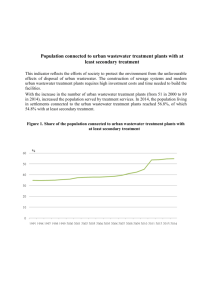
Standard Methods for the Examination of Water and Wastewater
advertisement

Standard Methods for the Examination of Water and Wastewater Part 9000 9010 MICROBIOLOGICAL EXAMINATION INTRODUCTION*#(1) The following sections describe procedures for making microbiological examinations of water samples to determine sanitary quality. The methods are intended to indicate the degree of contamination with wastes. They are the best techniques currently available; however, their limitations must be understood thoroughly. Tests for detection and enumeration of indicator organisms, rather than of pathogens, are used. The coliform group of bacteria, as herein defined, is the principal indicator of suitability of a water for domestic, industrial, or other uses. The cultural reactions and characteristics of this group of bacteria have been studied extensively. Experience has established the significance of coliform group density as a criterion of the degree of pollution and thus of sanitary quality. The significance of the tests and the interpretation of results are well authenticated and have been used as a basis for standards of bacteriological quality of water supplies. The membrane filter technique, which involves a direct plating for detection and estimation of coliform densities, is as effective as the multiple-tube fermentation test for detecting bacteria of the coliform group. Modification of procedural details, particularly of the culture medium, has made the results comparable with those given by the multiple-tube fermentation procedure. Although there are limitations in the application of the membrane filter technique, it is equivalent when used with strict adherence to these limitations and to the specified technical details. Thus, two standard methods are presented for the detection and enumeration of bacteria of the coliform group. It is customary to report results of the coliform test by the multiple-tube fermentation procedure as a Most Probable Number (MPN) index. This is an index of the number of coliform bacteria that, more probably than any other number, would give the results shown by the laboratory examination; it is not an actual enumeration. By contrast, direct plating methods such as the membrane filter procedure permit a direct count of coliform colonies. In both procedures coliform density is reported conventionally as the MPN or membrane filter count per 100 mL. Use of either procedure permits appraising the sanitary quality of water and the effectiveness of treatment processes. Because it is not necessary to provide a quantitative assessment of coliform bacteria for all samples, a qualitative, presence-absence test is included. Fecal streptococci and enterococci also are indicators of fecal pollution and methods for their detection and enumeration are given. A multiple-tube dilution and a membrane filter procedure are included. Methods for the differentiation of the coliform group are included. Such differentiation © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater generally is considered of limited value in assessing drinking water quality because the presence of any coliform bacteria renders the water potentially unsatisfactory and unsafe. Speciation may provide information on colonization of a distribution system and further confirm the validity of coliform results. Coliform group bacteria present in the gut and feces of warm-blooded animals generally include organisms capable of producing gas from lactose in a suitable culture medium at 44.5 ± 0.2°C. Inasmuch as coliform organisms from other sources often cannot produce gas under these conditions, this criterion is used to define the fecal component of the coliform group. Both the multiple-tube dilution technique and the membrane filter procedure have been modified to incorporate incubation in confirmatory tests at 44.5°C to provide estimates of the density of fecal organisms, as defined. Procedures for fecal coliforms and Escherichia coli include a 24-h multiple-tube test using A-1 medium, a 7-h rapid method, and chromogenic substrate coliform tests. This differentiation yields valuable information concerning the possible source of pollution in water, and especially its remoteness, because the nonfecal members of the coliform group may be expected to survive longer than the fecal members in the unfavorable environment provided by the water. The heterotrophic plate count may be determined by pour plate, spread plate, or membrane filter method. It provides an approximate enumeration of total numbers of viable bacteria that may yield useful information about water quality and may provide supporting data on the significance of coliform test results. The heterotrophic plate count is useful in judging the efficiency of various treatment processes and may have significant application as an in-plant control test. It also is valuable for checking quality of finished water in a distribution system as an indicator of microbial regrowth and sediment buildup in slow-flow sections and dead ends. Experience in the shipment of un-iced samples by mail indicates that noticeable changes may occur in type or numbers of bacteria during such shipment for even limited periods of time. Therefore, refrigeration during transportation is recommended to minimize changes, particularly when ambient air temperature exceeds 13°C. Procedures for the isolation of certain pathogenic bacteria and protozoa are presented. These procedures are tedious and complicated and are not recommended for routine use. Likewise, tentative procedures for enteric viruses are included but their routine use is not advocated. Examination of routine bacteriological samples cannot be regarded as providing complete information concerning water quality. Always consider bacteriological results in the light of information available concerning the sanitary conditions surrounding the sample source. For a water supply, precise evaluation of quality can be made only when the results of laboratory examinations are interpreted in the light of sanitary survey data. Consider inadequate the results of the examination of a single sample from a given source. When possible, base evaluation of water quality on the examination of a series of samples collected over a known and protracted period of time. Pollution problems of tidal estuaries and other bodies of saline water have focused attention on necessary modification of existing bacteriological techniques so that they may be used © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater effectively. In the following sections, applications of specific techniques to saline water are not discussed because the methods used for fresh waters generally can be used satisfactorily with saline waters. Methods for examination of the waters of swimming pools and other bathing places are included. The standard procedures for the plate count, fecal coliforms, and fecal streptococci are identical with those used for other waters. Procedures for Staphylococcus and Pseudomonas aeruginosa, organisms commonly associated with the upper respiratory tract or the skin, are included. Procedures for aquatic fungi and actinomycetes are included. Sections on rapid methods for coliform testing and on the recovery of stressed organisms are included. Because of increased interest and concern with analytical quality control, this section continues to be expanded. The bacteriological methods in Part 9000, developed primarily to permit prompt and rapid examination of water samples, have been considered frequently to apply only to routine examinations. However, these same methods are basic to, and equally valuable in, research investigations in sanitary bacteriology and water treatment. Similarly, all techniques should be the subject of investigations to establish their specificity, improve their procedural details, and expand their application to the measurement of the sanitary quality of water supplies or polluted waters. 9020 QUALITY ASSURANCE/QUALITY CONTROL*#(2) 9020 A. Introduction 1. General Considerations The growing emphasis on microorganisms in water quality standards and enforcement activities and their continuing role in research, process control, and compliance monitoring require the establishment and effective operation of a quality assurance (QA) program to substantiate the validity of analytical data. A laboratory quality assurance program is the integration of intralaboratory and interlaboratory quality control (QC), standardization, and management practices into a formal, documented program with clearly defined responsibilities and duties to ensure that the data are of the type, quality, and quantity required. The program must be practical and require only a reasonable amount of time or it will be bypassed. Generally, about 15% of overall laboratory time should be spent on different aspects of a quality assurance program. However, more time may be needed for more important analytical data, e.g., data for enforcement actions. When properly administered, a balanced, conscientiously applied QA program will optimize data quality without adversely affecting © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater laboratory productivity. Because microbiological analyses measure constantly changing living organisms, they are inherently variable. Some quality control tools used by chemists, such as reference standards, instrument calibration, and quality control charts, may not be available to the microbiologist. Because QA programs vary among laboratories as a result of differences in organizational mission, responsibilities, and objectives; laboratory size, capabilities, and facilities; and staff skills and training, this provides only general guidance. Each laboratory should determine the appropriate QA level for its purpose. 2. Guidelines for a Quality Assurance Program Develop a QA program to meet the laboratory’s specific needs and the planned use of the data. Emphasis on the use of data is particularly important where significant and costly decisions depend on analytical results. An effective QA program will confirm the quality of results and increase confidence in the data. a. Management responsibilities: Management must recognize the need for quality assurance, commit monetary and personnel resources, assume a leadership role, and involve staff in development and operation of the QA program. Management should meet with the laboratory supervisor and staff to develop and maintain a comprehensive program and establish specific responsibility for management, supervisors, and analysts. b. Quality assurance officer: In large laboratories, a QA officer has the authority and responsibility for application of the QA program. Ideally, this person should have a staff position reporting directly to upper management, not a line position. The QA officer should have a technical education, be acquainted with all aspects of laboratory work, and be familiar with statistical techniques for data evaluation. The QA officer is responsible for initiating the program, convincing staff of its value, and providing necessary information and training to the staff. Once the QA program is functioning, the coordinator conducts frequent (weekly to monthly) reviews with the laboratory supervisor and staff to determine the current status and accomplishments of the program and to identify and resolve problems. The QA officer also reports periodically to management to secure backing in actions necessary to correct problems that threaten data quality. c. Staff: Laboratory and field staffs participate with management in planning the QA program, preparing standard operating procedures, and most importantly, implementing the QC program in their daily tasks of collecting samples, conducting analyses, performing quality control checks, and calculating and reporting results. Because the staffs are the first to see potential problems, they should identify them and work with the supervisor to correct and avoid them. It is critical to the success of the QA program that staff understand and actively support it. 3. Quality Assurance Program Objectives The objectives of a QA program include providing data of known quality, ensuring a high quality of laboratory performance, maintaining continuing assessment of laboratory operations, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater identifying weaknesses in laboratory operations, detecting training needs, and improving documentation and recordkeeping. 4. Elements of a Quality Assurance Program Each laboratory should develop and implement a written QA plan describing the QA program and QC activities of the laboratory. The plan should address the following basic common aspects: a. Statement of objectives, describing the specific goals of the laboratory. b. Sampling procedures, including selection of representative sites and specified holding time and temperature conditions. If data may be subjected to litigation, use chain-of-custody procedures. c. Personnel policies, describing specific qualification and training requirements for supervisors and analysts. d. Equipment and instrument requirements, providing calibration procedures and frequency and maintenance requirements. e. Specifications for supplies, to ensure that reagents and supplies are of high quality and are tested for acceptability. f. Analytical methods, i.e., standardized methods established by a standards-setting organization and validated. Ideally, these laboratory methods have documented precision, bias, sensitivity, selectivity, and specificity. g. Analytical quality control measures, including such analytical checks as duplicate analyses, positive and negative controls, sterility checks, and verification tests. h. Standard operating procedures (SOPs), i.e., written statement and documentation of all routine laboratory operations. i. Documentation requirements, concerning data acquisition, recordkeeping, traceability, and accountability. j. Assessment requirements: 1) Internal audits of the laboratory operations, performed by the QA officer and supervisor. 2) On-site evaluations by outside experts to ensure that the laboratory and its personnel are following an acceptable QA program. 3) Performance evaluation studies, in which the QA officer works with the supervisor to incorporate unknown challenge samples into routine analytical runs and laboratories are encouraged to participate in state and national proficiency testing and accreditation programs. The collaborative studies confirm the abilities of a laboratory to generate acceptable data comparable to those of other laboratories and identify potential problems. k. Corrective actions: When problems are identified by the staff, supervisor, and/or QA coordinator, use standard stepwise procedures to determine the causes and correct them. Nonconformances identified by external laboratory evaluation are corrected, recorded, and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater signed off by the laboratory manager and QA officer. Detailed descriptions of quality assurance programs are available.1-4 The QC guidelines discussed in Section 9020B and Section 9020C are recommended as useful source material, but all elements need to be addressed in developing a QA program. 5. References 1. GASKIN, J.E. 1992. Quality Assurance in Water Quality Monitoring. Inland Water Directorate, Conservation & Protection, Ottawa, Ont., Canada. 2. RATLIFF, T.A., JR. 1990. The Laboratory Quality Assurance System. A Manual of Quality Procedures with Related Forms. Van Nostrand Reinhold, New York, N.Y. 3. GARFIELD, F.M. 1984. Quality Assurance Principles of Analytical Laboratories. Assoc. Official Analytical Chemists, Arlington, Va. 4. DUX, J.P. 1983. Quality assurance in the analytical laboratory. Amer. Lab. 26:54. 9020 B. Intralaboratory Quality Control Guidelines All laboratories have some intralaboratory QC practices that have evolved from common sense and the principles of controlled experimentation. A QC program applies practices necessary to minimize systematic and random errors resulting from personnel, instrumentation, equipment, reagents, supplies, sampling and analytical methods, data handling, and data reporting. It is especially important that laboratories performing only a limited amount of microbiological testing exercise strict QC. A listing of key QC practices is given in Table 9020:I. Other sources of QC practices are available.1-3 These practices and guidelines will assist laboratories in establishing and improving QC programs. Laboratories should address all of the QC guidelines discussed herein, but the depth and details may differ for each laboratory. 1. Personnel Microbiological testing should be performed by a professional microbiologist or technician trained in environmental microbiology whenever possible. If not, a professional microbiologist should be available for guidance. Train and evaluate the analyst in basic laboratory procedures. The supervisor periodically should review procedures of sample collecting and handling, media and glassware preparation, sterilization, routine analytical testing, counting, data handling, and QC techniques to identify and eliminate problems. Management should assist laboratory personnel in obtaining additional training and course work to advance their skills and career. 2. Facilities a. Ventilation: Plan well-ventilated laboratories that can be maintained free of dust, drafts, and extreme temperature changes. Whenever possible, laboratories should have air conditioning © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater to reduce contamination, permit more stable operation of incubators, and decrease moisture problems with media and instrumentation. b. Space utilization: Design and operate the laboratory to minimize through traffic and visitors, with a separate area for preparing and sterilizing media, glassware, and equipment. Use a vented laminar-flow hood for dispensing and preparing sterile media, transferring microbial cultures, or working with pathogenic materials. In smaller laboratories it may be necessary, although undesirable, to carry out these activities in the same room. c. Laboratory bench areas: Provide at least 2 m of linear bench space per analyst and additional areas for preparation and support activities. For stand-up work, typical bench dimensions are 90 to 97 cm high and 70 to 76 cm deep. For sit-down activities such as microscopy and plate counting, benches are 75 to 80 cm high. Specify bench tops of stainless steel, epoxy plastic, or other smooth, impervious surface that is inert and corrosion-resistant, has a minimum number of seams, and has adequate sealing of any crevices. Install even, glare-free lighting with about 1000 lux (100 ft-candles) intensity at the working surface. d. Walls and floors: Assure that walls are covered with a smooth finish that is easily cleaned and disinfected. Specify floors of smooth concrete, vinyl, asphalt tile, or other impervious, sealed washable surfaces. e. Work-area monitoring: Maintain high standards of cleanliness in work areas. Monitor air, at least monthly, with air density plates. The number of colonies on the air density plate test should not exceed 160/m2/15 min exposure (15 colonies/plate/15 min). Plate or the swab method1 can be used weekly or more frequently to monitor bench surface contamination. Although uniform limits for bacterial density have not been set, each laboratory can use these tests to establish a base line and take action on a significant increase. f. Laboratory cleanliness: Regularly clean laboratory rooms and wash benches, shelves, floors, and windows. Wet-mop floors and treat with a disinfectant solution; do not sweep or dry-mop. Wipe bench tops and treat with a disinfectant before and after use. Do not permit laboratory to become cluttered. 3. Laboratory Equipment and Instrumentation Verify that each item of equipment meets the user’s needs for precision and minimization of bias. Perform equipment maintenance on a regular basis as recommended by the manufacturer or obtain preventive maintenance contracts on autoclave, balances, microscopes, and other equipment. Directly record all quality control checks in a permanent log book. Use the following quality control procedures: a. Thermometer/temperature-recording instruments: Check accuracy of thermometers or temperature-recording instruments semiannually against a certified National Institute of Standards and Technology (NIST) thermometer or one traceable to NIST and conforming to NIST specifications. For general purposes use thermometers graduated in increments of 0.5°C or less. Maintain in water or glycerol for air incubators and refrigerators and glycerol for freezers © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and seal in a flask. For a 44.5°C water bath, use a submersible thermometer graduated to 0.2°C or less. Record temperature check data in a quality control log. Mark the necessary NIST calibration corrections on each thermometer and incubator, refrigerator, or freezer. When possible, equip incubators and water baths with temperature-recording instruments that provide a continuous record of operating temperature. b. Balances: Follow manufacturer’s instructions in operation and routine maintenance of analytical and top-loading balances. Balances should be serviced and recalibrated by a manufacturer technician annually or more often as conditions change or problems occur. In weighing 2 g or less, use an analytical balance with a sensitivity less than 1 mg at a 10-g load. For larger quantities use a pan balance with sensitivity of 0.1 g at a 150-g load. Wipe balance before use with a soft brush. Clean balance pans after use and wipe spills up immediately with a laboratory tissue. Inspect weights with each use and replace if corroded. Use only a plastic-tip forceps to handle weights. Check balance and working weights monthly against a set of reference weights (ANSI/ASTM Class 1 or NIST Class S) for accuracy, precision, and linearity.4 Record results. c. pH meter: Use a meter graduated in 0.1 pH units or less, that includes temperature compensation. Preferably use digital meters and commercial buffer solutions. With each use, standardize meter with two buffers that bracket the pH of interest and record. Date buffer solutions when opened and check monthly against another pH meter. Discard solution after each use and replace buffer supply before expiration date. For full details of pH meter use and maintenance, see Section 4500-H+. d. Water purification system: Commercial systems are available that include some combination of prefiltration, activated carbon, mixed-bed resins, and reverse-osmosis with final filtration to produce a reagent-grade water. The life of such systems can be extended greatly if the source water is pretreated by distillation or by reverse osmosis to remove dissolved solids. Such systems tend to produce the same quality water until resins or activated carbon are near exhaustion and quality abruptly becomes unacceptable. Some deionization components are available now that automatically regenerate the ion exchange resins. Do not store reagent water unless a commercial UV irradiation device is installed and is confirmed to maintain sterility. Monitor reagent water continuously or daily with a calibrated conductivity meter and analyze at least annually for trace metals. Replace cartridges at intervals recommended by the manufacturer based on the estimated usage and source water quality. Do not wait for column failure. If bacteria-free water is desired, include aseptic final filtration with a 0.22-µm-pore membrane filter and collect in a sterile container. Monitor treated water for contamination and replace the filter as necessary. e. Water still: Stills produce water of a good grade that characteristically deteriorates slowly over time as corrosion, leaching, and fouling occur. These conditions can be controlled with proper maintenance and cleaning. Stills efficiently remove dissolved substances but not dissolved gases or volatile organic chemicals. Freshly distilled water may contain chlorine and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater ammonia (NH3). On storage, additional NH3 and CO2 are absorbed from the air. Use softened water as the source water to reduce frequency of cleaning the still. Drain and clean still and reservoir according to manufacturer’s instructions and usage. f. Media dispensing apparatus: Check accuracy of volumes dispensed with a graduated cylinder at start of each volume change and periodically throughout extended runs. If the unit is used more than once per day, pump a large volume of hot reagent water through the unit to rinse between runs. Correct leaks, loose connections, or malfunctions immediately. At the end of the work day, break apparatus down into parts, wash, rinse with reagent water, and dry. Lubricate parts according to manufacturer’s instructions or at least once per month. g. Hot-air oven: Test performance monthly with commercially available Bacillus subtilis spore strips or spore suspensions. Monitor temperature with a thermometer accurate in the 160 to 180°C range and record results. Use heat-indicating tape to identify supplies and materials that have been exposed to sterilization temperatures. h. Autoclave: Record items sterilized, temperature, pressure, and time for each run. Optimally use a recording thermometer. Check and record operating temperature weekly with a minimum/maximum thermometer. Test performance with Bacillus stearothermophilus spore strips, suspensions, or capsules monthly. Use heat-indicating tape to identify supplies and materials that have been sterilized. i. Refrigerator: Maintain temperature at 1 to 4°C. Check and record temperature daily and clean monthly. Identify and date materials stored. Defrost as required and discard outdated materials quarterly. j. Freezer: Maintain temperature at −20°C to −30°C. Check and record temperature daily. A recording thermometer and alarm system are highly desirable. Identify and date materials stored. Defrost and clean semiannually; discard outdated materials. k. Membrane filtration equipment: Before use, assemble filtration units and check for leaks. Discard units if inside surfaces are scratched. Wash and rinse filtration assemblies thoroughly after use, wrap in nontoxic paper or foil, and sterilize. l. Ultraviolet lamps: Disconnect lamps monthly and clean bulbs with a soft cloth moistened with ethanol. Test lamps quarterly with an appropriate (short- or long-wave) UV light meter*#(3) and replace bulbs if output is less than 70% of the original. For short-wave lamps used in disinfecting work areas, expose plate count agar spread plates containing 200 to 300 organisms of interest, for 2 min. Incubate plates at 35°C for 48 h and count colonies. Replace bulb if count is not reduced 99%. CAUTION: Although short-wave (254-nm) UV light is known to be more dangerous than long-wave UV (365-nm), both types of UV light can damage eyes and skin and potentially are carcinogenic.5 Protect eyes and skin from exposure to UV light. (See Section 1090B .) m. Biohazard hood: Once per month expose plate count agar plates to air flow for 1 h. Incubate plates at 35°C for 48 h and examine for contamination. A properly operating biohazard hood should produce no growth on the plates. Disconnect UV lamps and clean monthly by © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater wiping with a soft cloth moistened with ethanol. Check lamps’ efficiency as specified above. Inspect cabinet for leaks and rate of air flow quarterly. Use a pressure monitoring device to measure efficiency of hood performance. Have laminar-flow safety cabinets containing HEPA filters serviced by the manufacturer. Maintain hoods as directed by the manufacturer. n. Water bath incubator: Verify that incubators maintain test temperature, such as 35 ± 0.5°C or 44.5 ± 0.2°C. Keep an appropriate thermometer (¶ 3a, above) immersed in the water bath; monitor and record temperature twice daily (morning and afternoon). For optimum operation, equip water bath with a gable cover. Use only stainless steel, plastic-coated, or other corrosion-proof racks. Clean bath as needed. o. Incubator (air, water jacketed, or aluminum block): Verify that incubators maintain appropriate test temperatures. Also, verify that cold samples are incubated at the test temperature for the required time. Check and record temperature twice daily (morning and afternoon) on the shelves in use. If a glass thermometer is used, submerge bulb and stem in water or glycerine to the stem mark. For best results use a recording thermometer and alarm system. Place incubator in an area where room temperature is maintained between 16 and 27°C (60 to 80°F). p. Microscopes: Use lens paper to clean optics and stage after each use. Cover microscope when not in use. Permit only trained technicians to use fluorescence microscope and light source. Monitor fluorescence lamp with a light meter and replace when a significant loss in fluorescence is observed. Log lamp operation time, efficiency, and alignment. Periodically check lamp alignment, particularly when the bulb has been changed; realign if necessary. Use known positive 4 + fluorescence slides as controls. 4. Laboratory Supplies a. Glassware: Before each use, examine glassware and discard items with chipped edges or etched inner surfaces. Particularly examine screw-capped dilution bottles and flasks for chipped edges that could leak and contaminate the analyst and the area. Inspect glassware after washing for excessive water beading and rewash if necessary. Make the following tests for clean glassware as necessary: 1) pH check—Because some cleaning solutions are difficult to remove completely, spot check batches of clean glassware for pH reaction, especially if soaked in alkali or acid. To test clean glassware for an alkaline or acid residue add a few drops of 0.04% bromthymol blue (BTB) or other pH indicator and observe the color reaction. BTB should be blue-green (in the neutral range). To prepare 0.04% bromthymol blue indicator solution, add 16 mL 0.01N NaOH to 0.1 g BTB and dilute to 250 mL with reagent water. 2) Test for inhibitory residues on glassware and plasticware—Certain wetting agents or detergents used in washing glassware may contain bacteriostatic or inhibiting substances that require 6 to 12 rinsings to remove all traces and insure freedom from residual bacteriostatic action. Perform this test annually and before using a new supply of detergent. If prewashed, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater presterilized plasticware is used, test it for inhibitory residues. Although the following procedure describes testing of petri dishes for inhibitory residue, it is applicable to other glass or plasticware. a) Procedure—Wash and rinse six petri dishes according to usual laboratory practice and designate as Group A. Wash six petri dishes as above, rinse 12 times with successive portions of reagent water, and designate as Group B. Rinse six petri dishes with detergent wash water (in use concentration), and air-dry without further rinsing, and designate as Group C. Sterilize dishes in Groups A, B, and C by the usual procedure. For presterilized plasticware, set up six plastic petri dishes and designate them as Group D. Prepare and sterilize 200 mL plate count agar and hold in a 44 to 46°C water bath. Prepare a culture of E. aerogenes known to contain 50 to 150 colony-forming units/mL. Preliminary testing may be necessary to achieve this count range. Inoculate three dishes from each test group with 0.1 mL and the other three dishes from each group with 1 mL culture. Analyze the four sets of six plates each, following heterotrophic plate count method (Section 9215B), and incubate at 35°C for 48 h. Count plates with 30 to 300 colonies and record results as CFU/ mL. b) Interpretation of results—Difference in averaged counts on plates in Groups A through D should be less than 15% if there are no toxic or inhibitory effects. Differences in averaged counts of less than 15% between Groups A and B and greater than 15% between Groups A and C indicate that the cleaning detergent has inhibitory properties that are eliminated during routine washing. Differences between B and D greater than 15% indicate an inhibitory residue. b. Utensils and containers for media preparation: Use utensils and containers of borosilicate glass, stainless steel, aluminum, or other corrosion-resistant material (see Section 9030). Do not use copper utensils. c. Dilution water bottles: Use scribed bottles made of nonreactive borosilicate glass or plastic with screwcaps containing inert liners. Clean before use. Disposable plastic bottles prefilled with dilution water are available commercially and are acceptable. Before use of each lot, check pH and volume and examine sterile bottles of dilution water for a precipitate; discard if present. Reclean bottles with acid if necessary, and remake the dilution water. If precipitate repeats, procure a different source of bottles. d. Reagent-grade water quality: The quality of water obtainable from a water purification system differs with the system used and its maintenance. See ¶ 3d and ¶ 3e above. Recommended limits for reagent water quality are given in Table 9020:II. If these limits are not met, investigate and correct or change water source. Although pH measurement of reagent water is characterized by drift, extreme readings are indicative of chemical contamination. e. Use test for evaluation of reagent water, media, and membranes: When a new lot of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater culture medium, membrane filters, or a new source of reagent-grade water is to be used make comparison tests, at least quarterly, of the current lot in use (reference lot) against the new lot (test lot). 1) Procedure—Use a single batch of control water (redistilled or distilled water polished by deionization), glassware, membrane filters, or other needed materials to control all variables except the one factor under study. Make parallel pour or spread plate or membrane filter plate tests on reference lot and test lot, according to procedures in Section 9215 and Section 9222. As a minimum, make single analyses on five different water samples positive for the target organism. Replicate analyses and additional samples can be tested to increase the sensitivity of detecting differences between reference and test lots. When conducting the use test on reagent water, perform the quantitative bacterial tests in parallel using a known high-quality water as a control water. Prepare dilution/rinse water and media with new source of reagent and control water. Test water for all uses (dilution, rinse, media preparation, etc.). 2) Counting and calculations—After incubation, compare bacterial colonies from the two lots for size and appearance. If colonies on the test lot plates are atypical or noticeably smaller than colonies on the reference lot plates, record the evidence of inhibition or other problem, regardless of count differences. Count plates and calculate the individual count per 1 mL or per 100 mL. Transform the count to logarithms and enter the log-transformed results for the two lots in parallel columns. Calculate the difference, d, between the two transformed results for each sample, including the + or − sign, the mean, and the standard deviation sd of these differences (see Section 1010B). Calculate Student’s t statistic, using the number of samples as n: These calculations may be made with various statistical software packages available for personal computers. 3) Interpretation—Use the critical t value, from a Student’s t table for comparison against the calculated value. At the 0.05 significance level this value is 2.78 for five samples (four degrees of freedom). If the calculated t value does not exceed 2.78, the lots do not produce significantly different results and the test lot is acceptable. If the calculated t value exceeds 2.78, the lots produce significantly different results and the test lot is unacceptable. If the colonies are atypical or noticeably smaller on the test lot or the Student’s t exceeds 2.78, review test conditions, repeat the test, and/or reject the test lot and obtain another one. f. Reagents: Because reagents are an integral part of microbiological analyses, their quality must be assured. Use only chemicals of ACS or equivalent grade because impurities can inhibit © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater bacterial growth, provide nutrients, or fail to produce the desired reaction. Date chemicals and reagents when received and when first opened for use. Make reagents to volume in volumetric flasks and transfer for storage to good-quality inert plastic or borosilicate glass bottles with borosilicate, polyethylene, or other plastic stoppers or caps. Label prepared reagents with name and concentration, date prepared, and initials of preparer. Include positive and negative control cultures with each series of cultural or biochemical tests. g. Dyes and stains: In microbiological analyses, organic chemicals are used as selective agents (e.g., brilliant green), as indicators (e.g., phenol red), and as microbiological stains (e.g., Gram stain). Dyes from commercial suppliers vary from lot to lot in percent dye, dye complex, insolubles, and inert materials. Because dyes for microbiology must be of proper strength and stability to produce correct reactions, use only dyes certified by the Biological Stain Commission. Check bacteriological stains before use with at least one positive and one negative control culture and record results. h. Membrane filters and pads: The quality and performance of membrane filters vary with the manufacturer, type, brand, and lot. These variations result from differences in manufacturing methods, materials, quality control, storage conditions, and application. 1) Membrane filters and pads for water analyses should meet the following specifications: a) Filter diam 47 mm, mean pore diam 0.45 µm. Alternate filter and pore sizes may be used if the manufacturer provides data verifying performance equal to or better than that of 47-mm-diam, 0.45-µm-pore size filter. At least 70% of filter area must be pores. b) When filters are floated on reagent water, the water diffuses uniformly through the filters in 15 s with no dry spots on the filters. c) Flow rates are at least 55 mL/min/cm2 at 25°C and a differential pressure of 93 kPa. d) Filters are nontoxic, free of bacterial-growth-inhibiting or stimulating substances, and free of materials that directly or indirectly interfere with bacterial indicator systems in the medium; ink grid is nontoxic. The arithmetic mean of five counts on filters must be at least 90% of the arithmetic mean of the counts on five agar spread plates using the same sample volumes and agar media. e) Filters retain the organisms from a 100-mL suspension of Serratia marcescens containing 1 × 103 cells. f) Water-extractables in filter do not exceed 2.5% after the membrane is boiled in 100 mL reagent water for 20 min, dried, cooled, and brought to constant weight. g) Absorbent pad has diam 47 mm, thickness 0.8 mm, and is capable of absorbing 2.0 ± 0.2 mL Endo broth. h) Pads release less than 1 mg total acidity calculated as CaCO3 when titrated to the phenolphthalein end point with 0.02N NaOH. i) If filter and absorbent pad are not sterile, they should not be degraded by sterilization at 121°C for 10 min. Confirm sterility by absence of growth when a membrane filter is placed on a © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater pad saturated with tryptone glucose extract broth or tryptone glucose extract agar and incubated at 35 ±0.5°C for 24 h. j) Some lots of membrane filters yield low recoveries, poor differentiation, or malformation of colonies due to toxicity, chemical composition, or structural defects.6 Perform the use test (¶ 4e) on new lots of filters. 2) Standardized tests: Standardized tests are available for evaluating retention, recovery, extractables, and flow rate characteristics of membrane filters.7 Some manufacturers provide information beyond that required by specifications and certify that their membranes are satisfactory for water analysis. They report retention, pore size, flow rate, sterility, pH, percent recovery, and limits for specific inorganic and organic chemical extractables. Although the standard membrane filter evaluation tests were developed for the manufacturers, a laboratory can conduct its own tests. To maintain quality control inspect each lot of membranes before use and during testing to insure they are round and pliable, with undistorted gridlines after autoclaving. After incubation, colonies should be well-developed with well-defined color and shape as defined by the test procedure. The gridline ink should not channel growth along the ink line nor restrict colony development. Colonies should be distributed evenly across the membrane surface. i. Culture media: Because cultural methods depend on properly prepared media, use the best available materials and techniques in media preparation, storage, and application. For control of quality, use commercially prepared media whenever available but note that such media may vary in quality among manufacturers and even from lot to lot from the same manufacturer. Order media in quantities to last no longer than 1 year. Use media on a first-in, first-out basis. When practical, order media in quarter pound (114 g) multiples rather than one pound (454 g) bottles, to keep the supply sealed as long as possible. Record kind, amount, and appearance of media received, lot number, expiration date, and dates received and opened. Check inventory quarterly for reordering. Store dehydrated media at an even temperature in a cool dry place, away from direct sunlight. Discard media that cake, discolor, or show other signs of deterioration. If expiration date is given by manufacturer, discard unused media after that date. A conservative time limit for unopened bottles is 2 years at room temperature. Compare recovery of newly purchased lots of media against proven lots, using recent pure-culture isolates and natural samples. Use opened bottles of media within 6 months. Dehydrated media are hygroscopic. Protect opened bottles from moisture. Close bottles as tightly as possible, immediately after use. If caking or discoloration of media occurs, discard media. Store opened bottles in a dessicator. 1) Preparation of media—Prepare media in containers that are at least twice the volume of the medium being prepared. Stir media, particularly agars, while heating. Avoid scorching or boil-over by using a boiling water bath for small batches of media and by continually attending to larger volumes heated on a hot plate or gas burner. Preferably use hot plate-magnetic stirrer © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater combinations. Label and date prepared media. Prepare media in reagent water. Measure water volumes and media with graduates or pipets conforming to NIST and APHA standards, respectively. Do not use blow-out pipets. After preparation and storage, remelt agar media in boiling water or flowing steam. Check and record pH of a portion of each medium after sterilization and cooling. Check pH of solid medium with a surface probe. Record results. Make minor adjustments in pH (<0.5 pH units) with 1N NaOH or HCl solution to the pH specified in formulation. If the pH difference is larger than 0.5 units, discard the batch and check preparation instructions and pH of reagent water to resolve the problem. Incorrect pH values may be due to reagent water quality, medium deterioration, or improper preparation. Review instructions for preparation and check water pH. If water pH is unsatisfactory, prepare a new batch of medium using water from a new source (see Section 9020B.3d and e). If water is satisfactory, remake medium and check; if pH is again incorrect, prepare medium from another bottle. Record pH problems in the media record book and inform the manufacturer if the medium is indicated as the source of error. Examine prepared media for unusual color, darkening, or precipitation and record observations. Consider variations of sterilization time and temperature as possible causes for problems. If any of the above occur, discard the medium. 2) Sterilization—Sterilize media at 121 to 124°C for the minimum time specified. A double-walled autoclave permits maintenance of full pressure and temperature in the jacket between loads and reduces chance for heat damage. Follow manufacturer’s directions for sterilization of specific media. The required exposure time varies with form and type of material, type of medium, presence of carbohydrates, and volume. Table 9020:III gives guidelines for typical items. Do not expose media containing carbohydrates to the elevated temperatures for more than 45 min. Exposure time is defined as the period from initial exposure to removal from the autoclave. Some currently available autoclave models are automatic and include features such as vertical sliding, self-sealing and opening doors, programmable sterilization cycles, and continuous multipoint monitoring of chamber temperature and pressure. These units also may incorporate solution cooling and vapor removal features. When sterilizer design includes heat exchangers and solution cooling features as part of a factory-programmed liquid cycle, strict adherence to the 45-min total elapsed time in the autoclave is not necessary provided that printout records verify normal cycle operation and chamber cooling during exhaust and vapor removal. Remove sterilized media from autoclave as soon as chamber pressure reaches zero, or, if a fully automatic model is used, as soon as the door opens. Do not reautoclave media. Check effectiveness of sterilization weekly by placing Bacillus stearothermophilus spore suspensions or strips (commercially available) inside glassware. Sterilize at 121°C for 15 min. Place in trypticase soy broth tubes and incubate at 55°C for 48 h. If growth of the autoclaved spores occurs after incubation at 55°C, sterilization was inadequate. A small, relatively inexpensive 55°C incubator is available commercially.†#(4) © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Sterilize heat-sensitive solutions or media by filtration through a 0.22-µm-pore-diam filter in a sterile filtration and receiving apparatus. Filter and dispense medium in a safety cabinet or biohazard hood if available. Sterilize glassware (pipets, petri dishes, sample bottles) in an autoclave or an oven at 170°C for 2 h. Sterilize equipment, supplies, and other solid or dry materials that are heat-sensitive, by exposing to ethylene oxide in a gas sterilizer. Use commercially available spore strips or suspensions to check dry heat and ethylene oxide sterilization. 3) Use of agars and broths—Temper melted agars in a water bath at 44 to 46°C until used but do not hold longer than 3 h. To monitor agar temperature, expose a bottle of water or medium to the same heating and cooling conditions as the agar. Insert a thermometer in the monitoring bottle to determine when the temperature is 45 to 46°C and suitable for use in pour plates. If possible, prepare media on the day of use. After pouring agar plates for streaking, dry agar surfaces by keeping dish slightly open for at least 15 min in a bacteriological hood to avoid contamination. Discard unused liquid agar; do not let harden or remelt for later use. Handle tubes of sterile fermentation media carefully to avoid entrapping air in inner tubes, thereby producing false positive reactions. Examine freshly prepared tubes to determine that gas bubbles are absent. 4) Storage of media—Prepare media in amounts that will be used within holding time limits given in Table 9020:IV. Protect media containing dyes from light; if color changes occur, discard the media. Refrigerate poured agar plates not used on the day of preparation. Seal agar plates with loose-fitting lids in plastic bags if held more than 2 d. Prepare broth media that will be stored for more than 2 weeks in screw-cap tubes, other tightly sealed tubes, or in loose-capped tubes placed in a sealed plastic bag or other tightly sealed container to prevent evaporation. Mark liquid level in several tubes and monitor for loss of liquid. If loss is 10% or more, discard the batch. If media are refrigerated, incubate overnight at test temperature before use and reject the batch if false positive responses occur. Prepared sterile broths and agars available from commercial sources may offer advantages when analyses are done intermittently, when staff is not available for preparation work, or when cost can be balanced against other factors of laboratory operation. Check performance of these media as described in ¶ 5 below. 5) Quality control of prepared media—Maintain in a bound book a complete record of each prepared batch of medium with name of preparer and date, name and lot number of medium, amount of medium weighed, volume of medium prepared, sterilization time and temperature, pH measurements and adjustments, and preparations of labile components. Compare quantitative recoveries of new lots with previously acceptable ones. Include sterility and positive and negative control culture checks on all media as described below. 5. Standard Operating Procedures (SOPs) SOPs are the operational backbone of an analytical laboratory. SOPs describe in detail all laboratory operations such as preparation of reagents, reagent water, standards, culture media, proper use of balances, sterilization practices, and dishwashing procedures, as well as methods of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater sampling, analysis, and quality control. The SOPs are unique to the laboratory. They describe the tasks as performed on a day-to-day basis, tailored to the laboratory’s own equipment, instrumentation, and sample types. The SOPs guide routine operations by each analyst, help to assure uniform operations, and provide a solid training tool. 6. Sampling a. Planning: Microbiologists should participate in the planning of monitoring programs that will include microbial analyses. They can provide valuable expertise on the selection of sampling sites, number of samples and analyses needed, workload, and equipment and supply needs. For natural waters, knowledge of the probable microbial densities, and the impact of season, weather, tide and wind patterns, known sources of pollution, and other variables, are needed to formulate the most effective sampling plan. b. Methods: Sampling plans must be specific for each sampling site. Prior sampling guidance can be only general in nature, addressing the factors that must be considered for each site. Sampling SOPs describe sampling equipment, techniques, frequency, holding times and conditions, safety rules, etc., that will be used under different conditions for different sites. From the information in these SOPs sampling plans will be drawn up. 7. Analytical Methods a. Method selection: Because minor variations in technique can cause significant changes in results, microbiological methods must be standardized so that uniform data result from multiple laboratories. Select analytical methods appropriate for the sample type from Standard Methods or other source of standardized methods and ensure that methods have been validated in a multi-laboratory study with the sample types of interest. b. Data objectives: Review available methods and determine which produce data to meet the program’s needs for precision, bias, specificity, selectivity, and detection limit. Ensure that the methods have been demonstrated to perform within the above specifications for the samples of interest. c. Internal QC: The written analytical methods should contain required QC checks of positive and negative control cultures, sterile blank, replicate analyses (precision), and a known quantitative culture, if available. d. Method SOPs: As part of the series of SOPs, provide each analyst with a copy of the analytical methods written in step-wise fashion exactly as they are to be performed and specific to the sample type, equipment, and instrumentation used in the laboratory. 8. Analytical Quality Control Procedures a. General quality control procedures: 1) New methods—Conduct parallel tests with the standard procedure and a new method to determine applicability and comparability. Perform at least 100 parallel tests across seasons of the year before replacement with the new method for routine use. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2) Comparison of plate counts—For routine performance evaluation, repeat counts on one or more positive samples at least monthly and compare the counts with those of other analysts testing the same samples. Replicate counts for the same analyst should agree within 5% and those between analysts should agree within 10%. See Section 9020B.10b for a statistical calculation of data precision. 3) Control cultures—For each lot of medium check analytical procedures by testing with known positive and negative control cultures for the organism(s) under test. See Table 9020:V for examples of test cultures. 4) Duplicate analyses—Perform duplicate analyses on 10% of samples and on at least one sample per test run. A test run is defined as an uninterrupted series of analyses. If the laboratory conducts less than 10 tests/week, make duplicate analyses on at least one sample each week. 5) Sterility checks—For membrane filter tests, check sterility of media, membrane filters, buffered dilution and rinse water, pipets, flasks and dishes, and equipment as a minimum at the end of each series of samples, using sterile reagent water as the sample. If contaminated, check for the source. For multiple-tube and presence-absence procedures, check sterility of media, dilution water, and glassware. To test sterility of media, incubate a representative portion of each batch at an appropriate temperature for 24 to 48 h and observe for growth. Check each batch of buffered dilution water for sterility by adding 20 mL water to 100 mL of a nonselective broth. Alternatively, aseptically pass 100 mL or more dilution water through a membrane filter and place filter on growth medium suitable for heterotrophic bacteria. Incubate at 35 ± 0.5°C for 24 h and observe for growth. If any contamination is indicated, determine the cause and reject analytical data from samples tested with these materials. Request immediate resampling and reanalyze. b. Precision of quantitative methods: Calculate precision of duplicate analyses for each different type of sample examined, for example, drinking water, ambient water, wastewater, etc., according to the following procedure: 1) Perform duplicate analyses on first 15 positive samples of each type, with each set of duplicates analyzed by a single analyst. If there is more than one analyst, include all analysts regularly running the tests, with each analyst performing approximately an equal number of tests. Record duplicate analyses as D1 and D2. 2) Calculate the logarithm of each result. If either of a set of duplicate results is <1, add 1 to both values before calculating the logarithms. 3) Calculate the range (R) for each pair of transformed duplicates as the mean (î ) of these ranges. See sample calculation in Table 9020:VI. 4) Thereafter, analyze 10% of routine samples in duplicate. Transform the duplicates as in ¶ 2) and calculate their range. If the range is greater than 3.27 R, there is greater than 99% probability that the laboratory variability is excessive. Determine if increased imprecision is acceptable; if not, discard all analytical results since the last precision check (see Table © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9020:VII). Identify and resolve the analytical problem before making further analyses. 5) Update the criterion used in ¶ 4) by periodically repeating the procedures of ¶s 1) through 3) using the most recent sets of 15 duplicate results. 9. Verification For the most part, the confirmation/verification procedures for drinking water differ from those for other waters because of specific regulatory requirements. a. Multiple-tube fermentation (MTF) methods: 1) Total coliform procedure (Section 9221B) a) Drinking water—Carry samples through confirmed phase only. Verification is not required. For QC purposes, if normally there are no positive results, analyze at least one positive source water quarterly to confirm that the media produce appropriate responses. For samples with a history of heavy growth without gas in presumptive-phase tubes, carry the tubes through the confirmed phase to check for false negative responses for coliform bacteria. Verify any positives for fecal coliforms or E. coli. b) Other water types—Verify by performing the completed MTF Test on 10% of samples positive through the confirmed phase. 2) Enzyme substrate coliform test (total coliform/E. coli) (Section 9223B) a) Drinking water—Verify at least 5% of total coliform positive results from enzyme substrate coliform tests by inoculating growth from a known positive sample and testing for lactose fermentation or for β-D-galactopyranosidase by the o-nitrophenyl-β-D-galactopyranoside (ONPG) test and indophenol by the cytochrome oxidase (CO) test. See Section 9225D for these tests. Coliforms are ONPG-positive and cytochrome-oxidase-negative. Verify E. coli using the EC MUG test (see Section 9221F). b) Other water types—Verify at least 10% of total coliform positive samples as in ¶ 2a above. 3) Fecal streptococci procedure—Verify as in 9230C.5. Growth of catalase-negative, gram-positive cocci on bile esculin agar at 35°C and in brain-heart infusion broth at 45°C verifies the organisms as fecal streptococci. Growth at 45°C and in 6.5% NaCl broth indicates the streptococci are members of the enterococcus group. 4) Include known positive and negative pure cultures as a QC check. b. Membrane filter methods: 1) Total coliform procedures a) Drinking water—Pick all, up to 5 typical and 5 atypical (nonsheen) colonies from positive samples on M-Endo medium and verify as in Section 9222B.5 f. Also verify any positives for fecal coliforms or E. coli. If there are no positive samples, test at least one known positive source water quarterly. b) Other water types—Verify positives monthly by picking at least 10 sheen colonies from a © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater positive water sample as in Section 9222B.5 f. Adjust counts based on percent verification. c) To determine false negatives, pick representative atypical colonies of different morphological types and verify as in Section 9222B.5 f. 2) Fecal coliform procedure a) Verify positives monthly by picking at least 10 blue colonies from one positive sample. Verify in lauryl tryptose broth and EC broth as in Section 9221B.3 and Section 9221E. Adjust counts based on percent verification. b) To determine false negatives, pick representative atypical colonies of different morphological types and verify as in Section 9221B.3 and Section 9221E. 3) Escherichia coli procedure a) Drinking water—Verify at least 5% of MUG-positive and MUG-negative results. Pick from well-isolated sheen colonies that fluoresce on nutrient agar with MUG (NA MUG), taking care not to pick up medium, which can cause a false positive response. Also verify nonsheen colonies that fluoresce. Verify by performing the citrate test and the indole test as described in Section 9225D, but incubate indole test at 44.5°C. E. coli are indole-positive and yield no growth on citrate. b) Other water types—Verify one positive sample monthly as in ¶ a) above. Adjust counts based on percentage of verification. 4) Fecal streptococci procedure—Pick to verify monthly at least 10 isolated esculin-positive red colonies from m-Enterococcus agar to brain heart infusion (BHI) media. Verify as described in Section 9230C. Adjust counts based on percentage of verification. 5) Enterococci procedures—Pick to verify monthly at least 10 well-isolated pink to red colonies with black or reddish-brown precipitate from EIA agar. Transfer to BHI media as described in Section 9230C. Adjust counts based on percentage of verification. 6) Include known positive and negative pure cultures as a quality control check. 9. Documentation and Recordkeeping a. QA plan: The QA program documents management’s commitment to a QA policy and sets forth the requirements needed to support program objectives. The program describes overall policies, organization, objectives, and functional responsibilities for achieving the quality goals. In addition, the program should develop a project plan that specifies the QC requirements for each project. The plan specifies the QC activities required to achieve the data representativeness, completeness, comparability, and compatibility. Also, the QA plan should include a program implementation plan that ensures maximum coordination and integration of QC activities within the overall program (sampling, analyses, and data handling). b. Sampling records: A written SOP for sample handling records sample collection, transfer, storage, analyses, and disposal. The record is most easily kept on a series of printed forms that prompt the user to provide all the necessary information. It is especially critical that this record be exact and complete if there is any chance that litigation may occur. Such record systems are © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater called chain of custody. Because laboratories do not always know whether analytical results will be used in future litigation, some maintain chain-of-custody on all samples. Details on chain of custody are available in Section 1060B and elsewhere.1 c. Recordkeeping: An acceptable recordkeeping system provides needed information on sample collection and preservation, analytical methods, raw data, calculations through reported results, and a record of persons responsible for sampling and analyses. Choose a format agreeable to both the laboratory and the customer (the data user). Ensure that all data sheets are signed and dated by the analyst and the supervisor. The preferable record form is a bound and page-numbered notebook, with entries in ink and a single line drawn through any change with the correction entered next to it. Keep records of microbiological analyses for at least 5 years. Actual laboratory reports may be kept, or data may be transferred to tabular summaries, provided that the following information is included: date, place, and time of sampling, name of sample collector; identification of sample; date of receipt of sample and analysis; person(s) responsible for performing analysis; analytical method used; the raw data and the calculated results of analysis. Verify that each result was entered correctly from the bench sheet and initialed by the analyst. If an information storage and retrieval system is used, double check data on the printouts. 10. Data Handling a. Distribution of bacterial populations: In most chemical analyses the distribution of analytical results follows the Gaussian curve, which has symmetrical distribution of values about the mean (see Section 1010B). Microbial distributions are not necessarily symmetrical. Bacterial counts often are characterized as having a skewed distribution because of many low values and a few high ones. These characteristics lead to an arithmetic mean that is considerably larger than the median. The frequency curve of this distribution has a long right tail, such as that shown in Figure 9020:1, and is said to display positive skewness. Application of the most rigorous statistical techniques requires the assumption of symmetrical distributions such as the normal curve. Therefore it usually is necessary to convert skewed data so that a symmetrical distribution resembling the normal distribution results. An approximately normal distribution can be obtained from positively skewed data by converting numbers to their logarithms, as shown in Table 9020:VIII. Comparison of the frequency tables for the original data (Table 9020:IX) and their logarithms (Table 9020:X) shows that the logarithms approximate a symmetrical distribution. b. Central tendency measures of skewed distribution: If the logarithms of numbers from a positively skewed distribution are approximately normally distributed, the original data have a log-normal distribution. The best estimate of central tendency of log-normal data is the geometric mean, defined as: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and that is, the geometric mean is equal to the antilog of the arithmetic mean of the logarithms. For example, the following means calculated from the data in Table 9020:VIII are drastically different. geometric mean and arithmetic mean Therefore, although regulations or tradition may require or cause microbiological data to be reported as the arithmetic mean or median, the preferred statistic for summarizing microbiological monitoring data is the geometric mean. An exception may be in the evaluation of data for risk assessment. The arithmetic mean may be a better measure for this purpose because it may generate a higher central tendency value and possibly provide a greater safety factor.8 c. ‘‘Less than’’ (<) values: There has always been uncertainty as to the proper way to include ‘‘less than’’ values in calculation and evaluation of microbiological data because such values cannot be treated statistically without modification. Proposed modifications involve © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater changing such numbers to zero, choosing values halfway between zero and the ‘‘less than’’ value, or assigning the ‘‘less than’’ value itself, i.e., changing <1 values to 1, 1/2, or 0. There are valid reasons for not including < values, whether modified or not. If the database is fairly large with just a few < values, the influence of these uncertain values will be minimal and of no benefit. If the database is small or has a relatively large number of < values, inclusion of modified < values would exert an undue influence on the final results and could result in an artificial negative or positive bias. Including < values is particularly inappropriate if the < values are <100, <1000, or higher because the unknown true values could be anywhere from 0 to 99, 0 to 999, etc. When < values are first noted, adjust or expand test volumes. The only exception to this caution would be regulatory testing with defined compliance limits, such as the <1/100 mL values reported for drinking water systems where the 100-mL volume is required. 11. References 1. BORDNER, R.H., J.A. WINTER & P.V. SCARPINO, eds. 1978. Microbiological Methods for Monitoring the Environment, Water and Wastes. EPA-600/8-78-017, Environmental Monitoring & Support Lab., U.S. Environmental Protection Agency, Cincinnati, Ohio. 2. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1995. Standard guide for good laboratory practices in laboratories engaged in sampling and analysis of water. D-3856-95, Annual Book of ASTM Standards, Vol. 11.01, American Soc. Testing & Materials, Philadelphia, Pa. 3. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1996. Standard practice for writing quality control specifications for standard test methods for water analysis. D-5847-96, Annual Book of ASTM Standards, Vol. 11.01, American Soc. Testing & Materials, West Conshohocken, Pa. 4. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1993. Annual Book of ASTM Standards, Vol. 14.02, General Methods and Instrumentation. E-319-86 (reapproved 1993), Standard Practice for Evaluation of Single-Pan Mechanical Balances, and E-898-88 (reapproved 1993), Standard Method of Testing Top-Loading, Direct-Reading Laboratory Scales and Balances. American Soc. Testing & Materials, Philadelphia, Pa. 5. SCHMITZ, S., C. GARBE, B. TEBBE & C. ORFANOS. 1994. Long wave ultraviolet radiation (UVA) and skin cancer. Hautarzt 45:517. 6. BRENNER, K. & C.C. RANKIN. 1990. New screening test to determine the acceptability of 0.45 µm membrane filters for analysis of water. Appl. Environ. Bacteriol. 56:54. 7. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1977. Annual Book of ASTM Standards. Part 31, Water. American Soc. Testing & Materials, Philadelphia, Pa. 8. HAAS, C.N. 1996. How to average microbial densities to characterize risk. Water Res. 30:1036. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9020 C. Interlaboratory Quality Control 1. Background Interlaboratory QC programs are a means of establishing an agreed-upon, common performance criteria system that will assure an acceptable level of data quality and comparability among laboratories with similar interests and/or needs. These systems may be volunteer, such as that for the cities in the Ohio River Valley Water Sanitation Commission (ORSANCO), or regulatory, such as the Federal Drinking Water Laboratory Certification Program (see below). Often, the term ‘‘accreditation’’ is used interchangeably with certification. Usually, interlaboratory quality control programs have three elements: uniform criteria for laboratory operations, external review of the program, and external proficiency testing. 2. Uniform Criteria Interlaboratory quality control programs begin as a volunteer or mandatory means of establishing uniform laboratory standards for a specific purpose. The participants may be from one organization or a group of organizations having common interests or falling under common regulations. Often one group or person may agree to draft the criteria. If under regulation, the regulating authority may set the criteria for compliance-monitoring analyses. Uniform sampling and analytical methods and quality control criteria for personnel, facilities, equipment, instrumentation, supplies, and data handling and reporting are proposed, discussed, reviewed, modified if necessary, and approved by the group for common use. Criteria identified as necessary for acceptable data quality should be mandatory. A formal document is prepared and provided to all participants. The QA/QC responsibilities of management, supervisors, and technical staff are described in 9020A. In large laboratories, a QA officer is assigned as a staff position but may be the supervisor or other senior person in smaller laboratories. After incorporation into laboratory operations and confirmation that the QA program has been adapted and is in routine use, the laboratory supervisor and the QA officer conduct an internal program review of all operations and records for acceptability, to identify possible problems and assist in their resolution. If this is done properly, there should be little concern that subsequent external reviews will find major problems. 3. External Program Review Once a laboratory has a QA program in place, management informs the organization and a qualified external QA person or team arranges an on-site visit to evaluate the QA program for acceptability and to work with the laboratory to solve any problems. An acceptable rating confirms that the laboratory’s QA program is operating properly and that the laboratory has the capability of generating valid defensible data. Such on-site evaluations are repeated and may be © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater announced or unannounced. 4. External Proficiency Testing Whenever practical, the external organization conducts formal performance evaluation studies among all participant laboratories. Challenge samples are prepared and sent as unknowns on a set schedule for analyses and reporting of results. The reported data are coded for confidentiality and evaluated according to an agreed-upon scheme. The results are summarized for all laboratories and individual laboratory reports are sent to participants. Results of such studies indicate the quality of routine analyses of each laboratory as compared to group performance. Also, results of the group as a whole characterize the performance that can be expected for the analytical methods tested. 5. Example Program In the Federal Drinking Water Laboratory Certification Program, public water supply laboratories must be certified according to minimal criteria and procedures and quality assurance described in the EPA manual on certification:1 criteria are established for laboratory operations and methodology; on-site inspections are required by the certifying state agency or its surrogate to verify minimal standards; annually, laboratories are required to perform acceptably on unknown samples in formal studies, as samples are available; the responsible authority follows up on problems identified in the on-site inspection or performance evaluation and requires corrections within a set period of time. Individual state programs may exceed the federal criteria. On-site inspections of laboratories in the present certification program show that primary causes for discrepancies in drinking water laboratories have been inadequate equipment, improperly prepared media, incorrect analytical procedures, and insufficiently trained personnel. 6. References 1. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1997. Manual for the Certification of Laboratories Analyzing Drinking Water, 4th ed. EPA-814B-92-002, U.S. Environmental Protection Agency, Cincinnati, Ohio. 9030 LABORATORY APPARATUS*#(5) 9030 A. Introduction This section contains specifications for microbiological laboratory equipment. For testing and maintenance procedures related to quality control, see Section 9020. 9030 B. Equipment Specifications © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Incubators Incubators must maintain a uniform and constant temperature at all times in all areas, that is, they must not vary more than ±0.5°C in the areas used. Obtain such accuracy by using a water-jacketed or anhydric-type incubator with thermostatically controlled low-temperature electric heating units properly insulated and located in or adjacent to the walls or floor of the chamber and preferably equipped with mechanical means of circulating air. Incubators equipped with high-temperature heating units are unsatisfactory, because such sources of heat, when improperly placed, frequently cause localized overheating and excessive drying of media, with consequent inhibition of bacterial growth. Incubators so heated may be operated satisfactorily by replacing high-temperature units with suitable wiring arranged to operate at a lower temperature and by installing mechanical air-circulation devices. It is desirable, where ordinary room temperatures vary excessively, to keep laboratory incubators in special rooms maintained at a few degrees below the recommended incubator temperature. Alternatively, use special incubating rooms well insulated and equipped with properly distributed heating units, forced air circulation, and air exchange ports, provided that they conform to desired temperature limits. When such rooms are used, record the daily temperature range in areas where plates or tubes are incubated. Provide incubators with open metal wire or perforated sheet shelves so spaced as to assure temperature uniformity throughout the chamber. Leave a 2.5-cm space between walls and stacks of dishes or baskets of tubes. Maintain an accurate thermometer, traceable to the National Institute of Standards and Technology (NIST), with the bulb immersed in liquid (glycerine, water, or mineral oil) on each shelf in use within the incubator and record daily temperature readings (preferably morning and afternoon). It is desirable, in addition, to maintain a maximum and minimum registering thermometer within the incubator on the middle shelf to record the gross temperature range over a 24-h period. At intervals, determine temperature variations within the incubator when filled to maximum capacity. Install a recording thermometer whenever possible, to maintain a continuous and permanent record of temperature. Ordinarily, a water bath with a gabled cover to reduce water and heat loss, or a solid heat sink incubator, is required to maintain a temperature of 44.5 ± 0.2°C. If satisfactory temperature control is not achieved, provide water recirculation. Keep water depth in the incubator sufficient to immerse tubes to upper level of media. 2. Hot-Air Sterilizing Ovens Use hot-air sterilizing ovens of sufficient size to prevent internal crowding; constructed to give uniform and adequate sterilizing temperatures of 170 ± 10°C; and equipped with suitable thermometers. Optionally use a temperature-recording instrument. 3. Autoclaves Use autoclaves of sufficient size to prevent internal crowding; constructed to provide uniform temperatures within the chambers (up to and including the sterilizing temperature of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 121°C); equipped with an accurate thermometer the bulb of which is located properly on the exhaust line so as to register minimum temperature within the sterilizing chambers (temperature-recording instrument is optional); equipped with pressure gauge and properly adjusted safety valves connected directly with saturated-steam supply lines equipped with appropriate filters to remove particulates and oil droplets or directly to a suitable special steam generator (do not use steam from a boiler treated with amines for corrosion control); and capable of reaching the desired temperature within 30 min. Confirm, by chemical or toxicity tests, that the steam supply has not been treated with amines or other corrosion-control chemicals that will impart toxicity. Use of a vertical autoclave or pressure cooker is not recommended because of difficulty in adjusting and maintaining sterilization temperature and the potential hazard. If a pressure cooker is used in emergency or special circumstances, equip it with an efficient pressure gauge and a thermometer the bulb of which is 2.5 cm above the water level. 4. Gas Sterilizers Use a sterilizer equipped with automatic controls capable of carrying out a complete sterilization cycle. As a sterilizing gas use ethylene oxide (CAUTION: Ethylene oxide is toxic—avoid inhalation, ingestion, and contact with the skin. Also, ethylene oxide forms an explosive mixture with air at 3-80% proportion.) diluted to 10 to 12% with an inert gas. Provide an automatic control cycle to evacuate sterilizing chamber to at least 0.06 kPa, to hold the vacuum for 30 min, to adjust humidity and temperature, to charge with the ethylene oxide mixture to a pressure dependent on mixture used, to hold such pressure for at least 4 h, to vent gas, to evacuate to 0.06 kPa, and finally, to bring to atmospheric pressure with sterile air. The humidity, temperature, pressure, and time of sterilizing cycle depend on the gas mixture used. Store overnight sample bottles with loosened caps that were sterilized by gas, to allow last traces of gas mixture to dissipate. Incubate overnight media sterilized by gas, to insure dissipation of gas. In general, mixtures of ethylene oxide with chlorinated hydrocarbons such as freon are harmful to plastics, although at temperatures below 55°C, gas pressure not over 35 kPa, and time of sterilization less than 6 h, the effect is minimal. If carbon dioxide is used as a diluent of ethylene oxide, increase exposure time and pressure, depending on temperature and humidity that can be used. Determine proper cycle and gas mixture for objects to be sterilized and confirm by sterility tests. 5. Optical Counting Equipment a. Pour and spread plates: Use Quebec-type colony counter, dark-field model preferred, or one providing equivalent magnification (1.5 diameters) and satisfactory visibility. b. Membrane filters: Use a binocular microscope with magnification of 10 to 15×. Provide daylight fluorescent light source at angle of 60 to 80° above the colonies; use low-angle lighting © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater for nonpigmented colonies. c. Mechanical tally. 6. pH Equipment Use electrometric pH meters, accurate to at least 0.1 pH units, for determining pH values of media. 7. Balances Use balances providing a sensitivity of at least 0.1 g at a load of 150 g, with appropriate weights. Use an analytical balance having a sensitivity of 1 mg under a load of 10 g for weighing small quantities (less than 2 g) of materials. Single-pan rapid-weigh balances are most convenient. 8. Media Preparation Utensils Use borosilicate glass or other suitable noncorrosive equipment such as stainless steel. Use glassware that is clean and free of residues, dried agar, or other foreign materials that may contaminate media. 9. Pipets and Graduated Cylinders Use pipets of any convenient size, provided that they deliver the required volume accurately and quickly. The error of calibration for a given manufacturer’s lot must not exceed 2.5%. Use pipets having graduations distinctly marked and with unbroken tips. Bacteriological transfer pipets or pipets conforming to APHA standards may be used. Do not pipet by mouth; use a pipet aid. Use graduated cylinders meeting ASTM Standards (D-86 and D-216) and with accuracy limits established by NIST where appropriate. 10. Pipet Containers Use boxes of aluminum or stainless steel, end measurement 5 to 7.5 cm, cylindrical or rectangular, and length about 40 cm. When these are not available, paper wrappings for individual pipets may be substituted. To avoid excessive charring during sterilization, use best-quality sulfate pulp (kraft) paper. Do not use copper or copper alloy cans or boxes as pipet containers. 11. Refrigerator Use a refrigerator maintaining a temperature of 1 to 4.4°C to store samples, media, reagents, etc. Do not store volatile solvents, food, or beverages in a refrigerator with media. Frost-free refrigerators may cause excessive media dehydration on storage longer than 1 week. 12. Temperature-Monitoring Devices Use glass or metal thermometers graduated to 0.5°C to monitor most incubators and refrigerators. Use thermometers graduated to 0.1°C for incubators operated above 40°C. Use © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater continuous recording devices that are equally sensitive. Verify accuracy by comparison with a NIST-certified thermometer, or equivalent. 13. Dilution Bottles or Tubes Use bottles or tubes of resistant glass, preferably borosilicate glass, closed with glass stoppers or screw caps equipped with liners that do not produce toxic or bacteriostatic compounds on sterilization. Do not use cotton plugs as closures. Mark graduation levels indelibly on side of dilution bottle or tube. Plastic bottles of nontoxic material and acceptable size may be substituted for glass provided that they can be sterilized properly. 14. Petri Dishes For the plate count, use glass or plastic petri dishes about 100 × 15 mm. Use dishes the bottoms of which are free from bubbles and scratches and flat so that the medium will be of uniform thickness throughout the plate. For the membrane filter technique use loose-lid glass or plastic dishes, 60 × 15 mm, or tight-lid dishes, 50 × 12 mm. Sterilize petri dishes and store in metal cans (aluminum or stainless steel, but not copper), or wrap in paper—preferably best-quality sulfate pulp (kraft)—before sterilizing. Presterilized petri dishes are commercially available. 15. Membrane Filtration Equipment Use filter funnel and membrane holder made of seamless stainless steel, glass, or autoclavable plastic that does not leak and is not subject to corrosion. Field laboratory kits are acceptable but standard laboratory filtration equipment and procedures are required. 16. Fermentation Tubes and Vials Use fermentation tubes of any type, if their design permits conforming to medium and volume requirements for concentration of nutritive ingredients as described subsequently. Where tubes are used for a test of gas production, enclose a shell vial, inverted. Use tube and vial of such size that the vial will be filled completely with medium, at least partly submerged in the tube, and large enough to make gas bubbles easily visible. 17. Inoculating Equipment Use wire loops made of 22- or 24-gauge nickel alloy*#(6) or platinum-iridium for flame sterilization. Use loops at least 3 mm in diameter. Sterilize by dry heat or steam. Single-service hardwood or plastic applicators also may be used. Make these 0.2 to 0.3 cm in diameter and at least 2.5 cm longer than the fermentation tube; sterilize by dry heat and store in glass or other nontoxic containers. 18. Sample Bottles For bacteriological samples, use sterilizable bottles of glass or plastic of any suitable size and shape. Use bottles capable of holding a sufficient volume of sample for all required tests and an adequate air space, permitting proper washing, and maintaining samples uncontaminated until © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater examinations are completed. Ground-glass-stoppered bottles, preferably wide-mouthed and of resistant glass, are recommended. Plastic bottles of suitable size, wide-mouthed, and made of nontoxic materials such as polypropylene that can be sterilized repeatedly are satisfactory as sample containers. Presterilized plastic bags, with or without dechlorinating agent, are available commercially and may be used. Plastic containers eliminate the possibility of breakage during shipment and reduce shipping weight. Metal or plastic screw-cap closures with liners may be used on sample bottles provided that no toxic compounds are produced on sterilization. Before sterilization, cover tops and necks of sample bottles having glass closures with aluminium foil or heavy kraft paper. 19. Bibliography COLLINS, W.D. & H.B. RIFFENBURG. 1923. Contamination of water samples with material dissolved from glass containers. Ind. Eng. Chem. 15:48. CLARK, W.M. 1928. The Determination of Hydrogen Ion Concentration, 3rd ed. Williams & Wilkins, Baltimore, Md. ARCHAMBAULT, J., J. CUROT & M.H. MCCRADY. 1937. The need of uniformity of conditions for counting plates (with suggestions for a standard colony counter). Amer. J. Pub. Health 27:809. BARKWORTH, H. & J.O. IRWIN. 1941. The effect of the shape of the container and size of gas tube in the presumptive coliform test. J. Hyg. 41:180. RICHARDS, O.W. & P.C. HEIJN. 1945. An improved dark-field Quebec colony counter. J. Milk Technol. 8:253. COHEN, B. 1957. The measurement of pH, titratable acidity, and oxidation-reduction potentials. In Manual of Microbiological Methods. Society of American Bacteriologists. McGraw-Hill Book Co., New York, N.Y. MORTON, H.E. 1957. Stainless-steel closures for replacement of cotton plugs in culture tubes. Science. 126:1248. MCGUIRE, O.E. 1964. Wood applicators for the confirmatory test in the bacteriological analysis of water. Pub. Health Rep. 79:812. BORDNER, R.H., J.A. WINTER & P.V. SCARPINO, eds. 1978. Microbiological Methods for Monitoring the Environment, Water and Wastes. EPA-600/8-78-017, Environmental Monitoring & Support Lab., U.S. Environmental Protection Agency, Cincinnati, Ohio. AMERICAN PUBLIC HEALTH ASSOCIATION. 1993. Standard Methods for the Examination of Dairy Products, 16th ed. American Public Health Assoc., Washington, D.C. 9040 WASHING AND STERILIZATION*#(7) Cleanse all glassware thoroughly with a suitable detergent and hot water, rinse with hot © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater water to remove all traces of residual washing compound, and finally rinse with laboratory-pure water. If mechanical glassware washers are used, equip them with influent plumbing of stainless steel or other nontoxic material. Do not use copper piping to distribute water. Use stainless steel or other nontoxic material for the rinse water system. Sterilize glassware, except when in metal containers, for not less than 60 min at a temperature of 170°C, unless it is known from recording thermometers that oven temperatures are uniform, under which exceptional condition use 160°C. Heat glassware in metal containers to 170°C for not less than 2 h. Sterilize sample bottles not made of plastic as above or in an autoclave at 121°C for 15 min. For plastic bottles loosen caps before autoclaving to prevent distortion. 9050 PREPARATION OF CULTURE MEDIA*#(8) 9050 A. General Procedures 1. Storage of Culture Media Store dehydrated media (powders) in tightly closed bottles in the dark at less than 30°C in an atmosphere of low humidity. Do not use them if they discolor or become caked and lose the character of a free-flowing powder. Purchase dehydrated media in small quantities that will be used within 6 months after opening. Additionally, use stocks of dehydrated media containing selective agents such as sodium azide, bile salts or derivatives, antibiotics, sulfur-containing amino acids, etc., of relatively current lot number (within a year of purchase) so as to maintain optimum selectivity. See also Section 9020. Prepare culture media in batches that will be used in less than 1 week. However, if the media are contained in screw-capped tubes they may be stored for up to 3 months. See Table 9020:IV for specific details. Store media out of direct sun and avoid contamination and excessive evaporation. Liquid media in fermentation tubes, if stored at refrigeration or even moderately low temperatures, may dissolve sufficient air to produce, upon incubation at 35°C, a bubble of air in the tube. Incubate fermentation tubes that have been stored at a low temperature overnight before use and discard tubes containing air. Fermentation tubes may be stored at approximately 25°C; but because evaporation may proceed rapidly under these conditions—resulting in marked changes in concentration of the ingredients—do not store at this temperature for more than 1 week. Discard tubes with an evaporation loss exceeding 1 mL. 2. Adjustment of Reaction State reaction of culture media in terms of hydrogen ion concentration, expressed as pH. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater The decrease in pH during sterilization will vary slightly with the individual sterilizer in use, and the initial reaction required to obtain the correct final reaction will have to be determined. The decrease in pH usually will be 0.1 to 0.2 but occasionally may be as great as 0.3 in double-strength media. When buffering salts such as phosphates are present in the media, the decrease in pH value will be negligible. Make tests to control adjustment to required pH with a pH meter. Measure pH of prepared medium as directed in Section 4500-H+. Titrate a known volume of medium with a solution of NaOH to the desired pH. Calculate amount of NaOH solution that must be added to the bulk medium to reach this reaction. After adding and mixing thoroughly, check reaction and adjust if necessary. The required final pH is given in the directions for preparing each medium. If a specific pH is not prescribed, adjustment is unnecessary. The pH of reconstituted dehydrated media seldom will require adjustment if made according to directions. Such factors as errors in weighing dehydrated medium or overheating reconstituted medium may produce an unacceptable final pH. Measure pH, especially of rehydrated selective media, regularly to insure quality control and media specifications. 3. Sterilization After rehydrating a medium, dispense promptly to culture vessels and sterilize within 2 h. Do not store nonsterile media. Sterilize all media, except sugar broths or broths with other specifications, in an autoclave at 121°C for 15 min after the temperature has reached 121°C. When the pressure reaches zero, remove medium from autoclave and cool quickly to avoid decomposition of sugars by prolonged exposure to heat. To permit uniform heating and rapid cooling, pack materials loosely and in small containers. Sterilize sugar broths at 121°C for 12 to 15 min. The maximum elapsed time for exposure of sugar broths to any heat (from time of closing loaded autoclave to unloading) is 45 min. Preferably use a double-walled autoclave to permit preheating before loading to reduce total needed heating time to within the 45-min limit. Presterilized media may be available commercially. 4. Bibliography BUNKER, G.C. & H. SCHUBER. 1922. The reaction of culture media. J. Amer. Water Works Assoc. 9:63. RICHARDSON, G.H., ed. 1985. Standard Methods for the Examination of Dairy Products, 15th ed. American Public Health Assoc., Washington, D.C. BALOWS, A., W.J. HAUSLER, JR., K.L. HERRMANN, H.D. ISENBERG & H.J. SHADOMY, eds. 1991. Manual of Clinical Microbiology, 5th ed. American Soc. Microbiology, Washington, D.C. 9050 B. Water 1. Specifications © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater To prepare culture media and reagents, use only distilled or demineralized reagent-grade water that has been tested and found free from traces of dissolved metals and bactericidal or inhibitory compounds. Toxicity in distilled water may be derived from fluoridated water high in silica. Other sources of toxicity are silver, lead, and various unidentified organic complexes. Where condensate return is used as feed for a still, toxic amines or other boiler compounds may be present in distilled water. Residual chlorine or chloramines also may be found in distilled water prepared from chlorinated water supplies. If chlorine compounds are found in distilled water, neutralize them by adding an equivalent amount of sodium thiosulfate or sodium sulfite. Distilled water also should be free of contaminating nutrients. Such contamination may be derived from flashover of organics during distillation, continued use of exhausted carbon filter beds, deionizing columns in need of recharging, solder flux residues in new piping, dust and chemical fumes, and storage of water in unclean bottles. Store distilled water out of direct sunlight to prevent growth of algae and turn supplies over as rapidly as possible. Aged distilled water may contain toxic volatile organic compounds absorbed from the atmosphere if stored for prolonged periods in unsealed containers. Good housekeeping practices usually will eliminate nutrient contamination. See Section 9020. 2. Bibliography STRAKA, R.P. & J.L. STOKES. 1957. Rapid destruction of bacteria in commonly used diluents and its elimination. Appl. Microbiol. 5:21. GELDREICH, E.E. & H.F. CLARK. 1965. Distilled water suitability for microbiological applications. J. Milk Food Technol. 28:351. MACLEOD, R.A., S.C. KUO & R. GELINAS. 1967. Metabolic injury to bacteria. II. Metabolic injury induced by distilled water or Cu++ in the plating diluent. J. Bacteriol. 93:961. 9050 C. Media Specifications The need for uniformity dictates the use of dehydrated media. Never prepare media from basic ingredients when suitable dehydrated media are available. Follow manufacturer’s directions for rehydration and sterilization. Commercially prepared media in liquid form (sterile ampule or other) also may be used if known to give equivalent results. See Section 9020 for quality-control specifications. The terms used for protein source in most media, for example, peptone, tryptone, tryptose, were coined by the developers of the media and may reflect commercial products rather than clearly defined entities. It is not intended to preclude the use of alternative materials provided that they produce equivalent results. NOTE—The term ‘‘percent solution’’ as used in these directions is to be understood to mean ‘‘grams of solute per 100 mL solution.’’ © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Dilution Water a. Buffered water: To prepare stock phosphate buffer solution, dissolve 34.0 g potassium dihydrogen phosphate (KH2PO4), in 500 mL reagent-grade water, adjust to pH 7.2 ± 0.5 with 1N sodium hydroxide (NaOH), and dilute to 1 L with reagent-grade water. Add 1.25 mL stock phosphate buffer solution and 5.0 mL magnesium chloride solution (81.1 g MgCl2⋅6H2O/L reagent-grade water) to 1 L reagent-grade water. Dispense in amounts that will provide 99 ± 2.0 mL or 9 ± 0.2 mL after autoclaving for 15 min. b. Peptone water: Prepare a 10% solution of peptone in distilled water. Dilute a measured volume to provide a final 0.1% solution. Final pH should be 6.8. Dispense in amounts to provide 99 ± 2.0 mL or 9 ± 0.2 mL after autoclaving for 15 min. Do not suspend bacteria in any dilution water for more than 30 min at room temperature because death or multiplication may occur. 2. Culture Media Specifications for individual media are included in subsequent sections. Details are provided where use of a medium first is described. 9060 9060 A. SAMPLES*#(9) Collection 1. Containers Collect samples for microbiological examination in nonreactive borosilicate glass or plastic bottles that have been cleansed and rinsed carefully, given a final rinse with deionized or distilled water, and sterilized as directed in Section 9030 and Section 9040. For some applications samples may be collected in presterilized plastic bags. 2. Dechlorination Add a reducing agent to containers intended for the collection of water having residual chlorine or other halogen unless they contain broth for direct planting of sample. Sodium thiosulfate (Na2S2O3) is a satisfactory dechlorinating agent that neutralizes any residual halogen and prevents continuation of bactericidal action during sample transit. The examination then will indicate more accurately the true microbial content of the water at the time of sampling. For sampling chlorinated wastewater effluents add sufficient Na2S2O3 to a clean sterile sample bottle to give a concentration of 100 mg/L in the sample. In a 120-mL bottle 0.1 mL of a 10% solution of Na2S2O3 will neutralize a sample containing about 15 mg/L residual chlorine. For drinking water samples, the concentration of dechlorinating agent may be reduced: 0.1 mL © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater of a 3% solution of Na2S2O3 in a 120-mL bottle will neutralize up to 5 mg/L residual chlorine. Cap bottle and sterilize by either dry or moist heat, as directed (Section 9040). Presterilized plastic bags or bottles containing Na2S2O3 are available commercially. Collect water samples high in metals, including copper or zinc (>1.0 mg/L), and wastewater samples high in heavy metals in sample bottles containing a chelating agent that will reduce metal toxicity. This is particularly significant when such samples are in transit for 4 h or more. Use 372 mg/L of the disodium salt of ethylenediaminetetraacetic acid (EDTA). Adjust EDTA solution to pH 6.5 before use. Add EDTA separately to sample bottle before bottle sterilization (0.3 mL 15% solution in a 120-mL bottle) or combine it with the Na2S2O3 solution before addition. 