcsB. Based on the assessment of either biofilm development or protein acetylation, we come to the conclusion that CobB and RcsB function in the same pathway. The order of the proteins, however, appears to be reversed depending on which phenotype we analyze. Most understandable is the prediction that CobB inhibits the activity of RcsB, which regulates protein acetylation. This fits with other evidence that our laboratory has obtained: RcsB becomes acetylated in vivo and some of that acetylation is sensitive to CobB (Hu et al., submitted). Fredericks C. E. et al (2006): Acetyl phosphate-sensitive regulation of flagellar biogenesis and capsular biosynthesis depends on the Rcs phosphorelay. Molecular Microbiology. 61(3), 734–747. Wolfe A. J. et al. (2003): Evidence that acetyl phosphate functions as a global signal during biofilm development. Molecular Microbiology. 48(4), 977–988. Lab: Alan Wolfe ________________ 116 BLAST XII _______________ Poster #66 ACETYL PHOSPHATE IS A POTENT REGULATOR OF BACTERIAL PROTEIN ACETYLATION Alan J. Wolfe1*, Birgit Schilling2, Bozena Zemaitaitis1, Linda Hu1, Bruno Lima1, Bradford Gibson2 1 Loyola University Chicago, Maywood, IL 60153 2 Buck Institute for Research on Aging, Novato, CA 94702 Reversible Nε-lysine (protein) acetylation of bacterial proteins is a previously unrecognized regulatory mechanism. Since the study of bacterial protein acetylation is in its infancy, many questions remain unanswered. The mechanisms responsible for most protein acetylations remain obscure, their effect on protein function remains unclear, and their global impact remains unquantified. Earlier studies provided evidence for this post-translational modification on about two hundred proteins in E. coli and S. enterica. We now report that that the acetylated proteome (‘acetylome’) is much larger than previously reported. Our preliminary experiments using a novel label-free quantitative mass spectrometry analysis identified 1515 unique acetylation sites on 541 E. coli proteins that function in diverse cellular processes, including signal transduction, transcription, quorum sensing, biofilm formation and pathogenesis. Some acetylated proteins include the response regulator RcsB, the global transcription factor CRP, and RNA polymerase. If only a fraction of the detected acetylations exert a significant impact on their protein’s function, then this post-translational modification would easily surpass phosphorylation as the primary regulatory mechanism. The Pta-AckA pathway links the central metabolite acetyl-CoA to ATP generation. The pathway intermediate acetyl phosphate has been shown to donate its phosphoryl group to two-component response regulators, e.g. CpxR and RcsB. We now propose that this pathway is a potent regulator of bacterial protein acetylation. Anti-acetyllysine Western immunoblot and quantitative mass spectrometry analyses showed hyperacetylation of mutants that lack AckA. In contrast, mutants that lack the entire Pta-AckA pathway are less acetylated like the WT parent. Since these pta ackA mutants differ from the ackA mutants by their retention of Pta and its ability to synthesize acP, these data argue that protein acetylation is regulated by Pta and/or acP. More surprising is the observation that the Pta-AckA pathway is a more potent regulator of protein acetylation than the known E. coli KAT and KDAC (YfiQ and CobB, respectively), as similar analyses of cells that lack these enzymes show more minor effects. Since similar effects are observed in other organisms including pathogens, we conclude that acetylation is both universal and relevant to public health. Lab: Alan Wolfe ________________ 117 BLAST XII _______________ Poster #67 A NOVEL CHEMORECEPTOR COUPLES DEGRADATION OF AROMATIC POLLUTANTS WITH CHEMOTAXIS Zhihong Xie, Jiangfeng Gong, Yongming Luo CAS and Shandong Provincial Key Laboratory of Coastal Environmental Processes, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, 264003, Yantai Shandong, PR China Pesticides and industrial wastes often contain aromatic constituents, and most of them are toxic to living organisms. The use of plants and bacterial to clean up in soil has gained increasing interest in past years. Rhizobia are motile alpha-proteobacteria that can establish a nitrogen-fixing symbiosis within the roots of leguminous plants. These bacteria show chemotaxis to aromatic constituents and soybean root exudates, such as organic acid, sugar, and amino acids, which is an initial and important step for establishing the symbiotic relationship with legumes. But the relationship between chemotaxis response and behavioral