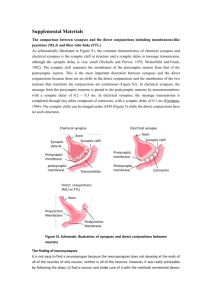
The sequence of events that underlie quantal transmission at central
advertisement

REVIEWS Nature Reviews Neuroscience | AOP, published online 18 July 2007; doi:10.1038/nrn2191 The sequence of events that underlie quantal transmission at central glutamatergic synapses John E. Lisman*, Sridhar Raghavachari‡ and Richard W. Tsien§ Abstract | The properties of synaptic transmission were first elucidated at the neuromuscular junction. More recent work has examined transmission at synapses within the brain. Here we review the remarkable progress in understanding the biophysical and molecular basis of the sequential steps in this process. These steps include the elevation of Ca2+ in microdomains of the presynaptic terminal, the diffusion of transmitter through the fusion pore into the synaptic cleft and the activation of postsynaptic receptors. The results give insight into the factors that control the precision of quantal transmission and provide a framework for understanding synaptic plasticity. *Brandeis University, Department of Biology, MS 008, 415 South Street, Waltham, Massachusetts 02454-9110, USA. ‡Duke University, Department of Neurobiology, Box 3209, Duke University Medical Center, Durham, North Carolina 27710, USA. § Stanford University School of Medicine, Department of Molecular & Cellular Physiology, Beckman Center, Room B100, 279 Campus Drive, Stanford, California 94305-5345, USA. Correspondence to J.E.L. e-mail: lisman@brandeis.edu doi:10.1038/nrn2191 Published online 18 July 2007 The basic properties of synaptic transmission were elucidated in pioneering work at the neuromuscular junction (NMJ). Studies showed that the arrival of the action potential at the presynaptic terminal activates voltage-dependent Ca2+ channels and that the resulting elevation of intracellular Ca2+ stimulates transmitter release1. Furthermore, it was shown that the amplitude of the postsynaptic response varies by an integral multiple of a stereotyped quanta. Later anatomical work2,3 provided evidence that synaptic vesicles are the basis of quantal transmission. Ca2+ stimulates the formation of a fusion pore between such vesicles and the presynaptic membrane. The neurotransmitter contained in the vesicle passes through this pore into the synaptic cleft, where it activates postsynaptic receptors. Computer simulations showed that the properties of this diffusion, together with the properties of the postsynaptic receptors, could satisfactorily account for the kinetics of the quantal response4. The basic steps in the overall process of transmission are illustrated in FIG.1. Chemical transmission at mammalian central synapses proved more difficult to study because of the synapses’ small size. However, methodological advances have now made it possible to study at least some central synapses with great precision. Much of this work has utilized two giant synapses, the Calyx of Held5 and the mossy fibre synapses in the hippocampal CA3 (Cornu Ammonis) region6. At these structures, it is possible to make simultaneous intracellular recordings from the pre- and postsynaptic sites. Such recordings (FIG. 2a) show that the synaptic delay between the arrival of an NATURE REVIEWS | NEUROSCIENCE action potential at the presynaptic terminal and the onset of the excitatory postsynaptic current (EPSC) is ~0.6 ms (~1 ms to EPSC midpoint). In this Review we discuss the sequence of molecular events that occur during this period, and how they determine the properties of the quantal response. Our focus will be on synapses that use the neurotransmitter glutamate, the most common excitatory transmitter in the CNS. We aim to convey how biophysical and molecular methods have provided a deep understanding of synaptic transmission, an understanding that will be vital for discovering the modifications of transmission that underlie learning and memory. Properties of Ca2+ elevation that trigger release Pioneering work on the squid giant synapse showed how action potentials stimulate the release of synaptic vesicles by triggering the opening of voltage-dependent Ca2+ channels in the presynaptic terminal7,8. Recent work has investigated this issue at vertebrate central glutamatergic synapses6; the results at mossy fibre synapses are shown in FIG.2a. In the first stage of a two-stage process, Ca2+ channels (primarily P/Q- and N-type9–13) open rapidly (0.1 ms) during the peak of the action potential (see BOX 1 for a description of Ca2+ channels). However, the Ca2+ current is small because the voltage is so positive that the electrochemical driving force is low. The second stage occurs during the descending phase of the action potential; the lowered voltage augments the driving force and thereby strongly increases the Ca2+ influx. This twostage process accounts for a substantial part (0.4 ms) of the synaptic delay at giant synapses (FIG. 2a). ADVANCE ONLINE PUBLICATION | 597 © 2007 Nature Publishing Group REVIEWS Depolarization opens Ca2+ channels Action potential in bouton generated by opening of Na+ channels + + + Presynaptic bouton + + + + + + + + + + + + + + + + ++ + Ca2+ + ++++ + + + + + ++ + + + ++ Ca2+ elevation occurs in microdomain Ca2+ binds to synaptotagmin causing opening of fusion pore Glutamate passes through fusion pore and diffuses in the cleft Ca2+ + + Dendritic spine + + + + + + + + + + + + + + Opening of AMPA channel generates EPSC Figure 1 | Steps in the process of chemical synaptic transmission. These steps occur in both vertebrates and invertebrates, at the neuromuscular junction and central synapses. Cartoons based on a drawing by J. A. Ernst and A. Brunger. Synapse The term synapse can be used either in a structural sense or to describe an entire connection. According to the structural definition, a synapse consists of a single presynaptic active zone and postsynaptic density, together with the specialized membranes and cleft in-between. Synapse diameter is between 0.2 and 1 micron. At most dendritic spines, there is a single such synapse. Giant synaptic connections have many structural synapses; the mossy fibre boutons in CA3 have over 10, whereas the Calyx of Held has approximately 50. Quanta The elementary building block of the EPSC, which contains an integral number of these events. Quantal size is derived from the distance between the peaks in the amplitude histogram of the EPSC, and equals the amplitude of the mEPSC at synapses where these events are uniquantal. Fusion pore Provides the passage from the interior of the vesicle into the synaptic cleft, through which neurotransmitter diffuses. Excitatory postsynaptic current (EPSC). The inward postsynaptic membrane current evoked by a single presynaptic action potential. It is measured using the voltage-clamp technique. Intriguingly, there are indications that the smaller synapses of the cerebellum may have found a onestage solution. At these synapses, substantial Ca 2+ entry occurs at the peak of the action potential14. This is perhaps because fast K+ channels prevent the peak of the action potential from becoming too positive, and thereby retain the Ca2+ driving force15. For such a solution to work, Ca2+ channels must open rapidly, and there is at least one example to show that this can occur16. Properties of the Ca2+ signal in the microdomain. As Ca2+ enters the presynaptic terminal, it can be effective immediately because only the concentration near the Ca2+ channels themselves has to rise, rather than that of the bulk cytoplasm. This is because the Ca2+ sensors that trigger vesicle release are strategically located within a ‘microdomain’ near the Ca2+ channels. The first support for this concept came from work on the giant synapse of the squid17,18, and indicated that the Ca2+ concentration could rise to 200 μM in the microdomain. An important advance came with the development of structural methods19–21 that localized the Ca2+ channels. At the NMJ, it was determined that vesicles are positioned within 20 nm of the Ca2+ channels — so close that the vesicle actually hinders diffusion of Ca2+ away from the channel. Strongly constrained simulations indicate that vesicle release is due to Ca2+ entry through just one or two nearby (within ~10–20nm) Ca2+ channels22. Recent work has analysed microdomain properties at central glutamatergic synapses23,24 and revealed a different picture. In particular, work in the laboratories of Bert Sakmann and Erwin Neher provided evidence about what occurs in the microdomains at the Calyx of Held. The release of synaptic vesicles was monitored during defined Ca2+ changes produced by uncaging methods. Once the relationship between Ca2+ levels and release rate was determined, it was used in reverse to predict the Ca2+ changes that trigger the release produced by 598 | ADVANCE ONLINE PUBLICATION an action potential. It was concluded that Ca2+ in the microdomain rises to 25 μM25 (or 10 μM using a different method26). The duration of this elevation is about 300 μs (FIG. 2b). In complementary experiments, the artificial production of such brief Ca2+ signals triggered release that satisfactorily mimicked those produced by an action potential27. Although much is now known about the structure of central synapses (FIG. 3), the distance between the Ca2+ channels and the vesicles has not been directly measured. The fact that Ca2+ channels can bind directly to vesicle proteins suggests that some close associations occur28–30. However, the influence of exogenous Ca2+ buffer on release is greater than would be expected if Ca2+ has to move only tens of nanometers before activating release. This suggests that the microdomain at central synapses is considerably larger than at the NMJ. Models that account for the buffer effects suggest that approximately ten Ca2+ channels influence release within a microdomain of ~200 nm31, a dimension only slightly smaller than the diameter of central synapses (FIG. 3). Both modelling32 and experimental results33 further suggest that the Ca2+ channels are clustered, resulting in Ca2+ gradients within the microdomain. A consequence of these gradients is that different vesicles will have different release probabilities. Importantly, the results of the studies on the Calyx of Held described above were obtained in young animals. A recent study34 found that the size of the microdomain is developmentally regulated. As animals mature, the effect of slow Ca2+ buffers on release diminishes, indicating a tighter spatial coupling between Ca2+ channels and sites of vesicle release, as has been found at the NMJ and squid giant synapse. Further progress in this field will probably require the localization of