3. Sampling Procedures When the sample is collected, leave ample air space in the bottle (at least 2.5 cm) to facilitate mixing by shaking, before examination. Collect samples that are representative of the water being tested, flush or disinfect sample ports, and use aseptic techniques to avoid sample contamination. Keep sampling bottle closed until it is to be filled. Remove stopper and cap as a unit; do not contaminate inner surface of stopper or cap and neck of bottle. Fill container without rinsing, replace stopper or cap immediately, and if used, secure hood around neck of bottle. a. Potable water: If the water sample is to be taken from a distribution-system tap without attachments, select a tap that is supplying water from a service pipe directly connected with the main, and is not, for example, served from a cistern or storage tank. Open tap fully and let water run to waste for 2 or 3 min, or for a time sufficient to permit clearing the service line. Reduce water flow to permit filling bottle without splashing. If tap cleanliness is questionable, choose another tap. If a questionable tap is required for special sampling purposes, disinfect the faucet (inside and outside) by applying a solution of sodium hypochlorite (100 mg NaOCl/L) to faucet before sampling; let water run for additional 2 to 3 min after treatment. Do not sample from leaking taps that allow water to flow over the outside of the tap. In sampling from a mixing faucet remove faucet attachments such as screen or splash guard, run hot water for 2 min, then cold water for 2 to 3 min, and collect sample as indicated above. If the sample is to be taken from a well fitted with a hand pump, pump water to waste for about 5 to 10 min or until water temperature has stabilized before collecting sample. If an outdoor sampling location must be used, avoid collecting samples from frost-proof hydrants. If there is no pumping machinery, collect a sample directly from the well by means of a sterilized bottle fitted with a weight at the base; take care to avoid contaminating samples by any surface scum. Other sterile sampling devices, such as a trip bailer, also may be used. In drinking water evaluation, collect samples of finished water from distribution sites selected to assure systematic coverage during each month. Carefully choose distribution system sample locations to include dead-end sections to demonstrate bacteriological quality throughout the network and to ensure that localized contamination does not occur through © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater cross-connections, breaks in the distribution lines, or reduction in positive pressure. Sample locations may be public sites (police and fire stations, government office buildings, schools, bus and train stations, airports, community parks), commercial establishments (restaurants, gas stations, office buildings, industrial plants), private residences (single residences, apartment buildings, and townhouse complexes), and special sampling stations built into the distribution network. Preferably avoid outdoor taps, fire hydrants, water treatment units, and backflow prevention devices. Establish sampling program in consultation with state and local health authorities. b. Raw water supply: In collecting samples directly from a river, stream, lake, reservoir, spring, or shallow well, obtain samples representative of the water that is the source of supply to consumers. It is undesirable to take samples too near the bank or too far from the point of drawoff, or at a depth above or below the point of drawoff. c. Surface waters: Stream studies may be short-term, high-intensity efforts. Select bacteriological sampling locations to include a baseline location upstream from the study area, industrial and municipal waste outfalls into the main stream study area, tributaries except those with a flow less than 10% of the main stream, intake points for municipal or industrial water facilities, downstream samples based on stream flow time, and downstream recreational areas. Dispersion of wastewaters into the receiving stream may necessitate preliminary cross-section studies to determine completeness of mixing. Where a tributary stream is involved, select the sampling point near the confluence with the main stream. Samples may be collected from a boat or from bridges near critical study points. Choose sampling frequency to be reflective of changing stream or water body conditions. For example, to evaluate waste discharges, sample every 4 to 6 h and advance the time over a 7- to 10-d period. To monitor stream and lake water quality establish sampling locations at critical sites. Sampling frequency may be seasonal for recreational waters, daily for water supply intakes, hourly where waste treatment control is erratic and effluents are discharged into shellfish harvesting areas, or even continuous. d. Bathing beaches: Sampling locations for recreational areas should reflect water quality within the entire recreational zone. Include sites from upstream peripheral areas and locations adjacent to drains or natural contours that would discharge stormwater collections or septic wastes. Collect samples in the swimming area from a uniform depth of approximately 1 m. Consider sediment sampling of the water-beach (soil) interface because of exposure of young children at the water’s edge. To obtain baseline data on marine and estuarine bathing water quality include sampling at low, high, and ebb tides. Relate sampling frequency directly to the peak bathing period, which generally occurs in the afternoon. Preferably, collect daily samples during the recognized bathing season; as a minimum include Friday, Saturday, Sunday, and holidays. When limiting sampling to days of peak recreational use, preferably collect a sample in the morning and the afternoon. Correlate bacteriological data with turbidity levels or rainfall over the watershed to make rapid assessment © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater of water quality changes. e. Sediments and biosolids: The bacteriology of bottom sediments is important in water supply reservoirs, in lakes, rivers, and coastal waters used for recreational purposes, and in shellfish-growing waters. Sediments may provide a stable index of the general quality of the overlying water, particularly where there is great variability in its bacteriological quality. Sampling frequency in reservoirs and lakes may be related more to seasonal changes in water temperatures and stormwater runoff. Bottom sediment changes in river and estuarine waters may be more erratic, being influenced by stormwater runoff, increased flow velocities, and sudden changes in the quality of effluent discharges. Microbiological examination of biosolids from water and wastewater treatment processes is desirable to determine the impact of their disposal into receiving waters, ocean dumping, land application, or burial in landfill operations. Collect and handle biosolids with less than 7% total solids using the procedures discussed for other water samples. Biosolids with more than 7% solids and exhibiting a ‘‘plastic’’ consistency or ‘‘semisolid’’ state typical of thickened sludges require a finite shear stress to cause them to flow. This resistance to flow results in heterogeneous distribution of biosolids in tanks and lagoons. Use cross-section sampling of accumulated biosolids to determine distribution of organisms within these impoundments. Establish a length-width grid across the top of the impoundment, and sample at intercepts. A thief sampler that samples only the solids layer may be useful. Alternatively use weighted bottle samplers that can be opened up at a desired depth to collect samples at specific locations. Processed biosolids having no free liquids are best sampled when they are being transferred. Collect grab samples across the entire width of the conveyor and combine into a composite sample. If solids are stored in piles, classification occurs. Exteriors of uncovered piles are subject to various environmental stresses such as precipitation, wind, fugitive dusts, and fecal contamination from scavengers. Consequently, surface samples may not reflect the microbiological quality of the pile. Therefore, use cross-section sampling of these piles to determine the degree of heterogeneity within the pile. Establish a length-width grid across the top of the pile, and sample intercepts. Sample augers and corers may prove to be ineffective for sampling piles of variable composition. In such cases use hand shovels to remove overburden. f. Nonpotable samples (manual sampling): Take samples from a river, stream, lake, or reservoir by holding the bottle near its base in the hand and plunging it, neck downward, below the surface. Turn bottle until neck points slightly upward and mouth is directed toward the current. If there is no current, as in the case of a reservoir, create a current artificially by pushing bottle forward horizontally in a direction away from the hand. When sampling from a boat, obtain samples from upstream side of boat. If it is not possible to collect samples from these situations in this way, attach a weight to base of bottle and lower it into the water. In any case, take care to avoid contact with bank or stream bed; otherwise, water fouling may occur. g. Sampling apparatus: Special apparatus that permits mechanical removal of bottle stopper below water surface is required to collect samples from depths of a lake or reservoir. Various © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater types of deep sampling devices are available. The most common is the ZoBell J-Z sampler,1 which uses a sterile 350-mL bottle and a rubber stopper through which a piece of glass tubing has been passed. This tubing is connected to another piece of glass tubing by a rubber connecting hose. The unit is mounted on a metal frame containing a cable and a messenger. When the messenger is released, it strikes the glass tubing at a point that has been slightly weakened by a file mark. The glass tube is broken by the messenger and the tension set up by the rubber connecting hose is released and the tubing swings to the side. Water is sucked into the bottle as a consequence of the partial vacuum created by sealing the unit at time of autoclaving. Commercial adaptations of this sampler and others are available. Bottom sediment sampling also requires special apparatus. The sampler described by Van Donsel and Geldreich2 has been found effective for a variety of bottom materials for remote (deep water) or hand (shallow water) sampling. Construct this sampler preferably of stainless steel and fit with a sterile plastic bag. A nylon cord closes the bag after the sampler penetrates the sediment. A slide bar keeps the bag closed during descent and is opened, thereby opening the bag, during sediment sampling. For sampling wastewaters or effluents the techniques described above generally are adequate; in addition see Section 1060. 4. Size of Sample The volume of sample should be sufficient to carry out all tests required, preferably not less than 100 mL. 5. Identifying Data Accompany samples by complete and accurate identifying and descriptive data. Do not accept for examination inadequately identified samples. 6. References 1. ZOBELL, C.E. 1941. Apparatus for collecting water samples from different depths for bacteriological analysis. J. Mar. Res. 4:173. 2. VAN DONSEL, D.J. & E.E. GELDREICH. 1971. Relationships of Salmonellae to fecal coliforms in bottom sediments. Water Res. 5:1079. 7. Bibliography PUBLIC HEALTH LABORATORY SERVICE WATER SUB-COMMITTEE. 1953. The effect of sodium thiosulphate on the coliform and Bacterium coli counts of non-chlorinated water samples. J. Hyg. 51:572. SHIPE, E.L. & A. FIELDS. 1956. Chelation as a method for maintaining the coliform index in water samples. Pub. Health Rep. 71:974. HOATHER, R.C. 1961. The bacteriological examination of water. J. Inst. Water Eng. 61:426. COLES, H.G. 1964. Ethylenediamine tetra-acetic acid and sodium thiosulphate as protective © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater agents for coliform organisms in water samples stored for one day at atmospheric temperature. Proc. Soc. Water Treat. Exam. 13:350. DAHLING, D.R. & B.A. WRIGHT. 1984. Processing and transport of environmental virus samples. Appl. Environ. Microbiol. 47:1272. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1992. Environmental Regulations and Technology Control of Pathogens and Vector Attraction in Sewage Sludge. EPA-625/R-92-013. Washington, D.C. 9060 B. Preservation and Storage 1. Holding Time and Temperature a. General: Start microbiological analysis of water samples as soon as possible after collection to avoid unpredictable changes in the microbial population. For most accurate results, ice samples during transport to the laboratory, if they cannot be processed within 1 h after collection. If the results may be used in legal action, employ special means (rapid transport, express mail, courier service, etc.) to deliver the samples to the laboratory within the specified time limits and maintain chain of custody. Follow the guidelines and requirements given below for specific water types. b. Drinking water for compliance purposes: Preferably hold samples at <10°C during transit to the laboratory. Analyze samples on day of receipt whenever possible and refrigerate overnight if arrival is too late for processing on same day. Do not exceed 30 h holding time from collection to analysis for coliform bacteria. Do not exceed 8 h holding time for heterotrophic plate counts. c. Nonpotable water for compliance purposes: Hold source water, stream pollution, recreational water, and wastewater samples below 10°C during a maximum transport time of 6 h. Refrigerate these samples upon receipt in the laboratory and process within 2 h. When transport conditions necessitate delays in delivery of samples longer than 6 h, consider using either field laboratory facilities located at the site of collection or delayed incubation procedures. d. Other water types for noncompliance purposes: Hold samples below 10°C during transport and until time of analysis. Do not exceed 24 h holding time. 2. Bibliography CALDWELL, E.L. & L.W. PARR. 1933. Present status of handling water samples—Comparison of bacteriological analyses under varying temperatures and holding conditions, with special reference to the direct method. Amer. J. Pub. Health 23:467. COX, K.E. & F.B. CLAIBORNE. 1949. Effect of age and storage temperature on bacteriological water samples. J. Amer. Water Works Assoc. 41: 948. PUBLIC HEALTH LABORATORY SERVICE WATER SUB-COMMITTEE. 1952. The effect of storage on the coliform and Bacterium coli counts of water samples. Overnight storage at room and refrigerator temperatures. J. Hyg. 50:107. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater PUBLIC HEALTH LABORATORY SERVICE WATER SUB-COMMITTEE. 1953. The effect of storage on the coliform and Bacterium coli counts of water samples. Storage for six hours at room and refrigerator temperatures. J. Hyg. 51:559. MCCARTHY, J.A. 1957. Storage of water sample for bacteriological examinations. Amer. J. Pub. Health 47:971. LONSANE, B.K., N.M. PARHAD & N.U. RAO. 1967. Effect of storage temperature and time on the coliform in water samples. Water Res. 1: 309. LUCKING, H.E. 1967. Death rate of coliform bacteria in stored Montana water samples. J. Environ. Health 29:576. MCDANIELS, A.E. & R.H. BORDNER. 1983. Effect of holding time and temperature on coliform numbers in drinking water. J. Amer. Water Works Assoc. 75:458. MCDANIELS, A.E. et al. 1985. Holding effects on coliform enumeration in drinking water samples. Appl. Environ. Microbiol. 50:755. 9211 RAPID DETECTION METHODS*#(10) 9211 A. Introduction There is a generally recognized need for methods that permit rapid estimation of the bacteriological quality of water. Applications of rapid methods may range from analysis of wastewater to potable water quality assessment. In the latter case, during emergencies involving water treatment plant failure, line breaks in a distribution network, or other disruptions to water supply caused by disasters, there is urgent need for rapid assessment of the sanitary quality of water. Ideally, rapid procedures would be reliable and have sensitivity levels equal to those of the standard tests routinely used. However, sensitivity of a rapid test may be compromised because the bacterial limit sought may be below the minimum bacterial concentration essential to rapid detection. Rapid tests fall into two categories, those involving modified conventional procedures and those requiring special instrumentation and materials. 9211 B. Seven-Hour Fecal Coliform Test (SPECIALIZED) This method1,2 is similar to the fecal coliform membrane filter procedure (see Section 9222D) but uses a different medium and incubation temperature to yield results in 7 h that generally are comparable to those obtained by the standard fecal coliform method. 1. Medium M-7 h FC agar: This medium may not be available in dehydrated form and may require preparation from the basic ingredients. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Proteose peptone No. 3 or polypeptone Yeast extract Lactose d-Mannitol Sodium chloride, NaCl Sodium lauryl sulfate Sodium desoxycholate Bromcresol purple Phenol red Agar Reagent-grade water 5.0 3.0 10.0 5.0 7.5 0.2 0.1 0.35 0.3 15.0 1 g g g g g g g g g g L Heat in boiling water bath. After ingredients are dissolved heat additional 5 min. Cool to 55 to 60°C and adjust pH to 7.3 ± 0.1 with 0.1N NaOH (0.35 mL/L usually required). Cool to about 45°C and dispense in 4- to 5-mL quantities to petri plates with tight-fitting covers. Store at 2 to 10°C. Discard after 30 d. 2. Procedure Filter an appropriate sample volume through a membrane filter, place filter on the surface of a plate containing M-7 h FC agar medium, and incubate at 41.5°C for 7 h. Fecal coliform colonies are yellow (indicative of lactose fermentation). 3. References 1. VAN DONSEL, D.J., R.M. TWEDT & E.E. GELDREICH. 1969. Optimum temperature for quantitation of fecal coliforms in seven hours on the membrane filter. Bacteriol. Proc. Abs. No. G46, p. 25. 2. REASONER, D.J., J.C. BLANNON & E.E. GELDREICH. 1979. Rapid seven hour fecal coliform test. Appl. Environ. Microbiol. 38:229. 9211 C. Special Techniques (SPECIALIZED) Special rapid techniques are summarized in Table 9211:I. Most are not sensitive enough for potable water quality measurement or are not specific. They may be useful in monitoring wastewater effluents and natural waters but require reagents not generally available, are tedious, or require special handling or incubation schemes incompatible with most water laboratory © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater schedules. Except for the colorimetric test, none are suitable for routine use but they may be used as research tools. The user should refer to the literature citations for the technique listed in the table for procedural details, conditions for use, and method limitations. Only the adenosine triphosphate (ATP) procedure (the firefly bioluminescence system), the colorimetric test to estimate total microbial density, and a radiometric fecal coliform procedure that uses a 14C-labeled substrate can be recommended. Correlate initial concentration of bacteria with ATP concentration by extracting ATP from serial dilutions of a bacterial suspension, or for the 14C radiometric method, standardize by determining the 14CO2 released by known concentrations of fecal coliform organisms in natural samples, not pure cultures. In using any rapid procedure, determine the initial bacterial density by using an appropriate procedure such as heterotrophic plate count (Section 9215) or total (Section 9221) or fecal (Section 9222) coliforms, and correlate with results from the special rapid technique. 1. Bioluminescence Test (Total Viable Microbial Measurement) The firefly luciferase test for ATP in living cells is based on the reaction between the luciferase enzyme, luciferin (enzyme substrate), magnesium ions, and ATP. Light is emitted during the reaction and can be measured quantitatively and correlated with the quantity of ATP extracted from known numbers of bacteria. When all reactants except ATP are in excess, ATP is the limiting factor. Addition of ATP drives the reactions, producing a pulse of light that is proportional to the ATP concentration. The assay is completed in less than 1 h.1-3 For monitoring microbial populations in water, the ATP assay is limited primarily by the need to concentrate bacteria from the sample to achieve the minimum ATP sensitivity level, which is 105 cells/mL. When combined with membrane filtration of a 1-L sample, ATP assay can provide the sensitivity level needed. 2. Radiometric Detection (Fecal Coliforms) In this test, 14CO2 is released from a 14C-labeled substrate.14 The technique permits presumptive detection of as few as 2 to 20 fecal coliform bacteria in 4.5 h. The test uses M-FC broth, uniformly labeled 14C-mannitol, and two-temperature incubation; 2 h at 35°C followed by 2.5 h at 44.5°C for fecal coliform specificity. Add labeled substrate at start of 44.5°C incubation. Use membrane filtration to concentrate organisms from sample and place membrane filter in M-FC broth in a sealable container. The 14CO2 released is trapped by exposure to Ba(OH)2-saturated filter paper disk. 14C activity is assayed by liquid scintillation spectrometry. Except for the use of the 14C-mannitol substrate and liquid scintillation spectrometry to count the activity of the 14CO2 released by the fecal coliforms, this procedure is similar to those given in Section 9222. 3. References © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. CHAPPELLE, E.W. & G.L. PICCIOLO. 1975. Laboratory Procedures Manual for the Firefly Luciferase Assay for Adenosine Triphosphate (ATP). NASA GSFC Doc. X-726-75-1, National Aeronautics & Space Admin., Washington, D.C. 2. PICCIOLO, G.L., E.W. CHAPPELLE, J.W. DEMING, R.R. THOMAS, D.A. NIBLE & H. OKREND. 1981. Firefly luciferase ATP assay development for monitoring bacterial concentration in water supplies. EPA-600/S2:81-014, U.S. Environmental Protection Agency, Cincinnati, Ohio; NTIS No. PB 88-103809/AS, National Technical Information Serv., Springfield, Va. 3. NELSON, W.H., ed. 1985. Instrumental Methods for Rapid Microbiological Analysis. VCH Publishers, Inc., Deerfield Beach, Fla. 4. SEITZ, W.R. & M.P. NEARY. 1974. Chemiluminescence and bioluminescence. Anal. Chem. 46:188A. 5. OLENIAZ, W.S., M.A. PISANO, M.H. ROSENFELD & R.L. ELGART. 1968. Chemiluminescent method for detecting microorganisms in water. Environ. Sci. Technol. 2:1030. 6. WHEELER, T.G. & M.C. GOLDSCHMIDT. 1975. Determination of bacterial cell concentrations by electrical measurements. J. Clin. Microbiol. 1:25. 7. SILVERMAN, M.P. & E.F. MUNOZ. 1979. Automated electrical impedance technique for rapid enumeration of fecal coliforms in effluents from sewage treatment plants. Appl. Environ. Microbiol. 37:521. 8. MUNOZ, E.F. & M.P. SILVERMAN. 1979. Rapid, single-step most-probable-number method for enumerating fecal coliforms in effluents from sewage treatment plants. Appl. Environ. Microbiol. 37:527. 9. FIRSTENBERG-EDEN, R. & G. EDEN. 1984. Impedance Microbiology. John Wiley & Sons, Inc., New York, N.Y. 10. WALLIS, C. & J.L. MELNICK. 1985. An instrument for the immediate quantification of bacteria in potable waters. Appl. Environ. Microbiol. 49:1251. 11. BITTON, G., R.J. DUTTON & J.A. FORAN. 1984. A new rapid technique for counting microorganisms directly on membrane filters. Stain Technol. 58:343. 12. SIERACKI, M.E., P.W. JOHNSON & J.M. SIEBURTH. 1985. Detection, enumeration, and sizing of planktonic bacteria by image-analyzed epifluoresence microscopy. Appl. Environ. Microbiol. 49:799. 13. MCCOY, W.F. & B.H. OLSON. 1985. Fluorometric determination of the DNA concentration in municipal drinking water. Appl. Environ. Microbiol. 49:811. 14. REASONER, D.J. & E.E. GELDREICH. 1978. Rapid detection of water-borne fecal coliforms by 14CO2 release. In A.N. Sharpe & D.S. Clark, eds. Mechanizing Microbiology. Charles C. Thomas, Publisher, Springfield, Ill. 15. MORAN, J.W. & L.D. WITTER. 1976. An automated rapid test for Escherichia coli in milk. J. Food Sci. 41:165. 16. MORAN, J.W. & L.D. WITTER. 1976. An automated rapid method for measuring fecal © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater pollution. Water Sewage Works 123:66. 17. TRINEL, P.A., N. HANOUNE & H. LECLERC. 1980. Automation of water bacteriological analysis: running test of an experimental prototype. Appl. Environ. Microbiol. 39:976. 18. WILKINS, J.R., G.E. STONER & E.H. BOYKIN. 1974. Microbial detection method based on sensing molecular hydrogen. Appl. Microbiol. 27:947. 19. WILKINS, J.R. & E.H. BOYKIN. 1976. Analytical notes—electrochemical method for early detection of monitoring of coliforms. J. Amer. Water Works Assoc. 68:257. 20. GRANA, D.C. & J.R. WILKINS. 1979. Description and field test results of an in situ coliform monitoring system. NASA Tech. Paper 1334, National Aeronautics & Space Admin., Washington, D.C. 21. NEWMAN, J.S. & R.T. O’BRIEN. 1975. Gas chromatographic presumptive test for coliform bacteria in water. Appl. Environ. Microbiol. 30:584. 22. WARREN, L.S., R.E. BENOIT & J.A. JESSEE. 1978. Rapid enumeration of faecal coliforms in water by a colorimetric β-galactosidase assay. Appl. Environ. Microbiol. 35:136. 23. JOUENNE, T., G.-A. JUNTER & G. CARRIERE. 1985. Selective detection and enumeration of fecal coliforms in water by potentiometric measurement of lipoic acid reduction. Appl. Environ. Microbiol. 50:1208. 24. TENCATE, J.W., H.R. BULER, A. STURK & J. LEVIN. 1985. Bacterial Endotoxins. Structure, Biomedical Significance, and Detection with the Limulus Amebocyte Lysate Test. Alan R. Liss, Inc., New York, N.Y. 25. JORGENSEN, J.H., J.C. LEE, G.A. ALEXANDER & H.W. WOLF. 1979. Comparison of Limulus assay, standard plate count, and total coliform count for microbiological assessment of renovated wastewater. Appl. Environ. Microbiol. 37:928. 26. JORGENSEN, J.H. & G.A. ALEXANDER. 1981. Automation of the Limulus amebocyte lysate test by using the Abbott MS-2 microbiology system. Appl. Environ. Microbiol. 41:1316. 27. TSUGI, K., P.A. MARTIN & D.M. BUSSEY. 1984. Automation of chromogenic substrate Limulus amebocyte lysate assay method for endotoxin by robotic system. Appl. Environ. Microbiol. 48:550. 28. ABSHIRE, R.L. 1976. Detection of enteropathogenic Escherichia coli strains in wastewater by fluorescent antibody. Can. J. Microbiol. 22:365. 29. ABSHIRE, R.L. & R.K. GUTHRIE. 1973. Fluorescent antibody techniques as a method for the detection of fecal pollution. Can. J. Microbiol. 19:201. 30. THOMASON, B.M. 1981. Current status of immunofluorescent methodology. J. Food Protect. 44:381. 9211 D. Coliphage Detection © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Coliphages are bacteriophages that infect and replicate in coliform bacteria and appear to be present wherever total and fecal coliforms are found. Correlations between coliphages and coliform bacteria in fresh water generally show that coliphages may be used to indicate the sanitary quality of water.1-5 Because coliphages are more resistant to chlorine disinfection than total or fecal coliforms, they may be a better indicator of disinfection efficiency than coliform bacteria.4 The quantitative relationship between coliphages and coliform bacteria in disinfected waters is different from that in natural fresh waters because of differences in their survival rates. 1. Materials and Culture Media a. Host culture: Escherichia coli C, ATCC No. 13706. b. Media: 1) Tryptic(ase) soy agar (TSA), to maintain E. coli C host stock cultures: Tryptone (pancreatic digest of casein) or equivalent 15.0 Soytone (soybean peptone) or equivalent 5.0 Sodium chloride, NaCl 5.0 Agar 15.0 Reagent-grade water 1 g g g g L pH should be 7.3 ± 0.1 at 25°C; if necessary, adjust pH with 0.1 or 1.0N NaOH or HCl. Heat to boiling to dissolve, then autoclave for 15 min at 121°C. For agar slants, dispense 5 to 8 mL in 16- × 125-mm screw-capped tubes before sterilizing; for plates, dispense 20 to 25 mL per petri dish after autoclaving and cooling to about 45°C. 2) Tryptic(ase) soy broth (TSB): Tryptone (pancreatic digest of casein), or equivalent Soytone (soybean peptone), or equivalent Dextrose Sodium chloride, NaCl Dipotassium hydrogen phosphate, K2HPO4 Reagent-grade water 17.0 3.0 2.5 5.0 2.5 g g g g g 1 L pH should be 7.3 ± 0.1 at 25°C; adjust with 0.1 or 1.0N NaOH or HCl, if necessary. Warm and agitate to dissolve completely. Dispense in appropriate volumes as needed; sterilize in autoclave for 15 min at 121°C. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 3) Modified tryptic(ase) soy agar (MTSA): To the ingredients of TSB, add ammonium nitrate, NH4NO3, 1.60 g; strontium nitrate, Sr(NO3)2, 0.21 g; and agar, 15 g. pH should be 7.3 ± 0.1 at 25°C; if necessary, adjust pH with 0.1 or 1.0N NaOH or HCl. Heat to boiling to dissolve, dispense 5.5 mL in 16- × 25-mm screw-capped tubes, and autoclave for 15 min at 121°C. 4) Glycerine: Add 10% (w/v) to tryptic(ase) soy broth before autoclave sterilization. 5) 2,3,5-triphenyl tetrazolium chloride (TPTZ), 1% (w/v) in ethanol. Add to MTSA tempered at 45 to 46°C to enhance plaque visibility. Prepare fresh weekly. 2. Procedure a. Frozen host preparation: Inoculate E. coli C from a stock agar slant (on TSA) into a tube(s) containing 10 mL TSB and 10% glycerine (w/v) and incubate overnight at 35°C. Then inoculate each tube into a flask containing 25 mL TSB plus 10% glycerine and incubate at 35 ± 0.5°C until an optical density of 0.5 at 520 nm is obtained (equivalent to about 1 × 109 E. coli C cells/mL). Measure optical density using a spectrometer. Zero spectrometer with sterile TSB plus 10% glycerine. Aseptically dispense 4.5-mL portions of cell suspension in sterile plastic test tubes, cap, chill to 9°C, and freeze at −20°C. Store for no longer than 6 weeks in non-frost-free freezer to reduce loss of frozen host culture viability. b. Assay procedure: The procedure is directly applicable to samples containing more than 5 coliphage/100 mL; if sample contains more than 1000 coliphage/100 mL, dilute sample 1:5 or 1: 10 with sterile distilled water before proceeding. Thaw tube(s) of frozen host E. coli C in 44.5°C water bath. Use one tube of host culture per sample. Add 1.0 mL of host E. coli C culture, 5 mL sample or dilution, and 0.08 mL TPTZ6 to each of four tubes of MTSA (melted and held at about 45°C). Mix thoroughly and pour into separate 100- × 15-mm labeled petri dishes, cover, and let agar gel. Incubate inverted plates at 35°C. Count plaques after incubating for 4 to 6 h. 3. Interpreting and Reporting Results Bacteriophage infect and multiply in sensitive bacteria. This results in lysis of the bacterial cells and a release of phage particles to infect adjacent cells. As the infected coliform bacteria are lysed, visible clear areas known as plaques develop in the lawn of confluent bacterial growth. Count plaques on each plate and record. Obtain the number of plaques/100 mL of sample by summing the plaques on the four plates and multiplying by 5. If a diluted sample has been used, additionally multiply by the reciprocal of the dilution factor. Based on coliphage counts, estimate total and fecal coliform numbers as shown below.4 Independently verify equations for specific types of samples and locations. Total coliforms: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater log y = 0.627 (log x) + 1.864 where: y = total coliforms/100 mL and x = coliphages/100 mL. Fecal coliforms: log y = 0.805 (log x) + 0.895 where: y = fecal coliforms/100 mL and x = coliphages/100 mL. 4. References 1. WENTZEL, R.S., P.E. O’NEILL & J.F. KITCHENS. 1982. Evaluation of coliphage detection as a rapid indicator of water quality. Appl. Environ. Microbiol. 43:430. 2. ISBISTER, J.D. & J.L. ALM. 1982. Rapid coliphage procedure for water treatment processes. In Proc. Amer. Water Works Assoc. Water Quality Technol. Conf., Seattle, Wash., Dec. 6–9, 1981. 3. ISBISTER, J.D., J.A. SIMMONS, W.M. SCOTT & J.F. KITCHENS. 1983. A simplified method for coliphage detection in natural waters. Acta Microbiol. Polonica 32:197. 4. KOTT, Y., N. ROZE, S. SPERBER & N. BETZER. 1974. Bacteriophages as viral pollution indicators. Water Res. 8:165. 5. KENNEDY, J.D., JR., G. BITTON & J.L. OBLINGER. 1985. Comparison of selective media for assay of coliphages in sewage effluent and lake water. Appl. Environ. Microbiol. 49:33. 6. HURST, C.J., J.C. BLANNON, R.L. HARDAWAY & W.C. JACKSON. 1994. Differential effect of tetrazolium dyes upon bacteriophage plaque assay titers. Appl. Environ. Microbiol. 60:3462. 9212 STRESSED ORGANISMS*#(11) 9212 A. Introduction 1. General Discussion Indicator bacteria, including total coliforms, fecal coliforms, and fecal streptococci, may become stressed or injured in waters and wastewaters. These injured bacteria are incapable of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater growth and colony formation under standard conditions because of structural or metabolic damage. As a result, a substantial portion of the indicator bacteria present, i.e., 10 to greater than 90%, may not be detected.1,2 These false negative bacteriological findings could result in an inaccurate definition of water quality and lead to the acceptance of a potentially hazardous condition resulting from contamination by resistant pathogens3 or the penetration of undetected indicator bacteria through treatment barriers.4 Stressed organisms are present under ordinary circumstances in treated drinking water and wastewater effluents, saline waters, polluted natural waters, and relatively clean surface waters. High numbers of injured indicator bacteria may be associated with partial or inadequate disinfection and the presence of metal ions or other toxic substances. These and other factors, including extremes of temperature and pH and solar radiation, may lead collectively to significant underestimations of the number of viable indicator bacteria. Publications support the health significance of injured coliform bacteria.2,5-7 These reports show that enteropathogenic bacteria are less susceptible than coliforms to injury under conditions similar to those in treated drinking water and wastewater, that injured pathogens retain the potential for virulence, and that they recover after being ingested. Hence, methods allowing for the enumeration of injured coliform bacteria yield more sensitive determinations of potential health risks. This conclusion is further supported by the observation that viruses and waterborne pathogens that form cysts also are more resistant than indicator bacteria to environmental stressors. 2. Sample Handling and Collection Certain laboratory manipulations following sample collection also may produce injury or act as a secondary stress to the organisms.2,8 These include excessive sample storage time, prolonged holding time (more than 30 min) of diluted samples before inoculation into growth media and of inoculated samples before incubation at the proper temperature, incorrect media formulations, incomplete mixing of sample with concentrated medium, and exposure to untempered liquefied agar media. Excessive numbers of nonindicator bacteria also interfere with detection of indicators by causing injury.9 3. References 1. MCFETERS, G.A., J.S. KIPPIN & M.W. LECHEVALLIER. 1986. Injured coliforms in drinking water. Appl. Environ. Microbiol. 51:1. 2. MCFETERS, G.A. 1990. Enumeration, occurrence, and significance of injured indicator bacteria in drinking water. In G.A. McFeters, ed. Drinking Water Microbiology: Progress and Recent Developments, p. 478. Springer-Verlag, New York. 3. LECHEVALLIER, M.W. & G.A. MCFETERS. 1985. Enumerating injured coliforms in drinking water. J. Amer. Water Works Assoc. 77:81. 4. BUCKLIN, K.E., G.A. MCFETERS & A. AMIRTHARAJAH. 1991. Penetration of coliforms through municipal drinking water filters. Water Res. 25:1013. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5. LECHEVALLIER, M.W., A. SINGH, D.A. SCHIEMANN & G.A. MCFETERS. 1985. Changes in virulence of waterborne enteropathogens with chlorine injury. Appl. Environ. Microbiol. 50:412. 6. SINGH, A. & G.A. MCFETERS. 1986. Repair, growth and production of heat-stable enterotoxin by E. coli following copper injury. Appl. Environ. Microbiol. 51:738. 7. SINGH, A., R. YEAGER & G.A. MCFETERS. 1986. Assessment of in vivo revival, growth, and pathogenicity of Escherichia coli strains after copper- and chlorine-induced injury. Appl. Environ. Microbiol. 52: 832. 8. MCFETERS, G.A., S.C. CAMERON & M.W. LECHEVALLIER. 1982. Influence of diluents, media and membrane filters on the detection of injured waterborne coliform bacteria. Appl. Environ. Microbiol. 43:97. 9. LECHEVALLIER, M.W. & G.A. MCFETERS. 1985. Interactions between heterotrophic plate count bacteria and coliform organisms. Appl. Environ. Microbiol. 49:1338. 4. Bibliography CLARK, H.F., E.E. GELDREICH, H.L. JETER & P.W. KABLER. 1951. The membrane filter in sanitary bacteriology. Pub. Health Rep. 66:951. MCKEE, J.E., R.T. MCLAUGHLIN & P. LESGOURGUES. 1958. Application of molecular filter techniques to the bacterial assay of sewage. III. Effects of physical and chemical disinfection. Sewage Ind. Wastes 30:245. ROSE, R.E. & W. LITSKY. 1965. Enrichment procedure for use with the membrane filter for the isolation and enumeration of fecal streptococci from water. Appl. Microbiol. 13:106. MAXCY, R.B. 1970. Non-lethal injury and limitations of recovery of coliform organisms on selective media. J. Milk Food Technol. 33:445. LIN, S.D. 1973. Evaluation of coliform tests for chlorinated secondary effluents. J. Water Pollut. Control Fed. 45:498. BRASWELL, J.R. & A.W. HOADLEY. 1974. Recovery of Escherichia coli from chlorinated secondary sewage. Appl. Microbiol. 28:328. STEVENS, A.P., R.J. GRASSO & J.E. DELANEY. 1974. Measurements of fecal coliform in estuarine water. In D.D. Wilt, ed., Proceedings of the 8th National Shellfish Sanitation Workshop, U.S. Dep. Health, Education, & Welfare, Washington, D.C. BISSONNETTE, G.K., J.J. JEZESKI, G.A. MCFETERS & D.S. STUART. 1975. Influence of environmental stress on enumeration of indicator bacteria from natural waters. Appl. Microbiol. 29:186. BISSONNETTE, G.K., J.J. JEZESKI, G.A. MCFETERS & D.S. STUART. 1977. Evaluation of recovery methods to detect coliforms in water. Appl. Environ. Microbiol. 33:590. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9212 B. Recovery Enhancement This section describes some general procedures and considerations regarding recovery of stressed indicator organisms. For chlorinated samples, insure that sufficient dechlorinating agent is present in the sample bottle (see Section 9060A.2). 1 Collect water samples with elevated concentrations of heavy-metal ions in a sample bottle containing a chelating agent2 (see Section 9060A.2) and minimize sample storage time (see Section 9060B). Use buffered peptone dilution water rather than buffered water (see Section 9050C.1) when preparing dilutions of samples containing heavy-metal ions. After making dilutions, inoculate test media within 30 min. Resuscitation of stressed or injured organisms is enhanced by inoculating samples and initially culturing organisms in an enriched, noninhibitory medium at a moderate temperature. Although no simple test is available to establish the presence of injured bacteria in a given sample, bacteria in water known to contain stressors such as disinfectants or heavy metals frequently will be injured.1,3 When multiple-tube fermentation test results consistently are higher than those obtained from parallel membrane filter tests, or there is other indication of suboptimal recovery, consider injury probable and use one or more of the following procedures. 1. Recovery of Injured Total Coliform Bacteria Using Membrane Filtration a. m-T7 agar: Use m-T7 agar4 in the procedure described for the membrane filter test (see Section 9222B). Proteose peptone No. 3 Yeast extract Lactose Tergitol 7 Polyoxyethylene ether W1 Bromthymol blue Bromcresol purple Agar Reagent-grade water 5.0 3.0 20.0 0.4 5.0 0.1 0.1 15.0 1 g g g mL g g g g L Adjust to pH 7.4 with 0.1N NaOH after sterilization at 121°C for 15 min. Aseptically add 1.0 µg penicillin G/mL when medium has cooled to about 45°C. After filtering sample place filter on m-T7 agar and incubate at 35°C for 22 to 24 h. Coliform colonies are yellow. Verify not less than 10% of coliform colonies by the procedure in Section 9222B.5 f. With some drinking water samples containing many non-coliform bacteria, confluent © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater growth occurs. To obtain reliable results, carefully distinguish target yellow colonies from background growth. b. Addition of sodium sulfite: The addition of sodium sulfite to some media (0.05 to 0.1%) can improve the detection of coliform bacteria following exposure to chloramine but not chlorine.5 Such modified medium is applicable to clean-water systems using chloramination or to chlorinated discharges such as wastewater effluent containing high levels of organic compounds. 2. Recovery of Injured Fecal Coliform Bacteria Using Membrane Filtration a. Enrichment-temperature acclimation: Use two-layer agar (M-FC agar with a nonselective overlay medium that does not contain glucose, i.e., tryptic soy agar) with a 2-h incubation at 35°C followed by 22 h at 44.5°C.6 Prepare the M-FC agar plate in advance but do not add the overlay agar more than 1 h before use. Alternatively, use a pre-enrichment in phenol red lactose broth incubated at 35°C for 4 h followed by M-FC agar at 44.5°C for 22 h.7 As a third option, prepare enrichment two-layer medium containing specific additives and incubate for 1.5 h at room temperature (22 to 26°C) followed by 35°C for 4.5 h and 44.5°C for 18 h.8 b. Temperature acclimation:9 Modify elevated temperature procedure by preincubation of M-FC cultures for 5 h at 35°C, followed by 18 ± 1 h at 44.5°C. Use a commercially available temperature-programmed incubator to make the change from 35 to 44.5°C after the 5 h preincubation period to eliminate inconvenience and provide a practical method of analysis. c. Deletion of suppressive agent:10 Eliminate rosolic acid from M-FC medium and incubate cultures at 44.5°C ± 0.2°C for 24 h. Fecal coliform colonies are intense blue on the modified medium and are distinguished from the cream, gray, and pale-green colonies typically produced by nonfecal coliforms. d. Alternative medium-temperature acclimation: Use m-T7 medium with an 8 h incubation at 37°C followed by 12 h at 44.5°C.11 e. Verification of stressed fecal coliform bacteria: Modifications of media and procedures may decrease selectivity and differentiation of fecal coliform colonies. Therefore, if any procedural modifications are used, verify not less than 10% of the blue colonies from a variety of samples. Use lauryl tryptose broth (Section 9221B) (35°C for 48 h) with transfer of gas-producing cultures to EC broth (Section 9221E) (44.5°C for 24 h). Gas production at 44.5°C confirms the presence of fecal coliforms. 3. Recovery of Stressed Fecal Streptococci Using Membrane Filtration Using bile broth medium yields fecal streptococcus recoveries comparable with multiple-tube fermentation tests.12 Preincubate membrane filters on an enrichment medium for 2 h at 35°C and follow by plating on m-Enterococcus agar (Section 9230) for 48 ± 2 h at 35°C. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Verification of stressed fecal streptococci—Verify not less than 10% of the colonies from a variety of samples using the confirmed test procedure given in Section 9230B.3. 4. References 1. MCFETERS, G.A. & A.K. CAMPER. 1983. Enumeration of coliform bacteria exposed to chlorine. In A.I. Laskin, ed. Advances in Applied Microbiology, Vol. 29, p. 177. 2. DOMEK, M.J., M.W. LECHEVALLIER, S.C. CAMERON & G.A. MCFETERS. 1984. Evidence for the role of metals in the injury process of coliforms in drinking water. Appl. Environ. Microbiol. 48:289. 3. LECHEVALLIER, M.W. & G.A. MCFETERS. 1985. Interactions between heterotrophic plate count bacteria and coliform organisms. Appl. Environ. Microbiol. 49:1338. 4. LECHEVALLIER, M.W., S.C. CAMERON & G.A. MCFETERS. 1983. New medium for the improved recovery of coliform bacteria from drinking water. Appl. Environ. Microbiol. 45:484. 5. WATTERS, S.K., B.H. PYLE, M.W. LECHEVALLIER & G.A. MCFETERS. 1989. Enumeration of E. cloacae after chlorine exposure. Appl. Environ. Microbiol. 55:3226. 6. ROSE, R.E., E.E. GELDREICH & W. LITSKY. 1975. Improved membrane filter method for fecal coliform analysis. Appl. Microbiol. 29:532. 7. LIN, S.D. 1976. Membrane filter method for recovery of fecal coliforms in chlorinated sewage effluents. Appl. Environ. Microbiol. 32: 547. 8. STUART, D.S, G.A. MCFETERS & J.E. SCHILLINGER. 1977. Membrane filter technique for quantification of stressed fecal coliforms in the aquatic environment. Appl. Environ. Microbiol. 34:42. 9. GREEN, B.L., E.M. CLAUSEN & W. LITSKY. 1977. Two-temperature membrane filter method for enumerating fecal coliform bacteria from chlorinated effluents. Appl. Environ. Microbiol. 33:1259. 10. PRESSWOOD, W.G. & D. STRONG. 1977. Modification of M-FC medium by eliminating rosolic acid. Amer. Soc. Microbiol. Abs. Annu. Meeting. ISSN-0067-2777:272. 11. LECHEVALLIER, M.W., P.E. JAKANOSKI, A.K. CAMPER & G.A. MCFETERS. 1984. Evaluation of m-T7 agar as a fecal coliform medium. Appl. Environ. Microbiol. 48:371. 12. LIN, S.D. 1974. Evaluation of fecal streptococci tests for chlorinated secondary effluents. J. Environ. Eng. Div., Proc. Amer. Soc. Civil Engr. 100:253. 9213 RECREATIONAL WATERS*#(12) © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9213 A. Introduction 1. Microbiological Indicators Recreational waters include freshwater swimming pools, whirlpools, and naturally occurring fresh and marine waters. Many local and state health departments require microbiological monitoring of recreational waters. Historically, the most common microbiological tests to assess sanitary quality have been heterotrophic counts and total and fecal coliform tests. Total coliform tests and heterotrophic counts usually are performed on treated waters and fecal coliform tests performed on untreated waters. Although detection of coliform bacteria in water indicates that it may be unsafe to drink, other bacteria have been isolated from recreational waters that may suggest health risks through body contact, ingestion, or inhalation. Other bacteria suggested as indicators of recreational water quality include Pseudomonas aeruginosa, fecal streptococci, enterococci, and staphylococci. Ideally, recreational water quality indicators are microorganisms for which densities in the water can be related quantitatively to potential health hazards resulting from recreational use, particularly where upper body orifices are exposed to water. The ideal indicator is the one with the best correlation between density and the health hazards associated with a given type of pollution. The most common potential sources of infectious agents in recreational waters include untreated or poorly treated municipal and industrial effluents or sludge, sanitary wastes from seaside residences, fecal wastes from pleasure craft, drainage from sanitary landfills, stormwater runoff, and excretions from animals. In addition, the source of infectious agents may be the aquatic environment itself. The potential health hazards from each of these sources are not equal. Exposure to untreated or inadequately treated human fecal wastes is considered the greatest health hazard. The presence of microbiological indicators in treated swimming pools or whirlpools indicate possible insufficient water exchange, disinfection, and maintenance. Bather density is a major factor in determining the probability of swimmer-associated illnesses with swimming pools, particularly when there is insufficient disinfection and water circulation. The bathers themselves may be the source of pollution by shedding organisms associated with the mouth, nose, and skin. 2. Infectious Diseases from Water Exposure In general, infections or disease associated with recreational water contact fall into two categories. The first group is gastroenteritis resulting from unintentional ingestion of water contaminated with fecal wastes. Enteric microorganisms that have been shown to cause gastroenteritis from recreational water contact include Giardia, Cryptosporidium, Shigella, Salmonella, E. coli 0157:H7, Hepatitis A, Coxsackie A and B, and Norwalk virus. Leptospirosis is not an enteric infection but also is transmitted through contact with waters contaminated with human or animal wastes. The second group or category of infections or disease is associated mainly with microorganisms that are indigenous to the environment, which include the following: Pseudomonas aeruginosa, Staphylococcus sp., Legionella sp., Naegleria fowleri, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Mycobacterium sp., and Vibrio sp. The illnesses or waterborne diseases caused by these organisms include dermatitis or folliculitis, otitis externa, Pontiac fever, granulomas, primary amebic meningoencephalitis (PAM), and conjunctivitis. Commonly occurring illnesses or infections associated with recreational water contact are dermatitis caused by Pseudomonas aeruginosa and otitis externa, ‘‘swimmer’s ear,’’ frequently caused by Pseudomonas aeruginosa and Staphylococcus aureus. 3. Microbiological Monitoring Limitations Routine examination for pathogenic microorganisms is not recommended except for investigations of water-related illness and special studies; in such cases, focus microbiological analyses on the known or suspected pathogen. Methods for several of these pathogens are given in Section 9260, Detection of Pathogenic Bacteria, Section 9510, Detection of Enteric Viruses, and Section 9711, Pathogenic Protozoa. Because some pathogenic organisms such as Giardia, Cryptosporidium, Mycobacterium, and Naegleria are more resistant to changes in environmental conditions than indicator bacteria, routine monitoring may not always reflect the risk of infection from these organisms. Described below are recommended methods for microbial indicators of recreational water quality. Consider the type(s) of water examined in selecting the microbiological method(s) or indicator(s) to be used. No single procedure is adequate to isolate all microorganisms from contaminated water. While bacterial indicators may not adequately reflect risk of viral, fungal, or parasitic infection from recreational waters, available technology limits monitoring for such organisms in routine laboratory operations. 4. Bibliography CABELLI, V.J. 1977. Indicators of recreational water quality. In Bacterial Indicators/Health Hazards Associated with Waters. STP 635, American Soc. Testing & Materials, Philadelphia, Pa. DUFOUR, A.P. 1986. Diseases caused by water contact. In Waterborne Diseases in the United States. CRC Press Inc., Boca Raton, Fla. MOE, C.L. 1996. Waterborne transmission of infectious agents. In Manual of Environmental Microbiology. American Soc. Microbiology, ASM Press, Washington, D.C. 9213 B. Swimming Pools 1. General Discussion a. Characteristics: A swimming pool is a body of water of limited size contained in a holding structure.1 The pool water generally is potable and treated with additional disinfectant but also may come from thermal springs or salt water. Modern pools have a recirculating system for filtration and disinfection. b. Monitoring requirements: 1) General—Monitor water quality in pools for changes in chemical and physical © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater characteristics that may result in irritation to the bather’s skin, eyes, and mucosal barriers or may adversely affect disinfection. Microorganisms of concern typically are those from the bather’s body and its orifices and include those causing infections of the eye, ear, upper respiratory tract, skin, and intestinal or genitourinary tracts. Water quality depends on the efficacy of disinfection, sanitary conditions, the number of bathers in the pool at any one time, and the total number of bathers per day. 2) Disinfected indoor pools—Swimming pools should be disinfected continuously when in use. Test swimming pool water for residual chlorine and pH when the pool is initially opened and at least three times/d. Collect samples from at least two locations for these determinations. Evaluate clarity of the swimming pool water before opening for the day and during periods of heavy usage.2 The heterotrophic plate count is the primary indicator of disinfection efficacy. Indicators of health risk include normal skin flora that are shed, such as Pseudomonas and Staphylococcus.3-6 These organisms account for a large percentage of swimming-pool-associated illnesses. In special circumstances Mycobacterium, Legionella, or Candida albicans may be associated with health risks related to recreational waters. Take samples for microbiological examination while the pool is in use. APHA recommends for public swimming pools that not more than 15% of the samples collected during any 30-d period shall have a heterotrophic plate count of 200/mL or show a positive confirmed total coliform test in any of five 10-mL portions of sample examined with the multiple-tube fermentation test or more than 1 total coliform/50 mL when the membrane filter test is used. Whenever swimming pool samples are examined for total staphylococci or Staphylococcus aureus, not more than 50 organisms/100 mL should be present.2 3) Disinfected outdoor pools—Fecal coliform bacteria and Pseudomonas species are the primary indicators of contamination from animal pets, rodents, stormwater runoff, and human sources. Supporting indicators include coliform bacteria, the heterotrophic plate count, and staphylococci. 4) Untreated pools—The primary indicator may be fecal coliform bacteria. Supporting indicators are those described for disinfected pools. Untreated pools are not recommended for recreational use due to increased health risks. 2. Samples a. Containers: Collect samples for bacteriological examination of swimming pool water as directed in Section 9060A. Use containers with capacities of 120 to 480 mL, depending on analyses to be made. Add sufficient sodium thiosulfate, Na2S2O3, to the sample to provide a concentration of approximately 100 mg/L in the sample. Do this by adding 0.1 mL of 10% solution of Na2S2O3 to a 120-mL bottle or 0.4 mL to a 480-mL bottle. After adding Na2S2O3, stopper or cap and sterilize container. b. Sampling procedure: Collect samples during periods of maximum bather load. Information on number of bathers may be helpful in subsequent interpretation of laboratory © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater results. Use sampling frequency consistent with state and local health regulations. Collect samples by carefully removing cap of a sterile sample bottle and holding bottle near the base at an angle of 45 deg. Fill in one slow sweep down through the water, with the mouth of the bottle always ahead of the hand. Avoid contamination of the sample by floating debris. Replace cap. Do not rinse bottle (i.e., retain sodium thiosulfate). For pools equipped with a filter, samples may be collected from sampling cocks provided in the return and discharge lines from the filter. Most bacteria shed by bathers are in body oils, saliva, and mucus discharges that occur near the surface; collect additional samples of the surface microlayer from the area in 1-m-deep water. Collect microlayer samples by plunging a sterile glass plate (approximately 20 cm by 20 cm) vertically through the water surface and withdrawing it upward at a rate of approximately 6 cm/s. Remove surface film and water layer adhering to both sides of plate with a sterile silicone rubber scraper and collect in a sterile glass bottle. Repeat until desired volume is obtained. To minimize microbial contamination, wrap glass plate and scraper in metal foil and sterilize by autoclaving before use. Wear sterile rubber or plastic gloves during sampling or hold glass plate with forceps, clips, or tongs. Determine residual chlorine or other disinfectant at poolside at the time of sample collection (see Section 4500-Cl.G). Residual disinfectant levels, chemical, and physical quality of pool water should be consistent with local, state, or APHA standards. The permissible bathing load should adhere to local, state, or APHA-recommended regulations. c. Sample storage: Analyze microbiological samples as soon as possible after collection (see Section 9060B). d. Sample volume: See Section 9222B.5. e. Sample dilution: If sample dilutions are required, use 0.1% peptone water or buffered dilution water as diluent to optimize recovery of stressed organisms (see Section 9222 for suggested sample volume). Because peptone water has a tendency to foam, avoid including air bubbles when pipetting to assure accurate measure. 3. Heterotrophic Plate Count Determine the heterotrophic plate count as directed in Section 9215. Use at least two plates per dilution. 4. Tests for Total Coliforms Determine total coliform bacteria as directed in Section 9221, Section 9222, or Section 9223. 5. Tests for Fecal Coliforms Test for fecal coliforms according to the multiple-tube fermentation technique (Section 9221), the membrane filter technique (Section 9222), or rapid methods (Section 9211). 6. Test for Staphylococci or Staphylococcus aureus © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater a. Baird-Parker agar base: Tryptone Beef extract Yeast extract Glycine Sodium pyruvate Lithium chloride Agar Reagent-grade water 10.0 5.0 1.0 12.0 10.0 5.0 20.0 1 g g g g g g g L Sterilize by autoclaving. Cool to 50°C and aseptically add 50 mL commercial egg yolk tellurite enrichment/L. Mix well. Final pH should be 7.0 ± 0.2. b. Procedure: Use membrane filter technique to prepare samples. Place membrane filter on Baird-Parker agar and incubate at 35 ± 0.5°C for 48 ± 4 h. Staphylococci typically form slate gray to jet black, smooth, entire colonies. If S. aureus is present egg yolk clearing may be observed if the membrane filter is raised from the medium. Verify some differentiated colonies with a commercial multi-test system or on the basis of such key characteristics as catalase reaction, coagulase production, aerobic and anaerobic acid production from certain carbohydrates, and typical microscopic morphology. 7. Test for Staphylococcus aureus Use a modified multiple-tube procedure. a. Media: 1) M-staphylococcus broth: Tryptone Yeast extract Lactose Mannitol Dipotassium hydrogen phosphate, K2HPO4 10.0 2.0 2.0 10.0 5.0 Sodium chloride, NaCl Sodium azide, NaN3 75.0 g 0.049 g Reagent-grade water 1 g g g g g L © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Sterilize by boiling for 4 min; pH should be 7.0 ± 0.2. For 10-mL inocula prepare and use double-strength medium. 2) Lipovitellenin-salt-mannitol agar: This medium may not be available in dehydrated form and may require preparation from the basic ingredients or by addition of egg yolk to a dehydrated base. Beef extract Polypeptone Sodium chloride, NaCl d-Mannitol Agar Phenol red Egg yolk Reagent-grade water 1.0 10.0 75.0 10.0 15.0 0.025 20.0 1 g g g g g g g L Sterilize by autoclaving; pH should be 7.4 ± 0.2. b. Procedure: Inoculate tubes of M-staphylococcus broth as directed in Section 9221. Incubate at 35 ± 1°C for 24 h. Hold original enrichment sample but streak from positive (turbid) tubes on plates of lipovitellenin-salt-mannitol agar and incubate at 35 ± 1°C for 48 h. Opaque (24 h), yellow (48 h) zones around the colonies are positive evidence of lipovitellenin-lipase activity (opaque) and mannitol fermentation (yellow). If the plate is negative, streak another plate from the original enrichment tube before discarding. Lipovitellenin-lipase activity has a 95% positive correlation with coagulase production. If necessary, confirm positive isolates as catalase-positive, coagulase-positive, fermenting mannitol, fermenting glucose anaerobically, yielding typical microscopic morphology, and gram-positive. 8. Tests for Pseudomonas aeruginosa Tests for P. aeruginosa are presented in Section 9213E and Section 9213F and include a membrane filter procedure and a multiple-tube technique. 9. Test for Streptococci or Enterococci Determine fecal streptococci or enterococci as described in Section 9230, and if necessary, perform additional biochemical tests to identify species. 10. References 1. CENTERS FOR DISEASE CONTROL. 1983. Swimming Pools—Safety and Disease Control © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. 3. 4. 5. 6. through Proper Design and Operation. DHHS—CDC No. 83-8319, Centers for Disease Control, Atlanta, Ga. AMERICAN PUBLIC HEALTH ASSOCIATION. 1981. Public Swimming Pools. Recommended Regulations for Design and Construction, Operation and Maintenance. American Public Health Assoc., Washington, D.C. SEYFRIED, P.L., R.S. TOBIN, N.E. BROWN & P.F. NESS. 1985. A prospective study of swimming-related illness. II. Morbidity and the microbiological quality of water. Amer. J. Pub. Health 75:1071. KLAPES, N.A. & D. VESLEY. 1988. Rapid assay for in situ identification of coagulase-positive staphylococci recovered by membrane filtration from swimming pool water. Appl. Environ. Microbiol. 52:589. COVERT, T.C. & P.V. SCARPINO. 1987. Comparison of Baird-Parker agar, Vogel-Johnson agar, and M-Staphylococcus broth for the isolation and enumeration of Staphylococcus aureus in swimming pool waters. Abstr. Annu. Meeting American Soc. Microbiology, Atlanta, Ga., American Soc. Microbiology, Washington, D.C. CHAROENCA, N. & R.S. FUJIOKA. 1995. Association of staphylococcal skin infections and swimming. Water Sci. Technol. 32:11. 11. Bibliography WORKING PARTY OF THE PUBLIC HEALTH LABORATORY SERVICE. 1965. A bacteriological survey of swimming baths in primary schools. Monthly Bull. Min. Health & Pub. Health Lab. Serv. 24:116. GUNN, B.A., W.E. DUNKELBERG, JR. & J.R. CRUTZ. 1972. Clinical evaluation of 2% LSM medium for primary isolation and identification of staphylococci. Amer. J. Clin. Pathol. 57:236. HATCHER, R.F. & B.C. PARKER. 1974. Investigations of Freshwater Surface Microlayers. VPI-SRRC-BULL 64. Virginia Polytechnic Inst. and State Univ., Blacksburg. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1985. Test Methods for Escherichia coli and Enterococci in Water by the Membrane Filter Procedure. EPA-600/4-85/076. HURST, C.J. 1991. Disinfection of drinking water, swimming-pool-water and treated sewage effluent. In S.S. Block. Disinfection, Sterilization and Preservation, 4th ed. Lea & Febiger, Philadelphia, Pa. 9213 C. Whirlpools 1. General Discussion a. Characteristics: A whirlpool is a shallow pool with a maximum water depth of 1.2 m; it has a closed-cycle water system, a heated water supply, and usually a hydrojet recirculation system. It may be constructed of plastic, fiberglass, redwood, or epoxy-lined surfaces. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Whirlpools are designed for recreational as well as therapeutic use and may accommodate one or more bathers. These pools usually are not cleaned, drained, and refilled after each use. They are located in homes, apartments, hotels, athletic facilities, rehabilitation centers, and hospitals. b. Monitoring requirements: Whirlpool-associated infections are common because of the inherent design and characteristics of whirlpools, which include high temperature, reduced disinfection efficacy, and increased organic material. All these factors contribute to favorable conditions for growth of microorganisms, especially Pseudomonas aeruginosa. Studies have shown that whirlpools can serve as a reservoir of Legionella pneumophila. Therefore, frequent testing for residual disinfectant levels and pH, along with scheduled maintenance, is necessary for safe whirlpool water quality.1-5 c. Microbiological indicators: The primary indicator of disinfection efficacy is P. aeruginosa, with total coliforms, heterotrophic plate count, and staphylococci as supporting indicators of water quality. The standard index of water quality, i.e., total coliforms, may be insufficient to judge the microbiological quality of whirlpool water. Pseudomonas aeruginosa is frequently isolated from whirlpool water that is coliform-negative.6 In the event of a whirlpool-associated outbreak, collect samples as close as possible to the time of the outbreak. Analyze for the suspected pathogen and P. aeruginosa. Methods for P. aeruginosa are described in Section 9213E and Section 9213F. d. Sample preservation: Examine samples as soon as possible after collection. See Section 9060B. 2. References 1. CENTERS FOR DISEASE CONTROL. 1981. Suggested Health and Safety Guidelines for Public Spas and Hot Tubs. DHHS-CDC #99-960. United States Government Printing Off., Washington, D.C. 2. SOLOMON, S.L. 1985. Host factors in whirlpool-associated Pseudomonas aeruginosa skin disease. Infect. Control 6:402. 3. HIGHSMITH, A.K., P.N. LEE, R.F. KHABBAZ & V.P. MUNN. 1985. Characteristics of Pseudomonas aeruginosa isolated from whirlpools and bathers. Infect. Control 6:407. 4. GROOTHUIS, D.G., A.H. HAVELAAR & H.R. VEENENDAAL. 1985. A note on legionellas in whirlpools. J. Appl. Bacteriol. 58:479. 5. HIGHSMITH, A.K. & M.S. FAVERO. 1985. Microbiological aspects of public whirlpools. Clin. Microbiol. Newsletter 7:9. 6. HALL, N. 1984. Whirlpools and Pseudomonas aeruginosa. UHL Lab Hotline 21:9. 3. Bibliography GELDREICH, E.E., A.K. HIGHSMITH & W.J. MARTONE. 1985. Public whirlpools—the epidemiology and microbiology of disease. Infect. Control 6:392. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9213 D. Natural Bathing Beaches 1. General Discussion a. Characteristics: A natural bathing beach is any area of a stream, lake, ocean, impoundment, or hot spring that is used for recreation. A wide variety of pathogenic microorganisms can be transmitted to humans through use of natural fresh and marine recreational waters contaminated by wastewater.1,2 These include enteric pathogens such as Salmonella, Shigella, enteroviruses, protozoa, multicellular parasites, and ‘‘opportunists’’ such as P. aeruginosa, Klebsiella sp., Vibrio sp., and Aeromonas hydrophila, which can multiply in recreational waters with sufficient nutrients. Other organisms of concern are those associated with the skin, mouth, or nose of bathers, such as Staphylococcus aureus and other organisms, e.g., nontuberculous mycobacteria and leptospira, and Naegleria sp..3-9 b. Monitoring requirements: Historically, fecal coliforms have been recommended as the indicator of choice for evaluating the microbiological quality of recreational waters. Many states have adopted use of this indicator in their water quality standards. Recent studies have demonstrated that E. coli and enterococci showed a stronger correlation with swimming-associated gastroenteritis than do fecal coliforms, and that both indicators were equally acceptable for monitoring fresh-water quality. For marine water, enterococci showed the strongest relationship of density to gastroenteritis. The recommended densities of these indicator organisms were calculated to approximate the degree of protection previously accepted for fecal coliforms. EPA-recommended water quality criteria are based on these findings.10 While the primary indicators of water quality are E. coli and enterococci, the enumeration of P. aeruginosa, Aeromonas hydrophila, and Klebsiella sp. in recreational waters may be useful in cases of discharge of pulp and paper wastes and effluents from textile finishing plants into receiving waters. 2. Samples a. Containers: Collect samples as directed in Section 9060A. The size of the container varies with the number and variety of tests to be performed. Adding Na2S2O3 to the bottle is unnecessary. b. Sampling procedure: Collect samples 0.3 m below the water surface in the areas of greatest bather load. Take samples over the range of environmental and climatic conditions, especially during times when maximal pollution can be expected, i.e., periods of tidal, current, and wind influences, stormwater runoff, wastewater treatment bypasses. See Section 9213B.2b for methods of sample collection and Section 9222 for suggested sample volumes. c. Sample storage: See Section 9060B. 3. Tests for Escherichia coli © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater a. Media: 1) mTEC agar:*#(13) Proteose peptone Yeast extract Lactose Sodium chloride, NaCl Dipotassium phosphate, K2HPO4 Monopotassium phosphate KH2PO4 Sodium lauryl sulfate Sodium desoxycholate Bromcresol purple Bromphenol red Agar Reagent-grade water 5.0 3.0 10.0 7.5 3.3 g g g g g 1.0 g 0.2 0.1 0.08 0.08 15.0 1 g g g g g L Sterilize by autoclaving; pH should be 7.3 ± 0.2. Pour 4 to 5 mL liquefied agar into culture dishes (50 × 10 mm). Store in refrigerator. 2) Urea substrate:* #(14) Urea Phenol red Reagent-grade water 2.0 g 10 mg 100 mL Adjust pH to between 3 and 4. Store at 2 to 8°C. Use within 1 week. b. Procedure: Filter sample through a membrane filter (see Section 9222), place membrane on mTEC agar, incubate at 35 ± 0.5°C for 2 h to rejuvenate injured or stressed bacteria, and then incubate at 44.5 ± 0.2°C for 22 h. Transfer filter to a filter pad saturated with urea substrate. After 15 min, count yellow or yellow-brown colonies, using a fluorescent lamp and a magnifying lens. E. coli produces yellow or yellow-brown colonies. Verify a portion of these differentiated colonies with a commercial multi-test system [see Section 9222B.5 f2)b)]. 4. Tests for Enterococci Perform tests for enterococci by the multiple-tube technique (Section 9230B) or membrane © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater filter technique (Section 9230C). 5. Tests for Pseudomonas aeruginosa Perform tests for P. aeruginosa as directed in Section 9213E and Section 9213F. Use the multiple-tube test with samples but note that the procedures may not be applicable to marine samples. 6. Tests for Salmonella/Shigella See Section 9260. 7. References 1. CABELLI, V.J. 1980. Health Effects Criteria for Marine Recreational Waters. EPA-600/1-80-031, U.S. Environmental Protection Agency, Research Triangle Park, N.C. 2. DUFOUR, A.P. 1984. Health Effects Criteria for Fresh Recreational Waters. EPA-600/1-84-004, U.S. Environmental Protection Agency, Research Triangle Park, N.C. 3. KESWICK, B.H., C.P. GERBA & S.M. GOYAL. 1981. Occurrence of enteroviruses in community swimming pools. Amer. J. Pub. Health 71: 1026. 4. DUTKA, B.J. & K.K. KWAN. 1978. Health indicator bacteria in water surface microlayers. Can. J. Microbiol. 24:187. 5. CABELLI, V.J., H. KENNEDY & M.A. LEVIN. 1976. Pseudomonas aeruginosa and fresh recreational waters. J. Water Pollut. Control Fed. 48: 367. 6. SHERRY, J.P., S.R. KUCHMA & B.J. DUTKA. 1979. The occurrence of Candida albicans in Lake Ontario bathing beaches. Can. J. Microbiol. 25:1036. 7. STEVENS, A.R., R.L. TYNDALL, C.C. COUTANT & E. WILLAERT. 1977. Isolation of the etiological agent of primary amoebic meningoencephalitis from artificially heated waters. Appl. Environ. Microbiol. 34:701. 8. WELLINGS, F.M., P.T. AMUSO, S.L. CHANG & A.L. LEWIS. 1977. Isolation and identification of pathogenic Naegleria from Florida lakes. Appl. Environ. Microbiol. 34:661. 9. N’DIAYE, A., P. GEORGES, A. N’GO & B. FESTY. 1985. Soil amoebas as biological markers to estimate the quality of swimming pool waters. Appl. Environ. Microbiol. 49:1072. 10. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1986. Ambient Water Quality Criteria for Bacteria—1986. EPA-440/5-84-002, U.S. Environmental Protection Agency, Washington, D.C. 8. Bibliography © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater OLIVIERI, V.P., C.W. DRUSE & K. KAWATA. 1977. Microorganisms in Urban Stormwater. EPA-600/2-77-087, U.S. Environmental Protection Agency, Cincinnati, Ohio. RICE, E.W., T.C. COVERT, D.K. WILD, D. BERMAN, S.A. JOHNSON & C.H. JOHNSON. 1993. Comparative resistance of Escherichia coli and Enterococci to chlorination. J. Environ. Health. A28:89. 9213 E. Membrane Filter Technique for Pseudomonas aeruginosa 1. Laboratory Apparatus See Section 9222B.1. 2. Culture Media a. M-PA agar: This agar may not be available in dehydrated form and may require preparation from the basic ingredients. L-lysine HCl Sodium chloride, NaCl Yeast extract Xylose Sucrose Lactose Phenol red Ferric ammonium citrate Sodium thiosulfate, Na2S2O3 Agar Reagent-grade water 5.0 5.0 2.0 2.5 1.25 1.25 0.08 0.8 6.8 15.0 1 g g g g g g g g g g L Adjust to pH 6.5 ± 0.1 and sterilize by autoclaving. Cool to 55 to 60°C; readjust to pH 7.1 ± 0.2 and add the following dry antibiotics per liter of agar base: sulfapyridine,*#(15) 176 mg; kanamycin,* 8.5 mg; nalidixic acid,* 37.0 mg; and cycloheximide,* 150 mg. After mixing dispense in 3-mL quantities in 50- × 12- mm petri plates. Store poured plates at 2 to 8°C. Discard unused medium after 1 month. b. Modified M-PA agar.†#(16) c. Milk agar (Brown and Scott Foster Modification): Mixture A: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Instant nonfat milk‡#(17) Reagent-grade water 100 500 g mL Nutrient broth Sodium chloride, NaCl Agar Reagent-grade water 12.5 2.5 15.0 500 g g g mL Mixture B: Separately prepare and sterilize Mixtures A and B; cool rapidly to 55°C; aseptically combine mixtures and dispense 20 to 25 mL per petri dish. 3. Procedure a. Presumptive tests: Filter 200 mL or less of natural waters or up to 500 mL of swimming pool waters through sterile membrane filters. Place each membrane on a poured plate of modified M-PA agar so that there is no air space between the membrane and the agar surface. Invert plates and incubate at 41.5 ± 0.5°C for 72 h. Typically, P. aeruginosa colonies are 0.8 to 2.2 mm in diameter and flat in appearance with light outer rims and brownish to greenish-black centers. Count typical colonies, preferably from filters containing 20 to 80 colonies. Use a 10- to 15-power magnifier as an aid in colony counting. b. Confirmation tests: Use milk agar to confirm a number of typical and atypical colonies. Make a single streak (2 to 4 cm long) from an isolated colony on a milk agar plate and incubate at 35 ± 1.0 °C for 24 h. P. aeruginosa hydrolyzes casein and produces a yellowish to green diffusible pigment. 4. Interpretation and Calculation of Density Confirmation is not routinely required. In the absence of confirmation, report results as the number of presumptive P. aeruginosa/100 mL. 5. Bibliography DRAKE, C.H. 1966. Evaluation of culture media for the isolation and enumeration of Pseudomonas aeruginosa. Health Lab. Sci. 3:10. BROWN, M.R.W. & J.H. SCOTT FOSTER. 1970. A simple diagnostic milk medium for Pseudomonas aeruginosa. J. Clin. Pathol. 23:172. LEVIN, M.A. & V.J. CABELLI. 1972. Membrane filter technique for enumeration of Pseudomonas aeruginosa. Appl. Microbiol. 24:864. DUTKA, B.J. & K.K. KWAN. 1977. Confirmation of the single-step membrane filter procedure for © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater estimating Pseudomonas aeruginosa densities in water. Appl. Environ. Microbiol. 33:240. BRODSKY, M.H. & B.W. CIEBIN. 1978. Improved medium for recovery and enumeration of Pseudomonas aeruginosa from water using membrane filters. Appl. Environ. Microbiol. 36:26. 9213 F. Multiple-Tube Technique for Pseudomonas aeruginosa 1. Laboratory Apparatus See Section 9221. 2. Culture Media a. Asparagine broth: This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Asparagine, DL Anhydrous dipotassium hydrogen phosphate, K2HPO4 3.0 g 1.0 g Magnesium sulfate, MgSO4⋅7H2O 0.5 g Reagent-grade water 1 L Adjust pH to 6.9 to 7.2 before sterilization. b. Acetamide broth: This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Acetamide Sodium chloride, NaCl Anhydrous dipotassium hydrogen phosphate, K2HPO4 10.0 g 5.0 g 1.39 g Anhydrous potassium dihydrogen phosphate, KH2PO4 0.73 g Magnesium sulfate, MgSO4⋅7H2O 0.5 g Dissolve 1.2 g phenol red in 100 mL 0.01N NaOH and add 1 mL/L of acetamide broth. Use phenol red stock solution within 1 year. Adjust pH to 7.1 to 7.3 before sterilization. Final pH should be 7.0 ± 0.2. Prepare acetamide broth as described above. If agar slants are preferred, prepare as described above but add 15 g agar/L, heat to dissolve agar, and dispense 8-mL quantities in 16-mm tubes. After autoclaving, incline tubes while cooling to provide a large slant surface. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 3. Procedure a. Presumptive test: Perform a five-tube multiple-tube test. Use 10 mL single-strength asparagine broth for inocula of 1 mL or less and 10 mL double-strength asparagine broth for 10-mL inocula. For swimming pools, higher dilutions may be necessary. Incubate inoculated tubes at 35 to 37°C. After 24 h and again after 48 h of incubation, examine tubes under long-wave ultraviolet light (black light) in a darkened room. Production of a green fluorescent pigment constitutes a positive presumptive test. b. Confirmed test: Confirm positive tubes by inoculating 0.1 mL of culture into acetamide broth or onto the surface of acetamide agar slants. Development of purple color (alkaline pH) within 24 to 36 h of incubation at 35 to 37°C is a positive confirmed test for Pseudomonas aeruginosa. c. Computing and reporting results: Refer to Table 9221:IV and to Section 9221D. 9215 HETEROTROPHIC PLATE COUNT*#(18) 9215 A. Introduction 1. Applications The heterotrophic plate count (HPC), formerly known as the standard plate count, is a procedure for estimating the number of live heterotrophic bacteria in water and measuring changes during water treatment and distribution or in swimming pools. Colonies may arise from pairs, chains, clusters, or single cells, all of which are included in the term ‘‘colony-forming units’’ (CFU). The final count also depends on interaction among the developing colonies; choose that combination of procedure and medium that produces the greatest number of colonies within the designated incubation time. To compare data, use the same procedure and medium. Three different methods and four different media are described. 2. Selection of Method a. Pour plate method: The pour plate method (9215B) is simple to perform and can accommodate volumes of sample or diluted sample ranging from 0.1 to 2.0 mL. The colonies produced are relatively small and compact, showing less tendency to encroach on each other than those produced by surface growth. On the other hand, submerged colonies often are slower growing and are difficult to transfer. A thermostatically controlled water bath is essential for tempering the agar, but even so, significant heat shock to bacteria from the transient exposure of the sample to 45 to 46°C agar may occur. b. Spread plate method: The spread plate method (9215C) causes no heat shock and all colonies are on the agar surface where they can be distinguished readily from particles and bubbles. Colonies can be transferred quickly, and colony morphology easily can be discerned © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and compared to published descriptions. However, this method is limited by the small volume of sample or diluted sample that can be absorbed by the agar: 0.1 to 0.5 mL, depending on the degree to which the prepoured plates have been dried. To use this procedure, maintain a supply of suitable predried, absorbent agar plates. c. Membrane filter method: The membrane filter method (9215D) permits testing large volumes of low-turbidity water and is the method of choice for low-count waters (< 1 to 10 CFU/ mL). This method produces no heat shock but adds the expense of the membrane filter. Further disadvantages include the smaller display area, the need to detect colonies by reflected light against a white background if colored filters or contrast stains are not used, possible damage to cells by excessive filtration pressures, and possible variations in membrane filter quality (see Section 9020B.4h). 3. Work Area Provide a level table or bench top with ample area in a clean, draft-free, well-lighted room or within a horizontal-flow laminar hood. Use table and bench tops having nonporous surfaces and disinfect before any analysis is made. 4. Samples Collect water as directed in Section 9060A. Initiate analysis as soon as possible after collection to minimize changes in bacterial population. The recommended maximum elapsed time between collection and analysis of samples is 8 h (maximum transit time 6 h, maximum processing time 2 h). When analysis cannot begin within 8 h, maintain sample at a temperature below 4°C but do not freeze. Maximum elapsed time between collection and analysis must not exceed 24 h. 5. Sample Preparation Mark each plate with sample number, dilution, date, and any other necessary information before examination. Prepare at least duplicate plates for each volume of sample or dilution examined. For the pour or spread plate methods use sterile glass (65 cm2) or presterilized disposable plastic (57 cm2) petri dishes. Thoroughly mix all samples or dilutions by rapidly making about 25 complete up-and-down (or back-and-forth) movements. Optionally, use a mechanical shaker to shake samples or dilutions for 15 s. 6. Media Compare new lots of media with current lot in use according to Section 9020B.4i. a. Plate count agar (tryptone glucose yeast agar): Use for pour and spread plate methods. This high-nutrient agar, widely used in the past, gives lower counts than R2A or NWRI agar. It is included for laboratories wishing to make comparisons of media or to extend the continuity of old data. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Tryptone Yeast extract Glucose Agar Reagent-grade water 5.0 2.5 1.0 15.0 1 g g g g L pH should be 7.0 ± 0.2 after autoclaving at 121°C for 15 min. b. m-HPC agar:†#(19) Use this high-nutrient medium only for the membrane filter method. Peptone Gelatin Glycerol Agar Reagent-grade water 20.0 25.0 10.0 15.0 1 g g mL g L Mix all ingredients except glycerol. Adjust pH to 7.1, if necessary, with 1N NaOH, heat slowly to boili ng to dissolve thoroughly, add glycerol, and autoclave at 121°C for 5 min.‡#(20) c. R2A agar: Use for pour, spread plate, and membrane filter methods. This low-nutrient agar gives higher counts than high-nutrient formulations. Yeast extract Proteose peptone No. 3 or polypeptone Casamino acids Glucose Soluble starch Dipotassium hydrogen phosphate, K2HPO4 0.5 0.5 0.5 0.5 0.5 0.3 Magnesium sulfate heptahydrate, MgSO4⋅7H2O 0.05 g Sodium pyruvate Agar Reagent-grade water 0.3 15.0 1 g g g g g g g g L Adjust pH to 7.2 with solid K2HPO4 or KH2PO4 before adding agar. Heat to dissolve agar and sterilize at 121°C for 15 min. d. NWRI agar (HPCA): Use for pour, spread plate, and membrane filter methods. This low-nutrient medium is likely to produce higher colony counts than high-nutrient media. It is not © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater currently available in dehydrated form and requires preparation from the basic ingredients; this makes its usage less desirable. Peptone Soluble casein K2HPO4 3.0 0.5 0.2 g g g MgSO4 0.05 g FeCl3 0.001 g Agar Reagent-grade water 15.0 1 g L Adjust pH to 7.2 before autoclaving for 15 min at 121°C. 7. Incubation For compliance monitoring purposes under U.S. EPA’s Surface Water Treatment Rule (40 CFR 141.74), provision on heterotrophic bacteria, incubate pour plates at 35°C for 48 h. Otherwise, select from among recommended times and temperatures for monitoring changes in water quality. The highest counts typically will be obtained from 5- to 7-d incubation at a temperature of 20 to 28°C. During incubation maintain humidity within the incubator so that plates will have no moisture weight loss greater than 15%. This is especially important if prolonged incubation is used. A pan of water placed at the bottom of the incubator may be sufficient but note that to prevent rusting or oxidation the inside walls and shelving should be of high-grade stainless steel or anodized aluminum. For long incubation in nonhumidified incubators, seal plates in plastic bags. 8. Counting and Recording a. Pour and spread plates: Count all colonies on selected plates promptly after incubation. If counting must be delayed temporarily, store plates at 5 to 10°C for no more than 24 h, but avoid this as routine practice. Record results of sterility controls on the report for each lot of samples. Use an approved counting aid, such as the Quebec colony counter, for manual counting. If such equipment is not available, count with any other counter provided that it gives equivalent magnification and illumination. Automatic plate counting instruments are available. These generally use a television scanner coupled to a magnifying lens and an electronics package. Their use is acceptable if evaluation in parallel with manual counting gives comparable results. In preparing plates, pipet sample volumes that will yield from 30 to 300 colonies/plate. The aim is to have at least one dilution giving colony counts between these limits, except as provided below. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Ordinarily, do not pipet more than 2.0 mL of sample; however, when the total number of colonies developing from 2.0 mL is less than 30, disregard the rule above and record result observed. With this exception, consider only plates having 30 to 300 colonies in determining the plate count. Compute bacterial count per milliliter by the following equation: If there is no plate with 30 to 300 colonies, and one or more plates have more than 300 colonies, use the plate(s) having a count nearest 300 colonies. Compute the count as above and report as estimated CFU per milliliter. If plates from all dilutions of any sample have no colonies, report the count as less than one (< 1) divided by the corresponding largest sample volume used. For example, if no colonies develop from the 0.01-mL sample volume, report the count as less than 100 (< 100) estimated CFU/mL. If the number of colonies per plate far exceeds 300, do not report result as ‘‘too numerous to count’’ (TNTC). If there are fewer than 10 colonies/cm2, count colonies in 13 squares (of the colony counter) having representative colony distribution. If possible, select seven consecutive squares horizontally across the plate and six consecutive squares vertically, being careful not to count a square more than once. Multiply sum of the number of colonies in 13 representative square centimeters by 5 to compute estimated colonies per plate when the plate area is 65 cm2. When there are more than 10 colonies/cm2, count four representative squares, take average count per square centimeter, and multiply by the appropriate factor to estimate colonies per plate. The factor is 57 for disposable plastic plates and 65 for glass plates. When bacterial counts on crowded plates are greater than 100 colonies/cm2, report result as greater than (>) 6500 divided by the smallest sample volume plated for glass plates or greater than (>) 5700 divided by the smallest sample volume plated for plastic plates. Report as estimated colony-forming units per milliliter. If spreading colonies (spreaders) are encountered on the plate(s) selected, count colonies on representative portions only when colonies are well distributed in spreader-free areas and the area covered by the spreader(s) does not exceed one-half the plate area. When spreading colonies must be counted, count each of the following types as one: a chain of colonies that appears to be caused by disintegration of a bacterial clump as agar and sample were mixed; a spreader that develops as a film of growth between the agar and bottom of petri dish; and a colony that forms in a film of water at the edge or over the agar surface. The last two types largely develop because of an accumulation of moisture at the point from which the spreader originates. They frequently cover more than half the plate and interfere with obtaining a reliable plate count. Count as individual colonies similar-appearing colonies growing in close proximity but not © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater touching, provided that the distance between them is at least equal to the diameter of the smallest colony. Count impinging colonies that differ in appearance, such as morphology or color, as individual colonies. If plates have excessive spreader growth, report as ‘‘spreaders’’ (Spr). When plates are uncountable because of missed dilution, accidental dropping, and contamination, or the control plates indicate that the medium or other material or labware was contaminated, report as ‘‘laboratory accident’’ (LA). b. Membrane filter method: Count colonies on membrane filters using a stereoscopic microscope at 10 to 15 × magnification. Preferably slant petri dish at 45° angle on microscope stage and adjust light source vertical to the colonies. Optimal colony density per filter is 20 to 200. If colonies are small and there is no crowding, a higher limit is acceptable. Count all colonies on the membrane when there are ≤ 2 colonies per square. For 3 to 10 colonies per square count 10 squares and obtain average count per square. For 10 to 20 colonies per square count 5 squares and obtain average count per square. Multiply average count per square by 100 and divide by the sample volume to give colonies per milliliter. If there are more than 20 colonies per square, record count as > 2000 divided by the sample volume. Report averaged counts as estimated colony-forming units. Make estimated counts only when there are discrete, separated colonies without spreaders. 9. Computing and Reporting Counts The term ‘‘colony-forming units’’ (CFU) is descriptive of the methods used; therefore, report all counts as colony-forming units. Include in the report the method used, the incubation temperature and time, and the medium. For example: CFU/mL, pour plate method, 35°C/48 h, plate count agar. To compute the heterotrophic plate count, CFU/mL, divide total number of colonies or average number (if duplicate plates of the same dilution) per plate by the sample volume. Record sample volumes used and number of colonies on each plate counted or estimated. When colonies on duplicate plates and/or consecutive dilutions are counted and results are averaged before being recorded, round off counts to two significant figures only when converting to colony-forming units. Avoid creating fictitious precision and accuracy when computing colony-forming units by recording only the first two left-hand digits. Raise the second digit to the next higher number when the third digit from the left is 5, 6, 7, 8, or 9; use zeros for each successive digit toward the right from the second digit. For example, report a count of 142 as 140 and a count of 155 as 160, but report a count of 35 as 35. 10. Analytical Bias Avoid inaccuracies in counting due to carelessness, damaged or dirty optics that impair vision, or failure to recognize colonies. Laboratory workers who cannot duplicate their own counts on the same plate within 5% and the counts of other analysts within 10% should discover © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater the cause and correct such disagreements. 9215 B. Pour Plate Method 1. Samples and Sample Preparation See Section 9215A.4 and Section 9215A.5. 2. Sample Dilution Prepare water used for dilution blanks as directed in Section 9050C. a. Selecting dilutions: Select the dilution(s) so that the total number of colonies on a plate will be between 30 and 300 (Figure 9215:1). For example, where a heterotrophic plate count as high as 3000 is suspected, prepare plates with 10−2 dilution. For most potable water samples, plates suitable for counting will be obtained by plating 1 mL and 0.1 mL undiluted sample and 1 mL of the 10−2 dilution. b. Measuring sample portions: Use a sterile pipet for initial and subsequent transfers from each container. If pipet becomes contaminated before transfers are completed, replace with a sterile pipet. Use a separate sterile pipet for transfers from each different dilution. Do not prepare dilutions and pour plates in direct sunlight. Use caution when removing sterile pipets from the container; to avoid contamination, do not drag pipet tip across exposed ends of pipets in the pipet container or across lips and necks of dilution bottles. When removing sample, do not insert pipets more than 2.5 cm below the surface of sample or dilution. c. Measuring dilutions: When discharging sample portions, hold pipet at an angle of about 45° with tip touching bottom of petri dish or inside neck of dilution bottle. Lift cover of petri dish just high enough to insert pipet. Allow 2 to 4 s for liquid to drain from 1-mL graduation mark to tip of pipet. If pipet is not a blow-out type, touch tip of pipet once against a dry spot on petri dish bottom. Less preferably, use a cotton-plugged blow-out-type pipet and gently blow out remaining volume of sample dilution. When 0.1-mL quantities are measured, let diluted sample drain from chosen reference graduation until 0.1 mL has been delivered. Remove pipet without retouching it to dish. Pipet 1 mL, 0.1 mL, or other suitable volume into sterile petri dish before adding melted culture medium. Use decimal dilutions in preparing sample volumes of less than 0.1 mL; in examining sewage or turbid water, do not measure a 0.1-mL inoculum of original sample, but prepare an appropriate dilution. Prepare at least two replicate plates for each sample dilution used. After depositing test portions for each series of plates, pour culture medium and mix carefully. Do not let more than 20 min elapse between starting pipetting and pouring plates. 3. Plating a. Melting medium: Melt sterile solid agar medium in boiling water or by exposure to flowing steam in a partially closed container, but avoid prolonged exposure to unnecessarily high temperatures during and after melting. Do not resterilize plating medium. If the medium is © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater melted in two or more batches, use all of each batch in order of melting, provided that the contents remain fully melted. Discard melted agar that contains precipitate. Maintain melted medium in a water bath between 44 and 46°C until used, preferably no longer than 3 h. In a separate container place a thermometer in water or medium that has been exposed to the same heating and cooling as the plating medium. Do not depend on the sense of touch to indicate proper medium temperature when pouring agar. Use plate count agar, R2A agar, or NWRI agar as specified in Section 9215A.6. Before using a new lot of medium test its suitability. b. Pouring plates: Limit the number of samples to be plated in any one series so that no more than 20 min (preferably 10 min) elapse between dilution of the first sample and pouring of the last plate in the series. Pour at least 10 to 12 mL liquefied medium maintained at 44 to 46°C into each dish by gently lifting cover just high enough to pour. Carefully avoid spilling medium on outside of container or on inside of dish lid when pouring. When pouring agar from flasks or tubes that have been held in a water bath, wipe with clean paper towel and flame the neck before pouring. As each plate is poured mix melted medium thoroughly with test portions in petri dish, taking care not to splash mixture over the edge, by rotating the dish first in one direction and then in the opposite direction, or by rotating and tilting. Let plates solidify (within 10 min) on a level surface. After medium solidifies, invert plates and place in incubator. c. Sterility controls: Check sterility of medium and dilution water blanks by pouring control plates for each series of samples. Prepare additional controls to determine contamination of plates, pipets, and room air. 4. Incubation See Section 9215A.7. 5. Counting, Recording, Computing, and Reporting See Section 9215A.8 and Section 9215A.9. 6. Bibliography BREED, R.S. & W.D. DOTTERER. 1916. The number of colonies allowable on satisfactory agar plates. Tech. Bull. 53, New York Agricultural Experiment Sta. BUTTERFIELD, C.T. 1933. The selection of a dilution water for bacteriological examinations. J. Bacteriol. 23:355; Pub. Health Rep. 48: 681. ARCHAMBAULT, J., J. CUROT & M.H. MCCRADY. 1937. The need of uniformity of conditions for counting plates (with suggestions for a standard colony counter). Amer. J. Pub. Health 27:809. RICHARDS, O.W. & P.C. HEIJN. 1945. An improved dark-field Quebec colony counter. J. Milk Technol. 8:253. BERRY, J.M., D.A. MCNEILL & L.D. WITTER. 1969. Effect of delays in pour plating on bacterial counts. J. Dairy Sci. 52:1456. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater GELDREICH, E.E., H.D. NASH, D.J. REASONER & R.H. TAYLOR. 1972. The necessity of controlling bacterial populations in potable waters: Community water supply. J. Amer. Water Works Assoc. 64:596. GELDREICH, E.E. 1973. Is the total count necessary? Proc. 1st Annu. AWWA Water Quality Technol. Conf., Dec. 3-4, 1973. Cincinnati, Ohio, p. VII-1. American Water Works Assoc., Denver, Colo. GINSBURG, W. 1973. Improved total count techniques. Proc. 1st Annu. AWWA Water Quality Technol. Conf., Dec. 3-4, 1973. Cincinnati, Ohio, p. VIII-1. American Water Works Assoc., Denver, Colo. DUTKA, B.J., A.S.Y. CHAU & J. COBURN. 1974. Relationship of heterotrophic bacterial indicators of water pollution and fecal sterols. Water Res. 8:1047. KLEIN, D.A. & S. WU. 1974. Stress: a factor to be considered in heterotrophic microorganism enumeration from aquatic environments. Appl. Microbiol. 37:429. GELDREICH, E.E., H.D. NASH, D.J. REASONER & R.H. TAYLOR. 1975. The necessity for controlling bacterial populations in potable waters: Bottled water and emergency water supplies. J. Amer. Water Works Assoc. 67:117. BELL, C.R., M.A. HOLDER-FRANKLIN & M. FRANKLIN. 1980. Heterotrophic bacteria in two Canadian rivers.—I. Seasonal variation in the predominant bacterial populations. Water Res. 14:449. MEANS, E.G., L. HANAMI, G.F. RIDGWAY & B.H. OLSON. 1981. Evaluating mediums and plating techniques for enumerating bacteria in water distribution systems. J. Amer. Water Works Assoc. 73: 585. AMERICAN PUBLIC HEALTH ASSOCIATION. 1993. Standard Methods for the Examination of Dairy Products, 16th ed. American Public Health Assoc., Washington, D.C. REASONER, D.J. & E.E. GELDREICH. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49:1. 9215 C. Spread Plate Method 1. Laboratory Apparatus a. Glass rods: Bend 4-mm-diam fire-polished glass rods, 200 mm in length, 45° about 40 mm from one end. Sterilize before using. b. Pipet, glass, 1.1 mL, with tempered, rounded tip. Do not use disposable plastic pipets. c. Turntable (optional).*#(21) d. Incubator or drying oven, set at 42°C, or laminar-flow hood. 2. Media See Section 9215A.6a, Section 9215A.6c, and Section 9215A.6d. If R2A agar is used best © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater results are obtained at 28°C with 7 d incubation; if NWRI is used, incubate at 20°C for 7 d. 3. Preparation of Plates Pour 15 mL of the desired medium into sterile 100 × 15 or 90 × 15 petri dishes; let agar solidify. Predry plates inverted so that there is a 2- to 3-g water loss overnight with lids on. See Figure 9215:2, Table 9215:I, or Figure 9215:3. Use predried plates immediately after drying or store for up to 2 weeks in sealed plastic bags at 4°C. For predrying and using plates the same day, pour 25 mL agar into petri dish and dry in a laminar-flow hood at room temperature (24 to 26°C) with the lid off to obtain the desired 2- to 3-g weight loss. See Figure 9215:3. 4. Procedure Prepare sample dilutions as directed in 9215B.2. a. Glass rod: Pipet 0.1 or 0.5 mL sample onto surface of predried agar plate. Using a sterile bent glass rod, distribute inoculum over surface of the medium by rotating the dish by hand or on a turntable. Let inoculum be absorbed completely into the medium before incubating. b. Pipet: Pipet desired sample volume (0.1, 0.5 mL) onto the surface of the predried agar plate while dish is being rotated on a turntable. Slowly release sample from pipet while making one to-and-fro motion, starting at center of the plate and stopping 0.5 cm from the plate edge before returning to the center. Lightly touch the pipet to the plate surface. Let inoculum be absorbed completely by the medium before incubating. 5. Incubation See Section 9215A.7. 6. Counting, Recording, Computing, and Reporting See Section 9215A.8 and Section 9215A.9. 7. Bibliography BUCK, J.D. & R.C. CLEVERDON. 1960. The spread plate as a method for the enumeration of marine bacteria. Limnol. Oceanogr. 5:78. CLARK, D.S. 1967. Comparison of pour and surface plate methods for determination of bacterial counts. Can. J. Microbiol. 13:1409. VAN SOESTBERGAN, A.A. & C.H. LEE. 1969. Pour plates or streak plates. Appl. Microbiol. 18:1092. CLARK, D.S. 1971. Studies on the surface plate method of counting bacteria. Can. J. Microbiol. 17:943. GILCHRIST, J.E., J.E. CAMPBELL, C.B. DONNELLY, J.T. PELLER & J.M. DELANEY. 1973. Spiral plate method for bacterial determination. Appl. Microbiol. 25:244. PTAK, D.M. & W. GINSBURG. 1976. Pour plate vs. streak plate method. Proc. 4th Annu. AWWA Water Quality Technol. Conf., Dec. 6-7, 1976. San Diego, Cal., p. 2B-5. American Water Works Assoc., Denver, Colo. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater DUTKA, B.J., ed. 1978. Methods for Microbiological Analysis of Waters, Wastewaters and Sediments. Inland Waters Directorate, Scientific Operation Div., Canada Centre for Inland Waters, Burlington, Ont. KAPER, J.B., A.L. MILLS & R.R. COLWELL. 1978. Evaluation of the accuracy and precision of enumerating aerobic heterotrophs in water samples by the spread method. Appl. Environ. Microbiol. 35:756. YOUNG, M. 1979. A modified spread plate technique for the determination of concentrations of viable heterotrophic bacteria. STP 673:41-51, American Soc. Testing & Materials, Philadelphia, Pa. GELDREICH, E.E. 1981. Current status of microbiological water quality criteria. ASM News 47:23. TAYLOR, R.H., M.J. ALLEN & E.E. GELDREICH. 1981. Standard plate count: A comparison of pour plate and spread plate methods. Proc. 9th Annu. AWWA Water Quality Technol. Conf., Dec. 6-9, 1981. Seattle, Wash., p. 223. American Water Works Assoc. Denver, Colo. 9215 D. Membrane Filter Method 1. Laboratory Apparatus See Section 9222B.1. 2. Media See Section 9215A.6. Use m-HPC agar, or alternatively R2A or NWRI agar. 3. Preparation of Plates Dispense 5-mL portions of sterile medium*#(22) into 50- × 9-mm petri dishes. Let solidify at room temperature. Prepared plates may be stored inverted in a plastic bag or tight container in a refrigerator, for no longer than 2 weeks. 4. Sample Size The volume to be filtered will vary with the sample. Select a maximum sample size to give 20 to 200 CFU per filter. 5. Procedure Filter appropriate volume through a sterile 47-mm, 0.45-µm, gridded membrane filter, under partial vacuum. Rinse funnel with three 20- to 30-mL portions of sterile dilution water. Place filter on agar in petri dish. 6. Incubation Place dishes in close-fitting box or plastic bag containing moistened paper towels. Incubate at 35 ± 0.5°C for 48 h if using m-HPC agar, or longer if using R2A medium, or at 20 to 28°C for © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5 to 7 d if using NWRI or R2A agar. Duplicate plates may be incubated for other time and temperature conditions as desired. 7. Counting, Recording, Computing, and Reporting See Section 9215A.8 and Section 9215A.9. Report as CFU/mL, membrane filter method, time, medium. 8. Bibliography CLARK, H.F., E.E. GELDREICH, H.L. JETER & P.W. KABLER. 1951. The membrane filter in sanitary bacteriology. Pub. Health Rep. 66:951. STOPERT, E.M., W.T. SOKOSKI & J.T. NORTHAM. 1962. The factor of temperature in the better recovery of bacteria from water by filtration. Can. J. Microbiol. 8:809. TAYLOR, R.H. & E.E. GELDREICH. 1979. A new membrane filter procedure for bacterial counts in potable water and swimming pool samples. J. Amer. Water Works Assoc. 71:402. CLARK, J.A. 1980. The influence of increasing numbers of non-indicator organisms upon the detection of indicator organisms by the membrane filter and presence-absence tests. Can. J. Microbiol. 20: 827. DUTKA, B.J., ed. 1981. Membrane Filtration, Applications, Techniques, and Problems. Marcel Dekker, Inc., New York, N.Y. and Basel, Switzerland. HOADLEY, A.W. 1981. Effect of injury on the recovery of bacteria on membrane filters. In B. J. Dutka, ed. Membrane Filtration, Applications, Techniques, and Problems, p. 413. Marcel Dekker, Inc., New York, N.Y. and Basel, Switzerland. 9216 DIRECT TOTAL MICROBIAL COUNT*#(23) 9216 A. Introduction Direct total cell counts of bacteria in water or wastewater usually exceed counts obtained from heterotrophic plate counts and most probable number methods because, unlike those procedures, direct counts preclude errors caused by viability-related phenomena such as selectivity of growth media, cell clumping, and slow growth rates. 9216 B. Epifluorescence Microscopic Method 1. General Discussion The epifluorescence microscopic method produces direct total cell counts with relative speed (20 to 30 min from time of sampling) and sensitivity. It does not permit differentiation of bacterial cells on the basis of taxonomy, metabolic activity, or viability, and it cannot be used to estimate the microbial biomass because of considerable variation in the volume of individual © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater cells. The method requires an experienced technician who can distinguish microbial cells from debris on the basis of morphology. The method consists of sample fixation for storage, staining with a chemical fluorochrome, vacuum filtration onto a nonfluorescing polycarbonate membrane, and enumeration by counting with an epifluorescence microscope. 2. Apparatus a. Microscope, vertical UV illuminator for epifluorescence with flat field 100× oil immersion objective lens, to give total magnification of at least 1000×. b. Counting graticule, ocular lens micrometer* calibrated with stage micrometer.*#(24) c. Filters,†#(25) including excitation filters (KP 490 and LP 455), beam splitter (LP 510), and barrier filter (LP 520 using mercury lamp, HBO 50). d. Blender or vortex mixer. e. Filtration unit, suitable for use with 25-mm-diam membrane filters. f. Membrane filters, polycarbonate,‡#(26) 25-mm-diam, 0.2-µm pore size (purchase nonfluorescent or prepare by soaking membrane in Irgalan black [2 g/L in 2% acetic acid] for 24 h, then rinse in water and air dry); cellulosic§#(27) 25-mm-diam, 5-µm pore size. g. Syringes, 3-mL, disposable, with disposable syringe filters, 0.2-µm pore size. h. Test tubes, glass, screw-capped, 13- × 125-mm. 3. Reagents a. Phosphate buffer: Dissolve 13.6 g KH2PO4 in water and dilute to 1 L. Adjust to pH 7.2 if necessary; filter through 0.2-µm membrane filter. b. Fixative, 5.0% (w/v) glutaraldehyde in phosphate buffer. Prepare fresh daily. c. Fluorochrome, 0.1% (w/v) acridine orangei#(28) in phosphate buffer. d. Immersion oil, low fluorescing.