variability in nodule bacteria is not clear. In this study, the functions of the methyl-accepting chemotaxis protein TLP5 in Azorhizobium caulinodans ORS571 is determined, since this protein were predicted to be important in chemotaxis and microbe-host interactions. By generating a tlp5 deleted mutant strain, we analyzed its behavior in chemotaxis, nodulation and degradation of 4-Hydroxybenzoate. We found that tlp5 was unaffected in its aerotaxis response but was affected in its chemotactic ability. We have found that TLP5 modulate the motility swimming bias of Azorhizobium cells and have demonstrated that TLP5 promotes competitive nodulation of the legumes and degrades the 4-Hydroxybenzoate. We therefore hypothesize that tlp5 is an energy sensing chemotaxis protein and represents a novel function of the chemoreceptor.This finding implies that the environmental cue(s) triggering chemotaxis of Azorhizobium cells towards the roots of legumes and facilitating degradation of pollutants in soil by root-nodule symbiosis. The findings obtained from this study will help to investigate novel regulatory mechanisms and catabolic activities that can be of great biotechnological interest for improving the microbial degradation of aromatic environmental pollutants. Further studies will be performed for better understanding of the mechanisms of Che-like pathways and their potential use in optimization of conditions for applications of Rhizobium species in bioremediation. Lab: Zhihong Xie _______________ 118 BLAST XII PARTICIPANT LIST 119 Alaa Abouelfetouh Loyola University Chicago Microbiology and Immunology 128 Washington Blvd Oak Park, IL 60302 alaa.abouelfetouh@pharmacy.alexu.edu.eg Christopher Adase Texas A&M University Biochemistry 3258 TAMU BSBE Room 303 College Station, TX 77843 Phone: (979) 845-1249 xpage@neo.tamu.edu Oshri Afanzar Weizmann Institute of Science Biological Chemistrey Rehovot 76100 Israel Phone: +972 8 934 3685 afanzar@gmail.com Abu Amar Al Mamun Baylor College of Medicine Molecular and Human Genetics One Baylor Plaza, MSC 225 Houston, TX 77030 Phone: (713) 798-6693 mamun@bcm.edu James Allen Oxford University Biochemsitry Parks Road New Biochemistry Oxford, Oxon OX1 3QU United Kingdom Phone: +44 1865 613 315 james.allen@wadh.ox.ac.uk Francois Anquez FOM Institute AMOLF P.O. Box 41883 Science Park 102 Amsterdam 1009 DB Netherlands Phone: +31 20 754 7278 anquez@amolf.nl Judith Armitage Oxford Biochemistry Department of Biochemistry Oxford OX1 3QU United Kingdom Phone: +44 1865 613 293 judith.armitage@bioch.ox.ac.uk Marie-Stephanie Aschtgen University of Wisconsin - Madison Department of Medical Microbiology and Immunology Microbial Sciences Building Room 5245 1550 Linden Drive Madison, WI 53706 Phone: (608) 262-5550 aschtgen@wisc.edu Sonia Bardy University of Wisconsin-Milwaukee Biological Sciences 3209 N Maryland Ave Milwaukee, WI 53211 Phone: (414) 229-6415 bardy@uwm.edu Szilvia Baron Weizmann Institute of Science Biological Chemistry Ulmann Building Room 17 Rehovot 76100 Israel Phone: +972 8 934 2702 szilvia.baron@weizmann.ac.il 120 Sarah Basford University of Nottingham Centre for Genetics and Genomics C15, Centre for Genetics and Genomics Medical School, QMC Nottingham NG7 2UH United Kingdom sbxsb@nottingham.ac.uk Morgan Beeby Imperial College London Life Sciences Division of Molecular Biosciences South Kensington Campus London SW7 2AZ United Kingdom mbeeby@imperial.ac.uk Stefan Behr Ludwig-Maximilians-University Grosshaderner Strasse 2-4 Martinsried - Munich, Bavaria D-82152 Germany Phone: +49 89 2180 74535 Fax: +49 89 2180 74520 stefan.behr@campus.lmu.de Wiebke Behrens Hannover Medical School Institute for Medical Microbiology Carl-Neuberg-Strasse 1 Hannover 30625 Germany Phone: +49 511 532 8028 ext. 8028 behrens.wiebke@mh-hannover.de Robert Belas University of Maryland Baltimore County Marine Biotechnology Institute of Marine & Environmental Technology 701 East Pratt Street Baltimore, MD 21202 Phone: (410) 234-8876 belas@umbc.edu Howard Berg Harvard University Molecular and Cellular Biology Harvard Biological Laboratories 16 Divinity Avenue, Room 3063 Cambridge, MA 02138 Phone: (617) 495-0924 Fax: (617) 496-1114 hberg@mcb.harvard.edu Indranil Biswas University of Kansas Medical Center 3901 Rainbow Blvd Kansas City, KS 66160 Phone: (913) 588-7019 Fax: (913) 588-7295 ibiswas@kumc.edu Bob Bourret University of North Carolina Microbiology & Immunology Chapel Hill, NC 27599-7290 Phone: (919) 966-2679 Fax: (919) 962-8103 bourret@med.unc.edu Caitlin Brennan University of Wisconsin 1550 Linden Drive, 5245 MSB Madison, WI 53706 Phone: (608) 