the Ca2+ channels at central synapses, and the elucidation of what a release site actually is. Specialized ‘triangular’ particles can be seen in the active zone34–37 (FIG. 3a), but the hypothesis that they organize vesicles for release remains to be tested. Recent work with tomographic www.nature.com/reviews/neuro © 2007 Nature Publishing Group 500 pA EPSC 1 nA 20 150 10 0 0 0 Synaptic delay 0.5 ms 1 2 3 d 12 [Glu] (mM) Ca2+ current 300 Local [Ca]i (μM) 100 mV c 30 Release rate Local [Ca]i Response (pA) b Action potential Release rate (vesicles per ms) a 6 mEPSC data Simulation 0 4 0.0 Time (ms) 0.3 Time (ms) 0.6 Open receptors Bound receptors Glutamate in hotspot 2 40 30 20 1 10 0 Number of receptors REVIEWS 0 0 0.1 0.2 0.3 0.4 0.5 Time (ms) Figure 2 | Timing of steps in transmission. a | Simultaneous recording of the membrane potential and currents from a mossy fibre presynaptic terminal and a CA3 hippocampal postsynaptic cell allows the determination of synaptic delay. The presynaptic Ca2+ current was determined by blocking all other conductances and imposing an action potential waveform by voltage clamp (at physiological temperature). After subtraction of the capacitative current, what remains is the Ca2+ current. b | The Ca2+ elevation and the vesicle release rate as a function of time after an action potential at the Calyx of Held (at room temperature). c | A mEPSC waveform recorded from a CA1 hippocampal dendrite is compared with a simulated (average) waveform based on a model of glutamate diffusion in the cleft and a kinetic model of the AMPA channel. d | Postrelease steps in the generation of the mEPSC. The glutamate concentration in the cleft (averaged over the hotspot of AMPA channel activation) rises rapidly after opening of the fusion pore. The number of receptors with at least one glutamate bound follows the glutamate elevation with little delay, but channels open with a substantial further delay due to the time required for conformational transitions. Simulations as in REF. 77. AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; CA, Cornu Ammonis; EPSC, excitatory postsynaptic current; mEPSC, miniature excitatory postsynaptic current. Part a reproduced with permission from REF. 6 © (2000) Cell Press. Part b reproduced with permission from Nature REF. 25 © (2000) Macmillan Publishers Ltd. Part c reproduced with permission from REF. 77 © (2004) American Physiological Society. Electrochemical driving force The difference between the Nernst equilibrium potential for an ion and the membrane voltage. The current through a channel is the product of the channel conductance and the electrochemical driving force. Uncaging methods The standard form of a compound is modified by chemical adducts, which can be cleaved by bright light to produce the standard form. One application of this technology allows rapid lightinduced Ca2+ elevation. Release site A site in the active zone where the vesicle is released. It remains unclear whether there is a structural basis for such sites, or whether release can occur anywhere in the active zone provided that the appropriate vesicle priming has occurred. analysis of hippocampal synapses frozen under high pressure is beginning to provide data relevant to this question. As shown in FIG. 3c , large synapses have several clusters of active zone material. These clusters contain the triangular particles mentioned above. Importantly, docked vesicles occur only within these clusters. Moreover, unidentified, specialized filaments (FIG. 3b) seem to guide vesicles to triangular particles. The clarification of the function of these structures will refine our understanding of what happens in the microdomain. However, regardless of what these findings show, the importance of the microdomain is clear: within it, Ca2+ channel opening leads to rapid (~50 μs) Ca2+ elevation32; after the channels close, diffusion out of the microdomain quickly reduces the Ca2+ signal31, thereby ensuring that there is no further activation of the Ca2+ sensor. The Ca2+ sensor and fusion pore Once Ca2+ elevation occurs in the microdomain, it triggers the release of vesicles with a delay of about 300 μs25,38 (FIG. 2b) (the delay will be shorter if Ca2+ levels are higher). The first reaction that occurs during this period is the binding of Ca2+ to the sensors that trigger vesicle release. Early work suggested that the sensor becomes activated only after the binding of many (four to five) Ca2+ ions39, something which has been confirmed using direct methods25,26,40. Importantly, during an action potential, only a fraction of the sensors become saturated, and this helps to account for why only a fraction (less than one-fifth) of the readily releasable pool of vesicles is released25. The time that it takes to load the sensors can be calculated to be approximately 200–300 μs (assuming that binding takes place near the diffusion limit (108 M–1 s–1) and that the Ca2+ concentration equals 30 μM). This leaves NATURE REVIEWS | NEUROSCIENCE only a short time (~100 μs) for the complex to actually produce a vesicle release25. The fact that the sensors are both unsaturated and nonlinear detectors makes the release highly sensitive to the Ca2+ concentration. This has important consequences for the modulation of transmission and shortterm synaptic plasticity. During such changes, the release produced by the action potential can be enhanced by residual Ca 2+ from a previous action potential, or by modulation of the voltage-dependent channels that shape the Ca2+ entry produced by an action potential24,41–43. The period of elevated release rate. Once Ca2+ fills the sites on the Ca2+ sensor to form an active complex, vesicle release can occur. Actual release occurs probabilistically until the concentration of the complex falls44,45. At the Calyx of Held, this period of elevated release lasts for approximately 300 μs; because the vesicle is released probablistically during this period, there is a jitter in the time of actual vesicle release (FIG. 2b). This jitter explains why the rising edge of the multiquantal EPSC is slower than that of the quantal event (FIG. 2a,c). The jitter compromises the temporal precision of the synapse, and one would expect it to be minimized. Consistent with this expectation, the release of Ca2+ from synaptotagmin (the probable Ca2+ sensor for fast vesicle release) is rapid46, making the lifetime of the active complex only slightly longer than the period of Ca2+ elevation. In some cells, the time-dependence of the release rate shows a long tail, producing what is termed ‘delayed release’. Although the basis of this effect is not completely known, a tail of Ca2+ elevation appears to be important. This elevation is due to the effect of endogenous Ca2+ buffers that have been loaded with Ca2+ during the action potential47. ADVANCE ONLINE PUBLICATION | 599 © 2007 Nature Publishing Group REVIEWS Box 1 | Voltage-dependent Ca2+ channels that control transmitter release Voltage-dependent Ca2+ channels are often classified according to their biophysical and pharmacological properties133–135. The channels that support mammalian neurotransmission (N-, P/Q- and R-type), among other functions, are different from those that dominate excitation – contraction or excitation –transcription coupling (L-type) or pacemaker activity (T-type and L-type). The presynaptic Ca2+ channels are exemplified by N-type channels, first discovered in recordings from the cell bodies of sensory neurons136 and later found to support neurotransmitter release from sympathetic neurons137 and cholinergic nerve terminals in the ciliary ganglion138,139. P/Q- and R-type channels were discovered in the early 1990s13,140–142, and also contribute in varying degrees to neurotransmission at various CNS synapses143-145, including the Calyx of Held146. P/Q-, N- and R-type channels are composed of pore-forming subunits known as CaV2.1, CaV2.2 and CaV2.3, respectively147; each is the specific target of a blocking toxin (ω-AgaIVA, ω-CTx-GVIA and SNX-482)148. Auxiliary subunits (α2-δ and β) contribute to proper cellular trafficking and the function of the pore-forming α1 subunits134,149. At most CNS synapses, neurosecretion is supported by more than one type of channel, but some synapses may be heavily dominated by a single channel type (for example, certain inhibitory neurons rely almost entirely on N-type). At some synapses, N-type channels develop early, but are gradually replaced by P/Q-type channels150. Why are there different kinds of presynaptic Ca2+ channel? Almost certainly, they are not a result of evolutionary pressure for different biophysical properties. Indeed, basic features such as extreme Ca2+ selectivity, high open channel flux rate and steeply voltage-dependent channel gating are similar for P/Q-, N- and R-type currents. In all cases, selectivity and permeation are conferred by a pore that is continually occupied by at least one Ca2+ ion, which prevents similarly sized and more abundant Na+ ions from rushing through151,152. Despite overall similarities, there are substantial differences in channel regulation, and this is likely to be the main reason for channel diversity. For example, P/Q- and N-type channels appear to differ in their susceptibility to modulation by G proteins in at least some systems153,154. The structural motifs in α1 subunits support various forms of regulation, for example, by G proteins and by protein kinases (see figure). Along with the release machinery itself, the presynaptic Ca2+ channels constitute a key convergence point for the regulation of neurosecretion. Several neurological diseases arise from genetic modifications of P/Q-, but not N-type, channels155; however, N-type channel-mediated neurotransmission is the target of therapeutic agents used to treat intractable pain156. CaM, calmodulin; CaMKII, Calmodulin kinase II; CBD, calmodulin binding domain; nCaBP, neuronal calcium-binding protein; PKC, protein kinase C; SNAREs, Soluble N-ethylmaleimide-sensitive factor attachment protein receptors; Gβγ, βγ subunit of G protein. I II III IV + 1 2 3 45 + + 1 23 45 + Outside + 1 2345 + 6 + 1 23 4 5 + 6 6 6 Inside C terminus N terminus P CaMKII CBD PKC Synaptotagmin as the Ca2+ sensor. There is now substantial evidence that the Ca2+ sensor for rapid vesicle release is synaptotagmin 1 (REF. 48); knockout of this protein in mice abolishes rapid transmission49. Other studies have shown that the C2B calcium binding domain of synaptotagmin I acts as the Ca2+ sensor for synchronized, rapid release of vesicles, while suppressing the slower, asynchronous component of vesicle release50,51. As mentioned above, the Ca2+ sensor acts as if it is cooperatively activated by the binding of four to five Ca2+ ions. The basis of this cooperativity remains unclear. The C2B domain on synaptotagmin has five Ca2+ binding sites, suggesting that cooperative properties within synaptotagmin itself may determine the cooperativity of release. Consistent with this, mutations in synaptotagmin that affect Ca2+ binding reduce this cooperativity52. However, important aspects of the release process are not understood. Vesicles contain fifteen synaptotagmin molecules53, but it is unclear how they work together to produce release. Also uncertain is how the cooperativity is influenced by other molecules54 and the interaction of multiple SNARE complexes55,56. Opening of the fusion pore by the activated sensor. Progress has been made in understanding the mechanism by which the Ca2+–synaptotagmin complex stimulates vesicle release. The vesicle is held at the synapse by the SNARE complex48 (FIG. 4a), some components of which (v-SNAREs) bind to the vesicle membrane, while others (t-SNAREs) bind to the plasma membrane. Recent structural and membrane labelling studies indicate that the vesicle may be primed for release by hemifusion36,57,58, a process in which the proximal leaflets of the plasma and vesicle membranes fuse (allowing transfer of membrane markers), without fusion of the distal leaflets (FIG. 4a). Fusion of the distal leaflets is thought to occur when the Ca2+–synaptotagmin complex displaces complexin from the SNARE complex. Complexin may function to clamp the hemifused state so that its release allows full fusion and the formation of the fusion pore59–62 (FIG. 4a). It has generally been thought that the fusion pore is lipid lined, but there are now some indications that it is protein-lined63. According to the latter view, synaptobrevin (a v-SNARE) and syntaxin (a t-SNARE) form a type of ion channel (FIG. 4a) that opens to form the fusion pore under the influence of the Ca2+–synaptotagmin complex64. Consistent with the electrostatic interactions that are characteristic of a standard ion channel, current through the fusion pore can be bidirectionally modified by changing the charge on what is thought to be the pore wall formed by syntaxin65. The controversy over whether the pore is lipid- or protein-lined only relates to the early stages of the process; in both scenarios a protein channel would no longer be involved if the process proceeded to complete fusion (FIG. 4a). SNAREs nCaBP’s/CaM Gβγ Figure courtesy of W. A. Catterall 600 | ADVANCE ONLINE PUBLICATION Conductance and modes of the fusion pore. The diameter of the fusion pore may seem like a technical detail, but it has surprisingly important consequences. Indeed, pore size may even have a role in the synaptic plasticity implicated in learning and memory (see BOX 2 ). www.nature.com/reviews/neuro © 2007 Nature Publishing Group REVIEWS Synaptic cleft a Active zone b c Presynaptic bouton Synaptic vesicle Docked vesicle d Hotspot Triangular particle Dendritic spine 250nm Postsynaptic density 0.5 μm Figure 3 | Synapse structure and the localization of activated AMPA channels. a | An electron micrograph of the hippocampal CA1 region, showing the presynaptic bouton, synaptic vesicles, active zone, synaptic cleft, postsynaptic density and spine. This is the first image of synapses in a slice preparation made by rapid freezing, a method that avoids the artefacts of fixation. The diameter of the active zone defines the synapse diameter. The presynaptic active zone has dense material that is coextensive with the postsynaptic density, but is more difficult to visualize. The picture shows a dense particle (triangular) in the active zone, which may be involved in organizing vesicle release. b | A tomographic reconstruction of the active zone and filaments (pink) leading to the triangular particle attached to the presynaptic membrane (white). These filaments may organize the process of bringing vesicles (yellow) to the membrane, where they become docked (blue). c | A tomographic reconstruction of the entire active zone showing the modular organization of dense active zone material (yellow) and docked vesicles (blue). d | Postsynaptic densities of hippocampal CA1 synapses reconstructed from serial sections of electron micrographs. The three shown are representative of the different sizes found. The red disc shows the size of the hotspot where most of the mEPSC is generated near a site of vesicle release77. This region is only a small fraction of the synapse area. The vesicle is assumed to be released at the centre of the red disc. Part a kindly provided by S. Marty, using methods from REF. 178. Parts b,c reproduced with permission from REF. 179 © (2007) Society for Neuroscience. Postsynaptic densities in part d downloaded from Synapse Web by K. M. Harris. CA, Cornu Ammonis. Impedance method A rapidly oscillating voltage is applied by voltage-clamp. The in-phase component of the resulting current is termed the ‘real’ component and can be related to patch conductance. The out-of-phase component is called the imaginary component and can be related to patch capacitance. The advantage of this method is that both conductance and capacitance can be measured simultaneously. Monte-Carlo simulation A simulation that involves keeping track of the position and state of each molecule. At each short time step the computer calculates the new position or state of each molecule according to the probability of each change. Measurements of pore properties using impedance methods were first made in non-neuronal cells66, and recently this method67 was used with patch recordings from the presynaptic membrane at the Calyx of Held (the secretory face was exposed by sucking away the postsynaptic cell). Using this method, the properties of each released vesicle could be determined from the change in patch capacitance and conductance (FIG. 4b). Changes in patch conductance provide information about the diameter of the fusion pore. Several important conclusions were drawn. First, the initial opening may involve either a small or large fusion pore. Second, some openings are followed by closure, a process termed ‘kissand-run’ which had been previously suggested by various methods68–75. In this mode the vesicle can be reused, whereas in the ‘full fusion’ mode, the vesicle membrane is added to the plasma membrane and a vesicle is subsequently reformed by endocytosis at extrasynaptic sites. An important finding was that kiss-and-run can occur even after large fusion pore formation, whereas the formation of small fusion pores is not necessarily followed by kiss-and-run. These results indicate that fusion pore size and the mode of vesicle recovery are not linked in an obligatory way. Although the existence of kiss-and-run is generally accepted, initially in endocrine cells and more recently in nerve terminals, the fraction of release events that use this mode is still uncertain. The functional significance of fusion pore size is discussed at the end of the next section. NATURE REVIEWS | NEUROSCIENCE Glutamate diffusion and AMPA channel activation When the fusion pore opens at glutamatergic synapses, glutamate (at an initial concentration within the vesicle of 200 mM76) diffuses through the pore and dilutes as it spreads throughout the cleft. There it encounters AMPA (α-amino-3-hydroxy-5-methylisoxazole-4propionic acid) channels at a density of ~1,000 per μm2 (REFS 77,78). Recent measurements indicate that diffusion in the cleft is only somewhat lower than the free value 79. Monte-Carlo computer simulations can be used to describe the diffusion of glutamate through the pore and in the cleft. If the properties of glutamate diffusion and the properties of the AMPA channel (as described by a kinetic model) are correctly understood, it should be possible to incorporate these properties into computer simulations and predict the amplitude and kinetics of miniature EPSCs (mEPSCs). These small spontaneous events generally arise from the spontaneous release of single vesicles (see below for a qualification). The development of a kinetic model of the AMPA channel (see BOX 3) has been based on the measurement of how channels in excised outside-out patches respond to ultra-fast, controlled applications of glutamate. Experiments of this kind revealed that micromolar levels of glutamate can bind to single subunits of the channel (there are four glutamate-binding subunits; see BOX 3) and cause the channel to adopt a long-lived desensitized state without opening. By contrast, high ADVANCE ONLINE PUBLICATION | 601 © 2007 Nature Publishing Group REVIEWS a Plasma membrane Hemifusion Vesicle Docked vesicle Vesicle SNARE complex Neurotransmitter Synaptobrevin II (v-SNARE) SNAP-25 50 aF Im Vesicle membrane Syntaxin (t-SNARE) b 10 pS 2s Re Cytoplasm 50 aF Im Plasma membrane Extracellular space Extracellular space Plasma membrane c Lipid-lined fusion pore EPSC (pA) Protein-lined fusion pore 5 pS Re 0 –6 1 nm; 50 pS 2 nm; 200 pS 4 nm; 800 pS 8 nm; 3,200 pS –12 0 1 2 3 4 Time (ms) Figure 4 | Vesicle fusion and fusion pores. a | Two potential mechanisms of formation of the fusion pore between the synaptic vesicle (upper membrane; pink) and the plasma membrane (lower membrane; green). In the top left panel the vesicle is in the ‘docked’ state in which it is held near the plasma membrane by the SNARE complex. In the top right panel, the vesicle and plasma membrane have their distal leaflets in a hemifused state that is primed for release. During the release process, a protein-lined pore (lower left panel) is formed by two of the SNARE proteins, syntaxin and synaptobrevin. This step may be reversible, or may be followed by a transition to a lipid-lined pore (bottom right panel). An alternative model is that fusion pore opening always involves the formation of a lipid-lined pore. b | Kiss-and-run events and full-fusion events observed by capacitance measurements at the presynaptic secretory face of the Calyx of Held. Real and imaginary components give information about the fusion pore conductance and membrane capacitance (in attoFarads) respectively. Top traces: the first event on the left-hand side is an increase in capacitance followed shortly thereafter by an equal decrease, indicating a kiss-and-run event. No conductance change is detectable (large fusion pores having conductance greater than 288 pS cannot be detected for technical reasons). The second event is longer and thus indicates full fusion. Bottom traces: kiss-and-run event in which a conductance change was detectable, indicating a small fusion pore; the average pore conductance of such events is 66 pS. c | The simulated average mEPSC depends on the size of the fusion pore (diameters and conductance are given). For the smallest pore, the resulting current would be too small to detect given the presence of noise, creating a ‘whispering’ form of silent synapse. Im, imaginary component of impedance; mEPSC, miniature excitatory postsynaptic current; Re, real component of impedance; SNAP, soluble N-ethylmaleimide-sensitive fusion protein attachment protein; SNARE, SNAP receptor. Part a modified with permission from REF. 63 © (2006) Annual Reviews. Part b modified with permission from Nature REF. 67 © (2006) Macmillan Publishers Ltd. Calculations in part c are from the model described in REF. 77. Kinetic model The kinetic model of an ion channel specifies the rate constants for the binding and unbinding of neurotransmitter to sites on the channel, and the rate of conformational transitions between various states that control channel opening and closing, as well as transitions to a desensitized state. levels (near mM) of glutamate induce rapid opening of the channel followed by desensitization. The results can be understood as a race between opening and desensitization; if only one glutamate binds, desensitization wins. The more glutamate that binds, the higher the probability of opening before desensitization and the higher the single channel conductance. BOX 3 gives further information about the kinetics of opening and desensitization and shows a recently derived structural model of the AMPA channel that provides insight into the mechanism of activation and desensitization. An important requirement for comparing predicted to actual mEPSCs is to have accurately measured mEPSCs. This is generally not simple to achieve because mEPSCs are generated in dendrites, and special procedures are required to record such events 602 | ADVANCE ONLINE PUBLICATION without electrotonic distortion 80,81. In the study by Smith et al.80, sucrose was applied to a small dendritic region and the resulting mEPSCs were recorded intradendritically from that region (measurements were taken in slices of the hippocampal CA1 region). This procedure minimized electrotonic distortion and revealed that the rise-time (0.1 ms) of the mEPSC is faster than previously suspected. Figure 2c shows the mEPSC recorded by this method, as well as the predictions of a Monte-Carlo computer simulation. It is clear that the amplitude and rise-time are adequately accounted for. Given that there were no freely adjustable parameters in the simulations, the agreement of theory and experiment argues that the main determinants of the mEPSC amplitude and rise-time are reasonably well understood. www.nature.com/reviews/neuro © 2007 Nature Publishing Group REVIEWS Box 2 | Experimental support for the importance of the mode of transmitter release Evidence for whispering synapses: partially silent synapses • At the Calyx of Held, a small fraction of release events have a low conductance fusion pore. These could give rise to the small fraction of mEPSCs that have small amplitudes and slow rise-times67. • EPSCs at the fly neuromuscular junction show a slow hump on the decay phase. The hump might be the result of prolonged release of transmitter from the vesicle due to a small fusion pore. Consistent with this, the hump is selectively reduced by a low affinity antagonist, indicating a low glutamate concentration75. • Inhibiting desensitization with cyclothiazide dramatically increases the frequency of mEPSCs without affecting the amount of glutamate released157 or the NMDAR-mediated EPSC (but see also REF. 158). One explanation is that the fusion pore conductance for many spontaneous release events is so low that AMPA channels preferentially desensitize without opening; if desensitization is blocked, the channels will open, accounting for the enhanced frequency of mEPSCs. In a variant of this hypothesis, synapses are modular and can be ‘partially silent’; some modules are AMPAsilent because they both release glutamate slowly and lack AMPA channels, whereas others are AMPA-functional because they release glutamate rapidly and contain AMPA channels. In cyclothiazide, the slowly released glutamate in silent modules can activate AMPA channels in nearby functional modules77,129. • At single synapses in culture, release was stimulated by an electrode that was placed near the terminal. Remarkably, some events produced only NMDA responses, whereas others had both AMPA and NMDA components159. These results can be explained by the heterogeneity in the speed of glutamate release from different vesicles at the same synapse160. • Acting through G protein βγ subunits, serotonin promotes kiss-and-run release, as determined by a fluorescent marker of vesicular turnover. This change is accompanied by a reduction in quantal size, suggesting that presynaptic inhibition can occur through the modulation of vesicle fusion properties161. Enhancement of fusion pore conductance may contribute to LTP • A low affinity antagonist results in less blockage of the EPSC after LTP induction than before LTP157. This indicates a higher concentration of glutamate in the synaptic cleft after LTP. The number of vesicles released, as judged by NMDARmediated EPSCs, was not increased. It was concluded that the increase in glutamate concentration in the cleft was the result of LTP increasing the fusion pore diameter. In a variant of this hypothesis, LTP both adds AMPA channels and increases the fusion pore diameter in silent modules, making them functional129. LTD-induced reduction in fusion pore conductance may contribute to reduced transmission • After LTD induction, there is an increase in the number of synaptic boutons that take up the fluorescent membrane marker FM1-43 into vesicles but do not release it during synaptic stimulation162. Because FM1-43 is lipophilic, it leaves a vesicle much more slowly than it enters. The reduced release after LTD induction may occur because the release mode has switched to kiss-and-run, thus allowing little time for the dye to exit the vesicle. AMPA, α-amino-3-hydroxy-5methylisoxazole-4-propionic acid; EPSC, excitatory postsynaptic current; LTD, long-term depression; LTP, long-term potentiation; mEPSC, miniature EPSC; NMDA, N-methyl-d-aspartate; NMDAR, NMDA receptor. mEPSC (miniature EPSC). An mEPSC is a spontaneously occurring synaptic event caused by spontaneous vesicle release. It is generally measured after blocking action potentials with tetrodotoxin to insure that there is no release due to spontaneous action potentials. The amplitude of mEPSCs is determined by AMPA channel density and is taken as a measure of postsynaptic processes in traditional quantal analysis. However, mEPSC amplitude can also be affected by vesicle glutamate concentration, multi-vesicular release and glutamate release mode. Outside-out patch A variant of the patch-clamp technique, in which a patch of plasma membrane covers the tip of the electrode. The outside of the membrane is exposed to bathing solution. Simulations of this kind can provide insight into the timing of events that occur between the opening of the fusion pore and the generation of AMPA channel current (FIG. 2d): peak glutamate elevation in the cleft occurs with a latency of ~80 μs; binding of glutamate to the channel occurs in ~10 μs; and the conformational transition of the channel to the open state requires ~100 μs. A hotspot of AMPA channel activation. Only a fraction of the average synapse area is actually involved in generating a quantal response. This is because there are large concentration gradients in the cleft, and the mM glutamate concentration necessary to open channels is only achieved near (within ~100 nm) the site of vesicle release. As a result, the 20 or so channels that open during a quantal event are mostly within a hotspot (with an area of 0.03 μm2) that is only ~25% of the area of the average CA1 synapse; this area is an even smaller fraction at large mushroom spines (FIG. 3d). These findings are consistent with previous evidence that the average quantal current is far below saturation82–84. Importantly, it follows that quantal size is not a good indicator of AMPA channel number (adding AMPA channels to enlarge the synapse will have little NATURE REVIEWS | NEUROSCIENCE effect). Rather, the main postsynaptic determinant of quantal size is AMPA channel density77,85. Fusion pore diameter affects AMPA channel activation. We now turn to the question of how the size of the fusion pore affects AMPA channel activation. This issue was recently analysed86, and FIG. 4c shows related calculations. If the fusion pore is at least 4 nm in diameter, the channels are efficiently activated. This activation is not sensitive to the exact pore diameter, because the diffusion bottleneck is the cleft itself. On the other hand, when the fusion pore is less than 2 nm in diameter, diffusion through the pore becomes limiting; indeed, at 1 nm, the concentration of glutamate in the cleft becomes so low that the mEPSC amplitude is insignificant compared with the background noise. However, even small pores generate a normal N-methyl-d-aspartate (NMDA) response, because these channels have a much higher glutamate affinity than AMPA channels. A synapse with small pores will therefore produce a ‘whispering’ form of ‘silent synapse’ (in contrast to synapses that are silent because they lack AMPA channels87). Experimental evidence for whispering synapses and the possible role of fusion pore modulation in synaptic plasticity is discussed in BOX 2. ADVANCE ONLINE PUBLICATION | 603 © 2007 Nature Publishing Group REVIEWS Box 3 | Structural and functional properties of AMPA channels a Resting Structural model of activation and desensitization AMPA channels are tetramers composed of combinations of channel subunits (GluR1–4) arranged in a dimer-ofdimers configuration163,164. Each subunit can bind glutamate; as illustrated in the figure part a, binding of glutamate (Glu) to the upper element is followed by an upward tilt of the lower element. The force of this tilt is conveyed to the channel through a linker region, causing the channel to open165–167. Desensitization occurs if glutamate binding causes a downward tilt of the binding domain; under these conditions, upward tilt of the lower element fails to produce channel opening168. Figure reproduced with permission from REF. 167 © (2006) Elsevier Sciences. Coefficient of variation (CV). The standard deviation divided by the mean. The CV is thus a convenient measure of the relative variability of a quantity. For instance, a CV of 0.2 would mean that most measurements were within plus or minus 20% of the