##(29) 4. Procedure Collect water samples as directed in Section 9060. Add 9.0 mL sample to test tube containing 1.0 mL fixative. Fixed samples can be stored at 4°C for up to 3 weeks without significant decrease in cell numbers. Disperse and dilute samples from mesotrophic or eutrophic sources to obtain reproducible results. Mix sample using blender or vortex mixer, then make tenfold dilutions in phosphate buffer as necessary. Clean water samples may not require dilution but larger sample volumes (>100 mL) may be required to obtain reliable counts. Place 1 mL sample or dilution on a nonfluorescent polycarbonate filter supported by a cellulosic membrane filter in filter holder. Using disposable sterile syringe filters, add 1 mL fluorochrome and wait 2 min, then add about 3 mL filtered phosphate buffer to promote more © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater even cell distribution. Alternatively, combine fluorochrome with sample in a small clean vial, let react, and add mixture to filter holder. Filter with vacuum (about 13 kPa). Wash with 2 mL phosphate buffer and filter. Remove polycarbonate filter with forceps and air dry for 1 to 2 min. The filter can be cut into quarter sections and saved if needed. Place dried filter on a drop of immersion oil on a clean glass microscope slide. Add a small drop of immersion oil to filter surface. Gently cover filter with a clean glass cover slip. Samples can be stored in the dark for several months without significant loss of fluorescence. Examine at least 10 randomly selected fields on the filter using the 100× oil immersion lens to establish that distribution of microbial cells is uniform and that individual cells can be enumerated (if not, dilute sample and repeat). Preferably count 10 to 50 cells per field. Count number of cells in at least 20 squares using the calibrated counting graticule. 5. Calculations Calculate the average number of cells per filter. Obtain effective filter area from specifications of filtration unit. Extrapolate to determine number of cells per milliliter of sample: Total cells/mL = (avg cells/square) × (squares/filter) × (dilution factor) / sample volume, mL. 6. Bibliography HOBBIE, J.E., R.J. DALEY & S. JASPER. 1977. Use of nuclepore filters for counting bacteria by fluorescence microscopy. Appl. Environ. Microbiol. 33:1225. SIERACK, M.E., P.W. JOHNSON & J.MCH. SIEBURTH. 1985. Detection, enumeration, and sizing of planktonic bacteria by image-analyzed epifluorescence microscopy. Appl. Environ. Microbiol. 49:799. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1987. Standard test method for enumeration of aquatic bacteria by epifluorescence microscopy counting procedure. ASTM D4455-85, Annual Book of ASTM Standards, Vol. 11.02, Water. American Soc. Testing & Materials, Philadelphia, Pa. 9217 ASSIMILABLE ORGANIC CARBON*#(30) 9217 A. Introduction 1. Significance Growth of bacteria in drinking water distribution and storage systems can lead to the deterioration of water quality, violation of water quality standards, and increased operating costs. Growth or regrowth results from viable bacteria surviving the disinfection process and utilizing nutrients in the water and biofilm to sustain growth.1 Factors other than nutrients that influence © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater regrowth include temperature,2 residence time in mains and storage units,3 and the efficacy of disinfection.4 Tests to determine the potential for bacterial regrowth focus on the concentration of nutrients.5-7 Not all organic compounds are equally susceptible to microbial decomposition; the fraction that provides energy and carbon for bacterial growth has been called labile dissolved organic carbon,8,9 biodegradable organic carbon (BDOC),7 or assimilable organic carbon (AOC).5 Easily measured chemical surrogates for AOC are not available now.10,11 As alternatives to chemical methods, bioassays have been proposed.5-7,12-14 In a bioassay, the growth of a bacterial inoculum to maximum density can be used to estimate the concentrations of limiting nutrients; the underlying assumptions of the AOC bioassay are that nitrogen and phosphorus are present in excess, i.e., that organic carbon is limiting, and that the bioassay organism(s) represent the physiological capabilities of the distribution system microflora. Various bioassay procedures use an inoculum of one to four species of bacteria5,12,13,15,16 growing in log phase or present in late stationary phase, or may use undefined bacteria attached to a sand substratum,7 suspended in the sample,6 or filtered from the sample and then resuspended.14 Incubation vessels vary as to material,17 size,18,19 closure,18 and cleaning procedure.5,18,19 Water to be tested for nutrient concentrations has been variously prepared.5,7,14 The AOC bioassay is an indirect or surrogate method, wherein nutrient concentrations are not measured directly, but colony-forming units (CFU) of the bioassay organism(s) are the test variable. Nutrient concentrations have been estimated directly from changes in dissolved organic carbon concentrations within the test vessel7 or indirectly from epifluorescence microscopic counts of the maximum number of bacterial cells grown,13,14 turbidity,14 or incorporation of tritiated thymidine into bacterial DNA.6,20 CFU densities, total cell densities, or bacterial production are converted to nutrient concentration by the growth yield of bacteria, defined as either the ratio between CFU or cells produced and organic carbon used, or biomass produced and organic carbon used.5,6 2. Selection of Method The method described below is a two-species bioassay using Pseudomonas fluorescens strain P-17 and Spirillum strain NOX (van der Kooij)10 that has been modified to reduce problems of bacterial and carbon contamination.18,19 It uses a defined inoculum and miniaturized incubation vessels, requires no specialized equipment, and has been related to the presence of coliforms in a drinking water distribution system.22 The two-species inoculum probably underestimates the total quantity of AOC, is consistently lower than BDOC estimates, and does not provide an estimate of refractory organic carbon.23 Critical aspects of the proposed method, including the preparation of the incubation vessel, test water, and inoculum, and enumeration of the test organisms, are transferable to alternate AOC assays that use a different defined inoculum. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater With an undefined bacterial inoculum, enumeration by the spread plate technique is not applicable; alternate response variables, such as changes in dissolved organic carbon (DOC) concentration, turbidity, epifluorescence microscopic counts, bacterial mortality, or bacterial growth, have been used.6,7,14 3. Sampling and Storage Follow precautions outlined in Section 9060A and Section 9060B for collecting and storing samples. Pasteurized and dechlorinated water samples probably can be held for several days without deterioration if properly sealed. Initiate the AOC assay as quickly as possible after pasteurization (see ¶ B.4c). 4. References 1. CHARACKLIS, W.G. 1988. Bacterial Regrowth in Distribution Systems. American Water Works Assoc. Research Foundation Research Rep., American Water Works Assoc., Denver, Colo. 2. FRANSOLET, G., G. VILLERS & W.J. MASSCHELEIN. 1985. Influence of temperature on bacterial development in waters. Ozone Sci. Eng. 7: 205. 3. MAUL, A., A.H. EL-SHAARAWI & J.C. BLOCK. 1985. Heterotrophic bacteria in water distribution systems. I. Spatial and temporal variation. Sci. Total Environ. 44:201. 4. LECHEVALLIER, M.W., C.D. CAWTHON & R.G. LEE. 1988. Factors promoting survival of bacteria in chlorinated water supplies. Appl. Environ. Microbiol. 54:649. 5. VAN DER KOOIJ, D., A. VISSER & W.A.M. HIJNEN. 1982. Determining the concentration of easily assimilable organic carbon in drinking water. J. Amer. Water Works Assoc. 74:540. 6. SERVAIS, P., G. BILLEN & M.C. HASCOET. 1987. Determination of the biodegradable fraction of dissolved organic matter in waters. Water Res. 21:445. 7. JORET, J.C., Y. LEVI, T. DUPIN & M. GILBERT. 1988. Rapid method for estimating bioeliminable organic carbon in water. In Proc. Annu. Conf. American Water Works Association, June 19–23, 1988, Orlando, Fla., p. 1715. American Water Works Assoc., Denver, Colo. 8. WETZEL, R.G. & B.A. MANNY. 1972. Decomposition of dissolved organic carbon and nitrogen compounds from leaves in an experimental hard-water stream. Limnol. Oceanogr. 17:927. 9. OGURA, N. 1975. Further studies on decomposition of dissolved organic matter in coastal seawater. Mar. Biol. 31:101. 10. VAN DER KOOIJ, D. 1988. Assimilable Organic Carbon (AOC) in Water. In The Search for a Surrogate. AWWA Research Foundation/KIWA Cooperative Research Rep. p. 311. American Water Works Assoc. Research Foundation, Denver, Colo. 11. KAPLAN, L.A. & T.L. BOTT. 1990. Nutrients for Bacterial Growth in Drinking Water: Bioassay Evaluation. EPA Project Summary, EPA-600/S2-89-030: 1-7. U.S. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. Environmental Protection Agency, Washington, D.C. KENNY, F.A., J.C. FRY & R.A. BREACH. 1988. Development and Operational Implementation of Modified and Simplified Method for Determination of Assimilable Organic Carbon (AOC) in Drinking Water. International Assoc. Water Pollution Research & Control, Brighton, U.K., pp. 1–5. NEDWELL, D.B. 1987. Distribution and pool sizes of microbially available carbon in sediment measured by a microbiological assay. Microbiol. Ecol. 45:47. WERNER, P. 1984. Investigations on the substrate character of organic substances in connection with drinking water treatment. Zentralbl. Bakt. Hyg. 180:46. VAN DER KOOIJ, D. & W.A.M. HIJNEN. 1983. Nutritional versatility of a starch utilizing Flavobacterium at low substrate concentrations. Appl. Environ. Microbiol. 45:804. VAN DER KOOIJ, D. & W.A.M. HIJNEN. 1984. Substrate utilization of an oxalate-consuming Spirillum species in relation to its growth in ozonated water. Appl. Environ. Microbiol. 47:551. COLBOURNE, J.S., R.M TREW & P.J. DENNIS. 1988. Treatment of water for aquatic bacterial growth studies. J. Appl. Bacteriol. 65:79. KAPLAN, L.A. & T.L. BOTT. 1989. Measurement of assimilable organic carbon in water distribution systems by a simplified bioassay technique. In Advances in Water Analysis and Treatment, Proc. 16th Annu. AWWA Water Quality Technology Conf., Nov. 13–17, 1988, St. Louis, Mo., p. 475. American Water Works Assoc., Denver, Colo. KAPLAN, L.A., T.L. BOTT & D.J. REASONER. 1993. Evaluation and simplification of the assimilable organic carbon nutrient bioassay for bacterial growth in drinking water. Appl. Environ. Microbiol. 59: 1532. MORIARTY, D.J.W. 1986. Measurement of bacterial growth rates in aquatic systems from rates of nucleic acid synthesis. In K.C. Marshall, ed. Advan. Microb. Ecol. 9:245. VAN DER KOOIJ, D., W.A.M. HIJNEN & J.C. KRUITHOF. 1989. The effects of ozonation, biological filtration and distribution on the concentration of easily assimilable organic carbon (AOC) in drinking water. Ozone Sci. Eng. 11:297. LECHEVALLIER, M.W., W.H. SHULZ & R.G. LEE. 1989. Bacterial nutrients in drinking water. In M.W. LeChevallier, B.H. Olson & G.A. McFeters, eds. Assessing and Controlling Bacterial Regrowth in Distribution Systems. American Water Works Assoc. Research Foundation Research Rep., American Water Works Assoc., Denver, Colo. PREVOST, M., D. DUCHESNE, J. COALLIER, R. DESJARDINS & P. LAFRANCE. 1990. Full-scale evaluation of biological activated carbon filtration for the treatment of drinking water. In Advances in Water Analysis and Treatment, Proc. 17th Annu. AWWA Water Quality Technology Conf., Nov. 12–16, 1989, Philadelphia, Pa., p. 147. American Water Works Assoc., Denver, Colo. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5. Bibliography VAN DER KOOIJ, D. 1979. Characterization and classification of fluorescent pseudomonads isolated from tap water and surface water. Antonie van Leeuwenhoek 45:225. VAN DER KOOIJ, D., A. VISSER & W.A.M. HIJNEN. 1980. Growth of Aeromonas hydrophila at low concentrations of substrates added to tap water. Appl. Environ. Microbiol. 39:1198. WERNER, P. 1981. Microbial studies on the chemical and biological treatment of ground water containing humic acid. Vom Wasser 57:157. OLSON, B.H. 1982. Assessment and implications of bacterial regrowth in water distribution systems. EPA Project Summary, EPA-600/S2-82-072:1-10. U.S. Environmental Protection Agency, Washington, D.C. RIZET, M., F. FIESSINGER & N. HOUEL. 1982. Bacterial regrowth in a distribution system and its relationship with the quality of the feed water: case studies. In Proc. Annu. Conf. American Water Works Association, May 16–20, 1982, Miami Beach, Fla., p. 1199. American Water Works Assoc., Denver, Colo. VAN DER KOOIJ, D., J.P. ORANJE & W.A.M. HIJNEN. 1982. Growth of Pseudomonas aeruginosa in tap water in relation to utilization of substrates at concentrations of a few micrograms per liter. Appl. Environ. Microbiol. 44:1086. CAMPER, A.K., M.W. LECHEVALLIER, S.C. BROADAWAY & G.A. MCFETERS. 1986. Bacteria associated with granular activated carbon particles in drinking water. Appl. Environ. Microbiol. 52:434. WENG, C., D.L. HOVEN & B.J. SCHWARTZ. 1986. Ozonation: An economic choice for water treatment. J. Amer. Water Works Assoc. 78(11):83. CARLUCCI, A.F., S.L. SHIMP & D.B. CRAVEN. 1987. Bacterial response to labile dissolved organic matter increases associated with marine discontinuities. Fed. European Microbiological Societies, Microbiol. Ecol. 45:211. LECHEVALLIER, M.W., T.M. BABCOCK & R.G. LEE. 1987. Examination and characterization of distribution system biofilms. Appl. Environ. Microbiol. 53:2714. THINGSTAD, T.F. 1987. Utilization of N, P, and organic C by heterotrophic bacteria. I. Outline of a chemostat theory with a consistent concept of maintenance metabolism. Marine Ecol. Progr. Ser. 35:99. ANSELME, C., I.H. SUFFET & J. MALLEVIALLE. 1988. Effects of ozonation on tastes and odors. J. Amer. Water Works Assoc. 80(10):45. FRANSOLET, G., A. DEPELCHIN, G. VILLERS, R. GOOSSENS & W.J. MASSCHELEIN. 1988. The role of bicarbonate in bacterial growth in oligotrophic waters. J. Amer. Water Works Assoc. 80(11):57. 9217 B. Pseudomonas fluorescens Strain P-17, Spirillum Strain NOX Method © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. General Discussion a. Principle: The AOC bioassay using Pseudomonas fluorescens strain P-17 and Spirillum strain NOX involves growth to a maximum density of a small inoculum in a batch culture of pasteurized test water. Pasteurization inactivates native microflora. The test organisms are enumerated by the spread plate method for heterotrophic plate counts (Section 9215C) and the density of viable cells is converted to AOC concentrations by an empirically derived yield factor for the growth of P-17 on acetate-carbon and NOX on oxalate-carbon as standards. The number of organisms at stationary phase is assumed to be the maximum number of organisms that can be supported by the nutrients in the sample and the yields on acetate carbon and oxalate carbon are assumed to equal the yield on naturally occurring AOC.1,2 b. Interferences: Untreated surface waters, especially those with high concentrations of suspended solids or high turbidity, can contain large numbers of spore-forming bacteria that may survive pasteurization, grow, and interfere with the enumeration of P-17 and NOX on spread plates. Such waters generally have high AOC concentrations and can be diluted with organic-free water amended with mineral salts or prefiltered through carbon-free filters. Potable waters that have been disinfected and carry a disinfectant residual will inhibit growth of the test organism unless the disinfectant is neutralized. Surface waters from reservoirs treated with copper sulfate also may be inhibitory unless a chelating agent is added to the sample,3 and lime-softened waters with elevated pH values may require pH adjustment. Any amendment to a sample requires a control for AOC contamination. c. Minimum detectable concentration: In theory, concentrations of less than 1 µg C/L can be detected. In practice, organic carbon contamination during glassware preparation and sample handling imposes a limit of detection of approximately 5 to 10 µg AOC/L. 2. Apparatus a. Incubation vessels: Organic-carbon-free borosilicate glass vials (45 mL capacity) with TFE-lined silicone septa. b. Incubator, set at 15 ± 0.5°C. c. Hot water bath capable of achieving and holding 70°C. d. Continuously adjustable pipet*#(31) capable of delivering between 10 and 100 µL. e. Erlenmeyer flask, 125-mL, with ground-glass stopper. f. Apparatus for preparing dilution water and making heterotrophic plate counts: See Section 9050C and Section 9215C. 3. Reagents a. Sodium acetate stock solution, 400 mg acetate-C/L: Dissolve 2.267 g CH3COONa⋅3H2O in 1 L organic-carbon-free, deionized water. Transfer to 45-mL vials, fill to shoulder, cap tightly, and autoclave. Although standard autoclave practice is to loosen caps, keep vials with septa © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater capped tightly for autoclaving. Store at 5°C in tightly capped vials. Solution may be held for up to 6 months. b. Sodium thiosulfate solution: Dissolve 30 g Na2S2O3 in 1 L deionized water. Transfer to 45-mL vials and autoclave as directed in ¶ 3a. c. Buffered water: See Section 9050C. d. R2A agar: See Section 9215A.6c. e. Sodium persulfate solution, 10% (w/v): Dissolve 100 g Na2S2O8 in 1 L deionized water. f. Organic-free water: See Section 5710B.3e. Alternatively, use HPLC-grade bottled water. g. Mineral salts solution: Dissolve 171 mg K2HPO4, 767 mg NH4Cl, and 1.444 g KNO3 in 1 L carbon-free water. Transfer to 45-mL vials and autoclave as directed in ¶ 3a. h. Cultures of strains P-17 (ATCC 49642) and NOX (ATCC 49643).†#(32) 4. Procedure a. Preparation of incubation vessels: Wash 45-mL vials with detergent, rinse with hot water, 0.1N HCl two times, and deionized water three times, dry, cap with foil, and heat to 550°C for 6 h. Soak TFE-lined silicone septa in a 10% sodium persulfate solution for 1 h at 60°C; rinse three times with carbon-free deionized water. Alternatively, use pre-cleaned water sampling vials4 or an equivalent AOC-free vial.‡#(33) Use same cleaning procedure for all glassware. b. Preparation of stock inoculum: Prepare individual turbid suspensions of P-17 and NOX by transferring growth from a slant culture on R2A agar into 2 to 3 mL filtered (0.2 µm), autoclaved sample. Use slant not older than 6 months. The autoclaved sample can be any water that supports growth of P-17 and NOX and is organic-carbon-limited. Neutralize chlorinated samples with sodium thiosulfate (42 µL/50 mL). Transfer 100 µL of suspension to 50 mL filtered, autoclaved sample in a sterile 125-mL ground-glass-stoppered erlenmeyer flask. Add 125 µL sodium acetate solution (suspension contains 1 mg acetate-C/L). Incubate at room temperature (≤ 25°C) until the viable cell count reaches the stationary phase. Organic-carbon limitation will insure complete utilization of acetate-C so that no AOC is transferred with the inoculum. The stationary phase is reached when the viable cell count, as measured by spread plates, reaches maximum value. Store stock cultures for not more than 6 months at 5°C. Before inoculating a bioassay vessel, make a viable count of the culture (spread plate) to determine the appropriate volume of inoculum to be added to each bioassay vessel. c. Preparation of incubation water: Collect samples directly into 10 45-mL vials. Use 9 vials for AOC measurement and 1 for growth control. Fill each vial to the neck (40 mL) within as short a time as possible. Place septa on the vials, TFE side down, and secure with open-topped screw caps. Alternatively, collect 500 mL sample in an organic-carbon-free vessel and pour into each vial. Neutralize samples containing disinfectant residuals with 33 µL sodium thiosulfate solution added to each vial or 0.5 mL per 500-mL sample. Preferably, collect an extra vial to check for residual chlorine after neutralization. In the laboratory, cap vials tightly and pasteurize © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater in 70°C water bath for 30 min. d. Inoculation and incubation: Cool, inoculate with 500 colony-forming units (CFU)/mL each of P-17 and NOX, either by injecting through the septum or by removing cap and using a carbon-free pipet. Plastic, sterile tips for continuously adjustable pipets are suitable. Use the following equation to calculate volume of inoculum: Hold vials at 15°C in the dark for 1 week. If a 15°C incubator is unavailable, incubate at room temperature not to exceed 25°C. Because incubation temperature influences growth yield, record and report temperature. Determine yields as directed below if an alternative temperature is used. e. Enumeration of test bacterium: On incubation days 7, 8, and 9 remove three vials from the incubator. Sample an individual vial on only 1 d. Shake vials vigorously for 1 min, remove 1 mL with a sterile pipet, and prepare a dilution series (see Section 9215B). Plate three dilutions (10–2, 10–3, and 10–4) in duplicate. Incubate plates at 25°C for 3 to 5 d and score the number of colonies of each strain. P-17 colonies appear on plates first; they are 3 to 4 mm in diameter with diffuse yellow pigmentation. NOX colonies are small (1- to 2-mm diam) white dots. It may be necessary to count P-17 and NOX colonies at different dilutions. Sample vials on three separate days to check whether maximum density has been reached. Day-to-day variations of between 11 and 16% of the mean for batch cultures of P-17 in stationary phase are typical.1 A consistent increase in cell densities of 20% or more over the 3-d period indicates that the cultures are not in stationary phase; repeat assay with longer incubation period. Alternatively, collect more samples (three for each additional sampling day) and prepare as in ¶ c above so that extended incubation can be used. A sharp population decrease of approximately 0.5 log over the 3 d is unusual, but may occur. If this happens repeat the assay. f. Determination of yield of P-17 and NOX: The yields of P-17 and NOX on model carbon compounds should be constant if organic carbon is limiting and the incubation temperature is kept constant. It is acceptable to use the previously derived empirical yield values of 4.1 × 106 CFU P-17/µg acetate-C, 1.2 × 107 CFU-NOX/µg acetate-C, and 2.9 × 106 CFU-NOX/µg oxalate-C at 15°C.5 However, the determination of a yield control provides an important check on both the bioassay (see also 6. Quality Control, below) and carbon limitation in the sample. 5. Calculation a. AOC concentration: Average viable count results for the 3 d and calculate concentration of AOC as the product of the mean of the viable counts and the inverse of the yield: µg AOC/L = [(mean P-17 CFU/mL)(1/yield) © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater +(mean NOX CFU/mL)(1/yield)](1000 mL/L) When the empirical yield factors5 are used, the equation becomes: µg AOC/L = [(mean P-17 CFU/mL)(µg acetate-C/4.1 × 106 CFU) +(mean NOX CFU/mL)(µg oxalate-C/2.9 × 106 CFU)] (1000 mL/L) or µg AOC/L = [(mean P-17 CFU/mL)(2.44 × 10–7 µg acetate-C/CFU) +(mean NOX CFU/mL)(3.45 × 10–7 µg oxalate-C/CFU)] (1000 mL/L) In practice, the densities of organisms vary during the stationary phase. Using average density over 3-d period provides a more accurate estimate of the real maximum density. Reporting AOC as µg C/L assumes that the yields on acetate and oxalate are equal to the yields on naturally occurring AOC. To permit data comparisons report incubation temperature, contribution of each species to AOC, and yield factors used. 6. Quality Control See Section 9020B for general quality control procedures. Quality control specific to the AOC bioassay includes testing the inoculum for purity and viability by plating a portion on R2A agar, testing the incubation vessel, inoculum, thiosulfate solution, and any supplemental procedure such as filtration or dilution for organic carbon contamination, testing the P-17 and NOX inocula for yield, and testing the sample for carbon limitation or inhibition of assay organisms. Test all deviations in procedure (see ¶ 6). To make these tests, use separate controls for blank, yield, and growth. The controls outlined below use a single vial and are meant as a trouble-shooting guide. Definitive determination, for example, that the yield is different from a published value or that a sample is inhibitory, requires replication and statistical analysis. a. Blank control: Dilute mineral salts solution 10:1 with carbon-free water. Follow procedures outlined above: Fill a vial to the shoulder with organic-carbon-free water, add 100 µL mineral salts and 100 µL sodium thiosulfate, pasteurize, inoculate with P-17/NOX, incubate, and enumerate growth. b. Yield control: Dilute sodium acetate or sodium oxalate solution 10:1 with carbon-free water, preparing 40 mg C/L working concentrations. Follow procedures outlined above: Fill a vial to the shoulder with carbon-free water, add 100 µL mineral salts, 100 µL sodium thiosulfate, and 100 µL sodium acetate or sodium oxalate working solution, pasteurize, inoculate with P-17/NOX, incubate, and enumerate growth. P-17, unlike NOX, will not grow with oxalate as sole carbon source (oxalate is considered a major by-product of ozonation). NOX growth in HPLC-grade water presumed to be organic carbon-free is to be expected. The yield control is a © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater quality control measurement and is not intended to provide a conversion factor for the calculation of AOC. c. Growth control: Use additional sample of test water collected with the nine AOC vials, (¶ 4c above) but amend with 100 µL diluted mineral salts and 100 µL of diluted acetate or oxalate solution per vial before pasteurization. As with other controls, inoculate with P-17/NOX, incubate, and enumerate growth. d. Yield calculations: If previously derived empirical yield values (see ¶ 4 f above) are not used, a conversion factor can be derived empirically by using pure cultures of P-17 and NOX. Mixed cultures of the organisms cannot be used and a separate blank control for each species is required. Convert density units to CFU/L by multiplying CFU/mL by 1000, and divide by 100 µg acetate or oxalate-C/L. Express yield as CFU P-17 or NOX/ µg acetate-C or oxalate-C. For P-17 and acetate-C, the equation is: e. Interpretation of growth control: Subtract densities of P-17 and NOX that grew in the sample amended with only thiosulfate from the densities of P-17 and NOX that grew in the growth control. Compare difference to the difference between yield and blank controls. If: (growth control − sample) = (yield control − blank control) Then: sample is carbon-limited and not inhibitory If: (growth control − sample) < (yield control − blank control) Then: sample is inhibitory to bioassay organism If: (growth control − sample) > (yield control − blank control) Then: sample is not carbon-limited f. Supplemental procedure check: When using such supplemental procedures as filtration, dilution, or chemical amendment check for carbon contribution to the AOC values. To test a procedure, use carbon-free water and blank control as a base line. Perform the supplemental procedure on additional carbon-free water and compare to densities of P-17 and NOX that grow in the blank control. 7. Precision and Bias © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater The P-17 bioassay performed in a single laboratory using 45-mL vials had a precision of ± 17.5% based on a total of 58 assays with 14 different samples.6 8. References 1. KAPLAN, L.A. & T.L. BOTT. 1989. Measurement of assimilable organic carbon in water distribution systems by a simplified bioassay technique. In Advances in Water Analysis and Treatment, Proc. 16th Annu. AWWA Water Quality Technology Conf., Nov. 13–17, 1988, St. Louis, Mo., p. 475. American Water Works Assoc., Denver, Colo. 2. VAN DER KOOIJ, D., A. VISSER & J.P. ORANJE. 1982. Multiplication of fluorescent pseudomonads at low substrate concentrations in tap water. Antonie van Leeuwenhoek 48:229. 3. LECHEVALLIER, M.W., W.H. SHULZ & R.G. LEE. 1989. Bacterial nutrients in drinking water. In M.W. LeChevallier, B.H. Olson & G.A. McFeters, eds. Assessing and Controlling Bacterial Regrowth in Distribution Systems. American Water Works Assoc. Research Foundation Research Rep., American Water Works Assoc., Denver, Colo. 4. KAPLAN, L.A. & T.L. BOTT. 1990. Modifications to simplify an AOC bioassay for routine use by utilities monitoring bacterial regrowth potential in water distribution systems. In Advances in Water Analysis and Treatment, Proc. 17th Annu. AWWA Water Quality Technology Conf., Nov. 12–16, 1989, Philadelphia, Pa., p. 1031. American Water Works Assoc., Denver, Colo. 5. VAN DER KOOIJ, D., W.A.M. HIJNEN & J.C. KRUITHOF. 1989. The effects of ozonation, biological filtration and distribution on the concentration of easily assimilable organic carbon (AOC) in drinking water. Ozone Sci. Eng. 11:297. 6. KAPLAN, L.A. & T.L. BOTT. 1990. Nutrients for bacterial growth in drinking water. Bioassay evaluation. EPA Project Summary, EPA-600/S2-89-030: 1-7. U.S. Environmental Protection Agency, Washington, D.C. 9. Bibliography KING, E.O., M.K. WARD & D.E. RANEY. 1954. Two simple media for the demonstration of pyocyanin and fluorescin. J. Lab. Clin. Med. 44: 301. MASON, J. & D.P. KELLY. 1988. Thiosulfate oxidation by obligately heterotrophic bacteria. Microbial Ecol. 15:123. 9221 MULTIPLE-TUBE FERMENTATION TECHNIQUE FOR MEMBERS OF THE COLIFORM GROUP*#(34) © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9221 A. Introduction The coliform group consists of several genera of bacteria belonging to the family Enterobacteriaceae. The historical definition of this group has been based on the method used for detection (lactose fermentation) rather than on the tenets of systematic bacteriology. Accordingly, when the fermentation technique is used, this group is defined as all facultative anaerobic, gram-negative, non-spore-forming, rod-shaped bacteria that ferment lactose with gas and acid formation within 48 h at 35°C. The standard test for the coliform group may be carried out either by the multiple-tube fermentation technique or presence-absence procedure (through the presumptive-confirmed phases or completed test) described herein, by the membrane filter (MF) technique (Section 9222) or by the enzymatic substrate coliform test (Section 9223). Each technique is applicable within the limitations specified and with due consideration of the purpose of the examination. Production of valid results requires strict adherence to quality control procedures. Quality control guidelines are outlined in Section 9020. When multiple tubes are used in the fermentation technique, results of the examination of replicate tubes and dilutions are reported in terms of the Most Probable Number (MPN) of organisms present. This number, based on certain probability formulas, is an estimate of the mean density of coliforms in the sample. Coliform density, together with other information obtained by engineering or sanitary surveys, provides the best assessment of water treatment effectiveness and the sanitary quality of source water. The precision of each test depends on the number of tubes used. The most satisfactory information will be obtained when the largest sample inoculum examined shows gas in some or all of the tubes and the smallest sample inoculum shows no gas in all or a majority of the tubes. Bacterial density can be estimated by the formula given or from the table using the number of positive tubes in the multiple dilutions (Section 9221C.2). The number of sample portions selected will be governed by the desired precision of the result. MPN tables are based on the assumption of a Poisson distribution (random dispersion). However, if the sample is not adequately shaken before the portions are removed or if clumping of bacterial cells occurs, the MPN value will be an underestimate of the actual bacterial density. 1. Water of Drinking Water Quality When drinking water is analyzed to determine if the quality meets the standards of the U.S. Environmental Protection Agency (EPA), use the fermentation technique with 10 replicate tubes each containing 10 mL, 5 replicate tubes each containing 20 mL, or a single bottle containing a 100-mL sample portion. When examining drinking water by the fermentation technique, process all tubes or bottles demonstrating growth with or without a positive acid or gas reaction to the confirmed phase (Section 9221B.2). Apply the completed test (Section 9221B.3) to not less than 10% of all coliform-positive samples per quarter. Obtain at least one positive sample per quarter. A positive EC broth (Section 9221E) or a positive EC MUG broth (Section 9221F) test result is © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater considered an alternative to the positive completed test phase. For the routine examination of public water supplies the object of the total coliform test is to determine the efficiency of treatment plant operation and the integrity of the distribution system. It is also used as a screen for the presence of fecal contamination. A high proportion of coliform occurrences in a distribution system may be attributed not to treatment failure at the plant or the well source, but to bacterial regrowth in the mains. Because it is difficult to distinguish between coliform regrowth and new contamination, assume all coliform occurrences to be new contamination unless otherwise demonstrated. 2. Water of Other than Drinking Water Quality In the examination of nonpotable waters inoculate a series of tubes with appropriate decimal dilutions of the water (multiples and submultiples of 10 mL), based on the probable coliform density. Use the presumptive-confirmed phase of the multiple-tube procedure. Use the more labor-intensive completed test (Section 9221B.3) as a quality control measure on at least 10% of coliform-positive nonpotable water samples on a seasonal basis. The object of the examination of nonpotable water generally is to estimate the density of bacterial contamination, determine a source of pollution, enforce water quality standards, or trace the survival of microorganisms. The multiple-tube fermentation technique may be used to obtain statistically valid MPN estimates of coliform density. Examine a sufficient number of samples to yield representative results for the sampling station. Generally, the geometric mean or median value of the results of a number of samples will yield a value in which the effect of sample-to-sample variation is minimized. 3. Other Samples The multiple-tube fermentation technique is applicable to the analysis of salt or brackish waters as well as muds, sediments, and sludges. Follow the precautions given above on portion sizes and numbers of tubes per dilution. To prepare solid or semisolid samples weigh the sample and add diluent to make a 10−1 dilution. For example, place 50 g sample in sterile blender jar, add 450 mL sterile phosphate buffer or 0.1% peptone dilution water, and blend for 1 to 2 min at low speed (8000 rpm). Prepare the appropriate decimal dilutions of the homogenized slurry as quickly as possible to minimize settling. 9221 B. Standard Total Coliform Fermentation Technique 1. Presumptive Phase Use lauryl tryptose broth in the presumptive portion of the multiple-tube test. If the medium has been refrigerated after sterilization, incubate overnight at room temperature (20°C) before use. Discard tubes showing growth and/or bubbles. a. Reagents and culture medium: 1) Lauryl tryptose broth: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Tryptose Lactose Dipotassium hydrogen phosphate, K2HPO4 20.0 g 5.0 g 2.75 g Potassium dihydrogen phosphate, KH2PO4 2.75 g Sodium chloride, NaCl Sodium lauryl sulfate Reagent-grade water 5.0 0.1 1 g g L Add dehydrated ingredients to water, mix thoroughly, and heat to dissolve. pH should be 6.8 ± 0.2 after sterilization. Before sterilization, dispense sufficient medium, in fermentation tubes with an inverted vial, to cover inverted vial at least one-half to two-thirds after sterilization. Alternatively, omit inverted vial and add 0.01 g/L bromcresol purple to presumptive medium to determine acid production, the indicator of a positive result in this part of the coliform test. Close tubes with metal or heat-resistant plastic caps. Make lauryl tryptose broth of such strength that adding 100-mL, 20-mL, or 10-mL portions of sample to medium will not reduce ingredient concentrations below those of the standard medium. Prepare in accordance with Table 9221:I. b. Procedure: 1) Arrange fermentation tubes in rows of five or ten tubes each in a test tube rack. The number of rows and the sample volumes selected depend upon the quality and character of the water to be examined. For potable water use five 20-mL portions, ten 10-mL portions, or a single bottle of 100 mL portion; for nonpotable water use five tubes per dilution (of 10, 1, 0.1 mL, etc.). In making dilutions and measuring diluted sample volumes, follow the precautions given in Section 9215B.2. Use Figure 9215:1 as a guide to preparing dilutions. Shake sample and dilutions vigorously about 25 times. Inoculate each tube in a set of five with replicate sample volumes (in increasing decimal dilutions, if decimal quantities of the sample are used). Mix test portions in the medium by gentle agitation. 2) Incubate inoculated tubes or bottles at 35 ± 0.5C. After 24 ± 2 h swirl each tube or bottle gently and examine it for growth, gas, and acidic reaction (shades of yellow color) and, if no gas or acidic reaction is evident, reincubate and reexamine at the end of 48 ± 3 h. Record presence or absence of growth, gas, and acid production. If the inner vial is omitted, growth with acidity signifies a positive presumptive reaction. c. Interpretation: Production of an acidic reaction or gas in the tubes or bottles within 48 ± 3 h constitutes a positive presumptive reaction. Submit tubes with a positive presumptive reaction to the confirmed phase (Section 9221B.2). The absence of acidic reaction or gas formation at the end of 48 ± 3 h of incubation © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater constitutes a negative test. Submit drinking water samples demonstrating growth without a positive gas or acid reaction to the confirmed phase (Section 9221B.2). An arbitrary 48-h limit for observation doubtless excludes occasional members of the coliform group that grow very slowly (see Section 9212). 2. Confirmed Phase a. Culture medium: Use brilliant green lactose bile broth fermentation tubes for the confirmed phase. Brilliant green lactose bile broth: Peptone Lactose Oxgall Brilliant green Reagent-grade water 10.0 10.0 20.0 0.0133 1 g g g g L Add dehydrated ingredients to water, mix thoroughly, and heat to dissolve. pH should be 7.2 ± 0.2 after sterilization. Before sterilization, dispense, in fermentation tubes with an inverted vial, sufficient medium to cover inverted vial at least one-half to two-thirds after sterilization. Close tubes with metal or heat-resistant plastic caps. b. Procedure: Submit all presumptive tubes or bottles showing growth, any amount of gas, or acidic reaction within 24 ± 2 h of incubation to the confirmed phase. If active fermentation or acidic reaction appears in the presumptive tube earlier than 24 ± 2 h, transfer to the confirmatory medium; preferably examine tubes at 18 ± 1 h. If additional presumptive tubes or bottles show active fermentation or acidic reaction at the end of a 48 ± 3- h incubation period, submit these to the confirmed phase. Gently shake or rotate presumptive tubes or bottles showing gas or acidic growth to resuspend the organisms. With a sterile loop 3.0 to 3.5 mm in diameter, transfer one or more loopfuls of culture to a fermentation tube containing brilliant green lactose bile broth or insert a sterile wooden applicator at least 2.5 cm into the culture, promptly remove, and plunge applicator to bottom of fermentation tube containing brilliant green lactose bile broth. Remove and discard applicator. Repeat for all other positive presumptive tubes. Incubate the inoculated brilliant green lactose bile broth tube at 35 ± 0.5°C. Formation of gas in any amount in the inverted vial of the brilliant green lactose bile broth fermentation tube at any time (e.g., 6 ± 1 h, 24 ± 2 h) within 48 ± 3 h constitutes a positive confirmed phase. Calculate the MPN value from the number of positive brilliant green lactose bile tubes as described in Section 9221C. c. Alternative procedure: Use this alternative only for polluted water or wastewater known to produce positive results consistently. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater If all presumptive tubes are positive in two or more consecutive dilutions within 24 h, submit to the confirmed phase only the tubes of the highest dilution (smallest sample inoculum) in which all tubes are positive and any positive tubes in still higher dilutions. Submit to the confirmed phase all tubes in which gas or acidic growth is produced only after 48 h. 3. Completed Phase To establish the presence of coliform bacteria and to provide quality control data, use the completed test on at least 10% of positive confirmed tubes (see Figure 9221:1). Simultaneous inoculation into brilliant green lactose bile broth for total coliforms and EC broth for fecal coliforms (see Section 9221E below) or EC-MUG broth for Escherichia coli may be used. Consider positive EC and EC-MUG broths elevated temperature (44.5°C) results as a positive completed test response. Parallel positive brilliant green lactose bile broth cultures with negative EC or EC-MUG broth cultures indicate the presence of nonfecal coliforms. a. Culture media and reagents: 1) LES Endo agar: See Section 9222B. Use 100- × 15-mm petri plates. 2) MacConkey agar: Peptone Proteose peptone Lactose Bile salts Sodium chloride, NaCl Agar Neutral red. Crystal violet Reagent-grade water 17 3 10 1.5 5 13.5 0.03 0.001 1 g g g g g g g g L Add ingredients to water, mix thoroughly, and heat to boiling to dissolve. Sterilize by autoclaving for 15 min at 121°C. Temper agar after sterilization and pour into petri plates (100 × 15 mm). pH should be 7.1 ± 0.2 after sterilization. 3) Nutrient agar: Peptone Beef extract Agar Reagent-grade water 5.0 3.0 15.0 1 g g g L © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Add ingredients to water, mix thoroughly, and heat to dissolve. pH should be 6.8 ± 0.2 after sterilization. Before sterilization, dispense in screw-capped tubes. After sterilization, immediately place tubes in an inclined position so that the agar will solidify with a sloped surface. Tighten screw caps after cooling and store in a protected, cool storage area. 4) Gram-stain reagents: a) Ammonium oxalate-crystal violet (Hucker’s): Dissolve 2 g crystal violet (90% dye content) in 20 mL 95% ethyl alcohol; dissolve 0.8 g (NH4)2C2O4⋅H2O in 80 mL reagent-grade water; mix the two solutions and age for 24 h before use; filter through paper into a staining bottle. b) Lugol’s solution, Gram’s modification: Grind 1 g iodine crystals and 2 g KI in a mortar. Add reagent-grade water, a few milliliters at a time, and grind thoroughly after each addition until solution is complete. Rinse solution into an amber glass bottle with the remaining water (using a total of 300 mL). c) Counterstain: Dissolve 2.5 g safranin dye in 100 mL 95% ethyl alcohol. Add 10 mL to 100 mL reagent-grade water. d) Acetone alcohol: Mix equal volumes of ethyl alcohol (95%) with acetone. b. Procedure: 1) Using aseptic technique, streak one LES Endo agar (Section 9222B.2) or MacConkey agar plate from each tube of brilliant green lactose bile broth showing gas, as soon as possible after the observation of gas. Streak plates in a manner to insure presence of some discrete colonies separated by at least 0.5 cm. Observe the following precautions when streaking plates to obtain a high proportion of successful isolations if coliform organisms are present: (a) Use a sterile 3-mm-diam loop or an inoculating needle slightly curved at the tip; (b) tap and incline the fermentation tube to avoid picking up any membrane or scum on the needle; (c) insert end of loop or needle into the liquid in the tube to a depth of approximately 0.5 cm; and (d) streak plate for isolation with curved section of the needle in contact with the agar to avoid a scratched or torn surface. Flame loop between second and third quadrants to improve colony isolation. Incubate plates (inverted) at 35 ± 0.5°C for 24 ± 2 h. 2) The colonies developing on LES Endo agar are defined as typical (pink to dark red with a green metallic surface sheen) or atypical (pink, red, white, or colorless colonies without sheen) after 24 h incubation. Typical lactose-fermenting colonies developing on MacConkey agar are red and may be surrounded by an opaque zone of precipitated bile. From each plate pick one or more typical, well-isolated coliform colonies or, if no typical colonies are present, pick two or more colonies considered most likely to consist of organisms of the coliform group, and transfer growth from each isolate to a single-strength lauryl tryptose broth fermentation tube and onto a nutrient agar slant. (The latter is unnecessary for drinking water samples.) If needed, use a colony magnifying device to provide optimum magnification when colonies are picked from the LES Endo or MacConkey agar plates. When transferring colonies, choose © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater well-isolated ones and barely touch the surface of the colony with a flame-sterilized, air-cooled transfer needle to minimize the danger of transferring a mixed culture. Incubate secondary broth tubes (lauryl tryptose broth with inverted fermentation vials inserted) at 35 ± 0.5°C for 24 ± 2 h; if gas is not produced within 24 ± 2 h reincubate and examine again at 48 ± 3 h. Microscopically examine Gram-stained preparations from those 24-h nutrient agar slant cultures corresponding to the secondary tubes that show gas. 3) Gram-stain technique—The Gram stain may be omitted from the completed test for potable water samples only because the occurrences of gram-positive bacteria and spore-forming organisms surviving this selective screening procedure are infrequent in drinking water. Various modifications of the Gram stain technique exist. Use the following modification by Hucker for staining smears of pure culture; include a gram-positive and a gram-negative culture as controls. Prepare separate light emulsions of the test bacterial growth and positive and negative control cultures on the same slide using drops of distilled water on the slide. Air-dry and fix by passing slide through a flame and stain for 1 min with ammonium oxalate-crystal violet solution. Rinse slide in tap water and drain off excess; apply Lugol’s solution for 1 min. Rinse stained slide in tap water. Decolorize for approximately 15 to 30 s with acetone alcohol by holding slide between the fingers and letting acetone alcohol flow across the stained smear until the solvent flows colorlessly from the slide. Do not over-decolorize. Counterstain with safranin for 15 s, rinse with tap water, blot dry with absorbent paper or air dry, and examine microscopically. Gram-positive organisms are blue; gram-negative organisms are red. Results are acceptable only when controls have given proper reactions. c. Interpretation: Formation of gas in the secondary tube of lauryl tryptose broth within 48 ± 3 h and demonstration of gram-negative, nonspore-forming, rod-shaped bacteria from the agar culture constitute a positive result for the completed test, demonstrating the presence of a member of the coliform group. 4. Bibliography MEYER, E.M. 1918. An aerobic spore-forming bacillus giving gas in lactose broth isolated in routine water examination. J. Bacteriol. 3:9. HUCKER, G.J. & H.J. CONN. 1923. Methods of Gram Staining. N.Y. State Agr. Exp. Sta. Tech. Bull. No. 93. NORTON, J.F. & J.J. WEIGHT. 1924. Aerobic spore-forming lactose fermenting organisms and their significance in water analysis. Amer. J. Pub. Health 14:1019. HUCKER, G.J. & H.J. CONN. 1927. Further Studies on the Methods of Gram Staining. N.Y. State Agr. Exp. Sta. Tech. Bull. No. 128. PORTER, R., C.S. MCCLESKEY & M. LEVINE. 1937. The facultative sporulating bacteria producing gas from lactose. J. Bacteriol. 33:163. COWLES, P.B. 1939. A modified fermentation tube. J. Bacteriol. 38:677. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater SHERMAN, V.B.D. 1967. A Guide to the Identification of the Genera of Bacteria. Williams & Wilkins, Baltimore, Md. GELDREICH, E.E. 1975. Handbook for Evaluating Water Bacteriological Laboratories, 2nd ed. EPA-670/9-75-006, U.S. Environmental Protection Agency, Cincinnati, Ohio. EVANS, T.M., C.E. WAARVICK, R.J. SEIDLER & M.W. LECHEVALLIER. 1981. Failure of the most-probable number technique to detect coliforms in drinking water and raw water supplies. Appl. Environ. Microbiol. 41:130. SEIDLER, R.J., T.M. EVANS, J.R. KAUFMAN, C.E. WAARVICK & M.W. LECHEVALLIER. 1981. Limitations of standard coliform enumeration techniques. J. Amer. Water Works Assoc. 73:538. GERHARDS, P., ed. 1981. Manual of Methods for General Bacteriology. American Soc. Microbiology, Washington, D.C. KRIEG, N.R. & J.G. HOLT, eds. 1984. Bergey’s Manual of Systematic Bacteriology, Vol 1. Williams & Wilkins, Baltimore, Md. GREENBERG, A.E. & D.A. HUNT, eds. 1985. Laboratory Procedures for the Examination of Seawater and Shellfish, 5th ed. American Public Health Assoc., Washington, D.C. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1989. National primary drinking water regulations: analytical techniques; coliform bacteria; final rule. Federal Register 54(135):29998 (July 17, 1989). 9221 C. Estimation of Bacterial Density 1. Precision of Fermentation Tube Test Unless a large number of sample portions is examined, the precision of the fermentation tube test is rather low. For example, if only 1 mL is examined in a sample containing 1 coliform organism/mL, about 37% of 1-mL tubes may be expected to yield negative results because of random distribution of the bacteria in the sample. When five tubes, each with 1 mL sample, are used under these conditions, a completely negative result may be expected less than 1% of the time. Consequently, exercise great caution when interpreting the sanitary significance of coliform results obtained from the use of a few tubes with each sample dilution, especially when the number of samples from a given sampling point is limited. 2. Computing and Recording of MPN To calculate coliform density, compute in terms of the Most Probable Number (MPN). The MPN values, for a variety of planting series and results, are given in Table 9221:II, Table 9221:III, and Table 9221:IV. Included in these tables are the 95% confidence limits for each MPN value determined. If the sample volumes used are those found in the tables, report the value corresponding to the number of positive and negative results in the series as the MPN/100 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater mL or report as total or fecal coliform presence or absence. The sample volumes indicated in Table 9221:II and Table 9221:III relate more specifically to finished waters. Table 9221:IV illustrates MPN values for combinations of positive and negative results when five 10-mL, five 1.0-mL, and five 0.1-mL volumes of samples are tested. When the series of decimal dilutions is different from that in the table, select the MPN value from Table 9221:IV for the combination of positive tubes and calculate according to the following formula: When more than three dilutions are used in a decimal series of dilutions, use the results from only three of these in computing the MPN. To select the three dilutions to be used in determining the MPN index, choose the highest dilution that gives positive results in all five portions tested (no lower dilution giving any negative results) and the two next succeeding higher dilutions. Use the results at these three volumes in computing the MPN index. In the examples given below, the significant dilution results are shown in boldface. The number in the numerator represents positive tubes; that in the denominator, the total tubes planted; the combination of positives simply represents the total number of positive tubes per dilution: Example a b c 1 mL 0.1 mL 0.01 mL 0.001 mL Combination of positives MPN Index /100 mL 5/5 5/5 4/5 1/5 2/5 2/5 0/5 0/5 0/5 0/5 5-2-0 5-4-2 0-1-0 5000 2200 20 5/5 0/5 In c, select the first three dilutions so as to include the positive result in the middle dilution. When a case such as that shown below in line d arises, where a positive occurs in a dilution higher than the three chosen according to the rule, incorporate it in the result for the highest chosen dilution, as in e: Example 1 mL 0.1 mL 0.01 mL 0.001 mL Combination of positives MPN Index /100 mL d e 5/5 5/5 3/5 3/5 1/5 2/5 1/5 0/5 5-3-2 5-3-2 1400 1400 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Example 1 mL 0.1 mL 0.01 mL 0.001 mL Combination of positives MPN Index /100 mL When it is desired to summarize with a single MPN value the results from a series of samples, use the geometric mean or the median. Table 9221:IV shows the most likely positive tube combinations. If unlikely combinations occur with a frequency greater than 1% it is an indication that the technique is faulty or that the statistical assumptions underlying the MPN estimate are not being fulfilled. The MPN for combinations not appearing in the table, or for other combinations of tubes or dilutions, may be estimated by Thomas’ simple formula: While the MPN tables and calculations are described for use in the coliform test, they are equally applicable to determining the MPN of any other organisms provided that suitable test media are available. 3. Bibliography MCCRADY, M.H. 1915. The numerical interpretation of fermentation tube results. J. Infect. Dis. 12:183. MCCRADY, M.H. 1918. Tables for rapid interpretation of fermentation-tube results. Pub. Health J. 9:201. HOSKINS, J.K. 1933. The most probable numbers of B. coli in water analysis. J. Amer. Water Works Assoc. 25:867. HOSKINS, J.K. 1934. Most Probable Numbers for evaluation of coli-aerogenes tests by fermentation tube method. Pub. Health Rep. 49:393. HOSKINS, J.K. & C.T. BUTTERFIELD. 1935. Determining the bacteriological quality of drinking water. J. Amer. Water Works Assoc. 27:1101. HALVORSON, H.O. & N.R. ZIEGLER. 1933–35. Application of statistics to problems in bacteriology. J. Bacteriol. 25:101; 26:331,559; 29:609. SWAROOP, S. 1938. Numerical estimation of B. coli by dilution method. Indian J. Med. Res. 26:353. DALLA VALLE, J.M. 1941. Notes on the most probable number index as used in bacteriology. Pub. Health Rep. 56:229. THOMAS, H.A., JR. 1942. Bacterial densities from fermentation tube tests. J. Amer. Water Works © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Assoc. 34:572. WOODWARD, R.L. 1957. How probable is the Most Probable Number? J. Amer. Water Works Assoc. 49:1060. MCCARTHY, J.A., H.A. THOMAS, JR. & J.E. DELANEY. 1958. Evaluation of the reliability of coliform density tests. Amer. J. Pub. Health 48: 1628. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1989. National primary drinking water regulations: analytical techniques; coliform bacteria; final rule. Federal Register 54(135):29998 (July 17, 1989). DE MAN, J.C. 1977. MPN tables for more than one test. European J. Appl. Microbiol. 4:307. 9221 D. Presence-Absence (P-A) Coliform Test The presence-absence (P-A) test for the coliform group is a simple modification of the multiple-tube procedure. Simplification, by use of one large test portion (100 mL) in a single culture bottle to obtain qualitative information on the presence or absence of coliforms, is justified on the theory that no coliforms should be present in 100 mL of a drinking water sample. The P-A test also provides the optional opportunity for further screening of the culture to isolate other indicators (fecal coliform, Aeromonas, Staphylococcus, Pseudomonas, fecal streptococcus, and Clostridium) on the same qualitative basis. Additional advantages include the possibility of examining a larger number of samples per unit of time. Comparative studies with the membrane filter procedure indicate that the P-A test may maximize coliform detection in samples containing many organisms that could overgrow coliform colonies and cause problems in detection. The P-A test is intended for use on routine samples collected from distribution systems or water treatment plants. When sample locations produce a positive P-A result for coliforms, it may be advisable to determine coliform densities in repeat samples. Quantitative information may indicate the magnitude of a contaminating event. 1. Presumptive Phase a. Culture media: 1) P-A broth: This medium is commercially available in dehydrated and in sterile concentrated form. Beef extract Peptone Lactose Tryptose Dipotassium hydrogen phosphate, K2HPO4 3.0 5.0 7.46 9.83 1.35 g g g g g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Potassium dihydrogen phosphate, KH2PO4 1.35 g Sodium chloride, NaCl Sodium lauryl sulfate Bromcresol purple Reagent-grade water 2.46 0.05 0.0085 1 g g g L Make this formulation triple (3×) strength when examining 100-mL samples. Dissolve the P-A broth medium in water without heating, using a stirring device. Dispense 50 mL prepared medium into a screw-cap 250-mL milk dilution bottle. A fermentation tube insert is not necessary. Autoclave for 12 min at 121°C with the total time in the autoclave limited to 30 min or less. pH should be 6.8 ± 0.2 after sterilization. When the PA medium is sterilized by filtration a 6× strength medium may be used. Aseptically dispense 20 mL of the 6× medium into a sterile 250-mL dilution bottle or equivalent container. 2) Lauryl tryptose broth: See Section 9221B.1. b. Procedure: Shake sample vigorously for 5 s (approximately 25 times) and inoculate 100 mL into a P-A culture bottle. Mix thoroughly by inverting bottle once or twice to achieve even distribution of the triple-strength medium throughout the sample. Incubate at 35 ± 0.5°C and inspect after 24 and 48 h for acid reactions. c. Interpretation: A distinct yellow color forms in the medium when acid conditions exist following lactose fermentation. If gas also is being produced, gently shaking the bottle will result in a foaming reaction. Any amount of gas and/or acid constitutes a positive presumptive test requiring confirmation. 2. Confirmed Phase The confirmed phase is outlined in Figure 9221:1. a. Culture medium: Use brilliant green lactose bile fermentation tubes (see Section 9221B.2). b. Procedure: Transfer all cultures that show acid reaction or acid and gas reaction to brilliant green lactose bile (BGLB) broth for incubation at 35 ± 0.5°C (see Section 9221B.2). c. Interpretation: Gas production in the BGLB broth culture within 48 ± 3 h confirms the presence of coliform bacteria. Report result as presence-absence test positive or negative for total coliforms in 100 mL of sample. 3. Completed Phase The completed phase is outlined in Section 9221B.3 and Figure 9221:1. 4. Bibliography WEISS, J.E. & C.A. HUNTER. 1939. Simplified bacteriological examination of water. J. Amer. Water Works Assoc. 31:707. CLARK, J.A. 1969. The detection of various bacteria indicative of water pollution by a © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater presence-absence (P-A) procedure. Can. J. Microbiol. 15:771. CLARK, J.A. & L.T. VLASSOFF. 1973. Relationships among pollution indicator bacteria isolated from raw water and distribution systems by the presence-absence (P-A) test. Health Lab. Sci. 10:163. CLARK, J.A. 1980. The influence of increasing numbers of nonindicator organisms upon the detection of indicator organisms by the membrane filter and presence-absence tests. Can. J. Microbiol. 26: 827. CLARK, J.A., C.A. BURGER & L.E. SABATINOS. 1982. Characterization of indicator bacteria in municipal raw water, drinking water and new main water samples. Can. J. Microbiol. 28:1002. JACOBS, N.J., W.L. ZEIGLER, F.C. REED, T.A. STUKEL & E.W. RICE. 1986. Comparison of membrane filter, multiple-fermentation-tube, and presence-absence techniques for detecting total coliforms in small community water systems. Appl. Environ. Microbiol. 51:1007. RICE, E.W., E.E. GELDREICH & E.J. READ. 1989. The presence-absence coliform test for monitoring drinking water quality. Pub. Health Rep. 104:54. 9221 E. Fecal Coliform Procedure Elevated-temperature tests for distinguishing organisms of the total coliform group that also belong to the fecal coliform group are described herein. Modifications in technical procedures, standardization of methods, and detailed studies of the fecal coliform group have established the value of this procedure. The test can be performed by one of the multiple-tube procedures described here or by membrane filter methods as described in Section 9222. The procedure using A-1 broth is a single-step method. The fecal coliform test (using EC medium) is applicable to investigations of drinking water, stream pollution, raw water sources, wastewater treatment systems, bathing waters, seawaters, and general water-quality monitoring. Prior enrichment in presumptive media is required for optimum recovery of fecal coliforms when using EC medium. The test using A-1 medium is applicable to source water, seawater, and treated wastewater. 1. Fecal Coliform Test (EC Medium) The fecal coliform test is used to distinguish those total coliform organisms that are fecal coliforms. Use EC medium or, for a more rapid test of the quality of shellfish waters, treated wastewaters, or source waters, use A-1 medium in a direct test. a. EC medium: Tryptose or trypticase Lactose Bile salts mixture or bile salts No. 3 Dipotassium hydrogen phosphate, K2HPO4 20.0 5.0 1.5 4.0 g g g g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Potassium dihydrogen phosphate, KH2PO4 1.5 g Sodium chloride, NaCl Reagent-grade water 5.0 g 1 L Add dehydrated ingredients to water, mix thoroughly, and heat to dissolve. pH should be 6.9 ± 0.2 after sterilization. Before sterilization, dispense in fermentation tubes, each with an inverted vial, sufficient medium to cover the inverted vial at least partially after sterilization. Close tubes with metal or heat-resistant plastic caps. b. Procedure: Submit all presumptive fermentation tubes or bottles showing any amount of gas, growth, or acidity within 48 h of incubation to the fecal coliform test. 1) Gently shake or rotate presumptive fermentation tubes or bottles showing gas, growth, or acidity. Using a sterile 3- or 3.5-mm-diam loop or sterile wooden applicator stick, transfer growth from each presumptive fermentation tube or bottle to EC broth (see Section 9221B.2). 2) Incubate inoculated EC broth tubes in a water bath at 44.5 ± 0.2°C for 24 ± 2 h. Place all EC tubes in water bath within 30 min after inoculation. Maintain a sufficient water depth in water bath incubator to immerse tubes to upper level of the medium. c. Interpretation: Gas production with growth in an EC broth culture within 24 ± 2 h or less is considered a positive fecal coliform reaction. Failure to produce gas (with little or no growth) constitutes a negative reaction. If multiple tubes are used, calculate MPN from the number of positive EC broth tubes as described in Section 9221C. When using only one tube for subculturing from a single presumptive bottle, report as presence or absence of fecal coliforms. 2. Fecal Coliform Direct Test (A-1 Medium) a. A-1 broth: This medium may be used for the direct isolation of fecal coliforms from water. Prior enrichment in a presumptive medium is not required. Lactose Tryptone Sodium chloride, NaCl Salicin Polyethylene glycol p-isooctylphenyl ether*#(35) Reagent-grade water 5.0 20.0 5.0 0.5 1.0 1 g g g g mL L Heat to dissolve solid ingredients, add polyethylene glycol p-isooctylphenyl ether, and adjust to pH 6.9 ± 0.1. Before sterilization dispense in fermentation tubes with an inverted vial sufficient medium to cover the inverted vial at least partially after sterilization. Close with metal or heat-resistant plastic caps. Sterilize by autoclaving at 121°C for 10 min. Store in dark at room © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater temperature for not longer than 7 d. Ignore formation of precipitate. Make A-1 broth of such strength that adding 10-mL sample portions to medium will not reduce ingredient concentrations below those of the standard medium. For 10-mL samples prepare double-strength medium. b. Procedure: Inoculate tubes of A-1 broth as directed in Section 9221B.1b1). Incubate for 3 h at 35 ± 0.5°C. Transfer tubes to a water bath at 44.5 ± 0.2°C and incubate for an additional 21 ± 2 h. c. Interpretation: Gas production in any A-1 broth culture within 24 h or less is a positive reaction indicating the presence of fecal coliforms. Calculate MPN from the number of positive A-1 broth tubes as described in Section 9221C. 3. Bibliography PERRY, C.A. & A.A. HAJNA. 1933. A modified Eijkman medium. J. Bacteriol. 26:419. PERRY, C.A. & A.A. HAJNA. 1944. Further evaluation of EC medium for the isolation of coliform bacteria and Escherichia coli. Amer. J. Pub. Health 34:735. GELDREICH, E.E., H.F. CLARK, P.W. KABLER, C.B. HUFF & R.H. BORDNER. 1958. The coliform group. II. Reactions in EC medium at 45°C. Appl. Microbiol. 6:347. GELDREICH, E.E., R.H. BORDNER, C.B. HUFF, H.F. CLARK & P.W. KABLER. 1962. Type distribution of coliform bacteria in the feces of warm-blooded animals. J. Water Pollut. Control Fed. 34:295. GELDREICH, E.E. 1966. Sanitary significance of fecal coliforms in the environment. FWPCA Publ. WP-20-3 (Nov.). U.S. Dep. Interior, Washington, D.C. ANDREWS, W.H. & M.W. PRESNELL. 1972. Rapid recovery of Escherichia coli from estuarine water. Appl. Microbiol. 23:521. OLSON, B.H. 1978. Enhanced accuracy of coliform testing in seawater by a modification of the most-probable-number method. Appl. Microbiol. 36:438. STRANDRIDGE, J.H. & J.J. DELFINO. 1981. A-1 Medium: Alternative technique for fecal coliform organism enumeration in chlorinated wastewaters. Appl. Environ. Microbiol. 42:918. 9221 F. Escherichia coli Procedure (PROPOSED) Escherichia coli is a member of the fecal coliform group of bacteria. This organism in water indicates fecal contamination. Enzymatic assays have been developed that allow for the identification of this organism. In this method E. coli are defined as coliform bacteria that possess the enzyme β-glucuronidase and are capable of cleaving the fluorogenic substrate 4-methylumbelliferyl-β-D-glucuronide (MUG) with the corresponding release of the fluorogen when grown in EC-MUG medium at 44.5°C within 24 ± 2 h or less. The procedure is used as a confirmatory test after prior enrichment in a presumptive medium for total coliform bacteria. This test is performed as a tube procedure as described here or by the membrane filter method as © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater described in Section 9222. The chromogenic substrate procedure (Section 9223) can be used for direct detection of E. coli. Tests for E. coli (using EC-MUG medium) are applicable for the analysis of drinking water, surface and ground water, and wastewater. E. coli is a member of the indigenous fecal flora of warm-blooded animals. The occurrence of E. coli is considered a specific indicator of fecal contamination and the possible presence of enteric pathogens. 1. Escherichia coli Test (EC-MUG medium) Use EC-MUG medium for the confirmation of E. coli. a. EC-MUG medium: Tryptose or trypticase Lactose Bile salts mixture or bile salts No. 3 Dipotassium hydrogen phosphate, K2HPO4 20.0 5.0 1.5 4.0 g g g g Potassium dihydrogen phosphate, KH2PO4 1.5 g Sodium chloride, NaCl 5.0 g 0.05 g 4-methylumbelliferyl-β-D-glucuronide (MUG) Reagent-grade water 1 L Add dehydrated ingredients to water, mix thoroughly, and heat to dissolve. pH should be 6.9 ± 0.2 after sterilization. Before sterilization, dispense in tubes that do not fluoresce under long-wavelength (366 nm) ultraviolet (UV) light. An inverted tube is not necessary. Close tubes with metal or heat-resistant plastic caps. b. Procedure: Submit all presumptive fermentation tubes or bottles showing growth, gas, or acidity within 48 ± 3 h of incubation to the E. coli test. 1) Gently shake or rotate presumptive fermentation tubes or bottles showing growth, gas, or acidity. Using a sterile 3- or 3.5-mm-diam metal loop or sterile wooden applicator stick, transfer growth from presumptive fermentation tube or bottle to EC-MUG broth. 2) Incubate inoculated EC-MUG tubes in a water bath or incubator maintained at 44.5 ± 0.2°C for 24 ± 2 h. Place all EC-MUG tubes in water bath within 30 min after inoculation. Maintain a sufficient water depth in the water-bath incubator to immerse tubes to upper level of medium. c. Interpretation: Examine all tubes exhibiting growth for fluorescence using a long-wavelength UV lamp (preferably 6 W). The presence of bright blue fluorescence is considered a positive response for E. coli. A positive control consisting of a known E. coli (MUG-positive) culture, a negative control consisting of a thermotolerant Klebsiella pneumoniae © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater (MUG-negative) culture, and an uninoculated medium control may be necessary to interpret the results and to avoid confusion of weak auto-fluorescence of the medium as a positive response. If multiple tubes are used, calculate MPN from the number of positive EC-MUG broth tubes as described in Section 9221C. When using only one tube or subculturing from a single presumptive bottle, report as presence or absence of E. coli. 2. Bibliography FENG, P.C.S. & P.A. HARTMAN. 1982. Fluorogenic assays for immediate confirmation of Escherichia coli. Appl. Environ. Microbiol. 43:1320. HARTMAN, P.A. 1989. The MUG (glucuronidase) test for E. coli in food and water. In A. Balows et al., eds., Rapid Methods and Automation in Microbiology and Immunology. Proc. 5th Intl. Symp. on Rapid Methods and Automation in Microbiology & Immunology, Florence, Italy, Nov. 4–6, 1987. SHADIX, L.C. & E.W. RICE. 1991. Evaluation of β-glucuronidase assay for the detection of Escherichia coli from environmental waters. Can. J. Microbiol. 37:908. 9222 MEMBRANE FILTER TECHNIQUE FOR MEMBERS OF THE COLIFORM GROUP*#(36) 9222 A. Introduction The membrane filter (MF) technique is highly reproducible, can be used to test relatively large sample volumes, and usually yields numerical results more rapidly than the multiple-tube fermentation procedure. The MF technique is extremely useful in monitoring drinking water and a variety of natural waters. However, the MF technique has limitations, particularly when testing waters with high turbidity or large numbers of noncoliform (background) bacteria. When the MF technique has not been used previously, it is desirable to conduct parallel tests with the method the laboratory is using currently to demonstrate applicability and comparability. 1. Definition As related to the MF technique, the coliform group is defined as those facultative anaerobic, gram-negative, non-spore-forming, rod-shaped bacteria that develop red colonies with a metallic (golden) sheen within 24 h at 35°C on an Endo-type medium containing lactose. Some members of the total coliform group may produce dark red, mucoid, or nucleated colonies without a metallic sheen. When verified these are classified as atypical coliform colonies. When purified cultures of coliform bacteria are tested, they produce negative cytochrome oxidase and positive β-galactosidase test reactions.†#(37) Generally, pink (non-mucoid), blue, white, or colorless colonies lacking sheen are considered noncoliforms by this technique. 2. Applications © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Turbidity caused by the presence of algae, particulates, or other interfering material may not permit testing of a sample volume sufficient to yield significant results. Low coliform estimates may be caused by the presence of high numbers of noncoliforms or of toxic substances. The MF technique is applicable to the examination of saline waters, but not wastewaters that have received only primary treatment followed by chlorination because of turbidity in high volume samples or wastewaters containing toxic metals or toxic organic compounds such as phenols. For the detection of stressed total coliforms in treated drinking water and chlorinated secondary or tertiary wastewater effluents, use a method designed for stressed organism recovery (see Section 9212B.1). A modified MF technique for fecal coliforms (Section 9212) in chlorinated wastewater may be used if parallel testing over a 3-month period with the multiple-tube fermentation technique shows comparability for each site-specific type of sample. The standard volume to be filtered for drinking water samples is 100 mL. This may be distributed among multiple membranes if necessary. However, for special monitoring purposes, such as troubleshooting water quality problems or identification of coliform breakthrough in low concentrations from treatment barriers, it may be desirable to test 1-L samples. If particulates prevent filtering a 1-L sample through a single filter, divide sample into four portions of 250 mL for analysis. Total the coliform counts on each membrane to report the number of coliforms per liter. Smaller sample volumes will be necessary for source or recreational waters and wastewater effluents that have much higher coliform densities. Statistical comparisons of results obtained by the multiple-tube method and the MF technique show that the MF is more precise (compare Table 9221:II and Table 9221:III with Table 9222:II). Data from each test yield approximately the same water quality information, although numerical results are not identical. 3. Bibliography CLARK, H.F., E.E. GELDREICH, H.L. JETER & P.W. KABLER. 1951. The membrane filter in sanitary bacteriology. Pub. Health Rep. 66:951. KABLER, P.W. 1954. Water examinations by membrane filter and MPN procedures. Amer. J. Pub. Health 44:379. THOMAS, H.A. & R.L. WOODWARD. 1956. Use of molecular filter membranes for water potability control. J. Amer. Water Works Assoc. 48: 1391. MCCARTHY, J.A., J.E. DELANEY & R.J. GRASSO. 1961. Measuring coliforms in water. Water Sewage Works 108:238. LIN, S. 1973. Evaluation of coliform test for chlorinated secondary effluents. J. Water Pollut. Control Fed. 45:498. MANDEL, J. & L.F. NANNI. 1978. Measurement evaluation. In S.L. Inhorn, ed. Quality Assurance Practices for Health Laboratories, p. 209. American Public Health Assoc., Washington, D.C. 9222 B. Standard Total Coliform Membrane Filter Procedure © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Laboratory Apparatus For MF analyses use glassware and other apparatus composed of material free from agents that may affect bacterial growth. a. Sample bottles: See Section 9030B.18. b. Dilution bottles: See Section 9030B.13. c. Pipets and graduated cylinders: See Section 9030B.9. Before sterilization, loosely cover opening of graduated cylinders with metal foil or a suitable heavy wrapping-paper substitute. Immediately after sterilization secure cover to prevent contamination. d. Containers for culture medium: Use clean borosilicate glass flasks. Any size or shape of flask may be used, but erlenmeyer flasks with metal caps, metal foil covers, or screw caps provide for adequate mixing of the medium contained and are convenient for storage. e. Culture dishes: Use sterile borosilicate glass or disposable, presterilized plastic petri dishes, 60 × 15 mm, 50 × 9 mm, or other appropriate size. Wrap convenient numbers of clean, glass culture dishes in metal foil if sterilized by dry heat, or suitable heavy wrapping paper when autoclaved. Incubate loose-lidded glass and disposable plastic culture dishes in tightly closed containers with wet paper or cloth to prevent moisture evaporation with resultant drying of medium and to maintain a humid environment for optimum colony development. Presterilized disposable plastic dishes with tight-fitting lids that meet the specifications above are available commercially and are used widely. Reseal opened packages of disposable dish supplies for storage. f. Filtration units: The filter-holding assembly (constructed of glass, autoclavable plastic, porcelain, or stainless steel) consists of a seamless funnel fastened to a base by a locking device or by magnetic force. The design should permit the membrane filter to be held securely on the porous plate of the receptacle without mechanical damage and allow all fluid to pass through the membrane during filtration. Discard plastic funnels with deep scratches on inner surface or glass funnels with chipped surfaces. Wrap the assembly (as a whole or separate parts) in heavy wrapping paper or aluminum foil, sterilize by autoclaving, and store until use. Alternatively expose all surfaces of the previously cleaned assembly to ultraviolet radiation (2 min exposure) for the initial sanitization before use in the test procedure, or before reusing units between successive filtration series. Field units may be sanitized by dipping or spraying with alcohol and then igniting or immersing in boiling water for 2 min. After submerging unit in boiling water, cool it to room temperature before reuse. Do not ignite plastic parts. Sterile, disposable field units may be used. For filtration, mount receptacle of filter-holding assembly on a 1-L filtering flask with a side tube or other suitable device (manifold to hold three to six filter assemblies) such that a pressure differential (34 to 51 kPa) can be exerted on the filter membrane. Connect flask to a vacuum line, an electric vacuum pump, a filter pump operating on water pressure, a hand aspirator, or other means of securing a pressure differential (138 to 207 kPa). Connect a flask of approximately the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater same capacity between filtering flask and vacuum source to trap carry-over water. g. Membrane filter: Use membrane filters (for additional specifications, see Section 9020) with a rated pore diameter such that there is complete retention of coliform bacteria. Use only those filter membranes that have been found, through adequate quality control testing and certification by the manufacturer, to exhibit: full retention of the organisms to be cultivated, stability in use, freedom from chemical extractables that may inhibit bacterial growth and development, a satisfactory speed of filtration (within 5 min), no significant influence on medium pH (beyond ± 0.2 units), and no increase in number of confluent colonies or spreaders compared to control membrane filters. Use membranes grid-marked in such a manner that bacterial growth is neither inhibited nor stimulated along the grid lines when the membranes with entrapped bacteria are incubated on a suitable medium. Preferably use fresh stocks of membrane filters and if necessary store them in an environment without extremes of temperature and humidity. Obtain no more than a year’s supply at any one time. Preferably use presterilized membrane filters for which the manufacturer has certified that the sterilization technique has neither induced toxicity nor altered the chemical or physical properties of the membrane. If membranes are sterilized in the laboratory, autoclave for 10 min at 121°C. At the end of the sterilization period, let the steam escape rapidly to minimize accumulation of water of condensation on filters. h. Absorbent pads consist of disks of filter paper or other material certified for each lot by the manufacturer to be of high quality and free of sulfites or other substances of a concentration that could inhibit bacterial growth. Use pads approximately 48 mm in diameter and of sufficient thickness to absorb 1.8 to 2.2 mL of medium. Presterilized absorbent pads or pads subsequently sterilized in the laboratory should release less than 1 mg total acidity (calculated as CaCO3) when titrated to the phenolphthalein end point, pH 8.3, using 0.02N NaOH and produce pH levels of 7 ± 0.2. Sterilize pads simultaneously with membrane filters available in resealable kraft envelopes, or separately in other suitable containers. Dry pads so they are free of visible moisture before use. See sterilization procedure described for membrane filters above and Section 9020 for additional specifications on absorbent pads. i. Forceps: Smooth flat forceps, without corrugations on the inner sides of the tips. Sterilize before use by dipping in 95% ethyl or absolute methyl alcohol and flaming. j. Incubators: Use incubators to provide a temperature of 35 ± 0.5°C and to maintain a humid environment (60% relative humidity). k. Microscope and light source: To determine colony counts on membrane filters, use a magnification of 10 to 15 diameters and a cool white fluorescent light source adjusted to give maximum sheen discernment. Optimally use a binocular wide-field dissecting microscope. Do not use a microscope illuminator with optical system for light concentration from an incandescent light source for discerning coliform colonies on Endo-type media. 2. Materials and Culture Media The need for uniformity dictates the use of commercial dehydrated media. Never prepare © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater media from basic ingredients when suitable dehydrated media are available. Follow manufacturer’s directions for rehydration. Store opened supplies of dehydrated media in a desiccator. Commercially prepared media in liquid form (sterile ampule or other) may be used if known to give equivalent results. See Section 9020 for media quality control specifications. Test each new medium lot against a previously acceptable lot for satisfactory performance as described in Section 9020B. With each new lot of Endo-type medium, verify a minimum 10% of coliform colonies, obtained from natural samples or samples with known additions, to establish the comparative recovery of the medium lot. Before use, test each batch of laboratory-prepared MF medium for performance with positive and negative culture controls. Check for coliform contamination at the beginning and end of each filtration series by filtering 20 to 30 mL of dilution or rinse water through the filter. If controls indicate contamination, reject all data from affected samples and request resample. a. LES Endo agar:*#(38) Yeast extract Casitone or trypticase Thiopeptone or thiotone Tryptose Lactose Dipotassium hydrogen phosphate, K2HPO4 1.2 3.7 3.7 7.5 9.4 3.3 g g g g g g Potassium dihydrogen phosphate, KH2PO4 1.0 g Sodium chloride, NaCl Sodium desoxycholate Sodium lauryl sulfate Sodium sulfite, Na2SO3 3.7 0.1 0.05 1.6 g g g g Basic fuchsin Agar Reagent-grade water 0.8 15.0 1 g g L Rehydrate product in 1 L water containing 20 mL 95% ethanol. Do not use denatured ethanol, which reduces background growth and coliform colony size. Bring to a near boil to dissolve agar, then promptly remove from heat and cool to 45 to 50°C. Do not sterilize by autoclaving. Final pH 7.2 ± 0.2. Dispense in 5- to 7-mL quantities into lower section of 60-mm glass or plastic petri dishes. If dishes of any other size are used, adjust quantity to give an equivalent depth of 4 to 5 m. Do not expose poured plates to direct sunlight; refrigerate in the dark, preferably in sealed plastic bags or other containers to reduce moisture loss. Discard unused medium after 2 weeks or sooner if there is evidence of moisture loss, medium © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater contamination, or medium deterioration (darkening of the medium). b. M-Endo medium:†#(39) Tryptose or polypeptone Thiopeptone or thiotone Casitone or trypticase Yeast extract Lactose Sodium chloride, NaCl Dipotassium hydrogen phosphate, K2HPO4 10.0 5.0 5.0 1.5 12.5 5.0 4.375 g g g g g g g Potassium dihydrogen phosphate, KH2PO4 1.375 g Sodium lauryl sulfate Sodium desoxycholate Sodium sulfite, Na2SO3 0.05 0.10 2.10 g g g 1.05 15.0 1 g g L Basic fuchsin Agar (optional) Reagent-grade water 1) Agar preparation—Rehydrate product in 1 L water containing 20 mL 95% ethanol. Heat to near boiling to dissolve agar, then promptly remove from heat and cool to between 45 and 50°C. Dispense 5- to 7-mL quantities into 60-mm sterile glass or plastic petri dishes. If dishes of any other size are used, adjust quantity to give an equivalent depth. Do not sterilize by autoclaving. Final pH should be 7.2 ± 0.2. A precipitate is normal in Endo-type media. Refrigerate finished medium in the dark and discard unused agar after 2 weeks. 2) Broth preparation—Prepare as above, omitting agar. Dispense liquid medium (at least 2.0 mL per plate) onto absorbent pads (see absorbent pad specifications, Section 9222B.1) and carefully remove excess medium by decanting the plate. The broth may have a precipitate but this does not interfere with medium performance if pads are certified free of sulfite or other toxic agents at a concentration that could inhibit bacterial growth. Refrigerated broth may be stored for up to 4 d. c. Buffered dilution rinse water: See Section 9050C.1. 3. Samples Collect samples as directed in Section 9060A and Section 9060B. 4. Coliform Definition Bacteria that produce a red colony with a metallic (golden) sheen within 24 h incubation at © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 35°C on an Endo-type medium are considered members of the coliform group. The sheen may cover the entire colony or may appear only in a central area or on the periphery. The coliform group thus defined is based on the production of aldehydes from fermentation of lactose. While this biochemical characteristic is part of the metabolic pathway of gas production in the multiple-tube test, some variations in degree of metallic sheen development may be observed among coliform strains. However, this slight difference in indicator definition is not considered critical to change its public health significance, particularly if suitable studies have been conducted to establish the relationship between results obtained by the MF and those obtained by the standard multiple-tube fermentation procedure. 5. Procedures a. Selection of sample size: Size of sample will be governed by expected bacterial density. In drinking water analyses, sample size will be limited only by the degree of turbidity or by the noncoliform growth on the medium (Table 9222:I). For regulation purposes, 100 mL is the official sample size. An ideal sample volume will yield 20 to 80 coliform colonies and not more than 200 colonies of all types on a membrane-filter surface. Analyze drinking waters by filtering 100 to 1000 mL, or by filtering replicate smaller sample volumes such as duplicate 50-mL or four replicates of 25-mL portions. Analyze other waters by filtering three different volumes (diluted or undiluted), depending on the expected bacterial density. See Section 9215B.2 for preparation of dilutions. When less than 10 mL of sample (diluted or undiluted) is to be filtered, add approximately 10 mL sterile dilution water to the funnel before filtration or pipet the sample volume into a sterile dilution bottle, then filter the entire dilution. This increase in water volume aids in uniform dispersion of the bacterial suspension over the entire effective filtering surface. b. Sterile filtration units: Use sterile filtration units at the beginning of each filtration series as a minimum precaution to avoid accidental contamination. A filtration series is considered to be interrupted when an interval of 30 min or longer elapses between sample filtrations. After such interruption, treat any further sample filtration as a new filtration series and sterilize all membrane filter holders in use. See Section 9222B.1 f for sterilization procedures and Section 9020B.3m and n for UV cleaning and safety guidelines. c. Filtration of sample: Using sterile forceps, place a sterile membrane filter (grid side up) over porous plate of receptacle. Carefully place matched funnel unit over receptacle and lock it in place. Filter sample under partial vacuum. With filter still in place, rinse the interior surface of the funnel by filtering three 20- to 30-mL portions of sterile dilution water. Alternatively, rinse funnel by a flow of sterile dilution water from a squeeze bottle. This is satisfactory only if the squeeze bottle and its contents do not become contaminated during use. Rinsing between samples prevents carryover contamination. Upon completion of final rinse and the filtration process disengage vacuum, unlock and remove funnel, immediately remove membrane filter with sterile forceps, and place it on selected medium with a rolling motion to avoid entrapment of air. If the agar-based medium is used, place prepared filter directly on agar, invert dish, and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater incubate for 22 to 24 h at 35 ± 0.5°C. If liquid medium is used, place a pad in the culture dish and saturate with at least 2.0 mL M-Endo medium and carefully remove excess medium by decanting the plate. Place prepared filter directly on pad, invert dish, and incubate for 22 to 24 h at 35 ± 0.5°C. Differentiation of some colonies from either agar or liquid medium substrates may be lost if cultures are incubated beyond 24 h. Insert a sterile rinse water sample (100 mL) after filtration of a series of 10 samples to check for possible cross-contamination or contaminated rinse water. Incubate the rinse water control membrane culture under the same conditions as the sample. For nonpotable water samples, preferably decontaminate filter unit after each sample (as described above) because of the high number of coliform bacteria present in these samples. Alternatively, use an additional buffer rinse of the filter unit after the filter is removed to prevent carryover between samples. d. Alternative enrichment technique: Place a sterile absorbent pad in the lid of a sterile culture dish and pipet at least 2.0 mL lauryl tryptose broth, prepared as directed in Section 9221B.1.a1), to saturate pad. Carefully remove any excess liquid from absorbent pad by decanting plate. Aseptically place filter through which the sample has been passed on pad. Incubate filter, without inverting dish, for 1.5 to 2 h at 35 ± 0.5°C in an atmosphere of at least 60% relative humidity. If the agar-based Endo-type medium is used, remove enrichment culture from incubator, lift filter from enrichment pad, and roll it onto the agar surface, which has been allowed to equilibrate to room temperature. Incorrect filter placement is at once obvious, because patches of unstained membrane indicate entrapment of air. Where such patches occur, carefully reseat filter on agar surface. If the liquid medium is used, prepare final culture by removing enrichment culture from incubator and separating the dish halves. Place a fresh sterile pad in bottom half of dish and saturate with at least 2.0 mL of M-Endo medium and carefully remove excess liquid from absorbent pad by decanting plate. Transfer filter, with same precautions as above, to new pad. Discard used enrichment pad. With either the agar or the liquid medium, invert dish and incubate for 20 to 22 h at 35 ± 0.5°C. Proceed to ¶ e below. e. Counting: To determine colony counts on membrane filters, use a low-power (10 to 15 magnifications) binocular wide-field dissecting microscope or other optical device, with a cool white fluorescent light source directed to provide optimal viewing of sheen. The typical coliform colony has a pink to dark-red color with a metallic surface sheen. Count both typical and atypical coliform colonies. The sheen area may vary in size from a small pinhead to complete coverage of the colony surface. Atypical coliform colonies can be dark red, mucoid, or nucleated without sheen. Generally pink, blue, white, or colorless colonies lacking sheen are considered noncoliforms. The total count of colonies (coliform and noncoliform) on Endo-type medium has no consistent relationship to the total number of bacteria present in the original sample. A high count of noncoliform colonies may interfere with the maximum development of coliforms. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Refrigerating cultures (after 22 h incubation) with high densities of noncoliform colonies for 0.5 to 1 h before counting may deter spread of confluence while aiding sheen discernment. Samples of disinfected water or wastewater effluent may include stressed organisms that grow relatively slowly and produce maximum sheen in 22 to 24 h. Organisms from undisinfected sources may produce sheen at 16 to 18 h, and the sheen subsequently may fade after 24 to 30 h. f. Coliform verification: Occasionally, typical sheen colonies may be produced by noncoliform organisms and atypical colonies (dark red or nucleated colonies without sheen) may be coliforms. Preferably verify all typical and atypical colony types. For drinking water, verify all suspect colonies by swabbing the entire membrane or pick at least five typical colonies and five atypical colonies from a given membrane filter culture. For waters other than drinking water, at a minimum, verify at least 10 sheen colonies (and representative atypical colonies of different morphological types) from a positive water sample monthly. See Section 9020B.8. Based on need and sample type, laboratories may incorporate more stringent quality control measures (e.g., verify at least one colony from each typical or atypical colony type from a given membrane filter culture, verify 10% of the positive samples). Adjust counts on the basis of verification results. Verification tests are listed below. 1) Lactose fermentation—Transfer growth from each colony or swab the entire membrane with a sterile cotton swab (for presence-absence results in drinking water samples) and place in lauryl tryptose broth; incubate the lauryl tryptose broth at 35 ± 0.5°C for 48 h. Gas formed in lauryl tryptose broth and confirmed in brilliant green lactose broth (Section 9221B.2 for medium preparation) within 48 h verifies the colony as a coliform. Simultaneous inoculation of both media for gas production is acceptable. Inclusion of EC broth inoculation for 44.5 ± 0.2°C incubation will provide information on the presence of fecal coliforms. Use of EC-MUG with incubation at 44.5 ± 0.2°C for 24 h will provide information on presence of E. coli. See Section 9222G for MF partition procedures. 2) Alternative coliform verifications—Apply this alternative coliform verification procedure to isolated colonies on the membrane filter culture. If a mixed culture is suspected or if colony separation is less than 2 mm, streak the growth to M-Endo medium or MacConkey agar to assure culture purity or submit the mixed growth to the fermentation tube method. a) Rapid test—A rapid verification of colonies utilizes test reactions for cytochrome oxidase (CO) and β-galactosidase. Coliform reactions are CO negative and β-galactosidase positive within 4 h incubation of tube culture or micro (spot) test procedure. b) Commercial multi-test systems—Verify the colony by streaking it for purification, selecting a well-isolated colony, and inoculating into a multi-test identification system for Enterobacteriaceae that includes lactose fermentation and/or β-galactosidase and CO test reactions. 6. Calculation of Coliform Density Compute the count, using membrane filters with 20 to 80 coliform colonies and not more than 200 colonies of all types per membrane, by the following equation: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater If no coliform colonies are observed, report the coliform colonies counted as ‘‘<1 coliform/100 mL.’’ For verified coliform counts, adjust the initial count based upon the positive verification percentage and report as ‘‘verified coliform count/100 mL.’’ a. Water of drinking water quality: While the EPA Total Coliform Rule for public water supply samples requires only a record of coliform presence or absence in 100-mL samples, it may be advisable to determine coliform densities in repeat sampling situations. This is of particular importance when a coliform biofilm problem is suspected in the distribution system. Quantitative information may provide an indication of the magnitude of a contaminating event. With water of good quality, the occurrence of coliforms generally will be minimal. Therefore, count all coliform colonies (disregarding the lower limit of 20 cited above) and use the formula given above to obtain coliform density. If confluent growth occurs, covering either the entire filtration area of the membrane or a portion thereof, and colonies are not discrete, report results as ‘‘confluent growth with (or without) coliforms.’’ If the total number of bacterial colonies, coliforms plus noncoliforms, exceeds 200 per membrane, or if the colonies are not distinct enough for accurate counting, report results as ‘‘too numerous to count’’ (TNTC) or ‘‘confluent,’’ respectively. For drinking water, the presence of coliforms in such cultures showing no sheen may be confirmed by either transferring a few colonies or placing the entire membrane filter culture into a sterile tube of brilliant green lactose bile broth. As an alternative, brush the entire filter surface with a sterile loop, applicator stick, or cotton swab and inoculate this growth to the tube of brilliant green lactose bile broth. If gas is produced from the brilliant green bile broth tube within 48 h at 35 ± 0.5°C, coliforms are present. For compliance with the EPA Total Coliform Rule, report confluent growth or TNTC with at least one detectable coliform colony (which is verified) as a total coliform positive sample. Report confluent growth or TNTC without detectable coliforms as invalid. For invalid samples, request a new sample from the same location within 24 h and select more appropriate volumes to be filtered per membrane, observing the requirement that the standard drinking water portion is 100 mL, or choose another coliform method that is less subject to heterotrophic bacterial interferences. Thus, to reduce interference from overcrowding, instead of filtering 100 mL per membrane, filter 50-mL portions through two separate © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater membranes, 25-mL portions through each of four membranes, etc. Total the coliform counts observed on all membranes and report as number per 100 mL. b. Water of other than drinking water quality: As with potable water samples, if no filter has a coliform count falling in the ideal range, total the coliform counts on all filters and report as number per 100 mL. For example, if duplicate 50-mL portions were examined and the two membranes had five and three coliform colonies, respectively, report the count as eight coliform colonies per 100 mL, i.e., Similarly, if 50-, 25-, and 10-mL portions were examined and the counts were 15, 6, and <1 coliform colonies, respectively, report the count as 25/100 mL, i.e., On the other hand, if 10-, 1.0-, and 0.1-mL portions were examined with counts of 40, 9, and <1 coliform colonies, respectively, select the 10-mL portion only for calculating the coliform density because this filter had a coliform count falling in the ideal range. The result is 400/100 mL, i.e., In this last example, if the membrane with 40 coliform colonies also had a total bacterial colony count greater than 200, report the coliform count as ≥400/100 mL. Report confluent growth or membranes with colonies too numerous to count as described in a above. Request a new sample and select more appropriate volumes for filtration or utilize the multiple-tube fermentation technique. c. Statistical reliability of membrane filter results: Although the precision of the MF technique is greater than that of the MPN procedure, membrane counts may underestimate the number of viable coliform bacteria. Table 9222:II illustrates some 95% confidence limits. These values are based on the assumption that bacteria are distributed randomly and follow a Poisson distribution. For results with counts, c, greater than 20 organisms, calculate the approximate 95% confidence limits using the following normal distribution equations: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 7. Bibliography FIFIELD, C.W. & C.P. SCHAUFUS. 1958. Improved membrane filter medium for the detection of coliform organisms. J. Amer. Water Works Assoc. 50:193. MCCARTHY, J.A. & J.E. DELANEY. 1958. Membrane filter media studies. Water Sewage Works 105:292. RHINES, C.E. & W.P. CHEEVERS. 1965. Decontamination of membrane filter holders by ultraviolet light. J. Amer. Water Works Assoc. 57: 500. GELDREICH, E.E., H.L. JETER & J.A. WINTER. 1967. Technical considerations in applying the membrane filter procedure. Health Lab. Sci. 4:113. WATLING, H.R. & R.J. WATLING. 1975. Note on the trace metal content of membrane filters. Water SA 1:28. LIN, S.D. 1976. Evaluation of Millipore HA and HC membrane filters for the enumeration of indicator bacteria. Appl. Environ. Microbiol. 32:300. STANDRIDGE, J.H. 1976. Comparison of surface pore morphology of two brands of membrane filters. Appl. Environ. Microbiol. 31:316. GELDREICH, E.E. 1976. Performance variability of membrane filter procedure. Pub. Health Lab. 34:100. GRABOW, W.O.K. & M. DU PREEZ. 1979. Comparison of m-Endo LES, MacConkey and Teepol media for membrane filtration counting of total coliform bacteria in water. Appl. Environ. Microbiol. 38:351. DUTKA, B.D., ed. 1981. Membrane Filtration Applications, Techniques and Problems. Marcel Dekker, Inc., New York, N.Y. EVANS, T.M., R.J. SEIDLER & M.W. LECHEVALLIER. 1981. Impact of verification media and resuscitation on accuracy of the membrane filter total coliform enumeration technique. Appl. Environ. Microbiol. 41: 1144. FRANZBLAU, S.G., B.J. HINNEBUSCH, T.M. KELLEY & N.A. SINCLAIR. 1984. Effect of noncoliforms on coliform detection in potable groundwater: improved recovery with an anaerobic membrane filter technique. Appl. Environ. Microbiol. 48:142. MCFETERS, G.A., J.S. KIPPIN & M.W. LECHEVALLIER. 1986. Injured coliforms in drinking water. Appl. Environ. Microbiol. 51:1. 