262-5550 cabrennan@wisc.edu Ariane Briegel California Institute of Technology Biology 1200 E. California Blvd. Mail code 114-96 Pasadena, CA 91125 Phone: (626) 395-8848 briegel@caltech.edu 121 Rebecca Calvo Indiana University Bloomington Biology 1001 E. Third St. Bloomington, IN 47405 rcalvo@indiana.edu Nyles Charon West Virginia Uninversity Microbiology, Immunology, and Cell Biology Health Sciences Center North Box 9177 Morgantown, WV 26506-9177 Phone: (304) 293-4170 Fax: (304) 293-7823 ncharon@hsc.wvu.edu Remy Colin Rowland Institute at Harvard 100 Edwin H. Land Bvd Cambridge, MA 02142 Phone: (617) 497-4715 colin@rowland.harvard.edu Kieran Collins UC Santa Cruz METX 1156 High Street Santa Cruz, CA 95064 Phone: (415) 613-3316 kdcollin@ucsc.edu Brian Crane Cornell University Chemistry and Chemical Biology G75 Chemistry Research Building Ithaca, NY 14853 Phone: (607) 254-8634 Fax: (607) 255-1248 bc69@cornell.edu Rachel Creager-Allen University of North Carolina Campus Box 7290 Chapel Hill, NC 27599 Phone: (919) 966-2679 rcreager@email.unc.edu Sean Crosson University of Chicago Biochemistry and Molecular Biology 929 E. 57th St. GCIS-W138 Chicago, IL 60637 Phone: (773) 834-1926 scrosson@uchicago.edu Rick Dahlquist Department of Chemistry & Biochemistry University of California Santa Barbara Santa Barbara, CA 93106 Phone: (805) 893-5326 dahlquist@chem.ucsb.edu Georges Dreyfus Univ. Nacional Autónoma de México (UNAM) Inst de Fisiología Celular Ap. Postal 70243 Mexico, Mexico City 04510 Mexico Phone: +52 55 5622 5618 Fax: +52 55 5622 5611 gdreyfus@ifc.unam.mx Ismael Duchesne Université Laval Physique 618 rue Fraser Quebec, QC G1S 1R7 Canada Phone: (418) 656-2131 ext. 3788 ismael.duchesne.1@ulaval.ca 122 Michael Eisenbach Weizmann Institute of Science Biological Chemistry PO Box 26 Rehovot 76100 Israel m.eisenbach@weizmann.ac.il Marc Erhardt Helmholtz Centre for Infection Research Infection Biologie of Salmonella Inhoffenstraße 7 Braunschweig, Niedersachsen 38124 Germany Phone: +49 531 6181 5700 marc.erhardt@helmholtz-hzi.de Lewis Evans University of Cambridge Pathology Tennis Court Road Cambridge, Cambridgeshire CB2 1QP United Kingdom Phone: +44 1223 333 733 le227@cam.ac.uk Joseph Falke Univ. Colorado Dept. of Chem. & Biochem. UCB 596 Univ. Colorado Boulder, CO 80309-0596 Phone: (303) 492-3503 falke@colorado.edu Milana Fraiberg Weizmann Institute of Science Biological chemistry P.O. Box 26 Rehovot 76100 Israel Rehovot 76100 Israel Phone: +972 8 934 2710 Fax: +972 8 947 2722 milana.fraiberg@weizmann.ac.il Carmen Friedrich Max Planck Institute for Terrestrial Microbiology Ecophysiology Karl-von-Frisch-Straße 10 Marburg 35043 Germany Phone: +49 6421 178 211 carmen.friedrich@mpi-marburg.mpg.de Hajime Fukuoka Tohoku University IMRAM Katahira 2-1-1 Aoba-Ku Sendai, Miyagi 980-8577 Japan Phone: +81 22 217 5804 Fax: +81 22 217 5804 f-hajime@tagen.tohoku.ac.jp Michael Galperin NCBI, NLM, National Institutes of Health Computational Biology Branch 8600 Rockville Pike, MSC3830 Bldg. 38A, Rm. 5N507 Bethesda, MD 20894 Phone: (301) 435-5910 Fax: (301) 435-7793 galperin@ncbi.nlm.nih.gov Susanne Gebhard Ludwig-Maximilans-University Munich Dept. Biology I, Microbiology Grosshaderner Str. 2-4 Planegg-Martinsried, Bavaria 82152 Germany Phone: +49 89 2180 74601 susanne.gebhard@bio.lmu.de Mizuki Gohara Nagoya Univercity Furo-cho, Chikusa-ku Nagoya, Aichi 4648602 Japan Phone: +81 52 789 2991 gouhara.mizuki@c.mbox.nagoya-u.ac.jp 123 Lindsie Goss Columbia University Microbiology & Immunology 100 Haven Ave T2-16B New York, NY 10032 lag2147@columbia.edu Xuesheng Han University of Utah Biology Department of Biology 257 South 1400 East Salt Lake City, UT 84112-0840 Phone: (801) 581-6307 xuesheng.han@utah.edu Rasika Harshey University of Texas at Austin Mol. Gen. & Microbiology 1 University Station A1000 Austin, TX 78712 Phone: (512) 471-6881 Fax: (512) 471-1218 rasika@uts.cc.utexas.edu Gerald Hazelbauer University of Missouri Biochemistry 117 Schweitzer Hall Columbia, MO 65211 Phone: (573) 882-4845 Fax: (573) 882-5635 hazelbauerg@missouri.edu Penelope Higgs Max-Planck-Institute for Terrestrial Microbiology Ecophysiology Karl-von-Frisch Strasse 10 Marburg, Hessen D35043 Germany Phone: +49 6421 178 301 Fax: +49 6421 178 309 higgs@mpi-marburg.mpg.de Geetha Hiremath-Mendez Hosei University Research Center for Micro-Nano Technology Koganei-shi, Kajino-cho, 3-7-2 Higashi kan E-3001 Tokyo 184-8584 Japan Phone: +42 387 7173 geetha.2225@gmail.com Basarab Hosu Harvard University Molecular and Cellular Biology 16 Divinity Avenue Bio Labs Buliding Room 3068 Cambridge, MA 02138 Phone: (617) 495-6127 Fax: (617) 