mean. Amplitude histogram An amplitude histogram of the EPSC is made by defining amplitude bins and then plotting the number of occurrences that fall within each bin as a function of amplitude. For responses comprising a variable number of elementary quantal units, the histogram should show multiple peaks at integral multiples of quantal size. In practice, the peaks become smeared because of the CV of quantal size and because of measurement noise. Kinetic model of an AMPA channel As illustrated in the state diagram of figure b, the channel undergoes a transition between states (horizontal transitions) when glutamate binds to each of the four subunits. The number of glutamates bound varies from left to right. From these close states, there may be conformational changes resulting in the open (O) or desensitized (D) states. The rate constant at physiological temperature for opening or desensitizing is shown, as is the average channel conductance. Both rate constant and channel conductance vary with the number of glutamates bound169,170. In this scheme, the opening rate increases threefold with each additional glutamate bound (multiplicative gating), in line with strong evidence for multiplicative gating in other channels171,172. With these factors known, the model parameters77 were generated by fitting responses to controlled glutamate applications to outside-out patches of CA1 dendritic membranes98. Early models of AMPA channel gating91,173–175 assumed only a single conductance state, reached upon the binding of two glutamate molecules (other ligand states were not considered). Consideration of multiple conductance levels and all liganded states is necessary to predict the 0.1 ms rise of mEPSCs77 at CA1 synapses, to predict the temperature dependence of EPSCs at the Calyx of Held176 and to account for the desensitization properties of AMPA channels177. AMPA, α-amino-3-hydroxy-5-methylisoxazole4-proprionic acid; CA, Cornu Ammonis; mEPSC, miniature excitatory postsynaptic current. How quantal is the quantal response? Neuronal computations often rely on small differences in the summed excitatory input from many synapses, and it is therefore important to minimize the variability of the responses from each contributing synapse. The elementary unit of transmission at the synapse is the quantal response. This term was developed at the NMJ to indicate that the EPSC is composed of stereotyped subunits, but the size of quantal responses is not always exactly the same. This variability is quantified by the coefficient of variation (CV), which is 0.3 at the NMJ. Variation of this magnitude still allows the quantal peaks in amplitude histograms to be detected. The extent of variability of the quantal response at central synapses is controversial. High CVs (~0.6) have been found at single synapses of hippocampal neurons in culture88 and in hippocampal slices89. However, analysis of amplitude histograms at other synapses indicates lower 604 | ADVANCE ONLINE PUBLICATION Bound 12.7 Å D1 Glu D2 31.9 Å Open Desensitized 22.0 Å 13.1 Å 14° 21° 14° 21° 26.5 Å 37.4 Å b 5pS 7pS 12pS O2 O3 O4 1200 R0 R1 3600 R2 1080 D1 R3 1440 D2 10800 D3 R4 1920 D4 CV values: a CV of 0.4 at the Calyx of Held, 0.22 at the CA3 mossy fibre synapses (FIG. 5a) and 0.23 at synapses on cerebellar granule cells 82,90,91. Most measurements of mEPSCs have been made from groups of synapses, but one study used a suction pipette to measure the current from a single synaptic spine (which generally contains a single synapse) and found a CV of 0.28 (REF. 81). Taken together, these lines of evidence leave little doubt that nature has found a way of making quantal variability low, at least at some central synapses. Origin of high CVs. How can the larger CVs (~0.6) be explained? It has been suggested that variation in vesicle volume (a CV of 0.3 measured in fast frozen material53) produces variation in vesicle glutamate content92–94, and that this, together with other factors (channel noise and intrasynaptic variation in AMPA channel density) accounts for high CV values. However, it is unclear how www.nature.com/reviews/neuro © 2007 Nature Publishing Group REVIEWS 40 20 c 80 60 Rise time (μs) b 50 Number of particles per synapse Number of events a 40 400 300 200 20 100 0 0 40 pA 1 ms 0 0 10 20 30 Peak EPSC (pA) 40 50 0 0.05 0.1 0.15 0.2 0.25 0.3 Synaptic area (μm2) 0 60 120 Amplitude (pA) Figure 5 | Determinants of quantal transmission a | A multi-peaked amplitude histogram of EPSCs evoked at a CA3 mossy fibre synapse. In this case (but not all), the data could be fitted as the sum of three Gaussian curves, with amplitudes corresponding to one, two or three released vesicles. The narrow half-width of the Gaussian indicates a low coefficient of variation (CV); after correction for noise, the CV of quantal size was 0.22. b | The AMPA receptor labelling of CA3 mossy fibre synapses, as determined by electron microscopy, immunogold labelling and reconstruction methods. Content varies linearly with synapse area, indicating that AMPA receptors are present at fixed density. c | Larger mEPSCs at CA1 hippocampal synapses have slower rise-times (inset shows mEPSCs). Responses were evoked by the application of sucrose to a local dendritic region and were recorded intradendritically from that region. CA, Cornu Ammonis; mEPSC, miniature excitatory postsynaptic current. Part a reproduced with permission from REF. 91 © (1993) Cambridge Univ. Press. Part b reproduced with permission from REF. 113 © (1998) Cell Press. Part c reproduced with permission from REF. 80 © (2003) Cambridge Univ. Press. the importance of glutamate content can be reconciled with the observation that larger mEPSCs have slower rise-times80,95,96 (FIG. 5c). If the larger responses are due to a higher glutamate content in the vesicles, it should be possible to reproduce the slower rise-time by artificially elevating vesicle glutamate. When this is done in simulations77 or experimentally97, mEPSCs do get larger but not slower (and they may even get faster). A different explanation of high CVs is that larger mEPSCs are generated by multiple vesicles. According to this view, the rise-time of multiquantal mEPSCs is slower because of jitter in the time of vesicle release. Consistent with this explanation, there have been many reports of mEPSC amplitude distributions that have more than one peak95,96,98–105. In some cases, the release of multiple vesicles appears to be due to spontaneous ‘sparks’ of Ca2+ coming from intracellular stores106–108. Notably, altering Ca2+ stores alters the amplitude of mEPSCs106,109–111. These results strongly suggest that in some cases the high CV of mEPSCs arises because the mEPSCs consist of a variable number of quantal responses. However, this need not be the only explanation; there are indications that the mechanisms responsible for low quantal variability (see next section) may simply not be present in young animals, and may instead develop over time95. Indeed, high CV and low CV synapses can coexist in the hippocampi of young animals89. Attaining a low CV. The fact that a low CV can be achieved is remarkable given the potential sources of variation112. Vesicles have to be made and filled in a reproducible way, variation in fusion pore properties must be minimized and the stochastic noise attributable to the gating of a small number of AMPA channels must be low. A low CV (0.22) at CA3 mossy fibre spines is particularly remarkable given that the recorded events are due to multiple synapses that vary considerably in their NATURE REVIEWS | NEUROSCIENCE size and AMPA channel content113 (FIG. 5b). However, this becomes more understandable in view of the fact that it is channel density (which is nearly constant), not channel number, that is the primary postsynaptic determinant of quantal size (see above). A major presynaptic determinant of quantal size is the number of glutamate transporters in the vesicle114, but it is not known how this number is regulated. A recent study surprisingly showed that quantal size is not related to vesicle volume, but that it does depend on vesicle glutamate content115. Much remains to be learned about how the size and content of synaptic vesicles is controlled in a way that minimizes the variability of quantal size. Binary or graded transmission? The NMJ, CA3 mossy fibre boutons and the Calyx of Held contain multiple synapses (active zones and postsynaptic densities), each of which can contribute to vesicle release. However, most excitatory connections in the CNS occur on single spines that contain only a single synapse (FIG. 3a). When considering the information transmission through such synapses, it is crucial to understand whether evoked transmission is univesicular, and therefore binary, or multivesicular, and therefore graded. Electrical recordings of transmission at identified mushroom spines have been difficult to achieve: the single existing recording at a large mushroom spine indicated multivesicular release on almost every trial; EPSCs were highly variable and the largest were much larger than mEPSCs116,117. Optical measurements of NMDA-mediated Ca2+ entry118 also provide evidence for multivesicular release. More evidence is provided by experiments using low-affinity antagonists; increasing multi-vesicular release (and thus glutamate concentration) reduces the inhibitory action of the competing antagonist119–121. At the Calyx of Held, high Ca2+ levels, generated by uncaging, release the readily releasable ADVANCE ONLINE PUBLICATION | 605 © 2007 Nature Publishing Group REVIEWS pool (1,500 vesicles) with a time-constant of 0.6 ms40. As there are ~500 synapses contributing122, each must release multiple vesicles (see also REF. 71). Related findings have been made at inhibitory synapses123. However, some studies indicate that release at certain synapses is always univesicular. Procedures that increase the probability of release produced no change in the size of successful responses (potency)124–126, contrary to what would be observed if multiple quanta sometimes summated. Similar constancy was found during depression89. Other experiments have shown that when release is enhanced, there is no reduction in the efficacy of low affinity antagonist, arguing against multiquantal transmission127. Much of this conflicting evidence has been obtained at hippocampal synapses and so cannot be attributed to regional variation. However, because of technical issues, different experiments probably sampled synapses of different sizes. One resolution may be that synapses are made of modules128,129; indeed, FIG. 3c could be interpreted as indicating a modular organization of release. Perhaps, then, each module contains only a single release site (possibly the triangular particle), which provides a structural basis for univesicular release. Small synapses may contain a single module, and thus be restricted to univesicular release, whereas larger synapses would be capable of multivesicular release. Consistent with this idea, the average synapse area at the Calyx of Held, where multivesicular release occurs, is 0.1 μm2 (REF. 122) — considerably larger than synapse areas at the cerebellar synapses (0.024 μm2), which show univesicular release130. This general idea is 1. Katz, B. Neural transmitter release: from quantal secretion to exocytosis and beyond. The Fenn Lecture. J. Neurocytol. 