9222 C. Delayed-Incubation Total Coliform Procedure Modification of the standard MF technique permits membrane shipment or transport after © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater filtration to a distant laboratory for transfer to another substrate, incubation, and completion of the test. This delayed-incubation test may be used where it is impractical to apply conventional procedures. It also may be used: (a) where it is not possible to maintain the desired sample temperature during transport; (b) when the elapsed time between sample collection and analysis would exceed the approved time limit; or (c) where the sampling location is remote from laboratory services. Independent studies using both fresh- and salt-water samples have shown consistent results between the delayed incubation and standard direct test. Determine the applicability of the delayed-incubation test for a specific water source by comparing with results of conventional MF methods. To conduct the delayed-incubation test, filter sample in the field immediately after collection, place filter on the transport medium, and ship to the laboratory. Complete the coliform determination in the laboratory by transferring the membrane to standard M-Endo or LES Endo medium, incubating at 35 ± 0.5°C for 20 to 22 h, and counting typical and atypical coliform colonies that develop. For drinking water samples collected for compliance with the EPA Total Coliform Rule, report the presence or absence of verified coliforms in 100-mL samples. Verify colonies as outlined previously in Section 9222B.5 f. Transport media are designed to keep coliform organisms viable and generally do not permit visible growth during transit time. Bacteriostatic agents in holding/preservative media suppress growth of microorganisms en route but allow normal coliform growth after transfer to a fresh medium. The delayed-incubation test follows the methods outlined for the total coliform MF procedure, except as indicated below. Two alternative methods are given, one using the M-Endo preservative medium and the other the M-ST holding medium. 1. Apparatus a. Culture dishes: Use disposable, sterile, plastic petri dishes (50 × 12 mm) with tight-fitting lids. Such containers are light in weight and are less likely to break in transit. In an emergency or when plastic dishes are unavailable, use sterile glass petri dishes wrapped in plastic film or similar material. See Section 9222B.1e for specifications. b. Field filtration units: See Section 9222B.1 f for specifications. Disinfect by adding methyl alcohol to the filtering chamber, igniting the alcohol, and covering unit to produce formaldehyde. Ultraviolet light disinfection also may be used in the field if an appropriate power source is available (115 V, 60 Hz). Glass or metal filtration units may be sterilized by immersing in boiling water for 2 min. Use a hand aspirator to obtain necessary vacuum. 2. Materials and Transport Media a. M-Endo methods: 1) M-Endo preservative medium: Prepare M-Endo medium as described in Section 9222B.2b. After cooling to below 45°C, aseptically add 3.84 g sodium benzoate (USP grade)/L © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater or 3.2 mL 12% sodium benzoate solution to 100 mL medium. Mix ingredients and dispense in 5to 7-mL quantities to 50- × 9-mm petri plates. Refrigerate poured plates. Discard unused medium after 96 h. 2) Sodium benzoate solution: Dissolve 12 g NaC7H5O2 in sufficient reagent water to make 100 mL. Sterilize by autoclaving or by filtering through a 0.22-µm pore size membrane filter. Discard after 6 months. 3) Cycloheximide:*#(40) Optionally add cycloheximide to M-Endo preservative medium. It may be used for samples that previously have shown overgrowth by fungi, including yeasts. Prepare by aseptically adding 50 mg cycloheximide/100 mL to M-Endo preservative medium. Store cycloheximide solution in refrigerator and discard after 6 months. Cycloheximide is a powerful skin irritant; handle with caution according to the manufacturer’s directions. b. M-ST method: M-ST holding medium: Sodium phosphate, monobasic, NaH2PO4⋅H2O 0.1 g Dipotassium hydrogen phosphate, KH2PO4 3.0 g Sulfanilamide Ethanol (95%) Tris (hydroxymethyl) aminomethane Reagent-grade water 1.5 10 3.0 1 g mL g L Dissolve ingredients by rehydrating in water. Sterilize by autoclaving at 121°C for 15 min. Final pH should be 8.6 ± 0.2. Dispense at least 2.0 mL to tight-lidded plastic culture dishes containing an absorbent pad and carefully remove excess liquid from pad by decanting plate. Store in refrigerator for use within 96 h. 3. Procedure a. Sample preservation and shipment: Place absorbent pad in bottom of sterile petri dish and saturate with selected coliform holding medium (see Section 9222C.2 above). Remove membrane filter from filtration unit with sterile forceps and roll it, grid side up, onto surface of medium-saturated pad. Protect membrane from moisture loss by tightly closing plastic petri dish. Seal loose-fitting dishes with an appropriate sealing tape to prevent membrane dehydration during transit. Place culture dish containing membrane in an appropriate shipping container and send to the laboratory for test completion. The sample can be held without visible growth for a maximum of 72 h on the holding/preservative medium. This usually allows use of the mail or a common carrier. Visible growth occasionally begins on the transport medium when high temperatures are encountered during transit. b. Transfer and incubation: At the laboratory, transfer membrane from holding medium on © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater which it was shipped to a second sterile petri dish containing M-Endo or LES Endo medium and incubate at 35 ± 0.5°C for 20 to 22 h. 4. Estimation of Coliform Density Proceed as in Section 9222B.6 above. Record times of collection, filtration, and laboratory examination, and calculate the elapsed time. Report elapsed time with coliform results. 5. Bibliography GELDREICH, E.E., P.W. KABLER, H.L. JETER & H.F. CLARK. 1955. A delayed incubation membrane filter test for coliform bacteria in water. Amer. J. Pub. Health 45:1462. PANEZAI, A.K., T.J. MACKLIN & H.G. COLES. 1965. Coli-aerogenes and Escherichia coli counts on water samples by means of transported membranes. Proc. Soc. Water Treat. Exam. 14:179. BREZENSKI, F.T. & J.A. WINTER. 1969. Use of the delayed incubation membrane filter test for determining coliform bacteria in sea water. Water Res. 3:583. CHEN, M. & P.J. HICKEY. 1986. Elimination of overgrowth in delayed-incubation membrane filter test for total coliforms by M-ST holding medium. Appl. Environ. Microbiol. 52:778. 9222 D. Fecal Coliform Membrane Filter Procedure Fecal coliform bacterial densities may be determined either by the multiple-tube procedure or by the MF technique. See Section 9225 for differentiation of Escherichia coli, the predominant fecal coliform. If the MF procedure is used for chlorinated effluents, demonstrate that it gives comparable information to that obtainable by the multiple-tube test before accepting it as an alternative. The fecal coliform MF procedure uses an enriched lactose medium and incubation temperature of 44.5 ± 0.2°C for selectivity. Because incubation temperature is critical, submerge waterproofed (plastic bag enclosures) MF cultures in a water bath for incubation at the elevated temperature or use an appropriate solid heat sink incubator or other incubator that is documented to hold the 44.5°C temperature within 0.2°C throughout the chamber, over a 24-h period. Areas of application for the fecal coliform method in general are stated in the introduction to the multiple-tube fecal coliform procedures, Section 9221E. 1. Materials and Culture Medium a. M-FC medium: The need for uniformity dictates the use of dehydrated media. Never prepare media from basic ingredients when suitable dehydrated media are available. Follow manufacturer’s directions for rehydration. Commercially prepared media in liquid form (sterile ampule or other) also may be used if known to give equivalent results. See Section 9020 for quality control specifications. M-FC medium: Tryptose or biosate 10.0 g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Proteose peptone No. 3 or polypeptone Yeast extract Sodium chloride, NaCl Lactose Bile salts No. 3 or bile salts mixture Aniline blue Agar (optional) Reagent-grade water 5.0 3.0 5.0 12.5 1.5 0.1 15.0 1 g g g g g g g L Rehydrate product in 1 L water containing 10 mL 1% rosolic acid in 0.2N NaOH.*#(41) Heat to near boiling, promptly remove from heat, and cool to below 50°C. Do not sterilize by autoclaving. If agar is used, dispense 5- to 7-mL quantities to 50- × 12-mm petri plates and let solidify. Final pH should be 7.4 ± 0.2. Refrigerate finished medium, preferably in sealed plastic bags or other containers to reduce moisture loss, and discard unused broth after 96 h or unused agar after 2 weeks. Test each medium lot against a previously acceptable lot for satisfactory performance as described in Section 9020B, by making dilutions of a culture of E. coli (Section 9020) and filtering appropriate volumes to give 20 to 60 colonies per filter. With each new lot of medium verify 10 or more colonies obtained from several natural samples, to establish the absence of false positives. For most samples M-FC medium may be used without the 1% rosolic acid addition, provided there is no interference with background growth. Such interference may be expected in stormwater samples collected during the first runoff (initial flushing) after a long dry period. Before use, test each batch of laboratory-prepared MF medium for performance with positive and negative culture controls. Check for coliform contamination at the beginning and end of each filtration series by filtering 20 to 30 mL of dilution or rinse water through filter. If controls indicate contamination, reject all data from affected samples and request resample. b. Culture dishes: Tight-fitting plastic dishes are preferred because the membrane filter cultures are submerged in a water bath during incubation. Place fecal coliform cultures in plastic bags or seal individual dishes with waterproof (freezer) tape to prevent leakage during submersion. Specifications for plastic culture dishes are given in Section 9222B.1e. c. Incubator: The specificity of the fecal coliform test is related directly to the incubation temperature. Static air incubation may be a problem in some types of incubators because of potential heat layering within the chamber, slower heat transfer from air to the medium, and the slow recovery of temperature each time the incubator is opened during daily operations. To meet the need for greater temperature control use a water bath, a heat-sink incubator, or a properly designed and constructed incubator shown to give equivalent results. A temperature tolerance of 44.5 ± 0.2°C can be obtained with most types of water baths that also are equipped with a gable © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater top for the reduction of water and heat losses. 2. Procedure a. Selection of sample size: Select volume of water sample to be examined in accordance with the information in Table 9222:III. Use sample volumes that will yield counts between 20 and 60 fecal coliform colonies per membrane. When the bacterial density of the sample is unknown, filter several volumes or dilutions to achieve a countable density. Estimate volume and/or dilution expected to yield a countable membrane and select two additional quantities representing one-tenth and ten times this volume, respectively. b. Filtration of sample: Follow the same procedure and precautions as prescribed under Section 9222B.5b above. c. Preparation of culture dish: Place a sterile absorbent pad in each culture dish and pipet at least 2.0 mL M-FC medium, prepared as directed above, to saturate pad. Carefully remove any excess liquid from culture dish by decanting the plate. Aseptically, place prepared filter on medium-impregnated pad as described in Section 9222B above. As a substrate substitution for the nutrient-saturated absorbent pad, add 1.5% agar to M-FC broth as described in Section 9222B above. d. Incubation: Place prepared dishes in waterproof plastic bags or seal, invert, and submerge petri dishes in water bath, and incubate for 24 ± 2 h at 44.5 ± 0.2°C. Anchor dishes below water surface to maintain critical temperature requirements. Place all prepared cultures in the water bath within 30 min after filtration. Alternatively, use an appropriate, accurate solid heat sink or equivalent incubator. e. Counting: Colonies produced by fecal coliform bacteria on M-FC medium are various shades of blue. Nonfecal coliform colonies are gray to cream-colored. Normally, few nonfecal coliform colonies will be observed on M-FC medium because of selective action of the elevated temperature and addition of rosolic acid salt reagent. Count colonies with a low-power (10 to 15 magnifications) binocular wide-field dissecting microscope or other optical device. f. Verification: Verify typical blue colonies and any atypical grey to green colonies as described in Section 9020 for fecal coliform analysis. Simultaneous inoculation at both temperatures is acceptable. 3. Calculation of Fecal Coliform Density a. General: Compute the density from the sample quantities that produced MF counts within the desired range of 20 to 60 fecal coliform colonies. This colony density range is more restrictive than the 20 to 80 total coliform range because of larger colony size on M-FC medium. Calculate fecal coliform density as directed in Section 9222B.6 above. Record densities as fecal coliforms per l00 mL. b. Sediment and biosolid samples: For total solid (dry weight basis) see Section 2540G. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Calculate fecal coliforms per gram dry weight for biosolid analysis as follows: where dilution and % dry solids are expressed in decimal form. Example 1: There were 22 colonies observed on the 1:10 000 dilution plate of a biosolid with 4% dry solids. If no filter has a coliform count falling in the ideal range (20 to 60), total the coliform counts on all countable filters and report as fecal coliforms per gram dry weight. Example 2: There were 18 colonies observed on the 1:10 000 dilution plate and 2 colonies observed on the 1:100 000 dilution plate of a biosolid sample with 4% dry solids. To compute a geometric mean of samples, convert coliform densities of each sample to log10 values. Determine the geometric mean for the given number of samples (usually seven) by averaging the log10 values of the coliform densities and taking the antilog of that value. 4. Bibliography GELDREICH, E.E., H.F. CLARK, C.B. HUFF & L.C. BEST. 1965. Fecal-coliform-organism medium for the membrane filter technique. J. Amer. Water Works Assoc. 57:208. ROSE, R.E., E.E. GELDREICH & W. LITSKY. 1975. Improved membrane filter method for fecal coliform analysis. Appl. Microbiol. 29:532. LIN, S.D. 1976. Membrane filter method for recovery of fecal coliforms in chlorinated sewage effluents. Appl. Environ. Microbiol. 32:547. PRESSWOOD, W.G. & D.K. STRONG. 1978. Modification of M-FC medium by eliminating rosolic acid. Appl. Environ. Microbiol. 36:90. GREEN, B.L., W. LITSKY & K.J. SLADEK. 1980. Evaluation of membrane filter methods for enumeration of faecal coliforms from marine waters. Mar. Environ. Res. 67:267. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater SARTORY, D.P. 1980. Membrane filtration faecal coliform determinations with unmodified and modified M-FC medium. Water SA 6:113. GRABOW, W.O.K., C.A. HILNER & P. COUBROUGH. 1981. Evaluation of standard and modified M-FC, MacConkey, and Teepol media for membrane filter counting of fecal coliform in water. Appl. Environ. Microbiol. 42:192. RYCHERT, R.C. & G.R. STEPHENSON. 1981. Atypical Escherichia coli in streams. Appl. Environ. Microbiol. 41:1276. PAGEL, J.E., A.A. QURESHI, D.M. YOUNG & L.T. VLASSOFF. 1982. Comparison of four membrane filter methods for fecal coliform enumeration. Appl. Environ. Microbiol. 43:787. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1992. Environmental Regulations and Technology. Control of Pathogens and Vector Attraction in Sewage Sludge. EPA-626/R-92-013, Washington, D.C. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1993. Standards for the Use or Disposal of Sewage Sludge: Final Rule. 40 CFR Part 257; Federal Register 58:9248, Feb. 19, 1993. 9222 E. Delayed-Incubation Fecal Coliform Procedure This delayed-incubation procedure is similar to the delayed-incubation total coliform procedure (Section 9222C). Use the delayed-incubation test only when the standard immediate fecal coliform test cannot be performed (i.e., where the appropriate field incubator is not available, or where, under certain circumstances, a specialized laboratory service is advisable to examine, confirm, or speciate the suspect colonies). Results obtained by this delayed method have been consistent with results from the standard fecal coliform MF test under various laboratory and field use conditions. However, determine test applicability for a specific water source by comparison with the standard MF test, especially for saline waters, chlorinated wastewaters, and waters containing toxic substances. To conduct the delayed-incubation test filter sample in the field immediately after collection, place filter on M-ST holding medium (see Section 9222C.2b below), and ship to the laboratory. Complete fecal coliform test by transferring filter to M-FC medium, incubating at 44.5°C for 24 ± 2 h, and counting fecal coliform colonies. The M-ST medium keeps fecal coliform organisms viable but prevents visible growth during transit. Membrane filters can be held for up to 3 d on M-ST holding medium with little effect on the fecal coliform counts. 1. Apparatus a. Culture dishes: See Section 9222C.1a for specifications. b. Field filtration units: See Section 9222C.1b. 2. Materials and Transport Medium a. M-ST medium: Prepare as described in Section 9222C.2b. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater b. M-FC medium: Prepare as described in Section 9222D.1a. 3. Procedure a. Membrane filter transport: Place an absorbent pad in a tight-lid plastic petri dish and saturate with M-ST holding medium. After filtering sample remove membrane filter from filtration unit and place it on medium-saturated pad. Use only tight-lid dishes to prevent moisture loss; however, avoid having excess liquid in the dish. Place culture dish containing membrane in an appropriate shipping container and send to laboratory. Membranes can be held on the transport medium at ambient temperature for a maximum of 72 h with little effect on fecal coliform counts. b. Transfer: At the laboratory remove membrane from holding medium and place it in another dish containing M-FC medium. c. Incubation: After transfer of filter to M-FC medium, place tight-lid dishes in waterproof plastic bags, invert, and submerge in a water bath at 44.5°C ± 0.2°C for 24 ± 2 h or use a solid heat sink or equivalent incubator. d. Counting: Colonies produced by fecal coliform bacteria are various shades of blue. Nonfecal coliform colonies are gray to cream-colored. Count colonies with a binocular wide-field dissecting microscope at 10 to 15 magnifications. e. Verification: Verify typical blue colonies and any atypical (grey to green) colonies as described in Section 9020 for fecal coliform analysis. 4. Estimation of Fecal Coliform Density Count as directed in Section 9222D.2e above and compute fecal coliform density as described in Section 9222D.3. Record time of collection, filtration, and laboratory examination, and calculate and report elapsed time. 5. Bibliography CHEN, M. & P.J. HICKEY. 1983. Modification of delayed-incubation procedure for detection of fecal coliforms in water. Appl. Environ. Microbiol. 46:889. 9222 F. Klebsiella Membrane Filter Procedure Klebsiella bacteria belong to the family Enterobacteriaceae and are included in the total coliform group. The outermost layer of Klebsiella bacteria consists of a large polysaccharide capsule, a characteristic that distinguishes this genus from most other bacteria in this family; this capsule provides some measure of protection from disinfectants. Klebsiella bacteria are commonly associated with coliform regrowth in large water supply distribution systems. Klebsiellae may be opportunistic pathogens that can give rise to bacteremia, pneumonia, urinary tract, and several other types of human infection. Approximately 60 to 80% of all Klebsiella from feces and from clinical specimens are positive in the fecal coliform test and are © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Klebsiella pneumoniae. Klebsiella bacteria also are widely distributed in nature, occurring in soil, water, grain, vegetation, etc. Wood pulp, paper mills, textile finishing plants, and sugar-cane processing operations contain large numbers of klebsiellae in their effluents (104 to 106), and Klebsiella sp. are often the predominant coliform in such effluents. Rapid quantitation may be achieved in the MF procedure by modifying M-FC agar base through substitution of inositol for lactose and adding carbenicillin or by using M-Kleb agar. These methods reduce the necessity for biochemical testing of pure strains. Preliminary verification of differentiated colonies is recommended. 1. Apparatus a. Culture dishes: See Section 9222B.1e for specifications. b. Filtration units: See Section 9222B.1 f. 2. Materials and Culture Medium a. Modified M-FC agar (M-FCIC agar): This medium may not be available in dehydrated form and may require preparation from the basic ingredients: Tryptose or biosate Proteose peptone No. 3 or polypeptone Yeast extract Sodium chloride, NaCl Inositol Bile salts No. 3 or bile salts mixture Aniline blue Agar Reagent-grade water 10.0 5.0 3.0 5.0 10.0 1.5 0.1 15.0 1 g g g g g g g g L Heat medium to boiling and add 10 mL 1% rosolic acid dissolved in 0.2N NaOH. Cool to below 45°C and add 50 mg carbenicillin.*#(42) Dispense aseptically in 5- to 7-mL quantities into 50- × 9-mm plastic petri dishes. Refrigerate until needed. Discard unused agar medium after 2 weeks. Do not sterilize by autoclaving. Final pH should be 7.4 ± 0.2. b. M-Kleb agar: Phenol red agar Adonitol Aniline blue 31.0 g 5.0 g 0.1 g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Sodium lauryl sulfate Reagent-grade water 0.1 g 1 L Sterilize by autoclaving for 15 min at 121°C. After autoclaving, cool to 50°C in a water bath; add 20 mL 95% ethyl alcohol (not denatured) and 0.05 g filter sterilized carbenicillin/L. Shake thoroughly and dispense aseptically into 50- × 9-mm plastic culture plates. The final pH should be 7.4 ± 0.2. Refrigerated medium can be held for 20 d at 4 to 8°C. 3. Procedure a. See Section 9222B.5 for selection of sample size and filtration procedure. Select sample volumes that will yield counts between 20 and 60 Klebsiella colonies per membrane. Place membrane filter on agar surface; incubate for 24 ± 2 h at 35 ± 0.5°C. Klebsiella colonies on M-FCIC agar are blue or bluish-gray. Most atypical colonies are brown or brownish. Occasional false positive occurrences are caused by Enterobacter species. Klebsiella colonies on M-Kleb agar are deep blue to blue gray, whereas other colonies most often are pink or occasionally pale yellow. Count colonies with a low-power (10 to 15 magnifications) binocular wide field dissecting microscope or other optical device. b. Verification: Verify Klebsiella colonies from the first set of samples from ambient waters and effluents and when Klebsiella is suspect in water supply distribution systems. Verify a minimum of five typical colonies by transferring growth from a colony or pure culture to a commercial multi-test system for gram-negative speciation. Key tests for Klebsiella are citrate (positive), motility (negative), lysine decarboxylase (positive), ornithine decarboxylase (negative), and urease (positive). A Klebsiella strain that is indole-positive, liquefies pectin, and demonstrates a negative fecal coliform response is most likely of nonfecal origin. 4. Bibliography DUNCAN, D.W. & W.E. RAZELL. 1972. Klebsiella biotypes among coliforms isolated from forest environments and farm produce. Appl. Microbiol. 24:933. STRAMER, S.L. 1976. Presumptive identification of Klebsiella pneumoniae on M-FC medium. Can. J. Microbiol. 22:1774. BAGLEY, S.T. & R.J. SEIDLER. 1977. Significance of fecal coliform-positive Klebsiella. Appl. Environ. Microbiol. 33:1141. KNITTEL, M.D., R.J. SEIDLER, C. EBY & L.M. CABE. 1977. Colonization of the botanical environment by Klebsiella isolates of pathogenic origin. Appl. Environ. Microbiol. 34:557. EDMONSON, A.S., E.M. COOK, A.P.D. WILCOCK & R. SHINEBAUM. 1980. A comparison of the properties of Klebsiella isolated from different sources. J. Med. Microbiol. (U.K.) 13:541. SMITH, R.B. 1981. A Critical Evaluation of Media for the Selective Identification and Enumeration of Klebsiella. M.S. thesis, Dep. Civil & Environmental Engineering, Univ. Cincinnati, Ohio. NIEMELA, S.I. & P. VAATANEN. 1982. Survival in lake water of Klebsiella pneumoniae © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater discharged by a paper mill. Appl. Environ. Microbiol. 44:264. GELDREICH, E.E. & E.W. RICE. 1987. Occurrence, significance, and detection of Klebsiella in water systems. J. Amer. Water Works Assoc. 79:74. DUNCAN, I.B.R. 1988. Waterborne Klebsiella and human disease. Toxicity Assess. 3:581. 9222 G. MF Partition Procedures 1. Escherichia coli Partition Methods a. Applications: Escherichia coli is a member of the fecal coliform group of bacteria; its presence is indicative of fecal contamination. Rapid quantitation and verification may be achieved with the MF procedure by transferring the membrane from a total-coliform- or fecal-coliform-positive sample to a nutrient agar substrate containing 4-methylumbelliferyl-β-D-glucuronide (MUG). In this method E. coli is defined as any coliform that produces the enzyme β-glucuronidase and hydrolyzes the MUG substrate to produce a blue fluorescence around the periphery of the colony. In the examination of drinking water samples, use this method to verify the presence of E. coli from a total-coliform-positive MF on Endo-type media. In the examination of wastewater and other nonpotable water samples, use this procedure to verify positive filters from mFC medium used in the fecal coliform MF procedure. b. Apparatus: 1) Culture dishes: See Section 9222B.1e. 2) Filtration units: See Section 9222B.1 f. 3) Forceps: See Section 9222B.1i. 4) Incubator: See Section 9222B.1 j. 5) Ultraviolet lamp, long wave (366 nm), preferably 6 W. 6) Microscope and light source: See Section 9222B.1k. c. Materials and culture medium: 1) Nutrient agar with MUG (NA-MUG): Peptone Beef extract Agar 4-methylumbelliferyl-β-D-glucuronide Reagent-grade water 5.0 3.0 15.0 0.1 g g g g 1 L Add dehydrated ingredients to reagent-grade water, mix thoroughly, and heat to dissolve. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Sterilize by autoclaving for 15 min at 121°C. Dispense aseptically into 50-mm plastic culture plates. The final pH should be 6.8 ± 0.2. Refrigerated prepared medium may be held for 2 weeks. 2) EC broth with MUG (EC-MUG): Tryptose or trypticase Lactose Bile salts mixture or bile salts No. 3 Dipotassium hydrogen phosphate, K2HPO4 20.0 5.0 1.5 4.0 g g g g Potassium dihydrogen phosphate, KH2PO4 1.4 g Sodium chloride, NaCl 5.0 g 0.1 g 4-methylumbelliferyl-β-D-glucuronide Reagent-grade water 1 L Add dehydrated ingredients to reagent-grade water, mix thoroughly and heat to dissolve. pH should be 6.9 ± 0.2 after sterilization. Before sterilization, dispense into culture tubes and cap with metal or heat-resistant plastic caps. d. Procedure: See Section 9222B.5 for selection of sample size and filtration procedure. For drinking water samples using Endo-type medium, count and record the metallic golden sheen colonies. Before transfer of the membrane, transfer a small portion of each target colony to the appropriate total coliform verification medium, using a sterile needle. See Section 9222B.5 for total coliform verification procedures. Alternatively, after transfer and incubation on NA-MUG, swab the surface growth on the filter and transfer to the appropriate total coliform verification medium. Aseptically transfer the membrane from the Endo-type medium to NA-MUG or EC-MUG medium. If differentiation of the total coliforms is desired using NA-MUG medium, mark each sheen colony with a fine-tipped marker or by puncturing a hole in the membrane adjacent to the colony with a sterile needle. Incubate NA-MUG at 35 ± 0.5°C for 4 h or EC-MUG at 44.5 ± 0.2 for 24 ±2 h. Observe individual colonies or tubes using a long-wave-length (366-nm) ultraviolet light source, preferably containing a 6-W bulb. The presence of a blue fluorescence in the tube, on the periphery (outer edge) of a colony, or observed from the back of the plate is considered a positive response for E. coli. Count and record the number of target colonies, if quantification is desired, or just record presence or absence of fluorescence. For nonpotable water samples, use mFC medium for initial isolation before transfer to NA-MUG or EC-MUG medium. The procedure is the same as the above, with the exception of the total coliform verification process. For the EC-MUG method, a positive control consisting of a known E. coli (MUG-positive) culture, a negative control consisting of a thermotolerant Klebsiella pneumoniae (MUG-negative) culture, and an uninoculated medium control may be necessary to interpret the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater results and to avoid confusion of weak autofluorescence of the medium as a positive response. See Section 9221F. 2. Fecal Coliform Partition Method a. Applications: Further partitioning of total coliforms from the original MF coliform-positive culture in a presence/absence search for fecal coliform in a drinking water sample may be achieved within 24 h. This procedure provides additional information from the original sample. b. Materials and culture medium: EC broth. See Section 9221E.1a. c. Procedure: See Section 9222B.5 for selection of sample size and filtration procedure. For drinking water samples using Endo-type media, count and record the metallic (golden) sheen colonies. Before transfer of membrane or swabbing of plate, transfer a small portion of each target colony to the appropriate total coliform verification media using a sterile needle (see Section 9222B.5 f). Use a sterile cotton swab to collect bacteria from the membrane surface, or pick discrete colonies with a 3-mm loop or sterile applicator stick, or transfer the entire membrane to inoculate a tube of EC medium. Incubate inoculated EC broth in a water bath at 44.5 ± 0.2°C for 24 ± 2 h. Place all EC tubes in water bath within 30 min after inoculation. Maintain a sufficient water depth in water bath incubator to immerse tubes to upper level of the medium. Gas production in an EC broth culture in 24 h or less is considered a positive response for fecal coliform bacteria. 3. Bibliography U.S. ENVIRONMENTAL PROTECTION AGENCY. 1989. Drinking Water; National Primary Drinking Water Regulations; Total Coliforms (Including Fecal Coliforms and E. coli); Final Rule. 40 CFR Parts 141 and 142. Federal Register 54:27544, June 29, 1989. MATES, A. & M. SHAFFER. 1989. Membrane filtration differentiation of E. coli from coliforms in the examination of water. J. Appl. Bacteriol. 67:343. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1991. National Primary Drinking Water Regulations; Analytical Techniques; Coliform Bacteria. 40 CFR Part 141, Federal Register 56:636, Jan. 8, 1991. MATES, A. & M. SHAFFER. 1992. Quantitative determination of Escherichia coli from coliforms and fecal coliforms in sea water. Microbios 71:27. SARTORY, D. & L. HOWARD. 1992. A medium detecting beta-glucuronidase for the simultaneous membrane filtration enumeration of Escherichia coli and coliforms from drinking water. Lett. Appl. Microbiol. 15:273. SHADIX, L.C., M.E. DUNNIGAN & E.W. RICE. 1993. Detection of Escherichia coli by the nutrient agar plus 4-methylumbelliferyl-β-D-glucuronide (MUG) membrane filter method. Can. J. Microbiol. 39: 1066. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9223 ENZYME SUBSTRATE COLIFORM TEST*#(43) 9223 A. Introduction The enzyme substrate test utilizes hydrolyzable substrates for the simultaneous detection of total coliform bacteria and Escherichia coli enzymes. When the enzyme technique is used, the total coliform group is defined as all bacteria possessing the enzyme β-D-galactosidase, which cleaves the chromogenic substrate, resulting in release of the chromogen. Escherichia coli are defined as bacteria giving a positive total coliform response and possessing the enzyme β-glucuronidase, which cleaves a fluorogenic substrate, resulting in the release of the fluorogen. The test can be used in either a multiple-tube, multi-well, or a presence-absence (single 100-mL sample) format. 1. Principle a. Total coliform bacteria: Chromogenic substrates, such as ortho-nitrophenyl-β-D-galactopyranoside (ONPG) or chlorophenol red-β-D-galactopyranoside (CPRG), are used to detect the enzyme β-D-galactosidase, which is produced by total coliform bacteria. The β-D-galactosidase enzyme hydrolyzes the substrate and produces a color change, which indicates a positive test for total coliforms at 24 h (ONPG) or 28 h (CPRG) without additional procedures. Noncoliform bacteria, such as Aeromonas and Pseudomonas species, may produce small amounts of the enzyme β-D-galactosidase, but are suppressed and generally will not produce a positive response within the incubation time unless more than 104 colony-forming units (CFU)/mL (106 CFU/100 mL) are present. b. Escherichia coli: A fluorogenic substrate, such as 4-methylumbelliferyl-β-D-glucuronide (MUG), is used to detect the enzyme β-glucuronidase, which is produced by E. coli. The β-glucuronidase enzyme hydrolyzes the substrate and produces a fluorescent product when viewed under long-wavelength (366-nm) ultraviolet (UV) light. The presence of fluorescence indicates a positive test for E. coli. Some strains of Shigella spp. also may produce a positive fluorescence response. Because Shigella spp. are overt human pathogens, this is not considered a detriment for testing the sanitary quality of water. 2. Applications The enzyme substrate coliform test is recommended for the analysis of drinking and source water samples. Formulations also are available for the analysis of marine waters. Initially, laboratories planning to use this procedure should conduct parallel quantitative testing (including seasonal variations) with one of the standard coliform tests to assess the effectiveness of the test for the specific water type being analyzed and to determine the comparability of the two techniques. This is particularly important when testing source waters. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Water samples containing humic or other material may be colored. If there is background color, compare inoculated tubes to a control tube containing only water sample. In certain waters, high calcium salt content can cause precipitation but this should not affect the reaction. Do not use the enzyme substrate test to verify presumptive coliform cultures or membrane filter colonies, because the substrate may be overloaded by the heavy inoculum of weak β-D-galactosidase-producing noncoliforms, causing false-positive results. 9223 B. Enzyme Substrate Test 1. Substrate Media Formulations are available commercially*#(44) in disposable tubes for the multiple-tube procedure, in disposable multi-wells†#(45) for the multi-well procedure, or in containers that will hold 100-mL samples for the presence-absence approach.* Appropriate preweighed portions of the reagent for mixing and dispensing into multiple tubes for 10-mL test portions or other containers for 100-mL samples also are available. The need for good quality assurance and uniformity requires the use of a commercial substrate medium. Avoid prolonged exposure of the substrate to direct sunlight. Store media according to directions and use before expiration date. Discard discolored media. 2. Procedure a. Multiple-tube procedure: Select the appropriate number of tubes per sample with predispensed media for the multiple-tube test and label. Follow manufacturer’s instructions for preparing serial dilutions for various formulations. Aseptically add 10 mL sample to each tube, cap tightly, and mix vigorously to dissolve. The mixture remains colorless with ONPG-based tests and turns yellow with the CPRG format. Some particles may remain undissolved throughout the test; this will not affect test performance. Incubate at 35 ± 0.5°C for period specified by substrate manufacturer. The procedure also can be performed by adding appropriate amounts of the substrate media to the sample, mixing thoroughly, and dispensing into five or ten sterile tubes. Incubate as stated for multiple-tube procedure. b. Multi-well procedure: The multi-well procedure is performed with sterilized disposable packets. Add sample to 100-mL container with substrate, shake vigorously, and pour into tray. The tray sealer dispenses the sample into the wells and seals the package. Incubate at 35 ± 0.5°C for period specified by substrate manufacturer. The MPN value is obtained from the table provided by the manufacturer. c. Presence-absence procedure (P/A): Aseptically add preweighed enzymatic medium to 100-mL sample in a sterile, transparent, nonfluorescent borosilicate glass or equivalent bottle or container. Optionally, add 100-mL sample to the enzymatic substrate in a sterile container provided by the manufacturer. Aseptically cap and mix thoroughly to dissolve. Incubate as © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater specified in manufacturer’s instructions. 3. Interpretation a. Total coliform bacteria: After the minimum proper incubation period, examine tubes or containers for the appropriate color change (Table 9223:I). ONPG is hydrolyzed by the bacterial enzyme to yield a yellow color. CPRG is hydrolyzed by the bacterial enzyme to yield a red or magenta color. If the color response is not uniform throughout the tube, mix by inversion before reading. Read manufacturer’s instructions for interpretation guidelines. Some manufacturers suggest comparing sample tubes against a color comparator available through the manufacturer. Samples are negative for total coliforms if no color is observed in ONPG tests or if the tube is yellow when CPRG is used. If a chromogenic response is questionable after 18 or 24 h for ONPG, incubate up to an additional 4 h. If response is negative after 28 h for CPRG, incubate up to an additional 20 h. If the chromogen intensifies, the sample is total-coliform positive; if it does not, the sample is negative. b. Escherichia coli: Examine positive total coliform tubes or containers for fluorescence using a long-wavelength (366-nm) ultraviolet lamp (preferably 6-W bulb). Compare each tube against the reference comparator available from a commercial source of the substrate. The presence of fluorescence is a positive test for E. coli. If fluorescence is questionable, incubate for an additional 4 h for ONPG tests and up to an additional 20 h for CPRG tests; intensified fluorescence is a positive test result. 4. Reporting If performing an MPN procedure, calculate the MPN value for total coliforms and E. coli from the number of positive tubes as described in Section 9221C. If using the presence-absence procedure, report results as total coliform and E. coli present or absent in 100-mL sample. 5. Quality Control Test each lot of media purchased for performance by inoculation with three control bacteria: Escherichia coli, a total coliform other than E. coli (e.g., Enterobacter cloacae), and a noncoliform. Also add a sterile water control. If the sterile water control exhibits faint fluorescence or faint positive coliform result, discard and use a new batch of substrate. Avoid using a heavy inoculum. If Pseudomonas is used as the representative noncoliform, select a nonfluorescent species. Incubate these controls at 35 ± 0.5°C as indicated above. Read and record results. Other quality-control guidelines are included in Section 9020. 6. Bibliography EDBERG, S.C., M.J. ALLEN, D.B. SMITH & THE NATIONAL COLLABORATIVE STUDY. 1988. National field evaluation of a defined substrate method for the simultaneous enumeration of total coliforms and Escherichia coli from drinking water: Comparison with the standard multiple tube fermentation method. Appl. Environ. Microbiol. 54:1595. EDBERG, S.C. & M.M. EDBERG. 1988. A defined substrate technology for the enumeration of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater microbial indicators of environmental pollution. Yale J. Biol. Med. 61:389. COVERT, T.C., L.C. SHADIX, E.W. RICE, J.R. HAINES & R.W. FREYBERG. 1989. Evaluation of the Autoanalysis Colilert test for detection and enumeration of total coliforms. Appl. Environ. Microbiol. 55:2443. EDBERG, S.C. & D.B. SMITH. 1989. Absence of association between total heterotrophic and total coliform bacteria from a public water supply. Appl. Environ. Microbiol. 55:380. EDBERG, S.C., M.J. ALLEN, D.B. SMITH & THE NATIONAL COLLABORATIVE STUDY. 1989. National field evaluation of a defined substrate method for the simultaneous detection of total coliforms and Escherichia coli from drinking water: Comparison with presenceabsence techniques. Appl. Environ. Microbiol. 55:1003. EDBERG, S.C., M.J. ALLEN, D.B. SMITH & N.J. KRIZ. 1990. Enumeration of total coliforms and Escherichia coli from source water by the defined substrate technology. Appl. Environ. Microbiol. 56:366. RICE, E.W., M.J. ALLEN & S.C. EDBERG. 1990. Efficacy of β-glucuronidase assay for identification of Escherichia coli by the defined-substrate technology. Appl. Environ. Microbiol. 56:1203. RICE, E.W., M.J. ALLEN, D.J. BRENNER & S.C. EDBERG. 1991. Assay for β-glucuronidase in species of the genus Escherichia and its application for drinking water analysis. Appl. Environ. Microbiol. 57:592. SHADIX, L.C. & E.W. RICE. 1991. Evaluation of β-glucuronidase assay for the detection of Escherichia coli from environmental waters. Can. J. Microbiol. 37:908. EDBERG, S.C., M.J. ALLEN & D.B. SMITH. 1991. Defined substrate technology method for rapid and simultaneous enumeration of total coliforms and Escherichia coli from water: Collaborative study. J. Assoc. Offic. Anal. Chem. 74:526. EDBERG, S.C., F. LUDWIG & D.B. SMITH. 1991. The Colilert® System for Total Coliforms and Escherichia coli. American Water Works Association Research Foundation, Denver, Colo. COVERT, T.C., E.W. RICE, S.A. JOHNSON, D. BERMAN, C.H. JOHNSON & P.M. MASON. 1992. Comparing defined-substrate coliform tests for the detection of Escherichia coli in water. J. Amer. Water Works Assoc. 84(5):98. MCCARTY, S.C., J.H. STANDRIDGE & M.C. STASIAK. 1992. Evaluating a commercially available defined-substrate test for recovery of chlorine-treated Escherichia coli. J. Amer. Water Works Assoc. 84(5): 91. PALMER, C.J., Y. TSAI, A.L. LANG & L.R. SANGERMANO. 1993. Evaluation of Colilert-marine water for detection of total coliforms and Escherichia coli in the marine environment. Appl. Environ. Microbiol. 59:786. CLARK, J.A. & A.H. SHAARAWI. 1993. Evaluation of commercial presence-absence test kits for detection of total coliforms, Escherichia coli, and other indicator bacteria. Appl. Environ. Microbiol. 59:380. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1994. National Primary and Secondary Drinking Water Regulation: Analytical methods for regulated drinking water contaminants; Final © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Rule. 40 CFR Parts 141 & 143; Federal Register 59:62456. MCFETERS, G.A., S.C. BROADWAY, B.H. PYLE, M. PICKETT & Y. EGOZY. 1995. Comparative performance of ColisureTM and accepted methods in the detection of chlorine-injured total coliforms and E. coli. Water Sci. Technol. 31:259. 9225 DIFFERENTIATION OF THE COLIFORM BACTERIA*#(46) 9225 A. Introduction Identification of bacteria that constitute the coliform group sometimes is necessary to determine the nature of pollution. It is of particular importance in reference to distinguishing the presence of Escherichia coli. Special procedures for detection of E. coli are given in Section 9221F, Section 9222G, and Section 9223. Differential tests for identification must be used with the knowledge that all strains taxonomically assigned to the coliform group do not conform necessarily to the coliform definition stated in this manual because they may not ferment lactose, or if they do, they may not produce gas. Furthermore, gram-negative bacteria other than coliforms ferment lactose and produce sheen (e.g., Aeromonas spp.) and not all strains of a species will react uniformly in media. Unusual strains (such as E. coli, inactive, Table 9225:I), mutants, and injured organisms may not give classical responses. The traditional ‘‘IMViC’’ tests (i.e., indole, methyl red, Voges-Proskauer, and citrate utilization) are useful for coliform differentiation, but do not provide complete identification. Additional biochemical tests often are necessary. Commercial kits for identification are available and may serve as economical alternatives to traditional differential media. Automated systems of identifying large numbers of isolates also are available. The significance of various coliform organisms in water has been and is a subject of considerable study. Collectively, the coliforms are referred to as indicator organisms. The genera Enterobacter, Klebsiella, Citrobacter, and Escherichia usually are represented in the majority of isolations made from raw and treated municipal water supplies. 9225 B. Culture Purification 1. Procedure A pure culture is essential for accurate identification. Obtain a pure culture by carefully picking a well-isolated colony that gives typical responses on an appropriate solid medium or membrane filter, and streaking on a tryptic soy or nutrient agar plate. Better distribution of colonies in the subculture is obtained if a portion of the picked colony is emulsified in peptone broth or physiological saline (0.85% w/v) and then streaked. When picking a colony from a primary culture on a selective medium, be aware that viable cells, which have not formed © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater colonies themselves, may surround the picked colony. Incubate the subculture at 35 ± 0.5°C for 24 h and test a single well-isolated colony by the Gram stain to confirm the sole presence of gram-negative, non-spore-forming rods (Section 9221B). Also determine that the culture is oxidase-negative (Section 9225D). Oxidase-positive, gram-negative, non-spore-forming rods are not coliform bacteria, but may be organisms such as Aeromonas, which is not regarded as an indicator of fecal pollution. Variation in organisms of the coliform group occurs occasionally and mixed reactions in differential media may indicate a pure culture undergoing variation. Persistent variations of reactions in differential media indicate a mixed culture caused by inadequate purification. 2. Bibliography PTAK, D.J., W. GINSBURG & B.F. WILLEY. 1974. Aeromonas, the great masquerader. Proc. AWWA Water Quality Technology Conf., Dallas, Tex., p. V-1. American Water Works Assoc., Denver, Colo. VAN DER KOOJ, D. 1988. Properties of aeromonads and their occurrence and hygienic significance in drinking water. Zentralbl. Bacteriol. Hyg. B 187:1. HARTMAN, P.A., B. SWAMINATHAN, M.S. CURIALE, R. FIRSTENBERG-EDEN, A.N. SHARPE, N.A. COX, D.Y.C. FUNG & M.C. GOLDSCHMIDT. 1992. Rapid methods and automation. In: C. Vanderzant & D.F. Splittstoesser, eds., Compendium of Methods for the Microbiological Examination of Foods, 3rd. ed. p.665. American Public Health Assoc., Washington, D.C. STAGER, C.E. & J.R. DAVIS. 1992. Automated systems for identification of microorganisms. Clin. Microbiol. Rev. 5:302. RICE, E.W., M.J. ALLEN, T.C. COVERT, J. LANGEWIS & J. STANDRIDGE. 1993. Identifying Escherichia species with biochemical test kits and standard bacteriological tests. J. Amer. Water Works Assoc. 85(2): 74. 9225 C. Identification 1. Definition Coliforms are defined here as facultative anaerobic, gram-negative non-spore-forming rods that ferment lactose with gas formation within 48 h at 35 °C or, as applied to the membrane filter method, produce a dark red colony with a metallic sheen within 24 h on an Endo-type medium containing lactose. However, anaerogenic (non-gas-producing) lactose-fermenting strains of Escherichia coli and coliforms that do not produce metallic sheen on Endo medium may be encountered. These organisms, as well as typical coliforms, can be considered indicator organisms, but they are excluded from the current definition of coliforms. More extensive testing may be required for proper identification. 2. Characteristics and Tests Coliforms belong to the bacterial taxonomic family Enterobacteriaceae. Table 9225:I © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater provides data on some of the biochemical reactions used for differentiating these organisms. Preparing differential media and reagents may not be as economical for many laboratories as using commercially prepared and prepackaged multiple-test kits, which reduce quality-control work. These commercial kits are simple to store and use, and give reproducible and generally accurate results. Periodically test reactions with known stock cultures of bacteria to assure accuracy and reproducibility of results. Make further tests if the kit provides equivocal results. 3. Bibliography KRIEG, N.R., ed. 1984. Bergey’s Manual of Systematic Bacteriology, Vol. I. Williams & Wilkins Co., Baltimore, Md. EDWARDS, P.R. & W.H. EWING. 1986. Identification of Enterobacteriaceae, 4th ed. Burgess Publ. Co., Minneapolis, Minn. 9225 D. Media, Reagents, and Procedures Commercially available media and reagents can reduce work and cost; however, include negative and positive controls with known stock cultures to assure accuracy and reliability. Detailed methods are available. Expected test results are shown in Table 9225:I. 1. Lactose, Sorbitol, and Cellobiose Fermentation Tests Suspend 16 g phenol red broth base and 5 g of the desired carbohydrate in 1 L reagent-grade water and stir to dissolve completely. Dispense in tubes to a depth of one-third tube length. To determine gas production place a small inverted vial (Durham tube) in the tubes of media at the time of preparation. Close tubes and sterilize at 121°C for 15 min. Store tubes in the dark (refrigeration preferred) and discard if evaporation exceeds 10% of the volume. To conduct a test, inoculate with a loopful of growth from a well-isolated colony or slant and incubate for 24 to 48 h at 35 ± 0.5°C. Carbohydrate fermentation (acid production) is indicated by a decrease in pH, resulting in a change in color of the pH indicator, phenol red, from red-orange to yellow (pH <6.6). Alternatively, for lactose fermentation, lauryl tryptose broth (Section 9221B) may be used. 2. ONPG Hydrolysis Numerous commercial test kits and disks for determining ONPG hydrolysis are available, or an ONPG-containing medium (Section 9222) can be used. Alternatively, prepare peptone water by dissolving 1 g peptone and 0.5 g NaCl in 100 mL reagent-grade water. Sterilize at 121°C for 15 min. Also prepare ONPG solution by dissolving 0.6 g o-nitrophenyl-β-D-galactopyranoside (ONPG) in 100 mL 0.01M Na2HPO4, sterilize by filtration, and store in the dark at 4 to 10°C. To prepare ONPG broth, aseptically combine 25 mL ONPG solution and 75 mL peptone water, dispense aseptically in 2.5-mL amounts in sterile 13- × 100-mm tubes, and store in the dark for up to 1 month at 4 to 10°C. Do not use the ONPG solution if it becomes yellow. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater To conduct the test, inoculate 0.5 mL ONPG broth with a heavy loopful of growth from a slant and incubate at 35 ± 0.5°C for up to 24 h. A yellow color, compared with an uninoculated tube or (preferably) a tube inoculated with an ONPG-negative culture, is a positive test. Interpret tests of yellow-pigmented organisms with caution. Do not use the enzyme substrate method (Section 9223) to test ONPG hydrolysis. 3. Indole Test Indole is a product of the metabolism of tryptophane. a. Reagents: 1) Medium: Use tryptophane broth. Dissolve 10.0 g tryptone or trypticase/L reagent-grade water. Dispense in 5-mL portions in test tubes and sterilize. 2) Test reagent: Dissolve 5 g p-dimethylaminobenzaldehyde in 75 mL isoamyl (or normal amyl) alcohol, ACS grade, and add 25 mL conc HCl. The reagent should be yellow. Some brands of p-dimethylaminobenzaldehyde are not satisfactory and some good brands become unsatisfactory on aging. The amyl alcohol solution should have a pH value of less than 6.0. Purchase both amyl alcohol and benzaldehyde in as small amounts as will be consistent with the volume of work to be done. b. Procedure: Inoculate 5-mL portions of medium from a pure culture and incubate at 35 ± 0.5°C for 24 ± 2 h. Add 0.2 to 0.3 mL test reagent and gently shake. Let stand for about 10 min and observe results. A dark red color in the amyl alcohol surface layer constitutes a positive indole test; the original color of the reagent, a negative test. An orange color probably indicates the presence of skatole, a breakdown product of indole. 4. Methyl Red Test The methyl red test measures the ability of organisms to produce stable acid end products from glucose fermentation. a. Reagents: 1) Medium: Use buffered glucose broth. Dissolve 7.0 g proteose peptone or equivalent peptone, 5.0 g glucose, and 5.0 g dipotassium hydrogen phosphate (K2HPO4) in 1 L reagent-grade water. Dispense in 5-mL portions in test tubes and sterilize by autoclaving at 121°C for 12 to 15 min, making sure that total time of exposure to heat is not longer than 30 min. 2) Indicator solution: Dissolve 0.1 g methyl red in 300 mL 95% ethyl alcohol and dilute to 500 mL with reagent-grade water. b. Procedure: Inoculate 10-mL portions of medium from a pure culture. Incubate at 35 ± 0.5°C for 5 d. To 5 mL of the culture add 5 drops methyl red indicator solution. Incubation for 48 h is adequate for most cultures, but do not incubate for less than 48 h. If test results are equivocal at 48 h repeat with cultures incubated for 4 or 5 d. In such cases © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater incubate duplicate cultures at 22 to 25°C. Testing of culture portions at 2, 3, 4, and 5 d may provide positive results sooner. Record a distinct red color as methyl-red-positive and a distinct yellow color as methyl-red-negative. Record a mixed shade as questionable and possibly indicative of incomplete culture purification. 5. Voges-Proskauer Test The Voges-Proskauer test measures the ability of organisms to produce a neutral end product (acetoin) from glucose fermentation. a. Reagents: 1) Medium: See ¶ 4a1) above. 2) Naphthol solution: Dissolve 5 g purified α-naphthol (melting point 92.5°C or higher) in 100 mL absolute ethyl alcohol. When stored at 5 to 10°C, this solution is stable for 2 weeks. 3) Potassium hydroxide, 7N: Dissolve 40 g KOH in 100 mL reagent-grade water. b. Procedure: Inoculate 5 mL medium and incubate for 48 h at 35 ± 0.5°C. To 1 mL of culture add 0.6 mL naphthol solution and 0.2 mL KOH solution. Shake well after the addition of each reagent. Development of a pink to crimson color at the surface within 5 min constitutes a positive test. Do not read after 10 min. Disregard tubes developing a copper color. 6. Simmons’ Citrate Test The citrate test measures the ability of bacteria to utilize citrate as the sole source of carbon. a. Medium: Use Simmons’ citrate agar. To make Simmons’ citrate agar, add 0.2 g MgSO4⋅7H2O, 1.0 g ammonium dihydrogen phosphate (NH4H2PO4), 1.0 g K2HPO4, 2.0 g sodium citrate dihydrate, 5.0 g NaCl, 15.0 g agar, and 0.08 g bromthymol blue to 1 L reagent-grade water. Tube for long slants. b. Procedure: Inoculate agar medium by the streak technique using a light inoculum. Incubate 48 h at 35 ± 0.5°C. Record growth on the medium with a blue color as a positive reaction; record absence of growth or color change as negative. 7. Motility Test The motility test measures whether an organism is motile in a semi-solid medium. a. Medium: Use motility test medium made by adding 3.0 g beef extract, 10.0 g peptone, 5.0 g NaCl, and 4.0 g agar to 1 L reagent-grade water. Adjust pH to 7.4, dispense in 3-mL portions in 13- × 100-mm tubes or 8-mL portions in 16- × 125-mm tubes, and sterilize. b. Procedure: Inoculate by stabbing into the center of the medium, using an inoculating needle, to a depth of 5 mm. Incubate for 1 to 2 d at 35°C. If negative, incubate an additional 5 d at 22 to 25°C. Diffuse growth through the medium from the point of inoculation is positive. In a negative test, growth is visible only along the stab line and the surrounding medium stays clear. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Alternatively, prepare the medium without agar and examine a young culture using the hanging drop slide technique for motile organisms. 8. Lysine and Ornithine Decarboxylase Tests This procedure tests the ability of bacteria to metabolize the amino acids lysine and ornithine. a. Reagents: 1) Media: Use a basal medium made according to the Moeller or Falkow methods. For the Moeller method, dissolve 5.0 g peptone (Orthana special, thiotone, or equivalent), 5.0 g beef extract, 0.625 mL bromcresol purple (1.6%), 2.5 mL cresol red (0.2%), 0.5 g glucose, and 5.0 mg pyridoxal in 1 L reagent-grade water and adjust to pH 6.0 to 6.5. For the Falkow method, dissolve 5.0 g peptone, 3.0 g yeast extract, 1.0 g glucose, and 1.0 mL bromcresol purple (1.6%) in 1 L reagent-grade water and adjust to pH 6.7 to 6.8. For either decarboxylase test divide into three portions: make no addition to the first portion, add enough L-lysine dihydrochloride to the second portion to make a 1% solution, and add L-ornithine dihydrochloride to the third to make 1% (for the Falkow method, add only 0.5% of the L-amino acid). After adding ornithine readjust pH of the medium to 6.0 ± 0.2. Dispense in 3- to 4-mL portions in screw-capped test tubes and sterilize by autoclaving at 121°C for 10 min. A floccular precipitate in the ornithine medium does not interfere with its use. 2) Mineral oil: Use mineral oil sterilized by autoclaving at 121°C for 30 to 60 min depending on the size of the container. b. Procedure: Lightly inoculate each of the three media, add a layer of about 10 mm thickness of mineral oil, and incubate at 37°C for up to 4 d. Examine tubes daily. A color change from yellow to violet or reddish-violet constitutes a positive decarboxylase test; a change to bluish gray indicates a weak positive; no color change or a yellow color represents a negative test. See Table 9225:I. 9. Oxidase Test The oxidase test determines the presence of oxidase enzymes. Coliform bacteria are oxidase-negative. a. Reagents: 1) Media: Use either nutrient agar or tryptic soy agar plates to streak cultures and produce isolated colonies. From these obtain the inoculum for oxidase testing on impregnated filter paper. Do not use any medium that includes a carbohydrate in its formulation. Use only tryptic soy agar if reagent is dropped on colonies. Tryptic soy agar: Tryptone Soytone Sodium chloride, NaCl 15.0 g 5.0 g 5.0 g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Agar Reagent-grade water 15.0 g 1.0 L pH should be 7.3 ± 0.2 after sterilization. 2) Tetramethyl p-phenylenediamine dihydrochloride, 1% aqueous solution, freshly prepared or refrigerated for no longer than 1 week. Impregnate a filter paper strip*#(47) with this solution. Alternatively, prepare a 1% solution of dimethyl p-phenylenediamine hydrochloride. Single-use reagent ampules, commercially available, are convenient and economical, but use them with caution. When the reagent is to be dropped directly on colonies, use tryptic soy agar plates because nutrient agar plates give inconsistent results; when smearing a portion of a picked colony on reagent-impregnated filter paper, do not transfer any medium with the culture material. b. Procedure: Remove some of a colony from agar plate with a platinum wire, a wooden or plastic applicator stick, or a glass rod and smear on the test strip. Do not use iron or other reactive wire because it will cause false positive reactions. A dark purple color that develops within 10 s is a positive oxidase test. Test positive and negative cultures concurrently. If the liquid reagent is used, drop it on colonies on the culture plate. Oxidase-positive colonies develop a pink color that successively becomes maroon, dark red, and finally, black. 10. Yellow Pigment Observe isolated colonies on nutrient agar slants and plates or plates of tryptic soy agar incubated at 35 ± 0.5°C for up to 48 h. Pigmentation often intensifies as time of incubation proceeds. 11. Bibliography MACFADDIN, J.F. 1976. Biochemical Tests for Identification of Medical Bacteria. Williams & Wilkins Co., Baltimore, Md. FARMER, J.J., III. 1985. Biochemical identification of new species and biogroups of Enterobacteriaceae. J. Clin. Microbiol. 21:46. WASHINGTON, J.A., ed. 1985. Laboratory Procedures in Clinical Microbiology, 2nd ed. Springer-Verlag, New York, N.Y. BALOWS, A.H., W.J. HAUSLER, JR., K.L. HERMANN, H.P. ISENBERG & H.J. SHADOMY, eds. 1991. Manual of Clinical Microbiology, 5th ed. American Soc. for Microbiology, Washington, D.C. 9230 FECAL STREPTOCOCCUS AND ENTEROCOCCUS GROUPS*#(48) 9230 A. Introduction © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Fecal Streptococcus Group The fecal streptococcus group consists of a number of species of the genus Streptococcus, such as S. faecalis, S. faecium, S. avium, S. bovis, S. equinus, and S. gallinarum. They all give a positive reaction with Lancefield’s Group D antisera1 and have been isolated from the feces of warm-blooded animals. In addition, S. avium sometimes reacts with Lancefield’s Group Q antisera. S. faecalis subsp. liquefaciens and S. faecalis subsp. zymogenes are differentiated based on the ability of these strains to liquefy gelatin and hemolyze red cells. However, the validity of these subspecies is questionable.2,3 The normal habitat of fecal streptococci is the gastrointestinal tract of warm-blooded animals. S. faecalis and S. faecium once were thought to be more human-specific than other Streptococcus species. Other species have been observed in human feces but less frequently.4 Similarly, S. bovis, S. equinus, and S. avium are not exclusive to animals, although they usually occur at higher densities in animal feces.5 Certain streptococcal species predominate in some animal species and not in others, but it is not possible to differentiate the source of fecal contamination based on speciation of fecal streptococci. The fecal streptococci have been used with fecal coliforms to differentiate human fecal contamination from that of other warm-blooded animals. Editions of Standard Methods previous to the 17th suggested that the ratio of fecal coliforms (FC) to fecal streptococci (FS) could provide information about the source of contamination. A ratio greater than four was considered indicative of human fecal contamination, whereas a ratio of less than 0.7 was suggestive of contamination by nonhuman sources. The value of this ratio has been questioned because of variable survival rates of fecal streptococcus group species. S. bovis and S. equinus die off rapidly, once exposed to aquatic environments, whereas S. faecalis and S. faecium tend to survive longer.6 Furthermore, disinfection of wastewaters appears to have a significant effect on the ratio of these indicators, which may result in misleading conclusions regarding the source of contaminants.7 The ratio is affected also by the methods for enumerating fecal streptococci. The KF membrane filter procedure has a false-positive rate ranging from 10 to 90% in marine and fresh waters.8-10 For these reasons, the FC/FS ratio cannot be recommended, and should not be used as a means of differentiating human and animal sources of pollution. 2. Enterococcus Group The enterococcus group is a subgroup of the fecal streptococci that includes S. faecalis, S. faecium, S. gallinarum, and S. avium. The enterococci are differentiated from other streptococci by their ability to grow in 6.5% sodium chloride, at pH 9.6, and at 10°C and 45°C. The enterococci portion of the fecal streptococcus group is a valuable bacterial indicator for determining the extent of fecal contamination of recreational surface waters. Studies at marine and fresh water bathing beaches indicated that swimming-associated gastroenteritis is related directly to the quality of the bathing water and that enterococci are the most efficient bacterial indicator of water quality.11,12 Water quality guidelines based on enterococcal density have been © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater proposed for recreational waters.13 For recreational fresh waters the guideline is 33 enterococci/100 mL while for marine waters it is 35/100 mL. Each guideline is based on the geometric mean of at least five samples per 30-d period during the swimming season. 3. Selection of Method The multiple-tube technique is applicable primarily to raw and chlorinated wastewater and sediments, and can be used for fresh and marine waters. The membrane filter technique also may be used for fresh and saline water samples, but is unsuitable for highly turbid waters. 4. References 1. SNEATH, P.H.A., N.S. MAIR, M.E. SHARPE & J.G. HOLT. eds. 1986. Bergey’s Manual of Systematic Bacteriology. Vol. 2, Williams & Wilkins, Baltimore, Md. 2. JACOB, A.E., G.J. DOUGLAS & S.J. HOBBS. 1973. Self-transferable plasmids determining the haemolysin and bacteriocin of Streptococcus faecalis var. zymogenes. J. Bacteriol. 121:863. 3. OLIVER, D.R., B.L. BROWN & D.B. CLEWELL. 1977. Characterization of plasmids determining haemolysin and bacteriocin production in Streptococcus faecalis 5952. J. Bacteriol. 130:948. 4. WATANABE, T., H. SHIMOHASHI, Y. KAWAI, & M. MUTAI. 1981. Studies on streptococci. I. Distribution of fecal streptococci in man. Microbiol. Immunol. 25:257. 5. THOMAS, C.D. & M.A. LEVIN. 1978. Quantitative analysis of group D streptococci. Abs. Annual Meeting, American Soc. Microbiology, p. 210. 6. FEACHAM, R. 1975. An improved role for faecal coliform to faecal streptococci ratios in the differentiation between human and non-human pollution sources. Water Res. 9:689. 7. ROSSER, P.A.E. & D.P. SARTORY. 1982. A note on the effect of chlorination of sewage effluents on faecal coliform to faecal streptococci ratios in the differentiation of faecal pollution sources. Water S.A. 8: 66. 8. FUJIOKA, R.S., A.A. UENO & O.T. NARIKAWA. 1984. Recovery of False Positive Fecal Streptococcus on KF Agar from Marine Recreational Waters. Tech. Rep. No. 168, Water Resources Research Center, Univ. Hawaii at Manoa, Honolulu. 9. OLIVIERI, V.P., C.W. KRUSE, K. KAWATA & J.E. SMITH. 1977. Microorganisms in Urban Stormwater. EPA-600/2-77-087, U.S. Environmental Protection Agency, Edison, N.J. 10. ERICKSEN, T.H., C. THOMAS & A. DUFOUR. 1983. Comparison of two selective membrane filter methods for enumerating fecal streptococci in freshwater samples. Abs. Annual Meeting, American Soc. Microbiology, p. 279. 11. CABELLI, V.J. 1983. Health Effects Criteria for Marine Waters. EPA-600/1-80-031, U.S. Environmental Protection Agency, Cincinnati, Ohio. 12. DUFOUR, A.P. 1984. Health Effects Criteria for Fresh Recreational Waters. EPA-600/1-84-004, U.S. Environmental Protection Agency, Cincinnati, Ohio. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 13. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1986. Ambient Water Quality Criteria for Bacteria—1986. EPA-440/5-84-002, U.S. Environmental Protection Agency, Washington, D.C. 9230 B. Multiple-Tube Technique 1. Materials and Culture Media a. Azide dextrose broth:1 Beef extract Tryptone or polypeptone Glucose Sodium chloride, NaCl Sodium azide, NaN3 4.5 15.0 7.5 7.5 0.2 g g g g g 1 L Peptone C Peptone B Yeast extract Bacteriological bile Sodium chloride, NaCl Sodium citrate Esculin Ferric ammonium citrate Sodium azide, NaN3 17.0 3.0 5.0 10.0 5.0 1.0 1.0 0.5 0.25 g g g g g g g g g Agar Reagent-grade water 15.0 1 g L Reagent-grade water pH should be 7.2 ± 0.2 at 25°C after sterilization. b. Pfizer selective enterococcus (PSE) agar:2 pH should be 7.1 ± 0.2 after sterilization. Hold medium for not more than 4 h at 45 to 50°C before plates are poured. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. Presumptive Test Procedure Inoculate a series of tubes of azide dextrose broth with appropriate graduated quantities of sample. Use sample of 10 mL portions or less. Use double-strength broth for 10-mL inocula. The portions used will vary in size and number with the sample character. Use only decimal multiples of 1 mL (see Section 9221 for suggested sample sizes). Incubate inoculated tubes at 35 ± 0.5°C. Examine each tube for turbidity at the end of 24 ± 2 h. If no definite turbidity is present, reincubate, and read again at the end of 48 ± 3 h. 3. Confirmed Test Procedure Subject all azide dextrose broth tubes showing turbidity after 24- or 48-h incubation to the confirmed test. Streak a portion of growth from each positive azide dextrose broth tube on PSE agar. Incubate the inverted dish at 35 ± 0.5°C for 24 ± 2 h. Brownish-black colonies with brown halos confirm the presence of fecal streptococci. Brownish-black colonies with brown halos may be transferred to a tube of brain-heart infusion broth containing 6.5% NaCl. Growth in 6.5% NaCl broth and at 45°C indicates that the colony belongs to the enterococcus group. 4. Computing and Recording of MPN Estimate fecal streptococci densities from the number of tubes in each dilution series that are positive on PSE agar. Similarly, estimate enterococci densities from the number of tubes in each dilution series containing streptococci that can grow in 6.5% NaCl broth. Compute the combination of positives and record as the most probable number (MPN). Refer to Section 9221D. 5. References 1. MALLMAN, W.L. & E.B. SELIGMANN. 1950. A comparative study of media for the detection of streptococci in water and sewage. Amer. J. Pub. Health 40:286. 2. ISENBERG, H.D., D. GOLDBERT & J. SAMPSON. 1972. Laboratory studies with a selective enterococcus medium. Health Lab. Sci. 9:289. 9230 C. Membrane Filter Techniques 1. Laboratory Apparatus See Section 9222B.1. 2. Materials and Culture Media a. mE agar for enterococci:1 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Peptone Sodium chloride, NaCl Yeast extract Esculin Actidione (cycloheximide) Sodium azide, NaN3 10.0 15.0 30.0 1.0 0.05 0.15 g g g g g g Agar Reagent-grade water 15.0 1 g L Heat to dissolve ingredients, sterilize, and cool in a water bath at 44 to 46°C. Mix 0.25 g nalidixic acid in 5 mL reagent-grade water, add a few drops of 0.1N NaOH to dissolve the antibiotic, and add to the basal medium. Add 0.15 g 2,3,5-triphenyl tetrazolium chloride and mix well to dissolve. Pour the agar into 9- × 50-mm petri dishes to a depth of 4 to 5 mm (approximately 4 to 6 mL), and let solidify. The final pH should be 7.1 ± 0.2. Store poured plates in the dark at 2 to 10°C. Discard after 30 d. (NOTE: This medium is recommended for culturing enterococci in fresh and marine recreational waters.) b. EIA substrate:1 Esculin Ferric citrate Agar Reagent-grade water 1.0 0.5 15.0 1 g g g L The pH should be 7.1 ± 0.2 before autoclaving. Heat to dissolve ingredients, sterilize, and cool in a water bath at 44 to 46°. Pour medium into 50-mm petri dishes to a depth of 4 to 5 mm (approximately 4 to 6 mL) and let solidify. Store poured plates in the dark at 2 to 10°C. Discard after 30 d. c. m Enterococcus agar for fecal streptococci:2 Tryptose Yeast extract Glucose Dipotassium phosphate, K2HPO4 Sodium azide, NaN3 20.0 5.0 2.0 4.0 g g g g 0.4 g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2,3,5-Triphenyl tetrazolium chloride Agar Reagent-grade water 0.1 g 10.0 g 1 L Heat to dissolve ingredients. Do not autoclave. Dispense into 9- × 50-mm petri plates to a depth of 4 to 5 mm (approximately 4 to 6 mL), and let solidify. Prepare fresh medium for each set of samples. (NOTE: This medium is recommended for Group D streptococci in fresh and marine waters.) d. Brain-heart infusion broth: Infusion of calf brain Infusion of beef heart Proteose peptone Glucose Sodium chloride, NaCl Disodium hydrogen phosphate, Na2HPO4 200 250 10.0 2.0 5.0 2.5 g g g g g g 1 L Reagent-grade water The pH should be 7.4 after sterilization. e. Brain-heart infusion agar: Add 15.0 g agar to the ingredients for brain-heart infusion broth. The pH should be 7.4 after sterilization. Tube for slants. f. Bile esculin agar:3 Beef extract Peptone Oxgall Esculin Ferric citrate Agar Reagent-grade water 3.0 5.0 40.0 1.0 0.5 15.0 1 g g g g g g L Heat to dissolve ingredients. Dispense 8 to 10 mL into tubes for slants or an appropriate volume into a flask for subsequent pouring into plates. Autoclave at 121°C for 15 min. Do not overheat because this may cause darkening of the medium. Cool to 44 to 46°C and slant the tubes or dispense 15 mL into 15- × 100-mm petri dishes. The final pH should be 6.6 ± 0.2 after © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater sterilization. Store at 4 to 10°C. 3. Procedures a. mE Method:1 1) Selection of sample size and filtration—Filter appropriate sample volumes through a 0.45-µm, gridded, sterile membrane to give 20 to 60 colonies on the membrane surface. Transfer filter to agar medium in petri dish, avoiding air bubbles beneath the membrane. 2) Incubation—Invert culture plates and incubate at 41°C ± 0.5°C for 48 h. 3) Substrate test—After 48 h incubation, carefully transfer filter to EIA medium. Incubate at 41°C ± 0.5°C for 20 min. 4) Counting—Pink to red enterococci colonies develop a black or reddish-brown precipitate on the underside of the filter. Count colonies using a fluorescent lamp and a magnifying lens. b. m Enterococcus method:2 1) Selection of sample size and filtration—See ¶ 3a. 2) Incubation—Let plates stand for 30 min, then invert and incubate at 35 ± 0.5°C for 48 h. 3) Counting—Count all light and dark red colonies as enterococci. Count colonies using a fluorescent lamp and a magnifying lens. 4. Calculation of Fecal Streptococci or Enterococci Density Compute density from sample quantities producing membrane filter counts within the desired 20- to 60-fecal streptococcus or enterococcus colony range. Calculate as in Section 9222B.6. Record densities as fecal streptococci or enterococci per 100 mL. 5. Verification Tests Pick selected typical colonies from a membrane and streak for isolation onto the surface of a brain-heart infusion agar plate. Incubate at 35°C ± 0.5°C for 24 to 48 h. Transfer a loopful of growth from a well-isolated colony on brain-heart infusion agar into a brain-heart infusion broth tube and to each of two clean glass slides. Incubate the brain-heart infusion broth at 35 ± 0.5°C for 24 h. Add a few drops of freshly prepared 3% hydrogen peroxide to the smear on a slide. The appearance of bubbles constitutes a positive catalase test and indicates that the colony is not a member of the fecal streptococcus group. If the catalase test is negative, i.e., no bubbles, make a Gram stain of the second slide. Fecal streptococci and enterococci are gram-positive, ovoid cells, 0.5 to 1.0 µm in diameter, mostly in pairs or short chains. Transfer a loopful of growth from the brain-heart infusion broth to each of the following media: bile esculin agar (incubate at 35 ± 0.5°C for 48 h); brain-heart infusion broth (incubate at 45 ± 0.5°C for 48 h); brain-heart infusion broth with 6.5% NaCl (incubate at 35 ± 0.5°C for 48 h). Growth of catalase-negative, gram-positive cocci on bile esculin agar and at 45°C in © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater brain-heart infusion broth verifies that the colony is of the fecal streptococcus group. Growth at 45°C and in 6.5% NaCl broth indicates that the colony belongs to the enterococcus group. 6. Serological Verification of Group D Fecal Streptococci An alternate verification test for Group D streptococci can be performed using the precipitin method of Lancefield.4 This test is highly specific for S. faecalis, S. faecium, S. avium, S. gallinarum, S. bovis, and S. equinus. a. Antigen preparation: Pick typical single colonies from the membrane filter and streak for isolation on brain-heart infusion agar or blood agar plates. Pick a well-isolated colony and inoculate into 30 to 50 mL of Todd-Hewitt broth.5 Incubate at 35°C for 24 h under aerobic conditions. Concentrate bacterial suspension by centrifuging (3000 × g for 5 min). Draw supernatant off and resuspend cells in 0.5 mL saline solution. Autoclave resuspended cells for 15 min at 121°C. Centrifuge the bacteria and decant clear supernatant fluid containing the group antigen. b. Capillary precipitin test: Antisera for this test may be obtained from commercial sources. Dip a 1.2- to 1.5-mm-OD capillary tube into antiserum and draw up about 1 cm of serum. Place a finger over upper end of tube so that no air will be drawn up and carefully wipe off excess antiserum. Dip tube into streptococcal antigen extract solution and draw up an equal volume of antigen. Carefully wipe off excess extract. Place a finger over upper end of tube and force lower end into plasticine to plug lower opening. Invert tube and place it in plasticine groove of a capillary holding rack. A positive test for Group D antigen is characterized by a white precipitate that appears at the antigen-antiserum interface within 15 min and usually by 5 min. If no reaction has occurred by 30 min, the test is negative. Examination of the tubes is more effective if they are read in a bright light against a dark background. Serological verification of Group D streptococci also can be done using commercially available agglutination tests.*#(49) The slide agglutination tests are simple and appear to be reliable. Group D streptococci are verified directly from isolated colonies on membrane filter plates or from broth culture tubes. To verify presumptive enterococci cultures, test also for salt tolerance (growth in 6.5% NaCl broth). 7. Identification of Individual Species within Fecal Streptococcus and Enterococcus Groups Table 9230:I shows some of the key biochemical reactions for identifying fecal streptococci, enterococci, and species within these two groups. 8. References 1. LEVIN, M.A., J.R. FISCHER & V.J. CABELLI. 1975. Membrane filter technique for enumeration of enterococci in marine waters. Appl. Microbiol 30:66. 2. SLANETZ, L.W. & C.H. BARTLEY. 1957. Numbers of enterococci in water, sewage and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 3. 4. 5. 6. 7. 8. 9. feces determined by the membrane filter technique with an improved medium. J. Bacteriol. 74:591. PHILLIPS, E. & P. NASH. 1985. Culture media. In E.H. Lennette, A. Ballows, W.J. Hausler, Jr. & H.J. Shadomy, eds. Manual of Clinical Microbiology, 5th ed. American Soc. Microbiology, Washington, D.C. LANCEFIELD, R.C. 1933. A serological differentiation of human and other groups of hemolytic streptococci. J. Exp. Med. 57:571. DIFCO LABORATORIES. 1984. Difco Manual, 10th ed. FACKLAM, R.R. & R.B. CAREY. 1985. Streptococci and aerococci. In E.H. Lennette, A. Ballows, W.J.Hausler, Jr. & H.J. Shadomy, eds. Manual of Clinical Microbiology, 5th ed. American Soc. Microbiology, Washington, D.C. GROSS, J.C., M.P. HOUGHTON & L.B. SENTERFIT. 1975. Presumptive speciation of Streptococcus bovis and other Group D streptococci from human sources by using arginine and pyruvate tests. J. Clin. Microbiol. 1:54. COWAN, S.T. & K.J. STEEL. 1965. Manual for the Identification of Medical Bacteria. Cambridge Univ. Press, Cambridge, England. BRIDGE, P.D. & P.H.A. SNEATH. 1983. Numerical taxonomy of streptococcus. J. Gen. Microbiol. 129:565. 9240 IRON AND SULFUR BACTERIA*#(50) 9240 A. Introduction The group of nuisance organisms collectively designated ‘‘iron and sulfur bacteria’’ is morphologically and physiologically heterogeneous, having in common the ability to transform or deposit significant amounts of iron or sulfur, usually in the form of objectionable slimes. However, iron and sulfur bacteria are not the sole producers of bacterial slimes and in some cases may be associated with slimes of other bacteria. The iron and sulfur bacteria may be filamentous or single-celled, autotrophic or heterotrophic, aerobic or anaerobic. The taxonomic position of these bacteria is very diverse. They are studied as iron and sulfur bacteria, because these elements and their transformations may be important in water treatment and distribution systems and may be especially bothersome in waters for industrial use, such as cooling and boiler waters. Iron bacteria may cause, or be associated with, fouling and plugging of wells. Also, the growth of these bacteria may result in consumer complaints of red water in distribution systems for potable water and sulfate-reducing bacteria may cause rusty water and tuberculation of pipes. These organisms also may cause odor, taste, frothing, color, and increases in turbidity in waters. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater The nutrient supply for iron and sulfur bacteria may be wholly or partly inorganic. Many of these bacteria can grow under oligotrophic conditions when attached to a substrate in flowing water. This seems quite important in the case of certain sulfur bacteria utilizing small amounts of hydrogen sulfide or in the case of organisms such as Gallionella, which obtain their energy from the oxidation of ferrous iron. Thiobacillus ferrooxidans contributes to the problem of acid mine drainage and can be identified by tests for transformation of ferrous to ferric iron or oxidation of reduced sulfur compounds under conditions of low pH. Temperature, light, pH, and oxygen supply are critical to the growth of iron and sulfur bacteria. Under different environmental conditions some bacteria may appear either as iron or as sulfur bacteria. 9240 B. Iron Bacteria 1. General Characteristics ‘‘Iron bacteria’’ are considered to be capable of metabolizing reduced iron present in their aqueous habitat and of depositing it in the form of hydrated ferric oxide on or in their mucilaginous secretions. A somewhat similar mechanism is used by bacteria that utilize manganese. The large amount of brown slime so produced will impart a reddish tinge and an unpleasant odor to drinking water and may render the supply unsuitable for domestic or industrial purposes. These bacteria obtain energy by the oxidation of iron from the ferrous to the ferric state; the ferric form is precipitated as ferric hydroxide [Fe(OH)3]. Iron may be obtained from the pipe itself if it is made of iron or from the water within it. Because of the energetics involved, the amount of ferric hydroxide deposited is very large in comparison with the enclosed cells. Some bacteria that do not oxidize ferrous iron may cause it to be dissolved or deposited indirectly. In their growth, either they liberate iron by utilizing organic radicals to which the iron is attached or they alter environmental conditions to permit the solution or deposition of iron. Under these conditions, less ferric hydroxide may be produced, but taste, odor, and fouling may be engendered. 2. Collection of Samples and Identification Examples of iron bacteria are shown in Figure 9240:1, Figure 9240:2, Figure 9240:3, Figure 9240:4, and Figure 9240:5. Identification of nuisance iron bacteria usually has been made on the basis of microscopic examination of the suspected material. Directly examine bulked activated sludge, masses of microbial growth in lakes, rivers, and streams, and slime growths in cooling-tower waters. Suspected development of iron bacteria in water wells or in distribution systems may require special efforts to secure samples useful for identification. Settle or centrifuge samples drawn directly from wells and examine sediment microscopically. Place a portion of sediment on a microscope slide, cover with a cover slip, and examine under a low-power microscope for filaments and iron-encrusted filaments. The material trapped by filters placed in front of back-surge valves often has yielded excellent specimens of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater iron bacteria. Water pumped from wells may be passed through a 0.45-µm membrane filter and the filter examined microscopically after drying and clearing with immersion oil applied directly to the membrane. Phase-contrast microscopes have made possible the examination of unstained culture material. Use india ink or lactophenol blue for staining when conventional light microscopy is used. Also, iron bacteria have been observed when the epifluorescence microscopic method (Section 9216B) was used. Continued heavy deposition of iron caused by the oxidation of ferrous iron by air or by other environmental changes often hides the sheaths or stalks of iron bacteria. The cells within the filaments often die and disintegrate and the filaments tend to be fragmented or crushed by the mass of the iron precipitate. To dissolve iron deposits place several drops of 1N HCl at one edge of cover slip and draw it under the cover slip by applying filter or blotting paper to the opposite edge. Reducing compounds such as sodium ascorbate also may be used to dissolve deposits and permit observation of cellular structure. To verify that the material is iron, add a solution of potassium ferrocyanide to a sample on a slide, cover, and draw 1N HCl under cover slip. A blue precipitate of Prussian blue will form as iron around cells or filaments is dissolved. Floating or hanging slide methods may be used to determine the presence of filamentous and other periphytic iron bacteria. In the floating method fix a glass microscope slide in a cork and let it float for 1 to 2 d on the surface of water taken from the source. Slides may be hung by means of string at various depths in the water for various times before being removed and observed microscopically for the presence of iron bacteria.1 A cultural method called the iron bacteria presence test has been developed for the detection of iron bacteria.2 Identify organisms by comparing with available drawings or photographs of iron bacteria.1-17 3. References 1. WOJCIK, W. & M. WOJCIK. 1986. Monitoring biofouling. In D.R. Cullimore, ed. Proc. International Symposium on Biofouled Aquifers: Prevention and Restoration. American Water Resources Assoc., Bethesda, Md. 2. CULLIMORE, D.R. & A.E. MCCANN. 1977. The identification, cultivation and control of iron bacteria in ground water. In F.A. Skinner & J.M. Shewan, eds. Aquatic Microbiology. Academic Press, New York, N.Y. 3. LUESCHOW, L.A. & K.M. MACKENTHUN. 1962. Detection and enumeration of iron bacteria in municipal water supplies. J. Amer. Water Works Assoc. 54:751. 4. STARKEY, R.L. 1945. Transformations of iron by bacteria in water. J. Amer. Water Works Assoc. 37:963. 5. STOKES, J.L. 1954. Studies on the filamentous sheathed iron bacterium Sphaerotilus natans. J. Bacteriol. 67:278. 6. KUCERA, S. & R.S. WOLFE. 1957. A selective enrichment method for Gallionella © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 7. 8. 9. 10. 11. ferruginea. J. Bacteriol. 74:344. WAITZ, S. & J.B. LACKEY. 1958. Morphological and biochemical studies on the organism Sphaerotilus natans. Quart. J. Fla. Acad. Sci. 21:335. WOLFE, R.S. 1958. Cultivation, morphology, and classification of the iron bacteria. J. Amer. Water Works Assoc. 50:1241. WOLFE, R.S. 1960. Observations and studies of Crenothrix polyspora. J. Amer. Water Works Assoc. 52:915. WOLFE, R.S. 1960. Microbial concentration of iron and manganese in water with low concentrations of these elements. J. Amer. Water Works Assoc. 52:1335. DONDERO, N.C., R.A. PHILIPS & H. HEUKELEKIAN. 1961. Isolation and preservation of cultures of Sphaerotilus. Appl. Microbiol. 9:219. 12. MULDER, E.G. 1964. IRON BACTERIA, PARTICULARLY THOSE OF THE Sphaerotilus-Leptothrix group, and industrial problems. J. Appl. Bacteriol. 27:151. 13. DRAKE, C.H. 1965. Occurrence of Siderocapsa treubii in certain waters of the Niederrhein. Gewasser Abwasser 39/40:41. 14. STALEY, J.T., M.P. BRYANT, N. PFENNING & J.G. HOLT, eds. 1989. Bergey’s Manual of Systematic Bacteriology, Volume 3. Williams & Wilkins, Baltimore, Md. 15. EDMONDSON, W.T., ed. 1959. Ward & Whipple’s Fresh Water Biology, 2nd ed. John Wiley & Sons, New York, N.Y. 16. SKERMAN, V.B.D. 1967. A Guide to the Identification of the Genera of Bacteria, 2nd ed. Williams & Wilkins, Baltimore, Md. 17. GHIORSE, W.C. 1984. Biology of iron and manganese depositing bacteria. Annu. Rev. Microbiol. 38:515. 9240 C. Sulfur Bacteria 1. General Characteristics The bacteria that oxidize or reduce significant amounts of inorganic sulfur compounds exhibit a wide diversity of morphological and biochemical characteristics. One group, the sulfate-reducing bacteria, consists mainly of single-celled forms that grow anaerobically and reduce sulfate, SO42−, to hydrogen sulfide, H2S. One member of this group, Desulfonema, is multicellular and exhibits gliding motility. A second group, the photosynthetic green and purple sulfur bacteria, grows anaerobically in the light and uses H2S as a hydrogen donor for photosynthesis. Members of a third colorless filamentous group are myxotrophic and utilize organic sources of carbon but may get their energy from the oxidation of reduced sulfur compounds. The sulfide is oxidized to sulfur or sulfate. A fourth group, the aerobic sulfur-oxidizers, oxidizes reduced sulfur compounds aerobically to obtain energy for © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater chemoautotrophic growth. The sulfur bacteria of most importance in the water and wastewater field are the sulfate-reducing bacteria, which include Desulfovibrio, and the single-celled aerobic sulfur-oxidizers of the genus Thiobacillus. The sulfate-reducing bacteria contribute greatly to tuberculations and galvanic corrosion of water mains and to taste and odor problems in water. Thiobacillus, by its production of sulfuric acid, has contributed to the destruction of concrete sewers and the acid corrosion of metals. 2. Collection of Samples and Identification Identification of nuisance sulfur bacteria usually has been made on the basis of microscopic examination of the suspected material. Examine samples of slimes suspended in waters, scrapings from exposed surfaces, or sediments directly. Three groups of sulfur bacteria may be recognized microscopically; green and purple sulfur bacteria; large, colorless filamentous sulfur bacteria; and large, colorless, nonfilamentous sulfur bacteria. The fourth group, consisting of sulfate-reducing bacteria and sulfur-oxidizing bacteria of the genus Thiobacillus, cannot be identified by appearance alone. a. Green and purple sulfur bacteria: 1) Green sulfur bacteria most frequently occur in waters containing H2S. They are small, ovoid to rod-shaped, nonmotile organisms, generally less than 1 µm in diameter, and with a yellowish-green color in masses. Sulfur globules are seldom if ever deposited within the cells. 2) Purple sulfur bacteria (Figure 9240:6) occur in waters containing H2S. They are large, generally stuffed with sulfur globules, and often so intensely pigmented as to make individual cells appear red. Large, dense, highly colored masses are detected easily by the naked eye. The presence of photosynthetic bacteria in concentrated masses can be confirmed by extracting the mass with ether and scanning the extract absorbance in the infrared region. Bacterial chlorophyll will absorb strongly in the range of 660 to 870 nm. b. Colorless filamentous sulfur bacteria: Colorless filamentous sulfur bacteria (Figure 9240:7, Figure 9240:8, and Figure 9240:9) occur in waters where both oxygen and H2S are present. They may form mats with a slightly yellowish-white appearance due to deposition of internal sulfur globules. They generally are large and may be motile with a characteristic gliding movement. Identify by comparing organisms with available photographs.1-4 c. Colorless nonfilamentous sulfur bacteria: Colorless, nonfilamentous sulfur bacteria (see Figure 9240:10, for example) usually are associated with decaying algae. They are extremely motile, ovoid to rod-shaped with sulfur globules and possible calcium carbonate deposits. They generally are very large. d. Colorless small sulfur bacteria and sulfate-reducing bacteria: The small single-celled bacteria, Thiobacillus spp., and the sulfate-reducing bacteria, such as Desulfovibrio, cannot be identified by direct microscopic examination. Thiobacillus types are small, colorless, motile, and rod-shaped and are found in an environment containing H2S. Sulfur globules are absent. Identify © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Thiobacillus types, Desulfovibrio, or other sulfate-reducing bacteria physiologically. 3. References 1. LACKEY, J.B. & E.W. LACKEY. 1961. The habitat and description of a new genus of sulfur bacterium. J. Gen. Microbiol. 26:28. 2. FAUST, L. & R.S. WOLFE. 1961. Enrichment and cultivation of Beggiatoa alba. J. Bacteriol. 81:99. 3. MORGAN, G.B. & J.B. LACKEY. 1965. Ecology of a sulfuretum in a semitropical environment. Z. Allg. Mikrobiol. 5:237. 4. STALEY, J.T., M.P. BRYANT, N. PFENNING & J.G. HOLT, eds. 1989. Bergey’s Manual of Systematic Bacteriology, Volume 3. Williams & Wilkins, Baltimore, Md. 9240 D. Enumeration, Enrichment, and Isolation of Iron and Sulfur Bacteria There are no good means of enumerating iron and sulfur bacteria other than the sulfate-reducing bacteria and the thiobacilli. Laboratory cultivation and isolation of pure cultures is difficult and successful isolation is uncertain. This is especially true of attempts to isolate filamentous bacteria from activated sludge or other sources where many different bacterial types are present. 1. Media a. Casitone-glycerol-yeast autolysate broth (CGY), for the Sphaerotilus group: This medium may not be available in dehydrated form and may require preparation from the basic ingredients. It may be solidified by adding 1.5% agar. Casitone Glycerol Yeast autolysate Reagent-grade water 5.0 10.0 1.0 1 g mL g L b. Isolation medium (iron bacteria): This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Glucose Ammonium sulfate, (NH4)2SO4 0.15 g 0.5 g Calcium nitrate, Ca(NO3)2 0.01 g Dipotassium hydrogen phosphate, K2HPO4 0.05 g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Magnesium sulfate, MgSO4⋅7H2O 0.05 g Potassium chloride, KCl Calcium carbonate, CaCO3 0.05 g 0.1 g Agar Cyanocobalamin Thiamine Reagent-grade water 10.0 0.01 0.4 1 g mg mg L c. Maintenance (SCY) medium (iron bacteria): This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Sucrose Casitone Yeast extract Trypticase soy broth without dextrose Agar Cyanocobalamin Thiamine Reagent-grade water 1.0 0.75 0.25 0.25 10.0 0.01 0.4 1 g g g g g mg mg L d. Mn agar No. 1: This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Manganous carbonate, MnCO3 2.0 g Beef extract Ferrous ammonium sulfate, Fe(NH4)2(SO4)2 1.0 150 g mg Sodium citrate Yeast extract Cyanocobalamin Agar Reagent-grade water 150 75 0.005 10.0 1 mg mg mg g L Prepare and sterilize the medium without cyanocobalamin. Separately sterilize cobalamin by filtration and add aseptically just before medium solidifies. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater e. Mn agar No. 2:1 Prepare fresh each time from basic ingredients: Manganous sulfate, MnSO4⋅H2O 10 Agar Natural water 15.0 g 1 L mg f. Iron oxidizing medium (Thiobacillus ferrooxidans): This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Basal salts: Ammonium sulfate, (NH4)2SO4 3.0 g Potassium chloride, KCl Dipotassium hydrogen phosphate, K2HPO4 0.10 g 0.50 g Magnesium sulfate, MgSO4⋅7H2O 0.50 g Calcium nitrate Ca(NO3)2 0.01 g H2SO4, 10N 1.0 mL Reagent-grade water 700 mL Ferrous sulfate, FeSO4⋅7H2O, 14.74% solution (w/v) 300 mL Energy source: Separately sterilize basal salts and energy source and combine when cool. Store in the refrigerator and discard after 2 weeks. A precipitate will form and the medium will be opalescent and green. The pH should be 3.0 to 3.6. g. Ferrous sulfide agar (Gallionella ferruginea): This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Agar layer: Ferrous sulfide, FeS (washed precipitate and liquid) Sodium sulfide, Na2S 500 mL 15.6 g Ferrous ammonium sulfate, Fe(NH4)2(SO4)2⋅6H2O 78.4 g Boiling reagent-grade water Agar (liquid) (30 g/L) 1 500 L mL © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Liquid overlay: Ammonium chloride, NH2Cl 1.0 g Dipotassium hydrogen phosphate, K2HPO4 0.5 g Magnesium sulfate, MgSO4⋅7H2O 0.2 g Calcium chloride, CaCl2 0.1 g Reagent-grade water 1 L Prepare FeS by reacting equal molar quantities of Na2S and Fe(NH4)2(SO4)2 in boiling reagent-grade water. Let precipitate settle from the hot solution in a completely filled and stoppered bottle. Wash precipitate four times by decanting supernatant and replacing with boiling water. Store FeS in a glass stoppered bottle completely filled with additional boiling water. Add equal volumes of FeS and 3% agar at 45°C. Prepare slants in screw-capped tubes. Prepare liquid overlay, bubble CO2 through it for 10 to 15 s, and add several milliliters to agar slant. Modifications to this medium and procedure are available.2-4 A variation of the basic medium requires adding 0.5 mL formalin (40% formaldehyde solution) to a screw-capped dilution bottle containing 10 mL FeS agar and 100 mL liquid overlay. This variation is said not to work in all cases.3,4 Add 0.001% bromthymol blue and 0.004% bromcresol purple to liquid overlay. h. Sulfate-reducing medium: This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Sodium lactate Beef extract Peptone Magnesium sulfate, MgSO4⋅7H2O 3.5 1.0 2.0 2.0 g g g g Sodium sulfate, Na2SO4 1.5 g Dipotassium hydrogen phosphate, K2HPO4 0.5 g Ferrous ammonium sulfate, Fe(NH4)2(SO4)2⋅6H2O 0.392 g Calcium chloride, CaCl2 0.10 g Sodium ascorbate Reagent-grade water 0.10 1 g L © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater pH should be 7.5 ± 0.3 after sterilization. Prepare medium excluding ferrous ammonium sulfate and sodium ascorbate, dispense in screw-capped test tubes, and sterilize. For use, the tubes must be completely filled; therefore, in a flask sterilize extra medium to be added to tubes for filling. On day of use, prepare separate solutions of ferrous ammonium sulfate (3.92 g/100 mL) and sodium ascorbate (1.00 g/100 mL), sterilize by filtration through a 0.45-µm membrane filter, and aseptically add 0.1 mL each solution/10 mL basal medium. i. Thiosulfate oxidizing medium (Thiobacillus thioparus): This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Sodium thiosulfate, Na2S2O3⋅5H2O 10.0 g Dipotassium hydrogen phosphate, K2HPO4 2.0 g Magnesium sulfate, MgSO4⋅7H2O 0.1 g Calcium chloride, CaCl2⋅2H2O 0.1 g Ammonium sulfate, (NH4)2SO4 0.1 g Ferric chloride, FeCl3⋅6H2O 0.02g Reagent-grade water 1 L pH should be 7.8 after sterilization. Separately sterilize Na2S2O3 and (NH4)2SO4 and add before use. If this medium is used to isolate Thiobacillus thioparus, check isolates to ensure they are autotrophs. j. Sulfur medium (Thiobacillus thiooxidans): This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Sulfur, elemental Potassium dihydrogen phosphate, KH2PO4 10.0 3.0 g g Magnesium sulfate, MgSO4⋅7H2O 0.5 g Ammonium sulfate, (NH4)2SO4 0.3 g Calcium chloride, CaCl2⋅2H2O 0.25 g Ferric chloride, FeCl3⋅6H2O 0.02 g Reagent-grade water 1 L pH should be 4.8 after sterilization. Weigh sulfur into 250-mL flasks using 1 g/flask. Add 100 mL medium to each flask and sterilize with intermittent steam (30 min for each of 3 consecutive d). © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater k. Media for Beggiatoa and myxotrophic strains of Thiothrix: 1) Extracted hay: Extract hay or grass at least five times by boiling in water for 30 min, with two rinses in cold water between each extraction. Dry the extracted hay, place 10 to 30 blades in a large test tube, and sterilize by autoclaving. 2) Basal medium Ammonium chloride, 4% solution Dipotassium hydrogen phosphate, K2HPO4, 1% solution 5 mL 1 mL Magnesium sulfate, MgSO4⋅7H2O, 1% solution 1 mL Calcium sulfate, CaSO4⋅2H2O, saturated solution 20 mL Trace elements [see ¶ 3) below] Reagent-grade water 5 mL 968 mL 3) Trace elements Reagent-grade water 920 mL 10 mL Zinc sulfate, ZnSO4⋅7H2O, 0.1% solution Manganous sulfate, MnSO4⋅4H2O, 0.02% solution 10 mL Copper sulfate, CuSO4⋅5H2O, 0.00005% solution 10 mL Boric acid, H3BO3, 0.1% solution 10 mL Cobalt nitrate, Co(NO3)2 or CoCl2⋅6H2O, 0.01% solution 10 mL Sodium molybdate, Na2MoO4⋅2H2O, 0.01% solution 10 mL EDTA solution, 2% EDTA with 7% ferrous sulfate, FeSO4⋅7H2O, with 1 mL conc HCl/100 mL 20 mL 4) MP agar: This medium may not be available in dehydrated form and should be made fresh before use. Basal medium Sodium acetate Sodium sulfide solution [see ¶ 5) below] Agar 1 0.1 3 15 L g mL g © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Adjust pH to between 7.0 and 7.5 and heat to dissolve agar. Sterilize by autoclaving for no more than 15 min and cool in a 45 to 50°C water bath. Add sodium sulfide solution immediately before pouring plates. If the medium is to be used for screw-capped tubes the sulfide may be added before autoclaving. 5) Sodium sulfide solution: Make up and separately autoclave a 10% solution of Na2S⋅9H2O. 6) MY agar: This medium may not be available in dehydrated form and should be made fresh before use. Basal medium Sodium acetate Nutrient broth powder (Difco) Yeast extract (Difco) Sodium sulfide solution [see ¶ 5) above] Agar 1 0.1 0.1 0.1 3 15 L g g g mL g Adjust pH to between 7.0 and 7.5 and heat to dissolve agar. Sterilize by autoclaving for no more than 15 min and cool in a 45 to 50°C water bath. Add sodium sulfide solution immediately before pouring plates. If the medium is to be used for screw-capped tubes the sulfide may be added before autoclaving. l. Media for heterotrophic strains of Thiothrix: Make the media 3) through 9) with solutions 1) and 2), plus indicated additives. After adding all ingredients, adjust pH to 7.2 to 7.5. For solid media add 12 g agar/L. 1) MSV Ammonium sulfate, (NH4)2SO4 0.5 g Dipotassium hydrogen phosphate, K2HPO4 0.11 g Potassium dihydrogen phosphate, KH2PO4 0.085 g Magnesium sulfate, MgSO4⋅7H2O 0.1 g Calcium chloride, CaCl2⋅2H2O 0.05 g Ferric chloride, FeCl3⋅H2O 0.002 g EDTA Vitamin mix Reagent-grade water 0.003 g 1 mL 1 L © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2) Vitamin mix Calcium pantothenate Niacin Biotin Cyanocobalamin Folic acid Pyridoxine p-aminobenzoic acid Cocarboxylase Inositol Thiamine Riboflavin Reagent-grade water 0.01 0.01 0.0005 0.0005 0.0005 0.01 0.01 0.01 0.01 0.01 0.01 100 g g g g g g g g g g g mL 3) AcS Sodium acetate Sodium sulfide, Na2S⋅9H2O 0.15 g 0.187 g MSV 1 Sucrose Sodium sulfide, Na2S⋅9H2O 0.15 g 0.187 g MSV 1 Glucose Sodium sulfide, Na2S⋅9H2O 0.15 g 0.187 g MSV 1 L 4) SS L 5) GS L 6) SUC © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Sodium succinate MSV 0.15 g 1 L 7) I Glucose MSV 0.15 g 1 L 8) S Sodium sulfide, Na2S⋅9H2O 0.187 g MSV 1 L 9) LT Sodium lactate Sodium thiosulfate, Na2S2O3 0.5 g 0.5 g MSV 1 L 2. Iron Bacteria a. Sphaerotilus-Leptothrix: 1) Iron bacteria, especially those belonging to the Sphaerotilus-Leptothrix group, thrive in media too dilute to support the proliferation of more rapidly growing organisms. One medium5 is partially selective for Sphaerotilus (BOD dilution water, Section 5210B, supplemented with 100 mg/L sodium lactate). Dispense 50 mL of this medium into French square bottles and autoclave at 69 kPa for 15 min. To inoculate sample add 25-mL portions of stream water or 1-, 5-, and 10-mL portions of settled wastewater or process liquor to duplicate bottles of medium. Incubate at 22 to 25°C for 5 d and observe for filamentous growth. Isolate pure cultures by picking a filament from the BOD-lactate broth and streaking on 0.05% meat extract agar. After incubating for 24 h at 25°C, pick typical curling filaments with the aid of a dissecting microscope and transfer to casitone-glycerol-yeast autolysate (CGY) broth. If a pellicle with no underlying turbidity develops in 2 to 3 d, transfer a filament to a CGY agar slant, incubate at 25°C until growth is visible, and store in a refrigerator. In addition, extracted alfalfa straw or pea straw may be used for enrichments.4 A detailed key for identifying filamentous microorganisms in complex mixtures such as © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater wastewater and activated sludge is available.6 2) Isolation and maintenance media have proven quite successful for identifying various groups of filamentous organisms, including iron bacteria.7 Prepare agar slants of these media and aseptically pipet 3 mL sterile, dechlorinated tap water onto surface of slants. Inoculate tubes and incubate at room temperature until turbid growth has developed in liquid layer. The cells will remain viable for 3 months in the refrigerator. 