496-1114 ghosu@mcb.harvard.edu Bo Hu IBM T.J. Watson Research Center 1101 Kitchawan Road, Route 134 Yorktown Heights, NY 10598 Phone: (914) 945-2857 bhu@us.ibm.com Isabelle Hug University of Basel Biozentrum Klingelbergstrasse 50/70 Basel CH-4056 Switzerland Phone: +41 61 267 2117 isa.hug@unibas.ch Kelly Hughes Utah Biology 257 South 1400 East Salt Lake City, UT 84112 Phone: (801) 587-3367 hughes@biology.utah.edu 124 Akihiko Ishijima Tohoku University Katahira 2-1-1,Aoba-ku Sendai 980-8577 Japan ishijima@tagen.tohoku.ac.jp Masahiro Ito Toyo University 1-1-1 Izumino, Itakura-machi Oura-gun, Gunma 3740193 Japan Phone: +81 27 682 9202 masahiro.ito@toyo.jp Beata Jakobczak Max Planck Institute for terrestrial microbiology Karl-von-Frisch-Straße 10 Marburg 35043 Germany Phone: +49 6241 178 222 beata.jakobczak@mpi-marburg.mpg.de Urs Jenal Biozentrum, University of Basel Infection Biology Biozentrum der Universität Basel Klingelbergstrasse 70 Basel, BS 4056 Switzerland Phone: +41 61 267 2135 Fax: +41 61 267 2118 urs.jenal@unibas.ch Grant Jensen Caltech Biology 1200 E California Blvd MC 114-96 Pasadena, CA 91125 Phone: (626) 395-8827 Fax: (626) 395-5730 jensen@caltech.edu Mark Johnson Loma Linda University Biochemistry and Microbiology AHBS 120 Loma Linda, CA 92350 Phone: (909) 558-4480 ext. 42765 Fax: (909) 558-4035 mjohnson@llu.edu Christopher Jones University of Oxford Biochemistry St. Peter's College New Inn Hall Street Oxford OX1 2DL United Kingdom Phone: +44 1865 613 315 christopher.jones2@spc.ox.ac.uk Christine Josenhans Medical University Hannover Institute for Medical Microbiology Carl-Neuberg-Strasse 1 Hannover 30625 Germany Phone: +49 511 532 4354 ext. 4354 Fax: +49 511 532 4354 josenhans.christine@mh-hannover.de Kirsten Jung Ludwig-Maximilians-Universitaet Muenchen Microbiology Biocenter Grosshaderner Str. 2-4 Martinsried, Bavaria 82152 Germany Phone: +49 89 2180 74500 jung@lmu.de Dan Kearns Indiana University Biology 1001 E. 3rd St Bloomington, IN 47405 Phone: (812) 856-2523 dbkearns@indiana.edu 125 Daniela Keilberg Max-Planck-Institute for Terrestrial Microbiology Karl-von-Frisch-Straße 10 Marburg, Hessen D-35043 Germany daniela.keilberg@mpi-marburg.mpg.de Shahid Khan Molecular Biology Consortium Cellular Biophysics MS6-2100 1 Cyclotron Road Berkeley, CA 94720 Phone: (312) 996-1216 khan@mbc-als.org Hyo Kyung Kim University of Texas, Austin 7630 Wood Hollow Dr. #272 Austin, TX 78731 Phone: (512) 963-3021 hkkim2009@gmail.com Yoshiaki Kinosita Gakushuin Physics Toshimaku Mejiro 1-5-1 Tokyo 1718588 Japan Phone: +81 3712 0033 12144002@gakushuin.ac.jp John Kirby University of Iowa Microbiology 51 Newton Road Iowa City, IA 52242 Phone: (319) 335-7818 Fax: (319) 335-9006 john-kirby@uiowa.edu Smiljka Kitanovic University of Utah Biology 257 South 1400 East 201 SB Salt Lake City, UT 84112 Phone: (801) 581-3592 smiljka_kitanovic@yahoo.com Anna Koganitsky Weizmann Institute of Science Biological Chemistry 234 Herzel St, Rehovot 76100 Israel Phone: +972 8 934 2701 anna.koganitsky@weizmann.ac.il Santosh Koirala University of Illinois Chemical and Biomolecular Engineering 600 S Mathews RAL210 Urbana, IL 61801 Phone: (217) 344-7528 koirala2@illinois.edu Seiji Kojima Nagoya University Division of Biological Science, Graduate school of Science Furo-cho, Chikusa-ku Nagoya, Aichi 464-8602 Japan Phone: +81-52-789-2992 Fax: +81-52-789-3001 z47616a@cc.nagoya-u.ac.jp Naomi Kreamer California Institue of Technology Biochemistry and Molecular BIophysics 1200 E California Blvd Pasadena, CA 91125 Phone: (626) 395-4856 nkreamer@gmail.com 126 Tino Krell Estación Experimental del Zaidín Albareda 1 Granada 18008 Spain tino.krell@eez.csic.es Ambroise Lambert Institut Pasteur-Paris 7 University 25-28 rue du Docteur Roux Paris 75015 France Phone: +33 1 40 61 34 06 alambert@pasteur.fr Jürgen Lassak Ludwig Maximillians University Munich Biology I; Microbiology Großhaderner Straße 2-4 Martinisried, Bavaria 82152 Germany Phone: +49 89 2180 74508 Fax: +49 89 2180 74520 juergen.lassak@lmu.de Milena Lazova FOM Institute AMOLF Systems Biology Sciencepark 104 Amsterdam, Noord Holland 1098 XG Netherlands Phone: +31 20 754 7302 lazova@amolf.nl Yi-Ying Lee University of Maryland, Baltimore County Department of Marine Biotechnology 701 E Pratt Street Baltimore, MD 21202 Phone: (410) 234-8877 yyinglee@umbc.edu Pushkar Lele Harvard University Molecular and Cellular Biology Harvard Biological Laboratories 16 Divinity Avenue, Room 3063 Cambridge, MA 02138 Phone: (617) 495-4217 lele@fas.harvard.edu Robert Levenson UC Santa Barbara Chemistry & Biochemistry 761 Birch Walk Apt J Goleta, CA 93117 Phone: (858) 342-5006 rlevenson@chem.ucsb.edu Chunhao Li SUNY at Buffalo Oral Biology 3435 Main St. Buffalo, NY 14214 Phone: (716) 829-6014 Fax: (716) 829-3942 cli9@buffalo.edu Tao Lin UT Medical School at Houston Pathology and Laboratory Medicine PO Box 20708, Room MSB 2.124 Houston, TX 77225-0708 Phone: (713) 500-5350 Fax: (713) 500-0730 tao.lin@uth.tmc.edu Jun Liu UT Houston Medical School Department of Pathology 6431 Fannin, MSB 2.228 Houston, TX 77030 Phone: (713) 500-5342 Jun.liu.1@uth.tmc.edu 127 Michael Manson Texas A&M University Biology 3258 TAMU College Station, TX 77840 mike@bio.tamu.edu Ana Martinez-del Campo Universidad Nacional Autonoma de Mexico Circuito Exterior S/N Ciudad Universitaria Coyoacan Mexico, Distrito Federal 04510 Mexico Phone: +52 55 5622 5618 acampo@email.ifc.unam.mx Shonna McBride Emory University Microbiology and Immunology 1510 Clifton Rd Rm 3022 Atlanta, GA 30322 Phone: (404) 727-6192 shonna.mcbride@emory.edu Regina McGrane Iowa State University Plant Pathology 103 14TH ST Ames, IA 50010 Phone: (641) 521-6759 rnickel@iastate.edu Jonathan McMurry Kennesaw State University Chemistry & Biochemistry 1000 Chastain Rd. MB #1203 Kennesaw, GA 30144 Phone: (770) 499-3238 jmcmurr1@kennesaw.edu Vishnu Menon Weizmann Institute of Science Ullman Building of Life Sciences Rehovot 76100 Israel Phone: +972 8 934 2702 menonvishnu@gmail.com David Milner The University of Nottingham Centre for Genetics and Genomics Queen's Medical Centre Nottingham NG7 2UH United Kingdom Phone: +44 115 823 0317 plxdsm@nottingham.ac.uk Makoto Miyata Osaka City University Department of Biology, Graduate School of Science 3-3-138 Sugimoto Sumiyoshi-ku Osaka 5588585 Japan Phone: +81 6 6605 3157 Fax: +81 6 6605 3158 miyata@sci.osaka-cu.ac.jp Andreas Möglich Humboldt-Universität zu Berlin Institut für Biologie Invalidenstrasse 42 Neubau Raum 304 Berlin 10115 Germany Phone: +49 30 2093 8850 andreas.moeglich@hu-berlin.de Michael Morse Brown University Physics 182 Hope St Providence, RI 02912 Phone: (207) 838-5885 michael_morse@brown.edu 128 Md Motaleb East Carolina University Microbiology and Immunology 600 Moye Blvd BT 116 Greenville, NC 27834 motalebm@ecu.edu Chakib Mouslim University of Utah Biology 257 S 1400 E, Department of biology Kelly Hughes lab Salt Lake City, UT 84112 Phone: (801) 585-6950 ymouslim@gmail.com Tarek Msadek Institut Pasteur Biology of Gram-Positive Pathogens, Department of Microbiology, Biology of Gram-Positive Pathogens 25 Rue du Dr. Roux Paris 75015 France Phone: +33 1 45 68 88 09 Fax: +33 1 45 68 89 38 tmsadek@pasteur.fr Sampriti Mukherjee Indiana University Bloomington 1001 E 3rd St Bloomington, IN 47405 Phone: (812) 856-2559 sampmukh@indiana.edu Daisuke Nakane Nagasaki University Biomedical Sciences 1-7-1 Sakamoto Nagasaki 852-8588 Japan Phone: +81 95 819 7649 jj20110031@cc.nagasaki-u.ac.jp Beiyan Nan UC Berkeley Molecular and Cell Biology 16 Barker Hall, UC Berkeley University of California, Berkeley Berkeley, CA 94720-3204 Phone: (510) 643-5457 nanbeiyan@gmail.com Masayoshi Nishiyama Kyoto University The HAKUBI Center Sakyo-ku Kyoto 606-8302 Japan Phone: +81 75 753 9828 mnishiyama@icems.kyoto-u.ac.jp Catherine Oikonomou Caltech 1200 E. California Blvd. Pasadena, CA 91125 Phone: (626) 395-8848 coiko@caltech.edu Manuel Osorio-Valeriano Universidad Nacional Autónoma de México Molecular Genetics Circuito Exterior S/N Ciudad Universitaria Coyoacán, 04510, México D.F. Mexico City, Distrito Federal 04510 Mexico Phone: +52 55 5622 5618 mosorio@email.ifc.unam.mx Karen Ottemann UC Santa Cruz Microbiology/Env Toxicology 1156 High Street METX Santa Cruz, CA 95064 Phone: (831) 459-3482 ottemann@ucsc.edu 129 Stephani Page University of North Carolina Biochemistry & Biophysics 116 Manning Drive CB# 7290 Durham, NC 27713 stephani_page@med.unc.edu Guillaume Paradis Laval University Physics, Engineering Physics and Optics 2610 d'Oviedo Quebec, QC G2B0G6 Canada Phone: (418) 656-2131 ext. 3788 guillaume.paradis.5@ulaval.ca John Parkinson University of Utah Biology 257 South 1400 East Salt Lake City, UT 84112 Phone: (801) 581-7639 parkinson@biology.utah.edu Jonathan Partridge University of Texas at Austin Molecular Genetics and Microbiology 2506 Speedway Stop A5000 Austin, TX 78712 Phone: (512) 471-6799 j.partridge@gmail.com Darysbel Perez Loma Linda University Microbiology and Molecular Genetics Lindsay Hall Box#284 Loma Linda, CA 92350 Phone: (909) 558-1000 ext. 42758 Fax: 909 558 4035 daperez@llu.edu Kene Piasta University of Colorado at Boulder Chemistry and Biochemistry Jennie Smoly Caruthers Biotechnology Building 596 UCB Boulder, CO 80309 Phone: (303) 