25, 677–686 (1996). 2. Heuser, J. E. et al. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. J. Cell Biol. 81, 275–300 (1979). 3. Heuser, J. E. & Reese, T. S. Structural changes after transmitter release at the frog neuromuscular junction. J. Cell Biol. 88, 564–580 (1981). 4. Stiles, J. R., Van Helden, D., Bartol, T. M. Jr, Salpeter, E. E. & Salpeter, M. M. Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle. Proc. Natl Acad. Sci. USA 93, 5747–5752 (1996). 5. Borst, J. G., Helmchen, F. & Sakmann, B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J. Physiol. 489, 825–840 (1995). 6. Geiger, J. R. & Jonas, P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron 28, 927–939 (2000). 7. Llinas, R., Steinberg, I. Z. & Walton, K. Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys. J. 33, 323–351 (1981). 8. Augustine, G. J., Charlton, M. P. & Smith, S. J. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J. Physiol. 367, 163–181 (1985). 9. Wu, L. G., Westenbroek, R. E., Borst, J. G., Catterall, W. A. & Sakmann, B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 19, 726–736 (1999). 10. Iwasaki, S., Momiyama, A., Uchitel, O. D. & Takahashi, T. Developmental changes in calcium channel types mediating central synaptic transmission. J. Neurosci. 20, 59–65 (2000). 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. consistent with the model of Bolshakov et al.131, which is based on their observation that the late phase of longterm potentiation, a process associated with synapse growth132, can convert synapses from uniquantal EPSCs and mEPSCs to multiquantal ones131. In summary, it seems safe to conclude that multivesicular release, though not universal, is common, and that analysis of information transmission at synapses should consider the enhancements made possible by graded release. Conclusion There is a tendency for scientists to become specialized, and the field of synaptic physiology is no different; there are presynaptic specialists and postsynaptic specialists. In this Review, we have attempted to bridge the cleft that divides these fields. We have given a brief overview of a broad range of topics in synaptic transmission, and tried to make clear the necessity of understanding presynaptic and postsynaptic events in an integrated way. We hope that the reader has gained an appreciation for the complexities of synaptic transmission, but also an appreciation of the tremendous progress that has been made. Although there remain aspects of the problem that are still quite mysterious, many aspects have been rigorously studied by adequate biophysical methods and can be accounted for by well-constrained computational models. The increasing understanding of the biophysics of synaptic transmission at central synapses will provide a sound framework for determining the molecular basis of transmission and for elucidating how synapses are modified during learning. Reid, C. A., Bekkers, J. M. & Clements, J. D. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci. 26, 683–687 (2003). Momiyama, A. & Takahashi, T. Development of inhibitory synaptic currents in rat spinal neurons. Ann. NY Acad. Sci. 707, 447–448 (1993). Mintz, I. M., Sabatini, B. L. & Regehr, W. G. Calcium control of transmitter release at a cerebellar synapse. Neuron 15, 675–688 (1995). Sabatini, B. L. & Regehr, W. G. Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384, 170–172 (1996). A study that provides a different view of the timing of steps leading to vesicle release. This study suggests that release can occur before the falling phase of the action potenital, making a much shorter synaptic delay possible. Sabatini, B. L. & Regehr, W. G. Timing of synaptic transmission. Annu. Rev. Physiol. 61, 521–542 (1999). McDonough, S. I., Mintz, I. M. & Bean, B. P. Alteration of P-type calcium channel gating by the spider toxin ω-Aga-IVA. Biophys. J. 72, 2117–2128 (1997). Llinas, R., Sugimori, M. & Silver, R. B. Microdomains of high calcium concentration in a presynaptic terminal. Science 256, 677–679 (1992). Serulle, Y., Sugimori, M. & Llinas, R. R. Imaging synaptosomal calcium concentration microdomains and vesicle fusion by using total internal reflection fluorescent microscopy. Proc. Natl Acad. Sci. USA 104, 1697–1702 (2007). Stanley, E. F. The calcium channel and the organization of the presynaptic transmitter release face. Trends Neurosci. 20, 404–409 (1997). Harlow, M. L., Ress, D., Stoschek, A., Marshall, R. M. & McMahan, U. J. The architecture of active zone material at the frog’s neuromuscular junction. Nature 409, 479–484 (2001). Pumplin, D. W., Reese, T. S. & Llinás, R. Are the presynaptic membrane particles the calcium channels? Proc. Natl Acad. Sci. USA 78, 7210–7213 (1981). 606 | ADVANCE ONLINE PUBLICATION 22. Shahrezaei, V., Cao, A. & Delaney, K. R. Ca2+ from one or two channels controls fusion of a single vesicle at the frog neuromuscular junction. J. Neurosci. 26, 13240–13249 (2006). 23. Ohana, O. & Sakmann, B. Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. J. Physiol. 513, 135–148 (1998). 24. Burnashev, N. & Rozov, A. Presynaptic Ca2+ dynamics, Ca2+ buffers and synaptic efficacy. Cell Calcium 37, 489–495 (2005). 25. Schneggenburger, R. & Neher, E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406, 889–893 (2000). An elegant study that measured the release attributable to defined Ca2+ changes in the presynaptic terminal. 26. Bollmann, J. H., Sakmann, B. & Borst, J. G. Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289, 953–957 (2000). 27. Bollmann, J. H. & Sakmann, B. Control of synaptic strength and timing by the release-site Ca2+ signal. Nature Neurosci. 8, 426–434 (2005). A clever strategy was used to mimic the brief Ca2+ elevation produced by an action potential and to study the resulting transmitter release. 28. Bennett, M. K., Calakos, N. & Scheller, R. H. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255–259 (1992). 29. Sheng, Z. H., Rettig, J., Cook, T. & Catterall, W. A. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature 379, 451–454 (1996). 30. Jarvis, S. E. & Zamponi, G. W. Masters or slaves? Vesicle release machinery and the regulation of presynaptic calcium channels. Cell Calcium 37, 483–488 (2005). 31. Meinrenken, C. J., Borst, J. G. & Sakmann, B. Local routes revisited: the space and time dependence of the Ca2+ signal for phasic transmitter release at www.nature.com/reviews/neuro © 2007 Nature Publishing Group REVIEWS 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. the rat calyx of Held. J. Physiol. 547, 665–689 (2003). Meinrenken, C. J., Borst, J. G. & Sakmann, B. Calcium secretion coupling at calyx of held governed by nonuniform channel-vesicle topography. J. Neurosci. 22, 1648–1667 (2002). Wadel, K., Neher, E. & Sakaba, T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron 53, 563–575 (2007). Fedchyshyn, M. J. & Wang, L. Y. Developmental transformation of the release modality at the calyx of Held synapse. J. Neurosci. 25, 4131–4140 (2005). Zhai, R. G. & Bellen, H. J. The architecture of the active zone in the presynaptic nerve terminal. Physiology (Bethesda) 19, 262–270 (2004). Zampighi, G. A. et al. Conical electron tomography of a chemical synapse: vesicles docked to the active zone are hemi-fused. Biophys. J. 91, 2910–2918 (2006). Phillips, G. R. et al. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron 32, 63–77 (2001). Felmy, F., Neher, E. & Schneggenburger, R. The timing of phasic transmitter release is Ca2+-dependent and lacks a direct influence of presynaptic membrane potential. Proc. Natl Acad. Sci. USA 100, 15200–15205 (2003). Dodge, F. A. Jr & Rahamimoff, R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193, 419–432 (1967). Wolfel, M. & Schneggenburger, R. Presynaptic capacitance measurements and Ca2+ uncaging reveal submillisecond exocytosis kinetics and characterize the Ca2+ sensitivity of vesicle pool depletion at a fast CNS synapse. J. Neurosci. 23, 7059–7068 (2003). Zucker, R. S. & Regehr, W. G. Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405 (2002). Bischofberger, J., Engel, D., Frotscher, M. & Jonas, P. Timing and efficacy of transmitter release at mossy fiber synapses in the hippocampal network. Pflugers Arch. 453, 361–372 (2006). Korogod, N., Lou, X. & Schneggenburger, R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J. Neurosci. 25, 5127–5137 (2005). Isaacson, J. S. & Walmsley, B. Counting quanta: direct measurements of transmitter release at a central synapse. Neuron 15, 875–884 (1995). Diamond, J. S. & Jahr, C. E. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15, 1097–1107 (1995). Millet, O., Bernado, P., Garcia, J., Rizo, J. & Pons, M. NMR measurement of the off rate from the first calcium-binding site of the synaptotagmin I C2A domain. FEBS Lett. 516, 93–96 (2002). Collin, T. et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J. Neurosci. 25, 96–107 (2005). Sudhof, T. C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547 (2004). Geppert, M. et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727 (1994). Nishiki, T. & Augustine, G. J. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+dependent neurotransmitter release. J. Neurosci. 24, 8542–8550 (2004). Nishiki, T. & Augustine, G. J. Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J. Neurosci. 24, 6127–6132 (2004). Fernandez-Chacon, R. et al. Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49 (2001). Takamori, S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006). Stewart, B. A., Mohtashami, M., Trimble, W. S. & Boulianne, G. L. SNARE proteins contribute to calcium cooperativity of synaptic transmission. Proc. Natl Acad. Sci. USA 97, 13955–13960 (2000). Hua, Y. & Scheller, R. H. Three SNARE complexes cooperate to mediate membrane fusion. Proc. Natl Acad. Sci. USA 98, 8065–8070 (2001). Montecucco, C., Schiavo, G. & Pantano, S. SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem. Sci. 30, 367–372 (2005). Xu, Y., Zhang, F., Su, Z., McNew, J. A. & Shin, Y. K. Hemifusion in SNARE-mediated membrane fusion. Nature Struct. Mol. Biol. 12, 417–422 (2005). 