3) Another good maintenance medium for cultivating the Sphaerotilus group is CGY.8 4) Leptothrix (Sphaerotilus discophorous) can be distinguished from Sphaerotilus natans by its ability to oxidize manganous ion. Use Mn agar No. 1 as the differential medium.9 Alternatively, Leptothrix may be grown by direct plating on Mn agar No. 2.1 b. Thiobacillus ferrooxidans: Although this organism also is a sulfur-oxidizing bacterium,10,11 its main importance has been in acid mine drainage. A medium suitable for enumeration of the MPN is available.12 Some oxidation of iron occurs during sterilization but the loss of ferrous iron is not appreciable. The medium has a precipitate (probably ferrous and ferric phosphates), is opalescent and green, has a pH of 3.0 to 3.6, and contains 9000 mg/L ferrous iron. Growth of the organism is manifested by a decrease in pH and an increase in concentration of oxidized iron. With practice and use of uninoculated controls, an increase of deep orange-brown color can be seen in positive enrichment tubes or flasks as compared to negative ones. Shake test-tube dilutions daily because these organisms are highly aerobic. c. Gallionella ferruginea: For cultivation of this organism use ferrous sulfide agar.13,14 Inoculate tubes with a drop of suspension of a suspected Gallionella deposit. Growth at room temperature usually occurs in 18 to 36 h and appears as a white deposit on sides of test tube. The ring of colonies occurring at a certain level reflects a balance between upward diffusion of ferrous ions and downward diffusion of oxygen molecules. Supplementation of ferrous sulfide agar with formalin to isolate pure cultures may be successful.1,4,15 d. Other iron bacteria: An acid-tolerant (pH 3.5 to 5.0) filamentous iron-oxidizing Metallogenium has been isolated with a medium16 containing (NH4)2SO4, 0.1%; CaCO3, 0.01%; MgSO4, 0.02%; K2HPO4, 0.001%; potassium acid phthalate, 0.4%; and 250 mg/L ferrous iron from an acidified FeSO4⋅7H2O, solution. Add 0.4% formalin to 100 mL of the isolating medium in a 250-mL erlenmeyer flask. For heterotrophic iron-precipitating bacteria17 use a ferric ammonium citrate medium consisting of: (NH4)2SO4, 0.5 g/L; NaNO3, 0.5 g/L; K2HPO4, 0.5 g/L; MgSO2⋅7H2O, 0.5 g/L; and ferric ammonium citrate, 10.0 g/L. Adjust pH to 6.6 to 6.8 and sterilize. To make the medium solid add 15 g agar/L. Alternatively grow iron bacteria by combining 20 mL liquid broth medium, 10 mL raw water or inoculum, and 3 g iron oxide. Incubate 48 to 72 h at 25°C on a wrist-action shaker to produce good, visible growth. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 3. Sulfur Bacteria a. Sulfate-reducing bacteria: 1) To enumerate sulfate-reducing bacteria such as Desulfovibrio, use a sulfate-reducing medium.18 Inoculate tubes and fill completely with sterile medium to create anaerobic conditions. For comparative purposes, incubate one or two uninoculated controls with each set of inoculated tubes. To sample volumes greater than 10 mL, pass sample through a 0.45-µm membrane filter and transfer filter to screw-cap test tube with medium. If sulfate-reducing bacteria are present, tubes will show blackening within 4 to 21 d of incubation at 20 to 30°C. 2) An agar medium suitable for growth and enumeration of sulfate-reducing bacteria also is available.19 The medium consists of trypticase soy agar, 4.0%, fortified with additional agar, 0.5%, to which is added 60% sodium lactate (0.4% v/v), hydrated magnesium sulfate, 0.2%, and ferrous ammonium sulfate, 0.2%. Adjust pH to 7.2 to 7.4 and sterilize. Medium should be clear and free from precipitate. Inoculate all plates within 1 or at most 4 h after agar hardens to prevent saturation with oxygen. To prevent moisture condensation on petri dish covers, replace covers with sterile absorbent tops until 10 to 15 min after agar hardens. Place uninverted plates in desiccator or Brewer jars and replace atmosphere with tank hydrogen or nitrogen by successive evacuation and gas replacement. Alternatively, use a disposable anaerobic generating system.*#(51) Incubate at room temperature (21 to 24°C) or at 28 to 30°C, the optimum temperature for these organisms. Growth and blackening around the colonies is typical of sulfate-reducing bacteria and may occur at any time between 2 and 21 d, although the usual time is 2 to 7 d. 3) Media suitable for enumeration or isolation of various species of sulfate-reducing bacteria are available.20-22 b. Photosynthetic purple and green sulfur bacteria: Because these organisms are so specialized and rarely cause problems in water and wastewater treatment processes, methods for their isolation and enumeration are not included here. In certain instances they can be beneficial because of their ability to oxidize hydrogen sulfide and thus reduce odor. An excellent review is available.23 Also, media formulations and methods for cultivating specific members of this group of bacteria are available.23 c. Thiobacillus spp.: The growth and physiology of different species of the single-celled sulfur-oxidizing bacteria of the genus Thiobacillus have been evaluated carefully.24,25 Media26 suitable for enumeration of Thiobacillus thioparus and Thiobacillus thiooxidans by an MPN technique are listed in Section 9240D.1. Inoculate medium and incubate for 4 to 5 d at 25 to 30°C. Growth of thiobacilli produces elemental sulfur, which sinks to the bottom with a coincident decrease in pH and turbidity of the medium. Chemical tests for formation of sulfate are necessary to confirm presence of Thiobacillus. d. Filamentous sulfur-oxidizing bacteria: 1) Beggiatoa—Beggiatoa exist in most aquatic habitats where sulfide and oxygen are both © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater present,27 including fresh and marine water, sediments, and wastewater systems. Because Beggiatoa is a multicellular bacterium there is no accurate method of determining the number of viable cells in a sample. To determine the population of Beggiatoa make a direct microscopic count of the number of filaments. Marine beggiatoas may be quite large, up to 100 µm or more in diameter.28 They have not been grown in the laboratory. However, small marine species (up to 5 µm diam) have been isolated.29 The media for isolating freshwater and marine beggiatoas differ slightly. a) Enrichment—Inoculate a tube of extracted hay, ¶ 1k1) above, with enough water and a little mud from the sample site to fill the tube to a depth of at least 8 cm. Incubate for at least 1 week and examine for the presence of ‘‘tufts’’ or ‘‘puff-balls’’ consisting of tangled filaments of Beggiatoa. Examine by phase-contrast microscopy for the presence of individual filaments.30 If no ‘‘puff-balls’’ are found, continue incubation for another week and repeat the examination. Continue examining the enrichment for up to 4 weeks before discarding it. b) Isolation—With sterilized fine-tipped forceps and using a dissecting microscope, transfer tufts of Beggiatoa from the enrichments to a small petri dish containing sterile basal medium,30 ¶ 1k2) above. Shake the tufts with the forceps to remove adherent bacteria and transfer tufts to a new petri dish with sterile basal medium. Continue until tufts have been washed at least five times. Transfer tufts to a ‘‘drying plate’’ containing basal medium and 1.6% agar for about 1 min to remove excess fluid, then transfer the tufts to the center of separate plates of either MY or MP medium,30,31 ¶s 1k3) and 4) above. Incubate plates at room temperature or below and examine with a dissecting microscope every 5 to 10 h for the presence of gliding filaments of Beggiatoa. Select filaments that have glided well away from the other filaments and appear to be uncontaminated and transfer them to separate plates of the same medium with a sterile, flattened wire, inoculating needle, or toothpick. Take a little agar with the filament to avoid drying the filament during transfer. Examine the first transfer plates every 5 to 10 h as before and transfer pure filaments to fresh media. 2) Thiothrix—Obligately myxotrophic Thiothrix have been isolated and characterized from sulfur springs and other bodies of flowing water that contain sulfide.31,32 Heterotrophic strains have been isolated and characterized from activated sludge wastewater treatment plants.33-35 While the techniques for the isolation of the myxotrophic and heterotrophic strains are similar, the isolation media differ. a) Isolation of myxotrophic Thiothrix—Collect tufts of the bacterium from rocks, water pipes, or other substrates. Using a dissecting microscope, pick up filaments with fine-tipped forceps, shake to remove contaminating bacteria, and transfer to sterile basal medium, ¶ 1k2) above. Repeat at least five times to try to obtain filaments with little or no contamination. With a sterile Pasteur pipet transfer separate drops to the edge of either MP or MY agar [¶s 1k3) and 4), above] petri dishes and tip the dishes so that the drops run from one side of the dish to the other. Draw off excess moisture with the pipet and incubate plates at room temperature or © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater below for about 48 h. Examine plates under a dissecting microscope for the appearance of typical filamentous colonies. With sterile toothpicks, pick colonies that are widely separated and transfer individually to fresh plates of the same medium. Streak transferred material with a wire loop. After about 48 h incubation, examine plates and restreak colonies that appear to be pure. b) Isolation of heterotrophic Thiothrix—There are two different procedures to use, depending on the concentration and size of the Thiothrix filaments in the sample.34 In one procedure, wash individual filaments or rosettes of large strains of Thiothrix several times in MSV broth, ¶ 1l 1) above, by transfer with a Pasteur pipet while observing with a dissecting microscope. After several washings transfer filaments to a small amount of MSV (1 to 3 mL) and plate on one or more of the following solid media, ¶s 1l 3)–9) above. If the filaments are small or scarce concentrate the sample by centrifuging. Then dilute 1:5 in MSV, sonicate at 30 W for 10 s, and wash three times by centrifugation at 1900 × g for 2 to 5 min. The supernatant contains free filaments that are used to inoculate one or more of the solid media, ¶s 1l 3)–9) above. 4. References 1. GHIORSE, W.C. 1984. Biology of iron- and manganese-depositing bacteria. Annu. Rev. Microbiol. 38:515. 2. HALLBRECK, E.L. & K. PEDERSON. 1986. The biology of Gallionella. In D.R. Cullimore, ed. Proc. International Symposium on Biofouled Aquifers: Prevention and Restoration. American Water Works Assoc., Denver, Colo. 3. STARR, M.P., H. STOLP, H.G. TRUPER, A. BALOWS & H.C. SCHLEGE. 1981. The Prokaryotes. A Handbook on Habitats, Isolation and Identification of Bacteria, Volume 1. Springer-Verlag, New York, N.Y. 4. STALEY, J.T., M.P. BRYANT, N. PFENNIG & J.G. HOLT, eds. 1989. Bergey’s Manual of Systematic Bacteriology, Volume 3. Williams & Wilkins, Baltimore, Md. 5. ARMBRUSTER, E.H. 1969. Improved technique for isolation and identification of Sphaerotilus. Appl. Microbiol. 17:320. 6. FARQUHAR, G.J. & W.C. BOYLE. 1971. Identification of filamentous microorganisms in activated sludge. J. Water Pollut. Control Fed. 43:604. 7. VANVEEN, W.L. 1973. Bacteriology of activated sludge, in particular the filamentous bacteria. Antonie van Leeuwenhoek (Holland) 39: 189. 8. DONDERO, N.C., R.A. PHILIPS & H. HEUKELEKIAN. 1961. Isolation and preservation of cultures of Sphaerotilus. Appl. Microbiol. 9:219. 9. MULDER, E.G. & W.L. VANVEEN. 1963. Investigations on the Sphaerotilus-Leptothrix group. Antonie van Leeuwenhoek (Holland) 29:121. 10. UNZ, R.F. & D.G. LUNDGREN. 1961. A comparative nutritional study of three chemoautotrophic bacteria: Ferrobacillus ferrooxidans, Thiobacillus ferrooxidans, and Thiobacillus thiooxidans. Soil Sci. 92: 302. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 11. MCGORAN, C.J.M., D.W. DUNCAN & C.C. WALDEN. 1969. Growth of Thiobacillus ferrooxidans on various substrates. Can. J. Microbiol. 15:135. 12. SILVERMAN, M.P. & D.C. LUNDGREN. 1959. Studies on the chemoautotrophic iron bacterium Ferrobacillus ferrooxidans. J. Bacteriol. 77:642. 13. KUCERA, S. & R.S. WOLFE. 1957. A selective enrichment method for Gallionella ferruginea. J. Bacteriol. 74:344. 14. WOLFE, R.S. 1958. Cultivation, morphology, and classification of the iron bacteria. J. Amer. Water Works Assoc. 50:1241. 15. NUNLEY, J.W. & N.R. KRIEG. 1968. Isolation of Gallionella ferruginea by use of formalin. Can. J. Microbiol. 14:385. 16. WALSH, F. & R. MITCHELL. 1972. A pH dependent succession of iron bacteria. Environ. Sci. Technol. 6:809. 17. CLARK, F.M., R.M. SCOTT & E. BONE. 1967. Heterotrophic, iron-precipitating bacteria. J. Amer. Water Works Assoc. 59:1036. 18. LEWIS, R.F. 1965. Control of sulfate-reducing bacteria. J. Amer. Water Works Assoc. 57:1011. 19. IVERSON, W.P. 1966. Growth of Desulfovibrio on the surface of agar media. Appl. Microbiol. 14:529. 20. LECHEVALIER, H.A. & D. PRAMER. 1970. The Microbes, 1st ed. J.B. Lippincott Co., Philadelphia, Pa. 21. MARA, D.D. & D.J.A. WILLIAMS. 1970. The evaluation of media used to enumerate sulphate reducing bacteria. J. Appl. Bacteriol. 33:543. 22. KREIG, N.R. & J.G. HOLT. 1986. Bergey’s Manual of Systematic Bacteriology, Volume 1. Williams & Wilkins. Baltimore, Md. 23. PFENNIG, N. 1967. Photosynthetic bacteria. Annu. Rev. Microbiol. 21: 285. 24. HUTCHINSON, M., K.I. JOHNSTONE & D. WHITE. 1965. The taxonomy of certain thiobacilli. J. Gen. Microbiol. 41:357. 25. HUTCHINSON, M., K.I. JOHNSTONE & D. WHITE. 1966. Taxonomy of the acidophilic thiobacilli. J. Gen. Microbiol. 44:373. 26. STARKEY, R.L. 1937. Formation of sulfide by some sulfur bacteria. J. Bacteriol. 33:545. 27. LACKEY, J.B., W.W. LACKEY & G.B. MORGAN. 1965. Taxonomy and ecology of the sulfur bacteria. Eng. Prog., Univ. Fla. Bull. Ser. 119, 19:3. 28. NELSON, D.C., C.O. WIRSEN & H.W. JANNASCH. 1989. Characterization of large, autotrophic Beggiatoa spp. abundant at hydrothermal vents of the Guaymas Basin. Appl. Environ. Microbiol. 55:2909. 29. NELSON, D.C., J.B. WATERBURY & H.W. JANNASCH. 1982. Nitrogen fixation and nitrate utilization by marine and freshwater Beggiatoa. Arch. Microbiol. 133:172. 30. STROHL, W.R. & J.M. LARKIN. 1978. Enumeration, isolation, and characterization of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 31. 32. 33. 34. 35. Beggiatoa from freshwater sediments. Appl. Environ. Microbiol. 36:755. LARKIN, J.M. 1980. Isolation of Thiothrix in pure culture and observation of a filamentous epiphyte on Thiothrix. Curr. Microbiol. 4: 155. LARKIN, J.M. & D.L. SHINABARGER. 1983. Characterization of Thiothrix nivea. Int. J. System. Bacteriol. 33:841. EIKELBOOM, D.H. 1975. Filamentous organisms observed in activated sludge. Water Res. 9:365. WILLIAMS, T.M. & R.F. UNZ. 1985. Isolation and characterization of filamentous bacteria present in bulking activated sludge. Appl. Microbiol. Technol. 22:273. WILLIAMS, T.M. & R.F. UNZ. 1985. Filamentous sulfur bacteria of activated sludge: characterization of Thiothrix, Beggiatoa, and Eikelboom type 021N strains. Appl. Environ. Microbiol. 49:887. 9250 DETECTION OF ACTINOMYCETES*#(52) 9250 A. Introduction 1. General Discussion Earthy-musty odors affect the quality and public acceptance of municipal water supplies in many parts of the world. They are among the naturally occurring odors that plant operators find most difficult to remove by conventional treatment. As early as 1929, it was assumed that these odors could be attributed to volatile metabolites formed during normal actinomycete development.1 Two such compounds, geosmin and 2-methylisoborneol, have been isolated2–8 and identified as the agents responsible for earthy-musty odor problems in surface water.8–10 Both, however, are produced also by some filamentous blue-green algae.11–15 Geosmin and 2-methylisoborneol have threshold odor concentrations well below the microgram-per-liter level. Thus, traces of these products are sufficient to impart a disagreeable odor to water or a muddy flavor to fish. In areas periodically plagued by this problem, it is prudent to enumerate actinomycetes. Identification of their relative abundance in a drinking water source can provide yet another means to assess water quality. The methods described are well-established techniques that have been used with success in the isolation and enumeration of actinomycetes related to public water supplies.16,17 Actinomycetes also have been recognized as a cause of disruptions in wastewater treatment. Massive growths are capable of producing thick foam in the activated sludge process.18,19 Of the general properties of actinomycetes, the most striking is their fungal-type morphology. Although actinomycetes were looked upon initially as fungi, later research revealed © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater that they were filamentous, branching bacteria.20 The actinomycetes are represented most commonly by saprophytic forms that have an extensive impact on the environment by decomposing and transforming a wide variety of complex organic residues. Widely distributed in nature, actinomycetes constitute a considerable proportion of the population of soil and lake and river muds. Most actinomycetes from which geosmin and 2-methylisoborneol have been identified are members of the genus Streptomyces, which is considered the most likely to be significant in water supply problems. 2. Samples a. Collection: Collect samples as directed in Section 9060A. b. Storage: Analyze samples as promptly after collection as possible. Store water samples below 10°C if they cannot be processed promptly. 3. References 1. ADAMS, B.A. 1929. Cladothrix dichotoma and allied organisms as a cause of an ‘‘indeterminate’’ taste in chlorinated water. Water & Water Eng. 31:327. 2. GERBER, N.N. & H.A. LECHEVALIER. 1965. Geosmin, an earthy-smelling substance isolated from actinomycetes. Appl. Microbiol. 13:935. 3. GERBER. N.N. 1968. Geosmin, from microorganisms, is trans-1,10-dimethyl-trans-9-decalol. Tetrahedron Lett. 25:2971. 4. MARSHALL, J.A. & A.R. HOCHSTETLER. 1968. The synthesis of (±)-geosmin and the other 1,10-dimethyl-9-decalol isomers. J. Org. Chem. 33:2593. 5. ROSEN, A.A., R.S. SAFFERMAN, C.I. MASHNI & A.H. ROMANO. 1968. Identity of odorous substances produced by Streptomyces griseoluteus. Appl. Microbiol. 16:178. 6. MEDSKER, L.L., D. JENKINS & J.F. THOMAS. 1969. Odorous compounds in natural waters: 2-exo-hydroxy-2-methylbornane, the major odorous compound produced by several actinomycetes. Environ. Sci. Technol. 3:476. 7. GERBER, N.N. 1969. A volatile metabolite of actinomycetes, 2-methylisoborneol. J. Antibiot. 22:508. 8. ROSEN, A.A., C.I. MASHNI & R.S. SAFFERMAN. 1970. Recent developments in the chemistry of odour in water: The cause of earthy/ musty odour. Water Treat. Exam. 19:106. 9. PIET, G.J., B.C.J. ZOETEMAN & A.J.A. KRAAYEVELD. 1972. Earthy-smelling substances in surface waters of the Netherlands. Water Treat. Exam. 21:281. 10. YURKOWSKI, M. & J.A.L. TABACHEK. 1974. Identification, analysis, and removal of geosmin from muddy-flavored trout. J. Fish. Res. Board Can. 31:1851. 11. SAFFERMAN, R.S., A.A. ROSEN, C.I. MASHNI & M.E. MORRIS. 1967. Earthy-smelling substances from a blue-green alga. Environ. Sci. Technol. 1:429. 12. MEDSKER, L.L., D. JENKINS & J.F. THOMAS. 1968. Odorous compounds in natural © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater waters. An earthy-smelling compound associated with blue-green algae and actinomycetes. Environ. Sci. Technol. 2: 461. 13. KIKUCHI, T., T. MIMURA, K. HARIMAYA, H. YANO, M. ARIMOTO, Y. MASADA & T. INOUE. 1973. Odorous metabolites of blue-green alga Schizothrix muelleri Nageli collected in the southern basin of Lake Biwa. Identification of geosmin. Chem. Pharm. Bull. 21:2342. 14. TABACHEK, J.L. & M. YURKOWSKI. 1976. Isolation and identification of blue-green algae producing muddy odor metabolites, geosmin and 2-methylisoborneol, in saline lakes in Manitoba. J. Fish. Res. Board Can. 33:25. 15. IZAGUIRRE, G., C.J. HWANG, S.W. KRASNER & M.J. MCGUIRE. 1982. Geosmin and 2-methylisoborneol from cyanobacteria in three water supply systems. Appl. Environ. Microbiol. 43:708. 16. SAFFERMAN, R.S. & M.E. MORRIS. 1962. A method for the isolation and enumeration of actinomycetes related to water supplies. Robert A. Taft Sanitary Engineering Center Tech. Rep. W62-10, U.S. Public Health Serv., Cincinnati, Ohio. 17. KUSTER, E. & S.T. WILLIAMS. 1964. Selection of media for isolation of Streptomyces. Nature 202:928. 18. LECHEVALIER, H.A. 1975. Actinomycetes of sewage-treatment plants. Environ. Protection Technol. Ser., EPA-600/2-75-031, U.S. Environmental Protection Agency, Cincinnati, Ohio. 19. LECHEVALIER, M.P. & H.A. LECHEVALIER. 1974. Nocardia amarae, sp. nov., an actinomycete common in foaming activated sludge. Int. J. Syst. Bacteriol. 24:278. 20. LECHEVALIER, H.A. & M.P. LECHEVALIER. 1967. Biology of actinomycetes. Annu. Rev. Microbiol. 21:71. 9250 B. Actinomycete Plate Count 1. General Discussion A plating method using a double-layer agar technique has been adapted for determining actinomycete density. Because only the thin top layer of the medium is inoculated with sample, surface colonies predominate and identification and counting of colonies is facilitated. 2. Preparation and Dilution Prepare and dilute samples as directed in Section 9215 or Section 9610. Dilutions up to 1:1000 (10–3) usually are suitable for raw water, while treated waters may be examined directly. For soil samples, use dilutions from 1:1000 (10–3) to 1:1 000 000 (10–6). 3. Medium © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Starch-casein agar: Soluble starch Casein Potassium nitrate, KNO3 10.0 g 0.3 g 2.0 g Sodium chloride, NaCl Dipotassium hydrogen phosphate, K2HPO4 2.0 g 2.0 g Magnesium sulfate, hydrate, MgSO4⋅7H2O g Calcium carbonate, CaCO3 Ferrous sulfate, hydrate, FeSO4⋅7H2O Agar Reagent-grade water 0.05 g 0.02 g 0.01 15.0 g 1 L No pH adjustment is required. Medium is used to prepare double-layer plates. Store medium for bottom layer in bulk or in tubes in 15-mL amounts. Store medium for surface layer in tubes in 17.0-mL amounts. 4. Procedure a. Plating: Prepare three plates for each dilution to be examined. Aseptically transfer 15 mL of sterile starch-casein agar to a petri dish and let agar solidify, thus forming the bottom layer. To a test tube containing 17.0 mL liquefied starch-casein agar at 45 to 48°C, add 2 mL of appropriately diluted sample and 1 mL of the antifungal antibiotic, cycloheximide,*#(53) prepared in reagent-grade water (1 mg/mL) and sterilized by autoclaving for 15 min at 121°C. Pipet 5 mL of inoculated agar over the hardened bottom layer with gentle swirling to obtain even distribution of the surface layer. b. Incubation: Invert and incubate at 28°C until no new colonies appear. Usually this requires 6 to 7 d. c. Counting: Plates suitable for counting contain 30 to 300 colonies. Identify actinomycetes by gross colony appearance. If necessary, verify by microscopic examination at a magnification of 50 to 100×, as shown in Figure 9250:1. Actinomycete colonies, because of filamentous growth, typically have a fuzzy colonial border. Table 9250:I lists the distinguishing characteristics commonly used to differentiate actinomycete from other bacterial colonies. Cycloheximide generally suppresses fungal growth; however, fungal colonies, if present, can be recognized by their wooly appearance. Microscopically, fungi reveal a considerably larger cell diameter than actinomycetes. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5. Calculation Report actinomycetes per milliliter of water or gram (dry weight) of soil. If three plates are used per sample, the average number of colonies on all plates (total number of colonies/3), times 2, times the reciprocal of the dilution (10/1, 100/1, 1000/1, etc.) equals the actinomycete colony count per milliliter of original sample. For solid or semisolid samples, correct for water content and report actinomycete colonies per gram, dry weight, of sample. 9260 DETECTION OF PATHOGENIC BACTERIA*#(54) 9260 A. Introduction 1. General Discussion One purpose of drinking water and wastewater treatment is to reduce the numbers of viable organisms to acceptable levels, and to remove or inactivate all pathogens capable of causing human disease. Despite the remarkable success of water treatment and sanitation programs in improving public health, sporadic cases and point-source outbreaks of waterborne diseases continue to occur. Water and wastewater may contain a wide variety of bacteria that are opportunistic or overt pathogens of animals and humans. Waterborne pathogens enter human hosts through intact or compromised skin, inhalation, ingestion, aspiration, and direct contact with the mucous membranes of the eye, ear, nose, mouth, and genitals. This section provides an introduction to the etiologic agents responsible for diseases transmitted by drinking and recreational waters in the U.S. Over 80 genera of bacteria that are nonpathogenic for humans have their natural habitat in water. In addition, some opportunistically pathogenic bacteria (Pseudomonas, Serratia, Acinetobacter, Chromobacterium, Achromobacter, Aeromonas, etc.) occur naturally in water. Other opportunists (Bacillus, Enterobacter, Klebsiella, Actinomyces, Streptomyces, etc.) are sometimes washed into water from their natural habitat in soil or on vegetative matter. Opportunistic pathogens also may be seeded from regrowth and biofilms in water treatment plants and distribution systems. Water contamination and disease transmission may result from conditions generated at overloaded and/or malfunctioning sanitary waste disposal and potable water treatment systems. In addition, common outdoor recreational activities such as swimming (including pools and hot tubs), boating, camping, and hiking, all place humans at risk of waterborne diseases from ingestion or direct contact with contaminated water.1 Outbreaks of gastroenteritis, pharyngoconjunctivitis, folliculitis, otitis, and pneumonia are associated with these recreational activities. Overcrowded parks and recreational areas contribute to the contamination of surface and groundwater. National statistics on outbreaks of waterborne diseases have been compiled in the U.S. since © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1920.2,3 Since 1971, the Centers for Disease Control and Prevention, the U.S. Environmental Protection Agency, and the Council of State and Territorial Epidemiologists have maintained a collaborative surveillance program on waterborne disease outbreaks of drinking water and recreational water origin.4 A summary of waterborne diseases in the U.S. has been published.5 Summary data from outbreaks reported through the national waterborne disease surveillance system for drinking water and recreation from 1985 to 1994 are shown in Table 9260:I. Laboratory diagnosis of infectious disease depends on isolation of the etiologic agent or demonstration of antibody response in the patient. Environmental microbiological examinations are conducted for compliance monitoring of the environment, to trouble-shoot problems in treatment plants and distribution systems, and in support of epidemiological investigations of disease outbreaks. Ideally, the public health microbiologist can contribute expertise in both clinical and environmental microbiology, thereby facilitating epidemiological investigations. When testing for pathogens in environmental samples, it usually is advisable to include analyses for indicator organisms. Besides coliform indicators (total coliform, fecal coliform, and E. coli), fecal streptococci, enterococci, Clostridium perfringens, and Aeromonas have been proposed as indicators of water quality. No single indicator provides assurance that water is pathogen-free. The choice of monitoring indicator(s) presupposes an understanding of the parameters to be measured and the relationship of the indicator(s) to the pathogen(s). Some bacterial pathogens, such as Pseudomonas, Aeromonas, Plesiomonas, Yersinia, Vibrio, Legionella, and Mycobacterium, may not correlate with coliform indicators. Traditional bacterial indicators also may not correlate with viruses or parasites in pristine waters or groundwaters, and they may be of limited utility in estuarine and marine waters. Nevertheless, tests for total and fecal bacteria and E. coli are useful, because it is rare to isolate bacterial enteric pathogens in the absence of fecal contamination. Other more general indicators also may be of value for assessing the potential for pathogen contamination and interpreting culture results. Heterotrophic plate count provides information about the total numbers of aerobic organotrophic bacteria and an indication of the total organic composition of the aquatic environment. Physicochemical factors, such as turbidity, pH, salinity, temperature, assimilable organic carbon, dissolved oxygen, biochemical oxygen demand, and ammonia may provide useful information about contamination or the potential of water to support bacterial growth. For treated waters, chlorine residual should be measured at the sample collection point. This section contains methods for Salmonella, Shigella, pathogenic E. coli, Campylobacter, Vibrio cholerae, Leptospira, Legionella, Yersinia entercolitica, Aeromonas, and Mycobacterium. Methods for isolation and enumeration of P. aeruginosa are found in Section 9213E and Section 9213F. Methods for other pathogens are found elsewhere.6 The methods outlined below may be used to analyze samples associated with disease outbreaks, or in other studies on the occurrence of pathogens in water and wastewater. Methods for recovery of bacterial pathogens from water and wastewater have not changed significantly in the past 30 years. The methods presented below are not standardized, and the procedures may © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater need modification to fit a particular set of circumstances. No single procedure is available for reliable detection of any pathogen or group of pathogens. Because the presence of pathogens is intermittent and the survival times in the environment are variable, routine examination of water and wastewater for pathogenic bacteria is not recommended. Even in outbreak situations, the recovery of pathogens from water and wastewater may be limited by lack of facilities, untrained personnel, inadequate methods, and high costs. 2. References 1. PITLIK, S., S.A. BERGER & D. HUMINER. 1987. Nonenteric infections acquired through contact with water. Rev. Infect. Dis. 9:54. 2. CRAUN, G.F., ed. 1986. Waterborne Diseases in the United States. CRC Press, Inc., Boca Raton, Fla. 3. LIPPY, E.C. & S.C. WALTRIP. 1984. Waterborne disease outbreaks— 1946–1980: A thirty-five year perspective. J. Amer. Water Works Assoc. 76:60. 4. KRAMER, M.H., B.L. HERWALDT, G.F. CRAUN, R.L. CALDERON & D.D. JURANEK. 1996. Waterborne diseases: 1993 and 1994. J. Amer. Water Works Assoc. 88:66. 5. KRAMER, M.H., B.L. HERWALDT, G.F. CRAUN, R.L. CALDERON & D.D. JURANEK. 1996. Surveillance for waterborne-disease outbreaks— United States, 1993–1994. Morbid. Mortal. Week. Rep. 45(SS-1):1. 6. MURRAY, P.R., E.J. BARON, M.A. PFALLER, F.C. TENOVER & R.H. YOLDEN, eds. 1995. Manual of Clinical Microbiology, 6th ed. American Soc. Microbiology Press, Washington, D.C. 9260 B. General Qualitative Isolation and Identification Procedures for Salmonella Rather than a specific protocol for Salmonella detection in water, a brief summary of methods suitable for recovery of these organisms is given. Methods currently available have been used in numerous field investigations to demonstrate Salmonella in both fresh and marine water environments. The occurrence of Salmonella in water is highly variable; there are limitations and variations in both the sensitivity and selectivity of accepted Salmonella isolation procedures for the detection of the more than 2300 Salmonella serotypes currently recognized. Thus, a negative result by any of these methods does not imply the absence of salmonellae, nor does it imply the absence of other pathogens. 1. Concentration Techniques Salmonella are ubiquitous in the environment and can be detected at low concentrations in most surface waters. These organisms are usually present in small numbers compared to coliforms; therefore, it is necessary to examine a relatively large sample to isolate the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater organisms.1 a. Swab technique: Prepare swabs from cheesecloth 23 cm wide, folded five times at 36-cm lengths, and cut lengthwise to within 10 cm from the head into strips approximately 4.5 cm wide. Securely wrap the uncut or folded end of each swab with 16-gauge wire for use in suspending the swab in water. Place the swabs in kraft-type bags and sterilize at 121°C for 15 min. Place swab just below the surface of the sampling location for 1 to 3 d.2,3 (Longer swab exposure will not increase entrapment of pathogens.) Gauze pads of similar thickness may be substituted. During sampling, particulate matter and microorganisms are concentrated from the water passing through or over the swab. After exposure, retrieve the swab, place it in a sterile plastic bag, ice, and send to the laboratory. Maximum storage-transit time allowable is 6 h. Do not transport swabs in enrichment media; ambient transport temperature may cause sufficient proliferation of competitive organisms to mask salmonellae. In the laboratory, place pad or portions of it in enrichment media. When flasks of enrichment medium containing iced swabs are to be incubated at 40 to 41°C, place flasks in a 44.5°C water bath for 5 min before incubation in an air incubator. b. Diatomaceous earth technique: Place an absorbent pad (not a membrane filter) on a membrane filter funnel receptacle, assemble funnel, and add 2.5 g sterile diatomaceous earth*#(55) to pack the funnel neck loosely. Apply vacuum and filter 2 L of sample. After filtration, disassemble funnel, divide resulting ‘‘plug’’ of diatomaceous earth and absorbent pad in half aseptically with a knife-edged, sterile spatula, and add to suitable enrichment media. Alternatively, place entire plug in enrichment medium. c. Large-volume sampler: Use a filter composed of borosilicate glass microfibers bonded with epoxy resin to examine several liters or more of sample, provided that sample turbidity does not limit filtration.4 The filter apparatus consists of a 2.5- × 6.4-cm cartridge filter and a filter holder.†#(56) Sterilize by autoclaving at 121°C for 15 min. Place sterile filter apparatus (connected in series with tubing to a 20-L water bottle reservoir and vacuum pump) in the 20-L sample container appropriately calibrated to measure volume of sample filtered. Apply vacuum and filter an appropriate volume. When filtration is complete, remove filter and place in a selective enrichment medium. d. Membrane filter technique: To examine low-turbidity water, filter several liters through a sterile 142-mm-diam membrane of 0.45-µm pore size.5 For turbid waters, precoat the filter: make 1 L of sterile diatomaceous earth suspension (5 g/L reagent-grade water) and filter about 500 mL. Without interrupting filtration, quickly add sample (1 L or more) to remaining suspension and filter. After filtration, place membrane in a sterile blender jar containing 100 mL sterile 0.1% (w/v) peptone water and homogenize at high speed for 1 min. Add entire homogenate to 100 mL double-strength selective enrichment medium. Alternatively, use multiple 47-mm-diam membrane filters to filter the sample. Immerse each membrane aseptically in 50 mL single-strength selective enrichment medium and incubate. Qualitative detection of Salmonella in suspect potable water also may be achieved successfully by further analysis of selected M-Endo MF cultures (from 100 mL sample volume) © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater that contain significant background growth and total coliforms.6 After completing routine coliform count, place entire filter with mixed growth into 10 mL tetrathionate broth (containing 1:50 000 brilliant green dye) for Salmonella enrichment before differential colony isolation on brilliant green agar. This unique approach requires no special large sample collections and can be an extension of the routine total coliform analysis. 2. Enrichment Selectively enrich the concentrated sample in a growth medium that suppresses growth of coliform bacteria. Sample enrichment is essential, because the pathogens usually are present in low numbers and solid selective media for colony isolation are somewhat toxic, even to pathogens. No single enrichment medium can be recommended that allows optimum growth of all Salmonella serotypes. Use two or more selective enrichment media in parallel for optimum detection. Elevated incubation temperatures including 40°, 41.5°, and 43°C and the addition of brilliant green dye to media help suppress background growth and may improve detection of Salmonella, but these modifications also suppress growth of some serotypes, including Salmonella typhi. a. Selenite cystine broth inhibits gram-positive and nonpathogenic enterobacteria while allowing for recovery of most species of Salmonella, including Salmonella typhi. Optimum incubation time for maximum recovery of Salmonella is 48 h at 35 to 37°C. Repeat streaking from tubes with turbidity several times during first day, and daily up to 5 d to increase potential recovery of all serotypes that may be present. Transfer 1 mL selenite broth culture to a fresh tube of same medium for continued incubation to enrich further Salmonella growth and enhance recovery of streak plates. b. Selenite-F broth allows for optimum recovery of most Salmonella species, including Salmonella typhi, after 24 h at 35 to 37°C. This increased recovery of Salmonella is accompanied by a slight decrease in selectivity when compared to selenite cystine. Most significantly, E. coli growth is not inhibited. Repeat streaking from tubes with turbidity several times during first day, and daily up to 5 d to increase potential recovery of all serotypes that may be present. Transfer 1 mL selenite broth culture to a fresh tube of same medium for continued incubation to enrich further Salmonella growth and enhance recovery of streak plates. c. Tetrathionate broth, incubated at 35°C, inhibits coliforms and Gram-positive bacteria, permitting selective enrichment of most Salmonella species, including S. typhi. It has been reported that tetrathionate broth is more selective for Salmonella than selenite-based media when incubated for 48 h at 43°C. While this formulation is highly selective, it is unable to inhibit Proteus mirabilis, which shows optimum growth. Growth of Proteus and Citrobacter can be inhibited with addition of brilliant green (see Section 9260B.3a). Incubation at 43°C and addition of brilliant green also will inhibit some species of Salmonella, including S. typhi. 3. Selective Growth Further separation of pathogens from the remaining nonpathogenic bacterial population is facilitated by proper choice of incubation temperature for primary enrichment followed by © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater secondary differentiation on selective solid media.7 These factors, incubation temperature, enrichment medium, and isolation medium, are interrelated. No one combination is optimum for recovery of all Salmonella serotypes. Method comparisons are encouraged to determine the best combination for a given circumstance. Solid media commonly used for enteric pathogen detection may be classed into three groups: (a) differential media with little or no inhibition toward nonpathogenic bacteria, such as EMB (containing sucrose); (b) selective media containing bile salts or sodium desoxycholate as inhibitors,8 such as MacConkey’s agar, desoxycholate agar, or xylose lysine desoxycholate (XLD) agar; and (c) selective media containing brilliant green dye, such as brilliant green agar or bismuth sulfite agar. Any medium selected must provide optimum suppression of coliforms while permitting good recovery of the pathogenic group. Great skill at screening for these pathogens is necessary because of the competing growth of various nonpathogens. Streaking duplicate plates, one heavily and one lightly, often aids in recognition of enteric pathogens in the presence of large numbers of interfering organisms. a. Brilliant green agar: Typical well-isolated Salmonella colonies grown on this medium are pinkish white with a red background. S. typhi and a few other species of Salmonella grow poorly because of the brilliant green dye content. Lactose-fermenters not subject to growth suppression will form greenish colonies or may produce other colorations. Occasionally, slow lactose-fermenters (Proteus, Citrobacter, and Pseudomonas) will produce colonies resembling those of a pathogen. Suppress spreading effect of pseudomonads by increasing agar concentration to 2%. In some instances, Proteus has been observed to ‘‘swarm’’; reduce this tendency by using agar plates dried to remove surface moisture. If suspect Salmonella colonies are not observed after 24 h incubation, reincubate for an additional 24 h to permit slow-growing or partially inhibited organisms to develop visible colonies. If typical colonies are not observed or if the streak plate is crowded, isolate in pure culture a few colonies for biochemical characterization. Non-lactose-fermenting colonies in close proximity to lactose-fermenting colonies may be masked. b. Bismuth sulfite agar (Wilson and Blair medium9): Luxuriant growth of many Salmonella species (including S. typhi) can be expected on this medium. Examine bismuth sulfite plates after 24 h incubation for suspect colonies; reincubate for 24 h to detect slow-growing strains. Typical colonies usually develop a black color, with or without a metallic sheen, and frequently this blackening extends beyond the colony to give a ‘‘halo’’ effect. A few species of Salmonella develop a green coloration; therefore, isolate some of these colony types when typical colonies are absent. As with brilliant green agar, typical colony coloration may be masked by numerous bordering colonies after 48 h incubation. A black color also is developed by other H2S-producing colonies, for example, Proteus and certain coliforms. c. Xylose lysine desoxycholate agar: Compared to brilliant green dye, sodium desoxycholate is only slightly toxic to fastidious Salmonella. Salmonella and Arizona organisms produce black-centered red colonies. Coliform bacteria, Proteus, and many Enterobacter produce yellow colonies. Optimum incubation time is 24 h. If plates are incubated longer, an alkaline reversion © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and subsequent blackening occur with H2S-positive nonpathogens (Citrobacter, P. vulgaris, and P. mirabilis). d. Xylose lysine brilliant green agar: This medium is especially good for Salmonella from marine samples. The brilliant green inhibits many Proteus, Enterobacter, and Citrobacter species. 4. Biochemical Reactions Many enteric organisms of little or no pathogenicity share certain major biochemical characteristics with Salmonella. The identification of pathogens by colony characteristics on selective solid media has limitations inherent in the biological variations of certain organisms and cannot be relied on for even tentative identification. Suspected colonies grown on selective solid media must be purified and further characterized by biochemical reactions; final verification is based on serological identification. Usually a large number of cultures will be obtained from the screening procedure. Commercially available differential media kits (see Section 9225) may be used as an alternative to Phases1, 2, and 3 described below, before serological confirmation. These kits give 95 to 98% agreement with conventional tests, although more significant tests will be necessary to achieve further differentiation among strains of Enterobacteriaceae. When such kits are not used, follow a sequential pattern of biochemical testing that will result in a greater saving of media and time for laboratory personnel.10 Phase 1—Preliminary screening, phenylalanine deaminase activity: Discard phenylalanine deaminase-positive cultures immediately as indicative of the Proteus group. In this test, spot isolates on phenylalanine agar and incubate for 24h at either 35 or 37°C. Phenylalanine deaminase activity is indicated by a green zone that develops around the colony after flooding of the plate with a 0.5M FeCl3 solution. Subject phenylalanine deaminase-negative cultures to the biochemical tests of Phase2. Phase 2—Biochemical tests: The tests used are: Medium Purpose of Test TSI Fermentation pattern: H2S production LIA Lysine decarboxylase activity, H2S production Urease production Urea broth Conformance to the typical biochemical patterns of the Salmonella determines whether to process cultures further (Phase3). Aberrant cultures may be encountered that do not conform to all the classical reactions attributed to each pathogenic group. In all cases, therefore, review © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater reactions as a whole and do not discard cultures on the basis of a small number of apparent anomalies. Phase 3—Fermentation reactions: Test fermentation reactions in dextrose, mannitol, maltose, dulcitol, xylose, rhamnose, and inositol broths to characterize further the biochemical capabilities of the isolates. This additional sorting reduces the possible number of positive cultures to be processed for serological confirmation. If the testing laboratory is equipped for serological confirmation (see 9260B.5.), this series of biochemical tests may be eliminated. 5. Genus Identification by Serological Techniques Upon completion of the recommended biochemical tests, inoculate the suspected Salmonella pure culture onto a brain-heart infusion agar slant and incubate for 18 to 24 h at 35 to 37°C. With wax pencil (china marker), divide an alcohol-cleaned glass slide into four sections. Prepare a dense suspension of test organism by suspending growth from an 18- to 24-h agar slant in 0.5 mL 0.85% NaCl solution. Place a drop of Salmonella ‘‘O’’ polyvalent antiserum in the first section and antiserum plus 0.85% NaCl in the second section. Using a clean inoculating loop, transfer a loopful of bacterial suspension to the third section containing 0.85% NaCl solution and to the fourth section containing 0.85% NaCl solution plus antiserum. Gently rock slide back and forth. If agglutination is not apparent in the fourth section at the end of 1 min, the test is negative. All other sections should remain clear. When biochemical reactions are characteristic of S. typhi and the culture reacts with ‘‘O’’ polyvalent antiserum, check other colonies from the same plate for Vi antigen reaction. If there is no agglutination with Salmonella Vi antiserum, the culture is not S. typhi. Identification of Salmonella serotypes requires determination of H antigens and phase of the organism as described by Edwards and Ewing.10 Isolates yielding biochemical reactions consistent for Salmonella and positive with polyvalent ‘‘O’’ antiserum may be identified as ‘‘Salmonella sp., serotype or bioserotype undetermined.’’ If species identification is necessary, send isolates confirmed as Salmonella by biochemical tests and polyvalent ‘‘O’’ antisera to reference laboratories for further analysis. 6. References 1. CHERRY, W.B., J.B. HANKS, B.M. THOMASON, A.M. MURLIN, J.W. BIDDLE & J.M. GROOM. 1972. Salmonellae as an index of pollution of surface waters. Appl. Microbiol. 24:334. 2. MOORE, B. 1948. The detection of paratyphoid carriers in towns by means of sewage examination. Mon. Bull. Mist. Health Pub. Health Lab. Serv. 7:241. 3. MOORE, B., E.L. PERRY & S.T. CHARD. 1952. A survey by the sewage swab method of latent enteric infection in an urban area. J. Hygiene 50:137. 4. LEVIN, M.A., J.R. FISCHER & V.J. CABELLI. 1974. Quantitative large-volume sampling technique. Appl. Microbiol. 28:515. 5. PRESNELL, M.W. & W.H. ANDREWS. 1976. Use of the membrane filter and a filter aid for concentrating and enumerating indicator bacteria and Salmonella from estuarine © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 6. 7. 8. 9. 10. waters. Water Res. 10:549. CANLAS, L. 1975. Personal communication. Guam Environmental Protection Agency, Agana, Guam. CHEN, H., A.D.E. FRASER & H. YAMAZAKI. 1993. Evaluation of the toxicity of Salmonella selective media for shortening the enrichment period. Int. J. Food Microbiol. 18:151. LEIFSON, E. 1935. New culture media based on sodium desoxycholate for the isolation of intestinal pathogens and for enumeration of colon bacilli in milk and water. J. Pathol. Bacteriol. 40:581. WILSON, W.J. & E.M. MCV. BLAIR. 1926. Combination of bismuth and sodium sulfite affording enrichment and selective medium for typhoid and paratyphoid groups of bacteria. J. Pathol. Bacteriol. 29:310. EDWARDS, P.R. & W.H. EWING. 1986. Identification of Enterobacteriaeceae, 4th ed. Elsevier Science Publ. Co., Inc., New York, N.Y. 7. Bibliography MULLER, G. 1947. Der Nachweis von Keimer der Typhus-Paratyphusgruppe in Wasser. H.H. Nolke Verlag, Hamburg, Germany. GREENBERG, A.E., R.W. WICKENDEN & T.W. LEE. 1957. Tracing typhoid carriers by means of sewage. Sewage Ind. Wastes 29:1237. MCCOY, J.H. 1964. Salmonella in crude sewage, sewage effluent, and sewage polluted natural waters. In Int. Conf. Water Pollut. Res., 1st, London, 1962. Vol. 1:205. MacMillan, New York, N.Y. BREZENSKI, F.T., R. RUSSOMANNO & P. DEFALCO, JR. 1965. The occurrence of Salmonella and Shigella in post-chlorinated and nonchlorinated sewage effluents and receiving waters. Health Lab. Sci. 2:40. SPINO, D.E. 1966. Elevated temperature technique for the isolation of Salmonella from streams. Appl. Microbiol. 14:591. GALTON, M.M., G.K. MORRIS & W.T. MARTIN. 1968. Salmonella in foods and feeds. Review of isolation methods and recommended procedures. Public Health Serv. Bur. Disease Prevention & Environmental Control, National Center for Disease Control, Atlanta, Ga. BREZENSKI, F.T. & R. RUSSOMANNO. 1969. The detection and use of Salmonella in studying polluted tidal estuaries. J. Water Pollut. Control Fed. 41:725. MORINIGO, M.A., M.A. MUNOZ, E. MARTINEZ-MANZANARES, J.M. SANCHEZ & J.J. BORREGO. 1993. Laboratory study of several enrichment broths for the detection of Salmonella spp. particularly in relation to water samples. J. Appl. Bacteriol. 74:330. U.S. FOOD AND DRUG ADMINISTRATION. 1995. Bacteriological and Analytical Manual, 8th ed. Assoc. Official Analytical Chemists International, Gaithersburg, Md. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9260 C. Immunofluorescence Identification Procedure for Salmonella The direct fluorescent antibody (FA) technique is a rapid and effective means of detecting salmonellae in fresh- and seawater samples. It may be used as a screening technique to provide rapid results for large numbers of samples, such as those from recreational or shellfish-harvesting waters. Positive FA tests are presumptive evidence for the presence of Salmonella. Because of potential cross-reactivity of antibodies, positive FA results should be confirmed by other methods. Sample volumes used depend on the degree of contamination. Where gross pollution is present, use smaller samples. When background information is absent, analyze a 2-L sample, using the diatomaceous earth concentration technique. 1. Apparatus for Fluorescence Microscopy Standard fluorescent antibody microscopy equipment may be obtained separately or in a package containing the essential instrumentation (a-f): a. Light microscope with microscope stand. b. Light source, providing energy in the short-wavelength region of the spectrum. A high-pressure mercury 200-W arc enclosed in a quartz envelope, a 75- to 150-W xenon high-pressure lamp, or a low-voltage 100-W quartz halogen lamp may satisfy this requirement. A significant portion of the energy should be emitted in the ultraviolet and blue region of the spectrum. c. Power pack to provide constant voltage and wattage output for the selected lamp. d. Basic filters including heat-absorbing filter (KG-1 or KG-2, or equivalent): red-absorbing filter (BG-38 or equivalent); exciter filter (BG-12 or equivalent, BG-12 being also a blue filter); and barrier filter (OG-1 or blue-absorbing filter). New interference excitation filters (KP500 or equivalent) having very high transmission in the blue portion of the spectrum (490 nm) are available. Barrier or suppression filters used with these have a sharp cutoff at 500 to 510 nm. e. Optics: The fluorescence microscope must have high-quality optics. A 100 × objective with an iris diaphragm to reduce the numerical aperture (N.A.) for dark-field work is essential. Because the N.A. is similar for all 100 × objectives (1.25 to 1.30), base selection on desire for a flat-field (plano) lens. f. Cardioid dark-field condenser for illuminating specimen: A 95 × oil immersion objective with build-in iris diaphragm is desirable. True dark-field illumination can be achieved only if the objective N.A. is smaller than the condenser N.A., i.e., of the illuminating cone of light. (Difference in N.A. between objective and condenser should be at least 0.05.) Reduce N.A. of an oil immersion objective by using the built-in diaphragm or by putting a funnel stop onto the objective. g. FA pre-cleaned micro slides, 7.6- × 2.5-cm, 0.8- to 1.0-mm thickness. h. Cover glass for FA slides, No. 1 1/2, 0.16- to 0.19-mm thickness. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater i. Staining assembly consisting of dish, cover, and slide rack with handle. Five dishes are required; for Kirkpatrick’s fixative, 95% ethanol, first PBS rinse, second PBS rinse, and reagent water. j. Moist chamber used to incubate slides containing smears with added conjugate. A simple chamber consists of water-saturated toweling with a culture dish bottom (150 by 20 mm) placed over the wet toweling. 2. Reagents a. Nondrying immersion oil, Type A (low fluorescence, PCB-free).*#(57) b. FA Kirkpatrick fixative, consisting of 60 mL absolute ethanol, 30 mL chloroform, and 10 mL formaldehyde.†#(58) c. Phosphate-buffered saline (PBS): Add 10 g buffer‡#(59) to 1000 mL freshly prepared distilled water. Stir until the powder dissolves completely. Adjust with 0.2N NaOH to pH 8.0. d. FA mounting fluid: Use standardized reagent-grade glycerine adjusted to pH 9.0 with 0.2N NaOH and intended for mounting slides to be viewed with the FA microscope. e. Reagent (laboratory pure) water: Use double-distilled water from an all-glass still or other high-quality analytical-grade laboratory water. f. FA Salmonella panvalent conjugate is a fluorescein-conjugated anti-Salmonella globulin.§#(60) To rehydrate, add 5 mL reagent water to a vial or conjugate. Determine working dilution (see ¶ 5e). Store unused rehydrated conjugate in a freezer, preferably at −60°C. Avoid repeated freezing and thawing. g. Zn-CdS: Ag phosphor particle.i#(61) 3. Concentration Technique Place an absorbent pad on a membrane filter funnel and add sufficient sterile diatomaceous earth##(62) to pack funnel neck loosely. Filter 2 L of sample. Rinse funnel with 50 to 100 mL sterile phosphate-buffered dilution water or 0.1% peptone water. Disassemble funnel and remove resulting ‘‘plug’’ of diatomaceous earth and the absorbent pad. Repeat with a second 2-L sample. 4. Enrichment Immerse one plug and absorbent pad in a flask containing 300 mL selenite cystine broth. Immerse second plug and absorbent pad in a flask containing 300 mL tetrathionate broth supplemented with 3 mL 1:1000 aqueous solution of brilliant green dye and 3 mg l-cystine. Incubate at either 35 or 37°C for 24 h. 5. Fluorescent Antibody Reaction and Analysis a. Prepare spot plates of brilliant green agar (BGA) and xylose lysine brilliant green (XLBG) agar by placing 1 drop (about 0.01 mL, delivered with a wire or sterile plastic loop) of the enrichment medium (selenite cystine or tetrathionate broth) at each of four separate points on the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater agar surface.1 Space drops on agar plate so that FA microscope slide will cover two inoculation points. This is essential because glass slide impression smears of the inoculated points will be made after incubation of plates. b. Incubate BGA and XLBG plates at 37 ± 0.5°C for 2.5 to 3 h. After incubation, micro CFUs will develop. Make impression smears by taking a clean FA microscope glass slide and placing it over two inoculated points on the medium. Press down lightly, being careful not to move glass slide horizontally. Do not apply too much pressure, because it will cause movement of the slide and collection of additional agar. Repeat this process for the other two inoculation points and for inoculation points on second agar medium. Prepare a total of four FA slides in this manner. c. Air-dry smears and fix for 2 min in Kirkpatrick’s fixative. Rinse slides briefly in 95% ethanol and let air dry. Do not blot. d. Cover fixed smears with 1 drop of Salmonella panvalent conjugate. Before use, dilute commercial conjugate and determine appropriate working dilution. Most batches are effective at a 1:4 dilution but this will vary with the type of fluorescence equipment used, light source, alignment, magnification, cultures, etc. Determine working dilution (titer) of each lot of conjugate. e. To determine conjugate titer use a known 18- to 24-h Salmonella culture grown in veal infusion broth and make smears on FA glass slide. Dilute conjugate and treat as outlined in c and d above. For example, if the following results are obtained: Dilution of Conjugate Fluorescence 1:2 1:4 1:6 1:8 1:10 4+ 4+ 4+ 2+ 1+ use the second highest dilution giving 4+ fluorescence. In the above example use a 1:4 dilution of conjugate. Diluting conjugate insures minimum cross-reactivity. Prepare fresh diluted conjugate daily. f. After covering each smear with 1 drop of appropriate dilution of conjugate, place slides in a moist chamber to prevent evaporation of staining reagent. After 30 min wash away excess reagent by dipping slides into phosphate-buffered saline (pH 8.0). Place slides in second bath of buffered saline for 10 min. Remove, rinse in distilled water, and drain dry. Do not blot. g. Place a small drop of mounting fluid (pH 9.0) on the smear and cover with a No. 1 1/2 cover slip. Seal edges of cover slip with clear fingernail polish. Examine sealed slides within a few hours while fluorescence is of optimum intensity. Examine under a fluorescence microscope © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater unit fitted with appropriate filters. h. Include a positive control slide with each set of samples. This checks conjugate reactivity and FA equipment generally. 6. Recording and Interpreting Results The intensity of organisms fluorescing in any given field is important in assessing positive Salmonella smears. If the majority of cells present fluoresce (4+ or 3+) the smear is positive. Carefully scrutinize smears showing only a few scattered fluorescing cells. Critical examination of cellular morphology may distinguish between these cells and salmonellae. The degree of fluorescence is the criterion on which positivity is based. Consider weakly fluorescing cells (2+ and 1+) negative. Confirm all positive FA results by cultural techniques (see Section 9260B). Reaction Positive Positive Negative Negative Negative Description Brilliant yellow-green fluorescence, cells sharply outlined Bright yellow-green fluorescence, cells sharply outlined with dark center Dull yellow-green fluorescence, cells not sharply outlined Faint green fluorescence discernible in dense areas, cells not outlined No fluorescence Fluorescence Intensity 4+ 3+ 2+ 1+ 0 7. Quantitative Immunofluorescence Microspectrofluorometric Microscopy To make such analyses use a system consisting of analyzing and illumination sections. The analyzing section includes an eyepiece monochromator assembly and a photomultiplier-photometer. The eyepiece uses a beam splitter that reflects to the monochromator and the observer’s eye, allowing for simultaneous visual observation and quantitative analysis of the yellow-green fluorescence intensity. The photometer package provides meter readout in milliamperes so that visual observation of fluorescence can be correlated with objective reading. Microspectrofluorometry can be done with a conventional fluorescence microscope. 8. Reference 1. KATZ, I.J. & F.T. BREZENSKI. 1973. Detection of Salmonella by fluorescent antibody. U.S. Environmental Protection Agency, Edison, N.J. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9. Bibliography SCHULTE, S.J., J.S. WITZEMAN & W.M. HALL. 1968. Immunofluorescent screening for Salmonella in foods: comparison with culture methods. J. Amer. Org. Agr. Chem. 51:1334. THOMASON, B.M. & J.G. WALLS. 1971. Preparation and testing of polyvalent conjugates for F.A. detection of Salmonellae. Appl. Microbiol. 22:876. THOMASON, B.M. 1971. Rapid detection of Salmonella microcolonies by fluorescent antibody. Appl. Microbiol. 22:1064. 9260 D. Quantitative Salmonella Procedures This procedure describes one approach for estimating Salmonella density in water samples. Other methods have been described in the literature and a comparative study is recommended to select the best quantitative method for any given application. The following procedure must be modified for use with solid or semisolid samples. Because of the high ratio of coliform bacteria to pathogens, large samples (1 L or more) are required. Any concentration method in Section 9260B.1 may be used but preferably concentrate the sample by the membrane filter technique (Section 9260B.1d). After blending the membrane with 100 mL sterile 0.1% (w/v) peptone water, use a quantitative MPN procedure by proportioning homogenate into a five-tube, three-dilution multiple-tube procedure using either selenite cystine, selenite-F, or tetrathionate broth as the selective enrichment medium (See Section 9260B.3). Incubate for 24 h as specified or required for the enrichment medium used and streak from each tube to plates of brilliant green and xylose lysine desoxycholate agars. Incubate for 24 h at 35°C. Select from each plate at least one, and preferably two to three, colonies suspected of being Salmonella, inoculate a slant each of triple sugar iron (TSI) and lysine iron (LIA) agars, and incubate for 24 h at 35°C. Test cultures giving a positive reaction for Salmonella by serological techniques (see Section 9260B.5). From the combination of Salmonella negative and positive tubes, calculate the MPN/1.0 L of original sample (see Section 9221E). 9260 E. Shigella Shigellosis is an acute gastrointestinal disease of humans, caused by four species or serogroups of the genus Shigella, S. dysentariae (Group A), S. flexneri (Group B), S. boydii (Group C), and S. sonnei (Group D). Shigellae invade the intestinal mucosa, producing dysentery characterized by abdominal pain, fever, and diarrhea. The infectious dose for Shigella spp. is low, and most cases result from person-to-person transmission. When outbreaks occur, they are usually associated with fecal contamination of foods and, less frequently, water. The shigellosis case rate has gradually risen in the U.S. over the past 30 years from 6 cases/100 000 population in 1965 to 12 cases/100 000 population in 1995.1 In the U.S., S. sonnei (66.5%) is the most common cause of shigellosis, followed by S. flexneri (16.4%), S. boydii (1.1%), and S. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater dysentariae (0.5%). The serogroup is not reported for 15.5% of cases. Shigellosis is most common among children. Outbreaks from direct transmission have been reported in schools, day-care centers, and institutions providing custodial care. Waterborne outbreaks are associated with fecal contamination together with inadequate chlorination of private or noncommunity water supplies, as the result of cross-connections between wastewater and potable water lines, and from exposure to fecally contaminated recreational waters. Shigellae are sensitive to chlorination at normal levels, and they do not compete favorably with other organisms in the environment. Their survival time is measured in hours and days, and is a function of the extent of pollution, as well as physical conditions such as temperature and pH. Shigellae survive up to 4 d in river water. However, the time required to establish a laboratory diagnosis by culture of patient specimens (1 to 2 d) makes it improbable that shigellae can be recovered from an environmental source unless there is a continuous source of contamination such as wastewater seepage. Shigellae can survive in a viable but nonculturable state after 21 d.2 The public health significance of nonculturable shigellae in the environment is unknown. Methods for the reliable quantitative recovery of shigellae from the environment are not yet available. Culture of shigellae is usually either not attempted or unsuccessful. Methods that have resulted in isolation of Shigella include membrane filtration3,4 and centrifugation5,6 with or without subsequent broth enrichment. Recently, the polymerase chain reaction (PCR) has shown promise for detection of shigellae in environmental samples.7-9 1. Sampling and Storage Collect a water sample in a sterile 1-L container. Collect soil, sediment, sludge, or other samples in plastic bags*#(63) or glass or plastic bottles. Hold samples at 2 to 8°C until they are processed. Process samples as soon as possible after collection. 2. Enrichment Choose a selective enrichment medium to minimize accumulation of volatile acid by-products derived from growth of potentially antagonistic bacteria. Selenite F broth has been used successfully to recover shigellae from water and sand.5,6 While GN broth facilitates better recovery of shigellae from stools than Selenite F broth, the only reported attempt to use GN broth as an enrichment for membrane filters for isolation of shigellae failed to recover the organism.10 Alternatively, use reduced-strength nutrient medium adjusted to pH 8.0 (0.15 g tryptic soy broth, added directly to the sample). During outbreak investigations, the enrichment medium may be made selective by incorporation of antibiotics to which the clinical isolates have shown resistance, such as tetracycline and streptomycin at concentrations of 150 µg/mL.11 3. Membrane Filter Procedure This procedure is suitable for low-turbidity potable and surface waters with low © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater concentrations of coliform bacteria. Filter 100-mL to 1-L samples through 0.45-µm pore size membranes and place filters face up on the surface of XLD or MacConkey agar plates; incubate plates at 35°C overnight. Where growth is confluent, sweep growth from plate and inoculate GN or Selenite F broth enrichments; incubate for 6 h and streak onto MacConkey and XLD plates for colony isolation. Pick colorless colonies (lactose nonfermenters) from membrane or plates to TSI and LIA slants; incubate overnight at 35°C. For biochemical reactions and serological grouping, see ¶ 5 below. 4. Centrifugation Procedure This procedure is suitable for surface waters, wastewater, and sediments. Centrifuge 200- to 250-mL water samples at 1520 × g for 15 min and pour off all but last 2 mL of supernatant. Resuspend pellet and add 8 mL Selenite F or GN broth. Incubate suspension for 24 h at 35°C. Mix suspension and inoculate one loopful to each of several MacConkey and XLD plates. Streak plates for isolation and incubate overnight at 35°C. Examine plates for colorless colonies, and pick suspect colonies to TSI and LIA slants; incubate at 35°C overnight. For biochemical reactions and serological grouping, see ¶ 5 below. For solid samples (sediments, soil, sludge, etc.) suspend 10 g sample in 100 mL Selenite F or GN broth and mix thoroughly. Incubate suspension overnight at 35°C. Resuspend sediment and streak one loopful onto each of several MacConkey and XLD agar plates; incubate overnight at 35°C. Pick colorless colonies to TSI and LIA slants, and proceed as above. For biochemical reactions and serological grouping, see ¶ 5 below. 5. Biochemical Identification and Serological Grouping Examine the TSI and LIA slants for the reactions shown in Table 9260:II. Cultures that are presumptively identified as Shigella spp. are serogrouped by a slide agglutination test using polyvalent and group specific antisera. Refer cultures to a public health reference laboratory if molecular typing is desirable for outbreak-related strains. 6. References 1. CENTERS FOR DISEASE CONTROL AND PREVENTION. 1996. Summary of notifiable diseases, United States 1995. Morbid. Mortal. Week. Rep. 44:1. 2. COLWELL, R.R., P.R. BRAYTON, D.J. GRIMES, D.B. ROSZAK, S.A. HUQ & L.M. PALMER. 1985. Viable but non-culturable Vibrio cholerae and related pathogens in the environment: implications for release of genetically engineered microorganisms. Bio/Technology 3:817. 3. DANIELSSON, D. & G. LAURELL. 1968. A membrane filter method for the demonstration of bacteria by the fluorescent antibody technique. Acta. Path. Microbiol. Scand. 72:251. 4. LINDELL, S.S. & P. QUINN. 1973. Shigella sonnei isolated from well water. Appl. Microbiol. 26:424. 5. CODY, R.M. & R.G. TISCHER. 1965. Isolation and frequency of occurrence of Salmonella © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and Shigella in stabilization ponds. J. Water Pollut. Control Fed. 37:1399. 6. DABROWSKI, J. 1982. Isolation of the Shigella genus bacteria from the beach sand and water of the bay of Gdansk. Biul. Inst. Med. Morskiej. 33:49. 7. BEJ, A.K., J.L. DICESARE, L. HAFF & R.M. ATLAS. 1991. Detection of Escherichia coli and Shigella spp. in water by using the polymerase chain reaction and gene probes for uid. Appl. Environ. Microbiol. 57:1013. 8. ISLAM, M.S., M.K. HASAN, M.A. MIAH, G.C. SUR, A. FELSENSTEIN, M. VENKATESAN, R.B. SACK & M.J. ALGERT. 1993. Use of the polymerase chain reaction and fluorescent-antibody methods for detecting viable but nonculturable Shigella dysenteriae Type 1 in laboratory microcosms. Appl. Environ. Microbiol. 59:536. 9. SETHABUTR, O., P. ECHEVERRIA, C.W. HOGE, L. BODHIDATTA & C. PITARANGSI. 1994. Detection of Shigella and enteroinvasive Escherichia coli by PCR in the stools of patients with dysentery in Thailand. J. Diarrh. Dis. Res. 12:265. 10. MAKINTUBEE, S., J. MALLONEE & G. ISTRE. 1987. Shigellosis outbreak associated with swimming. Amer. J. Pub. Health 77:166. 11. ROSENBERG, M.L., K.K. HAZLET, J. SCHAEFER, J.G. WELLS & R.C. PRUNEDA. 1976. Shigellosis from swimming. J. Amer. Water Works Assoc. 236:1849. 9260 F. Pathogenic Escherichia coli Escherichia coli is a normal inhabitant of the human digestive tract; however, some E. coli cause diarrheal diseases in humans.1 These pathogenic E. coli are classed into five groups: enterotoxigenic (ETEC), enterohemorrhagic (EHEC), enteroinvasive (EIEC), enteropathogenic (EPEC), and the newly recognized group called enteroadherent-aggregative E. coli (EA-AggEC) for its aggregative or ‘‘stacked-brick’’-like adherence to cultured mammalian cells.2 Pathogenic E. coli can be grouped on the basis of serology but, because they are classed on the basis of distinct pathogenic factors, definitive identification requires the determination of the characteristic virulence properties associated with each group. These include: plasmid-mediated cell invasion, plasmid-mediated colonization and enteroadherence factors, production of several potent cytotoxins, hemolysins, as well as heat-labile and stable enterotoxins.3 Although pathogenic E. coli have most often been implicated in foodborne illness, several major waterborne outbreaks have been reported.4 Outbreaks have involved both water supplies5-7 and recreational waters.8,9 Some E. coli pathogens have a low infectious dose. 1. Examination Procedures The pathogenic E. coli groups are phenotypically diverse; hence, no standard microbiological methods have been developed for these pathogens. Unlike typical E. coli, some pathogenic groups like EIEC do not ferment lactose3; hence, coliform methods based on lactose © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater fermentation are not suitable for detection of EIEC. Also, many fecal coliform confirmation or enrichment procedures use elevated incubation temperature, which is inhibitory to the growth of EHEC.10 Elevated temperatures and sodium lauryl sulfate used in lauryl tryptose broth (LTB) for MPN analysis also have been found to cause the loss of plasmid, which encodes many of the virulence-associated factors.11 Pathogenic E. coli that ferment lactose and are not affected by elevated temperatures still can be presumptively distinguished from non-E. coli by the MPN fecal coliform procedure (9221E) or the fecal coliform membrane filter method (9222D) followed by serotyping and virulence analysis. These methods, as well as methods from other sources,12 also have been modified to detect specific pathogenic groups. Regardless of the method, however, when testing for pathogenic E. coli, first identify isolates as E. coli either by conventional biochemical testing or by using commercially available biochemical identification kits (see Section 9260B.4) before serotyping and assaying for the virulence factors associated with the respective pathogenic groups. a. EHEC O157:H7: The following procedure is a modification of the standard total coliform fermentation technique (9221B) for detecting E. coli O157:H7 in water.13 Inoculate a 100-mL sample into 50 mL 3× lauryl tryptose broth (LTB) and incubate at 35°C for 24 h. Serially dilute the sample, spread plate (0.1 mL) onto sorbitol MacConkey agar (SMAC)*#(64) and incubate at 35°C for 18 to 24 h. EHEC O157:H7 form colorless colonies because they do not ferment, or are slow fermenters of, sorbitol. Pick ten sorbitol-negative colonies, transfer individually into LTB-MUG (4-methylumbelliferone glucuronide; 0.1 g/L)14 and incubate at 35°C for 18 to 24 h. EHEC O157:H7 ferment lactose, but do not have β-glucuronidase activity to hydrolyze MUG, so cultures will appear gas-positive and nonfluorescent. Assay these for positive glutamate decarboxylase activity,13 then identify biochemically as E. coli. Larger volumes of sample also may be examined by the following procedure modified from a procedure for detecting O157: H7 in food.15 This procedure has not been tested for use in water analysis; however, it has been used extensively to detect O157: H7 bacteria in apple juice. Centrifuge 200 mL sample at 10 000 × g for 10 min. Resuspend pellet in 225 mL EHEC enrichment broth (EEB) and incubate at 35°C for 6 h. Spread plate 0.1 mL from EEB and a 1:10 dilution of EEB onto tellurite cefixime SMAC (TC SMAC). Both EEB and TC SMAC contain antibiotics to reduce growth of normal flora bacteria; therefore, they are best suited for highly contaminated samples. Incubate EEB sample and TC SMAC at 35°C for 18 to 24 h. Observe TC SMAC plates for isolated, colorless colonies. If none are evident, serially dilute the overnight EEB sample and replate onto TC SMAC. Test colorless colonies for positive indole reaction and identify biochemically as E. coli before serotyping and virulence analysis for the Shiga toxin genes. b. EPEC, ETEC, EIEC: With the exception of EIEC, use either the MPN fecal coliform procedure (9221E) or the fecal coliform membrane filter method (9222D) for presumptive isolation of these pathogenic E. coli groups from water. Alternatively plate presumptive positive © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater samples onto selective media, such as LES Endo and MacConkey (MAC) agars (see Section 9221B.3, Completed Phase). In food analysis, L-EMB agar also has been used. For EIEC, which ferment lactose slowly or not at all, the MPN method is not useful; however, the membrane filter method (9222D) can be used. In food testing for EIEC, Hektoen agar (HE), Salmonella-Shigella (SS) agar and MAC are used for selective plating, but HE and MAC appear less inhibitory and are best suited for the isolation of EIEC10. In the analysis of each pathogenic E. coli group, preferably pick 10 typical (lactose-positive) and 10 atypical (lactose-negative) colonies for biochemical identification. Identify all isolates as E. coli before serological typing and analysis for the group-specific virulence factors. 2. Serotyping For definitive identification, serotype for the O:H antigens any isolates presumptively identified as pathogenic E. coli by microbiological methods. Polyvalent antisera are available commercially, but only for the common serotypes. Several anti-O157 and anti-H7 latex agglutination kits are available for typing O157:H7 isolates. Serotype information also is essential for epidemiological investigations. 3. Virulence Analysis The pathogenic potential of an E. coli isolate can be determined only by testing for its distinctive virulence properties. A simple antibody-bound latex agglutination kit and several enzyme linked immunosorbent assay kits are available for testing Shiga cytotoxins of EHEC†#(65). An agglutination kit also is available for testing labile and stable enterotoxins of ETEC,‡#(66) but analysis of other virulence factors may require bioassays using animal models, tissue cultures, or other antibody and nucleic-acid-based molecular methods. A partial listing of commercially available assays and media for pathogenic E. coli is available.12 Most of the assays are specific for EHEC O157:H7 and introduced only recently for food analysis; hence, few have been evaluated by collaborative studies. 4. References 1. ORSKOV, F. & ORSKOV, I. 1992. Escherichia coli serotyping and disease in man and animals. Can. J. Microbiol. 38:699. 2. VIAL, P.A., R. ROBINS-BROWNE, H. LIOR, V. PRADO, J.B. KAPER, J.P. NATARO, D. MENEVAL, A.-E.-D. ELSAYED & M.M. LEVINE. 1988. Characterization of enteroadherent-aggregative Escherichia coli, a putative agent of diarrheal disease. J. Infect. Dis. 158:70. 3. LEVINE, M.M. 1987. Escherichia coli that cause diarrhea: enterotoxigenic, enteropathogenic, enteroinvasive, enterohemorrhagic and enteroadherent. J. Infect. Dis. 155:377. 4. FENG, P. 1995. Escherichia coli serotype O157:H7: novel vehicles of infection and emergence of phenotypic variants. Emerging Infec. Dis. 2:47. 5. SCHROEDER, S.A., J.R. CALDWELL, T.M. VERNON, P.C. WHITE, S.I. GRANGER & J.V. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater BENNETT. 1968. A waterborne outbreak of gastroenteritis in adults associated with Escherichia coli. Lancet 1:737. 6. ROSENBERG, M.L., J.P. KOPLAN, I.K. WACHSMUTH, J.G. WELLS, E.J. GANGAROSA, R.L. GUERRANT & D.A. SACK. 1977. Epidemic diarrhea at Crater Lake from enterotoxigenic Escherichia coli. Ann. Intern. Med. 86:714. 7. SWERDLOW, D.L., B.A. WOODRUFF, R.C. BRADY, P.M. GRIFFIN, S. TIPPEN, H.D. DONNELL, E. GELDREICH, B.J. PAYNE, A. MEYER, J.G. WELLS, K.D. GREENE, M. BRIGHT, N.H. BEAN & P.A. BLAKE. 1992. A waterborne outbreak in Missouri of Escherichia coli O157:H7 associated with bloody diarrhea and death. Ann. Intern. Med. 117:812. 8. KEENE, W.E., J.M. MCANULTY, F.C. HOESLY, L.P. WILLIAMS, K. HEDBERG, G.L. OXMAN, T.J. BARRETT, M.A. PFALLER & D.W. FLEMING. 1994. A swimming-associated outbreak of hemorrhagic colitis caused by Escherichia coli O157:H7 and Shigella sonnei. N. England J. Med. 331:579. 9. BREWSTER, D.H., M.I. BROWNE, D. ROBERTSON, G.L. HOUGHTON, J. BIMSON & J.C.M. SHARP. 1994. An outbreak of Escherichia coli O157 associated with a children’s paddling pool. Epidemiol. Infect. 112:441. 10. DOYLE, M.P. & V.V. PADHYE. 1989. Escherichia coli. In M.P. Doyle, ed. Foodborne Bacterial Pathogens. Marcel Dekker, Inc., N.Y. 11. HILL, W.E. & C.L. CARLISLE. 1981. Loss of plasmids during enrichment for Escherichia coli. Appl. Environ. Microbiol. 41:1046. 12. U.S. FOOD AND DRUG ADMINISTRATION. 1995. Bacteriological Analytical Manual, 8th ed. Assoc. Official Analytical Chemists International, Gaithersburg, Md. 13. RICE, E.W., C.H. JOHNSON & D.J. REASONER. 1996. Detection of Escherichia coli O157:H7 in water from coliform enrichment cultures. Lett. Appl. Microbiol. 23:179. 14. FENG, P. & P.A. HARTMAN. 1982. Fluorogenic assay for immediate confirmation of Escherichia coli. Appl. Environ. Microbiol. 43:1320. 15. HITCHINS, A.D., P. FENG, W.D. WATKINS, S.R. RIPPEY & L.A. CHANDLER. 1995. Escherichia coli and the coliform bacteria. In Bacteriological Analytical Manual, 8th ed. Assoc. Official Analytical Chemists International, Gaithersburg, Md. 9260 G. Campylobacter jejuni Campylobacters are commonly found in the normal gastrointestinal and genitourinary flora of wild animals, birds, and domestic animals including sheep, cattle, swine, goats, and chickens.1 Campylobacter infections often are acquired by the fecal oral route, often as zoonoses through exposure to infected animals. Large outbreaks have resulted from contaminated milk, uncooked meat or fowl, and contaminated water systems.2 Campylobacter has been reported to be the most common cause of bacterial enteritis worldwide.3 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Waterborne transmission of Campylobacter has resulted from drinking untreated surface water, contamination of groundwater with surface water, faulty disinfection, and contamination by wild bird feces.4 In remote mountain areas, the infection has been associated with drinking surface water from cold mountain streams.5 Occurrence of campylobacters in surface water is variable and appears to be seasonally dependent, with lowest levels occurring in summer. Survival in surface water is affected by both temperature and sunlight.6 Between 1978 and 1986, 57 outbreaks of campylobacteriosis were reported, including 11 waterborne outbreaks, 7 of which occurred in community water supplies. 1. Water Collection and Filtration Method Collect large-volume water samples in sterile 10-L plastic containers. Process samples immediately after collection or store at 4°C and process as soon as possible. Filter one to several liters of the water through a 0.45- or 0.22-µm-pore-size, 47-mm-diam, cellulose nitrate membrane filter. Remove filter and place face down on selective medium (see isolation section). Incubate microaerophilically at 42°C for 24 h. Remove filter from the plate and place it face down on another selective plate. Incubate both plates at 42°C for up to 5 d.7 For turbid water pre-filtration is necessary. Use a stainless steel filtration device with a 1.5-L reservoir.*#(67) Assemble with the following filter sequence: Place a 142-mm, 3.0-µm filter on the screen inside reservoir with a 124-mm prefilter on top. In the bottom tubing adapter place a 47-mm, 1.2-µm filter. Then place Swinnex filter holders in parallel with a 47-mm, 0.65-µm filter in the upstream filter holder and a 47-mm, 0.45-µm filter in the downstream holder. Add 1 L sample to the reservoir, seal, and apply pressure of about 350 kPa. After filtration, remove the 0.45-µm pore-size filter and culture on selective plate medium as described above. 2. Isolation Campylobacter isolation requires use of selective media containing antimicrobial agents, microaerophilic atmosphere (5% O2, 10% CO2, and 85% N2), and 42°C incubation temperature, to suppress the growth of most common bacteria.8 The thermophilic campylobacters (C. jejuni, C. coli, C. lari, and C. upsaliensis) grow well at 42°C. However, other campylobacters (C. jejuni subsp. doylei and C. fetus) do not grow well at 42°C; incubate plates at both 37°C and 42°C for optimal isolation of these bacteria.9 Microaerophilic conditions can be provided by using commercially available systems and equipment.†#(68) Several selective media for plating campylobacters are commercially available. Skirrow’s medium contains blood agar base with lysed horse blood, trimethoprim, vancomycin, and polymixin B. Campy-BAP contains Brucella agar base with sheep blood, trimethoprim, vancomycin, polymixin B, amphotericin B, and cephalothin (to which some campylobacters are sensitive). Butzler’s medium contains thioglycollate agar with sheep blood, bacitracin, novobiocin, cycloheximide, and cefazolin. Preston’s medium contains Campylobacter agar base with horse blood, cycloheximide, rifampicin, trimethoprim, and polymyxin B. Other media, such as Campylobacter blood-free selective medium and Campylobacter charcoal differential agar, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater can be used to isolate campylobacters.10 Use of enrichment broth will improve recovery of campylobacters. Several enrichment media, such as Campylobacter broth, Campy-thio broth, Gifu anaerobe-modified semisolid medium, and Preston medium, are used to enhance recovery of campylobacters.9 Add 10 mL water sample to 10 mL Campylobacter enrichment broth tubes in duplicate, and incubate cultures at 37°C and 42°C for 8 h or overnight. Pre-enrichment of water sample in a selective enrichment broth for 4 h at 37°C may be important for recovery of stressed cells of C. jejuni that show less tolerance to elevated growth temperatures. For pre-enrichment of water sample, add 10 mL water to 10 mL enrichment medium and incubate culture for 4 h at 37°C, then transfer the cultures to another incubator at 42°C for overnight incubation.11,12 C. jejuni may be induced to a nonculturable state in water, and it is not clear whether pre-enrichment or enrichment will facilitate isolation of these bacteria.13 Use of a decreased substrate concentration enhances metabolic activity in nonculturable campylobacters from water.14 3. Identification a. Culture examination: Examine Campylobacter plates at 24 and 48 h for characteristic colonies, which can range from flat, spreading colonies that cover the entire surface of the plate, to very small, convex, translucent colonies. Colony colors range from gray to yellowish or pinkish. b. Microscopy identification: Campylobacter spp. do not stain well by the conventional Gram stain. If safranin is used as a counterstain, apply it for 2 to 3 min; carbol fuchsin is a better alternative. Even 24-h cultures of campylobacters appear pleomorphic in stained smears, and cells range from small Gram-negative rods and coccoid forms to longer rods that may show an ‘‘S’’ or seagull shape, and long spirals, particularly from older cultures.15 c. Motility test: Campylobacter normally are motile by a single polar flagellum at one or both ends. Suspend cells in Mueller-Hinton or nutrient broth, and observe motility using phase microscopy or brightfield microscopy with reduced illumination. Do not use saline or distilled water because they may inhibit motility.8 Young cells are 0.2 to 0.8 µm wide by 1.5 to 5 µm long, curved or spiral, and motile with darting or corkscrew-like motion.16 d. Biochemical tests: Despite numerous studies, campylobacters remain relatively difficult to rapidly identify, classify, and type biochemically.17 Campylobacters do not ferment or oxidize carbohydrates, and they are inert in most biochemical media used to characterize bacterial isolates.18 Although no standard methods for the characterization of campylobacters have been published, oxidase, catalase, nitrite and nitrate reduction, H2S production, hippurate hydrolysis, resistance to various agents, temperature tolerances, and growth requirements are among the common phenotypic tests used to characterize campylobacters.3 4. Serological Identification Tests © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Commercially available kits‡#(69) for serotyping campylobacters are available. These kits use latex particles coated with polyvalent immunoglobulins for several Campylobacter species. They are designed for rapid presumptive identification of the thermophilic, enteropathogenic Campylobacter species (C. jejunei, C. coli, and C. lari); use in accordance with manufacturer’s instructions.19 Other techniques that are not widely available in all laboratories include lectin agglutination, cellular fatty acid profiles, nucleic acid probes, polymerase chain reaction, and other genomic methods that can be used in reference and research laboratories for detection and identification of campylobacters.3 5. References 1. RYAN, K.J. 1990. Vibrio and Campylobacter. In J.C. Sherris, ed. Medical Microbiology: An Introduction to Infectious Diseases. Elsevier, New York, N.Y. 2. BARON, E.J., R.S. CHANG, D.H. HOWARD, J.N. MILLER & J.A. TURNER, eds. 1994. Medical Microbiology: A Short Course. Wiley-Liss, New York, N.Y. 3. ON, S.L.W. 1996. Identification methods for campylobacters, helicobacters, and related organisms. Clin. Microbiol. Rev. 9:405. 4. TAUXE, R.V. 1992. Epidemiology of Campylobacter jejuni infections in the United States and other industrialized nations. In I. Nachamkin, M.J. Blaser & L.S. Tompkins, eds. Campylobacter jejuni: Current Status and Future Trends. American Soc. Microbiology, Washington, D.C. 5. TAYLOR, D.N., K.T. MCDERMOTT, J.R. LITTLE, J.G. WELLS & M.J. BLASER. 1983. Campylobacter enteritis from untreated water in the Rocky Mountains. Ann. Intern. Med. 99:38. 6. VOGT, R.L., H.E. SOURS, T. BARRETT, R.A. FELDMAN, R.J. DICKINSON & L. WITHERELL. 1982. Campylobacter enteritis associated with contaminated water. Ann. Intern. Med. 96:292. 7. PEARSON, A.D., M. GREENWOOD, T.D. HEALING, D. ROLLINS, M. SHAHAMAT, J. DONALSON & R.R. COLWELL. 1993. Colonization of broiler chickens by waterborne Campylobacter jejuni. Appl. Environ. Microbiol. 59:987. 8. ISENBERG, H.D., ed. 1992. Clinical Microbiology Procedures Handbook. Vol. 1. American Soc. Microbiology, Washington, D.C. 9. GOOSSENS, H. & J.P. BUTZLER. 1992. Isolation and identification of Campylobacter spp. In I. Nachamkin, M.J. Blaser & L.S. Tompkins, eds. Campylobacter jejuni: Current Status and Future Trends. American Soc. Microbiology, Washington, D.C. 10. PARKS, L.C., ed. 1993. Handbook of Microbiological Media. CRC Press, Boca Raton, Fla. 11. HUMPHREY, T.J. 1989. An appraisal of the efficacy of preenrichment for the isolation of Campylobacter jejuni from water and food. J. Appl. Bacteriol. 66:119. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 12. HUMPHREY, T.J. 1986. Techniques for the optimum recovery of cold injured Campylobacter jejuni from milk or water. J. Appl. Bacteriol. 61:125. 13. ROLLINS, D.M. & R.R. COLWELL. 1986. Viable but nonculturable stage of Campylobacter jejuni and its role in survival in the natural aquatic environment. Appl. Environ. Microbiol. 52:531. 14. ROLLINS, D.M. 1987. Characterization of Growth, Decline, and the Viable but Nonculturable State of Campylobacter jejuni. Ph.D dissertation, Univ. Maryland, College Park. 15. KAPLAN, R.L. & A.S. WEISSFELD. 1994. Campylobacter, Helicobacter and related organisms. In B.J. Howard et al., eds. Clinical and Pathogenic Microbiology, 2nd ed. Mosby, St. Louis, Mo. 16. BEUCHAT, L.R. 1986. Methods for detecting and enumerating Campylobacter jejuni and Campylobacter coli in poultry. Poultry Sci. 65:2192. 17. DUBREUIL, J.D., M. KOSTRZYNSKA, S.M. LOGAN, L.A. HARRIS, J.W. AUSTIN & T.J. TRUST. 1990. Purification, characterization, and localization of a protein antigen shared by thermophilic campylobacters. J. Clin. Microbiol. 28:1321. 18. CARDARELLI-LEITE, P., K. BLOM, C.M. PATTON, M.A. NICHOLSON, A.G. STEIGERWALT, S.B. HUNTER, D.J. BRENNER, T.J. BARRETT & B. SWAMINATHAN. 1996. Rapid identification of Campylobacter species by restriction fragment length polymorphism analysis of a PCR-amplified fragment of the gene coding for 16S-rRNA. J. Clin. Microbiol. 34:62. 19. HODINKA, R.L. & P.H. GILLIGAN. 1988. Evaluation of the Campyslide agglutination test for confirmatory identification of selected Campylobacter species. J. Clin. Microbiol. 26:47. 9260 H. Vibrio cholerae Vibrio cholerae is the causative agent of cholera, a waterborne illness with symptoms ranging from mild to severe and potentially fatal diarrheal disease.1,2 This is a well-defined species on the basis of biochemical tests and DNA studies, but the serotypes within the species can be quite diverse in their ability to produce infection. The O1 serogroup is associated with epidemic and pandemic cholera, especially in developing countries. The current (seventh) pandemic has affected over 100 countries, including the United States, with over one million reported cases and 10 000 deaths.3 The newly identified O139 Bengal serogroup4 also is capable of producing epidemic cholera. In contrast, the great majority of non-O1/non-O139 strains, which are more common in the environment, do not produce cholera toxin, and are not associated with epidemic cholera. However, these strains occasionally are associated with potentially fatal extra-intestinal infections. V. cholerae occurs as part of the normal microflora in estuarine areas, with non-O1/non-O139 strains being much more common than are O1 strains. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Concentration Techniques Levels of V. cholerae in natural waters and sewage usually are quite low. Thus, methods of concentration or enrichment usually are employed. One method for isolating V. cholerae O1 from contaminated waters is placement of Moore swabs in flowing wastewater for periods up to 1 week, followed by placement into enrichment media at a 1:1 (weight/volume) ratio.5 2. Enrichment Procedures Samples are enriched in alkaline peptone broth (1% peptone, 1% NaCl, pH 8.4), using appropriate concentration of broth relative to sample volume. Incubate enrichment cultures for 6 to 8 h at 35°C, then streak a loopful of the enrichment broth onto thiosulfate-citrate-bile salts-sucrose (TCBS) agar and incubate these plates at 35°C for 18 to 24 h.6 Other enrichment and plating media have been reviewed.7,8 3. Selective Growth Suspected V. cholerae colonies appear yellow, a result of sucrose fermentation. A variety of other sucrose-fermenting vibrios also appear on TCBS, however, including V. fluvialis, V. furnissii, V. alginolyticus, V. metschnikovii, V. cincinnatiensis, and V. carchariae.2 4. Presumptive Tests to Differentiate V. cholerae The following key tests are used to identify V. cholerae: Test Reaction Gram-negative rod Cytochrome oxidase Glucose fermented (no gas) Growthin nutrient broth: No NaCl added 8% NaCl added + + + Arginine dihydrolase − + + Ornithine decarboxylase ONPG hydrolysis + − After isolation on TCBS, streak presumptive V. cholerae isolates to a nonselective medium, such as trypticase soy agar, containing a minimum of 0.5% NaCl. 5. Classification of Isolates as V. cholerae The tests listed below may be used for a more extensive phenotypic characterization of V. cholerae.7 To determine the serogroup, use agglutination assays. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Test ONPG Nitrate reduction Indole 0/129 sensitivity: 10 mg 150 mg Swarming Luminescence Thornley’s arginine dihydrolase Lysine decarboxylase Ornithine decarboxylase Growth at 42°C Growth at % NaCl: 0% 3% 6% 8% 10% Voges-Proskauer reaction Gas from glucose fermentation Fermentation to acid: L-Arabinose m-Inositol D-Mannose Sucrose Enzyme production: Alginase Amylase Chitinase Gelatinase Lipase Utilization as sole source of carbon: Reaction + + + + + − v* − + + + + + v − − v − − − v + − + + + + © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Test Reaction γ-Aminobutyrate Cellobiose − L-Citruline − Ethanol D-Gluconate − + D-Glucuronate − L-Leucine − Putrescine − + − Sucrose D-Xylose − * v = variable, differs for strains within the species. 6. Serological Identification Slide agglutination with polyvalent antisera can be used to identify the serogroups of V. cholerae. Polyvalent antiserum for V. cholerae O1 is available commercially.*#(70) The O1 serogroup can be further divided into two primary serotypes, Ogawa and Inaba. 7. Biotypes of Serogroup O1 V. cholerae V. cholerae can be divided into two biotypes or biovars, classical and El Tor, which differ in several characteristics. The El Tor biotype currently is the most important biotype. Biovar Test7 Classical El Tor Hemolysis of sheep erythrocytes − v* Voges-Proskauer reaction − + Chicken erythrocyte agglutination − + Antibiotic sensitivity: Polymyxin B (50 IU) + − Lysis No lysis No lysis Lysis Bacteriophage susceptibility: Mukerjee classical phage IV Mukerjee El Tor phage 5 * v = different reaction within the serovar. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 8. Other Procedures Environmental samples also may be examined by fluorescent-antibody techniques, but the number of V. cholerae cells in aquatic samples is generally quite low.7 Nucleic acid probes are not routinely used for the identification of V. cholerae, although DNA probes are extremely useful in determining which strains of this species contain the cholera toxin gene.2 This distinction is especially important in examining environmental isolates of V. cholerae because the great majority of these strains lack the cholera toxin gene. 9. References 1. KAPER, J.B., J.G. MORRIS, JR. & M.M. LEVINE. 1995. Cholera. Clin. Microbiol. Rev. 8:48. 2. OLIVER, J.D. & J.B. KAPER. 1997. Vibrio species. In M.P. Doyle, L.R. Beuchat & T.J. Montville, eds. Fundamentals of Food Microbiology. American Soc. Microbiology, Washington, D.C. 3. CENTERS FOR DISEASE CONTROL. 1995. Update: Vibrio cholerae O1— Western hemisphere, 1991–1994, and V. cholerae O139—Asia, 1994. Morbid. Mortal. Week. Rep. 44:215. 4. ALBERT, M.J. 1994. Vibrio cholerae O139 Bengal. J. Clin. Microbiol. 32:2345. 5. BARRETT, T.J., A. BLAKE, G.K. MORRIS, N.D. PUHR, H.B. BRADFORD & J.G. WELLS. 1980. Use of Moore swabs for isolating Vibrio cholerae from sewage. J. Clin. Microbiol. 11:385. 6. SPECK, M.L., ed. 1984. Compendium of Methods for the Microbiological Examination of Foods, 2nd ed. American Public Health Assoc., Washington, D.C. 7. WEST, P.A. & R.R. COLWELL. 1984. Identification and classification of Vibrionaceae—an overview. In R.R. Colwell, ed. Vibrios in the Environment. John Wiley & Sons, New York, N.Y. 8. KAYSNER, C.A. & W.E. HILL. 1994. Toxigenic Vibrio cholerae O1 in food and water. In I.K. Wachsmuth, P.A. Blake & O. Olsvik, eds. Vibrio cholerae and Cholera: Molecular to Global Perspectives. ASM Press, Washington, D.C. 9260 I. Leptospira Leptospira spp. are motile, aerobic spirochetes that require fatty acids for growth.1 Serum or polysorbate enrichments must be incorporated into artificial media, and some pathogenic strains may require CO2 upon initial isolation. Leptospires are divided into two groups, based on their pathogenicity and growth characteristics. The saprophytic leptospires are assigned to the Biflexa Complex, and the pathogenic leptospires make up the Interrogens Complex. Pathogenic strains have an optimal growth temperature of 28 to 30°C, and they grow over a pH range from 5.2 to © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 7.7. Saprophytic strains prefer a growth temperature between 5 and 10°C below pathogenic strains. Leptospires prefer alkaline conditions, and they persist longest in warm, moist environments protected from sunlight. Under favorable temperature and pH conditions, leptospires survive for 3 to 5 d in damp soil and up to 10 d in natural waters. They survive for 12 to 14 h in undiluted wastewater, up to 3 d in aerated wastewater, and up to 4 weeks in sterile tapwater at pH 7. Nonpathogenic leptospires are ubiquitous, and they have been isolated from municipal water supplies.2 Generally, pathogenic leprospires require an animal host and do not survive and propagate in the environment. Leptospirosis is a worldwide zoonotic disease of wild animals.3 Reservoirs of leptospires in wildlife include deer, foxes, raccoons, skunks, opossums, muskrats, and rodents. Domestic animals harboring leptospires include horses, cattle, goats, pigs, and sheep. Dogs may become infected but cats are spared. Humans are incidental hosts. Humans acquire leptospirosis (Weil’s disease) directly from animals, and from occupational or recreational exposure to urine-contaminated water4-6 or environmental surfaces. Swimming and other water sports,7 travel to tropical areas with occupational or recreational exposure to surface waters,8 and natural disasters that affect sewer systems and runoff9,10 increase risk of the disease. Outbreaks of leptospirosis associated with drinking water are extremely unusual, and are invariably caused by contamination of domestic water reservoirs with urine of infected rodents.11 Leptospirosis ranges from mild nonspecific febrile illness to severe or fatal renal, hepatic, or meningeal disease.12,13 Leptospires enter through imperfections in the skin, through mucous membranes, or by ingestion of contaminated water. Urine of infected animals and humans may contain 106 to 108 organisms/ mL. Leptospires may be shed into the environment up to 3 months after clinical recovery from disease. Diagnosis of disease in animals and humans usually is based upon serology, darkfield examination of urine sediments, examination of histopathological stains, or culture of the organism from urine or tissues. Recently polymerase chain reaction (PCR) methods have been introduced for diagnosis and typing of leptospires. While leptospirosis remains relatively common in tropical regions of the world, only 40 to 120 cases/year have been reported in the U.S. over the past 30 years. Leptospirosis was dropped from the list of notifiable diseases in 1994. Leptospires are recovered from environmental sources with great difficulty.14-17 Because both saprophytic and pathogenic strains of leptospires may be recovered from environmental samples, their presence has no public health significance apart from an epidemiological context. 