492-3597 kene.piasta@colorado.edu German Pinas University of Utah Biology 257 S 1400 E 201 SB Salt Lake City, UT 84112 Phone: (801) 581-6307 g.pinas@utah.edu Steven Porter University of Exeter Biosciences Biosciences, Geoffrey Pope Building Stocker Road Exeter, Devon EX4 4QD United Kingdom Phone: +44 1392 722 172 s.porter@exeter.ac.uk Birgit Prüβ North Dakota State University Veterinary and Microbiological Sciences 1523 Centennial Blvd. Fargo, ND 58108 Phone: (701) 231-7848 Birgit.Pruess@ndsu.edu Simon Rainville Laval University Physics Pavillon optique photonique 2375, rue de la Terrasse Quebec, QC G1V 0A6 Canada Phone: (418) 656-2131 ext. 12511 Fax: (418) 656-2623 simon.rainville@phy.ulaval.ca 130 Christopher Rao University of Illinois Chemical Engineering 211 Roger Adams Lab 600 S Mathews Ave Urbana, IL 61801 Phone: (217) 244-2247 chris@scs.uiuc.edu S. James Remington University of Oregon Molecular Biology 297 Klamath Hall 1229 University of Oregon Eugene, OR 97403 remington@molbio.uoregon.edu Anna Roujeinikova Monash University Microbiology Building 76 Monash University Clayton, N/A 3800 Australia Phone: +613 9902 9194 Anna.Roujeinikova@monash.edu Keiko Sato Nagasaki University 1-7-1 Sakamoto Nagasaki 852-8588 Japan Phone: +81 95 819 7649 satou@nagasaki-u.ac.jp Florian Schubot Virginia Tech Biological Sciences Life Science I, Room 125 Washington Street Blacksburg, VA 24061 Phone: (540) 231-2393 fschubot@vt.edu Matthew Sears Texas A&M University Biology 3258 TAMU College Station, TX 77843 Phone: (979) 845-1249 matthewrsears@gmail.com Larry Shimkets University of Georgia Microbiology 527 Biological Sciences Athens, GA 30602 Phone: (706) 542-2681 Fax: (706) 542-2674 shimkets@uga.edu Joshua Shrout University of Notre Dame 156 Fitzpatrick Hall Notre Dame, IN 46556 Phone: (574) 631-1726 joshua.shrout@nd.edu Ruth Silversmith University of North Carolina Microbiology and Immunology Room 804 Mary Ellen Jones Chapel Hill, NC 27599 Phone: (919) 966-2679 Fax: (919) 962-8103 silversr@med.unc.edu Ria Sircar Cornell University Chemistry & Chemical Biology Physical Sciences Building, Room # 360 Cornell University Ithaca, NY 14853 Phone: (607) 255-0752 rs693@cornell.edu 131 Matthew Smith University of Oxford Biochemistry OCISB New Biochemistry South Parks Road Oxford OX1 3QU United Kingdom Phone: +44 1865 613 317 matthew.smith@chem.ox.ac.uk Lotte Søgaard-Andersen Max Planck Institute for Terrestrial Microbiology Karl-von-Frisch. Str. Marburg 35043 Germany sogaard@mpi-marburg.mpg.de Claudia Studdert Universidad Nacional de Mar del Plata Instituto de Investigaciones Biológicas Funes 3250 Mar del Plata, Buenos Aires 7600 Argentina Phone: +54 223 475 3030 ext. 12 Fax: +54 223 475 3150 studdert@mdp.edu.ar Syed Sultan East Carolina University Microbiology & Immunology BT-115, 600 Moye Blvd Greenville, NC 27834 Phone: (252) 744-3128 sultans@ecu.edu Ching Wooen Sze University at Buffalo Oral Biology 316 Foster Hall 3435 Main Street Buffalo, NY 14214 Phone: (716) 829-6329 chingsze@buffalo.edu Hendrik Szurmant The Scripps Research Institute Molecular and Experimental Medicine 10550 N Torrey Pines Rd MEM-116 La Jolla, CA 92037 Phone: (858) 784-7904 Fax: (858) 784-7966 szurmant@scripps.edu Yuhei Tahara Osaka City University Biology 3-3-138 Sugimoto Sumiyoshi-ku Osaka 558-8585 Japan Phone: +81 6 6605 2575 Fax: +81 6 6605 3158 tahara@sci.osaka-cu.ac.jp Hirotaka Tajima Nagoya University Chikusa-ku, Furo-cho Nagoya, Aichi Prefecture 464-8603 Japan Phone: +81 52 789 2717 tajima@mein.nagoya-u.ac.jp Norihiro Takekawa Nagoya University Division of Biological Science Furo-cho, Chikusa-ku Nagoya, Aichi 464-8602 Japan Phone: +81 52 789 3543 takekawa.norihiro@d.mbox.nagoya-u.ac.jp Akihiro Tanaka Osaka City University Biology 3-3-138 Sugimoto Sumiyoshi-ku Osaka Osaka 558-8585 Japan Phone: +81 6 6605 2575 Fax: +81 6 6605 3158 atanaka@sci.osaka-cu.ac.jp 132 Lynmarie Thompson University of Massachusetts Chemistry LGRT 122 Amherst, MA 01003 Phone: (413) 545-0827 thompson@chem.umass.edu Yuhai Tu IBM Research 1101 Kitchawan Rd./Rt. 134 Yorktown Heights, NY 10598 yuhai@us.ibm.com Isil Tulum Osaka City University Grad.Sch.of Sci. Depart. of Bio. Cell Function Lab Sugimoto, Sumiyoshi-ku Osaka 558-8585 Japan Phone: +81 6 6605 2575 Fax: +81 6 6605 3158 iciltlm@gmail.com Ady Vaknin Hebrew University Givat Ram Jerusalem 91904 Israel Phone: +972 50 250 2873 avaknin@phys.huji.ac.il Barry Wanner Purdue University BioSci 915 West State Street BioSci Lilly Hall West Lafayette, IN 47907 Phone: (765) 494-8034 blwanner@purdue.edu Kylie Watts Loma Linda University Microbiology and Molecular Genetics Div. of Microbiology