58. Reese, C., Heise, F. & Mayer, A. Trans-SNARE pairing can precede a hemifusion intermediate in intracellular membrane fusion. Nature 436, 410–414 (2005). 59. Giraudo, C. G., Eng, W. S., Melia, T. J. & Rothman, J. E. A clamping mechanism involved in SNARE-dependent exocytosis. Science 313, 676–680 (2006). 60. Tang, J. et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187 (2006). This paper provides insights into the steps that make the vesicle primed for release. 61. Schaub, J. R., Lu, X., Doneske, B., Shin, Y. K. & McNew, J. A. Hemifusion arrest by complexin is relieved by Ca2+–synaptotagmin I. Nature Struct. Mol. Biol. 13, 748–750 (2006). Direct measurements of membrane mixing provide unique insights into the fusion process. 62. Zimmerberg, J., Akimov, S. A. & Frolov, V. Synaptotagmin: fusogenic role for calcium sensor? Nature Struct. Mol. Biol. 13, 301–303 (2006). 63. Jackson, M. B. & Chapman, E. R. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu. Rev. Biophys. Biomol. Struct. 35, 135–160 (2006). An excellent review of the issues regarding the mechanism by which the fusion pore is generated. 64. Han, X., Wang, C. T., Bai, J., Chapman, E. R. & Jackson, M. B. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289–292 (2004). 65. Han, X. & Jackson, M. B. Electrostatic interactions between the syntaxin membrane anchor and neurotransmitter passing through the fusion pore. Biophys. J. 88, L20–L22 (2005). 66. Almers, W. Exocytosis. Annu. Rev. Physiol. 52, 607–624 (1990). 67. He, L., Wu, X. S., Mohan, R. & Wu, L. G. Two modes of fusion pore opening revealed by cell-attached recordings at a synapse. Nature 444, 102–105 (2006). A technical breakthrough allows direct measurements of vesicle fusion and fusion pore formation at the secretory face of a central synapse. 68. Chen, X., Wang, L., Zhou, Y., Zheng, L. H. & Zhou, Z. ‘Kiss-and-run’ glutamate secretion in cultured and freshly isolated rat hippocampal astrocytes. J. Neurosci. 25, 9236–9243 (2005). 69. Klyachko, V. A. & Jackson, M. B. Capacitance steps and fusion pores of small and large-dense-core vesicles in nerve terminals. Nature 418, 89–92 (2002). 70. Harata, N. C., Aravanis, A. M. & Tsien, R. W. Kiss-andrun and full-collapse fusion as modes of exoendocytosis in neurosecretion. J. Neurochem. 97, 1546–1570 (2006). 71. Harata, N. C., Choi, S., Pyle, J. L., Aravanis, A. M. & Tsien, R. W. Frequency-dependent kinetics and prevalence of kiss-and-run and reuse at hippocampal synapses studied with novel quenching methods. Neuron 49, 243–256 (2006). 72. Aravanis, A. M., Pyle, J. L. & Tsien, R. W. Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423, 643–647 (2003). 73. Ceccarelli, B., Hurlbut, W. P. & Mauro, A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J. Cell Biol. 57, 499–524 (1973). 74. Gandhi, S. P. & Stevens, C. F. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423, 607–613 (2003). 75. Pawlu, C., DiAntonio, A. & Heckmann, M. Postfusional control of quantal current shape. Neuron 42, 607–618 (2004). 76. Burger, P. M. et al. Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3, 715–720 (1989). 77. Raghavachari, S. & Lisman, J. E. Properties of quantal transmission at CA1 synapses. J. Neurophysiol. 92, 2456–2467 (2004). This paper used Monte-Carlo simulations to show how the glutamate released from a vesicle diffuses in the cleft and activates AMPA channels. 78. Tanaka, J. et al. Number and density of AMPA receptors in single synapses in immature cerebellum. J. Neurosci. 25, 799–807 (2005). 79. Nielsen, T. A., DiGregorio, D. A. & Silver, R. A. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron 42, 757–771 (2004). This paper uses a clever combination of experimental manipulation and modelling to discern the diffusion coefficient of the NATURE REVIEWS | NEUROSCIENCE 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. neurotransmitter glutamate in the synpaptic cleft. This is an important parameter in determining the amplitude of synaptic responses. Smith, M. A., Ellis-Davies, G. C. & Magee, J. C. Mechanism of the distance-dependent scaling of Schaffer collateral synapses in rat CA1 pyramidal neurons. J. Physiol. 548, 245–258 (2003). Forti, L., Bossi, M., Bergamaschi, A., Villa, A. & Malgaroli, A. Loose-patch recordings of single quanta at individual hippocampal synapses. Nature 388, 874–878 (1997). A direct measurement of the quantal response at putatively single synapses showing that quantal responses have small variability. Silver, R. A., Cull-Candy, S. G. & Takahashi, T. NonNMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J. Physiol. 494, 231–250 (1996). Liu, G., Choi, S. & Tsien, R. W. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron 22, 395–409 (1999). McAllister, A. K. & Stevens, C. F. Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc. Natl Acad. Sci. USA 97, 6173–6178 (2000). Franks, K. M., Bartol, T. M. Jr. & Sejnowski, T. J. A Monte Carlo model reveals independent signaling at central glutamatergic synapses. Biophys. J. 83, 2333–2348 (2002). Choi, S., Klingauf, J. & Tsien, R. W. Fusion pore modulation as a presynaptic mechanism contributing to expression of long-term potentiation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 695–705 (2003). A review of experiments pointing to the importance of the mode of vesicle release in AMPA channel activation. Experiments suggesting a role for this process in synaptic plasticity are also reviewed. Liao, D., Hessler, N. A. & Malinow, R. Activation of postsynaptically silent synapses during pairinginduced LTP in CA1 region of hippocampal slice. Nature 375, 400–404 (1995). Bekkers, J. M., Richerson, G. B. & Stevens, C. F. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc. Natl Acad. Sci. USA 87, 5359–5362 (1990). Hanse, E. & Gustafsson, B. Quantal variability at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J. Physiol. 531, 467–480 (2001). Sahara, Y. & Takahashi, T. Quantal components of the excitatory postsynaptic currents at a rat central auditory synapse. J. Physiol. 536, 189–197 (2001). Jonas, P., Major, G. & Sakmann, B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J. Physiol. 472, 615–663 (1993). Franks, K. M., Stevens, C. F. & Sejnowski, T. J. Independent sources of quantal variability at single glutamatergic synapses. J. Neurosci. 23, 3186–3195 (2003). Karunanithi, S., Marin, L., Wong, K. & Atwood, H. L. Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J. Neurosci. 22, 10267–10276 (2002). Schikorski, T. & Stevens, C. F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 17, 5858–5867 (1997). Wall, M. J. & Usowicz, M. M. Development of the quantal properties of evoked and spontaneous synaptic currents at a brain synapse. Nature Neurosci. 1, 675–682 (1998). Ulrich, D. & Luscher, H. R. Miniature excitatory synaptic currents corrected for dendritic cable properties reveal quantal size and variance. J. Neurophysiol. 69, 1769–1773 (1993). Yamashita, T., Ishikawa, T. & Takahashi, T. Developmental increase in vesicular glutamate content does not cause saturation of AMPA receptors at the calyx of held synapse. J. Neurosci. 23, 3633–3638 (2003). Magee, J. C. & Cook, E. P. Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nature Neurosci. 3, 895–903 (2000). This paper combines local dendritic recording and focal sucrose application to measure the mEPSC with minimal dendritic filtering. This provides the most constrained measurement of the time-course of the mEPSC. ADVANCE ONLINE PUBLICATION | 607 © 2007 Nature Publishing Group REVIEWS 99. Bornstein, J. C. Spontaneous multiquantal release at synapses in guinea-pig hypogastric ganglia: evidence that release can occur in bursts. J. Physiol. 282, 375–398 (1978). 100. Paulsen, O. & Heggelund, P. The quantal size at retinogeniculate synapses determined from spontaneous and evoked EPSCs in guinea-pig thalamic slices. J. Physiol. 480, 505–511 (1994). 101. Paulsen, O. & Heggelund, P. Quantal properties of spontaneous EPSCs in neurones of the guinea-pig dorsal lateral geniculate nucleus. J. Physiol. 496, 759–772 (1996). 102. Min, M. Y. & Appenteng, K. Multimodal distribution of amplitudes of miniature and spontaneous EPSPs recorded in rat trigeminal motoneurones. J. Physiol. 494, 171–182 (1996). 103. Dennis, M. J., Harris, A. J. & Kuffler, S. W. Synaptic transmission and its duplication by focally applied acetylcholine in parasympathetic neurons in the heart of the frog. Proc. R. Soc. Lond. B Biol. Sci. 177, 509–539 (1971). 104. Liu, G. & Feldman, J. L. Quantal synaptic transmission in phrenic motor nucleus. J. Neurophysiol. 68, 1468–1471 (1992). 105. Edwards, F. A., Konnerth, A. & Sakmann, B. Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study. J. Physiol. 430, 213–249 (1990). 106. Llano, I. et al. Presynaptic calcium stores underlie largeamplitude miniature IPSCs and spontaneous calcium transients. Nature Neurosci. 3, 1256–1265 (2000). 107. Melamed, N., Helm, P. J. & Rahamimoff, R. Confocal microscopy reveals coordinated calcium fluctuations and oscillations in synaptic boutons. J. Neurosci. 13, 632–649 (1993). 108. Emptage, N. J., Reid, C. A. & Fine, A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29, 197–208 (2001). 109. Simkus, C. R. & Stricker, C. The contribution of intracellular calcium stores to mEPSCs recorded in layer II neurones of rat barrel cortex. J. Physiol. 545, 521–535 (2002). 110. Galante, M. & Marty, A. Presynaptic ryanodinesensitive calcium stores contribute to evoked neurotransmitter release at the basket cell–Purkinje cell synapse. J. Neurosci. 23, 11229–11234 (2003). 111. Sharma, G. & Vijayaraghavan, S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38, 929–939 (2003). 112. Jack, J. J., Larkman, A. U., Major, G. & Stratford, K. J. Quantal analysis of the synaptic excitation of CA1 hippocampal pyramidal cells. Adv. Second Messenger Phosphoprotein Res. 29, 275–299 (1994). 113. Nusser, Z. et al. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21, 545–559 (1998). 114. Wilson, N. R. et al. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J. Neurosci. 25, 6221–6234 (2005). 115. Wu, X. S. et al. The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J. Neurosci. 27, 3046–3056 (2007). 116. Conti, R. & Lisman, J. The high variance of AMPA receptor- and NMDA receptor-mediated responses at single hippocampal synapses: evidence for multiquantal release. Proc. Natl Acad. Sci. USA 100, 4885–4890 (2003). 117. Conti, R. & Lisman, J. A large sustained Ca2+ elevation occurs in unstimulated spines during the LTP pairing protocol but does not change synaptic strength. Hippocampus 12, 667–679 (2002). 