1. Sample Collection Collect water samples of 100 mL to 1 L in sterile containers for transport to the laboratory at ambient temperature within 72 h of collection. Multiple samples from each sample site usually are required for successful isolation because finding leptospires in 10 to 20% of samples of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater surface waters receiving farm runoff is considered a high yield. Leptospires find their ecological niche at the interface between sediment and shallow water. Gently agitate the water to bring some of the sediment to the surface of shallow bodies of water to improve the probability of recovering organisms.18 For soil samples, collect 10 to 20 g of soil in sterile bottles or plastic bags. Use a small, tightly sealed container to protect sample from drying. A small amount of sterile deionized water may be added to soil samples to prevent drying. 2. Sample Processing Centrifuge a portion of a water sample at 5000 × g for 10 min and examine sediment by darkfield microscopy for leptospires. Their presence indicates that conditions are favorable for leptospire survival, but does not differentiate saprophytic from pathogenic forms. In the laboratory, thoroughly mix soil samples with three volumes of sterile deionized water and let coarse particulate material settle by gravity. Process remaining suspension as a water sample. Leptospira can pass through 0.22-µm membrane filters (¶ a below); this ability has been exploited to separate them from other bacteria in environmental samples and in mixed cultures. Similarly, guinea pig inoculation (¶ b below) has been used as a biological filter for isolation of leptospires from contaminated samples. a. Filtration method: Filter surface water samples through filter paper*#(71) to remove coarse debris before membrane filtration. Occasionally, samples may have to be passed through a series of prefilters of decreasing pore sizes (8-µm, 4-µm, 1-µm, 0.65-µm, and 0.45-µm) to prevent clogging of the final 0.22-µm filter. b. Animal inoculation method: Filter water through a 0.45-µm membrane filter and inoculate 1 to 3 mL intraperitoneally into weanling guinea pigs. After 3 to 6 d, inject a small amount of sterile saline and withdraw fluid for darkfield examination. If leptospires are seen, perform a cardiac puncture to obtain blood for inoculation of culture media. If no leptospira are seen by darkfield examination, record rectal temperatures daily until a fever spike indicates infection, then repeat the darkfield examination of peritoneal fluid for leptospires. Exsanguinate guinea pigs at 4 weeks and save serum for serological tests. Culture blood, kidney, and brain of guinea pigs with serological evidence of infection. Details of the method are described elsewhere.19 3. Culture Cultures of environmental samples usually will be contaminated with other bacteria unless the samples are filtered through a 0.22-µm membrane filter before inoculation. Filtration also may be used to isolate leptospires from mixed cultures, by direct filtration or another method.20 Unless sample filtration is used in conjunction with selective media or animal inoculation, a culture contamination rate of 60 to 80% is not uncommon. The amount of sample cultured will depend on the amount of particulate material in the sample. Generally, culture sample volumes from a few drops to 3.5 mL. a. Culture media: Pathogenic leptospires have been cultured in liquid, semisolid, and solid media, but not all pathogenic strains will grow on solid media. Optimal pH of culture media is © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 7.2 to 7.4 and optimal incubation temperature is 30°C. Leptospires are sensitive to detergents, so keep glassware free of detergent residues. When using serum enrichments in culture media, use serum free of antibody to leptospires. Bovine serum albumin shows manufacturer and lot variations; test new batches for their ability to support growth of leptospires. Modifications of the Ellinghausen-McMullough formulation (EMJH) that incorporate bovine serum albumin fraction V and polysorbates are used as serum replacements.21-24 EMJH base is available commercially. Neomycin is used in culture media at concentrations between 5 and 25 µg/mL to inhibit competing microflora, but it may be toxic to some strains.25 5-fluorouracil is used at 100 or 200 µg/mL in culture media, but it too is toxic for some strains, particularly at concentrations above 100 µg/mL.26 b. Culture methods: 1) Direct culture method—To recover leptospires from surface waters, place a few drops of water in EMJH liquid medium and incubate overnight at 30°C. Filter inoculated medium through a 0.22-µm membrane filter into a sterile tube and reincubate at 30°C for up to 6 weeks. 2) Dilution method—When samples may contain reasonable numbers of organisms in the presence of inhibitors or competing microflora, prepare 10-fold dilutions in duplicate, and inoculate 0.1 mL undiluted sample and each dilution into EMHJ medium. One tube of each pair may be made selective by addition of a single 30-µg neomycin antimicrobial susceptibility disk to the media before incubation. Incubate cultures at 20 to 30°C for up to 4 months. 3) Animal inoculation method—Add 1 to 2 drops of heart blood from infected guinea pigs to each of three to five tubes of EMJH medium. Incubate cultures at 20°C for up to 4 months. c. Culture examination: Leptospires usually are detected in cultures of environmental samples within 7 to 14 d; however, incubate and examine cultures weekly for 6 weeks before discarding them as negative. Observe tubes for a lightly turbid ring of growth just below the surface of the medium. This band of maximum turbidity at the zone of optimal oxygen tension is referred to as Dinger’s ring. Remove a drop of the culture weekly for darkfield examination and prepare subcultures if motile leptospires are observed. Generally, saprophytic leptospires grow at lower temperatures, and form rings closer to the surface of culture media than pathogenic serovars. Cultures remain viable in semisolid media for at least 8 weeks at room temperature. 4. Identification Experience and skill are required to differentiate artifacts from leptospires by darkfield microscopy. The biochemical tests previously thought to differentiate between pathogenic and saprophytic serovars do not reliably predict pathogenicity of leptospires, and they are not recommended. Leptospira are identified to serogroup by the microscopic agglutination test using reference antisera. Identification to serovar requires use of adsorbed antisera that are available only in reference laboratories. Over 200 serotypes of Leptospira are known. 5. References 1. FAINE, S. 1992. The genus Leptospira. In A. Balows, H.G. Truper, M. Dworkin, W. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. 3. 4. 5. 6. 7. 8. 9. 10. Harder & K.H. Schleifer, eds. The Prokaryotes, Vol. IV. Springer-Verlag, New York, N.Y. HENRY, R.A. & R.C. JOHNSON. 1978. Distribution of the genus Leptospira in soil and water. Appl. Envrion. Microbiol. 35:492. MICHNA, S.W. 1970. Leptospirosis. Vet. Record 86:484. ANDERSON, D.C., D.S. FOLLAND, M.D. FOX, C.M. PATTON & A.F. KAUFMANN. 1978. Leptospirosis: a common-source outbreak due to leptospires of the grippotyphosa serogroup. Amer. J. Epidemiol. 107:538. COGGINS, W.J. 1962. Leptospirosis due to Leptospira pomona: an outbreak of nine cases. J. Amer. Med. Assoc. 181:1077. VENKATARAMAN, K.S. & S. NEDUNCHELLIYAN. 1992. Epidemiology of an outbreak of leptospirosis in man and dog. Comp. Immun. Microbiol. Infect. Dis. 15:243. SHAW, R.D. 1992. Kayaking as a risk factor for leptospirosis. Missouri Med. 89:354. VAN CREVEL, R., P. SPEELMAN, C. GRAVEKAMP & W.J. TERPSTRA. 1994. Leptospirosis in travelers. Clin. Infect. Dis. 19:132. FUORTES, L. & M. NETTLEMAN. 1994. Leptospirosis: a consequence of the Iowa flood. Iowa Med. 84:449. KAT, A.R., S. MANEA & D.M. SASAKI. 1991. Leptospirosis on Kauai: investigation of a common source waterborne outbreak. Amer. J. Pub. Health 81:1310. 11. CACCIAPUOTI, B., L. CICERONI, C. MAFFEI, F. DI STANISLAO, P. STRUSI, L. CALEGARI, R. LUPIDI, G. SCALISE, G. CAGNONI & G. RENGA. 1987. A waterborne outbreak of leptospirosis. Amer. J .Epidemiol. 126:535. 12. HEATH, C.W., A.D. ALEXANDER & M.M. GALTON. 1965. Leptospirosis in the United States (concluded). Analysis of 483 cases in man, 1949–1961. N. England J. Med. 272:915. 13. HEATH, C.W., A.D. ALEXANDER & M.M. GALTON. 1965. Leptospirosis in the United States. Analysis of 483 cases in men, 1949–1961. N. England J. Med. 273:857. 14. ALEXANDER, A.D., H.G. STOENNER, G.E. WOOD & R.J. BYRNE. 1962. A new pathogenic Leptospira, not readily cultivated. J. Bacteriol. 83:754. 15. BAKER, M.F. & H.J. BAKER. 1970. Pathogenic Leptospira in Malaysian surface waters I. A method of survey for Leptospira in natural waters and soils. Amer. J. Trop. Med. Hyg. 19:485. 16. DIESCH, S.L. & W.F. MCCULLOCH. 1966. Isolation of pathogenic leptospires from water used for recreation. Pub. Health Rep. 81:299. 17. GILLESPIE, W.H., S.G. KENZY, L.M. RINGEN & F.K. BRACKEN. 1957. Studies on bovine leptospirosis. III. Isolation of Leptospira pomona from surface water. Amer. J. Vet. Res. 18:76. 18. BRAUN, J.L., S.L. DIESCH & W.F. MCCULLOCH. 1968. A method for isolating leptospires from natural surface waters. Can. J. Microbiol. 14:1011. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 19. FAINE, S. 1982. Guidelines for the control of leptospirosis. WHO offset publ. No. 67. World Health Organization, Geneva, Switzerland. 20. SMIBERT, R.M. 1965. A technique for the isolation of leptospirae from contaminating microorganisms. Can. J. Microbiol. 11:743. 21. ELLINGHAUSEN, H.C., JR. & W.G. MCCULLOUGH. 1965. Nutrition of Leptospira pomona and growth of 13 other serotypes: a serum-free medium employing oleic albumin complex. Amer. J. Vet. Res. 26:39. 22. ELLINGHAUSEN, H.C., JR. & W.G. MCCULLOUGH. 1965. Nutrition of Leptospira pomona and growth of 13 other serotypes: fraction of oleic albumin complex and a medium of bovine albumin and polysorbate 80. Amer. J. Vet. Res. 26:45. 23. TURNER, L.H. 1970. Leptospirosis III. Trans. Roy. Soc. Trop. Med. Hyg. 64:623. 24. ADLER, B., S. FAINE, W.L. CHRISTOPHER & R.J. CHAPPEL. 1986. Development of an improved selective medium for isolation of leptospires from clinical material. Vet. Microbiol. 12:377. 25. MYERS, D.M. & V.M. VARELA-DIAZ. 1973. Selective isolation of leptospiras from contaminated material by incorporation of neomycin to culture media. Appl. Microbiol. 25:781. 26. JOHNSON, R.C. & P. ROGERS. 1964. 5-fluorouracil as a selective agent for growth of leptospirae. J. Bacteriol. 87:422. 9260 J. Legionella The Legionellaceae have been implicated in outbreaks of disease occurring since 1947.1 Two forms of disease are recognized: a pneumonic form called Legionnaires’ Disease and a nonpneumonic form called Pontiac fever. The first species was isolated following the historic outbreak associated with the Legionnaires’ Convention in Philadelphia, Pa., in 1976. Epidemiological findings and animal studies have shown that the organism is transmitted via the airborne route2 and is ubiquitous in moist environments. The reservoirs for most outbreaks have been either contaminated air conditioning cooling tower water or contaminated potable water distribution systems.3,4 Legionella species also have been isolated in non-disease-related circumstances from a wide variety of aquatic environments such as lakes, streams, reservoirs, and sewage.5,6 The organisms are able to survive for prolonged periods in laboratory distilled and tap water.7 The Legionellaceae are composed of a single genus, Legionella, and more than 35 different species.8 The organisms are Gram-negative, aerobic, non-spore-forming bacteria. They are 0.5 to 0.7 µm wide and 2 to 20 µm long. They possess polar, subpolar, and/or lateral flagella. With the exception of L. oakridgensis, all require cysteine and iron salts for growth. Although Legionella originally were isolated in guinea pigs and embryonated hen’s eggs, it © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater has been shown that plating directly on artificial media is more sensitive than animal inoculation for L. pneumophila.9 The most widely used medium is an ACES (N-2-acetamideo-2-aminoethanesulfonic acid) buffered (pH 6.9) charcoal yeast extract (BCYE) agar supplemented with cysteine, ferric pyrophosphate, and optimally, alpha-ketoglutarate (BCYE-alpha).10 No one medium will be optimal for the recovery of Legionella from every environmental site; thus different selective media with various antibiotic combinations in a BCYE base may be necessary.10-12 Also, pretreating samples with hydrochloric acid-potassium chloride, pH 2.2, is useful for eliminating non-Legionella organisms.13 The two most commonly used selective media are GPVA medium (BCYE-alpha supplemented with glycine anisomycin, vancomycin, and polymyxin B) and CCVC medium (BCYE-alpha supplemented with polymyxin B, cephalothin, vancomycin, and cycloheximide). The GPVA medium is less inhibitory to some Legionella species. Use CCVC medium in combination with a less selective medium. Recovery of legionellae from environmental water samples sometimes is difficult. Legionellae may take up to a week to grow on plate media, and even with acid pretreatment and the addition of antibiotics to the medium, faster-growing organisms may overgrow legionellae. In addition, other organisms, including Pseudomonas spp., secrete into surrounding media bacterial products that can inhibit Legionella growth.14 Rapid methods for detecting Legionella utilizing direct fluorescent antibody staining (DFA) or polymerase chain reaction technology (PCR) also are available and may be more sensitive than culture-based assays.15-17 DFA can be quantitative, but may be subject to interference due to cross-reactivity with other organisms.18 The PCR method is semiquantitative. Both DFA and PCR may not distinguish between viable and nonviable bacteria. 1. Sample Collection Collect water samples from the littoral zone or from cooling towers, condenser coils, storage tanks, showers, water taps, etc. In most instances, a 1-L water sample is sufficient. Larger volumes of water (1 to 10 L)6 may be needed in water having low bacterial counts. In addition to collecting water samples, it may be useful to swab various fixtures (e.g., shower heads) and plate directly on selective media. Transport samples to the laboratory in insulated containers. Refrigerate samples that cannot be processed immediately. Treat chlorinated water with sodium thiosulfate (see Section 9060A.2). 2. Immunofluorescence Procedure Centrifuge 100 mL at 3500 × g for 30 min at room temperature and reconstitute the sedimented material in 6 to 10 mL filter-sterilized (0.2-mm filter) water from sample. Prepare smears for DFA by filling two 1.5-cm circles on a microscope slide with the concentrate. Air-dry sample smears, gently heat-fix, treat with 10% formalin for 10 min, rinse with phosphate-buffered saline (pH 7.6), and react with specific fluorescent antibodies.6,19 The DFA procedure lacks specificity20 and cannot determine viability. Some environmental bacteria (i.e., © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Pseudomonas spp. and Xanthomonas-Flavobacterium group) cross-react with the Legionella DFA reagents. To determine whether organisms are viable, use secondary staining with a tetrazolium dye.18 Confirm Legionella using direct isolation procedures. 3. Media and Reagents a. Buffered charcoal yeast extract alpha base:19 Norit SG charcoal Yeast extract ACES buffer Ferric pyrophosphate, soluble L-cysteine, HCl⋅H2O 2.0 10.0 10.0 0.25 0.4 g g g g g Agar Potassium alpha-ketoglutarate Reagent-grade water 17.0 1.0 1.0 g g L Dissolve yeast extract, agar, charcoal, glycine, and alphaketoglutarate in approximately 850 mL water; boil. Dissolve 10g ACES buffer in 100mL warm water, adjust pH to 6.9 with 1N KOH and add. Autoclave 15min at 121°C. Cool to 50°C. Dissolve 0.4g cysteine and 0.25g ferric pyrophosphate in 10mL of water each and filter sterilize separately (0.22µm). After base has cooled, add cysteine, ferric pyrophosphate, and dyes in that order. Adjust pH to 6.9 with sterile 1N KOH and dispense. b. GPVA medium:11,12*#(72) Glycine Polymyxin B 0.3 % 100 units/mL Vancomycin 5 µg/mL Anisomycin 80 µg/mL To cooled BCYE-alpha base with glycine, add filter-sterilized antibiotics and mix. Adjust pH to 6.9 with sterile 1N KOH and dispense. c. CCVC medium11†#(73) Cephalothin 4 µg/ml © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Colistin Vancomycin Cycloheximide 16 µg/mL 0.5 µg/mL 80 µg/mL To cooled BCYE-alpha base add filter-sterilized antibiotics and mix. Adjust to pH 6.9 with sterile 1N KOH and dispense. d. Acid treatment reagent,11 pH 2.0 (0.2M KCl/HCl): Solution A—0.2M KCl (14.9 g/L in distilled water). Solution B—0.2M HCl (16.7 mL/L 10N HCl in distilled water). Mix 18 parts of Solution A with 1 part of Solution B. Check pH against a pH 2.0 standard buffer. Dispense into screw-cap tubes in 1.0-mL volumes and sterilize by autoclaving. e. Alkaline neutralizer reagent11 (0.1N KOH): Stock solution—0.1N KOH (6.46 g/L in deionized water). Dilute 10.7 mL of stock solution with deionized water to 100 mL. Dispense into screw-cap tubes in convenient volumes and sterilize by autoclaving. The pH of d and e combined in equal volumes should be 6.9. 4. Sample Preparation a. Low-bacterial-count water: Concentrate water that has a low total bacterial count either by filtration11 or continuous-flow centrifugation.21 Filter samples through sterile 47-mm filter funnel assemblies containing a 0.2-µm porosity polycarbonate filter.‡#(74) After filtration, immediately remove the filter aseptically and place it in a 50-mL centrifuge tube or similar-size vessel containing 10 mL sterile tap water or phosphate buffer. If more than one filter is required to concentrate a sample, combine them. b. High-bacterial-count water: Process water that has a high total bacterial count directly. Place 10 mL sample in a 50-mL centrifuge tube or similar-size vessel containing 10 mL of sterile tap water or phosphate buffer. c. Sample dispersion: Disperse organisms from filter or aggregates by mixing with a vortex mixer (3 × 30 s). d. Plating: Plate acid-treated and non-acid-treated samples on two types of BCYE: plain and selective with antibiotics. 1) No acid treatment—Inoculate three plates each of BCYE-alpha and selective BCYE-alpha (GPVA or CCVC) with 0.1 mL of suspension. Spread with a sterile smooth glass rod. Save remainder of specimen for acid treatment and store at 4°C. 2) Acid treatment—Place 1.0 mL of suspension in a sterile 13 × 100-mm screw-capped tube containing 1.0 mL acid treatment reagent and mix. Final pH of mixture should be approximately 2.2. Let stand for 15 min at room temperature, neutralize by adding 1.0 mL alkaline neutralizer reagent, and mix. Inoculate 0.1 mL onto three plates each of BCYE-alpha and selective © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater BCYE-alpha (GPVA or CCVC) and spread with a sterile smooth glass rod. 3) Incubation—Incubate all plates at 35°C in a humidified atmosphere (>50%) for up to 10 d. A candle jar or humidified CO2 incubator (2 to 5% CO2) is acceptable. e. Total bacterial count examination: Determine the adequacy of processing for each high-bacterial-count water. Some samples may require dilution, concentration, or animal inoculation. If the total count of the acid-treated sample exceeds 300 colonies on BCYE selective medium, make a further 10-fold dilution of the sample stored at 4°C. Repeat acid-treatment and plating. If the total count of the non-acid-treated sample is less than 30 colonies on BCYE agar, concentrate and treat the collected water as previously described for low-bacterial-count water. 5. Examination of Cultures of Legionellae With the aid of a dissecting microscope, examine all cultures daily after 48 h incubation for the presence of opaque bacterial colonies that have a ‘‘ground-glass’’ appearance. Place plates with Legionella-like colonies in a biological safety cabinet equipped with a burner, a bacteriological needle, and a loop. Aseptically pick each suspect colony onto BCYE-alpha agar and a BCYE agar plate prepared without L-cysteine. Streak the inoculated portion of each plate with a sterile loop to provide areas of heavy growth and incubate for 24 h. Reincubate plates without growth an additional 24 h. Plates demonstrating growth on only BCYE-alpha agar are presumptive for Legionella. Confirm Legionella by slide agglutination or direct immunofluorescence. If these confirmatory techniques are not available, send subcultures of the presumptive legionellae to a reference laboratory for further identification. Because there are many serotypes in some species, especially L. pneumophila, investigation of environmental sites as possible reservoirs of epidemic-causing strains may be useful.22 Effective investigatory techniques include monoclonal antibody subtyping, electrophoretic isoenzyme analysis, restriction endonuclease tests, and plasmid analysis. 6. Polymerase Chain Reaction Procedure A test kit utilizing the polymerase chain reaction (PCR) is available commercially§#(75) and has been used successfully in an epidemiological investigation of an outbreak of Pontiac fever.23 Perform tests according to manufacturer’s instructions. The kit provides sample processing reagents, PCR primers, detection strips, and positive and negative controls. Specific probes allow for the detection of twenty-five Legionella species as well as specific detection of Legionella pneumophila. The test is semiquantitative, based on a colorimetric comparison to control strips equivalent to 103 cells/mL. 7. References 1. MCDADE, J.E., C.C. SHEPARD, D.W. FRASIER, T.R. TSAI, M.A. REDUS, W.T. DOWDLE & THE LABORATORY INVESTIGATION TEAM. 1977. Legionnaires’s Disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N. England J. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Med. 297:1197. 2. BERENDT, R.F., et al. 1980. Dose-response of guinea pigs experimentally infected with aerosols of Legionella pneumophila. J. Infect. Dis. 141:186. 3. FLIERMANS, C.B., W.B. CHERRY, L.H. ORRISON, S.J. SMITH, D.L. TISON & D.H. POPE. 1981. Ecological distribution of Legionella pneumophila. Appl. Environ. Microbiol. 41:9. 4. TOBIN, J.O.H., R.A. SWAN & C.L.R. BARTLETT. 1981. Isolation of Legionella pneumophila from water systems: methods and preliminary results. Brit. Med. J. 282:515. 5. CHERRY, W.B., G.W. GORMAN, L.H. ORRISON, C.W. MOSS, A.G. STEIGERWALT, H.W. WILKINSON, S.E. JOHNSON, R.M. MCKINNEY & D.J. BRENNER. 1982. Legionella jordanis: a new species of Legionella isolated from water and sewage. J. Clin. Microbiol. 15:290. 6. FLIERMANS, C.B., W.B. CHERRY, L.H. ORRISON & L. THACKER. 1979. Isolation of Legionella pneumophila from nonepidemic related aquatic habitats. Appl. Environ. Microbiol. 37:1239. 7. SKALIY, P. & H.V. MCEACHERN. 1979. Survival of the Legionnaires’s Disease bacterium in water. Ann. Intern. Med. 90:662. 8. BRENNER, D.J., A.G. STEIGERWALT, G.W. GORMAN, H.W. WILKINSON, W.F. BIBB, M. HACKEL, R.L. TYNDALL, J. CAMPBELL, J.C. FEELEY, W.L. THACKER, P. SKALIY, W.T. MARTIN, B.J. BRAKE, B.S. FIELDS, H.W. MCEACHERN & L.K. CORCORAN. 1985. Ten new species of Legionella. Int. J. System. Bacteriol. 35:50. 9. FEELEY, J.C., R.J. GIBSON, G.W. GORMAN, N.C. LANGFORD, J.K. RASHEED, D.C. MACEL & W.B. BAINE. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10:437. 10. EDELSTEIN, P.H. 1982. Comparative studies of selective media for isolation of Legionella pneumophila from potable water. J. Clin. Microbiol. 16:697. 11. GORMAN, G.W., J.M. BARBAREE & J.C. FEELEY. 1983. Procedures for the Recovery of Legionella from Water. Developmental Manual, Centers for Disease Control, Atlanta, Ga. 12. WADOWSKY, R.M. & R.B. YEE. 1981. Glycine-containing selective medium for isolation of Legionellaceae from environmental specimens. Appl. Environ. Microbiol. 42:768. 13. BOPP, C.A., J.W. SUMNER, G.K. MORRIS & J.G. WELLS. 1981. Isolation of Legionella spp. from environmental water samples by low-pH treatment and use of selective medium. J. Clin. Microbiol. 13:714. 14. PASZKO-KOLVA, C., P.A. HACKER, M.A. STEWART & R.L. WOLFE. 1993. Inhibitory effect of heterotrophic bacteria on the cultivation of Legionella dumoffi. In J.M. Barbaree, R.F. Breiman & A.P. Dufour, eds. Legionella: Current Status and Emerging Perspectives. ASM Press, Washington, D.C. 15. PALMER, C.J., Y. TSAI, C. PASZKO-LOLVA, C. MAYER & L.R. SANGERMANO. 1993. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Detection of Legionella species in sewage and ocean water by polymerase chain reaction, direct fluorescent-antibody, and plate culture methods. Appl. Environ. Microbiol. 59:3618. 16. PALMER, C.J., G.F. BONILLA, B. ROLL, C. PASZKO-KOLVA, L.R. SANGERMANO & R.S. FUJIOKA. 1995. Detection of Legionella species in reclaimed water and air with the Enviroamp Legionella PCR kit and direct fluorescent antibody staining. Appl. Environ. Microbiol. 61:407. 17. WILLIAMS, H.N., C. PASZKO-KOLVA, M. SHAHAMAT, C.J. PALMER, C. PETTIS & J. KELLEY. 1996. Molecular techniques reveal high prevalence of Legionella in dental units. J. Amer. Dental Assoc. 127:1188. 18. FLIERMANS, C.B., R.J. SORACCO & D.H. POPE. 1981. Measure of Legionella pneumophila activity in situ. Curr. Microbiol. 6:89. 19. JONES, G.L. & G.A. HEBERT. 1979. Legionnaires—the disease, the bacterium and methodology. U.S. Dep. Health, Education, & Welfare, Centers for Disease Control, Atlanta, Ga. 20. EDELSTEIN, P.H., R.M. MCKINNEY, R.D. MEYER, M.A.C. EDELSTEIN, C.J. KRAUSE & S.M. FINEGOLD. 1980. Immunologic diagnosis of Legionnaires’ Disease: cross reactions with anaerobic and microaerophilic organisms and infections caused by them. J. Infect. Dis. 141:652. 21. VOSS, L., K.S. BUTTON, M.S. RHEINS & O.H. TUOVINEN. 1984. Sampling methodology for enumeration of Legionella spp. in water distribution systems. In C. Thornsberry, A. Balows, J.C. Feeley & W. Jakubowski, eds. Legionella, Proc. 2nd International Symposium. American Soc. Microbiology, Washington, D.C. 22. BARBAREE, J.M., G.W. GORMAN, W.T. MARTIN, B.S. FIELDS & W.E. MORRILL. 1987. Protocol for sampling environmental sites for Legionellae. Appl. Environ. Microbiol. 53:1454. 23. MILLER, L.A., J.I. BEEBE, J.C. BUTLER, W. MARTIN, R. BENSON, R.E. HOFFMAN & B.S. FIELDS. 1993. Use of polymerase chain reaction in epidemiological investigations of Pontiac fever. J. Infect. Dis. 168:769. 8. Bibliography CENTERS FOR DISEASE CONTROL, NATIONAL INSTITUTE OF ALLERGY AND INFECTIOUS DISEASES & WORLD HEALTH ORGANIZATION. 1979. International Symposium on Legionnaire’s Disease. Ann. Intern. Med. 90:489. BLACKMAN, J.A., F.W. CHANDLER, W.B. CHERRY, A.C. ENGLAND, J.C. FEELEY, M.D. HICKLIN, R.M. MCKINNEY & H.W. WILKINSON. 1981. Legionellosis. Amer. J. Pathol. 103:427. DUFOUR, A. & W. JAKUBOWSKI. 1982. Drinking water and Legionnaire’s Disease. J. Amer. Water Works Assoc. 74:631. THORNSBERRY, C., A. BALOWS, J.C. FEELEY & W. JAKUBOWSKI, eds. 1984. Legionella, Proc. 2nd © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater International Symposium. American Soc. Microbiology, Washington, D.C. 9260 K. Yersinia enterocolitica Yersinia enterocolitica is a gram-negative bacterium that can cause acute gastroenteritis and can be found in water in cold or temperate areas of the United States. Many wild, domestic, and farm animals are reservoirs of this organism, including wild animals associated with water habitats (beavers, minks, muskrats, nutrias, otters, and racoons).1,2 The organism can grow at temperatures as low as 4°C with a generation time of 3.5 to 4.5 h if at least trace amounts of organic nitrogen are present.3 Most environmental strains of Y. enterocolitica and the closely related species, Y. kristensenii, Y. frederiksenii, and Y. intermedia, generally are considered nonpathogenic, but disease outbreaks have been associated with environmental sources. Some strains lacking classic virulence markers also may be associated with disease.4 Y. enterocolitica has become recognized worldwide as an important human pathogen and in several countries it is nearly as common as Salmonella and Campylobacter as a leading cause of acute or chronic enteritis.5 Y. enterocolitica usually is associated with sporadic cases of gastroenteritis in the U.S.; however, epidemiologic investigations suggest that the predominant pathogenic serotype isolated in the U.S. has been changing.4 Y. enterocolitica serogroup O:3 has replaced O:8 as the most common species recovered from patients, reflecting the same pattern seen in other parts of the world.4,5 Two reported incidents of waterborne gastroenteritis possibly caused by Yersinia occurred during the period 1971 to 1978.3,6,7 Yersinia has been isolated from untreated surface and ground waters in the Pacific Northwest, New York, and other regions of North America, with highest isolations during the colder months.8-10 Concentrations have ranged from 3 to 7900 CFU/100 mL. Laboratory tests used to isolate and enumerate yersiniae do not discriminate between pathogenic and nonpathogenic strains. Yersinia isolations do not correlate with levels of total and fecal coliforms or total plate count bacteria.9 There is little information on Yersinia survival in natural waters and water treatment processes. In studies of Yersinia in chlorinated-dechlorinated secondary effluent and receiving (river) water, the organism was isolated in 27% of the effluent samples, 9% of the upstream samples, and 36% of the downstream samples.11 Mean total and fecal coliform reductions in effluent chlorination were 99.93 and 99.95%, respectively. In a survey of untreated and treated (chlorination or filtration plus chlorination) drinking water supplies, Yersinia was found in 14.0 and 5.7% of the samples, respectively.9 Of water samples with less than 2.2 coliforms/100 mL, 15.9% were Yersinia-positive. Yersinia isolation did not correlate with presence of total or fecal coliforms in this study. Another study confirmed that E. coli also is not a good indicator for Yersinia in water and that Y. enterocolitica O:3 strains harboring a virulence plasmid have enhanced resistance to chlorine compared to non-virulent strains.12 Because of the existence of animal reservoirs, widespread occurrence and persistence of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Yersinia in natural and treated water in at least some geographic areas, the evidence for possible waterborne outbreaks, and the lack of definitive information on its reduction by treatment processes, this pathogen is of potential importance in drinking water. 1. Concentration and Cultivation A membrane filter method for enumerating and isolating Yersinia enterocolitica is available.13 The method may be used for examining large volumes of low-turbidity water and for presumptively identifying the organism without transferring colonies to multiple confirmatory media. Filter sample through a membrane filter (see Section 9260B.1d). Place membrane filter on a cellulose pad saturated with m-YE recovery broth. Incubate for 48 h at 25°C. Aseptically transfer the membrane to a lysine-arginine agar substrate and incubate anaerobically at 35°C. After 1 h, puncture a hole in the membrane next to each yellow to yellow-orange colony with a needle, transfer the membrane to a urease-saturated absorbent pad, and incubate at 25°C for 5 to 10 min. Immediately count all distinctly green or deep bluish-purple colonies. The green or bluish colonies are sorbitol-positive, lysine- and arginine-negative, and urease-positive. They may be presumptively identified as Y. enterocolitica or a closely related Yersinia species. Additional biochemical testing is necessary to determine species. Reasonably simple tests have been described to screen isolates for pathogenicity.14 Comprehensive biochemical and serological characterization or the use of molecular methods is necessary to confirm virulence, but these methods are not generally available. 2. References 1. WETZLER, T.F. & J. ALLARD. 1977. Yersinia enterocolitica from trapped animals in Washington State. Paper presented at International Conf. Disease in Nature Communicable to Man. Yellow Bay, Mont. 2. WETZLER, T.F., J.T. REA, G. YUEN & W. TURNBERG. 1978. Yersinia enterocolitica in waters and wastewaters. Paper presented at 106th Annual Meeting, American Public Health Assoc., Los Angeles, Calif. 3. HIGHSMITH, A.K., J.C. FEELEY, P. SKALIY, J.G. WELLS & B.T. WOOD. 1977. The isolation and enumeration of Yersinia enterocolitica from well water and growth in distilled water. Appl. Environ. Microbiol. 34:745. 4. BISSETT, M.J., C. POWERS, S.L. ABBOTT & J.M. JANDA. 1990. Epidemiologic investigations of Yersinia enterocolitica and related species: sources, frequency, and serogroup distribution. J. Clin. Microbiol. 28:910. 5. FENWICK, S.G. & M.D. MCCARTY. 1995. Yersinia enterocolitica is a common cause of gastroenteritis in Auckland. N. Zealand Med. J. 108:269. 6. EDEN, K.V., M.L. ROSENBERG, M. STOOPLER, B.T. WOOD, A.K. HIGHSMITH, P. SKALIY, J.G. WELLS & J.C. FEELEY. 1977. Waterborne gastrointestinal illness at a ski-resort—isolation of Yersinia enterocolitica from drinking water. Pub. Health Rep. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 92:245. 7. KEET, E. 1974. Yersinia enterocolitica septicemia. N.Y. State J. Med. 74:2226. 8. HARVEY, S., J.R. GREENWOOD, M.J. PICKETT & R.A. MAH. 1976. Recovery of Yersinia enterocolitica from streams and lakes of California. Appl. Environ. Microbiol. 32:352. 9. WETZLER, T.F., J.R. REA, G.J. MA & M. GLASS. 1979. Non-association of Yersinia with traditional coliform indicators. In Proc. Annu. Meeting American Water Works Assoc., American Water Works Assoc., Denver, Colo. 10. SHAYEGANI, M., I. DEFORGE, D.M. MCGLYNN & T. ROOT. 1981. Characteristics of Yersinia enterocolitica and related species isolated from human, animal, and environmental sources. J. Clin. Microbiol. 14:304. 11. TURNBERG, W.L. 1980. Impact of Renton Treatment Plant effluent upon the Green-Duwamish River. Masters Thesis, Univ. Washington, Seattle. 12. LUND, D. 1996. Evaluation of E. coli as an indicator for the presence of Campylobacter jejuni and Yersinia enterocolitica in chlorinated and untreated oligotrophic lake water. Water Res. 30:1528. 13. BARTLEY, T.D., T.J. QUAN, M.T. COLLINS & S.M. MORRISON. 1982. Membrane filter technique for the isolation of Yersinia enterocolitica. Appl. Environ. Microbiol. 43:829. 14. FARMER, J.J., G.P. CARTER, V.L. MILLER, S. FALKOW & I.W. WACHSMUTH. 1992. Pyrazinamidase, CR-MOX agar, salicin fermentation-esculin hydrolysis, and d-xylose fermentation for identifying pathogenic serotypes of Yersinia enterocolitica. J. Clin. Microbiol. 30:2589. 3. Bibliography HIGHSMITH, A.K., J.C. FEELEY & G.K. MORRIS. 1977. Yersinia enterocolitica: a review of the bacterium and recommended laboratory methodology. Health Lab. Sci. 14:253. BOTTONE, E.J. 1977. Yersinia enterocolitica: a panoramic view of a charismatic microorganism. CRC Crit. Rev. Microbiol. 5:211. YANKO, W.A. 1993. Occurrence of Pathogens in Distribution and Marketing Municipal Sludges. National Technical Information Serv. Rep. PB88-154273-AS, Springfield, Va. 9260 L. Aeromonas (PROPOSED) 1. Introduction Aeromonas spp. are natural inhabitants of aquatic environments worldwide. These Gram-negative, facultatively anaerobic, glucose-fermenting organisms have been isolated from groundwater, treated drinking water, surface waters, wastewater, sludge, and sediment. Their populations are seasonal in all natural waters, with the highest numbers present in warmer © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater months. Aeromonads cause serious diseases of aquatic animals and represent an economic threat to the aquaculture industry. The motile aeromonads have emerged as a serious microbial threat to human populations, especially the immunocompromised.1 As a result of recent taxonomic studies, Aeromonas bacteria have been removed from the family Vibrionaceae and established as the sole genus of the new family Aeromonadaceae. The genus Aeromonas comprises 14 recognized and 2 proposed DNA hybridization groups with 13 named phenospecies and 4 unnamed genospecies. The extreme difficulty of phenotypically differentiating aeromonads and the unavailability of DNA hybridization techniques in most laboratories have lead clinical microbiologists to report aeromonads as A. hydrophila, A. sobria, or A. caviae, according to a published classification scheme.2 Environmental microbiologists usually combine all motile, mesophilic aeromonads into the Aeromonas hydrophila complex, or simply report isolates as A. hydrophila. These practices obscure understanding of the medical and public health significance of aeromonads isolated from clinical specimens, environmental samples, and public water supplies; identification of Aeromonas isolates according to established taxonomic principles is preferable.3 While no waterborne outbreaks of gastroenteritis attributed to aeromonads have implicated public drinking water supplies in the U.S., this does not mean that none have occurred. The epidemiologic association between ingestion of untreated well water and subsequent Aeromonas gastrointestinal illness has been widely documented. Numerous cases and outbreak investigations of water- and food-transmitted illnesses caused by aeromonads have been reported.4 Outbreaks of gastroenteritis caused by aeromonads have occurred in custodial care institutions, nursing homes, and day-care centers. Aeromonas contamination of drinking water has been documented as a cause of travelers’ diarrhea.5 For many years, Aeromonas have been considered nuisance organisms by environmental microbiologists because they were reported to interfere with coliform multiple tube fermentation (MTF) methods. While aeromonads comprise 12% of bacteria isolated from drinking water by presence-absence methods, no data have demonstrated inhibition of coliform organisms by aeromonads in drinking water. Slight turbidity of LTB tubes, with or without a small bubble of gas in the inverted tube, is suggestive of aeromonads. When the MTF method is used for drinking water samples, cultures producing turbidity at 35°C that remain clear at 44.5°C are suggestive of aeromonads. The presence of aeromonads can be verified by subculturing a loopful of turbid broth to a MacConkey plate and screening colorless colonies for gelatinase and oxidase production. No data are available to support invalidation of coliform MTF tests based on turbidity of tubes in the absence of gas production. The ecology of mesophilic aeromonads in aquatic environments, including water treatment plants and distribution systems, has been reviewed.6 The Netherlands and the Province of Quebec have established drinking water standards for Aeromonas at 20 CFU/100 mL for water leaving the treatment plant, and 200 CFU/100 mL for distribution system water. Canada has established an Aeromonas MCL of 0 (zero) for bottled water. A resuscitation method for © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater recovery of aeromonads in bottled water has been published.7 The ability to isolate, enumerate, and identify aeromonads from water and wastewater sources is important because of their role in causing human and animal disease, their ability to colonize treatment plants and distribution systems, and their presence and distribution as alternative indicators of the trophic state of waters. The diversity of aeromonads in drinking water plants and distribution systems was shown by several investigators.8-10 Many media and methods have been proposed for the isolation and enumeration of aeromonads.11,12 The methods presented below represent a compromise, because no single enrichment method, isolation medium, or enumeration method is capable of recovering all aeromonads present in a water sample. The methods were chosen on the basis of reproducibility of results, objectivity of interpretation, availability of materials, and specificity of the method for detection of aeromonads in the presence of other heterotrophic bacteria. Consult the literature for additional methods for use in special circumstances.13 2. Sample Collection Collect water samples in sterile screw-capped glass or plastic bottles or plastic bags.*#(76) Sample volumes of 200 mL to 1 L are sufficient for most analyses. For chlorinated waters, add sodium thiosulfate (see Section 9060A.2). The potentially toxic effect of heavy metals is neutralized by adding EDTA (see Section 9060A.2). Transport samples to the laboratory at 2 to 8°C within 8 h. Samples for presence-absence analyses may be transported at ambient temperatures within 24 h. Grab samples are most common. Moore swabs (see 9260B.1a) have been used for wastewater sampling, and Spira bottles have been used for tapwater sampling.13 Both of these methods are used in conjunction with enrichment in 1% alkaline peptone water (APW), pH 8.6.13 Place sediment and sludge samples in bottles or bags and submit in same way as water samples. 3. Enrichment Methods Do not use enrichment methods for ecological studies because the predominant strain(s) will overgrow other organisms. Reserve enrichments for presence-absence tests for aeromonads in drinking water, foods, stools, or for monitoring aeromonad populations in wastewater or marine environments, where organisms may be present in low numbers or require resuscitation due to injury from exposure to inimical agents or hostile physical environments. For isolation of aeromonads from clear water samples, filter through 0.45 µm membrane filters, place filters in a bottle with 10 mL APW, incubate overnight at 35°C, and inoculate to plating media for isolation. Optimally, to sample clear water intended for drinking, filter a volume of water through a mini-capsule filter†#(77), decant residual water from inlet, plug ends with sterile rubber stoppers, and fill filter with APW, pH 8.6, through syringe port. Incubate filter at 35°C for 6 h or overnight and streak loopfuls of broth onto selective and differential plating media.14 4. Enumeration Methods © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater a. Spread plates: Enumerate samples expected to contain predominantly aeromonads in high numbers (sludge, sediments, wastewater effluents, polluted surface waters, etc.) directly by spreading 0.1-mL portions of decimal dilutions on ampicillin dextrin agar (ADA)15-17 plates. Incubate plates at 35°C overnight and count bright yellow colonies 1 to 1.5 mm in diameter. Presumptively identify colonies using the screening methods below. b. Membrane filtration (MF): Enumerate aeromonads in drinking water samples or other low-turbidity waters by using MF procedures with ADA medium and incubating aerobically overnight at 35°C. Filter sample volumes equivalent to 1 mL, 10 mL, and 100 mL. To achieve a countable plate (1 to 30 colonies), prepare decimal dilutions when aeromonads are present in high numbers. Count bright yellow colonies, 1 to 1.5 mm in diameter, and pick to screening media. c. Multiple-tube fermentation tests (MTF): Multiple-tube fermentation tests using APW, pH 8.6, or trypticase soy broth (TSB) containing ampicillin at 30 µg/mL (TSB30) have been applied to foods; however, they have not been used for enumeration of aeromonads in water samples. Some aeromonads are sensitive to ampicillin and will not grow in TSB30 medium. ADA without agar has been used to enumerate aeromonads in drinking water.8 Use MTF methods only for clean samples such as groundwater or treated drinking water samples, because the effect of competing microflora present in surface waters on recovery of aeromonads in broth media has not been studied adequately. Similarly, the correlation between MTF population estimates and other enumeration methods has not been examined adequately for matrices other than foods. 5. Screening Tests Pick 3 to 10 colonies resembling aeromonads on differential and selective plating media or membrane filters and stab-inoculate into deeps of Kaper’s multi-test medium19 or one tube each of triple sugar iron (TSI) agar and lysine iron agar (LIA). Incubate cultures at 30°C for 24 h. Perform a spot oxidase test on growth taken from the LIA slant. Do not test for oxidase on growth from TSI slants, MacConkey agar, or other selective or differential media, because acid production interferes with the oxidase reaction. Reactions of enteric bacteria on TSI and LIA media are shown in Table 9260:III. When Kaper’s medium is used instead of TSI/LIA slants, colonies may be picked and inoculated onto sheep blood agar plates; incubate at 35°C overnight to provide growth for the oxidase test and to record hemolysin production. Cultures are identified presumptively using Kaper’s medium according to the characteristics shown in Table 9260:IV. If species identification is desirable, submit presumptively identified Aeromonas cultures to a reference laboratory. Cultures with potential public health or regulatory significance may be subtyped using various molecular methods to determine clonality for outbreak investigations and trouble-shooting of treatment plant or distribution system problems.1 6. References 1. JANDA, J.M. & S.L. ABBOTT. 1996. Human Pathogens. In B. Austin, M. Altwegg, P. Gosling & S.W. Joseph, eds. The Genus Aermonas, p. 151. John Wiley & Sons, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Chichester, U.K. 2. POPOFF, M. & M. VERON. 1976. A taxonomic study of the Aeromonas hydrophila-Aeromonas punctata group. J. Gen. Microbiol. 94:11. 3. CARNAHAN, A.M. & M. ALTWEGG. 1996. Taxonomy. In B. Austin, M. Altwegg, P. Gosling & S.W. Joseph, eds. The Genus Aeromonas, p. 1. John Wiley & Sons, Chichester, U.K. 4. JOSEPH, S.W. 1996. Aeromonas gastrointestinal disease: a case study in causation?. In B. Austin, M. Altwegg, P. Gosling & S.W. Joseph, eds. The Genus Aeromonas, p. 311. John Wiley & Sons, Chichester, U.K. 5. HANNINEN, M.L., S. SALMI, L. MATTILA, R. TAIPALINEN & A. SIITONEN. 1995. Association of Aeromonas spp. with travellers’ diarrhoea in Finland. J. Med. Microbiol. 42:26. 6. HOLMES, P., L.M. NICCOLLS & D.P. SARTORY. 1996. The ecology of mesophilic Aeromonas in the aquatic environment. In B. Austin, M. Altwegg, P. Gosling & S.W. Joseph, eds. The Genus Aeromonas, p. 127. John Wiley & Sons, Chichester, U.K. 7. WARBURTON, D.W., J.K. MCCORMICK & B. BOWEN. 1993. Survival and recovery of Aeromonas hydrophila in water: development of methodology for testing bottled water in Canada. Can. J. Microbiol. 40:145. 8. HANNINEN, M.-L. & A. SIITONEN. 1995. Distribution of Aeromonas phenospecies and genospecies among strains isolated from water, foods or from human clinical samples. Epidemiol. Infect. 115:39. 9. HUYS, G., I. KERSTERS, M. VANCANNEYT, R. COOPMAN, P. JANSSEN & K. KERSTERS. 1995. Diversity of Aeromonas sp. in Flemish drinking water production plants as determined by gas-liquid chromatographic analysis of cellular fatty acid methyl esters (FAMEs). J. Appl. Bacteriol. 78:445. 10. MOYER, N.P., G.M. LUCCINI, L.A. HOLCOMB, N.H. HALL & M. ALTWEGG. 1992. Application of ribotyping for differentiating aeromonads isolated from clinical and environmental sources. Appl. Environ. Microbiol. 58:1940. 11. GAVRIEL, A. & A.J. LAMB. 1995. Assessment of media used for selective isolation of Aeromonas spp. Lett. Appl. Microbiol. 21:313. 12. JEPPESEN, C. 1995. Media for Aeromonas spp., Plesiomonas shigelloides and Pseudomonas spp. from food and environment. Int. J. Food Microbiol. 26:25. 13. MOYER, N.P. 1996. Isolation and enumeration of aeromonads. In B. Austin, M. Altwegg, P. Gosling & S.W. Joseph, eds. The Genus Aeromonas, p. 39. John Wiley & Sons, Chichester, U.K. 14. MOYER, N.P., G. MARTINETTE, J. LUTHY-HOTTENSTEIN & M. ALTWEGG. 1992. Value of rRNA gene restriction patterns of Aeromonas spp. for epidemiological investigations. Curr. Microbiol. 24:15. 15. HANDFIELD, M., P. SIMARD & R. LETARTE. 1996. Differential media for quantitative © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 16. 17. 18. 19. recovery of waterborne Aeromonas hydrophila. Appl. Environ. Microbiol. 62:3544. HAVELAAR, A.H., M. DURING & J.F. VERSTEEGH. 1987. Ampicillin-dextrin agar medium for the enumeration of Aeromonas species in water by membrane filtration. J. Appl. Bacteriol. 62:279. HAVELAAR, A.H. & M. VONK. 1988. The preparation of ampicillin dextrin agar for the enumeration of Aeromonas in water. Lett. Appl. Microbiol. 7:169. ALTWEGG, M. 1996. Subtyping methods for Aeromonas species. In B. Austin, M. Altwegg, P. Gosling & S.W. Joseph, eds. The Genus Aeromonas, p. 109. John Wiley & Sons, Chichester, U.K. KAPER, J., R.J. SEIDLER, H. LOCKMAN & R.R. COLWELL. 1979. Medium for the presumptive identification of Aeromonas hydrophila and Enterobacteriaceae. Appl. Environ. Microbiol. 38:1023. 9260 M. Mycobacterium (PROPOSED) The genus Mycobacterium comprises over 70 characterized species that are non-motile spore-forming, aerobic, acid-fast bacilli measuring 0.2 to 0.6 × 1 to 10 µm. Most organisms in this genus are saprophytes, but some species are capable of causing disease in humans. The primary pathogens in this group include Mycobacterium tuberculosis and Mycobacterium leprae, the causative agents of tuberculosis and leprosy, respectively. Recently there has been an increase in the incidence of disease caused by nontuberculosis mycobacteria, probably related to the increasing numbers of immunocompromised patients.1-3 In the genus Mycobacterium, the most important opportunistic pathogens include M. avium-intracellulare, M. kansasii, M. marinum, and M. simiae, which are capable of causing disease when the immune system is compromised. Some of the common hosts and environmental reservoirs of Mycobacteria are shown in Table 9260:V. Because of the complex nature of the cell wall, which is rich in lipids and therefore has a hydrophobic surface, this genus is resistant to many common disinfectants. As a result, several members of this genus are becoming important waterborne pathogens in the immunocompromised population. Mycobacteria also are acid-fast and extremely slow-growing. Some species such as M. avium-intracellulare require from 3 to 8 weeks to form colonies on culture media. Mycobacterium avium and Mycobacterium intracellulare exhibit overlapping properties, making speciation extremely difficult. As a result, these two pathogens are grouped together and called M. avium-intracellulare or refered to as the MAC complex. Organisms from this group are ubiquitous in the environment and have been isolated from potable water systems, including those in hospitals4-6 as well as from soil and dairy products. This pathogen causes a chronic pulmonary disease in immunocompetent hosts that is clinically and pathologically indistinguishable from tuberculosis; it also causes disseminated disease in immunocompromised © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater hosts. The primary route of transmission is believed to be through ingestion, but increasing numbers of cases originate in the respiratory tract, indicating an aerosol route of transmission. 1. Sample Collection and Concentration Mycobacteria typically constitute a minority of the microflora, especially in finished waters, and require sample concentration. Collect water samples in sterile 1-L polypropylene containers. For finished, disinfected waters, add 1 mL 10% sodium thiosulfate solution/L water collected. Transport samples to laboratory immediately after collection. If samples cannot be analyzed immediately, store at 4°C and begin analysis within 24 h of sampling. 2. Screening Water Samples by Direct Fluorescent Assay Before committing the sample to a lengthy culture incubation, survey for acid-fast bacteria by using a combination solution of Auramine-Rhodamine (A-R) fluorescent dye.7*#(78) Auramine and Rhodamine nonspecifically bind to mycolic acids and resist decolorization by acid alcohol.8 Filter a minimum of 500 mL finished water or 100 mL source water (depending on turbidity), through a sterile 0.45-µm-porosity, 47-mm-diam black filter. Aseptically transfer filter to a sterile polypropylene 50-mL tube and add 5 mL of buffered dilution water. Resuspend organisms from filter by vortexing for 2 min. Aspirate suspension and aseptically transfer to a sterile 15-mL polypropylene centrifuge tube. Centrifuge suspension at 5000 g for 10 min and discard all but about 0.5 mL of supernatant. Resuspend pellet by vortexing. Transfer 100 mL of the concentrate to a clean glass slide and air-dry and heat-fix at 60 to 70°C for 2 h or overnight. Primary stain the smear with A-R (15 min), decolorize with acid-alcohol†#(79) for 2 to 3 min, rinse with deionized water, apply secondary potassium permanganate counterstain (no longer than 2 to 4 min), rinse, and let air-dry. Examine smear at 100 × and 400 × with a microscope fitted with a BG-12 or 5113 primary filter with a OG-1 barrier filter. Acid-fast organisms will stain yellow-orange on a black background. To confirm for acid-fastness, apply a traditional acid-fast stain (Ziehl-Nielsen with Kenyon modification) directly to the prepared smear following the A-R stain. For wastewater or highly turbid source waters, collect a 10-mL subsample and transfer to a sterile polypropylene 15-mL tube. Centrifuge at 5000 g for 10 min and discard all but about 0.5 mL of supernatant. Follow slide preparation procedure and staining as above. 3. Decontamination and Culture Methods Mycobacteria grow very slowly on laboratory media. Therefore, eliminate from the sample naturally occurring organisms that can out-compete and overgrow the mycobacteria. Various isolation and identification methods have been described for the recovery of mycobacteria, especially in the hospital environment.4-6 Decontamination of the sample concentrate is required for the selection for mycobacteria before culture. In addition, the matrix may affect the success of the recovery of mycobacteria. Several methods (a through c below) are detailed for recovering mycobacteria from water samples; determine which method performs best with the matrix to be © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater examined. a. Filter 500-mL water sample through a sterile 0.45-µm-porosity, 47-mm-diam filter. Aseptically transfer filter to a sterile polypropylene 50-mL tube. Add 5 mL sterile distilled water and resuspend organisms off the filter by shaking with two 5-mm glass beads for 1 h on a mechanical shaker.9 Add a 3% sodium lauryl sulfate, 1% NaOH solution.10 Spread portions of this suspension onto a selective agar medium as described in ¶ 4 below. b. Filter 500-mL water sample through sterile 0.45-µm-porosity, 47-mm-diam filter. Aseptically transfer filter to a sterile polypropylene 50-mL tube. Add 5 mL sterile distilled water and resuspend organisms off the filter by shaking with glass beads for 5 min on a mechanical shaker. Add 10 mL 1M NaOH for 20 min followed by centrifugation at 8600 g at 4°C for 15 min. Discard supernatant and add 5 mL 5% oxalic acid for 20 min. Re-centrifuge, discard supernatant, and add 30 mL sterile distilled water to neutralize. Centrifuge again, and resuspend in 0.7 mL distilled water.11 Use portions of this material for selective growth (¶ 4 below). c. Add 20 mL 0.04% (w/v) cetylpridinium chloride (CPC) to 500-mL water sample and leave at room temperature for approximately 24 h. Filter sample and wash filter with 500 mL sterile water.12 A study of decontamination methods for the isolation of mycobacteria from drinking water samples found a CPC concentration of 0.005% (w/v) to yield the highest isolation rate and lowest contamination rate for the water examined.13 4. Selective Growth Culture all samples in duplicate. After sample decontamination, either spread portions of the concentrates or use sterile forceps to place filters on selective media. One common egg-based medium that successfully isolates mycobacteria from environmental concentrates is Lowenstein-Jensen agar. An agar-based medium containing cycloheximide (7H10) is a general growth medium for mycobacteria as well. Place plates in humid chambers or gas-permeable bags to prevent dehydration, and incubate at 37°C. Additional plates also can be incubated at 30°C in a humidified chamber to detect mycobacteria that grow optimally at lower temperatures. Examine plates periodically during a 3- to 8-week incubation period. Count suspect colonies (acid-fast coccobacilli) and subculture to a tube of 7H9 broth. After 5 d, remove subsamples and stain with Ziehl-Nielsen stain with Kenyon modification. Subculture coccobacillary acid-fast organisms further onto 7H10 plates. Conduct phenotypic tests (Table 9260:VI) as a first step toward identification. If biochemical tests do not allow speciation, use other methods, such as fatty acid profile by HPLC or GLC, serological typing, and/or molecular tests such as DNA probes, and RFLP, which have been used for rapid detection of a limited number of species.14 Although phenotypic tests have been the standard for species identification, there are several inherent problems in this approach. First, because initial identification of mycobacteria can take 3 to 8 weeks, observing biochemical changes entails additional time for the isolates (especially those of nontuberculosis mycobacteria) to metabolize specific substrates or to exhibit certain characteristics. Second, phenotypic traits are not stable; thus some species of mycobacteria are © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater untypable by conventional methods. One approach that can successfully speciate Mycobacterium is sequencing amplified rDNA.15 The method produces objective results in 2 d, gives reproducible results (due to the stability of the 16S rRNA), and can identify new species. These techniques have been used in clinical diagnostic laboratories, and now are available in some full-service environmental testing laboratories. 5. References 1. GOOD, R.C. Opportunistic pathogens in the genus Mycobacterium. 1985. Annu. Rev. Microbiol. 39:347. 2. CARSON, L.A., L.A. BLAND, L.B. CUSICK, M.S. FAVERO, G.A. BOLAN, A.L. REINGOLD & R.C. GOOD. 1988. Prevalence of nontuberculous mycobacteria in water samples of hemodialysis centers. Appl. Environ. Microbiol. 54:3122. 3. DUMOULIN, G.C. & K.D. STOTTMEIR. 1986. Waterborne mycobacteria: an increasing threat to health. ASM News 52: 525. 4. DUMOULIN, G.C., K.D. STOTTMEIR, P.A. PELLETIER, A.Y. TSANG & J. HEDLEY-WHITE. 1988. Concentration of Mycobacterium avium by hospital water systems. J. Amer. Med. Assoc. 260:1599. 5. POWELL, B.L. & J.E. STEADHAM. 1981. Improved technique for isolation of Mycobacterium kansasii from water. J. Clin. Microbiol. 13:969. 6. CARSON, L.A., L.B. CUSICK, L.A. BLAND & M.S. FAVERO. 1988. Efficiency of chemical dosing methods for isolating nontuberculous mycobacteria from water supplies of dialysis centers. Appl. Environ. Microbiol. 54:1756. 7. NOLTE, F.S. & B. METCHOCK. 1995. Mycobacterium. In P.R. Murray, E.J. Baron, M.A. Pfaller, F.C. Tenover & R.H. Yolken, eds. Manual of Clinical Microbiology. American Soc. Microbiology Press, Washington, D.C. 8. CHAPIN, K. 1995. Clinical Miscroscopy. In P.R. Murray, E.J. Baron, M.A. Pfaller, F.C. Tenover & R.H. Volken, eds. Manual of Clinical Microbiology. American Soc. Microbiology Press, Washington, D.C. 9. KAMALA, T., C.N. PARAMASIVAN, D. HERBERT, P. VENKATESAN & R. PRABHAKAR. 1994. Evaluation of procedures for isolation of nontuberculous mycobacteria from soil and water. Appl. Environ. Microbiol. 60:1021. 10. ENGEL, H.W.B., L.G. BERWALD & A.H. HAVELAAR. 1980. The occurrence of Mycobacterium kansasii in tapwater. Tubercle 61:21. 11. IIVANANINEN, E.K., P.J. MARTIKAINEN, P.K. VAANANEN & M.-L.KATILA. 1993. Environmental factors affecting the occurrence of mycobacteria in brook waters. Appl. Environ. Microbiol. 59:398. 12. DUMOULIN, G.C. & K.D. STOTTMEIR. 1978. Use of cetylpyridinium chloride in the decontamination of water culture of mycobacteria. Appl. Environ. Microbiol. 36:771. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 13. SCHULZE-ROBBECKE, R., A. WEBER & R. FISCHEDER. 1991. Comparison of decontamination methods for the isolation of mycobacteria from drinking water samples. J. Microbiol. Methods 14:177. 14. ANDREW, P.W. & G.J. BOULNOIS. 1990. Early days in the use of DNA probes for Mycobacterium tuberculosis and Mycobacterium avium complexes. In A.J.L. Macario & E.C. de Macario, eds. Gene Probes for Bacteria. Academic Press, San Diego, Calif. 15. ROGALL, T., T. FLOHR & E.C. BOTTGER. 1990. Differentiation of Mycobacterium species by direct sequencing of amplified DNA. J. Gen. Microbiol. 136:1915. 6. Bibliography TSUKAMURA, M. 1981. A review of the methods of identification and differentiation of mycobacteria. Rev. Infect. Dis. 3:841. GOOD, R.C. 1985. Opportunistic pathogens in the genus Mycobacterium. Annu. Rev. Microbiol. 39:347. ICHIYAMA, S. & K. SHIMOKATA. 1988. The isolation of Mycobacterium avium complex from soil, water, and dusts. Microbiol. Immunol. 32:733. BROADLEY, S.J., P.A. JENKINS, J.R. FURR & A.D. RUSSELL. 1991. Anti-mycobacterial activity of biocides. Lett. Appl. Microbiol. 13:118. FISCHEDER, R., R. SCHULZE-ROBBECKE & A. WEBER. 1991. Occurrence of mycobacteria in drinking water samples. Zentralbl. Hyg. Umweltmed. 192:154. JENKINS, P. A. 1991. Mycobacteria in the environment. J. Appl. Bacteriol. 70:137. SCHULZE-ROBBECKE, R., B. JANNING & R. FISCHEDER. 1992. Occurrence of mycobacteria in biofilm samples. Tubercle Lung Dis. 73:141. COLLINS, J. & M. YATES. 1994. Mycobacteria in water. J. Appl. Bacteriol. 1984. 57:193. JENSEN, P.A. 1997. Airborne Mycobacterium spp. In C.J. Hurst, G.R. Knudsen, M.J. McInerney, L.D. Stetzenbach & M.V. Walter, eds. Manual of Environmental Microbiology. American Soc. Microbiology Press, Washington, D.C. 9510 DETECTION OF ENTERIC VIRUSES*#(80) 9510 A. Introduction 1. Occurrence Viruses excreted with feces or urine from any species of animal may pollute water. Especially numerous, and of particular importance to health, are the viruses that infect the gastrointestinal tract of man and are excreted with the feces of infected individuals. These viruses are transmitted most frequently from person to person by the fecal-oral route. However, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater they also are present in domestic sewage which, after various degrees of treatment, is discharged to either surface waters or the land. Consequently, enteric viruses may be present in sewage-contaminated surface and ground waters that are used as sources of drinking water. The viruses known to be excreted in relatively large numbers with feces include polioviruses, coxsackieviruses, echoviruses, and other enteroviruses, adenoviruses, reoviruses, rotaviruses, the hepatitis A (infectious hepatitis) virus(es), and the Norwalk-type agents that can cause acute infectious nonbacterial gastroenteritis. With the possible exception of hepatitis A, each group or subgroup consists of a number of different serological types; thus more than 100 different human enteric viruses are recognized.1-4 In temperate climates enteroviruses occur at peak levels in sewage during the late summer and early fall. However, hepatitis A virus (HAV), Norwalk-type viruses, and rotaviruses may be important exceptions because the incidence of the diseases due to these viruses increases in the colder months. Quantitative information on seasonal patterns of occurrence in water and wastewater of these latter viruses is lacking because they cannot be assayed readily with conventional cell culture techniques. The Norwalk-type viruses have not been cultivated in any cell cultures, although immunochemical assay methods have been developed to detect them as antigens.5,6 Human rotaviruses and HAV have been cultivated recently in cell cultures, but the techniques are difficult and require concomitant use of immunoassays such as immunofluorescence to detect virus growth or gene probes.7-11 Viruses are not normal flora in the intestinal tract; they are excreted only by infected individuals, mostly infants and young children. Infection rates vary considerably from area to area, depending on sanitary and socioeconomic conditions. Viruses usually are excreted in numbers several orders of magnitude lower than those of coliform bacteria. Because enteric viruses multiply only within living, susceptible cells, their numbers cannot increase in sewage. Sewage treatment, dilution, natural inactivation, and water treatment further reduce viral numbers. Thus, although large outbreaks of waterborne viral disease may occur when massive sewage contamination of a water supply takes place,12 waterborne transmission of viral infection and disease in technologically advanced nations depends on whether minimal quantities of viruses are capable of producing infections. It has been demonstrated that infection can be produced experimentally by a very few virus units,13 although the risk of infection increases with increasing ingested doses.14 The risk of infection incurred by an individual in a community with a water supply containing a very few virus units has not been determined. Risk analysis has suggested that significant risk of infection could result from low numbers of enteric viruses present in a drinking water supply.15 The percentage of individuals who develop clinical illness may be as low as 1% for poliovirus and as great as 97% for hepatitis A.15 Most recognized waterborne virus disease outbreaks in the U.S. have been caused by obvious sewage contamination of untreated or inadequately treated private and semipublic water supplies. Virus disease outbreaks in community water supply systems usually are caused by contamination through the distribution system.16 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. Testing for Viruses The routine examination of water and wastewater for enteric viruses is not recommended now. However, in special circumstances such as wastewater reclamation, disease outbreaks, or special research studies, it may be prudent or essential to conduct virus testing. Such testing should be done only by competent and specially trained water virologists having adequate facilities. Laboratories planning to concentrate viruses from water and wastewater should do so with the clear understanding that the available methodology has important limitations.17 Even the most current methods for concentrating viruses from water still are being researched and continue to be modified and improved. The efficiency of a virus concentration method may vary widely depending on water quality. Furthermore, none of the available virus detection methods have been tested adequately with representatives from all of the virus groups of public health importance. Most virus concentration methods have achieved adequate virus recoveries with water or wastewater samples that have been contaminated experimentally with known quantities of a few specific enteric viruses. Although method effectiveness in field trials is difficult to evaluate, some virus concentration methods have been used successfully to recover naturally occurring enteric viruses. Some of these methods require large equipment for sample processing and virus assay and identification procedures usually require cell culture and related virology laboratory facilities. Detecting viruses in water through recovery of infectious virus requires three general steps: (a) collecting a representative sample, (b) concentrating the viruses in the sample, and (c) identifying and estimating quantities of the concentrated viruses. Particular problems associated with the detection of viruses of public health interest in the aquatic environment are: (a) the small size of virus particles (about 20 to 100 nm in diameter), (b) the low virus concentrations in water and the variability in amounts and types that may be present, (c) the inherent instability of viruses as biological entities, (d) the various dissolved and suspended materials in water and wastewater that interfere with virus detection procedures, and (e) the present limitations of virus estimation and identification methods. 3. Selection of Concentration Method The densities of enteric viruses in water and wastewater usually are so low that virus concentration is necessary, except possibly for raw sewage in certain areas or seasons.18 Numerous methods for concentrating waterborne enteric viruses have been proposed, tested under laboratory conditions with experimentally contaminated samples, and in some cases used to detect viruses under field conditions.19,20 Virus concentration methods often are capable of processing only limited volumes of water of a given quality. In selecting a virus concentration method consider the probable virus density, the volume limitations of the concentration method for that type of water, and the presence of interfering constituents. A sample volume less than 1 L and possibly as small as a few milliliters may suffice for recovery of viruses from raw or primary treated sewage. For drinking water and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater other relatively nonpolluted waters, the virus levels are likely to be so low that hundreds or perhaps thousands of liters must be sampled to increase the probability of virus detection. Three different techniques used to concentrate viruses from water are described herein: adsorption to and elution from microporous filters (Methods B and C); aluminum hydroxide adsorption-precipitation (Method D); and polyethylene glycol (PEG) hydroextraction-dialysis (Method E).19,20 A separate technique (Method F) for recovering viruses from solids in small volumes of water also is described. Virus concentration by adsorption to and elution from microporous filters can be used for both small volumes of wastewater and large volumes of natural and finished waters. The aluminum hydroxide adsorption-precipitation and PEG hydroextraction-dialysis methods are impractical for processing large fluid volumes. However, they are suitable for concentrating viruses from wastewater or other waters having relatively high virus densities and for second-step concentration (reconcentration) of viruses in primary eluates obtained by processing large sample volumes through microporous filters. 4. Recovery Efficiencies In examining a particular water include a preliminary evaluation of virus recovery efficiency. To do this add a known quantity of one or more test virus types to the required volume of sample, process the sample by the concentration method, and assay the concentrate for test viruses to determine virus recovery efficiency. Ideally, such seeded samples should be used whenever field samples are processed. If seeded samples are used concurrently with field samples, take appropriate steps, including disinfection and sterilization and the use of aseptic technique, to prevent accidental contamination of samples. 5. References 1. RAO, V.C. & J.M. MELNICK. 1986. Environmental Virology. American Soc. Microbiology, Washington, D.C. 2. FEACHEM, R.G., D.J. BRADLEY, H. GARELICK & D.D. MARA. 1983. Sanitation and Disease. Health Aspects of Excreta and Wastewater Management. John Wiley & Sons, New York, N.Y. 3. WILLIAMS, F.P. & E.W. AKIN. 1986. Waterborne gastroenteritis. J. Amer. Water Works Assoc. 78:34. 4. FEACHEM, R., H. GARELICK & J. SLADE. 1981. Enteroviruses in the environment. Trop. Dis. Bull. 78:185. 5. BLACKLOW, N.R. & G. CUKOR. 1980. Viral gastroenteritis agents, Chap. 90 in E.H. Lennette, A. Balows, W.J. Hausler, Jr. & J.P. Truant, eds. Manual of Clinical Microbiology, 3rd ed. American Soc. Microbiology, Washington, D.C. 6. KAPIKIAN, A.Z., R.H. YOLKEN, H.B. GREENBERG, R.G. WYATT, A.R. KALICA, R.M. CHANOCK & H.W. KIM. 1979. Gastroenteritis viruses. In E.H. Lennette & N.J. Schmidt, eds. Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infections. American Public Health Assoc., Washington, D.C. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 7. SOBSEY, M.D., S.E. OGLESBEE, D.A. WAIT & A.I. CUENEA. 1984. Detection of hepatitis A in drinking water. Water Sci. Technol. 17: 23. 8. SMITH, E.M. & C.P. GERBA. 1984. Development of a method for detection of human rotavirus in water. Appl. Environ. Microbiol. 43: 1440. 9. SATO, K., Y. INABA, T. SHINOZAKI, R. FUJII & M. MATUMOTO. 1981. Isolation of human rotavirus in cell cultures. Arch. Virol. 69:155. 10. HEJKAL, T.W., E.M. SMITH & C.P. GERBA. 1984. Seasonal occurrence of rotavirus in sewage. Appl. Environ. Microbiol. 47:588. 11. JIANG, X., M.K. ESTES & T.G. METCALF. 1987. Detection of hepatitis A virus by hybridization with single-stranded RNA probes. Appl. Environ. Microbiol. 53:2487. 12. MELNICK, J.L. 1957. A water-borne urban epidemic of hepatitis. In Hepatitis Frontiers. Little, Brown & Co., Boston, Mass. 13. WARD, R.L., D.I. BERNSTEIN & E.C. YOUNG. 1986. Human rotavirus studies in volunteers: Determination of infectious dose and serological response to infection. J. Infect. Dis. 154:871. 14. AKIN, E. 1981. A review of infective dose data for enteroviruses and other enteric microorganisms in human subjects. In Microbial Health Considerations of Soil Disposal of Domestic Wastewaters. EPA-600/9-83-017, U.S. Environmental Protection Agency, Washington, D.C. 15. GERBA, C.P. & C.N. HAAS. 1988. Assessment of risks associated with enteric viruses in contaminated drinking water. In J.J. Lichtenberg, J.A. Winter, C.I. Weber & L. Frankin, eds. Chemical and Biological Characterization of Sludges, Sediments, Dredge Spoils, and Drilling Muds. ASTM STP 976. American Soc. Testing & Materials, Philadelphia, Pa. 16. CRAUN, G.F. 1986. Waterborne Disease in the United States. CRC Press, Boca Raton, Fla. 17. SOBSEY, M.D. 1982. Quality of currently available methodology for monitoring viruses in the environment. Environ. Internat. 7:39. 18. BURAS, N. 1976. Concentration of enteric viruses in wastewater and effluent: A two year survey. Water Res. 10:295. 19. SOBSEY, M.D. 1976. Methods for detecting enteric viruses in water and wastewater. In G. Berg, H.L. Bodily, E.H. Lennette, J.L. Melnick & T.G. Metcalf, eds. Viruses in Water. American Public Health Assoc., Washington, D.C. 20. GERBA, C.P. & S.M. GOYAL. 1982. Methods in Environmental Virology. Marcel Dekker, New York. 9510 B. Virus Concentration from Small Sample Volumes by Adsorption to © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and Elution from Microporous Filters 1. General Discussion Viruses can be concentrated from aqueous samples by reversibly adsorbing them to microporous filters and then eluting them from the filters in a small liquid volume.1 The virus-containing sample is pressure-filtered through microporous filters having large surface areas to which viruses adsorb, presumably by both electrostatic and hydrophobic interactions.2 Two general types of adsorbent filters are available: electronegative (negative surface charge) and electropositive (positive surface charge). The former filters are composed of either cellulose esters or fiberglass with organic resin binders. They adsorb viruses most efficiently in the presence of multivalent cations such as Al3+ and Mg2+ and/or at low pH, usually pH 3.5. The latter filters are composed of either fiberglass or cellulose and a positively charged organic, polymeric resin. They adsorb viruses efficiently over a wide pH range without added polyvalent salts. If the sample is neutral or acidic, it can be processed with these filters without chemical conditioning. Electropositive filters have given virus recoveries comparable to those with electronegative filters.3-5 They have been used in field studies,6,7 and were evaluated with a variety of virus types8-13 and waters. Adsorbed viruses usually are eluted from the surfaces of microporous filters by pressure-filtering a small volume of eluent fluid through the filters in situ. The eluent is either a slightly alkaline proteinaceous fluid such as beef extract or a more alkaline buffer such as glycine-NaOH, pH 10.5 to 11.5. If glycine-NaOH is used as eluent, preferably use pH 10.5 because of the greater likelihood of virus inactivation at the higher pH.14,15 Microporous filter methods suffer from three main limitations. Sample suspended matter tends to clog the adsorbent filter, thereby limiting the volume that can be processed and possibly interfering with the elution process.16 Dissolved and colloidal organic matter in some waters can interfere with virus adsorption to filters, presumably by competing with viruses for adsorption sites,17-19 and they also can interfere with virus elution. Finally, viruses adsorbed to suspended matter may be removed in any clarification procedure applied before virus adsorption. These solids-associated viruses are lost from the sample unless special efforts are made to recover the solids and process them for viruses.16 A method for recovering solids-associated viruses from small volumes of water and wastewater is given in Section 9510F. Despite these limitations, virus concentration by adsorption to and elution from microporous filters is a most promising technique for detecting viruses. 2. Equipment and Apparatus a. Adsorbent filter holder, 47-, 90-, or 142-mm diam, equipped with pressure relief valve. b. Pressure vessel, 12- or 20-L capacity. c. Positive pressure source up to about 400 kPa with regulator: laboratory air line, air pump, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater or cylinder of compressed air or nitrogen gas. d. Autoclavable vinyl plastic tubing with plastic or metal connectors (quick-disconnect type), for connecting positive pressure source, pressure vessel, and filter holder in series. e. pH meter. f. Beakers, 50- to 500-mL. g. Laboratory balance. h. Graduated cylinders, 25- to 100-mL. i. Pipets, 1-, 5-, and 10-mL. 3. Materials a. Electronegative virus adsorbent filter: Use either: 1) Cellulose nitrate filter, 0.45-µm porosity.*#(81) 2) Fiberglass-acrylic resin filter, 0.45-µm porosity.†#(82) Filter media available commercially only as flat sheets can be cut to the desired disk diameter with scissors. b. Electropositive virus adsorbent filter: Use either: 1) Surface modified cellulose and filter aid disk depth-filter.‡#(83) 2) Surface modified cellulose and filter aid thin-sheet medium, 0.20-µm porosity.§#(84) c. Prefilter: Use one or more cellulose nitrate or fiberglass-acrylic resin filters or equivalent, with porosities greater than 0.45 µm to prevent clogging of the virus adsorbent filter by suspended matter. Place prefilters on top of the 0.45-µm-porosity virus adsorbent filter in the same filter holder. 4. Reagents a. Hydrochloric acid, HCl, 0.1, 1.0, and 10N. b. Sodium hydroxide, NaOH, 0.1, 1.0, and 10N. c. Aluminum chloride, AlCl3⋅6H2O, 0.15N, or magnesium chloride, MgCl2⋅6H2O, 5N (necessary only for electronegative filters). d. Sodium thiosulfate, Na2S2O3⋅5H2O, 0.5% (w/v). e. Sodium chloride, 0.14N, pH 3.5: Dissolve 8.18 g in 1 L reagent-grade water and adjust to pH 3.5 with HCl (necessary only for electronegative filters). f. Virus eluent: Use either: 1) Glycine-NaOH, pH 10.5 or 11.5: Prepare 0.05M glycine solution, autoclave, and adjust to pH 10.5 or 11.5 with 1 to 10N NaOH. Add phenol red, 0.0005%, as a pH indicator. 2) Beef extract, 3%, pH 9.0: Dissolve 30 g beef extract paste or 24 g beef extract powder in 1000 mL reagent-grade water, adjust to pH 9.0 with 1 to 10N NaOH, and sterilize by autoclaving. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater g. Glycine-HCl, pH 1.5: Prepare 0.05M glycine solution, autoclave, and adjust to pH 1.5 with 1 to 10N HCl. Add phenol red, 0.0005%, as a pH indicator. h. Nutrient broth, 10X, pH 7.5: Dissolve 8.0 g nutrient broth in 90 mL reagent-grade water, adjust to pH 7.5, dilute to 100 mL with reagent-grade water, and sterilize by autoclaving. i. Antibiotics: Use either: 1) Penicillin-streptomycin, 10X: Contains 5000 IU penicillin/mL and 5000 µg streptomycin/mL. Use commercially available form or prepare by dissolving powdered sodium or potassium penicillin-G and streptomycin sulfate in reagent-grade water and sterilizing by filtration. Store frozen. 2) Gentamycin-kanamycin, 100X: Contains 5000 µg/mL each of gentamycin (base) and kanamycin (base). Prepare by combining aseptically equal volumes of commercially available sterile gentamycin and kanamycin solutions, 10 000 µg/mL, respectively, or by dissolving powdered gentamycin sulfate and kanamycin sulfate in reagent-grade water and sterilizing by filtration. Store refrigerated or frozen. j. Hanks balanced salt solution, 10X: Use commercially available form or prepare following a standard protocol.20 k. Sodium hypochlorite, 5.25% available chlorine (household bleach). 5. Procedure a. Sterilization of apparatus, materials, and reagents: Most reagents, virus adsorbent filters, filter holders, tubing, and labware can be sterilized by autoclaving or made virus-free by streaming steam. To sterilize filters load into their holders; if several filters are to be placed in one holder, place filter with smallest porosity on the bottom with progressively larger filters on top. Do not use an automatic drying cycle when autoclaving virus adsorbent filters. Sterilize apparatus and material that cannot be autoclaved or treated with streaming steam by treating with 10-mg/L free chlorine solution, pH 7.0, for 30 min and rinse or flush with 50-mg/ L sterile Na2S2O3 solution. Do not treat adsorbent filters with chlorine. Use aseptic technique during all virus concentration operations to prevent extraneous microbial contamination. b. Sample size and choice of filter size: Sample size and, hence, filter diameter depend partly on water quality and the probable virus concentration. Single-stage microporous filter adsorption-elution methods have been used to recover viruses from 100 mL raw sewage on 47-mm-diam filters21 and from 3.8 to 4.6 L secondary and tertiary sewage effluent on 90- or 142-mm-diam filters.18,21,22 Based on the diameter and solids-holding characteristics of the filters, the scale and volume capacity of the apparatus and materials, and the quality of the samples, the practical limits for sample size are 20, 8, and 2 L for 142-, 90-, and 47-mm-diam filters, respectively. c. Choice of filter type: Virus adsorption to electropositive filters decreases above pH 8 and pH adjustment below this value may be necessary for optimal virus adsorption.4 Virus recovery © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater from raw sewage may be less than with electronegative filters.10 d. Sample collection and storage: Collect samples aseptically in sterile containers. If they contain residual chlorine, immediately add Na2S2O3 solution to give a final concentration of 50 mg/L. Process samples as soon as possible after collection; do not hold samples for more than 2 h at up to 25°C or 48 h at 2 to 10°C. Do not freeze samples unless they cannot be processed within 48 h; then freeze and store at −70°C or less. e. Sample processing of electronegative filters: Adjust sample to pH 3.5 and 0.0015N AlCl3 or to between pH 6.0 and 3.5 and 0.1N MgCl2. Make sample adjustments either in a pressure vessel or in another appropriate container. Mix sample vigorously during addition of 1.0 or 0.1N HCl and AlCl3 solution (1 part solution to 100 parts sample) or MgCl2 solution (1 part solution to 50 parts sample). Because AlCl3 is an acid salt, it may decrease sample pH slightly. Do not let sample pH fall below 3.0. Place sample in a pressure vessel connected to a source of positive pressure and connect pressure vessel outlet to inlet of virus adsorbent filter holder. With pressure relief valve on filter holder opened, apply a slight positive pressure to purge air from filter holder. When sample just begins to flow from pressure relief valve, quickly close valve and continue filtration at a rate not exceeding 28 mL/min/cm2 of filter area (about 130, 250, and 4000 mL/min for 47-, 90-, and 142-mm-diam filters, respectively). After filtering entire sample let positive pressure source purge excess fluid from filter holder. Wash filters with 0.14N NaCl to remove excess Al3+ or Mg2+ from virus adsorbent filter. Use about 1.5 mL NaCl solution/cm2 filter area (25, 100, and 240 mL for 47-, 90-, and 142-mm-diam filters, respectively). Place wash solution in a pressure vessel connected to filter holder inlet, use positive pressure to filter solution through virus adsorbent filter, discard filtrate, and let positive pressure purge virus adsorbent filter of excess wash solution. Elute viruses from filters with a recommended eluent. Use about 0.45 mL eluent/cm2 filter surface area (about 7.5, 28, and 71 mL for 47-, 90-, and 142-mm-diam filters, respectively). With pressure relief valve on filter holder open, add eluent to filter holder so that it completely covers filter surface. When eluent begins to discharge from pressure relief valve, quickly close valve. If pH 11.5 glycine-NaOH is the eluent, place a sterile beaker under filter outlet and apply positive pressure so that filtrate flows slowly from filter holder outlet. Collect filtrate in sterile beaker and, when filtrate no longer flows, slowly increase pressure to force retained fluid from filters. Quickly check eluate (filtrate) pH. If it is less than 11.0, elute with additional pH 11.5 glycine-NaOH until an eluate with a pH ≥ 11.0 is obtained. Immediately after checking pH, adjust eluate to a pH between 9.5 and 7.5 with pH 1.5 glycine-HCl or 0.1N HCl while mixing vigorously. Complete elution and eluate pH adjustment to 7.5 to 9.5 in 5 min or less to avoid the possibility of appreciable virus inactivation. If pH 10.5 glycine-NaOH is the eluent, proceed as with pH 11.5 glycine-NaOH, but pass the eluate through the filters a total of five times. For each elution, collect the filtrate, readjust to pH © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 10.5 with 1.0 or 0.1N NaOH, and then pass through the filter. After the fifth elution, adjust filtrate to pH 7.4 with glycine-HCl, pH 1.5, or 0.1N HCl. If 3% beef extract, pH 9.0, is the eluent, place a sterile beaker under filter outlet, apply a slight positive pressure to eluent-containing filter holder so that filtrate flows slowly from the outlet, and collect filtrate. Slowly increase pressure to force additional retained fluid from filters. Measure eluate volume and add 1/10 of the measured volume each of penicillin-streptomycin or gentamycin-kanamycin, Hanks balanced salt solution, and 10X nutrient broth (add last item to glycine eluates only). Adjust sample to pH 7.4 with glycine-HCl or 0.1N HCl while mixing vigorously. Store at either 4 or −70°C, depending on the time until virus assay. Maximum storage at 4°C is 48 h. f. Processing of electropositive filters: Processing for electropositive filters is identical to that for electronegative filters except that addition of Al3+ and Mg2+ and sample pH adjustments are unnecessary; because Al3+ and Mg2+ are not added, it is not necessary to wash filters with 0.14N NaOH before elution. If sample pH is greater than 8.0, adjust to less than pH 8 by adding 1.0 or 0.1N HCl. 6. References 1. FARRAH, S.R., C.P. GERBA, C. WALLIS & J.L. MELNICK. 1976. Concentration of viruses from large volumes of tapwater using pleated membrane filters. Appl. Environ. Microbiol. 31:221. 2. FARRAH, S.R., D.O. SHAH & L.O. INGRAM. 1981. Effects of chaotropic and antichaotropic agents on the elution of poliovirus adsorbed to membrane filters. Proc. Nat. Acad. Sci. U.S. 18:1229. 3. SOBSEY, M.D. & B.L. JONES. 1979. Concentration of poliovirus from tap water using positively charged microporous filters. Appl. Environ. Microbiol. 37:588. 4. SOBSEY, M.D. & J.S. GLASS. 1980. Poliovirus concentration from tap water with electropositive adsorbent filters. Appl. Environ. Microbiol. 40:201. 5. SOBSEY, M.D., R.S. MOORE & J.S. GLASS. 1981. Evaluating adsorbent filter performance for enteric virus concentrations in tap water. J. Amer. Water Works Assoc. 73:542. 6. CHANG, L.T., S.R. FARRAH & G. BITTON. 1981. Positively charged filters for virus recovery from wastewater treatment plant effluents. Appl. Environ. Microbiol. 42:921. 7. HEJKAL, T.W., B. KESWICK, R.L. LABELLE, C.P. GERBA, Y. SANCHEZ, G. DREESMAN, B. HAFKIN & J.L. MELNICK. 1982. Viruses in a community water supply associated with an outbreak of gastroenteritis and infectious hepatitis. J. Amer. Water Works Assoc. 74:318. 8. SCHLAAK, M., E. TISCHER & J.M. LOPEZ. 1983. Evaluation of current procedures for the concentration of viruses in water. Zentralbl. Bakteriol. Microbiol. Hyg. I. Abt. Orig. B 177:127. 9. GUTTMAN-BASS, N. & R. ARMON. 1983. Concentration of Simian rotavirus SA-11 from © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. tap water by membrane filtration and organic flocculation. Appl. Environ. Microbiol. 45:850. ROSE, J.B., S.N. SINGH, C.P. GERBA & L.M. KELLEY. 1984. Comparison of microporous filters for concentration of viruses from wastewater. Appl. Environ. Microbiol. 45:989. RAPHAEL, R.A., S.A. SATTAR & V.S. SPRINGTHROPE. 1985. Rotavirus concentration from raw water using positively charged filters. J. Virol. Methods 11:131. NUPEN, E.M. & B.W. BATEMAN. 1985. The recovery of viruses from drinking water by means of an in-line electropositive filter. Water Sci. Technol. 17:63. TORANZOS, G.A. & C.P. GERBA. 1989. An improved method for the concentration of rotaviruses from large volumes of water. J. Virol. Methods 24:131. SOBSEY, M.D., J.S. GLASS, R.J. CARRICK, R.R. JACOBS & W.A. RUTALA. 1980. Evaluation of the tentative standard method for enteric virus concentration from large volumes of tap water. J. Amer. Water Works Assoc. 72:292. SOBSEY, M.D., J.S. GLASS, R.R. JACOBS & W.A. RUTALA. 1980. Modification of the tentative standard method for improved virus recovery efficiency. J. Amer. Water Works Assoc. 72:350. WELLINGS, F.M., A.L. LEWIS & C.W. MOUNTAIN. 1976. Demonstration of solids-associated virus in wastewater and sludge. Appl. Environ. Microbiol. 31:354. FARRAH, S.R., S.M. GOYAL, C.P. GERBA, C. WALLIS & P.T.B. SHAFFER. 1976. Characteristics of humic acid and organic compounds concentrated from tapwater using the aquella virus concentrator. Water Res. 10:897. WALLIS, C. & J.L. MELNICK. 1967. Concentration of viruses from sewage by adsorption on Millipore membranes. Bull. World Health Org. 36:219. SOBSEY, M.D., C. WALLIS, M. HENDERSON & J.L. MELNICK. 1973. Concentration of enteroviruses from large volumes of water. Appl. Microbiol. 26:529. SCHMIDT, N.J. 1979. Tissue culture technics for diagnostic virology. In E.H. Lennette & N.J. Schmidt, eds. Diagnostic Procedures for Viral and Rickettsial Infections, 5th ed. American Public Health Assoc., Washington, D.C. RAO, V.C., U. CHANDORKAR, N.U. RAO, P. KUMARAN & S.B. LAKHE. 1972. A simple method for concentrating and detecting viruses in wastewater. Water Res. 6:1565. GERBA, C.P., S.R. FARRAH, S.M. GOYAL, C. WALLIS & J.L. MELNICK. 1978. Concentration of enteroviruses from large volumes of tap water, treated sewage, and seawater. Appl. Environ. Microbiol. 35:540. 9510 C. Virus Concentration from Large Sample Volumes by Adsorption to and Elution from Microporous Filters 1. General Discussion © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater This section describes a two-stage process for concentrating viruses from large sample volumes. Viruses in eluate volumes too large to be conveniently and economically assayed directly in cell cultures, such as those obtained from processing large volumes of water through cartridge or large disk filters, can be concentrated further (reconcentrated) by several alternative methods. Viruses in proteinaceous eluates can be reconcentrated by either ‘‘organic flocculation,’’1,2 aluminum hydroxide adsorption-precipitation (Section 9510D), or polyethylene glycol hydroextraction-dialysis (Section 9510E). These reconcentration techniques can be used for both proteinaceous and organic buffer eluates from all types of water. Organic flocculation, now used widely, involves precipitating viruses by acidifying eluates to pH 3.5, recovering the precipitate by centrifugation, and then resuspending it in a small volume of alkaline buffer.1 Additionally, viruses in nonproteinaceous eluates such as glycine-NaOH can be reconcentrated by adsorption to and elution from small microporous filters. The eluate is adjusted to pH and ionic conditions for optimum virus adsorption, filtered through a secondary adsorbent, and adsorbed viruses are eluted with a small volume of eluent. This procedure can be used only for reconcentrating primary eluates obtained from processing drinking water and other highly finished waters because of potential interfering substances likely to be present in primary eluates from natural and less finished waters. Figure 9510:1 shows the alternative microporous filter adsorption-elution and reconcentration methods. For general information on microporous filter techniques, see Section 9510B.1. 2. Equipment and Apparatus a. Apparatus for first-stage concentration (Figure 9510:2): 1) First-stage virus adsorbent filter holder. 2) Chemical additive system. Use either: a) Fluid proportioner with four feed pumps (quadraplex) and a mixing chamber.*#(85) b) Venturi-type proportioning injector†#(86) with plastic or metal connectors (quick-disconnect type) and a length of vinyl tubing for the chemical feed line.3 To feed two separate additives, attach a ‘‘Y’’ or ‘‘T’’ connector and two lengths of vinyl tubing to the chemical feed port, or alternatively, use two separate proportioning injectors. It may be necessary to use a bypass system with the injector to prevent loss of chemical feed due to back pressure from the water line.4 This bypass system consists of ‘‘T’’ pipe fittings on the injector inlet and outlet ports connected by a length of flexible hose with an in-line shut-off/control valve (see Figure 9510:2). Proportioning injectors available commercially will process water at flow rates of 3 to 33 L/min with water-to-chemical feed ratios between 10 to 1 and 1110 to 1. Select equipment and operating conditions providing a water-to-chemical feed ratio of 100 to 1. 3) Water flow meter. 4) Pressure gauge, 0 to 400 kPa. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5) Vinyl plastic tubing, autoclavable, with plastic or metal connectors (quick-disconnect type). 6) Pressure relief valve (optional). 7) Carboys, 20- to 50-L, or similar containers. 8) Positive pressure source up to 400 kPa with regulator: laboratory air line, positive pressure pump, or cylinder of compressed air or nitrogen gas. 9) Pump (if source water is not under pressure). b. pH meter. c. Laboratory balance. d. Beakers, 2- or 4-L. e. Pressure vessel, 4-L. f. Graduated cylinders, 1- and 2-L. g. Pipets, 1-, 5-, and 10-mL. h. Centrifuge with rotor and buckets for 250- to 500-mL-capacity bottles.‡#(87) i. Centrifuge bottles, 250- to 500-mL. 3. Materials a. First-stage electronegative virus adsorbent filters: Use one of the following: 1) 293-mm-diam, 8.0- and 1.2-µm-porosity cellulose nitrate filter series.§#(88) 2) 17.8-cm-long, 8.0-µm-porosity fiberglass-epoxy filter tube.i#(89) 3) 25.4-cm-long, 0.25- or 0.45-µm-porosity fiberglass-acrylic resin pleated filter cartridge.##(90) b. Second-stage electronegative virus adsorbent filters: 47-mm-diam, 3.0-, 0.45-, and 0.25-µm-porosity fiberglass-acrylic resin filter series. Use to reconcentrate highly finished water samples only.##(91) c. First-stage electropositive adsorbent filters: Use one of the following: 1) 293-mm-diam surface modified cellulose and filter aid filters.**#(92) 2) 25-cm-long, 0.20-µm-porosity surface modified thin-sheet media pleated filter cartridge.††#(93) 4. Reagents a. Hydrochloric acid, HCl, 0.06, 1,‡‡#(94) and 6N. b. Sodium hydroxide, NaOH, 10N. c. Aluminum chloride, AlCl3⋅6H2O, 0.15 and 6N.‡‡ d. Magnesium chloride, MgCl2⋅6H2O, 10N.‡‡ © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater e. Sodium thiosulfate, Na2S2O3⋅5H2O, 0.5% (w/v). f. Sodium hypochlorite, 5.25% available chlorine (household bleach). g. Eluent: Use either: 1) Glycine-NaOH, pH 10.5 or 11.5: See Section 9510B.4 f1). Use within 2 h of pH adjustment. 2) Beef extract, 3%, pH 9.0.§§#(95) See Section 9510B.4 f2). h. Eluate neutralizing solution: Use either: 1) Glycine-HCl, pH 1.5: Prepare 0.05M glycine solution and adjust to pH 1.5 with 6N HCl. Add phenol red, 0.0005%, as a pH indicator. Use within 2 h of pH adjustment. 2) HCl, 1.0N. i. Nutrient broth, 10X, pH 7.5: Dissolve 8.0 g nutrient broth in 90 mL distilled water, adjust to pH 7.5 with 10N NaOH, dilute to 100 mL with distilled water, and sterilize by autoclaving. j. Disodium phosphate, 0.45N: Dissolve 40.2 g Na2HPO4⋅7H2O in 1 L distilled water and sterilize by autoclaving. k. Antibiotics: See Section 9510B.4i. l. Sodium chloride, 0.14N: Dissolve 8.18 g NaCl in 1 L distilled water (necessary only with electronegative filters). m. Hanks balanced salt solution, 10X: See Section 9510B.4 j. 5. Procedure When using electronegative filters, follow ¶s a–f below for production of primary eluate. When using electropositive filters, first see ¶ g for procedural modifications. a. Sterilization of apparatus, materials, and reagents: See Section 9510B.5a. b. Sample size: For drinking water use a minimum sample of 400 L, although 2000 L or more may have to be processed to detect viruses at a concentration of 1 to 2 infectious units/400 L. c. Preparation of feed solutions for electronegative filters: Use an HCl additive solution to adjust sample pH to 3.5 for virus adsorption to filters. If acidification to pH 3.5 is inadequate for obtaining maximum virus adsorption, add either AlCl3 or MgCl2 solution. When only HCl is used, prepare additive solution as follows: Determine concentration of HCl additive solution by titrating a 1-L sample of dechlorinated water to pH 3.5 with 0.06N HCl and noting volume required. The volume, in milliliters, of titrant required is equal to the volume of 6N HCl needed/L distilled water for making the additive solution. Make at least 5 L additive solution for 400 L of sample. When AlCl3 is used to enhance virus adsorption use pH 3.5 and a final concentration of added AlCl3 of 0.0015N. Because AlCl3 is an acid salt, titrate a 1-L sample to about pH 4.0 with 0.06N HCl, add AlCl3 to a concentration of 0.0015N and continue titration to pH 3.5, noting © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater volume of titrant used. Prepare additive solution by adding titrant volume (mL) of 6.0N HCl/L of 0.15N AlCl3. When MgCl2 is used to enhance virus adsorption, use a pH between 3.5 and 6.0 and a final concentration of added MgCl2 of 0.1N. To prepare additive solution titrate a 1-L sample to desired pH with 0.06N HCl as previously described and note volume of titrant used. Add the titrant volume of 6.0N HCl/L 10N MgCl2 to make the additive solution. d. Preparation of chemical additive system: 1) When using a fluid proportioner, operate at a pressure of 100 to 700 kPa and a water flow rate of 4 to 40 L/min. Adjust each of the four chemical additive pumps of the proportioner for a ratio of 1 to 200 (1 part chemical additive to 200 parts water). Use two pumps, operating reciprocally, for each additive so that the overall dilution for each additive is 1 to 100. One additive is either HCl, HCl-AlCl3 or HCl-MgCl2; the other additive, 0.5% Na2S2O3, is needed only when processing samples containing chlorine. Place lines from the two pumps of each additive solution into the additive containers and manually operate the pump metering rods to fill feed lines and purge them of air. Connect fluid proportioner to source water and operate briefly without a virus adsorbent in place. Sample conditioned water from proportioner outlet and check pH. The pH should be 3.5 ± 0.3. When using a Venturi-type proportioning injector, connect injector assembly to water source and to adsorbent filter inlet, and place additive feed line(s) into additive container(s). Position valve on injector outlet to drain line position (away from adsorbent filter). Begin flow of sample. Adjust screw-operated control valve on chemical feed of proportioning injector until water collected from drain line is at the desired pH as measured with a pH meter. If Na2S2O3 is used to neutralize chlorine, check to insure that chlorine is absent. Connect virus concentrator assembly to source water by attaching concentrator inlet hose to valved outlet of a pressurized water source or to outlet of a water pump, the inlet of which has been placed in the source water. Operate for several minutes without a virus adsorbent in place to purge the unit of chlorine solution. Collect a sample from outlet of meter to insure absence of chlorine. e. First-stage concentration: After preparing concentration apparatus and additive solutions and checking conditioned water for proper pH and absence of chlorine, attach a virus adsorbent filter to outlet of chemical additive system. Attach water meter and effluent hose to virus adsorbent outlet. Record initial meter reading and add to this value the desired volume to be processed plus an additional 1 or 2% (to account for volume of either 1 or 2 additive solutions, respectively). This gives meter reading at which sampling is to be stopped. Turn on water and start a timer (or record starting time). Shortly after filtration begins collect a sample from filter outlet and check for absence of chlorine and for appropriate pH value. Also check flow rate. Do not use a flow rate above 40 L/min. Recheck pH and chlorine residual several times during sample processing, or monitor continuously. When desired volume has been processed, turn water off. Purge filter holder of excess water with positive pressure from an air or nitrogen gas source. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater f. Washing and virus elution: If AlCl3 or MgCl2 has been used, wash excess Al3+ or Mg2+ from filter with 4 L 0.14N NaCl. Omit washing if only HCl was used. Place wash solution in a 4-L pressure vessel and pass through filter with positive pressure. Purge filter of excess wash solution with positive pressure and discard entire filtrate. Using aseptic technique, elute virus from filter as soon as possible in the field or after returning to the laboratory. If filter holders with adsorbed viruses must be returned to the laboratory, seal filter holder openings, place filter holder in a sterile plastic bag, and chill. Use pH 10.5 or 11.5 glycine-NaOH or 3% beef extract, pH 9.0, to elute viruses from first-stage adsorbent filters. Because some viruses are inactivated when pH 11.5 glycine-NaOH is used, alternatively elute with pH 10.5 glycine-NaOH or 3% beef extract, pH 9.0.1,3,4 To elute, place eluent in a pressure vessel. Use minimum eluent volumes of 1 L and 300 mL for cartridge and 293-mm-diam disk filters, respectively. To elute with pH 11.5 glycine-NaOH, connect pressure vessel to inlet of filter holder and with pressure relief valve on filter holder open, apply a small positive pressure to the system so that eluent fills void volume of filter holder. When eluent begins to discharge from pressure relief valve, quickly close it. Filter remaining eluent slowly through filter within 1 to 2 min and collect filtrate (eluate) in a sterile 2or 4-L beaker. When filtrate no longer appears, slowly increase pressure to force additional fluid from filter. If using pH 11.5 glycine-NaOH eluent, immediately check filtrate pH and if it is less than 11.0, elute with 1 L more of pH 11.5 glycine-NaOH. Immediately after checking pH, adjust filtrate to a pH between 7.5 and 9.5 with pH 1.5 glycine-HCl while mixing vigorously. Complete elution and pH adjustment to 7.5 to 9.5 in 5 min or less to avoid possibility of appreciable virus inactivation. To elute with pH 10.5 glycine-NaOH, use either batch or continuous-flow eluent recirculation. For the batch method, begin elution as with pH 11.5 glycine-NaOH. Collect the filtrate, measure pH, and readjust to pH 10.5 with 1.0 or 0.1N NaOH while mixing vigorously. Then, using this eluate, elute filters four more times, readjusting filtrate to pH 10.5 before each elution. After the fifth elution, adjust filtrate to pH 7.4 with pH 1.5 glycine-HCl or 1.0N HCl while mixing vigorously. Alternatively, elute with pH 10.5 glycine-NaOH by continuous recirculation. Place eluent in a sterile beaker. Attach short lengths of sterile vinyl or rubber tubing to inlet and outlet openings of filter holder and place free ends of tubing in eluent beaker; slip midsection of filter inlet tubing into a peristaltic or roller pump. Open pressure relief valve on filter holder and operate pump at slow speed so that eluent fills void volume of filter holder. When eluent begins to discharge from pressure relief valve, quickly close it. Increase pump speed so that eluent recirculates through filter assembly and beaker at a minimum flow rate of 100 mL/min. After 5 min recirculation, remove filter inlet tube from beaker and pump remaining fluid from filter assembly. Connect filter inlet to positive pressure source to force additional eluent from filter. Adjust eluate to pH 7.4 with pH 1.5 glycine-HCl or 1.0N HCl while mixing vigorously. To elute with 3% beef extract, pH 9.0, follow the procedure described above for pH 11.5 glycine-NaOH. Adjust collected filtrate to pH 7.4 with pH 1.5 glycine-HCl or 1N HCl while © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater mixing vigorously. The 5 min time limit to complete elution with pH 11.5 glycine-NaOH is not necessary when beef extract is used. g. Sample processing of electropositive filters: Processing for electropositive filters is identical to that for electronegative filters except that addition of Al3+ and Mg2+ and sample pH adjustments are unnecessary; because Al3+ and Mg2+ are not added, it is not necessary to wash filters with 0.14N NaOH before elution. If the sample pH is greater than 8.0, adjust to less than pH 8 by adding 1.0 or 0.1N HCl. h. Reconcentration of primary eluates: Further concentrate (reconcentrate) viruses in primary eluates either by organic flocculation, Al(OH)3 adsorption-precipitation (Section 9510D), polyethylene glycol hydroextraction-dialysis (Section 9510E), or adsorption to and elution from microporous filters. The latter technique can be used only for glycine or other organic buffer eluates. To concentrate (reconcentrate) viruses in glycine eluates by adsorption to and elution from filters, adjust to pH 3.5 with pH 1.5 glycine-HCl and add AlCl3 to a final concentration of 0.0015N while mixing vigorously. Transfer sample to a 4-L pressure vessel. Filter through a 47-mm-diam 3.0-, 0.45-, and 0.25-µm-porosity fiberglass-acrylic resin filter series at a flow rate of no more than 130 mL/min and discard filtrate. Rinse filters with 25 mL 0.14N NaCl to remove excess Al3+. Pipet NaCl solution directly into filter inlet or place in a small pressure vessel connected to the inlet. Use positive pressure to pass NaCl solution through filter and discard filtrate. Elute adsorbed viruses from filter with 7-mL portions of either pH 10.5 or 11.5 glycine-NaOH or 3% beef extract, pH 9.0. Pipet 7 mL eluent directly into filter holder inlet or into a small pressure vessel connected to filter inlet and connect to a positive pressure source. Carefully apply positive pressure so that eluate flows slowly from filter outlet into a sterile container. When filtrate no longer flows from outlet, increase pressure to force retained fluid from filters. If using pH 11.5 glycine-NaOH, measure eluate pH and immediately adjust to pH between 7.5 and 9.5 with pH 1.5 glycine-HCl. Repeat this elution procedure with another 7-mL portion of pH 11.5 glycine-NaOH. Complete reconcentration within 5 min. If neither eluate portion had a final pH of 11.0 or more, repeat elution procedure with additional 7-mL portions of pH 11.5 glycine-NaOH until an eluate portion has a pH of at least 11.0. Combine all eluates. If using pH 10.5 glycine-NaOH, elute five successive times with 7-mL volumes of eluent. After each elution, readjust eluate to pH 10.5 with 0.1N NaOH while mixing vigorously. After the fifth elution, adjust eluate to pH 7.4 with pH 1.5 glycine-HCl or 0.1N HCl while mixing vigorously. If using 3% beef extract, pH 9.0, elute with two 7-mL volumes, combine filtrates, and adjust to pH 7.4 if necessary. Measure total eluate volume. For glycine eluates, add 1/10th the measured sample volume of 10X Hanks balanced salt solution and 10X nutrient broth. To all eluates add appropriate volumes of antibiotics (1/10th volume penicillin-streptomycin or 1/100th volume gentamycin-kanamycin, or both). Store at 4 or −70°C, depending on time until virus assay. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Further concentrate viruses in beef extract eluates by precipitation at pH 3.5 (organic flocculation). Viruses in glycine eluates also can be reconcentrated by this technique by first supplementing them with beef extract to a final concentration of 1 to 3%. Use sterile beef extract paste (about 80% beef extract) or sterile 20% beef extract solution made from powder to bring glycine eluates to the desired beef extract concentration. While mixing vigorously, adjust eluate to pH 3.5 by adding 1N HCl dropwise. Continue to mix at slow speed for 30 min and centrifuge at 3000 × g for 10 min. Decant and discard supernatant. With vigorous mixing, resuspend sediment in 1/20 the initial sample volume of 0.45N Na2HPO4. Add antibiotics (1/10 final sample volume penicillin-streptomycin, 1/100 final sample volume gentamycin-kanamycin, or both) and while mixing vigorously adjust to pH 7.4 with 1.0 or 0.1N NaOH. Check electrical conductivity of sample. If conductivity is > 13 000 µmhos, dialyze sample against Hanks balanced salt solution before assay. Store at 4 or −70°C, depending on time until virus assay. 6. References 1. KATZENELSON, E., B. FATTAL & T. HOSTOVESKY. 1976. Organic flocculation: an efficient second-step concentration method for the detection of viruses in tapwater. Appl. Environ. Microbiol. 32:638. 2. BITTON, G., B.N. FELDBERG & S.R. FARRAH. 1979. Concentration of enteroviruses from seawater and tap water by organic flocculation using non-fat dry milk and casein water. Air Soil Pollut. 10:187. 3. PAYMENT, P. & M. TRUDEL. 1980. A simple low cost apparatus for conditioning large volumes of water for virological analysis. Can. J. Microbiol. 26:548. 4. PAYMENT, P. & M. TRUDEL. 1981. Improved method for the use of proportioning injectors to condition large volumes of water for virological analysis. Can. J. Microbiol. 27:455. 9510 D. Virus Concentration by Aluminum Hydroxide Adsorption-Precipitation 1. General Discussion Viruses can be concentrated from small volumes of water, wastewater, and adsorbent filter eluates by precipitation with aluminum hydroxide.1-4 This process probably involves both electrostatic interactions between the negatively charged virus surface and the positively charged aluminum hydroxide [Al(OH)3] surfaces and coordination of the virus surface by hydroxo-aluminum complexes.5 Viruses are adsorbed to an Al(OH)3 precipitate that is either added to the sample or formed in the sample from a soluble aluminum salt and a base such as sodium carbonate (Na2CO3) or sodium hydroxide (NaOH). Viruses are allowed to adsorb to the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Al(OH)3 precipitate and the virus-containing precipitate is collected by filtration or centrifugation. The recovered precipitate may be inoculated directly into laboratory hosts for virus assay or the viruses are eluted from the precipitate with an alkaline buffer or a proteinaceous solution before virus assay. The major limitations of this method are that sample size is limited to perhaps a few liters, soluble organic matter can interfere with virus adsorption, and virus recovery from the precipitate may be incomplete. Virus adsorption may be improved by forming the Al(OH)3 precipitate in the sample instead of adding it preformed. Although virus adsorption can be maximized by using large amounts of Al(OH)3, the adsorbed viruses become more difficult to elute. Therefore, some intermediate amount of Al(OH)3 is used to achieve maximum virus recovery. Also, Al(OH)3 is a relatively nonspecific adsorbent so that other substances may be concentrated with viruses. The presence of such impurities may cause the concentrated sample to be toxic for the cell cultures normally used for virus assay. Several modifications of the Al(OH)3 adsorption-precipitation procedure have been used to concentrate viruses from water, wastewater, and eluates from adsorbent filters. Initially, preformed Al(OH)3 precipitates were made by adding Na2CO3 to AlCl3 solutions and the Al(OH)3 precipitate was resuspended in 0.15N NaCl. This was added to the wastewater and the mixture was stirred gently for 1 h or more to allow viruses to adsorb to the precipitate. The precipitate was recovered by filtration, resuspended in cell culture media, and inoculated into cell cultures.3,4 More recent procedural modifications include: (a) Al(OH)3 precipitate formation within the sample,1,2,6-8 (b) recovery of the Al(OH)3 precipitate by centrifugation followed by elution of viruses from the precipitate with alkaline eluents,1,2,6,7 and (c) a large-volume method in which the precipitate is formed in the sample and collected on a cartridge filter, and viruses are eluted from the precipitate on the filter with alkaline eluent.9 The method described here is for relatively small sample volumes and uses Al(OH)3 that is either preformed or generated within the sample. The latter modification is preferable because some viruses are not adsorbed efficiently by preformed precipitates.10 2. Equipment and Apparatus a. Centrifuge, with rotor and buckets, capable of operating at about 1900 × g. b. Centrifuge bottles and tubes. c. Beaker, 100-mL or larger. d. pH meter. e. Magnetic stirrer and stirring bars or alternative mixing device. f. Graduated cylinders, 100-mL or larger. g. Pipets, 1-, 5-, and 10-mL. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater h. Laboratory balance. i. Vacuum-type filter holder or Buchner filter funnel,*#(96) 47-mm diam or larger. j. Filter flask.*#(97) k. Spatula,*#(98) flat blade, metal or autoclavable plastic. l. Vacuum source,*#(99) vacuum pump or laboratory vacuum line. 3. Materials Filter:* Fiberglass-acrylic resin filter†#(100) or microporous filter, 0.45-µm porosity,‡#(101) 47-mm diam or larger. To prevent virus adsorption, filter 0.1% polyoxyethylene sorbitan monooleate solution (¶ 4h) through the filters, using about 1 mL solution/cm2 of filter surface area. Rinse filter with distilled water, using about 10 mL/cm2 of filter surface area. Sterilize treated filters by autoclaving. 4. Reagents a. Hydrochloric acid, HCl, 0.1 and 1.0N. b. Sodium hydroxide, NaOH, 0.1 and 1.0N. c. Sodium carbonate, Na2CO3, 4N§#(102). d. Aluminum chloride, AlCl3, 0.075N§ or 0.9N. e. Sodium chloride, NaCl, 0.14N. f. Beef extract, 3%, pH 7.4: Dissolve 3 g beef extract paste or 2.4 g beef extract powder in 90 mL distilled water, adjust to pH 7.4 with 1.0 or 0.1N NaOH, dilute to 100 mL with distilled water, and sterilize by autoclaving. g. Antibiotics: Use either: 1) Penicillin-streptomycin, 10X, containing 5000 IU penicillin/mL and 5000 µg streptomycin/mL. Available commercially or prepare by dissolving powdered sodium or potassium penicillin-G and streptomycin sulfate in distilled water and sterilizing by filtration. 2) Gentamycin-kanamycin, 100X, containing 5000 µg/mL each of gentamycin base and kanamycin base (Section 9510B.4i). h. Polyoxyethylene sorbitan monooleate,i#(103) 0.1% (ν/ν) in distilled water. 5. Procedure a. Sterilization of apparatus, materials, and reagents: See Section 9510B.5a. b. Preparation of preformed Al(OH)3 precipitate: While mixing 100 mL 0.075N AlCl3 at room temperature, slowly add 4N Na2CO3 solution to form precipitate and adjust to pH 7.2. Continue mixing for 15 min and, if necessary, add more Na2CO3 to maintain pH 7.2. Centrifuge at 1100 × g for 15 min and discard supernatant. Resuspend sediment in 0.14N NaCl and recentrifuge. Discard supernatant, resuspend sediment in 0.14N NaCl, and sterilize by © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater autoclaving. Cool, centrifuge again, decant supernatant, and resuspend Al(OH)3 sediment in 50 mL sterile 0.14N NaCl. Store at 4°C. c. Sample size, collection, and storage: Process samples of no more than several liters because the method is too cumbersome and time-consuming for larger volumes. See Section 9510B.5d for sample collection and storage procedures. d. Sample processing: Do not prefilter sample11,12 because substantial virus losses can occur. Adjust sample to pH 6.0 with 1.0 or 0.1N HCl while mixing vigorously. Form Al(OH)3 precipitate in sample by adding 1 part 0.9N AlCl3 solution to 100 parts sample to give a final 0.009N Al3+ concentration. Check sample pH and re-adjust to 6.0 with 1.0 or 0.1N NaOH or HCl, if necessary. Mix slowly for 15 min at room temperature. Alternatively, use preformed Al(OH)3 precipitate by adding 1 part stock Al(OH)3 suspension/100 parts sample and mix slowly for 2 h at 4 to 10°C to allow for virus adsorption. Collect virus-containing Al(OH)3 precipitate by centrifugation or filtration. To collect precipitate by centrifugation, centrifuge at 1700 × g for 15 to 20 min, discard supernatant, and resuspend sediment in 1/1000 to 1/20 original sample volume of 3% beef extract, pH 7.4. To collect precipitate by filtration, vacuum filter sample through a treated filter (¶ 3 above) held in a vacuum-type filter holder or Buchner funnel, using additional filters if filter clogs before entire sample is filtered. Carefully scrape precipitate from filter(s) with a sterile spatula and resuspend in 1/1000 to 1/20 original sample volume of 3% beef extract, pH 7.4. Regardless of collection method, vigorously mix the Al(OH)3 beef extract suspension and, if necessary, adjust to pH 7.4 with 0.1N HCl or NaOH. Continue mixing for a total of 10 min. Centrifuge at 1900 × g for 30 min. Decant supernatant, add 1/10 the volume of the concentrate of penicillin-streptomycin solution or 1/100 volume of gentamycin-kanamycin and store at 4 or −70°C. 6. References 1. PAYMENT, P., C.P. GERBA, C. WALLIS & J.L. MELNICK. 1976. Methods for concentrating viruses from large volumes of estuarine water on pleated membranes. Water Res. 10:893. 2. FARRAH, S.R., S.M. GOYAL, C.P. GERBA, C. WALLIS & J.L. MELNICK. 1977. Concentration of enteroviruses from estuarine water. Appl. Environ. Microbiol. 33:1192. 3. WALLIS, C. & J.L. MELNICK. 1967. Concentration of viruses on aluminum hydroxide precipitates. In G. Berg, ed. Transmission of Viruses by the Water Route. Interscience Publ., New York, N.Y. 4. WALLIS, C. & J.L. MELNICK. 1967. Virus concentration on aluminum and calcium salts. Amer. J. Epidemiol. 85:459. 5. COOKSON, J.T., JR. 1974. The chemistry of virus concentration by chemical methods. Develop. Ind. Microbiol. 15:160. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 6. LYDHOLM, B. & A.L. NIELSEN. 1979. Methods for detection of virus in wastewater applied to samples from small scale treatment systems. Water Res. 14:169. 7. SELNA, M.W. & R.P. MIELE. 1977. Virus sampling in wastewater-field experiences. J. Environ. Eng. Div., Proc. Amer. Soc. Civil Eng. 103:693. 8. DOBBERKAU, H.J., R. WALTER & S. RUDIGER. 1981. Methods for virus concentration from water. In M. Goddard & M. Butler, eds. Viruses and Wastewater Treatment. Pergamon Press, New York, N.Y. 9. FARRAH, S.R., C.P. GERBA, C. WALLIS & J.L. MELNICK. 1978. Concentration of poliovirus from tapwater onto membrane filters with aluminum chloride at ambient pH levels. Appl. Environ. Microbiol. 35:624. 10. FARRAH, S.R., G.M. GOYAL, C.P. GERBA, R.H. CONKLIN & E.M. SMITH. 1978. Comparison between adsorption of poliovirus and rotavirus by aluminum hydroxide and activated sludge flocs. Appl. Environ. Microbiol. 35:360. 11. SOBSEY, M.D., C.P. GERBA, C. WALLIS & J.L. MELNICK. 1977. Concentration of enteroviruses from large volumes of turbid estuary water. Can. J. Microbiol. 23:770. 12. HOMMA, A., M.D. SOBSEY, C. WALLIS & J.L. MELNICK. 1973. Virus concentration from sewage. Water Res. 7:945. 9510 E. Hydroextraction-Dialysis with Polyethylene Glycol 1. General Discussion Polyethylene glycol (PEG) hydroextraction is an ultrafiltration process in which the sample is placed in a cellulose dialysis bag and exposed to PEG, a hygroscopic material. Water and microsolutes leave the sample by passing across the semipermeable dialysis membrane into the hygroscopic PEG.1 Viruses and other macrosolutes, including PEG, cannot cross the dialysis membrane. The sample volume in the dialysis bag is reduced by water loss to the PEG, thereby concentrating viruses and other macrosolutes. The viruses retained in the dialysis bag are recovered by opening the bag, collecting the remaining sample, and eluting any viruses possibly adsorbed to the inner walls of the bag with a small volume of slightly alkaline proteinaceous solution such as 3% beef extract, pH 9.0. The collected concentrate and eluate are combined and assayed for viruses. The main limitations of this method are that only small samples (less than 1 L) can be processed conveniently, virus elution from the walls of the dialysis bag may be incomplete unless the elution is done painstakingly, and other macrosolutes in the sample that are concentrated with viruses may interfere with virus assays by being cytotoxic. Initial investigations of this method reported low and highly variable virus recoveries from wastewater.2,3 The type of dialysis tubing and eluent solution as well as the thoroughness of the elution step have been found to influence virus recovery efficiency. More recently, with © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater modified procedures, efficient and consistent virus recoveries have been obtained from wastewater and from adsorbent filter eluates.4,5 2. Equipment and Apparatus a. Beakers, 100-mL or larger. b. Graduated cylinders, 100-mL or larger. c. Dialysis tubing clamps.*#(104) d. Pan, approximately 30 × 30 × 12 cm, autoclavable. e. Magnetic stirrer and stirring bars or alternative mixing device. f. Centrifuge, with rotor and buckets, capable of operating at about 1900 × g. g. pH meter. h. Pipets, 1-, 5-, and 10-mL. i. Tape roller†#(105) or similar device to aid in washing the inside walls of dialysis bags with eluting fluid. j. Ultrasonic disruptor-emulsifier,‡#(106) probe type, capable of generating 100 W of acoustical output. 3. Materials a. Dialysis tubing, seamless, regenerated cellulose, 4.8-nm average pore diameter.§#(107) b. Polyethylene glycol (PEG),i#(108) dry flakes. 4. Reagents See Section 9510D.4. 5. Procedure a. Sterilization of apparatus, materials, and reagents: See Section 9510B.5a. Do not sterilize PEG. b. Sample size, collection, and storage: Process samples of no more than a few hundred milliliters. See Section 9510B.5d for sample collection and storage procedures. c. Preparation of dialysis tubing: Cut a length of dialysis tubing long enough to accommodate entire sample. Close one end with a clamp. Do not tie knots to close dialysis tubing. Fill tubing bag with distilled water, sterilize by autoclaving, and let cool. d. Sample processing: Aseptically remove dialysis bag from distilled water and drain. Fill bag with sample and close open end with a second clamp. Place bag in a pan containing a 5-cm layer of PEG, making sure that bag does not touch pan walls. Cover tubing with an additional 5 cm PEG and store at 4°C (for about 18 h) until sample volume has been reduced to no more than a few milliliters. (If PEG 6000 is used the process time is reduced to 4 to 6 h.) Although sample may be allowed to dewater completely, do not let it remain in this state. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Remove dialysis bag from PEG and quickly wash PEG from outside of bag with sterile distilled water. Remove clamp from one end of bag and carefully collect sample concentrate. Add about 1/200 to 1/20 the original sample volume of 3% beef extract, pH 9.0, and clamp closed. Thoroughly wash inside walls of bag with beef extract by rubbing fluid from one end to the other several times using either fingers or a roller device. Remove clamp from one end of bag and collect fluid, kneading or squeezing to recover the last traces. Add recovered fluid to previously collected sample concentrate. Adjust to pH 7.5 with 1.0 or 0.1N HCl while mixing vigorously. To disperse solids-associated viruses in sample, stir overnight (about 18 h) in the cold (about 4°C) or treat with ultrasonics at 100 W for 1 to 2 min. Prevent sample temperature from rising above 37°C during ultrasonic treatment by chilling in an ice bath. Centrifuge at 1900 × g for 30 min. Decant supernatant, add 1/10 the volume of the concentrate of penicillin-streptomycin solution or 1/100 volume of gentamycin-kanamycin, and store at 4 or −70°C. 6. References 1. SOBSEY, M.D. 1976. Methods for detecting enteric viruses in water and wastewater. In G. Berg, H.L. Bodily, E.H. Lennette, J.L. Melnick & T.G. Metcalf, eds. Viruses in Water. American Public Health Assoc., Washington, D.C. 2. CLIVER, D.O. 1967. Detection of enteric viruses by concentration with polyethylene glycol. In G. Berg., ed. Transmission of Viruses by the Water Route. Interscience Publ., New York, N.Y. 3. SHUVAL, H.I., S. CYMBALISTA, B. FATTAL & N. GOLDBLUM. 1967. Concentration of enteric viruses in water by hydro-extraction and two-phase separation. In G. Berg, ed. Transmission of Viruses by the Water Route. Interscience Publ., New York, N.Y. 4. WELLINGS, F.M., A.L. LEWIS, C.W. MOUNTAIN & L.V. PIERCE. 1975. Demonstration of virus in groundwater after effluent discharge onto soil. Appl. Microbiol. 29:751. 5. RAMIA, S. & S.A. SATTAR. 1979. Second-step concentration of viruses in drinking and surface waters using polyethylene glycol hydroextraction. Can. J. Microbiol. 25:587. 9510 F. Recovery of Viruses from Suspended Solids in Water and Wastewater 1. General Discussion Viruses in the aquatic environment often are associated with solids or particulate matter, either adsorbed to particulate surfaces or embedded within the solid.1-3 Both freely suspended and solids-associated viruses are concentrated from water by the methods described above. There is evidence that solids-associated viruses are not eluted efficiently from adsorbent filters or from Al(OH)3 precipitates and organic flocs. Recovery of solids-associated viruses by microporous filter methods employing in-situ elution is inconsistent.2 Solids-associated viruses © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater on adsorbent filters are eluted more efficiently by disrupting filters in elution fluid than by in-situ elution,2 but this is cumbersome and time-consuming, especially for large-diameter disk filters and cartridge filters. For small volumes of water and wastewater, solids-associated viruses can be recovered expediently by separating the solids by centrifuging, decanting the supernatant, and eluting viruses from the solids by resuspending in a small volume of eluent.4 Viruses in the supernatant can be concentrated by one of the procedures described in Section 9510B, Section 9510C, Section 9510D, or Section 9510E. Viruses eluted from the resuspended solids are separated from the solids by centrifuging and are assayed directly or concentrated further by organic flocculation.5,6 Major limitations of these methods are incomplete virus elution and poor virus recoveries due to interferences from sample constituents. 2. Equipment and Apparatus a. Centrifuge, with rotor and buckets for 250- to 1000-mL-capacity bottles, capable of operating at about 1250 × g. b. Centrifuge bottles, 250- to 1000-mL. c. pH meter. d. Laboratory balance. e. Graduated cylinder, 250-mL or larger. f. Beaker, 250-mL or larger. g. Sample bottles, 250-mL or larger. h. Magnetic stirrer and stirring bars, or alternative mixing device. i. Pipets, 1-, 5-, and 10-mL. 3. Reagents a. Hydrochloric acid, HCl, 0.1 and 1.0N. b. Sodium hydroxide, NaOH, 0.1 and 1.0N. c. Eluent: Dissolve 10 g beef extract, 1.34 g disodium phosphate heptahydrate, Na2HPO4⋅7H2O, and 0.12 g citric acid in 90 mL distilled water, adjust to pH 7.0 with 1N HCl or NaOH, dilute to 100 mL with distilled water, and sterilize by autoclaving. d. Antibiotics: See Section 9510B.4i. 4. Procedure a. Sterilization of apparatus, materials, and reagents: See Section 9510B.5a. b. Sample size, collection, and storage: Collect and process samples of no more than 10 L, depending on capacity of centrifuge. See Section 9510B.5d for sample collection and storage procedures. c. Sample processing: Aseptically transfer 250- to 1000-mL sample volumes to centrifuge © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater bottles and centrifuge at 1250 × g for 20 min. Decant and pool supernatants for subsequent processing for viruses by one of the methods for water or wastewater described previously. Elute viruses from the sedimented solids by resuspending in eluent. Use 40 mL eluent per quantity of sediment from 250 mL of original sample. Pool resuspended sediments from multiple centrifuge bottles in a sterile beaker. Alternatively, keep resuspended sediments from small numbers of centrifuge bottles in the bottles and process them individually. While vigorously mixing with a magnetic stirrer, adjust to pH 7.0 by slowly adding 1N NaOH or HCl, if necessary. Reduce mixing speed and continue mixing for 30 min. During this period, check sample pH and readjust to pH 7.0 as necessary. As an alternative to mixing for 30 min, sonicate samples at 100 W for 15 min in a rosette cooling cell maintained at 4°C. Return sample to centrifuge bottles. Centrifuge at 1250 × g and 4°C for 15 min, collect supernatant for subsequent assay or further concentration, and discard the sediment. If desired, further concentrate viruses from this supernatant by organic flocculation (see Section 9510E). For supernatants that will be assayed directly for viruses with no further concentration, adjust to pH 7.4, add 1/10 the volume of sample of penicillin-streptomycin or 1/100 volume of gentamycin-kanamycin and store at 4 or −70°C. 5. References 1. SCHAUB, S.A. & B.P. SAGIK. 1975. Association of enteroviruses with natural and artificially introduced colloidal solids in water and infectivity of solids-associated virions. Appl. Microbiol. 30:212. 2. WELLINGS, F.M., A.L. LEWIS & C.W. MOUNTAIN. 1976. Viral concentration techniques for field sample analysis. In L.B. Baldwin, J.M. Davidson & J.F. Gerber, eds. Virus Aspects of Applying Municipal Waste to Land. Univ. Florida, Gainesville. 3. WELLINGS, F.M., A.L. LEWIS & C.W. MOUNTAIN. 1974. Virus survival following wastewater spray irrigation of sandy soils. In J.F. Malina, Jr. & B.P. Sagik, eds. Virus Survival in Water and Wastewater Systems. Univ. Texas, Austin. 4. BERG, G. & D.R. DAHLING. 1980. Method for recovering viruses from river water solids. Appl. Environ. Microbiol. 39:850. 5. GERBA, C.P. 1982. Detection of viruses in soil and aquatic sediments. In C.P. Gerba & S.M. Goyal, eds. Methods in Environmental Virology. Marcel Dekker, Inc., New York, N.Y. 6. FARRAH, S.R. 1982. Isolation of viruses associated with sludge particles. In C.P. Gerba & S.M. Goyal, eds. Methods in Environmental Virology. Marcel Dekker, Inc., New York, N.Y. 9510 G. Assay and Identification of Viruses in Sample Concentrates © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Storage of Sample Concentrates Because it often is impossible to assay sample concentrates immediately, store them at room temperature (about 25°C) for up to 2 h or at refrigerator temperatures (4 to 10°C) for up to 48 h to minimize virus losses. Freeze samples requiring storage longer than 48 h at −70°C or less. Do not freeze samples at −10 to −20°C because extensive inactivation of some enteric viruses may occur. Store sample concentrates from finished waters in separate freezers or physically separated from other virus-containing material in common freezers. 2. Decontamination of Sample Concentrates Sample concentrates, especially those from wastewater, are likely to be contaminated with bacteria and fungi that can overgrow cell cultures and interfere with virus detection and assay. Do not decontaminate by centrifugation or filtration because virus losses are likely to occur. For many samples, especially those from finished waters, contamination is controlled adequately by antibiotics such as penicillin-streptomycin or gentamycin-kanamycin that are added immediately after the sample is obtained. To provide additional protection against fungal contamination, add amphotericin B or nystatin at concentrations of 2.5 and 50 µg/mL, respectively.1 If penicillin-streptomycin or gentamycin-kanamycin are inadequate, use one or more additional antibiotics such as aureomycin, neomycin, or polymyxin B. To maximize the antibiotic effects, incubate samples for 1 to 3 h at 25 to 37°C after adding the antibiotics. Bacterial destruction is further enhanced by freezing at −70°C after incubation with antibiotics. Keep samples frozen until assayed for viruses. To determine if antibiotic treatment has been effective, plate a small subsample on a general-purpose medium such as plate count agar by the spread plate technique and incubate at 37°C for 24 to 48 h. If extensive bacterial contamination persists after antibiotic treatment, treat with chloroform. Add 1/10 volume of sample of chloroform (CHCl3) and mix vigorously for 30 min at room temperature or homogenize 1 to 2 min at 4 to 10°C. For phase separation, centrifuge at ≥ 1000 × g or store overnight in a refrigerator. Separate sample (upper layer) from CHCl3 (bottom layer) by aspirating with a pipet and bubble with filter-sterilized air for about 15 min to remove dissolved CHCl3. It may be necessary to place sample in a sterile, shallow container and expose it to the atmosphere in a sterile air environment (laminar air flow clean bench or biological safety cabinet) for up to several hours to remove remaining traces of CHCl3. Do not use ether to decontaminate samples because of the hazard of explosion or fire. 3. Laboratory Facilities and Host Systems for Virus Assay Because viruses are obligate, intracellular parasites, they grow (multiply) only in living host cells. This ability to multiply in, and thereby destroy, their host cells is the basis for virus detection and assay. The two major host cell systems for human enteric viruses are whole animals (usually mice) and mammalian cell cultures of primate origin. A complete description of facilities, equipment, materials, and methods for conducting virus © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater assays is beyond the scope of this book; see standard handbooks on virology and cell culture.1-4 Virus assay is beyond the capability of most water and wastewater microbiology laboratories. It should be done only by a trained virologist working in specially equipped virology laboratory facilities. Take particular care to prevent samples or inoculated hosts from becoming contaminated with viruses from other sources and to prevent virus cross-contamination arising from sample concentrates or inoculated hosts. Process and handle samples in a Class II Type I biological safety cabinet5 or in a ‘‘sterile’’ room or cubicle. The use of such cabinets or facilities is mandatory for testing drinking water or other finished water samples. There is no single, universal host system for all enteric viruses. Some enteric viruses, notably hepatitis A virus, human rotaviruses, and Norwalk-type gastroenteritis viruses, cannot be assayed routinely in any convenient laboratory host systems. However, most of the known enteric viruses can be detected by using two or more cell culture systems and perhaps suckling mice. The latter previously were considered essential for the detection of group A coxsackie-viruses, but recent studies indicate that the RD cell line may be nearly as sensitive as suckling mice for the isolation of these viruses as well as other enteroviruses.6,7 In general, the more different host systems used, the greater the enteric virus recovery rate. However, the number of different host systems used is limited by practical and economic considerations. There have been numerous comparative studies on relative sensitivities of various cell culture systems for enteric virus detection,6-26 but no systematic, comprehensive study has been reported for enteric virus recoveries from water and wastewater. Primary or secondary human embryonic kidney (HEK) cell cultures appear to be the single most sensitive host system for enteric virus isolations, but they are becoming increasingly more difficult to obtain regularly and, when available from commercial sources, they are expensive. Primary or secondary African green, cynomolgus, or rhesus monkey or baboon kidney cells are sensitive hosts for many enteroviruses and reoviruses, but are not particularly suitable for recovering adenoviruses or group A coxsackieviruses. BGM, a continuous line derived from African green monkey kidney cells, may be comparable in sensitivity to primary monkey kidney cells for enteric virus recovery.7,18,21,25,26 A number of other continuous cell lines as well as human fetal diploid cell strains have been evaluated for enteric virus recoveries. Some human fetal diploid cell strains give virus isolation rates comparable to primary monkey kidney cells, but plentiful supplies of specific human fetal diploid cell strains are not readily available and many are difficult to maintain. Furthermore, each different cell strain must be characterized for virus susceptibility. Most continuous cell lines generally are less effective than primary cells, but comparable isolation rates for some enteric virus groups have been obtained with Hep-211 and HeLa17,25 cells. Assay the entire sample concentrate for enteric viruses, using at least two different host systems and dividing entire sample equally among the hosts. Preferably use primary (or secondary) HEK cells with either primary (or secondary) monkey kidney or BGM cells for the recovery of most enteroviruses, adenoviruses, and reoviruses. Additional use of either suckling mice or RD cells provides for enhanced recovery of group A coxsackieviruses. Different host © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater systems may be substituted for these if it is demonstrated that they have equivalent sensitivity. 4. Virus Quantitation Procedures for Sample Concentrates a. Advantages and disadvantages of different quantitation procedures: Virus assays in suckling mice or other animals are quantal assays and in cell cultures they can be done either by quantal (most probable number or 50% endpoint) or enumerative (plaque) methods. Selection between cell culture assay methods depends on the sample and the choice between achieving either maximum virus sensitivity or maximum precision and accuracy in estimating virus concentration. The plaque technique generally is more precise and accurate than the quantal assay because relatively large numbers of individual infectious units can be counted directly as discrete, localized areas of infection (plaques). Quantal assays are more sensitive than monolayer plaque assays, but are less sensitive than an agar cell suspension plaque assay.26 Because virus plaques are discrete areas of infection arising from a single infectious virus unit, it is relatively easy to recover viruses from individual plaques and then to inoculate them into additional cell cultures to obtain a pure virus culture for identification. However, large proportions of so-called ‘‘false-positive’’ plaques that do not confirm as virus-positive when material from these plaques is further passaged in cell cultures have been reported.27,28 Whether this problem is due to nonviral, plaque-like areas of cytotoxicity from the sample or to technical inability to passage viruses successfully from the initial plaques remains uncertain.27,28 The use of specific plaque assay conditions for optimizing the recovery of certain enteric virus groups may preclude efficient recovery of other enteric groups requiring different plaque assay conditions. Furthermore, some viruses, such as adenoviruses, do not form plaques efficiently under any conditions. Cytotoxicity due to water or wastewater constituents in sample concentrates is difficult to control in plaque assay systems because the agar overlay medium is difficult to remove and replace. A potential limitation of quantal assays is the possibility that two or more different virus types will be inoculated into the same cell culture and thus produce a simple positive culture. This not only results in an underestimation of virus concentration but also requires separation of the individual virus types by further passage in cell culture. Such mixed cultures may go undetected unless virus isolates are identified serologically. Recent results indicate that mixed positive cultures are encountered rarely when samples are divided into small portions for inoculation into a series of replicate cell cultures.7,25 Cytotoxicity due to constituents of sample concentrates usually can be controlled in quantal assay cell cultures by replacing the culture medium before the cells die. b. Cell culture procedures for virus isolation and assay: To assay sample concentrates in cell cultures by quantal or plaque methods, drain the medium from newly confluent cultures. To reduce toxicity, rinsing with buffered saline solution may be helpful. Inoculate with unit volumes of sample. Use no more than 0.06 mL sample/cm2 of cell layer surface, e.g., maximum volumes of 1.0, 3.0, and 6.0 mL in cell culture flasks with areas of 25, 75, and 150 cm2, respectively.29 If © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater samples are expected to contain such large quantities of viruses that it would be difficult to make reliable estimates of concentration, inoculate cell cultures with small sample volumes or dilutions of concentrates. Allow viruses to adsorb to cells for 2 h at 37 ± 0.5°C. Redistribute inoculum over the cell layer manually every 15 min or keep cultures on a mechanical rocker during the adsorption period. Add liquid maintenance medium to cultures for quantal assays or agar-containing medium for plaque assays. Invert plaque assay cultures so that cell (agar) side of culture faces up and incubate at 37°C. Microscopically examine quantal assay cultures for the appearance of cytopathic effects (CPE) daily during the first 3 d and then periodically for a total of at least 14 d. Do not change cell culture medium unless cytotoxicity or cell deterioration occurs. Freeze cultures developing CPE at −70°C when more than 75% of the cells become involved. After 14 or more days, freeze at −70°C all remaining cultures, including those remaining negative for CPE as well as controls. Thaw cultures and clarify culture fluid-cell lysate by slow-speed centrifugation or filtration through sterile 0.22- or 0.45-µm porosity filters. Inoculate clarified material from each initial (first-passage) culture into a second (second-passage) culture by transferring 20% of the total initial culture into newly confluent cell cultures of the same type. Microscopically examine second-passage cultures for development of CPE periodically over a period of 14 or more days. Consider second-passage cultures developing CPE as confirmed virus-positive. Freeze and store at −70°C for virus identification. Discard as negative any virus cultures negative for CPE after this second incubation period of 14 or more days. Periodically examine plaque assay cultures for appearance of plaques over a 14-d period. Mark and tally plaques as they appear. Transfer viruses from each plaque directly to at least two newly confluent, liquid-medium cell cultures of the same type27 before plaques become too large and grow together or before the entire cell layer deteriorates. Do not store material obtained from plaques before transfer to new cell cultures, as this may result in loss of virus titer and unsuccessful transfers. Microscopically examine these second-passage cultures periodically over 14 d for development of CPE. Freeze cultures developing CPE at −70°C for virus identification. c. Virus isolation and assay in mice: To detect group A and B coxsackieviruses in mice, inoculate samples into animals no older than 24 h using standard procedures.2,3,8 Use either the intracerebral or intraperitoneal route, inoculating 0.02 and 0.05 mL, respectively. Observe mice daily over a 14-d period for development of weakness, tremors, and either flaccid (due to group A coxsackieviruses) or spastic (due to group B coxsackieviruses) paralysis. Sacrifice animals developing symptoms, and using sterile technique, prepare 20% tissue suspensions in Hanks balanced salt solution of the entire skinned, eviscerated torso or just the brain and legs. Store suspensions at −70°C until used for further passage and identification. For second passage in mice, follow general procedures used for the initial inoculations. However, making a second passage in cell cultures is preferable to making a second passage in mice because it is easier to do subsequent virus identification by neutralization tests. d. Estimating virus concentration: Determining the amount of virus in a sample concentrate depends on the assay used. If a sample concentrate is assayed in cell cultures by the plaque © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater technique, count all plaques and calculate the virus concentration, expressed as plaque-forming units (PFU). If a sample concentrate is assayed by the quantal method, estimate the virus concentration by the most probable number (MPN) method and express as most probable number of infectious units (MPNIU), or by a 50% end point method and express as 50% infectious or lethal dose (ID50 or LD50).2,4,30-32 If the undiluted sample concentrate or a single sample dilution was inoculated into a series of replicate cell cultures (or mice), calculate the MPNIU from the number of confirmed CPE-negative cultures (or mice), q, per total number of cultures (or mice) inoculated, n, according to the formula MPN = −ln(q/n) If more than one sample dilution was inoculated into cell cultures (or mice), calculate the MPNIU from the formula developed by Thomas:33 where: P = total number of positive cultures (or mice) from all dilutions, N = total mL sample inoculated for all dilutions, and Q = total mL sample in all negative cultures (or mice). In using this formula, exclude from the computation all dilutions containing only positive cultures (or mice). For MPN values obtained from a single sample dilution, the 95% confidence interval is based on the standard error of the binomial distribution when more than 30 cultures (or mice) are inoculated or from the confidence coefficient table of Crow32,34 when 30 or fewer cultures (or mice) are inoculated. Make 50% end-point estimates arithmetically by either the Reed-Muench or Karber method.2,4 These methods require results from several equally spaced sample dilutions, preferably with about the same number of dilutions above and below the 50% end point, and may not be useful for sample concentrates containing relatively low virus levels. e. Identification of virus isolates: Identify enteric viruses isolated from sample concentrates by standard serological techniques, although preliminary identification of genus (enterovirus, reovirus, or adenovirus) sometimes can be made on the basis of information obtained from the isolation procedure. Enteric viruses recovered in suckling mice are likely to be either group A or B coxsackieviruses. For enteric viruses isolated in cell cultures, preliminary identification of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater genus often can be made from the characteristic appearance of cytopathic effects (CPE) in infected cell cultures. Confirm preliminary identification of suspected adenovirus and reovirus isolates by detecting their respective group specific antigens by complement fixation tests using clarified, second-passage cell-culture lysate as the antigen. Identify specific reovirus serotypes by hemagglutination-inhibition (HI) or neutralization (Nt) tests. Adenovirus serotypes can be separated into four groups on the basis of their ability (or inability) to hemagglutinate rhesus monkey or rat erythrocytes.2,3,8 Except for type 18, the first 28 numbered adenoviruses can be identified as to specific serotype by HI. Alternatively, identify all adenovirus serotypes by Nt tests using either individual type-specific antisera or intersecting antisera pools. Also identify specific enterovirus serotypes by neutralization tests in cell cultures using intersecting pools of hyperimmune sera.2,3,8,35 Use mice for Nt tests for group A and B coxsackieviruses only if the virus isolates fail to propagate in cell cultures.36 Because polioviruses often are the most prevalent enteroviruses in water and wastewater, test enterovirus isolates for neutralization by an antisera pool against the three types of poliovirus before making neutralization tests with intersecting antisera pools. 5. References 1. PAUL, J. 1975. Cell and Tissue Culture, 5th ed. Churchill Livingstone, New York, N.Y. 2. LENNETTE, E.H. & N.J. SCHMIDT, eds. 1979. Diagnostic Procedures for Viral and Rickettsial Infections, 5th ed. American Public Health Assoc., Washington, D.C. 3. LENNETTE, E.H., A. BALOWS, W.J. HAYSLER & J.P. TRUANT, eds. 1980. Manual of Clinical Microbiology, 3rd ed. American Soc. Microbiology, Washington, D.C. 4. FRESHNEY, R.I. 1987. Culture of Animal Cells. A Manual of Basic Technique, 2nd ed. Alan R. Liss, Inc., New York, N.Y. 5. U.S. PUBLIC HEALTH SERVICE. 1976. Guidelines for Research Involving Recombinant DNA Molecules. Appendix D-I, Biological Safety Cabinets. National Inst. Health, Bethesda, Md. 6. SCHMIDT, N.J., H.H. HO & E.H. LENNETTE. 1975. Propagation and isolation of group A coxsackieviruses in RD cells. J. Clin. Microbiol. 2:183. 7. SCHMIDT, N.J., H.H. HO, J.L. RIGGS & E.H. LENNETTE. 1978. Comparative sensitivity of various cell culture systems for isolation of viruses from wastewater and fecal samples. Appl. Environ. Microbiol. 36:480. 8. HSIUNG, G.D. 1982. Diagnostic Virology, 3rd ed. Yale Univ. Press, New Haven, Conn. 9. KELLY, S., J. WINSSER & W. WINKELSTEIN. 1957. Poliomyelitis and other enteric viruses in sewage. Amer. J. Pub. Health 47:72. 10. KELLY, S. & W.W. SANDERSON. 1962. Comparison of various tissue cultures for the isolation of enteroviruses. Amer. J. Pub. Health 52: 455. 11. PAL, S.R., J. MCQUILLIN & P.S. GARDNER. 1963. A comparative study of susceptibility of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. primary monkey kidney cells, Hep 2 cells and HeLa cells to a variety of faecal viruses. J. Hyg., Camb. 61:493. LEE, L.H., C.A. PHILLIPS, M.A. SOUTH, J.L. MELNICK & M.D. YOW. 1965. Enteric virus isolations in different cell cultures. Bull. World Health Org. 32:657. SCHMIDT, N.J., H.H. HO & E.H. LENNETTE. 1965. Comparative sensitivity of human fetal diploid kidney cell strains and monkey kidney cell cultures for isolation of certain human viruses. Amer. J. Clin. Pathol. 43:297. BERQUIST, K.R. & G.J. LOVE. 1966. Relative efficiency of three tissue culture systems for the primary isolation of viruses from feces. Health Lab. Sci. 3:195. HERRMANN, E.C. 1967. The usefulness of human fibroblast cell lines for the isolation of viruses. Amer. J. Epidemiol. 85:200. FAULKNER, R.S. & C.E. VAN ROOYEN. 1969. Studies on surveillance and survival of viruses in sewage in Nova Scotia. Can. J. Pub. Health 60:345. LUND, E. & C.E. HEDSTROM. 1969. A study on sampling and isolation methods for the detection of virus in sewage. Water Res. 3:823. SHUVAL, H., B. FATTAL, S. CYMBALISTA & N. GOLDBLUM. 1969. The phase-separation method for the concentration and detection of viruses in water. Water Res. 3:225. SCHMIDT, N.J. 1972. Tissue culture in the laboratory diagnosis of virus infections. Amer. J. Clin. Pathol. 57:820. COONEY, M.K. 1973. Relative efficiency of cell cultures for detection of viruses. Health Lab. Sci. 4:295. DAHLING, D.R., G. BERG & D. BERMAN. 1974. BGM: A continuous cell line more sensitive than primary rhesus and African green kidney cells for the recovery of viruses from water. Health Lab. Sci. 11:275. SCHMIDT, N.J., H.H. HO & E.H. LENNETTE. 1976. Comparative sensitivity of the BGM cell line for the isolation of enteric viruses. Health Lab. Sci. 13:115. HATCH, M.H. & G.E. MARCHETTI. 1971. Isolation of echoviruses with human embryonic lung fibroblast cells. Appl. Microbiol. 22:736. RUTALA, W.A., D.F. SHELTON & D. ARBITER. 1977. Comparative sensitivities of viruses to cell cultures and transport media. Amer. J. Clin. Pathol. 67:397. IRVING, L.G. & F.A. SMITH. 1981. One-year survey of enteroviruses, adenoviruses and reoviruses isolated from effluent at an activated-sludge purification plant. Appl. Environ. Microbiol. 41:51. MORRIS, R. & W.M. WAITE. 1980. Evaluation of procedures for recovery of viruses from water—II detection systems. Water Res. 14: 795. LEONG, L.Y.C., S.J. BARRETT & R.R. TRUSSELL. 1978. False-positives in testing of secondary sewage for enteric viruses. Abs. Annu. Meeting, American Soc. Microbiology, Washington, D.C. KEDMI, S. & B. FATTAL. 1981. Evaluation of the false-positive enteroviral plaque © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 29. 30. 31. 32. 33. 34. 35. 36. phenomenon occurring in sewage samples. Water Res. 15:73. PAYMENT, P. & M. TRUDEL. 1985. Influence of inoculum size, incubation temperature, and cell culture density on virus detection in environmental samples. Can. J. Microbiol. 31:977. CHANG, S.L., G. BERG, K.A. BUSCH, R.E. STEVENSON, N.A. CLARKE & P.W. KABLER. 1958. Application of the Most Probable Number method for estimating concentrations of animal viruses by tissue culture technique. Virology 6:27. CHANG, S.L. 1965. Statistics of the infective units of animal viruses. In G. Berg, ed. Transmission of Viruses by the Water Route. Interscience Publ., New York, N.Y. SOBSEY, M.D. 1976. Field monitoring techniques and data analysis. In L.B. Baldwin, J.M. Davidson & J.F. Gerber, eds. Virus Aspects of Applying Municipal Waste to Land. Univ. Florida, Gainesville. THOMAS, H.A., JR. 1942. Bacterial densities from fermentation tube tests. J. Amer. Water Works Assoc. 34:572. CROW, E.L. 1956. Confidence intervals for a proportion. Biometrika 43:423. MELNICK, J.L., V. RENNICK, B. HAMPIL, N.J. SCHMIDT & H.H. HO. 1973. Lyophilized combination pools of enterovirus equine antisera: Preparation and test procedures for the identification of field strains of 42 enteroviruses. Bull. World Health Org. 48:263. MELNICK, J.L., N.J. SCHMIDT, B. HAMPIL & H.H. HO. 1977. Lyophilized combination pools of enterovirus equine antisera: Preparation and test procedures for the identification of field strains of 19 group A coxsackievirus serotypes. Intervirology 8:1720. 9610 DETECTION OF FUNGI*#(109) 9610 A. Introduction 1. Significance Fungi, including yeasts and filamentous species or molds, are ubiquitously distributed, achlorophyllous, heterotrophic organisms with organized nuclei and usually with rigid walls. They may be found wherever nonliving organic matter occurs, although some species are pathogenic and others are parasitic. In spring water near the source, the number of fungus spores usually is minimal. Unpolluted stream water has relatively large numbers of species representing the true aquatic fungi (species possessing flagellated zoospores and gametes), aquatic Hyphomycetes, and soil fungi. Moderately polluted water may carry cells or spores of the three types; however, it has fewer true aquatic fungi and aquatic Hyphomycetes, and soil fungi are more numerous. Heavily polluted water has large numbers of soil fungi. The group designated as © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater soil fungi includes yeast-like fungi, many species of which have been isolated from polluted waters. The association between fungal densities and organic loading suggests that fungi may be useful indicators of pollution. Unfortunately, no single species or group of fungi has been identified as important in this role. There may be some exceptional special cases; for example, the principal distinction between the yeasts Candida lambica and C. krusei is the ability to use pentose sugars. Because the former species grows well on pentoses, it could be used as an indicator of pulp and paper mill wastes, which are high in such sugars. Certain species of yeasts and filamentous fungi are characteristic of warmer waters and may be useful indicators of thermal pollution. Because fungi possess broad enzymatic capabilities, they can degrade actively most complex natural substances and certain synthetic compounds, including some pesticides. Most fungi are aerobic or microaerophilic, although a few species show limited anaerobic metabolism and a very few are capable of totally anaerobic growth.1 Some species do not require light. 2. Occurrence and Survival Fungi are present in, and have been recovered from, diverse, remote, and extreme aquatic habitats including lakes, ponds, rivers, streams, estuaries, marine environments, wastewaters, sludge, rural and urban stormwater runoff, well waters, acid mine drainage, asphalt refineries, jet fuel systems, and aquatic sediments. a. Fungi in potable water: Fungi have been found in potable water2-7 and on the inner surface of distribution system pipes.8 Either they survive water treatment or they enter the system after treatment and remain viable. Tuberculate macroconidia of Histoplasma capsulatum9 can pass through a 0.75-m rapid sand filter. Plain sedimentation or alum flocculation and settling removed 80 to 99% of the spores. If these relatively large (8- to 14-µm) globose, tuberculate macroconidia pass through treatment, it is not surprising that other fungi, typically with smaller spores, are found in treated water. Having survived treatment or having been introduced after treatment, fungal spores can remain viable for extended periods of time. Pathogenic fungi have been stored effectively in sterile distilled water for relatively long periods.10 Spores of H. capsulatum, stored in raw Ohio River water and sterile tap water remained highly infective for mice after 400 d.11 Tastes and odors in potable water are associated with the presence of procaryotic organisms such as bacteria, actinomycetes, and cyanobacteria. However, fungi may be involved.6,7 Propagules from 19 genera of filamentous fungi have been isolated from a chlorinated surface water system and a nonchlorinated groundwater distribution system.2 The mean number of colony-forming units (CFU) was 18/100 mL in the groundwater system and 34/100 mL in the surface system. In Finland,3 fungi were isolated from rivers, lakes, and ponds supplying nine communities with sand-filtered water, three with artificially recharged groundwater, of which two used © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater chemical coagulation, and three with chemically coagulated and disinfected water. Mesophilic fungi were common in all raw water samples; however, thermotolerant fungi were more abundant in river than in lake water. Chemical coagulation and disinfection proved far more efficient in removing fungi than sand filtration and disinfection. Aspergillus fumigatus was the most common fungus. Five chlorinated groundwater systems4 in the U.S. yielded an average count per positive sample of about 5.5 CFU/100 mL. In France5 yeasts were recovered from 50% of 38 samples and filamentous fungi from 81%. Except for Aspergillus fumigatus3,4, A. flavus,4,5 and A. niger,4 the fungi isolated from potable water usually are not considered to be medically important. Fungus infections may be significant for individuals with compromised immune systems. Most of the fungi are common soil saprobes. b. Fungi in recreation waters: Some fungi pathogenic to humans may be expected in recreational waters such as pools and beaches and in accompanying washing facilities such as shower stalls. Trichophyton mentagrophytes, the cause of tinea pedis or athlete’s foot, has been isolated12 from the wooden flooring of a shower stall. Seven species of pathogenic and potentially pathogenic fungi were isolated from 361 samples of beach sand in Hawaii.13 Beach sand on the German Baltic coast, in Portugal, and on the Adriatic coast yielded Epidermophyton.14 c. Survival on chlorination: An unidentified yeast was isolated from other organisms that survived chlorination of wastewater effluents.15 This yeast survived 1 mg free chlorine/L for 20 min in contrast with E. coli, which failed to survive 5 min contact with 0.03 mg free chlorine/L. The amount of chlorine for fungus control is known at least for C. albicans. It has been shown16 that cells of C. albicans were inactivated effectively with 4 mg chlorine/L in 30 min when the initial cell count was 105 cells/mL. In an Illinois study with C. parapsilosis,17,18 a commonly isolated yeast known to cause health problems in the tropics, greater amounts of chlorine were required to inactivate the organism than coliform bacteria. Mechanisms of inactivation by chlorine on assimilative stages of yeasts and other microorganisms have been suggested.19 Fungal cells, especially conidia, can survive much higher doses of chlorine than coliform bacteria,20 including a 10-min exposure to 10 mg chlorine/L when the initial spore count was approximately 106/mL. 3. Growth Patterns and Identification In water there are two basic patterns of fungal growth. True aquatic fungi produce zoospores or gametes that are motile by means of flagella, either of the whiplash or tinsel type. Some fungi, particularly the Trichomycetes (fungi that inhabit the hind gut of certain worms, mosquito larvae, etc.), have amoeboid stages. Aquatic fungi typically are collected by exposing suitable baits (solid foodstuffs) in the habitat being examined or in a sample within the laboratory. Relatively little work on these fungi in polluted water has been done in the U.S. They have been © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater studied more extensively in polluted waters in England, Germany, and Japan. The second fungal growth form is nonmotile in all stages of the life cycle. Growth and reproduction usually are asexual (anamorphic). Three growth processes have been recognized: (a) filamentous growth with blastic spores or spores produced in special structures; (b) filamentous growth with the filaments breaking up in an arthric (fragmenting) manner to form separate spores called arthroconidia as in Geotrichum and related genera; and (c) single-celled growth produced on each parent cell, called budding, typical of the yeasts. Identification of fungi, which are considerably larger than bacteria, is dependent on colonial morphology on a solid medium, growth and reproduction morphology, and, for yeasts, physiological activity in laboratory cultures. Increasing numbers of fungi usually indicate increasing organic loadings in water or soil. Large numbers of similar fungi suggest excessive organic load while a highly diversified mycobiota indicates populations adjusted to the environmental organics. Despite their wide occurrence, little attention has been given to the presence and ecological significance of fungi in aquatic habitats. The relevance of fungi and their activities in water is emphasized by increasing knowledge of their pathogenicity for humans, animals, and plants; their role as food or energy sources; their activity in natural purification processes; and their function in sediment formation. A survey of the literature of fungi occurring in water, wastewater, and related organically polluted substrata listed 984 species21: 133 species were assigned to the Mastigomycotina (fungi with flagellated zoospores); 79 to the Zygomycotina, mostly mucoraceous fungi; 161 to the Ascomycotina, including perfect (teleomorphic) states of some of those assigned to the Fungi Imperfecti; 18 to the Basidiomycotina, including perfect states of several yeasts; and 593 to the Deuteromycotina or Fungi Imperfecti. Of the total, 133 species were zoosporic, 131 species were yeast-like, and 718 species were filamentous. Most zoosporic species were recovered from mildly polluted or unpolluted waters; of the remaining species fewer than half were recovered in numbers large enough to indicate membership in a population for even a brief period of time. The significance of fungi in both aquatic and terrestrial environments has been discussed in detail.22-38 Quantitative enumeration of fungi is not equivalent to that of unicellular bacteria because a fungal colony may develop from a single cell (spore), an aggregate of cells (a cluster of spores or a single multi-celled spore), or from a mycelial or pseudomycelial fragment (containing more than one viable cell). It is assumed that each fungal colony developing in laboratory culture originates from a single colony-forming unit (CFU), which may or may not be a single cell. 4. References 1. TABAK, H. & W.B. COOKE. 1968. Growth and metabolism of fungi in an atmosphere of nitrogen. Mycologia 60:115. 2. NAGY, L.A. & B.H. OLSON. 1982. The occurrence of filamentous fungi in water distribution systems. Can. J. Microbiol. 28:667. 3. NIEMI, R.M., S. KUNTH & K. LUNDSTROM. 1982. Actinomycetes and fungi in surface © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. waters and potable water. Appl. Environ. Microbiol. 43:378. ROSENZWEIG, W.D., H. MINNIGH & W.O. PIPES. 1986. Fungi in potable water distribution systems. J. Amer. Water Works Assoc. 78: 53. HINZELIN F. & J.C. BLOCK. 1985. Yeast and filamentous fungi in drinking water. Environ. Tech. Letters 6:101. BURMAN, N.P. 1965. Symposium on consumer complaints. 4. Taste and odour due to stagnation and local warming in long lengths of piping. Proc. Soc. Water Treat. Exam. 14:125. BAYS, L.R., N.P. BURMAN & W.M. LEWIS. 1970. Taste and odour in water supplied in Great Britain. A survey of the present position and problems for the future. Proc. Soc. Water Treat. Exam. 19:136. NAGY, L.A. & B.H. OLSON. 1985. Occurrence and significance of bacteria, fungi, and yeasts associated with distribution pipe surfaces. Proc. American Water Works Assoc., Water Quality Technology Conf., p. 213. METZLER, D.F., C. RITTER & R.L. CULP. 1956. Combined effect of water purification processes on the removal of Histoplasma capsulatum from water. Amer. J. Pub. Health 46:1571. CASTELLANI, A. 1963. The cultivation of pathogenic fungi in sterile distilled water. Commentarii 1(10):1. COOKE, W.B. & P.W. KABLER. 1953. The survival of Histoplasma capsulatum in water. Lloydia 16:252. AJELLO, L. & M.E. GETZ. 1954. Recovery of dermatophytes from shoes and shower stalls. J. Invest. Derm. 22:17. KISHIMOTO, R.A. & G.E. BAKER. 1969. Pathogenic and potentially pathogenic fungi isolated from beach sands and selected soils of Oahu, Hawaii. Mycologia 61:539. MULLER, G. 1973. Occurrence of dermatophytes in the soils of European beaches. Sci. Total Environ. 2:116. ENGELBRECHT, R.S., D.H. FOSTER, E.O. GREENING & S.H. LEE. 1974. New microbial indicators of waste water efficiency. Environ. Protect. Technol. Ser. No. 670/2-73-082. JONES, J. & J.A. SCHMITT. 1978. The effect of chlorination on the survival of cells of Candida albicans. Mycologia 70:684. ENGELBRECHT, R.S. & C.N. HAAS. 1977. Acid-fast bacteria and yeasts as disinfection indicators: Enumeration methodology. Proc. American Water Works Assoc. Water Quality Technology Conf. 1977, p. 1. HAAS, C.N. & R.S. ENGELBRECHT. 1980. Chlorine dynamics during inactivation of coliforms, acid-fast bacteria, and yeasts. Water Res. 14:1749. HAAS, C.N. & R.S. ENGELBRECHT. 1980. Physiological alterations of vegetative microorganisms resulting from chlorination. J. Water Pollut. Control Fed. 52:1976. ROSENZWEIG, D.W., H.A. MINNIGH & W.O. PIPES. 1983. Chlorine demand and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. inactivation of fungal propagules. Appl. Environ. Microbiol. 45:182. COOKE, W.B. 1986. The Fungi of ‘‘Our Mouldy Earth’’. Beihefte zur Nova Hedwigia 85:1. BARLOCHER, F. & B. KENDRICK. 1976. Hyphomycetes as intermediates of energy flow in streams. In E. B. Jones, ed. Recent Advances in Aquatic Mycology. Elek Science, London, England. COOKE, W.B. 1961. Pollution effects on the fungus population of a stream. Ecology 42:1. COOKE, W.B. 1965. The enumeration of yeast populations in a sewage treatment plant. Mycologia 57:696. COOKE, W.B. 1970. Our Mouldy Earth. FWPCA Res. Contract Ser. Publ. No. CWR, Cincinnati, Ohio. COOKE, W.B. 1971. The role of fungi in waste treatment. CRC Critical Rev. Environ. Control. 1:581. COOKE, W.B. 1976. Fungi in sewage. In E.B.G. Jones, ed. Recent Advances in Aquatic Mycology. Elek Science, London, England. COOKE, W.B. 1979. The Ecology of Fungi. CRC Press, Boca Raton, Fla. DICK, M.W. 1971. The ecology of Saprolegniales in the lentic and littoral muds with a general theory of fungi in the lake ecosystem. J. Gen. Microbiol. 65:325. HARLEY, J.L. 1971. Fungi in ecosystems. J. Appl. Ecol. 8:627. MEYERS, S.P., D.G. AHEARN & W.L. COOK. 1970. Mycological studies of Lake Champlain. Mycologia 62:504. NOELL, J. 1973. Slime-inhabiting geofungi in a polluted stream (winter/spring). Mycologia 65:57. PARK, D. 1972. Methods of detecting fungi in organic detritus in water. Trans. Brit. Mycol. Soc. 58:281. QURESHI, A.A. & B.J. DUTKA. 1974. A preliminary study on the occurrence and distribution of geofungi in Lake Ontario near the Niagara River. Proc. 17th Conf. Great Lakes Research, International Soc. Great Lakes Research. SHERRY, J.P. & A.A. QURESHI. 1986. Isolation and enumeration of fungi using membrane filtration. In B.J. Dutka, ed. Membrane Filtration Applications, Techniques, and Problems. Marcel Dekker, New York, N.Y. SIMARD, R.E. 1971. Yeasts as an indicator of pollution. Marine Poll. Bull. 12:123. SPARROW, F.K. 1968. Ecology of fresh water fungi. In G.C. Ainsworth & A.S. Sussman, eds. The Fungi, An Advanced Treatise. Vol. 3. The Fungal Population. Academic Press, New York, N.Y. TOMLINSON, T.G. & I.L. WILLIAMS. 1975. Fungi, In C.R. Curds & H.A. Hawkes, eds. Ecological Aspects of Used-Water Treatment. Vol. 1. The Organisms and their © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Ecology. Academic Press, New York, N.Y. 5. Bibliography EMERSON, R. 1958. Mycological organization. Mycologia 50:589. COOKE, W.B. 1958. CONTINUOUS SAMPLING OF TRICKLING FILTER POPULATIONS. I. PROCEDURES. Sewage Ind. Wastes 30:21. COOKE, W.B. & A. HIRSCH. 1958. Continuous sampling of trickling filter populations. II. Populations. Sewage Ind. Wastes 30:139. COOKE, W.B. 1959. Trickling filter ecology. Ecology 40:273. SPARROW, F.K. 1959. Fungi. (Ascomycetes, Phycomycetes); including W.W. Scott, Key to genera, Fungi Imperfecti (Aquatic Hyphomycetes only). In W.T. Edmondson, ed. Ward & Whipple’s Fresh Water Biology, 2nd ed. John Wiley & Sons, New York, N.Y. COOKE, W.B. 1963. A Laboratory Guide to Fungi in Polluted Waters, Sewage, and Sewage Treatment Systems, Their Identification and Culture. USPHS Publ. 999-WP-1, Cincinnati, Ohio. FULLER, M.S. & R.O. PAYTON. 1964. A new technique for the isolation of aquatic fungi. BioScience 14:45. WILLOUGHBY, L.C. & V.G. COLLINS. 1966. A study of fungal spores and bacteria in Blelham Tarn and its associated streams. Nova Hedwigia 12:150. COOKE, W.B. & G.S. MATSUURA. 1969. Distribution of fungi in a waste stabilization pond system. Ecology 50:689. BROCK, T.D. 1970. Biology of Microorganisms. Prentice-Hall, Englewood Cliffs, N.J. COOKE, W.B. 1970. Fungi in the Lebanon sewage treatment plant and in Turtle Creek, Warren Co., Ohio. Mycopathol. Mycol. Appl. 42:89. PATERSON, R.A. 1971. Lacustrine fungal communities. In J. Cairns, ed. Structure and Function of Microbial Communities. Symposium, American Microscopical Soc., Burlington, Vt. 1969. Virginia Polytechnic Inst. & State Univ. Res. Div. Mono. 3:209. JONES, E.B.G. 1971. Aquatic Fungi. In C. Booth, ed. Methods in Microbiology 4:335. Academic Press, New York, N.Y. FARR, D.F. & R.A. PATERSON. 1974. Aquatic fungi in rivers: Their distribution and response to pollutants. VPI-WRRC Bull. 68, Virginia Water Resources Research Center, Virginia Polytechnic Inst. & State Univ., Blacksburg. GARETH JONES, E.B., ed. 1976. Recent Advances in Aquatic Mycology. Elek Science, London, England. FULLER, M.S., ed. 1978. Lower Fungi in the Laboratory. Palfrey Contrib. in Botany 1:1-212. Athens, Ga. ALEXOPOULOS, C.J. & C.W. MIMS. 1979. Introductory Mycology, 3rd ed. John Wiley & Sons, New York, N.Y. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9610 B. Pour Plate Technique 1. Samples a. Containers: Collect samples as directed in Section 9060A. Alternatively, use cylindrical plastic vials with snap-on caps. These vials usually are sterile as received. Transport them in an upright position to minimize the chance of leakage and discard after use. b. Storage: Hold samples for not more than 24 h. If analysis is not begun promptly after sample collection, refrigerate. 2. Media For counting, neopeptone-glucose-rose bengal-aureomycin® agar is the usual medium of choice, although experience may indicate that Czapek agar (for Aspergillus, Penicillium, and related fungi) and yeast extract-malt extract-glucose agar or Diamalt agar (for yeasts) may be preferable. For inventory, use neopeptone-glucose agar. a. Neopeptone-glucose-rose bengal aureomycin® agar: Add 5.0 g neopeptone, 10.0 g glucose, 3.5 mL rose bengal solution (1 g/100 mL reagent-grade water), and 20.0 g agar to 1 L reagent-grade water. Because this medium is used for making pour plates, prepare and store basal agar either in bulk, or more conveniently, in tubes in 10-mL amounts. Sterilize by autoclaving; the final pH should be about 6.5. Separately prepare a solution of chlortetracycline or tetracycline (1.0 g water-soluble antibiotic/150 mL reagent-grade water) and refrigerate. Before use, sterilize by filtration. To complete the medium, add 0.05 mL sterile solution to 10 mL melted basal agar at about 45°C. This medium may not be available in dehydrated form and may require preparation from the basic ingredients. Dehydrated Cooke’s rose bengal agar may be used in place of the agar base. This medium is useful for isolating a broad spectrum of fungal species. b. Czapek (or Czapek-Dox) agar: Dissolve 30.0 g sucrose, 3.0 g sodium nitrate (NaNO3), 1.0 g dipotassium hydrogen phosphate (K2HPO4), 0.5 g magnesium sulfate (MgSO4), 0.5 g potassium chloride (KCl), 0.01 g ferrous sulfate (FeSO4), and 15.0 g agar in 1 L reagent-grade water. The pH should be 7.3 after sterilization. This medium is useful for isolating species of Aspergillus, Penicillium, Paecilomyces, and some other fungi with similar physiological requirements. c. Yeast extract-malt extract-glucose agar: Dissolve 3.0 g yeast extract, 3.0 g malt extract, 5.0 g neopeptone (or equivalent), 10.0 g glucose, and 20.0 g agar in 1 L reagent-grade water. No adjustment of pH is required. This medium is useful for isolating yeasts. d. Diamalt agar: Dissolve 150 g diamalt and 20.0 g agar in 1 L reagent-grade water. No adjustment of pH is required. The medium will be turbid but filtration is unnecessary. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater This medium is useful in the purification of yeast isolates and for study of yeast species in various specified tests. e. Neopeptone-glucose agar: Dissolve 5.0 g neopeptone (or equivalent), 10.0 g glucose, and 20.0 g agar in 1 L reagent-grade water. The pH should be about 6.5 after sterilization. (This medium is known also as Emmons’ Sabouraud Agar or Emmons’ Sabouraud Dextrose Agar.) This medium is useful for maintaining stock cultures. It is comparable to neopeptone-glucose-rose bengal aureomycin® agar but contains neither rose bengal nor an antibiotic. 3. Procedure As many as 40 samples can be analyzed simultaneously by a single analyst by the following procedure; however, 20 samples represents the optimum number. a. Preparation and dilution: To a sterile 250-mL erlenmeyer flask add 135 mL sterile reagent-grade water and 15 mL sample to obtain a 1:10 sample dilution. Use a sterile measuring device for each sample, or, less preferably, rinse the measure with sterile reagent-grade water between samples. Mix sample well before withdrawing the 15-mL portion. Shake flask on a rotary shaker at about 120 to 150 oscillations/min for about 30 min or transfer flask contents to a blender jar, cover and blend at low speed for 1 min or high speed for 30 s. Preferably use a sterile blender jar and appurtenances for each sample or wash jar thoroughly between samples and rinse with sterile water. Further dilutions may be made by adding 45 mL sterile water to 5 mL of a 1:10 diluted suspension. For stream water samples a dilution of 1:10 usually is adequate. Dilute samples with large amounts of organic material, such as sediments, to 1:100 or 1:1000. Dilute stream bank or soil samples to 1:1000 or 1:10 000. b. Plating: Prepare five plates for each dilution to be examined. To use neopeptone-glucose-rose bengal-aureomycin® agar, aseptically transfer 10 mL of medium at 45°C to a 9-cm petri dish. Add 1 mL of appropriate sample dilution and mix thoroughly by tilting and rotating dish (see plating procedure under heterotrophic plate count, Section 9215). Alternatively add to petri dish 1 mL sample, 0.05 mL antibiotic solution, and 10 mL liquefied agar medium at 43 to 45°C. Solidify agar as rapidly as possible. (In arid areas use more medium to prevent dehydration during incubation.) c. Incubation: Stack plates but do not invert. Incubate at room conditions of temperature (20 to 24°C) and lighting but avoid direct sunlight. Examine and count plates after 3, 5, and 7 d. d. Counting and inventory: The fungus plate count will provide the basis for rough quantitative comparisons among samples; the inventory will give relative importance of at least the more readily identifiable species or genera. In preparing plates, use sample portions that will give about 50 to 60 colonies on a plate. Determine this volume by trial and error. When first examining a new habitat plate at least two sample dilutions. Estimates of up to 300 colonies may be made, but discard more crowded plates. The medium containing rose bengal tends to produce discrete colonies and permits © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater slow-growing organisms to develop. The inventory includes the direct identification of fungi based on colonial morphology and the counting of colonies assignable to various species or genera. When discrete colonies cannot be identified and identification is important, use a nichrome wire with its tip bent in an L-shape to pick from each selected colony and streak on a slant of neopeptone-glucose agar (¶ 2e). If five plates are used per sample, the average number of colonies on all plates (total number of colonies counted/5) times the reciprocal of the dilution (10/1, 100/1, 1000/1, etc.) equals the fungus colony count per milliliter of original sample. For solid or semisolid samples, use a correction for the water content to report fungus colonies per gram dry weight. Determine water content by drying paired 15-mL portions of original sample at 100°C overnight; the difference between wet and dry weights is the amount of water lost from the sample. 9610 C. Spread Plate Technique The spread plate technique is an alternative procedure for obtaining quantitative data on colony-forming units. 1. Samples See Section 9610B.1. 2. Media Use any of the following media. Aureomycin®-rose bengal-glucose-peptone agar (ARGPA) (e) and streptomycin-terramycin®-malt extract agar (STMEA) () are useful in analyzing sewage and polluted waters.1 a. Neopeptone-glucose-rose bengal aureomycin® agar: See Section 9610B.2a. b. Czapek (or Czapek-Dox) agar: See Section 9610B.2b. c. Yeast extract-malt extract-glucose agar: See Section 9610B.2c. d. Diamalt agar: See Section 9610B.2d. e. Aureomycin®-rose bengal-glucose-peptone agar: Dissolve 10.0 g glucose, 5.0 g peptone, 1.0 g potassium dihydrogen phosphate (KH2PO4), 0.5 g magnesium sulfate (MgSO4⋅7H2O), 0.035 g rose bengal, and 20.0 g agar in 800 mL reagent-grade water and sterilize. Dissolve 70.0 mg aureomycin® hydrochloride in 200 mL reagent-grade water, sterilize by filtration, and add to the cooled (42 to 45°C) agar base. The pH should be 5.4. Pour 25-mL portions into sterile petri dishes (100 × 15 mm) and let agar harden. Poured plates may be held up to 4 weeks at 4°C. f. Streptomycin-terramycin®-malt extract agar: Dissolve 30.0 g malt extract, 5.0 g peptone, and 15.0 g agar in 800 mL reagent-grade water and sterilize. Dissolve 70.0 mg each of streptomycin and terramycin® in separate 100-mL portions reagent-grade water, sterilize by filtration, and add to the cooled (42 to 45°C) agar base. The pH should be 5.4. Pour about 20-mL portions into sterile petri dishes (60 × 15 mm) and let agar harden. Poured plates may be held up © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater to 4 weeks at 4°C. 3. Procedure a. Preparation and dilution: See Section 9215A.5 and Section 9610B.3a. Make dilutions with buffered water (Section 9050C.1) and select dilutions that yield 20 to 150 colonies per plate. b. Plating: Pre-dry plates separately with lids off in a laminar-flow hood at room temperature and about 30% relative humidity for 1 to 1.5 h. Prepare at least three plates, or five plates if data are to be analyzed statistically, per sample or dilution. Using a sterile pipet, transfer 1.0 mL of sample or dilution onto surface of a pre-dried agar plate. Spread sample over entire agar surface using a sterile L-shaped glass rod or use a mechanical device to rotate plate and ensure proper sample distribution. c. Incubation: With dish covers on, let plates dry at room temperature, invert plates, and incubate at 15°C for 7 d in an atmosphere of high humidity (90 to 95%). Alternatively incubate at 20°C for 5 to 7 d. Slow-growing fungi may not produce noticeable colonies until 6 or 7 d. d. Counting and recording: Using a Quebec colony counter, count all colonies on each selected plate. If counting must be delayed temporarily, hold plates at 4°C for not longer than 24 h. Depending on colony size, plates with as many as 150 colonies can be counted but the optimal maximum number is 100 colonies. Record results as colony-forming units (CFU)/100 mL original sample. For solid or semisolid samples report CFU/g wet or dry, preferably dry. If three or more plates are used per sample, use average number of colonies times the reciprocal of the dilution (see Section 9610B) to give colony count. If no plates have colonies, record count as < 1 for the highest dilution. If there are more than 150/plate, record as Too Numerous To Count (TNTC) but indicate a count of > 150 for the appropriate dilution. If colonies are crowded and overlapping with spreaders, record as ‘‘obscured’’ (OBSC) and repeat analysis with higher dilution or earlier observations. 4. Reference 1. EL-SHAARAWI, A., A.A. QURESHI & B.J. DUTKA. 1977. Study of microbiological and physical parameters in Lake Ontario adjacent to the Niagara River. J. Great Lakes Res. 3:196. 9610 D. Membrane Filter Technique For general information on the membrane filter technique and apparatus needed see Section 9222. However, except for comparisons of different manufacturers’ membranes, no critical tests have been reported for membrane filters for fungal isolation efficiency. Media components, pH levels, and antibiotics have been used in routine plating procedures. It appears that the reported procedures are satisfactory. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 1. Samples See Section 9610B.1 and Section 9060A. 2. Media Use unmodified or modified aureomycin®-rose bengal-glucose-peptone agar (MARGPA) or modified streptomycin-terramycin®-malt extract agar (MSTMEA).1 These media are prepared identically to the unmodified media described in 9610C.2e and f except that the concentration of each antibiotic is increased from 70 mg/L to 200 mg/L. Dispense media in portions of 5 to 7 mL in glass or plastic petri dishes (60 × 15 mm); plastic dishes with tight-fitting lids are preferred. 3. Procedure a. Preparation and dilution: See Section 9215A.5 and Section 9610B.3a. Select dilutions to yield 20 to 100 colonies per membrane. b. Filtration: Filter appropriate volumes of well-shaken sample or dilution, in triplicate, through membrane filters with pore diameter of 0.45 or 0.8 µm. See Section 9222. c. Incubation: Transfer filters to dishes, invert dishes or not, and incubate at 15°C for 5 d in a humid atmosphere. Alternatively incubate at 20°C for 3 d, or longer depending on fungi present. d. Counting and recording: Using a binocular dissecting microscope at a magnification of 10×, count all colonies on each selected plate. If counting must be delayed temporarily, hold plates at 4°C for not longer than 24 h. Ideal plates have 20 to 80 colonies per filter. See Section 9610C. 4. Reference 1. QURESHI, A.A. & B.J. DUTKA. 1978. Comparison of various brands of membrane filter for their ability to recover fungi from water. Appl. Environ. Microbiol. 32:445. 9610 E. Technique for Yeasts Of the total number of fungal colonies obtained from polluted waters, as many as 50% may be yeast colonies. Solid media such as those described above do not permit growth of all yeasts; thus, a quantitative enrichment technique may be useful in addition to the plate count (see also Fungi Pathogenic to Humans, Section 9610H). 1. Media For enrichment, use yeast nitrogen base-glucose broth; for isolation, use yeast extract-malt extract-glucose agar or diamalt agar. a. Yeast nitrogen base-glucose broth: Dissolve 13.4 g yeast nitrogen base in 1 L reagent-grade water; sterilize by filtration. Prepare 500 mL each of 2% and 40% aqueous glucose solutions and sterilize separately by filtration. To make final medium, aseptically add to © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater a sterile 250-mL erlenmeyer flask 25 mL yeast nitrogen base solution and 25 mL of either 2% or 40% glucose solutions to make 1% or 20% final glucose concentrations. Stopper flask with a gauze-wrapped cotton stopper and store until used. b. Yeast extract-malt extract-glucose agar: See Section 9610B.2c. c. Diamalt agar: See Section 9610B.2d. 2. Procedure a. Sample preparation and dilution: Prepare as directed in Section 9610B. b. Enrichment: In 250-mL erlenmeyer flasks prepare one flask each of yeast nitrogen base medium containing 1% and 20% glucose. Inoculate with 1 mL of appropriate sample dilution and incubate at room temperature on a rotary shaker operating at 120 to 150 oscillations/min for at least 64 h. Shaken cultures are necessary to prevent overgrowth by filamentous fungi. c. Isolation: Remove flasks from shaker and let settle 4 to 5 h. Yeast cells, if present, will settle to the bottom, bacteria and filamentous fungi will remain in suspension, and filamentous fungi will float on the surface or will be attached to the glass surface at or above the meniscus. With a nichrome wire loop remove a loopful of sediment at the sediment-supernatant interface from a tilted flask and smear-streak on yeast extract-malt extract-glucose agar. Use three plates per flask. Incubate at room temperature but out of direct sunlight for 2 to 3 d. It is not necessary to invert dishes. To obtain pure cultures, pick from reasonably isolated colonies and restreak on the same medium or on diamalt agar plates. Obtain pure cultures of as many different colonies as can be recognized. d. Counting: It is impossible to obtain a meaningful plate count after this type of enrichment isolation. If it is assumed that one cell in the original sample will produce one or more colonies on the plates after enrichment, it can be stated that yeasts, or specific types of yeasts, occur at a minimal number dependent on the highest positive dilution. The reciprocal of this dilution is the indicated number of yeasts in the sample. 3. Bibliography LODDER, J., ed. 1970. Th e Yeasts, A Taxonomic Study, 2nd ed. North Holland Publ. Co., Amsterdam. BUCK, J.D. 1975. Distribution of aquatic yeasts—effect of inoculation temperature and chloramphenicol concentration on isolation. Mycopathologia 56:73. 9610 F. Zoosporic Fungi 1. Occurrence and Significance Most fungi found in lacustrine (lake) and lotic (river) habitats that reproduce asexually by motile, uniflagellate spores and have determinate growth of the fungal body belong to the class Chytridiomycetes. Fungi with indeterminate growth, asexual reproduction by motile, biflagellate © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater spores, and sexual reproduction involving oogonia and antheridia, are members of the class Oomycetes. A reduction in numbers of species of both classes tends to occur in polluted areas of rivers but more species of Oomycetes than of Chytridiomycetes can be found in polluted situations. Species of the Oomycete genera Saprolegnia (notably S. ferax) and Leptomitus appear to be more tolerant than other forms. Bioassay studies indicate that Oomycetes are more tolerant to zinc, cyanide, and mannitol than are Chytridiomycetes. The latter appear to be more tolerant to treatment with surfactants than do the Oomycetes. Some Chytridiomycetes may parasitize planktonic and other algae. In the case of epidemic fungal infections of phytoplankton species, the activities of fungi may affect the composition of phytoplankton communities by delaying the time of algal maxima and by reducing the population of certain algae so that other phytoplankters will replace the infected algal populations. In the case of nonepidemic infections, fungi may not influence algal populations; instead, they may infect only phytoplankters during periods of decline and thus only hasten decomposition of the algae. Filamentous Oomycetes, particularly members of the Saprolegniaceae and Pythiaceae, are found in virtually all types of freshwater habitats and damp-to-wet soils. Most of the nearly 250 species involved occur as saprobes on dead and decaying organic matter such as insect exuviae, algae, and submerged vascular plant remains. A few occur as parasites of algae, aquatic invertebrates, fish, and vascular plants; none are associated with human disease. Rarely do any of these fungi develop in sufficient numbers to be observed or collected directly. Consequently, various techniques have been devised for their collection and isolation. 2. Sampling and Baiting Collect samples in sterile 35-mL plastic vials, refrigerate, and start analysis within 6 to 8 h. Place each sample in a sterile plate (20 × 100 mm) and dilute with 10 to 15 mL sterile reagent-grade water. Add three to four split hemp seed halves (Cannabis sativa), or whole seeds of mustard (Brassica) or sesame (Sesamum) as bait to each culture. Incubate at 18 to 23°C and examine daily for fungal growth on the bait. As growth becomes evident, usually within 72 h, remove the infected bait, wash it thoroughly with water from a wash bottle, and transfer to a fresh plate of water containing two to three halves of hemp or other seed. Genera may be identified from spore arrangement within the sporangium and the manner in which spores are released. Specific determination requires microscopic examination of the sexual reproductive structures. To collect the few naturally occurring parasites or pathogens, place the host organisms in a plate containing sterile water and hemp seed. 3. Isolation Although most filamentous Oomycetes can be cultivated on plain cornmeal agar, selective media for isolating Saprolegnia from fresh water have been developed.1 Obtain axenic cultures by drawing spores into a micropipet as they emerge from the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater sporangium. Less preferably, use hyphal tips, but note that several different genera and species frequently occur on a single piece of bait. Transfer the spore suspension or hyphal tip to a plate of cornmeal agar. When growth on the agar has occurred, remove bacteria-free hyphal tips aseptically by cutting out a small block of agar. Transfer to fresh medium or water. If growth is not free from contamination after one transfer, make additional transfers to insure pure cultures. Other methods have been outlined.2 4. Dilution Plating Make serial dilutions with sterile reagent-grade water (1:100 000 to 1:700 000) and spread 1 mL over surface of a freshly prepared cornmeal agar plate. Remove each developing colony and transfer to water for identification. This method permits numerical estimation and determination of composition of the Oomycete community but requires at least 10 plates. 5. References 1. HO, H.H. 1975. Selective media for the isolation of Saprolegnia spp. from fresh water. Can. J. Microbiol. 21:1126. 2. SEYMOUR, R.L. 1970. The Genus Saprolegnia. Nova Hedwigia Beihefte 19:1. 6. Bibliography WILLOUGHBY, L.G. 1962. The occurrence and distribution of reproductive spores of Saprolegniales in fresh water. J. Ecol. 50:733. 9610 G. Aquatic Hyphomycetes 1. Occurrence and Significance Freshwater Hyphomycetes are a very specialized group of conidial fungi that usually occur on partially decayed, submerged leaves and occasionally wood of angiosperms. The mycelium, which is branched and septate, ramifies through the leaf tissue, especially in petioles and veins. The conidiophores project into the water and the conidia that usually develop are liberated under water. Mature conidia also can be found in the surface foam of most rivers, streams, and lakes. The conidia of the majority of these fungi are hyaline, thin-walled, and either tetraradiately branched, that is, with four divergent arms, or sigmoid (S-shaped) with the curvature in more than one plane. A special feature of the conidia is that while suspended in water, even for long periods, they do not germinate. However, if they come to rest on a solid surface, germ tubes are produced within a few hours. The size and morphology of these spores make them potentially more prominent in plankton analysis work than the spores of other fungi. Ecological investigations of freshwater Hyphomycetes have been limited to substrate, habitat, dispersal, and their role in the enhancement of leaf substrates as food for aquatic invertebrates. The most common substrates of these organisms are submerged, decaying leaves © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater of angiosperms such as alder (Alnus), oak (Quercus), hazelnut (Corylus), elm (Ulmus), maple (Acer), chestnut (Castanea), blackberry (Rubus), ash (Fraxinus), and willow (Salix). Submerged gymnosperm leaves usually are free of aquatic Hyphomycetes. The usual habitat of these fungi is well-oxygenated water, such as alpine brooks, mountain streams, and fast-flowing rivers. However, they also have been found in slow-running, often contaminated, rivers, stagnant or temporary pools, melting snow, and soil. There is often an increase in the numbers of species and individuals of aquatic Hyphomycetes from autumn until spring, with a decline between April and June. 2. Sample Collection and Storage For most freshwater environments, collect foam or partially decayed, submerged, angiosperm leaves in sterile bottles. Refrigerate sample until it is examined. 3. Sample Treatment and Analysis Wash the leaf samples in sterile distilled water and place one to three leaves in a sterile petri dish about 1 cm deep containing sterile pond, river, or lake water. Incubate at room temperature. Within 1 to 2 d, the mycelium and conidia develop. Conidiophores and conidia can be observed with a dissecting microscope on any portion of a leaf surface, but most frequently are seen on petioles and veins. When released, the conidia either remain suspended in the water or settle to the dish bottom. Using a dissecting microscope, pick up single conidia with a micropipet. Transfer each conidium to a microscope slide in a drop of water for identification. The conidium may be transferred with a sterile needle to a plate of 2% malt extract agar for colony production. Search for conidia in foam samples with a dissecting microscope and isolate single conidia as described above. Submerge mycelial plugs from stock culture isolates of aquatic Hyphomycetes in autoclaved pond water in deep petri dishes; conidiogenesis usually occurs within 2 to 10 d. Conidia in all stages of development can be preserved on slides with lactophenol mounting medium in which either acid fuchsin or cotton blue is dissolved and sealed with clear fingernail polish. To permit good adherence of the nail polish, avoid excessive amounts of mounting medium. 9610 H. Fungi Pathogenic to Humans 1. Occurrence and Significance Routine isolations of fungi from polluted streams and wastewater treatment plants usually have yielded relatively few species pathogenic to human and other higher animals. Geotrichum candidum, an arthroconidium-producing fungus, for which there is, in the United States, presumptive evidence of an association with disease, is isolated almost universally. When its teleomorphic stage (an ascus) develops, it is known as Endomyces candidus. Rhinocladiella mansonii, now called Exophiala mansonii, a causal agent of one form of chromomycosis, usually in the tropics, is equally widespread. It has been listed also as Phialophora jeanselmei and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Trichosporium heteromorphum. Aspergillus fumigatus, a causal agent of pulmonary aspergillosis, is commonly isolated. Pseudallescheria (Petriellidium, Allescheria) boydii is a causal agent of eumycotic mycetomas and other eumycotic conditions grouped1 under the heading ‘‘Pseudallescheriasis.’’ Infection may follow from a puncture wound with contaminated materials or breathing such contaminated materials as sprays at wastewater treatment plants or contaminated air. It usually is recovered in its anamorphic state, Scedosporium (Monosporium) apiospermum. The presence of these fungi in stream water probably represents soil runoff because virtually all zoopathogenic fungi exist saprobically in soil as their natural reservoir. Other zoopathogenic fungi occasionally are recovered in low frequencies from streams, polluted or not. Another fungus, the yeast Candida albicans, can be recovered in varying numbers from wastewater treatment plant effluents, streams receiving such effluents, and recreational waters. In humans this fungus is usually a commensal organism, like Geotrichum candidum, coexisting in harmony with its host organism; up to 80% of normal, healthy adults have detectable levels of C. albicans in their feces, while about 35% harbor it in their oral cavities in the absence of any overt disease. A very large proportion of the female population has vaginal candidiasis in varying degrees of severity. The presence of C. albicans in raw wastewater, wastewater treatment plant effluent, or contaminated water is not surprising. C. albicans has been isolated from these habitats on routine media heavily supplemented with antibacterial drugs, but not on media or with techniques described in Section 9610B or Section 9610E. It also has been isolated from estuarine and marine habitats on a maltose-yeast nitrogen base-chloramphenicol-cycloheximide medium. 2. Identification of C. albicans C. albicans can be identified among the white and pink yeasts growing on an 0.8-µm black membrane filter on maltose-yeast nitrogen base-chloramphenicol-cycloheximide medium. From each colony, inoculate a 0.5-mL portion of calf or human blood serum, incubate at 37°C for 2 to 3 h, transfer a drop or two to a slide, and examine microscopically for the production of germ tubes from a majority of the cells. Of the white yeasts, only C. albicans produces these short hyphae from the parent cell within 2 to 3 h incubation.2 3. References 1. RIPPON, J. 1982. Medical Mycology. W.B. Saunders Co., Philadelphia, Pa. 2. BUCK, J.D. & B.M. BUBACIS. 1978. Membrane filter procedure for enumeration of Candida albicans in natural waters. Appl. Environ. Microbiol. 35:237. 4. Bibliography PAGAN, E.F. 1970. Isolation of human pathogenic fungi from river water. Ph.D. dissertation, Botany Dep., Ohio State Univ., Columbus. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9711 PATHOGENIC PROTOZOA*#(110) 9711 A. Introduction 1. Significance Pathogenic intestinal protozoa are significant problems in drinking water supplies. These organisms cause diarrhea or gastroenteritis of varying severity; numerous outbreaks have occurred. During the 1970s, waterborne outbreaks due to Giardia lamblia were noted with increasing frequency, especially in communities using unfiltered surface water sources.1 By the mid 1980s, waterborne outbreaks due to Cryptosporidium parvum began to appear, and in 1993, this organism was responsible for the largest waterborne outbreak in U.S. history.2 Recreational waterborne outbreaks also have been reported.3 Cryptosporidium can cause a severe diarrhea that is self-limiting in immunocompetent individuals but may be prolonged and life-threatening in the immunocompromised.4 Microscopic antibody-based methods generally have been used for detecting and quantifying the environmentally resistant cyst stages of the pathogenic intestinal protozoa in water samples. Using these methods, surveys have demonstrated the wide distribution and occurrence of Giardia cysts and Cryptosporidium oocysts in raw and treated water supplies.5-7 Recently, another protozoan intestinal pathogen, Cyclospora cayetanensis, has been associated with waterborne8 and foodborne illness.9 Another group of protozoan organisms, Microsporidia, while not yet associated with a waterborne outbreak, appear to be widely distributed in nature. Microsporidia have caused intestinal illness and conjunctivitis, primarily in the immunocompromised.10 Although Cyclospora have been found in drinking water,11 the methods for detecting both Cyclospora and Microsporidia in environmental samples are developmental. Giardia and Cryptosporidium occur in domestic and feral animals as well as in humans. The environment may become contaminated through direct deposit of human and animal feces or through sewage and wastewater discharges to receiving water. Ingestion of water containing these organisms may cause disease. 2. References 1. CRAUN, G.F. 1990. Waterborne giardiasis. In E.A. Meyer, ed. Giardiasis. Elsevier, New York, N.Y. 2. MACKENZIE, W.R., N.J. HOXIE, M.E. PROCTOR, M.S. GRADUS, K.A. BLAIR, D.E. PETERSON, J.J. KAZMIERCZAK, D.G. ADDISS, K.R. FOX, J.B. ROSE & J.P. DAVIS. 1994. A massive outbreak in Milwaukee of Cryptosporidium infection transmitted through the public water supply. N. England J. Med. 331(3):161. 3. ROSE, J.B., J.T. LISLE & M. LECHEVALLIER. 1997. Waterborne cryptosporidiosis: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 4. 5. 6. 7. 8. 9. 10. 11. incidence, outbreaks and treatment strategies. In R. Fayer, ed. Cryptosporidium and Cryptosporidiosis. CRC Press, Boca Raton, Fla. UNGAR, B.L.P. 1994. Cryptosporidium and cryptosporidiosis. In S. Broder, T.C. Merigan, Jr. & D. Bolognesi, eds. Textbook of AIDS Medicine. Williams & Wilkins, Baltimore, Md. LECHEVALLIER, M.W., W.D. NORTON & R.G. LEE. 1991. Occurrence of Giardia and Cryptosporidium spp. in surface water supplies. Appl. Environ. Microbiol. 57:2610. LECHEVALLIER, M.W., W.D. NORTON & R.G. LEE. 1991. Giardia and Cryptosporidium spp. in filtered drinking water supplies. Appl. Environ. Microbiol. 57:2617. ROSE, J.B., C.P. GERBA & W. JAKUBOWSKI. 1991. Survey of potable water supplies for Cryptosporidium and Giardia. Environ. Sci. Technol. 25:1393. SOAVE, R. & W.D. JOHNSON, JR. 1995. Cyclospora: conquest of an emerging pathogen. Lancet 345:667. CENTERS FOR DISEASE CONTROL AND PREVENTION. 1996. Update: outbreaks of Cyclospora cayetanensis infection—United States and Canada, 1996. Morbid. Mortal. Weekly Rep. 45:611. WEBER, R., R.T. BRYAN, D.A. SCHWARTZ & R.L. OWEN. 1994. Human microsporidial infection. Clin. Microbiol. Rev. 7:426. RABOLD, J.G., C.W. HOGE, D.R. SHLIM, C. KEFFORD, R. RAJAH & P. ECHEVERRIA. 1994. Cyclospora outbreak associated with chlorinated drinking water. Lancet 344:1360. 9711 B. Detection and Enumeration Methods 1. General Discussion Methods for the simultaneous detection and enumeration of Giardia cysts and Cryptosporidium oocysts in water have appeared in previous editions of Standard Methods. These methods were developed to assist in the investigation of suspected waterborne disease outbreaks. They were applied subsequently to occurrence and distribution studies and to the determination of drinking water treatment effectiveness. The need for quantitative methods for regulatory purposes1 resulted in method evaluation studies that underscored the deficiencies of the existing antibody-based immunofluorescence methods.2 These deficiencies included requiring analysts with a high degree of experience, lengthy analysis time, high expense, lack of specificity, erratic efficiency, low precision, and difficulty in determining viability.3 The regulatory requirements for precise, sensitive, and quantitative methods for these organisms have stimulated interest and research on methods for their detection. No method is included in this edition of Standard Methods because the methods for Giardia and Cryptosporidium are evolving rapidly. A regulatory method based on the widely used © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater immunofluorescence assay is available.4 In addition, a draft method that addresses some of the limitations of the regulatory method has been published.5 The reader with a need to analyze water samples for pathogenic intestinal protozoa is advised to consult the current literature for the latest methodology. 2. References 1. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1996. Monitoring requirements for public drinking water supplies: Cryptosporidium, Giardia, viruses, disinfection byproducts, water treatment plant data and other information requirements. 40 CFR Part 141; Federal Register 61: 24353. 2. Development of Performance Evaluation (PE) Sample Preparation Protocols for Giardia Cysts and Cryptosporidium Oocysts. 