and Molecular Genetics AHBS 102 Loma Linda, CA 92350 Phone: (909) 558-1000 ext. 83394 Fax: 909 558 4035 kwatts@llu.edu Ben Webb Virginia Tech Biological Sciences Life Sciences 1 Washington St. Blacksburg, VA 24060 Phone: (571) 426-9433 mercury1@vt.edu Laurence Wilson The Rowland Institute at Harvard 100 Edwin H Land Boulevard Cambridge, MA 02142 Phone: (617) 497-4643 wilson@rowland.harvard.edu Alan Wolfe Loyola University Chicago Microbiology and Immunology 2160 South First Avenue Maywood, IL 60153 Phone: (708) 216-5814 awolfe@lumc.edu Chui Ching Wong National University of Singapore Mechanobiology Institute 5A Engineering Drive 1 T-Lab Building, #09-01 Singapore 117411 Singapore Phone: +65 8188 7475 mbiwong@nus.edu.sg 133 Liang Wu Iowa State University Department of Plant Pathology and Microbiology 207 Science I Iowa State University Ames, IA 50010 Phone: (515) 294-3198 liangwu@iastate.edu Kristin Wuichet Max Plank Institute for Terrestrial Microbiology Karl von Frisch Str. 10 Marburg, Hesse 35043 Germany kristin.wuichet@mpi-marburg.mpg.de Elsio Wunder Yale School of Public Health EMD 60 College Street, LEPH 607 New Haven, CT 06510 Phone: (203) 737-6412 elsio.wunder@yale.edu Zhihong Xie Yantai Institute of Coastal Zone Research,Chinese Academy of Sciences Chinese Academy of Sciences 17 Chunhui Rd, Laishan District Yantai, Shandong 264003 China Phone: +86 535 210 9183 Fax: +86 535 210 9000 zhxie@yic.ac.cn Hiroki Yamamoto Osaka City University Biology 3-3-138 Sugimoto Sumiyoshi-ku Osaka Japan Osaka 558-8585 Japan Phone: +81 6 6605 2575 Fax: +81 6 6605 3158 hiroki-y@sci.osaka-cu.ac.jp Xiaowei Zhao University of Texas Health Science Center at Houston Department of Pathology and Laboratory Medicine 6431 Fannin Houston, TX 77054 Phone: (713) 500-5245 xiaowei.zhao@uth.tmc.edu Shiwei Zhu University of Wisconsin-Milwaukee Division of Biological Science 3209 N. Maryland Ave. Milwaukee, WI 53211 Phone: (414) 229-2910 zhu6@uwm.edu Yongtao Zhu Nagoya University Furo-cho, Chikusa-ku Nagoya, Aichi 464-8602 Japan Phone: +81 52 789 2993 zhushiwei23@hotmail.com David Zusman University of California Molecular and Cell Biology 16 Barker Hall #3204 Berkeley, CA 94720-3204 Phone: (510) 642-2293 Fax: (510) 642-7038 zusman@berkeley.edu 134 BLAST STAFF Tarra Bollinger Molecular Biology Consortium 835 S. Wolcott Ave. (M/C 790) Chicago, IL 60612 Phone: (312) 996-1216 Fax: (312) 413-2952 tbolli1@uic.edu Peggy O’Neill Molecular Biology Consortium 835 S. Wolcott Ave. (M/C 790) Chicago, IL 60612 Phone: (312) 996-1216 Fax: (312) 413-2952 oneill@uic.edu 135 INDEX 136 Participant’s Name, Abstract page(s), Contact Information Page – PI Lab A Abouelfetouh, Alaa, 116, 120 - Wolfe, Alan Adase, Christopher, 88, 120 - Manson, Michael Afanzar, Oshri, 120 - Eisenbach, Michael Al Mamun, Abu Amar, 105, 120 - Rosenberg, Susan Allen, James, 52, 120 - Armitage, Judith Anquez, Francois, 115, 120 - Tom, Shimizu Armitage, Judith, 120 - Armitage, Judith Aschtgen, Marie-Stephanie, 106, 120 - Ruby, Edward B Bardy, Sonia, 54, 120 - Bardy, Sonia Baron, Szilvia, 120 - Eisenbach, Michael Basford, Sarah, 109, 121 - Sockett, Liz Beeby, Morgan, 2, 121 - Beeby, Morgan Behr, Stefan, 79, 121 - Jung, Kirsten Behrens, Wiebke, 78, 121 - Josenhans, Christine Belas, Robert, 56, 121 - Belas, Robert Berg, Howard, vi, 121 - Berg, Howard Biswas, Indranil, 121 - Biswas, Indranil Bollinger, Tarra, ii, 135 - Matsumura, Philip Bourret, Bob, ii, 58, 121 - Bourret, Robert Brennan, Caitlin, 31, 121 - Ruby, Edward Briegel, Ariane, 16, 121 - Jensen, Grant C Calvo, Rebecca, 81, 122 - Kearns, Dan Charon, Nyles, 122 - Charon, Nyles Colin, Remy, 122 - Wilson, Laurence Collins, Kieran, 122 - Ottemann, Karen Crane, Brian, ii, 122 - Crane, Brian Creager-Allen, Rachel, 10, 122 - Bourret, Robert Crosson, Sean, ii, 61, 122 - Crosson, Sean D Dahlquist, Rick, 122 - Dahlquist, Rick Dreyfus, Georges, 122 - Dreyfus, Georges Duchesne, Ismael, 103, 122 - Rainville, Simon E Eisenbach, Michael, 123 - Eisenbach, Michael Erhardt, Marc, 65, 123 - Erhardt, Marc Evans, Lewis, 3, 123 - Hughes, Colin F Falke, Joseph, ii, 66, 123 - Falke, Joseph Fraiberg, Milana, 4, 123 - Eisenbach, Michael Friedrich, Carmen, 111, 123 - Søgaard-Andersen, Lotte Fukuoka, Hajime, 5, 123 - Ishijima, Akihiko 137 Participant’s Name, Abstract page(s), Contact Information Page – PI Lab G Galperin, Michael, 123 - Galperin, Michael Gebhard, Susanne, 46, 123 - Gebhard, Susanne Gohara, Mizuki, 70, 123 - Homma, Michio Goss, Lindsie, 11, 124 - Dworkin, Jonathan H Han, Xuesheng, 99, 124 - Parkinson, John S. Harshey, Rasika, ii, 124 - Harshey, Rasika Hazelbauer, Gerald, 124 - Hazelbauer, Gerald