118. Oertner, T. G., Sabatini, B. L., Nimchinsky, E. A. & Svoboda, K. Facilitation at single synapses probed with optical quantal analysis. Nature Neurosci. 5, 657–664 (2002). This paper uses two-photon calcium imaging to infer that multivesicular release can occur at single synapses. 119. Wadiche, J. I. & Jahr, C. E. Multivesicular release at climbing fiber–Purkinje cell synapses. Neuron 32, 301–313 (2001). 120. Foster, K. A., Kreitzer, A. C. & Regehr, W. G. Interaction of postsynaptic receptor saturation with presynaptic mechanisms produces a reliable synapse. Neuron 36, 1115–1126 (2002). 121. Christie, J. M. & Jahr, C. E. Multivesicular release at Schaffer collateral–CA1 hippocampal synapses. J. Neurosci. 26, 210–216 (2006). 122. Satzler, K. et al. Three-dimensional reconstruction of a calyx of Held and its postsynaptic principal neuron in 123. 124. 125. 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. 146. the medial nucleus of the trapezoid body. J. Neurosci. 22, 10567–10579 (2002). Biro, A. A., Holderith, N. B. & Nusser, Z. Quantal size is independent of the release probability at hippocampal excitatory synapses. J. Neurosci. 25, 223–232 (2005). Stevens, C. F. & Wang, Y. Facilitation and depression at single central synapses. Neuron 14, 795–802 (1995). Bolshakov, V. Y. & Siegelbaum, S. A. Regulation of hippocampal transmitter release during development and long-term potentiation. Science 269, 1730–1734 (1995). Chen, G., Harata, N. C. & Tsien, R. W. Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc. Natl Acad. Sci. USA 101, 1063–1068 (2004). Silver, R. A., Lubke, J., Sakmann, B. & Feldmeyer, D. High-probability uniquantal transmission at excitatory synapses in barrel cortex. Science 302, 1981–1984 (2003). Shapira, M. et al. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron 38, 237–252 (2003). Lisman, J. & Raghavachari, S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Science’s STKE [online] http://stke. sciencemag.org/cgi/content/full/sigtrans;2006/356/ re11 (2006). Cathala, L., Holderith, N. B., Nusser, Z., DiGregorio, D. A. & Cull-Candy, S. G. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nature Neurosci. 8, 1310–1318 (2005). Bolshakov, V. Y., Golan, H., Kandel, E. R. & Siegelbaum, S. A. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3–CA1 synapses in the hippocampus. Neuron 19, 635–651 (1997). Ostroff, L. E., Fiala, J. C., Allwardt, B. & Harris, K. M. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35, 535–545 (2002). Tsien, R. W., Ellinor, P. T. & Horne, W. A. Molecular diversity of voltage-dependent Ca2+ channels. Trends Pharmacol. Sci. 12, 349–354 (1991). Catterall, W. A. Structure and function of voltagegated sodium and calcium channels. Curr. Opin. Neurobiol. 1, 5–13 (1991). Dunlap, K., Luebke, J. I. & Turner, T. J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18, 89–98 (1995). Nowycky, M. C., Fox, A. P. & Tsien, R. W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–443 (1985). Hirning, L. D. et al. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science 239, 57–61 (1988). Stanley, E. F. & Goping, G. Characterization of a calcium current in a vertebrate cholinergic presynaptic nerve terminal. J. Neurosci. 11, 985–993 (1991). Stanley, E. F. Single calcium channels on a cholinergic presynaptic nerve terminal. Neuron 7, 585–591 (1991). Llinás, R., Sugimori, M., Hillman, D. E. & Cherksey, B. Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci. 15, 351–355 (1992). Sather, W. A. et al. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel α1 subunits. Neuron 11, 291–303 (1993). Randall, A. & Tsien, R. W. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J. Neurosci. 15, 2995–3012 (1995). Takahashi, T. & Momiyama, A. Different types of calcium channels mediate central synaptic transmission. Nature 366, 156–158 (1993). Luebke, J. I., Dunlap, K. & Turner, T. J. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron 11, 895–902 (1993). Wheeler, D. B., Randall, A. & Tsien, R. W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111 (1994). Forsythe, I. D. Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS in vitro. J. Physiol. 479, 381–387 (1994). 608 | ADVANCE ONLINE PUBLICATION 147. Ertel, E. A. et al. Nomenclature of voltage-gated calcium channels. Neuron 25, 533–535 (2000). 148. Olivera, B. M., Miljanich, G. P., Ramachandran, J. & Adams, M. E. Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annu. Rev. Biochem. 63, 823–867 (1994). 149. Campbell, K. P., Leung, A. T. & Sharp, A. H. The biochemistry and molecular biology of the dihydropyridine-sensitive calcium channel. Trends Neurosci. 11, 425–430 (1988). 150. Scholz, K. P. & Miller, R. J. Developmental changes in presynaptic calcium channels coupled to glutamate release in cultured rat hippocampal neurons. J. Neurosci. 15, 4612–4617 (1995). 151. Tsien, R. W., Hess, P., McCleskey, E. W. & Rosenberg, R. L. Calcium channels: mechanisms of selectivity, permeation, and block. Annu. Rev. Biophys. Biophys. Chem. 16, 265–290 (1987). 152. Sather, W. A. & McCleskey, E. W. Permeation and selectivity in calcium channels. Annu. Rev. Physiol. 65, 133–159 (2003). 153. Zhou, Y. D., Turner, T. J. & Dunlap, K. Enhanced G protein-dependent modulation of excitatory synaptic transmission in the cerebellum of the Ca2+ channelmutant mouse, tottering. J. Physiol. 547, 497–507 (2003). 154. Colecraft, H. M., Patil, P. G. & Yue, D. T. Differential occurrence of reluctant openings in G-protein-inhibited N- and P/Q-type calcium channels. J. Gen. Physiol. 115, 175–192 (2000). 155. Pietrobon, D. Calcium channels and channelopathies of the central nervous system. Mol. Neurobiol. 25, 31–50 (2002). 156. Miljanich, G. P. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 11, 3029–3040 (2004). 157. Choi, S., Klingauf, J. & Tsien, R. W. Postfusional regulation of cleft glutamate concentration during LTP at ‘silent synapses’. Nature Neurosci. 3, 330–336 (2000). 158. Montgomery, J. M., Pavlidis, P. & Madison, D. V. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron 29, 691–701 (2001). 159. Renger, J. J., Egles, C. & Liu, G. A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron 29, 469–484 (2001). 160. Krupa, B. & Liu, G. Does the fusion pore contribute to synaptic plasticity? Trends Neurosci. 27, 62–66 (2004). 161. Photowala, H., Blackmer, T., Schwartz, E., Hamm, H. E. & Alford, S. G protein βγ-subunits activated by serotonin mediate presynaptic inhibition by regulating vesicle fusion properties. Proc. Natl Acad. Sci. USA 103, 4281–4286 (2006). 162. Zakharenko, S. S., Zablow, L. & Siegelbaum, S. A. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35, 1099–1110 (2002). 163. Nakagawa, T., Cheng, Y., Sheng, M. & Walz, T. Three-dimensional structure of an AMPA receptor without associated stargazin/TARP proteins. Biol. Chem. 387, 179–187 (2006). 164. Madden, D. R. The structure and function of glutamate receptor ion channels. Nature Rev. Neurosci. 3, 91–101 (2002). 165. Armstrong, N., Sun, Y., Chen, G. Q. & Gouaux, E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395, 913–917 (1998). 166. Armstrong, N. & Gouaux, E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181 (2000). 167. Sun, Y. et al. Mechanism of glutamate receptor desensitization. Nature 417, 245–253 (2002). 168. Armstrong, N., Jasti, J., Beich-Frandsen, M. & Gouaux, E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell 127, 85–97 (2006). A structural study of AMPA channels that provides clear insight into the mechanism of desensitization. 169. Rosenmund, C., Stern-Bach, Y. & Stevens, C. F. The tetrameric structure of a glutamate receptor channel. Science 280, 1596–1599 (1998). An important paper demonstrating the tetrameric structure of AMPA channels and the fact that different channel occupancy by glutamate leads to different single-channel conductance. www.nature.com/reviews/neuro © 2007 Nature Publishing Group REVIEWS 170. Smith, T. C. & Howe, J. R. Concentration-dependent substate behavior of native AMPA receptors. Nature Neurosci. 3, 992–997 (2000). 171. Ruiz, M. & Karpen, J. W. Opening mechanism of a cyclic nucleotide-gated channel based on analysis of single channels locked in each liganded state. J. Gen. Physiol. 113, 873–895 (1999). 172. Ruiz, M. L. & Karpen, J. W. Single cyclic nucleotidegated channels locked in different ligand-bound states. Nature 389, 389–392 (1997). 173. Raman, I. M. & Trussell, L. O. The mechanism of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor desensitization after removal of glutamate. Biophys. J. 68, 137–146 (1995). 174. Hausser, M. & Roth, A. Estimating the time course of the excitatory synaptic conductance in neocortical pyramidal cells using a novel voltage jump method. J. Neurosci. 17, 7606–7625 (1997). 175. Diamond, J. S. & Jahr, C. E. Transporters buffer synaptically released glutamate on a submillisecond time scale. J. Neurosci. 17, 4672–4687 (1997). 176. Postlethwaite, M., Hennig, M. H., Steinert, J. R., Graham, B. P. & Forsythe, I. D. Acceleration of AMPA receptor kinetics underlies temperature-dependent changes in synaptic strength at the rat calyx of Held. J. Physiol. 579, 69–84 (2007). 177. Robert, A. & Howe, J. R. How AMPA receptor desensitization depends on receptor occupancy. J. Neurosci. 23, 847–858 (2003). 178. Rostaing, P. et al. Analysis of synaptic ultrastructure without fixative using high-pressure freezing and tomography. Eur. J. Neurosci. 24, 3463–3474 (2006). 179. Siksou, L. et al. Three-dimensional architecture of presynaptic terminal cytomatrix. J. Neurosci. 27, 6868–6877 (2007). Acknowledgements The authors would like to thank M. Jackson, J. Zimmerberg, A. Silver, S. Marty, L.-G. Wu, Z. Nusser, R. Schneggenburger, G. Fain and E. Marder for useful discussions. J.E.L. has been supported by the US National Institutes of Health (NIH) grants R01 NS27337 and R01 NS50944 as part of the NATURE REVIEWS | NEUROSCIENCE Collaborative Research in Computational Neuroscience Program. R.W.T. has been supported by NIH grants NS24067 and MH64070 and generous support from the Mathers Foundation. S.R. has been supported by the National Science Foundation grant 0642000. Competing interests statement The authors declare no competing financial interests. DATABASES The following terms in this article are linked online to: Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query. fcgi?db=gene Cav2.1 | Cav2.2 | Cav2.3 FURTHER INFORMATION John Lisman’s homepage: http://www.bio.brandeis.edu/lismanlab/ Synapse Web: http://synapse-web.org/ Access to this links box is available online. ADVANCE ONLINE PUBLICATION | 609 © 2007 Nature Publishing Group