Higgs, Penelope, 69, 124 - Higgs, Penelope Hiremath-Mendez, Geetha, 80, 124 - Kawagishi, Ikuro Hosu, Basarab, 57, 124 - Berg, Howard Hu, Bo, 39, 124 - Tu, Yuhai Hug, Isabelle, 76, 124 - Jenal, Urs Hughes, Kelly, viii, 73, 74, 124 - Hughes, Kelly I Ishijima, Akihiko, 125 - Ishijima, Akihiko Ito, Masahiro, 75, 125 - Ito, Masahiro J Jakobczak, Beata, 110, 125 - Søgaard-Andersen, Lotte Jenal, Urs, ii, 125 - Jenal, Urs Jensen, Grant, 125 - Jensen, Grant Johnson, Mark, 125 - Johnson, Mark Jones, Christopher, 17, 125 - Armitage, Judith Josenhans, Christine, ii, 125 - Josenhans, Christine Jung, Kirsten, ii, 125 - Jung, Kirsten K Kearns, Dan, 125 - Kearns, Dan Keilberg, Daniela, 12, 126 - Søgaard-Andersen, Lotte Khan, Shahid, 41, 126 - Khan, Shahid Kim, Hyo Kyung, 68, 126 - Harshey, Rasika Kinosita, Yoshiaki, 95, 126 - Nishizaka, Takayuki Kirby, John, ii, 83, 126 - Kirby, John Kitanovic, Smiljka, 98, 126 - Parkinson, John S. Koganitsky, Anna, 126 - Eisenbach, Michael Koirala, Santosh, 104, 126 - Rao, Christopher Kojima, Seiji, 72, 126 - Homma, Michio Kreamer, Naomi, 94, 126 - Newman , Dianne Krell, Tino, 30, 127 - Krell, Tino L Lambert, Ambroise, 100, 127 - Picardeau, Mathieu Lassak, Jürgen, 47, 127 - Jung, Kirsten Lazova, Milena, 18, 127 - Shimizu, Tom Lee, Yi-Ying, 24, 127 - Belas, Robert Lele, Pushkar, 6, 127 - Berg, Howard 138 Participant’s Name, Abstract page(s), Contact Information Page – PI Lab Levenson, Robert, 62, 127 - Dahlquist, Frederick Li, Chunhao, 85, 127 - Li, Chunhao Lin, Tao, 43, 127 - Norris, Steven Liu, Jun, 86, 87, 127 - Liu, Jun M Manson, Michael, ii, 128 - Manson, Michael Martinez-del Campo, Ana, 63, 128 - Dreyfus, Georges McBride, Shonna, 13, 128 - McBride, Shonna McGrane, Regina, 55, 128 - Beattie, Gwyn McMurry, Jonathan, 128 - McMurry, Jonathan Menon, Vishnu, 128 - Eisenbach, Michael Milner, David, 108, 128 - Sockett, Liz Miyata, Makoto, 128 - Miyata, Makoto Möglich, Andreas, 33, 128 - Möglich, Andreas Morse, Michael, 34, 128 - Tang, Jay Motaleb, Md, 129 - Motaleb, Md Mouslim, Chakib, 48, 129 - Hughes, Kelly Msadek, Tarek, viii, 14, 129 - Msadek, Tarek Mukherjee, Sampriti, 25, 129 - Kearns, Daniel N Nakane, Daisuke, 26, 129 - Nakayama, Koji Nan, Beiyan, 7, 129 - Zusman, David Nishiyama, Masayoshi, 44, 129 - Nishiyama, Masayoshi O Oikonomou, Catherine, 129 - Jensen, Grant O'Neill, Peggy, ii, 135 - Matsumura, Philip Osorio-Valeriano, Manuel, 64, 129 - Dreyfus, Georges Ottemann, Karen, ii, 96, 129 - Ottemann, Karen P Page, Stephani, 60, 130 - Bourret, Robert Paradis, Guillaume, 102, 130 - Rainville, Simon Parkinson, John S, ii, 19, 130 - Parkinson, John S. Partridge, Jonathan, 8, 130 - Harshey, Rasika Perez, Darysbel, 77, 130 - Taylor, Barry Piasta, Kene, 20, 130 - Falke, Joseph Piñas, Germán, 97, 130 - Parkinson, John S. Porter, Steven, vi, 101, 130 - Porter, Steven Prüβ, Birgit, viii, 49, 130 - Prüβ, Birgit R Rainville, Simon, 130 - Rainville, Simon Rao, Christopher, ii, 35, 131 - Rao, Christopher Remington, S. James, 131 - Remington, S. James Roujeinikova, Anna, 40, 131 - Roujeinikova, Anna S Sato, Keiko, 84, 131 - Nakayama, Koji 139 Participant’s Name, Abstract page(s), Contact Information Page – PI Lab Schubot, Florian, 107, 131 - Schubot, Florian Sears, Matthew, 32, 131 - Manson, Michael Shimkets, Larry, vii, 131 - Shimkets, Larry Shrout, Joshua, 27, 131 - Shrout, Joshua Silversmith, Ruth, 59, 131 - Bourret, Robert Sircar, Ria, 42, 131 - Crane, Brian Smith, Matthew, 53, 132 - Armitage, Judith Søgaard-Andersen, Lotte, 132 - Søgaard-Andersen, Lotte Studdert, Claudia, 113, 114, 132 - Studdert, Claudia Sultan, Syed, 93, 132 - Motaleb, Md Sze, Ching Wooen, 50, 132 - Li, Chunhao Szurmant, Hendrik, 15, 132 - Szurmant, Hendrik T Tahara, Yuhei, 28, 132 - Miyata, Makoto Tajima, Hirotaka, 67, 132 - Fukuda, Toshio Takekawa, Norihiro, 71, 132 - Homma, Michio Tanaka, Akihiro, 90, 132 - Miyata, Makoto Thompson, Lynmarie, 21, 133 - Thompson, Lynmarie Tu, Yuhai, vii, 133 - Tu, Yuhai Tulum, Isil, 92, 133 - Miyata, Makoto V Vaknin, Ady, 22, 133 - Vaknin, Ady W Wanner, Barry, 133 - Wanner, Barry Watts, Kylie, ii, 23, 133 - Watts, Kylie Webb, Ben, 36, 133 - Scharf, Birgit Wilson, Laurence, 37, 133 - Wilson, Laurence Wolfe, Alan, ii, 117, 133 - Wolfe, Alan Wong, Chui Ching, 82, 133 – Chiam, Keng-Hwee Wu, Liang, 29, 134 - Beattie, Gwyn Wuichet, Kristin, 112, 134 - Søgaard-Andersen, Lotte Wunder, Elsio, 45, 134 - Ko, Albert X Xie, Zhihong, 118, 134 - Xie, Zhihong Y Yamamoto, Hiroki, 91, 134 - Miyata, Makoto Z Zhao, Xiaowei, 9, 134 - Liu, Jun Zhu, Shiwei, 38, 134 - Homma, Michio Zhu, Yongtao, 89, 134 - McBride, Mark Zusman, David, 134 - Zusman, David 140