1996. EPA Contract No. 68-C3-0365, available from EPA Water Docket, 202/ 260-3027. 3. JAKUBOWSKI, W., S. BOUTROS, W. FABER, R. FAYER, W. GHIORSE, M. LECHEVALLIER, J. ROSE, S. SCHAUB, A. SINGH & M. STEWART. 1996. Status of environmental methods for Cryptosporidium. J. Amer. Water Works Assoc. 88(9):107. 4. FOUT, G.S., F.W. SCHAEFER, J.W. MESSER, D.R. JAHLING & R.E. STETLER. 1996. ICR Microbial Laboratory Manual. EPA 600/R-95-178, U.S. Environmental Protection Agency, Washington, D.C. [available online] 5. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1997. Method 1622: Cryptosporidium in Water by Filtration/IMS/FA (draft). EPA-821/R-97-023, U.S. Environmental Protection Agency, Washington, D.C. [available online] Figures Figure 9020:1. Frequency curve (positively skewed distribution). Figure 9215:1. Preparation of dilutions. Figure 9215:2. Drying weight loss of 15-mL agar plates stored separately, inverted with lids on. Source: Unpublished data. Water Purification Lab., Chicago Dep. Water. Figure 9215:3. Weight loss of 25-mL agar plates (100 × 15 mm) dried separately in a laminar-flow hood at room temperature (24 to 26°C), relative humidity (30 to 33%), and air velocity 0.6 m/s. Source: Unpublished data. Alberta Environmental Centre, Vegreville, Alta. Figure 9221:1. Schematic outline of presumptive, confirmed, and completed phases for total coliform detection. Figure 9240:1. Filaments of Crenothrix polyspora showing variation of size and shape of cells within the sheath. Note especially the multiple small round cells, or ‘‘conidia,’’ found in one of the filaments. This distinctive feature is the reason for the name polyspora. Young © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater growing colonies usually are not encrusted with iron or manganese. Older colonies often exhibit empty sheaths that are heavily encrusted. Cells may vary considerably in size: Rod-shaped cells average 1.2 to 2.0 µm in width by 2.4 to 5.6 µm in length; coccoid cells of ‘‘conidia’’ average 0.6 µm in diameter. Figure 9240:2. Filaments of Sphaerotilus natans, showing cells within the filaments and some free ‘‘swarmer’’ cells. Filaments show false branching and areas devoid of cells. Individual cells within the sheath may vary in size, averaging 0.6 to 2.4 µm in width by 1.0 to 12.0 µm in length; most strains are 1.1 to 1.6 µm wide by 2.0 to 4.0 µm long. Figure 9240:3. Laboratory culture of Gallionella ferruginea, showing cells, stalks excreted by cells, and branching of stalks where cells have divided. A precipitate of inorganic iron on and around the stalks often blurs the outlines. Cells at tip of stalk average 0.4 to 0.6 µm in width by 0.7 to 1.1 µm in length. Figure 9240:4. Mixture of fragments of stalks of Gallionella ferruginea and inorganic iron-manganese precipitate found in natural samples from wells. Fragmented stalks appear golden yellow to orange when examined under the microscope. Figure 9240:5. Single-celled iron bacterium Siderocapsa treubii. Cells are surrounded by a deposit of ferric hydrate. Individual cells average 0.4 to 1.5 µm in width by 0.8 to 2.5 µm in length. Figure 9240:6. Photosynthetic purple sulfur bacteria. Large masses of cells have brown-orange to purple color—may appear chalky if there is a large amount of sulfur within the cells. Left: cells of Chromatium okenii (5.0 to 6.5 µm wide by 8 to 15 µm long) containing sulfur globules. Right: Thiospirillum jenense (3.5 to 4.5 µm wide by 30 to 40 µm long); cell contains sulfur globules and polar flagellum is visible. Figure 9240:7. Colorless filamentous sulfur bacteria: Beggiatoa alba trichomes, containing globules of sulfur. Filaments are composed of a linear series of individual rod-shaped cells that may be visible when not obscured by light reflecting from sulfur granules. Trichomes are 2 to 15 µm in diameter and may be up to 1500 µm long; individual cells, if visible, are 4.0 to 16.0 µm long. Figure 9240:8. Thiothrix nivea in a rosette formation. Filaments are 1 to 1.5 µm in diameter and of varying length. Individual cells are 1 to 1.5 µm wide and 2 to 4 µm long. Figure 9240:9. Colorless filamentous sulfur bacteria: portion of a colony, showing branching of the mucoid filament, identified as Thiodendron mucosum.1 Because the name Thiodendron previously had been used in bacterial taxonomy, its use here is illegitimate and this organism remains unnamed. Individual cells (1.0 to 2.5 µm wide by 3 to 9 µm long) have been found within the jelly-like material of the filaments. The long axis of the cells runs parallel to the long axis of the filaments. Figure 9240:10. Colorless nonfilamentous sulfur bacteria: dividing cell of Thiovolum majus, containing sulfur globules. Cells may measure 9 to 17 µm in width by 11 to 18 µm in © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater length and are generally found in nature in a marine littoral zone rich in organic matter and hydrogen sulfide. Figure 9250:1. Bacterial colonies—typical colony type vs. actinomycete colony type, 50×. Left: A typical bacterial colony characterized by a smooth mucoid appearance and a relatively distinct smooth border. Right: An actinomycete colony characterized by the mass of branching filaments that result in the fuzzy appearance of its border and by the dull powdery appearance of the spore-laden, aerial hyphae. Figure 9510:1. Two-stage microporous filter adsorption-elution method for concentrating viruses from large volumes of water with electronegative filters. Figure 9510:2. Schematic of apparatus for first-stage concentration with negatively charged filters. Tables TABLE 9020:I. KEY QUALITY CONTROL PRACTICES Further Information in Section 9020B, ¶ Item Action Frequency Reagent water Bench surface Air in workplace Thermometers Balances and weights Balances pH meter Monitor quality Monitor for contamination Monitor bacterial density Check accuracy Check accuracy Service and recalibrate Standardize Check against another meter Check volume accuracy See Table 9020:II Weekly 2e Monthly 2e Semiannually 3a Monthly 3b Annually 3b Each use 3c Monthly 3c Each use 3f Check performance Check performance Check temperature Check temperature Defrost Check for leaks and surface scratches Test with UV meter Monthly Each use Daily Daily Semiannually Each use 3g 3h 3i 3j 3j 3k Quarterly 3l Media-dispensing apparatus Hot-air oven Autoclave Refrigerator Freezer Membrane filtration equipment UV lamps © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Further Information in Section 9020B, ¶ Item Action Frequency Biohazard hood Monitor air and UV lamps Inspect for airflow Check temperature Clean optics and stage Inspect for cleanliness, chips, and etching Check pH Conduct inhibitory residue test Check pH and volume Check pH and appearance Check performance Perform duplicate analyses Repeat counts Monthly Quarterly Twice daily Each use Each use 3m 3m 3n and o 3p 4a Each batch Annually Each use Each use Weekly Weekly Monthly 4a1) 4a2) 4c 4i1) 4i2) 8a4) 8a2) Incubator Microscope Glassware Dilution water bottles Media Autoclave Plate counts TABLE 9020:II. QUALITY OF REAGENT WATER USED IN MICROBIOLOGY TESTING Test Chemical tests: Conductivity pH Total organic carbon Heavy metals, single (Cd, Cr, Cu, Ni, Pb, and Zn) Heavy metals, total Ammonia/organic nitrogen Total chlorine residual Bacteriological tests: Heterotrophic plate count (See Section 9215) Use test (see 4e) Monitoring Frequency Maximum Acceptable Limit Continuously or with each use With each use Monthly Annually* >0.5 megohms resistance or <2 µmhos/cm at 25°C 5.5–7.5 <1.0 mg/L <0.05 mg/L Annually* Monthly Monthly or with each use <0.10 mg/L <0.10 mg/L <0.01 mg/L Monthly < 1000 CFU/mL Quarterly and for a new source Student’s t ≤ 2.78 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater * Or more frequently if there is a problem. TABLE 9020:III. TIME AND TEMPERATURE FOR AUTOCLAVE STERILIZATION Material Time at 121°C* Membrane filters and pads Carbohydrate-containing media (lauryl tryptose, BGB broth, etc.) Contaminated materials and discarded cultures Membrane filter assemblies (wrapped), sample collection bottles (empty) Buffered dilution water, 99 mL in screw-cap bottle Rinse water, volume > 100 mL 10 min 12–15 min 30 min 15 min 15 min Adjust for volume * Except for media, times are guidelines; check for sterility. TABLE 9020:IV. HOLDING TIMES FOR PREPARED MEDIA Medium Holding Time Membrane filter (MF) broth in screw-cap flasks at 4°C MF agar in plates with tight-fitting covers at 4°C Agar or broth in loose-cap tubes at 4°C Agar or broth in tightly closed screw-cap tubes or other sealed containers Poured agar plates with loose-fitting covers in sealed plastic bags at 4°C Large volume of agar in tightly closed screw-cap flask or bottle at 4°C 96 h 2 weeks 2 weeks 3 months 2 weeks 3 months TABLE 9020:V. CONTROL CULTURES FOR MICROBIOLOGICAL TESTS Control Culture Group Total coliforms Positive Escherichia coli Negative Staphylococcus aureus © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Control Culture Group Positive Negative Fecal coliforms Enterobacter aerogenes E. coli Escherichia coli Fecal streptococci E. coli Enterococcus faecalis Enterococci S. faecalis Pseudomonas sp. E. aerogenes Streptococcus faecalis E. aerogenes Staphylococcus aureus E. coli S. mitis/salivarius TABLE 9020:VI. CALCULATION OF PRECISION CRITERION Duplicate Analyses Sample No. Logarithms of Counts Range of Logarithms (Rlog) D1 D2 L1 L2 (L1 − L2) 1 2 3 89 38 58 71 34 67 1.9494 1.5798 1.7634 1.8513 1.5315 1.8261 0.0981 0.0483 0.0627 ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ 14 15 ⋅ ⋅ 7 110 6 121 ⋅ 0.8451 2.0414 ⋅ 0.7782 2.0828 ⋅ 0.0669 0.0414 Calculations: 1) ∑ of Rlog = 0.0981 + 0.0483 + 0.0627 + . . . + 0.0669 + 0.0414= 0.718 89 3) Precision criterion = 3.27 î = 3.27 (0.0479) = 0.1566 TABLE 9020:VII. DAILY CHECKS ON PRECISION OF DUPLICATE COUNTS* Duplicate Analyses Logarithms of Counts © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater TABLE 9020:VII. DAILY CHECKS ON PRECISION OF DUPLICATE COUNTS* Duplicate Analyses Date of Analysis 8/29 8/30 8/31 D1 Logarithms of Counts L1 L2 Range of Logarithms Acceptance of Range† 1.8513 2.0414 1.8633 1.8129 2.0828 1.6990 0.0383 0.0414 0.1643 A A U D2 71 65 110 121 73 50 * Precision criterion = (3.27î) = 0.1566. † A = acceptable; U = unacceptable. TABLE 9020:VIII. COLIFORM COUNTS AND THEIR LOGARITHMS MPN Coliform Count No./100 mL log MPN 11 27 36 48 80 85 120 130 136 161 317 601 760 1020 3100 1.041 1.431 1.556 1.681 1.903 1.929 2.079 2.114 2.134 2.207 2.501 2.779 2.881 3.009 3.491 x = 442 xg = antilog 2.1825 = 152 TABLE 9020:IX. COMPARISON OF FREQUENCY OF MPN DATA © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater TABLE 9020:IX. COMPARISON OF FREQUENCY OF MPN DATA Class Interval Frequency (MPN) 0 to 400 400 to 800 800 to 1200 1200 to 1600 1600 to 2000 2000 to 2400 2400 to 2800 2800 to 3200 11 2 1 0 0 0 0 0 TABLE 9020:X. COMPARISON OF FREQUENCY OF LOG MPN DATA Class Interval Frequency (log MPN) 1.000 to 1.300 1.300 to 1.600 1.600 to 1.900 1.900 to 2.200 2.200 to 2.500 2.500 to 2.800 2.800 to 3.100 3.100 to 3.400 3.400 to 3.700 1 2 1 5 1 2 2 0 1 TABLE 9211:I. SPECIAL RAPID TECHNIQUES Microbial Group Nonspecific microflora Rapid Method Bioluminescence Chemiluminescence Impedance Colorimetric Epifluorescence/fluorometric Test Time h Sensitivity cells/mL 1 1 3–12 0.02 <1–several 100 000 500 000 100 000 10 000 — © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Microbial Group Test Time h Sensitivity cells/mL 4–5 10–13 1–7 6–12 9–12 8–20 3.5–15 2–20 0.01–500 000 1 000 000 200–100 000 >50 5–130 000 0.1–>10 000 000 2 2–3 500–3000 — Rapid Method Fecal coliforms Radiometric Glutamate decarboxylase Electrochemical Impedance Gas chromatographic assay Colorimetric Potentiometric Gram-negative bacteria Limulus assay Fluorescent antibody TABLE 9215:I. EFFECT OF TEMPERATURE OF DRYING ON WEIGHT LOSS OF 15-ML AGAR PLATES STORED SEPARATELY* Time for Plates to Lose 1 to 4 g of Water (Avg. for 5 Plates) h Plates Inverted with Lids On Plates Inverted with Lids Removed Temp. °C 1g 2g 3g 4g 1g 2g 3g 4g 24 37 50 60 32 17 6 4 64 35 12 8 95 51 18 12 125 67 24 16 3.7 1.7 0.7 — 7.0 3.5 1.3 — 10.5 5.3 1.9 — 14.0 7.0 2.7 — * Referenced in Canada Centre for Inland Waters Manual, Burlington, Ont. TABLE 9221:I. PREPARATION OF LAURYL TRYPTOSE BROTH Inoculum mL Amount of Medium in Tube mL Volume of Medium + Inoculum mL Dehydrated Lauryl Tryptose Broth Required g/L © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Inoculum mL Amount of Medium in Tube mL Volume of Medium + Inoculum mL Dehydrated Lauryl Tryptose Broth Required g/L 1 10 10 20 100 100 100 10 or more 10 20 10 50 35 20 11 or more 20 30 30 150 135 120 35.6 71.2 53.4 106.8 106.8 137.1 213.6 TABLE 9221:II. MPN INDEX AND 95% CONFIDENCE LIMITS FOR VARIOUS COMBINATIONS OF POSITIVE AND NEGATIVE RESULTS WHEN FIVE 20-ML PORTIONS ARE USED No. of Tubes Giving Positive Reaction Out of 5 of 20 mL Each MPN Index/ 100 mL 0 1 2 3 4 5 <1.1 1.1 2.6 4.6 8.0 >8.0 95% Confidence Limits (Approximate) Lower Upper 0 0.05 0.3 0.8 1.7 4.0 3.0 6.3 9.6 14.7 26.4 Infinite TABLE 9221:III. MPN INDEX AND 95% CONFIDENCE LIMITS FOR VARIOUS COMBINATIONS OF POSITIVE AND NEGATIVE RESULTS WHEN TEN 10-ML PORTIONS ARE USED No. of Tubes Giving Positive Reaction Out of 10 of 10 mL Each 0 1 MPN Index/ 100 mL <1.1 1.1 95% Confidence Limits (Approximate) Lower Upper 0 0.03 3.0 5.9 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater No. of Tubes Giving Positive Reaction Out of 10 of 10 mL Each MPN Index/ 100 mL 2 3 4 5 6 7 8 9 10 2.2 3.6 5.1 6.9 9.2 12.0 16.1 23.0 >23.0 95% Confidence Limits (Approximate) Lower Upper 0.26 8.1 0.69 10.6 1.3 13.4 2.1 16.8 3.1 21.1 4.3 27.1 5.9 36.8 8.1 59.5 13.5 Infinite TABLE 9221:IV. MPN INDEX AND 95% CONFIDENCE LIMITS FOR VARIOUS COMBINATIONS OF POSITIVE RESULTS WHEN FIVE TUBES ARE USED PER DILUTION (10 ML, 1.0 ML, 0.1 ML) 95% Confidence Limits 95% Confid Limits Combination of Positives MPN Index/ 100 mL Lower Upper Combination of Positives MPN Index/ 100 mL 0-0-0 0-0-1 0-1-0 0-2-0 <2 2 2 4 — 1.0 1.0 1.0 — 10 10 13 4-2-0 4-2-1 4-3-0 4-3-1 4-4-0 22 26 27 33 34 9.0 12 12 15 16 1-0-0 1-0-1 1-1-0 1-1-1 1-2-0 2 4 4 6 6 1.0 1.0 1.0 2.0 2.0 11 15 15 18 18 5-0-0 5-0-1 5-0-2 5-1-0 5-1-1 5-1-2 23 30 40 30 50 60 9.0 10 20 10 20 30 Lower © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 95% Confidence Limits Combination of Positives MPN Index/ 100 mL 2-0-0 2-0-1 2-1-0 2-1-1 2-2-0 2-3-0 4 7 7 9 9 12 3-0-0 3-0-1 3-1-0 3-1-1 3-2-0 3-2-1 8 11 11 14 14 17 4-0-0 4-0-1 4-1-0 4-1-1 4-1-2 Upper Combination of Positives 1.0 2.0 2.0 3.0 3.0 5.0 17 20 21 24 25 29 5-2-0 5-2-1 5-2-2 5-3-0 5-3-1 5-3-2 50 70 90 80 110 140 20 30 40 30 40 60 3.0 4.0 4.0 6.0 6.0 7.0 24 29 29 35 35 40 5-3-3 5-4-0 5-4-1 5-4-2 5-4-3 5-4-4 170 130 170 220 280 350 80 50 70 100 120 160 38 45 46 55 63 5-5-0 5-5-1 5-5-2 5-5-3 5-5-4 5-5-5 240 300 500 900 1600 ≥1600 100 100 200 300 600 — Lower 13 17 17 21 26 95% Confid Limits 5.0 7.0 7.0 9.0 12 MPN Index/ 100 mL Lower TABLE 9222:I. SUGGESTED SAMPLE VOLUMES FOR MEMBRANE FILTER TOTAL COLIFORM TEST Volume (X) To Be Filtered mL Water Source Drinking water Swimming pools Wells, springs Lakes, reservoirs Water supply intake Bathing beaches 100 50 10 1 0.1 X X X X X X X X X X X X X X 0.01 0.001 0.0001 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Volume (X) To Be Filtered mL Water Source 100 50 River water Chlorinated sewage Raw sewage 10 1 0.1 0.01 0.001 X X X X X X X X X X 0.0001 X TABLE 9222:II. CONFIDENCE LIMITS FOR MEMBRANE FILTER COLIFORM RESULTS USING 100-ML SAMPLE 95% Confidence Limits Number of Coliform Colonies Counted Lower Upper 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 0.0 0.1 0.2 0.6 1.0 1.6 2.2 2.8 3.4 4.0 4.7 5.4 6.2 6.9 7.7 8.4 9.2 9.9 10.7 11.5 12.2 3.7 5.6 7.2 8.8 10.2 11.7 13.1 14.4 15.8 17.1 18.4 19.7 21.0 22.3 23.5 24.8 26.0 27.2 28.4 29.6 30.8 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater TABLE 9222:III. SUGGESTED SAMPLES VOLUMES FOR MEMBRANE FILTER FECAL COLIFORM TEST Volume (X) To Be Filtered mL Water Source 100 50 Lakes, reservoirs Wells, springs Water supply intake Natural bathing waters Sewage treatment plant Farm ponds, rivers Stormwater runoff Raw municipal sewage Feedlot runoff Sewage sludge X X X X X X 10 1 X X X X X X X X 0.1 X X X X X 0.01 0.001 0.0001 X X X X X X X X X TABLE 9223:I. COLOR CHANGES FOR VARIOUS MEDIA Substrate Total Coliform Positive ONPG-MUG Yellow CPRG-MUG Red or magenta E. coli Positive Negative Result Blue fluorescence Blue fluorescence Colorless/no fluorescence Yellow/no fluorescence TABLE 9225:I. BIOCHEMICAL REACTIONS OF KEY SPECIES OF THE FAMILY ENTEROBACTERIACEAE* Perc Species Citrobacter diversus Citrobacter freundii Lactose Fermentation 35 50 ONPG Hydrolysis 96 95 Indole Production Methyl Red 99 5 100 100 VogesProskauer 0 0 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Perc Species Enterobacter aerogenes Enterobacter agglomerans Enterobacter cloacae Escherichia coli Escherichia coli, inactive Escherichia fergusonii Escherichia hermannii Escherichia vulneris Hafnia alvei Klebsiella oxytoca Klebsiella ozaenae Klebsiella pneumoniae Klebsiella rhinoscleromatis Serratia fonticola Serratia marcescens Lactose Fermentation ONPG Hydrolysis Indole Production Methyl Red VogesProskauer 95 40 93 95 25 0 45 15 5 100 30 98 0 97 2 100 90 99 95 45 83 98 100 90 100 80 99 0 100 95 0 20 0 98 80 98 99 0 0 99 0 0 0 0 1 5 50 5 99 95 100 100 100 40 20 98 10 100 100 20 98 70 100 0 0 0 0 0 85 95 0 98 0 9 98 * Modified after Farmer, J.J. III, 1985. Clinical identification of new species and biogroups of Enterobacteriaceae. J. Clin. Microbiol. 21:46. † Reactions that become positive after 2 d are not considered. TABLE 9230:I. SELECTED KEY BIOCHEMICAL CHARACTERISTICS OF THE STREPTOCOCCUS SPECIES WITHIN THE FECAL STREPTOCOCCUS AND ENTEROCOCCUS GROUPS* Fecal Streptococcus Group Enterococcus Group Test S. faecalis S. faecium S. avium S. gallinarum S. bovis − + + − + + − + + − + + − + + + + + + + + + + + Growth at 10°C + + + + − Pyruvate utilization7 + − − − − Phosphatase activity8 + − + + − Catalase 40% Bile Esculin6 Growth at 45°C Growth in 6.5% NaCl − © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation S Standard Methods for the Examination of Water and Wastewater Fecal Streptococcus Group Enterococcus Group Test S. faecalis S. faecium S. avium S. gallinarum S. bovis Arginine hydrolysis8 + + − − L-Sorbose fermentation9 − − −d + − − Lactose fermentation8 + + + + + n-Acetyl-β-glucoseaminidase activity9 −d − − + − Starch6 − − − − + Arabinose6 − + + − − S * + = 90% or more of strains are positive − = 90% or more of strains are negative d = reactions variable TABLE 9250:I. GENERAL MACROSCOPIC PROPERTIES OF BACTERIAL COLONIES ON SOLID MEDIUM Characteristic Typical Colony Type Actinomycete Colony Type* Appearance Shiny or opalescent Texture Degree of adherence to solid medium Edge of colony Soft Weak When young it is composed of hyphae, b some species these may later fragme Substrate and surface hyphae have n distinctive color. As the colony mature fluffy aerial hyphae that carry spores f and give to colonies of different speci various colors and sometimes a chalk appearance. Soluble pigments, either melanin or brightly colored type, that diffuse into the medium, also are com Strong and leathery Strong Regular, continuous, and not different from colony as a whole Irregular, intermittent, slightly less dense colony as a whole, and of hyphal appearance * Actinomycetes are authentic bacteria by all modern criteria, except for their hyphal character and mode of spore formation. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater TABLE 9260:I. SUMMARY DATA FROM WATERBORNE BACTERIAL DISEASE OUTBREAKS, 1985–94 Type of Water Variable Drinking water Total outbreaks Agent: Shigella Campylobacter Salmonella E. coli O157:H7 System: Noncommunity Community Individual Source: Well Lake Spring Cistern Cause: Untreated groundwater Distribution system deficiency Treatment deficiency Unknown Total outbreaks Agent: Pseudomonas Shigella Legionella Leptospira E. coli O157:H7 Location: Hotel/motel Recreational water Number 21 12 6 2 1 10 8 3 17 2 1 1 9 7 4 1 71 44 17 6 2 2 23 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Type of Water Variable Number Outdoor recreation area (surface water) Home Spa or public swimming pool Resort Apartment complex/condominum Source: Whirlpool/hot tub Lake/pond Swimming pool Stream 21 14 5 4 4 47 20 3 1 TABLE 9260:II. REACTIONS OF COMMON BACTERIA ON TSI AND LIA MEDIA Organism Shigella TSI* LIA* K/A− K/Ag+ K/A− K/A+ A/Ag− A/Ag+ or K/Ag+ A/Ag+ K/K− R/A+ K/A+ A/Ag− K/A− Aeromonas A/A− K/A− Yersinia A/A− or K/A− K/A− Plesiomonas K/A− K/A− Salmonella Escherichia Proteus Citrobacter Enterobacter * Fermentation reactions = slant/butt; H2S production = + or −; K = alkaline,A = acid, R = red (deaminase reaction); g = gas produced. TABLE 9260:III. REACTIONS OF ENTERIC BACTERIA ON TSI AND LIA MEDIA Organism TSI Reactions* LIA Reactions* Shigella K/A− K/Ag+ K/A− K/K+ Salmonella © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Organism Escherichia Proteus Citrobacter Enterobacter TSI Reactions* LIA Reactions* A/Ag− A/Ag+ or K/Ag+ A/Ag+ K/A− R/A+ K/A+ A/Ag− K/A− A/A− K/A− A/A− or K/A− K/A− A/Ag− K/A− Aeromonas Yersinia Klebsiella * Fermentation reactions = slant/butt, H2S production = + or −, K = alkaline,A = acid, R = red (deaminase reaction), g = gas produced. TABLE 9260:IV. REACTIONS OF AEROMONAS AND ENTERIC BACTERIA ON KAPER’S MEDIUM Fermentation Pattern* Motility H 2S Indole Aeromonas hydrophila K/A + − + Klebsiella pneumoniae A/A − − Klebsiella oxytoca A/A − − − + Escherichia coli K/K or K/A K/K, K/A, A/K or A/A − + + Salmonella spp. + or − + K/K, K/N or N/N + − R/K or R/A + + or − − + K/K, K/N or N/N − + + or − K/K or K/A − + K/K, K/N, or N/N + − − Organism Enterobacter spp. Proteus spp. Yersinia enterocolitica Citrobacter spp. Serratia spp. − − * K = alkaline; A = acid; N = neutral; R = red (deamination reaction). TABLE 9260:V. MYCOBACTERIA OF WATERBORNE OR UNKNOWN ORIGIN Mycobacterium Species Environmental Contaminant Reservoir M. kansasii M. marinum M. simiae Rarely Rarely No Water, swine, cattle Fish, water Primates, possibly water © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Mycobacterium Species Environmental Contaminant Reservoir M. scrofulaceum M. szulgai M. avium-intracellulare M. xenopi M. ulcerans M. fortuitum M. chelonae Possibly No Possibly Possibly No Yes Yes Soil, water, foodstuffs Unknown Soil, water, swine, cattle, birds Water Unknown Soil, water, animals, marine life Soil, water, animals, marine life TABLE 9260:VI. PHENOTYPIC CHARACTERISTICS OF CLINICALLY SIGNIFICANT ENVIRONMENTAL MYCOBACTERIA* Mycobacterium Species Nitrate Reduction Hydrolysis of Polyoxyethylen Sorbitan Monooleate† Growth Rate Pigmentation Urease M. kansasii M. marinum S S P P ± + + − + + M. simiae S P ± − − M. scrofulaceum S S ± M. szulgai M. xenopi S S S/P S + − + − ± − − − M. avium-intracellulare S N − − M. ulcerans S N − ± M. fortuitum M. chelonae R R N N + + − + − ± ± − * S = slow (3 to 8 weeks), R = rapid (7 d or longer), P = photochromogenic, S = scotochromogenic, N = nonphotochromogenic, S/P = scotochromogenic at 37°C andphotochromogenic at 24°C. † Tween 80. 10010 INTRODUCTION*#(111) Physical and chemical characteristics of water bodies affect the abundance, species © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater composition, stability, productivity, and physiological condition of aquatic organism populations. Biological methods used for assessing water quality include the collection, counting, and identification of aquatic organisms; biomass measurements; measurements of metabolic activity rates; measurements of the toxicity, bioconcentration, and bioaccumulation of pollutants; and processing and interpretation of biological data. Information from these methods may serve one or more of the following purposes: 1. To explain the cause of color, turbidity, odor, taste, or visible particulates in water; 2. To aid in the interpretation of chemical analyses, for example, in relating the presence or absence of certain biological forms to oxygen deficiency or supersaturation in natural waters; 3. To identify the source of a water that is mixing with another water; 4. To explain the clogging of pipes, screens, or filters, and to aid in the design and operation of water and wastewater treatment plants; 5. To determine optimum times for treatment of surface water with algicides and to monitor treatment effectiveness; 6. To determine the effectiveness of drinking water treatment stages and to aid in determining effective chlorine dosage within a water treatment plant; 7. To identify the nature, extent, and biological effects of pollution; 8. To indicate the progress of self-purification in bodies of water; 9. To aid in determining the condition and effectiveness of unit processes and biological wastewater treatment methods in a wastewater treatment plant; 10. To document short- and long-term variability in water quality caused by natural phenomena and/or human activities; 11. To provide data on the status of an aquatic system on a regular basis; 12. To correlate the biological mass or components with water chemistry or conditions. The specific nature of a problem and the reasons for collecting samples will dictate which communities of aquatic organisms will be examined and which sampling and analytical techniques will be used. The following communities of aquatic organisms are considered in specific sections that follow: 1. PLANKTON (Section 10200): A community of plants (phytoplankton) and animals (zooplankton), usually drifting or suspended in water, nonmotile or insufficiently motile to overcome transport by currents. In fresh water they generally are small or microscopic in size; in the marine or estuarine environment, larger forms are observed more frequently. 2. PERIPHYTON (Section 10300): A community of microscopic plants and animals associated with the surfaces of submersed objects. Some are attached, some move about. Many of the protozoa and other minute invertebrates and algae found in the plankton also occur in the periphyton. 3. MACROPHYTON (Section 10400): The larger plants of all types. They are sometimes attached to the bottom (benthic), sometimes free-floating, sometimes totally submersed, and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater sometimes partly emergent. Complex types usually have true roots, stems, and leaves; the macroalgae are simpler but may have stem- and leaf-like structures. 4. MACROINVERTEBRATES (Section 10500): The invertebrates defined here are those retained by the US Standard No. 30 sieve. They are generally bottom-dwelling organisms (benthos). 5. FISH (Section 10600): Vertebrates of diverse morphology, ecology, and behavior, inhabiting (and generally limited to) aquatic systems. They have fins and gills. 6. AMPHIBIANS, AQUATIC REPTILES, BIRDS, AND MAMMALS: These vertebrates also may be affected directly or indirectly by spills or other discharges of pollutants and may be useful in monitoring the presence of toxic substances or long-term changes in water quality. Discussion of these organisms is not included. Large numbers of bacteria and fungi are present in the plankton and periphyton and constitute an essential element of the total aquatic ecosystem. Although their interactions with living and dead organic matter profoundly affect the larger aquatic organisms, techniques for their investigation are not included herein (see Part 9000). Field observations are indispensable for meaningful biological interpretations, but many biological factors cannot be evaluated directly in the field. These must be analyzed as field data or field samples within the laboratory. Because the significance of the analytical result depends upon the representativeness of the sample, attention is given to field methods as well as to associated laboratory procedures. Before sampling begins, clearly define study objectives. For example, the frequency of a repetitive sampling program may vary from hourly, for a detailed study of diel variability, to every third month (quarterly) for a general assessment of seasonal conditions, depending on objectives. The scope of the study must be adjusted to limitations in personnel, time, and budget. Before the development of a study plan, examine historic data for the study area and conduct a literature search of work by previous investigators. Whenever practicable, biologists should collect their own samples. Much of the value of an experienced biologist lies in personal observations of conditions in the field and in the ability to recognize signs of environmental changes as reflected in the various aquatic communities. The primary orientation of Part 10000 is toward field collection and associated laboratory analyses to aid in determining the status of aquatic communities under field conditions and to aid in interpreting the influence of past and present environmental conditions. The methods selected are necessary for the appraisal of water quality. Principal emphasis is on methods and equipment, rather than on interpretation or application of results. The complex interrelationships existing in an aquatic environment often require many different field and laboratory procedures; consequently, frequent cross-references between sections have been made. Many other types of studies may be, and are being, conducted that are oriented more toward laboratory research. Such laboratory studies will develop further basic knowledge of community and/ or organism responses under controlled conditions and will aid in predicting effects of future changes in environmental conditions on the aquatic communities. However, such studies © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater are not within the scope of this book. 10200 PLANKTON*#(112) 10200 A. Introduction The term ‘‘plankton’’ refers to those microscopic aquatic forms having little or no resistance to currents and living free-floating and suspended in natural waters. Planktonic plants, ‘‘phytoplankton,’’ and planktonic animals, ‘‘zooplankton,’’ are covered in this section. The phytoplankton (microscopic algae) occur as unicellular, colonial, or filamentous forms. Many are photosynthetic and are grazed upon by zooplankton and other aquatic organisms. Other organisms occurring in the same environment are dealt with elsewhere: zoosporic fungi in Section 9610F; aquatic hyphomycetes in Section 9610G; and bacteria in Part 9000. The zooplankton in fresh water comprise principally protozoans, rotifers, cladocerans, and copepods; a greater variety of organisms occurs in marine waters. 1. Significance Plankton, particularly phytoplankton, long have been used as indicators of water quality.1-4 Some species flourish in highly eutrophic waters while others are very sensitive to organic and/or chemical wastes. Some species develop noxious blooms, sometimes creating offensive tastes and odors5 or anoxic or toxic conditions resulting in animal deaths or human illness.6 The species assemblage of phytoplankton and zooplankton also may be useful in assessing water quality.7 Because of their short life cycles, plankters respond quickly to environmental changes, and hence their standing crop and species composition are more likely to indicate the quality of the water mass in which they are found. They strongly influence certain nonbiological aspects of water quality (such as pH, color, taste, and odor), and in a very practical sense, they are a part of water quality. Certain taxa often are useful in determining the origin or recent history of a given water mass. Because of their transient nature, and often patchy distribution, however, the utility of plankters as water quality indicators may be limited. Information on plankton as indicators is interpreted best in conjunction with concurrently collected, physicochemical and other biological data. Planktonic organisms predominate in ponds, lakes, and oceans. Potamoplankton develop in large rivers with slow-moving waters that approach lentic conditions. Because their origin can be uncertain and the duration of their exposure to pollutants unknown, plankters generally are less valuable as water quality indicators in lotic than in lentic environments. 2. References 1. PALMER, C.M. 1969. A composite rating of algae tolerating organic pollution. J. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. 3. 4. 5. 6. 7. Phycol. 5:78. PALMER, C.M. 1963. The effect of pollution on river algae. Bull. N.Y. Acad. Sci. 108:389. RAWSON, D.S. 1956. Algal indicators of trophic lake types. Limnol. Oceanogr. 1:18. STOERMER, E.F. & J.J. YANG. 1969. Plankton Diatom Assemblages in Lake Michigan. Spec. Rep. No. 47, Great Lakes Research Div., Univ. Michigan, Ann Arbor. PRESCOTT, G.W. 1968. The Algae: A Review. Houghton Mifflin Co., Boston, Mass. CARMICHAEL, W., ed. 1981. The Water Environment, Algal Toxins and Health. Plenum Press, New York, N.Y. GANNON, J.E. & R.S. STEMBERGER. 1978. Zooplankton (especially crustaceans and rotifers) as indicators of water quality. Trans. Amer. Microsc. Soc. 97:16. 10200 B. Sample Collection 1. General Considerations The frequency and location of sampling is dictated by the purpose of the study.1 Locate sampling stations as near as possible to those selected for chemical and bacteriological sampling to insure maximum correlation of findings. Establish a sufficient number of stations in as many locations as necessary to define adequately the kinds and quantities of plankton in the waters studied. The physical nature of the water (standing, flowing, or tidal) will influence greatly the selection of sampling stations. The use of sampling sites selected by previous investigators usually will assure the availability of historical data that will lead to a better understanding of current results and provide continuity in the study of an area. In stream and river work, locate stations upstream and downstream from suspected pollution sources and major tributary streams and at appropriate intervals throughout the reach under investigation. If possible, locate stations on both sides of the river because lateral mixing of river water may not occur for great distances downstream. In a similar manner, investigate tributary streams suspected of being polluted but take care in the interpretation of data from a small stream because much of the plankton may be periphytic in origin, arising from scouring of natural substrates by the flowing water. Plankton contributions from adjacent lakes, reservoirs, and backwater areas, as well as soil organisms carried into the stream by runoff, also can influence data interpretation. The depth from which water is discharged from upstream stratified reservoirs also can affect the nature of the plankton. Because water of rivers and streams usually is well mixed vertically, subsurface sampling, i.e., the upper meter or a composite of two or more strata, often is adequate for collection of a representative sample. There may be problems caused by stratification due to thermal discharges or mixing of warmer or colder waters from tributaries and reservoirs. Always sample in the main channel of a river and avoid sloughs, inlets, or backwater areas that reflect local habitats rather © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater than river conditions. In rivers that are mixed vertically and horizontally, measure plankton populations by examining periodic samples collected at midstream 0.5 to 1 m below the surface. If it can be determined or correctly assumed that the plankton distribution is uniform and normal, use a scheme of random sampling to accomodate statistical testing. Include both random selection of sampling sites and transects as well as the random collection of samples at each selected site. On the other hand, if it is known or assumed that plankton distribution is variable or patchy, include additional sampling sites, collect composite samples, and increase sample replication. Use appropriate statistical tests to determine population variability. In sampling a lake or reservoir use a grid network or transect lines in combination with random procedures. Take a sufficient number of samples to make the data meaningful. Sample a circular lake basin at strategic points along a minimum of two perpendicular transects extending from shore to shore; include the deepest point in the basin. Sample a long, narrow basin at several points along a minimum of three regularly spaced parallel transects that are perpendicular to the long axis of the basin, with the first near the inlet and the last near the outlet. Sample a large bay along several parallel transects originating near shore and extending to the lake proper. Because many samples are required to appraise completely the plankton assemblage, it may be necessary to restrict sampling to strategic points, such as the vicinity of water intakes and discharges, constrictions within the water body, and major bays that may influence the main basin. In lakes, reservoirs, and estuaries where plankton populations can vary with depth, collect samples from all major depth zones or water masses. The sampling depths will be determined by the water depth at the station, the depth of the thermocline or an isohaline, or other factors. In shallow areas of 2 to 3 m depth, subsurface samples collected at 0.5 to 1 m may be adequate. In deeper areas, collect samples at regular depth intervals. In estuaries sample above and below the pyncocline. Depth intervals for sampling vary for estuaries of different sizes and depths, but use depths representative of the vertical range. Composite sampling above and below the pyncocline often is used. In marine sampling, the intent and scope of the study will determine the collection extent. Over the continental shelf, take samples at stations approximately equidistant from the shore seaward. Take a vertical series from surface to near bottom at each station, gradually adding more stations across the shelf. It is important to sample the entire vertical range over a continental shelf. Benthic grab samples may be taken to collect dormant resting cells or cysts. Beyond the shelf in pelagic waters, sample in the photic zone from the surface to the thermocline for phytoplankton and to deeper depths for zooplankton. Sampling depths vary, but often are at 10- to 25-m intervals above the thermocline, then at 100- to 200-m intervals below the thermocline to 1000 m, and thereafter at 500- to 1000-m intervals. Samples usually are referred to as ‘‘surface’’ or ‘‘depth’’ (subsurface) samples. The latter are samples taken from some stated depth, whereas surface samples may be interpreted as samples collected as near the water surface as possible. A ‘‘skimmed’’ sample of the surface film plankton (neuston)2 can be revealing; however, ordinarily do not include a disproportionate © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater quantity of surface film in a surface sample because a neustonic flora3 as well as plankton often are trapped on top or at the surface film together with pollen, dust, and other detritus. Various methods have been used for sampling surface organisms. Sampling frequency depends on the intent of the study as well as the range of seasonal fluctuations, the immediate meteorological conditions, adequacy of equipment, and availability of personnel. Select a sampling frequency at some interval shorter than community turnover time. This requires consideration of life-cycle length, competition, predation, flushing, and current displacement. Frequent plankton sampling is desirable because of normal temporal variability and migratory character of the plankton community. Daily vertical migrations occur in response to sunlight, and random horizontal migrations or drifts are produced by winds, shifting currents, and tides. Ideally, collect daily samples and, when possible, sample at different times during the day and at different depths. When this is not possible, weekly, biweekly, monthly, or even quarterly sampling still may be useful for determining major population changes. In river, stream, and estuarine regions subject to tidal influence, expect fluctuations in plankton composition over a tidal cycle. A typical sampling pattern at a station within an estuary includes a vertical series of samples taken from the surface, across the pyncocline, to near bottom, collected at 3-h intervals, over at least two complete tidal cycles. Once a characteristic pattern is recognized the sampling routine may be modified. A useful series of monographs on oceanographic methodology has been published.4-7 Representative taxonomic references for estuarine and marine phytoplankton include diatoms,8-11 dinoflagellates,12-14 coccolithophores,15 and cyanophyceae16 (cyanobacteria). 2. Sampling Procedures Once sampling locations, depths, and frequency have been determined, prepare for field sampling. Label sample containers with sufficient information to avoid confusion or error. On the label indicate date, cruise number, sampling station, study area (river, lake, reservoir), type of sample, and depth. Use waterproof labels. When possible, enclose collection vessels in a protective container to avoid breakage. If samples are to be preserved immediately after collection, add preservative to container before sampling. Sample size depends on type and number of determinations to be made; the number of replicates depends on statistical design of the study and statistical analyses selected for data interpretation. Always design a study around an objective with a statistical approach rather than fit statistical analyses to data already collected. In a field record book note sample location, depth, type, time, meteorological conditions, turbidity, water temperature, salinity, and other significant observations. Engineer’s field notebooks with waterproof paper are very suitable. Field data are invaluable when analytical results are interpreted and often help to explain unusual changes caused by the variable character of the aquatic environment. Collect coincident samples for chemical analyses to help define environmental variations having a potential effect on plankton. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater a. Phytoplankton: In oligotrophic waters or where phytoplankton densities are expected to be low collect a sample of up to 6 L. For richer, eutrophic waters collect a sample of 0.5 to 1 L. Because of their small size, nannoplankton and picoplankton can pass through collection nets, making nets unsuitable for most phytoplankton sampling. For qualitative and quantitative evaluations collect whole (unfiltered and unstrained) water samples with a water collection bottle consisting of a cylindrical tube with stoppers at each end and a closing device. Lower the open sampler to the desired depth and close by dropping a weight, called a messenger, which slides down the supporting wire or cord and trips the closing mechanism. If possible, obtain composite samples from several depths or pool samples from one depth from several casts. The most commonly used samplers that operate on this principle are the Kemmerer,17 Van Dorn18 (Figure 10200:1), Niskin, and Nansen samplers. Because these samplers collect whole water samples, all size classes of phytoplankton are collected. Different size categories of phytoplankton can be separated by subsequently filtering these whole water samples through netting of the appropriate mesh size. Select appropriate mesh sizes for concentrating the various size categories of phytoplankton typical of the aquatic system under study.19,20 The Van Dorn usually is the preferred sampler for standing crop, primary productivity, and other quantitative determinations because its design offers no inhibition to free flow of water through the cylinder. In deep-water situations, the Niskin bottle is preferred. It has the same design as the Van Dorn sampler except that the Niskin sampler can be cast in a series on a single line for simultaneous sampling at multiple depths with the use of auxiliary messengers. Because the triggering devices of these samplers are very sensitive, avoid rough handling. Always lower the sampler into the water; do not drop. Kemmerer and Van Dorn samplers have capacities of 0.5 L or more. Polyethylene or polyvinyl chloride sampling devices are preferred to metal samplers because the latter liberate metallic ions that may contaminate the sample. Use polyethylene or glass sample storage bottles. Metallic ion contamination can lead to significant errors when algal assays or productivity measurements are made. For shallow waters use the Jenkins surface mud sampler,21 one of the bottle samplers modified so that it is held horizontally,22 or an appropriate bacteriological sampler.23 For greater speed of collection and to obtain large, accurately measured quantities of organisms, use a pump. Diaphragm and peristaltic pumps are less damaging to organisms than centrifugal pumps.24 Centrifugal pump impellers can damage organisms as can passage through the hose.25 Lower a weighted hose, attached to a suction pump, to the desired depth, and pump water to the surface. The pump is advantageous because it supplies a homogeneous sample from a given depth or an integrated sample from the surface to a particular depth. If a centrifugal pump is used, draw samples from the line before they reach the impeller. For samples to be analyzed for organochlorine compounds use TFE tubing. To examine live samples fill containers partially and store in a refrigerator or ice chest in the dark, or preferably, hold at ambient temperature. Examine specimens promptly after collection. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater If it is impossible to examine living material or if phytoplankton are to be counted later, preserve the sample. For a sample that will be preserved, fill the container completely. The most suitable phytoplankton preservative is Lugol’s solution, which can be used for most forms including the naked flagellates. Unfortunately, acidic Lugol’s solution (or formalin) dissolves the coccoliths of Coccolithophores, which are common in estuarine and marine waters. Lugol’s solution: To preserve samples with Lugol’s solution add 0.3 mL Lugol’s solution to 100 mL sample and store in the dark. For long-term storage add 0.7 mL Lugol’s solution per 100 mL sample and buffered formaldehyde to a minimum of 2.5% final concentration after 1 h. Prepare Lugol’s solution by dissolving 20 g potassium iodide (KI) and 10 g iodine crystals in 200 mL distilled water containing 20 mL glacial acetic acid.26 Utermohl’s27 modification of Lugol’s solution results in a neutral or slightly alkaline solution. Prepare modified Lugol’s solution by dissolving 10 g KI and 5 g iodine crystals in 20 mL distilled water, then adding 50 mL distilled water in which 5 g anhydrous sodium acetate has been dissolved. This allows preservation of Coccolithophores, but would be less effective for other flagellates. Other acceptable preservatives are: Formalin: To preserve samples with formalin, add 40 mL buffered formalin (20 g sodium borate, Na2B2O4, + 1 L 37% formaldehyde) to 1 L of sample immediately after collection. In estuarine and marine collections, adjust pH to at least 7.5 with sodium borate for samples containing Coccolithophores. Merthiolate: To preserve samples with merthiolate add 36 mL merthiolate solution to 1 L of sample and store in the dark. Prepare merthiolate solution by dissolving 1.0 g merthiolate, 1.5 g sodium borate, and 1.0 mL Lugol’s solution in 1 L distilled water. Merthiolate-preserved samples are not sterile, but can be kept effectively for 1 year, after which time formalin must be added.28 ‘‘M3’’ fixative: Prepare by dissolving 5 g KI, 10 g iodine, 50 mL glacial acetic acid, and 250 mL formalin in 1 L distilled water (dissolve the iodide in a small quantity of water to aid in solution of the iodine). Add 20 mL fixative to 1 L sample and store in the dark. Glutaraldehyde: Preserve samples by adding neutralized glutaraldehyde to yield a final concentration of 1 to 2%. Other commonly used preservatives include 95% alcohol, and 6-3-1 preservative, (6 parts water, 3 parts 95% alcohol, and 1 part formalin). Use equal volumes of preservative and sample. To retain color in preserved plankton, store samples in the dark or add 1 mL saturated copper sulfate (CuSO4) solution/L. Most preservatives distort and disrupt certain cells,29,30 especially those of delicate forms such as Euglena, Cryptomonas, Synura, Chromulina, and Mallamonas. Lugol’s iodine solution usually is least damaging for these phytoflagellates. To become familiar with live specimens and preservation-caused distortions, use reference collection from biological supply houses or consult experienced co-workers. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater b. Zooplankton: The choice of sampler depends on the type of zooplankton, the kind of study (distribution, productivity, etc.) and the body of water being investigated. Zooplankton populations invariably are distributed in a patchy way, making both sampling and data interpretation difficult. For collecting microzooplankton (20 to 200 µm) such as protozoa, rotifers, and immature microcrustacea, use the bottle samplers described for phytoplankton. The small zooplankters usually are sufficiently abundant to yield adequate samples in 5- to 10-L bottles; however, composite samples over depth and time are recommended. Water bottle samplers are suitable especially for discrete-depth samples. If depth-integrated samples are desired, use pumps or nets. The larger and more robust microzooplankters (e.g., loricate forms and crustacea) may be concentrated by passing the whole water through a 20-µm mesh net. If quantitative estimates of other nonloricate, delicate forms are required, do not screen. Fix 0.5 to 5 L of whole water for enumeration of these forms. Bottle samplers usually are unsuitable for collecting larger zooplankton, such as mature microcrustacea, that, unlike the smaller forms, are much less numerous and are sufficiently agile to avoid capture. Although comparatively large water volumes, and consequently adequate numbers of microcrustacea, can be sampled with a pump, avoidance by larger, more agile zooplankters at the pump head can cause sampling error. Consequently, larger trap samplers or nets are the preferred collection methods. The Juday trap31 operates on the same principle as the water bottle samplers but is generally larger (10 L). The larger size makes the Juday trap more suitable for collecting zooplankters, especially larger copepods. However, it is awkward to use and its 10-L capacity is inadequate for oligotrophic lakes or other water bodies with few zooplankters. Because it is constructed of metal it is unsuited if heavy metals analyses are required. The Schindler-Patalas trap32 (Figure 10200:2) usually is preferred to the Juday trap because it is constructed of clear acrylic plastic and is transparent. It can be lowered into the water with minimal disturbance and is suitable for collecting larger zooplankters. Models of 10- to 12-L capacity are available but the 30-L size is preferred. It has no mechanical closing mechanism and thus is convenient for cold-weather sampling when mechanical devices tend to malfunction. Like the Juday trap, it can be fitted with nets of various mesh sizes, but the No. 20 mesh net is used most often. Plankton nets are preferred to bottles and traps for sampling where plankters are few or where only qualitative data or a large biomass is needed for analysis. Because they were designed originally for qualitative sampling, modifications are required for quantitative work. The mesh size, type of material, orifice size, length, hauling method, type of tow, and volume sampled will depend on the particular needs of the study.33,34 Type of netting and mesh size determine filtration efficiency, clogging tendencies, velocity, drag, and the condition of the sample after collection. Silk, formerly the common mesh material in plankton nets, is not recommended because of shrinkage of mesh openings and rotting with age. Nylon monofilament mesh is preferred because of its mesh size accuracy and durability. Nylon nets of different mesh © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater sizes still are labelled by the silk rating system: characteristics of commonly used nylon plankton nets are listed in Table 10200:I. Finer mesh sizes clog more readily than coarser mesh; a compromise must be made between mesh size small enough to retain desired organisms effectively and a size large enough to preclude a serious clogging problem. If clogging occurs, reduce its effects by decreasing the length of tow. The maximum volume, VM, of water that can be filtered through a net during a vertical tow can be estimated with the formula, where: r = radius of net orifice and d = depth to which net is lowered. This volume is a maximum because clogging of the net’s meshes by phytoplankton and other particles and, for fine netting, even the netting itself can cause some water to be diverted from the net’s path.35,36 Keep net towing distance as short as practical to alleviate clogging. If the net has a pronounced green or brown color after towing, clogging probably has occurred. To estimate sampling volume, VA, mount a calibrated flow meter midway between the net rims and mouth center (the meter is mounted off-center to avoid flow reduction associated with the towing bridle).37 Equip meter with lock mechanisms to prevent it turning in reverse or while in air. Record flow-meter readings before and after collecting sample. Calculate filtration efficiency, E, from: If E is less than about 0.8, substantial clogging has occurred. Take steps to increase efficiency. Clogging not only decreases the volume filtered, but also leads to biased samples because filtration efficiency is nonuniform during the tow.34 Various types of plankton nets are shown in Figure 10200:3. Simple conical nets have been used for many years with little modification in design or improvement in accuracy. Their major source of error is that the filtration characteristics of conical nets usually are unknown. Filtration efficiency in No. 20 mesh cone nets ranges from 40 to 77%. To improve efficiency, place a porous cylinder collar or nonporous truncated cone in front of the conical portion of the net. The Juday net exemplifies a commonly used net with a truncated cone. For good filtration characteristics the ratio of filtering area of net to orifice area should be at least 3:1. Bridles attaching the net to the towing line also adversely influence filtration efficiency and increase turbulence in front of the net, thereby increasing the potential for net avoidance by larger zooplankters. The tandem, Bongo net design (Figure 10200:3) reduces these influences and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater permits duplicate samples to be collected simultaneously. Three types of tows are used: vertical, horizontal, and oblique. Vertical tows are preferred to obtain an integrated water column sample. To make a vertical tow, lower the weighted net to a given depth, then raise vertically at an even speed of 0.5 m/s. In small water bodies haul the net hand over hand with a steady, unhurried motion approximating the speed of 0.5 m/s. In large bodies where long net hauls and vessel drifting are expected, use a davit, meter wheel, angle indicator, and winch. Attach a 3- to 5-kg weight to hold the net down. Determine depth of the net by multiplying the length of the extended wire by the cosine of the wire’s angle with the vertical direction. Maintain wire angle as close to the vertical as possible by controlling the boat’s speed null against the wind drift, or wherever feasible, do vertical hauls from an anchored boat. Vertical and oblique tows collect a composite sample, whereas horizontal tows collect a sample at a discrete depth. Oblique tows usually are preferred over vertical tows in shallow water or wherever a longer net tow is required. For oblique tows, lower the net or sampler to some predetermined depth and then raise at a constant rate as the boat moves forward. Oblique tows do not necessarily sample a true angle from the bottom to the surface. Under best conditions the pattern is somewhat sigmoid due to boat acceleration and slack in the tow line. Horizontal tows usually are used to obtain depth distribution information on zooplankton. Although a variety of horizontal samplers is available (see Figure 10200:4), use the Clarke-Bumpus sampler38 for quantitative collection of zooplankton because of its built-in flowmeter and opening-closing device. For horizontal tows use a boat equipped as above and determine sampler depth as above. Lower sampler to preselected depth, open, tow at that depth for 5 to 10 min, then close and raise it. A variety of zooplankton sampling methods can be used in flowing water. The method of choice depends largely on flow velocity. Properly weighted bottles, traps and pump hoses, and nets can be used in medium- to slow-flowing waters. In turbulent, well-mixed waters, collect surface water by bucket and filter it through the appropriate mesh size. Select sample size based on concentration of zooplankters. Give plankton nets proper care and maintenance. Do not let particulate matter dry on the net because it can significantly reduce size of mesh apertures and increase frequency of clogging. Wash net thoroughly with water after each use. Periodically clean with a warm soap solution. Because nylon net material is susceptible to deterioration from abrasion and sunlight, guard against unnecessary wear and store in the dark. Traps and nets do not work well in shallow areas with growths of aquatic vegetation. To obtain an integrated sample for the entire water column in such areas, use a length of light-weight rubber or polyethylene tubing with netting attached over one end and a rope on the other.39 Attach netting by tape or rubber bands that will stay in place in water, but can be removed easily after sampling. Use tubing of 5- to 10-cm diam and long enough to reach from the surface to the bottom. Lower the open end (the end with the rope attached) until it almost touches the bottom. Then pull this end up using the rope and keep the covered end above the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater water surface. When the open end is out of the water, let the end with the netting fall back into the water, pull the tubing into the boat, open end first, and let the water in the tube drain out through the netting. When the zooplankton has been concentrated in a small volume, just above the netting, remove the netting over a container and catch the concentrated sample. Wash netting and end of tubing into the container to assure that all the zooplankton is collected. This method is not limited to areas with aquatic vegetation. It provides an excellent method of obtaining an integrated sample from any shallow area. In standing waters, collect tow samples by filtering 1 to 5 m3 of water. Preserve zooplankton samples with 70% ethanol or 5% buffered formalin. Ethanol preservative is preferred for materials to be stained in permanent mounts or stored. Formalin may be used for the first 48 h of preservation with subsequent transfer to 70% ethanol. Formalin preservative may cause distortion of pleomorphic forms such as protozoans and rotifers. Make formalin in sucrose-saturated water to minimize carapace distortion and loss of eggs in crustaceans, especially cladocerans.40 Bouin’s fixative produces reasonable results for soft-bodied microzooplankton.41 This fixative is picric acid saturated in calcium carbonate-buffered formaldehyde containing 5% (v/v) acetic acid. Dilute Bouin’s fixative 1:19 with the sample. Because rapid fixation is necessary, pour the sample onto the fixative or inject fixative rapidly into the sample. Use a narcotizing agent such as carbonated water, menthol-saturated water, or neosynephrine to prevent or reduce contraction or distortion of organisms, especially rotifers, cladocerans, and many marine invertebrates.42,43 Adding a few drops of detergent prevents clumping of preserved organisms. Preserve samples as soon as most animal movement has ceased, usually within a half hour of narcotization. To prevent evaporation, add 5% glycerin to the concentrated sample. In turbid samples, differentiate animal and detrital material by adding 0.04% rose bengal stain, which intensely stains the carapace (shell) of zooplankters and is a good general cytoplasmic stain. 3. References 1. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1982. Handbook for Sampling and Sample Preservation of Water and Wastewater. EPA-600/4-82-029. 2. PARKER B.C. & R.F. HATCHER. 1974. Enrichment of surface freshwater microlayers with algae. J. Phycol. 10:185. 3. TAGUCHI, S. & K. NAKAJIMA. 1971. Plankton and seston in the sea surface of three inlets of Japan. Bull. Plankton Soc. Japan 18:20. 4. UNITED NATIONS EDUCATIONAL, SCIENTIFIC AND CULTURAL ORGANIZATION. 1966. Determination of Photosynthetic Pigments in Sea-water. Monogr. Oceanogr. Methodol. No. 1. United Nations Educational, Scientific & Cultural Org., Paris. 5. UNITED NATIONS EDUCATIONAL, SCIENTIFIC AND CULTURAL ORGANIZATION. 1968. Zooplankton Sampling. Monogr. Oceanogr. Methodol. No. 2. United Nations Educational, Scientific & Cultural Org., Paris. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 6. UNITED NATIONS EDUCATIONAL, SCIENTIFIC AND CULTURAL ORGANIZATION. 1973. A Guide to the Measurement of Marine Primary Production under Some Special Conditions. Monogr. Oceanogr. Methodol. No. 3. United Nations Educational, Scientific & Cultural Org., Paris. 7. SOURNIA, A., ed. 1978. Phytoplankton Manual. Monogr. Oceanogr. Methodol. No. 6. United Nations Educational, Scientific & Cultural Org., Paris. 8. CUPP, E.E. 1943. Marine plankton diatoms of the west coast of North America. Bull. Scripps Inst. Oceanogr. 5:1. 9. HUSTEDT, F. 1927–66. Die Kieselalgen Deutschlands, Osterreichs und der Schweiz mit Berucksichtigung der Ubrigen Lander Europas Sowie der Angrenzenden Meeresgebiete. In L. Rabenhorst, Kryptogamen-Flora. Vol. 7: Teil 1 (1927–30); Teil 2 (1931–59); Teil 3 (1961–66). Akademie Verlag, Leipzig, Germany. 10. LEBOUR, M.V. 1930. The Planktonic Diatoms of Northern Seas. Ray Soc., London. 11. HENDEY, N.I. 1964. An introductory account of the smaller algae of British coastal waters, V. Bacillariophyceae (Diatoms). Fish. Invest. Min. Agr. Fish. Food (G.B.), Ser. IV:1. 12. DODGE, J.D. 1975. The prorocentrales (Dinophyceae), II. Revision of the taxonomy within the genus Prorocentrum. Bot. Limnol. Soc. 71: 103. 13. LEBOUR, M.V. 1925. The Dinoflagellates of Northern Seas. Marine Biological Assoc. United Kingdom, Plymouth. 14. SCHILLER, J. 1931–37. Dinoflagellatae (Peridineae) in monographischer Behandlung. In L. Rabenhorst, Kryptogamen-Flora. Vol. 10; Teil 1 (1931–33); Teil 2 (1935–37). Akademie Verlag, Leipzig, Germany. 15. SCHILLER, J. 1930. Coccolithineae. In L. Rabenhorst, Kryptogamen-Flora. Vol. 10, p. 89. Akademie Verlag, Leipzig, Germany. 16. GEITLER, L. 1932. Cyanophyceae von Europa unter Berucksichtigung der anderen Kontinente. In L. Rabenhorst, Kryptogamen-Flora. Vol. 14, p. 1. Akademie Verlag, Leipzig, Germany. 17. WELCH, P.S. 1948. Limnological Methods. Blakiston Co., Philadelphia, Pa. 18. STRICKLAND, J.D.H. & T.R. PARSONS. 1968. A Practical Manual of Sea Water Analysis. Fish. Res. Board Can. Bull. No. 167. Queen’s Printer, Ottawa, Ont. 19. DUSSART, B.M. 1965. Les differentes categories de plancton. Hydrobiologia 26:72. 20. SIEBURTH, J.MCN., V. SMETACEK & J. LENZ. 1978. Pelagic ecosystem structure: Heterotrophic compartments of plankton and their relationship to plankton size fractions. Limnol. Oceanogr. 23: 1256. 21. MORTIMER, C.H. 1942. The exchange of dissolved substances between mud and water in lakes. J. Ecol. 30:147. 22. VOLLENWEIDER, R.A. 1969. A Manual on Methods for Measuring Primary Production in Aquatic Environments. IBP Handbook No. 12. Blackwell Scientific Publ., Oxford, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater England. 23. GELDREICH, E.E., H.D. NASH, D.F. SPINO & D.J. REASONER. 1980. Bacterial dynamics in a water supply reservoir: a case study. J. Amer. Water Works Assoc. 72:31. 24. BEERS, J.R. 1978. Pump sampling. In A. Sournia, ed. Phytoplankton Manual. United Nations Educational, Scientific and Cultural Org., Paris. 25. EXTON, R.J., W.M. HOUGHTON, W. ESAIAS, L.W. HAAS & D. HAYWARD. 1983. Spectral differences and temporal stability of phycoerythrin fluorescence in estuaries and coastal waters due to the domination of labile cryptophytes and stable cyanobacteria. Limnol. Oceanogr. 28:1225. 26. EDMONDSON, W.T., ed. 1959. Freshwater Biology, 2nd ed. John Wiley & Sons, New York, N.Y. 27. UTERMOHL, H. 1958. Zur Vervollkommung der quantitativen Phytoplankton-Methodik. Int. Ver. Theoret. Angewand. Limnol., Commun. No. 9. 28. WEBER, C.I. 1968. The preservation of phytoplankton grab samples. Trans. Amer. Microsc. Soc. 87:70. 29. PAERL, H.W. 1984. An evaluation of freeze fixation as a phytoplankton preservation method for microautoradiography. Limnol. Oceanogr. 29:417. 30. SILVER, M.W. & P.J. DAVOLL. 1978. Loss of 14C activity after chemical fixation of phytoplankton: Error source for autoradiography and other productivity measurements. Limnol. Oceanogr. 23:362. 31. JUDAY, C. 1916. Limnological apparatus. Trans. Wis. Acad. Sci. 18: 566. 32. SCHINDLER, D.W. 1969. Two useful devices for vertical plankton and water sampling. J. Fish. Res. Board Can. 26: 1948. 33. SCHWOERBEL, J. 1970. Methods of Hydrobiology. Pergamon Press, Toronto, Ont. 34. TRANTER, D.J., ed. 1980. Reviews on Zooplankton Sampling Methods. United Nations Educational, Scientific & Cultural Org., Switzerland. 35. GANNON, J.E. 1980. Towards improving the use of zooplankton in water quality surveillance of the St. Lawrence Great Lakes. Proc. 1st Biol. Surveillance Symp., 22nd Conf. Great Lakes Research Can. Tech. Rep. Fish. Aquat. Sci. 976, p. 87. 36. ROBERTSON, A. 1968. Abundance, distribution, and biology of plankton in Lake Michigan with the addition of a Research Ships of Opportunity project. Spec. Rep. No. 35, Great Lakes Research Div., Univ. Michigan, Ann Arbor. 37. EVANS, M.S. & D.W. SELL. 1985. Mesh size and collection characteristics of 50-cm diameter conical plankton nets. Hydrobiologia 122: 97. 38. CLARKE, G.L. & D.F. BUMPUS. 1940. The Plankton Sampler: An Instrument for Quantitative Plankton Investigations. Spec. Publ. No. 5, Limnological Soc. America. 39. PENNAK, R.W. 1962. Quantitative zooplankton sampling in littoral vegetation areas. Limnol. Oceanog. 7:487. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 40. HANEY, J.F. & D.J. HALL. 1973. Sugar-coated Daphnia; A preservation technique for Cladocera. Limnol. Oceanogr. 18:331. 41. COATS, D.W. & J.F. HEINBOKEL. 1982. A study of reproduction and other life cycle phenomena in plankton protists using an acridine orange fluorescence technique. Mar. Biol. 67:71. 42. GANNON, J.E. & S.A. GANNON. 1975. Observations on the narcotization of crustacean zooplankton. Crustaceana 28(2):220. 43. STEEDMAN, H.F. 1976. Narcotizing agents and methods. In H.F. Steedman, ed. Zooplankton Fixation and Preservation. Monogr. Oceanogr. Methodol. No. 4. United Nations Educational, Scientific & Cultural Org., Paris. 10200 C. Concentration Techniques The organisms contained in water samples sometimes must be concentrated in the laboratory before analysis. Three techniques for concentrating phytoplankton, namely, sedimentation, membrane filtration, and centrifugation, are described below. A special technique for zooplankton also is given. 1. Sedimentation Sedimentation is the preferred method of concentration because it is nonselective (unlike filtration) and nondestructive (unlike filtration or centrifugation), although many of the picoplankton, the smaller nannoplankton, and actively swimming flagellates (in unpreserved samples) may not settle completely. The volume concentrated varies inversely with the abundance of organisms and is related to sample turbidity. It may be as small as 1 mL for use with an inverted microscope or as large as 1 L for general phytoplankton and zooplankton enumeration. Allow 1 h settling/mm of column depth. For a treated sample (10 mL liquid detergent/L) allow about 0.5 h settling/mm depth.1 The sample may be concentrated in a series of steps by quantitatively transferring the sediment from the initial container to sequentially smaller ones. Use cylindrical settling chambers with thin, clear glass bottoms. Fill settling chambers without forming a vortex, keep them vibration-free, and move them carefully to avoid nonrandom distribution of settled matter. Carefully siphon or decant the supernatants to obtain the desired final volume (5 mL for diatom mounts). Store the concentrated sample in a closed, labeled glass vial. 2. Membrane Filtration The filtration method permits use of high magnification for enumerating small plankters including flagellates and cyanobacteria. However, delicate forms such as ‘‘naked’’ flagellates are distorted by even gentle filtration. When populations are dense and the content of detritus is © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater high, the filter clogs quickly and silt may crush the organisms or obscure them from view. Pour a measured volume of well-mixed sample into a funnel equipped with a membrane filter having a pore diameter of 0.45 µm. Apply a vacuum of less than 50 kPa to the filter until about 0.5 cm of sample remains on filter. Break vacuum, then apply low vacuum (about 12 kPa) to remove remaining water but not to dry the filter. For samples with a low phytoplankton and silt content the method does not require counting of individual plankters to assemble enumeration data and it increases the probability of observing less abundant forms.2 Samples also may be concentrated on a filter, inverted onto a microscope slide, and quick-frozen, permitting the removal of the filter and transfer of plankton to the slide.3,4 3. Centrifugation Plankton can be concentrated by batch or continuous centrifugation. Centrifuge batch samples at 1000 g for 20 min. The Foerst continuous centrifuge is no longer recommended as a quantitative device but it may be desirable to continue its use in existing programs to assure continuity with previously collected data. Although centrifugation accelerates sedimentation, it may damage fragile organisms. 4. Zooplankton Concentration Zooplankton samples often need to be concentrated in the field, especially when large water bottles or pump methods of sampling are used. Moreover, samples obtained by nets or other methods sometimes need to be concentrated further for storage or preparation for examination. When only small volume reductions are needed, pour sample back into the bucket of traps or nets. In processing large volumes of water as with pump sampling, use larger plankton buckets or funnels with greater water volume retention and filtration surface area. Construct a filter funnel similar to that shown in Figure 10200:5 of clear acrylic plastic or other suitable material.5 The volume of the apparatus and the mesh size depend on volume of water to be filtered and size of organisms to be retained. The mesh size of the filter funnel normally is the same as that of the net or other field sampling device. 5. References 1. FURET, J.E. & K. BENSON-EVANS. 1982. An evaluation of the time required to obtain sedimentation of fixed algal particles prior to enumeration. Brit. Phycol. J. 17:253. 2. MCNABB, C.D. 1960. Enumeration of freshwater phytoplankton concentrated on the membrane filter. Limnol. Oceanogr. 5:57. 3. HEWES, C.D. & O. HOLM-HANSEN. 1983. A method for recovering nanoplankton from filters for identification with the microscope: The filter-transfer-freeze (FTF) technique. Limnol. Oceanogr. 28:389. 4. HEWES, C.D., F.M.H. REID & O. HOLM-HANSEN. 1984. The quantitative analysis of nanoplankton: A study of methods. J. Plankton Res. 6:601. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5. LIKENS, G.E. & J.J. GILBERT. 1970. Notes on quantitative sampling of natural populations of planktonic rotifers. Limnol. Oceanogr. 15:816. 10200 D. Preparing Slide Mounts 1. Phytoplankton Semi-Permanent Wet Mounts Agitate the settled sample concentrate and withdraw a subsample with an accurately calibrated pipet. Clean pipet regularly. To prepare wet mounts transfer 0.1 mL to a glass slide, place a cover slip over the sample, and ring the cover slip with an adhesive such as clear nail polish to prevent evaporation. For semipermanent mounts, add a few drops of glycerin to the slide. As the sample ages the water evaporates, leaving the organisms imbedded in the glycerin. If the cover slip is ringed with adhesive, the slide can be retained for a few years if stored in the dark. 2. Phytoplankton Permanent Mounts a. Membrane filter mounts: Place two drops of immersion oil on a labeled slide. Immediately after filtering place the filter on top of the oil with a pair of forceps and add two drops of oil on top of the filter. The oil impregnates the filter and makes it transparent. Impregnation time is 24 to 48 h. This procedure can be completed in 1 to 2 h by applying heat (70°C). Once the filter has cleared, place a few additional drops of oil on it and cover with a cover slip. The mounted filter is now ready for microscopic examination. Alternatively, mount membrane filters in mounting medium.*#(113) Immerse filters in 1-propanol to displace residual water and transfer to xylol for several minutes to clear filters. Place a section of filter or entire filter on a microscope slide with the mounting medium, cover with a cover glass, and dry at low temperature.1 b. Sedimented slide mounts: Two techniques are available for making permanent, resin mounts of natural phytoplankton that has been deposited by sedimentation on a microscope slide or cover glass and dehydrated by ethanol vapor substitution.2,3 3. Diatom Mounts Samples concentrated for diatom analysis by settling or centrifugation may contain dissolved materials, such as marine salts, formalin, and detergents, that will leave interfering residues. Wash well with distilled water before slide preparation. Transfer several drops of washed concentrate by means of a large-bore disposable pipet or large-bore dropper to a cover glass on a hot plate warmed enough to increase the evaporation rate but not enough to cause boiling (use a large-bore pipet or dropper to prevent possible selective filtration, thus exclusion, of larger forms or those forming colonies or chains). If the cleaned material is very concentrated, improve distribution of diatoms by adding the drops to a cover glass already flooded with distilled water. Evaporate to dryness. Repeat addition and evaporation until a sufficient quantity of sample has been transferred to the cover glass, but avoid producing a residue so dense that © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater organisms cannot be recognized. If in doubt about the density, examine under a compound microscope. After evaporation, incinerate the residue on the cover glass on a hot plate at 300 to 500°C; alternatively, use a muffle furnace. This usually requires 20 to 45 min. Mount as described below. Treat samples concentrated for diatom analysis by membrane filtration as described by Patrick and Reimer.4 Mix equal volumes of conc nitric acid (HNO3) and sample. CAUTION: When working with conc HNO3 wear safety goggles and an acid-resistant apron and gloves, and work under a hood. Add a few grains of potassium dichromate (K2Cr2O7)5 to facilitate digestion of the filter and cellular organic matter. Add more dichromate if solution color changes from yellow to green. Place sample on a hot plate and boil down to approximately one-third the original volume. Alternatively, let treated sample stand overnight. This cleaning process destroys organic matter and leaves only diatom shells (frustules). Cool, wash with distilled water, and mount as described above. Transfer cleaned frustules to a cover glass and dry as described above. Place a drop of mounting medium in the center of a labeled slide. Use 25- by 75-mm slides with frosted ends. Using a suitable high-refractive-index microscopic mounting medium assures permanent, easily handled mounts for examination under oil immersion. Heat the slide to near 90°C for 1 to 2 min before applying the heated cover slip with its sample residue to hasten evaporation of solvent in the mounting medium. Remove the slide to a cool surface and, during cooling (5 to 10 s), apply firm but gentle pressure to the cover glass with a broad, flat instrument. 4. Zooplankton Mounts For zooplankton analyses, withdraw a 5-mL subsample from the concentrate and dilute or concentrate further as necessary. Transfer sample to a counting cell or chamber (see below) for analysis as a wet mount. Use polyvinyl lactyl phenol†#(114) for preparing semipermanent zooplankton mounts. The mounts are good for about a year, after which time the clearing agent causes deterioration of organisms. For long-term storage ring cover slip with clear lacquer (fingernail polish) to retard mountant crystallization. For permanent mounting, other mountants are available.‡#(115) For the protozoan portion of the microzooplankton, a protargol staining procedure6 not only provides a permanent mount but also reveals the cytological details often necessary for identification. This procedure is qualitative and is especially important in taxonomic studies of the ciliated protozoa. 5. References 1. MILLIPORE FILTER CORPORATION. 1966. Biological examination of water, sludge and bottom materials. Millipore Techniques, Water Microbiology, p. 25. 2. SANFORD, G.R., A. SANDS & C.R. GOLDMAN. 1962. A settle-freeze method for concentrating phytoplankton in quantitative studies. Limnol. Oceanogr. 14:790. 3. CRUMPTON, W.G. & R.G. WETZEL. 1981. A method for preparing permanent mounts of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater phytoplankton for critical microscopy and cell counting. Limnol. Oceanogr. 26:976. 4. PATRICK, R. & C.W. REIMER. 1967. The Diatoms of the United States. Vol. 1. Monogr. 13, Philadelphia Acad. Natur. Sci. 5. HOHN, M.H. & J. HELLERMAN. 1963. The taxonomy and structure of diatom populations for three eastern North American rivers using three sampling methods. Trans. Amer. Microsc. Soc. 62:250. 6. SMALL, E.B. & D.H. LYNN. 1985. Phylum Ciliophora Doflein, 1901. In J.J. Lee, S.H. Hunter & E.C. Bovee, eds. An Illustrated Guide to the Protozoa. Soc. Protozoology, Lawrence, Kansas. 10200 E. Microscopes and Calibrations 1. Compound Microscope Use either a standard or an inverted compound microscope for algal identification and enumeration. Equip either type with a mechanical stage capable of moving all parts of a counting cell past the objective lens. Standard equipment is a set of 10× or 12.5× oculars and 10×, 20×, 40×, and 100× objectives. Use objectives to provide adequate working distance for the counting chamber. Magnification requirements vary with the plankton fraction being investigated, the type of microscope, counting chamber used, and optics. With standard objectives, the Sedgwick-Rafter chamber limits magnification to approximately 200× and the Palmer-Maloney cell limits magnification to approximately 500×. Inverted microscopes are limited in resolution by their optics. The useful upper limit of magnification for any objective is 1000 times the numerical aperture (NA). Above this magnification, no greater detail can be resolved. Use combinations of oculars, intermediate magnifiers, and objectives to obtain the greatest magnification without exceeding the useful limit of magnification. When the limit is exceeded, empty magnification results. Empty magnification occurs where the image is larger but no greater resolution is achieved. Optics providing contrast enhancement such as phase contrast or differential interference contrast are useful. 2. Stereoscopic Microscope The stereoscopic microscope is essentially two complete microscopes assembled into a binocular instrument to give a stereoscopic view and an erect rather than an inverted image. Use this microscope for the study and counting of large plankters such as mature microcrustacea. Include 10× to 15× paired oculars in combination with 1× to 8× objectives. This combination of optics bridges the gap between the hand lens and the compound microscope and provides magnification ranging from 10× to 120×. Alternatively, use a good-quality zoom-type instrument with comparable magnification. 3. Inverted Compound Microscope © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater The inverted compound microscope often is used routinely for plankton counting in many laboratories.1-3 This instrument is unique in that the objectives are below a movable stage and the illumination comes from above, thus permitting viewing of organisms that have settled to the bottom of a chamber. Place samples in a cylindrical settling chamber having a thin, clear glass bottom. Chambers of various capacities are available; the appropriate size depends on the density of organisms. After a suitable period of settling (see Section 10200C.1), count organisms in the settling chamber. The major advantage of the inverted microscope is that by a simple rotation of the nosepiece a specimen can be examined (or counted) directly in the settling chamber at any desired magnification. Although not recommended, oil immersion objectives have some useful applications. No preparation or manipulation other than settling is required. Generally, examine a preserved sample. Techniques are available for samples with an abundance of organisms that tend to float.4 4. Epifluorescence Microscope An epifluorescence microscope may be either standard or inverted. It uses incident light to excite electrons in intracellular compounds, such as pigments or absorbed stains, with the energy emitted during electron return to the ground state being measured as fluorescent light. The technique has been applied to the microscopic identification of chlorophyll-containing cells (autotrophs) and nonpigmented heterotrophic plankton; fluorescent stains such as primulin or proflavin also have been used to differentiate nannoplanktonic primary and secondary producers.5-7 Excitation and emission wavelengths are unique for each pigment and stain and require distinct light filter combinations and light sources. Select the filter combinations for the particular application. Epifluorescence microscopy is particularly useful for the enumeration of picoplankton and heterotrophic flagellate populations common to most aquatic systems. Concentrate samples by membrane filtration. Use epifluorescence microscopy as a complementary procedure to standard light microscope counting techniques. 5. Microscope Calibration Microscope calibration is essential. The usual equipment for calibration is a Whipple grid (ocular micrometer, reticle, or reticule) placed in an eyepiece of the microscope and a stage micrometer that has a standardized, accurately ruled scale on a glass slide. The Whipple disk (Figure 10200:6) has an accurately ruled grid subdivided into 100 squares. One square near the center is subdivided further into 25 smaller squares. The outer dimensions of the grid are such that with a 10× objective and a 10× ocular, it delimits an area of approximately 1 mm2 on the microscope stage. Because this area may differ from one microscope to another, carefully calibrate the Whipple grid for each microscope. With the ocular and stage micrometers parallel and in part superimposed, match the line at the left edge of the Whipple grid with the zero mark on the stage micrometer scale (Figure 10200:7). Determine the width of the Whipple grid image to the nearest 0.01 mm from the stage © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater micrometer scale. Should the width of the image of the Whipple grid be exactly 1 mm (1000 µm), the larger squares will be 1/10 mm (100 µm) on a side and each of the smaller squares 1/50 mm (20 µm). When the microscope is calibrated at higher magnifications, the entire scale on the stage micrometer will not be seen; make measurements to the nearest 0.001 mm. Additional details for calibration are available.8 6. References 1. WETZEL, R.G. & G.E. LIKENS. 1991. Limnological Analyses, 2nd ed. Springer-Verlag, New York, N.Y. 2. LUND, J.W.G., C. KIPLING & E.D. LECREN. 1958. The inverted microscope method of estimating algal numbers and the statistical basis of estimations by counting. Hydrobiologia 11:143. 3. SICKO-GOAD, L. & E.F. STOERMER. 1984. The need for uniform terminology concerning phytoplankton cell size fractions and examples of picoplankton from the Laurentian Great Lakes. J. Great Lakes Res. 10:90. 4. REYNOLDS, C.S. & G.H.M. JAWORSKI. 1978. Enumeration of natural Microcystis populations. Brit. Phycol. J. 13:269. 5. DAVIS, P.G. & J. MCN. SIEBURTH. 1982. Differentiation of phototrophic and heterotrophic nanoplankton populations in marine waters by epifluorescence microscopy. Ann. Inst. Oceanogr. 58:249. 6. CARON, D.A. 1983. Techniques for enumeration of heterotrophic and phototrophic nanoplankton, using epifluorescence microscopy, and comparison with other procedures. Appl. Environ. Microbiol. 46:491. 7. SHERR, E.B. & B.F. SHERR. 1983. Double-staining epifluorescence techniques to assess frequency of dividing cells and bacteriovory in natural populations of heterotrophic microprotozoa. Appl. Environ. Microbiol. 46:1388. 8. JACKSON, H.W. & L.G. WILLIAMS. 1962. Calibration and use of certain plankton counting equipment. Trans. Amer. Microsc. Soc. 81:96. 10200 F. Phytoplankton Counting Techniques 1. Counting Units Some phytoplankton are unicellular while others are multicellular (colonial). The variety of configurations poses a problem in enumeration. For example, should a four-celled colony of Scenedesmus (Plate 32) be reported as one colony or four individual cells? Listed below are suggestions for reporting: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater suggestions for reporting: Enumeration Method Total cell count Natural unit count1 (clump count) Areal standard unit count*#(116) Counting Unit Reporting Unit One cell One organism (any unicellular organism or natural colony) Cells/mL Units/mL 400 µm2 Units/mL Making a total cell count is time-consuming and tedious, especially when colonies consist of thousands of individual cells. The natural unit or clump is the most easily used system; however, it is not necessarily accurate because sample handling and preserving may dislodge cells from the colony. The unit method also may not be quantitatively accurate nor reflect abundance of biomass or biovolume. Whatever method is chosen, identify it in reporting results. If the distribution of organisms is random and the population fits a Poisson distribution, the counting error may be estimated.2 For example, the approximate 95% confidence limits, as a percentage of the number of units counted (N), equals: Thus, if 100 units are counted, the 95% confidence limits approximate ± 20%. For a count of 400 units, the limits are about 10%. 2. Counting Procedures To enumerate plankton use a counting cell or chamber that limits the volume and area for ready calculation of population densities. When counting with a Whipple grid, establish a convention for tallying organisms lying on an outer boundary line. For example, in counting a ‘‘field’’ (entire Whipple square), designate the top and left boundaries as ‘‘no-count’’ sides, and the bottom and right boundaries as ‘‘count’’ sides. Thus, tally every plankter touching a ‘‘count’’ side from the inside or outside but ignore any touching a ‘‘no-count’’ side. If significant numbers of filamentous or other large forms cross two or more boundaries of the grid, count them separately at a lower magnification and include their number in the total count. To identify organisms use standard bench references (see Section 10900). © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Do not count dead cells or broken diatom frustules. Tally empty centric and pennate diatoms separately as ‘‘dead centric diatoms’’ or ‘‘dead pennate diatoms’’ for use in converting the diatom species proportional count to a count per milliliter. Magnification is important in phytoplankton identification and enumeration. Although magnifications of 100× to 200× are useful for counting large organisms or colonies, much higher magnifications often are required. It is useful to categorize techniques for phytoplankton counting according to the magnifications provided. a. Low-magnification (up to 200×) methods: The Sedgwick-Rafter (S-R) cell is a device commonly used for plankton counting because it is easily manipulated and provides reasonably reproducible data when used with a calibrated microscope equipped with an eyepiece measuring device such as the Whipple grid. The greatest disadvantage associated with the cell is that objectives providing high magnification cannot be used. As a result, the S-R cell is not appropriate for examining nannoplankton. The S-R cell is approximately 50 mm long by 20 mm wide by 1 mm deep. The total area of the bottom is approximately 1000 mm2 and the total volume is approximately 1000 mm3 or 1 mL. Carefully check the exact length and depth of the cell with a micrometer and calipers before use. 1) Filling the cell—Before filling the S-R cell with sample, place the cover glass diagonally across the cell and transfer sample with a large-bore pipet (Figure 10200:8). Placing cover slip in this manner will help prevent formation of air bubbles in cell corners. The cover slip often will rotate slowly and cover the inner portion of the S-R cell during filling. Do not overfill because this would yield a depth greater than 1 mm and produce an invalid count. Do not permit large air spaces caused by evaporation to develop in the chamber during a lengthy examination. To prevent formation of air spaces, occasionally place a small drop of distilled water on edge of cover glass. Before counting let the S-R cell stand for at least 15 min to settle plankton. Count plankton on the bottom of the S-R cell. Some phytoplankton, notably some blue-green algae or motile flagellates in unpreserved samples, may not settle but rise to the underside of the cover slip. When this occurs, count these organisms and add to total of those counted on the cell bottom to derive total number of organisms. Count algae in strips or fields. 2) Strip counting—A ‘‘strip’’ the length of the cell constitutes a volume approximately 50 mm long, 1 mm deep, and the width of the total Whipple grid. The number of strips to be counted is a function of the precision desired and the number of units (cells, colonies, or filaments) per strip. Derive number of plankton in the S-R cell from the following: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: C= L= D= W= S= number of organisms counted, length of each strip (S-R cell length), mm, depth of a strip (S-R cell depth), mm, width of a strip (Whipple grid image width), mm, and number of strips counted. Multiply or divide number of cells per milliliter by a correction factor to adjust for sample dilution or concentration. 3) Field counting—On samples containing many plankton (10 or more plankters per field), make field counts rather than strip counts. Count plankters in random fields each consisting of one Whipple grid. The number of fields counted will depend on plankton density and statistical accuracy desired (see Section 10200F.1). Calculate the number of plankton per milliliter as follows: where: C= A= D= F= number of organisms counted, area of a field (Whipple grid image area), mm2, depth of a field (S-R cell depth), mm, and number of fields counted. Multiply or divide the number of cells per milliliter by a correction factor to adjust for sample dilution or concentration. b. Intermediate magnification (low to 500×) methods: The Palmer-Maloney (P-M) nannoplankton cell3 is designed specifically for nannoplankton enumeration. It has a circular chamber with a 17.9-mm diam, 0.4-mm depth, and 0.1-mL volume. The shallow depth permits use of 40 to 45× objectives with sufficient working distance. The principal disadvantage of the P-M cell is that these magnifications (400 to 450×) often are insufficient for nannoplankton identification and enumeration. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Because a relatively small sample portion is examined in the P-M cell do not use it unless the sample contains a dense population (10 or more plankters per field). Such a small sample portion from a less dense population causes serious underestimation of density. Introduce sample with a pipet into one of the 2- by 5-mm channels on the side of the chamber with the cover slip in place. After a 10-min settling period count the plankters in random fields, with the number of fields depending on density and variety of plankton and the statistical accuracy desired. Strips may be counted in this or any other circular cell by measuring the effective diameter and counting two perpendicular strips that cross at the center. Calculate the number per milliliter as follows: where: C= A= D= F= number of organisms counted, area of a field (Whipple grid image), mm2, depth of a field (P-M cell depth), mm, and number of fields counted. Multiply or divide the number of cells per milliliter by a correction factor to adjust for sample dilution or concentration. Another readily available chamber is the standard medical hemacytometer used for enumerating blood cells. It has a ruled grid machined into a counting plate and is fitted with a ground-glass cover slip. The grid is divided into 1-mm2 divisions; the chamber is 0.1 mm deep. Introduce sample by pipet and view under 450× magnification. Count all cells within the grid. The chamber comes from the manufacturer with a detailed instruction sheet containing directions on calculations and proper usage. A disadvantage to these counting cells is that the sample must have a very high plankton density to yield statistically reliable data. c. High-magnification methods: Examination of phytoplankton at high magnification requires the use of oil immersion objectives. Suitable procedures include using inverted microscope chambers, membrane filter mounts, sedimented slide mounts, the Lackey drop method, and diatom mounts. 1) Inverted microscope counts—Prepare a sample for examination by filling the settling chamber. After the desired settling time (see Section 10200C.1), transfer the chamber to the microscope stage. Count perpendicular strips across the center of the bottom cover glass. Strip counts may be made by using a Whipple grid or special counting oculars that have a pair of adjustable parallel hairs and a single cross hair. Determine the width of the strip with a stage micrometer and tally organisms as they pass the single cross hair that functions as a reference © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater point. Hold strip width constant for any series of samples. Alternatively examine random nonoverlapping fields until at least 100 units of the dominant species are counted. For highest accuracy, particularly because algae distribution may be nonuniform, count the entire chamber floor. Alternatively, make a random field-minimum count to attain a precision level of at least 85%.4 where: C= At = L= W= S= V= number of organisms counted, total area of bottom of settling chamber, mm2, length of a strip, mm, width of a strip (Whipple grid image width), mm, number of strips counted, and volume of sample settled, mL. where: A f = area of a field (Whipple grid image area), mm2, F = number of fields counted, and other terms are as defined above. 2) Membrane filter mounts—Concentrate sample as directed in Section 10200C.2 and prepare membrane filter as directed in Section 10200D.2a. Examine samples, concentrated on unlined membrane filters and mounted in oil as described above. Count enough random fields to ensure desired level of statistical accuracy (see Section 10200F.1). Select magnification level and size of microscope field (quadrat) such that the most abundant species appear in at least 70% but not more than 90% of microscopic fields examined (80% is optimum). Adjust microscope field size by using part or all of the Whipple grid. Examine 30 random microscope fields and record number of fields in which each species occurred. Report results as organisms per milliliter, calculated as follows: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: N= Q= V= D= density (organisms/field) from Table 10200:II, number of fields per filter, milliliters filtered, and dilution factor (0.96 for 4% formalin preservative). 3) Sedimented slide mounts—Examine mounts prepared as directed in Section 10200D.2b. 4) Lackey drop method—The Lackey drop (microtransect) method5 is a simple method of obtaining counts of considerable accuracy with samples containing a dense plankton population. It is similar to the S-R strip count. Prepare slides as directed in Section 10200D.1. Oil immersion objectives can be used with the semipermanent slides. Count organisms in enough strips to ensure desired level of statistical accuracy (see Section 10200F.1). Calculate number of organisms per milliliter as follows: where: C = number of organisms counted, At = area of cover slip, mm2, As = area of one strip, mm2, S = number of strips counted, and V = volume of sample under the cover slip, mL. 5) Diatom mounts—Prepare samples as directed in Section 10200D.3. For diatom species proportional count, examine diatom samples under oil immersion at a magnification of at least 900×. Scan lateral strips the width of the Whipple grid until at least 250 cells are counted. Available time and accuracy required dictate the number of cells to be counted. Determine percentage abundance of each species from tallied counts and calculate counts per milliliter of each species by multiplying percent abundance by total live and dead diatom count obtained from the plankton counting chamber. For greater accuracy distinguish between living and dead diatoms at the species level. 6) Phytoplankton staining technique—Staining algae permits differentiation between ‘‘live’’ © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and ‘‘dead’’ diatoms.6 This permits enumerating total phytoplankton in a single sample without sacrificing detailed diatom taxonomy. It also results in permanent reference slides. The procedure is most useful when diatoms are major components of phytoplankton and it is important to distinguish between living and dead diatoms. Preferably preserve samples in Lugol’s solution or alternatively in formalin (see Section 10200B.3). For analysis thoroughly mix the sample and filter a portion through a 47-mm-diam membrane filter (pore diam 0.45 or 0.65 µm). Use a vacuum of 16 to 20 kPa and never let sample dry. Add 2 to 5 mL aqueous acid fuchsin solution (dissolve 1 g acid fuchsin in 100 mL distilled water to which 2 mL glacial acetic acid has been added; filter) to the filter and let stand for 20 min. After staining, filter sample, wash briefly with distilled water, and filter again. Administer successive rinses of 50%, 90%, and 100% propanol to the sample while filtering. Soak for 2 min in a second 100% propanol wash, filter, and add xylene. At least two washes are required; let the final one soak 10 min before filtering. Trim the xylene-soaked filter and place on a microscope slide on which there are several drops of mounting medium.†#(117) Apply several more drops of medium to top of filter and install a cover glass. Carefully squeeze out excess mounting medium. Make the final mount permanent by lacquering the edges of the cover glass. Count organisms using the most appropriate magnification. ‘‘Live’’ diatoms typically are red while ‘‘dead’’ ones are unstained. Oil immersion is necessary for species identifications of diatoms and many other algae. Count either strips or random fields and calculate plankton densities per milliliter: where: C= At = Ac = V= number of organisms counted, total area of effective filter before trimming and mounting, area counted (strips or fields), and volume of sample filtered, mL. 3. References 1. INGRAM, W.M. & C.M. PALMER. 1952. Simplified procedures for collecting, examining, and recording plankton in water. J. Amer. Water Works Assoc. 44:617. 2. STRICKLAND, J.D.H. & T.R. PARSONS. 1968. A Practical Manual of Sea Water Analysis. Fish. Res. Board Can. Bull. No. 167. Queen’s Printer, Ottawa, Ont. 3. PALMER, C.M. & T.E. MALONEY. 1954. A New Counting Slide for Nannoplankton. Spec. Publ. No. 21, American Soc. Limnology & Oceanography. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 4. SOURNIA, A., ed. 1978. Phytoplankton Manual. Monogr. Oceanogr. Methodol. No. 6. United Nations Educational, Scientific & Cultural Org., Paris. 5. LACKEY, J.B. 1938. The manipulation and counting of river plankton and changes in some organisms due to formalin preservation. Pub. Health Rep. 53:2080. 6. OWEN, B.B., JR., M. AFZAL & W.R. CODY. 1978. Staining preparations for phytoplankton and periphyton. Brit. Phycol. J. 13:155. 10200 G. Zooplankton Counting Techniques 1. Subsampling Count entire samples having low zooplankton numbers (<200 zooplankters) without subsampling. However, most zooplankton samples will contain more organisms than can be enumerated practically; therefore, use a subsampling procedure. Before subsampling, remove and enumerate all large uncommon organisms such as fish larvae in fresh water or coelenterates, decapods, fish larvae, etc., in salt water. Subsample by the pipet or splitting method. In the pipet method, adjust sample to a convenient volume in a graduated cylinder or Imhoff cone. Concentrating the plankton by using a rubber bulb and clear acrylic plastic tube with fine mesh netting fitted on the end is convenient and accurate (Figure 10200:9). For picoplankton and the smaller microzooplankton, use sedimentation techniques described for concentrating phytoplankton. Transfer sample to a beaker or other wide-mouth vessel for subsampling with a Hensen-Stempel or similar wide-bore pipet. Gently stir sample completely and randomly with the pipet and quickly withdraw 1 to 5 mL. Transfer to a suitable counting chamber. Alternatively, subsample by splitting with any of a number of devices of which the Folsom plankton splitter1 is best known (Figure 10200:10). Level splitter before using. Place sample in the splitter and divide into subsplits. Rinse splitter into the subsamples. Repeat until a workable number (200 to 500 individuals) is obtained in a subsample. Exercise care to provide unbiased splits. Even when using the Folsom splitter unbiased subsamples cannot be unquestioningly assumed;2 therefore, count animals in several subsamples from the same sample to verify that the splitter is unbiased and to determine the sampling error introduced by using it. Another method permits abundance estimates of more equivalent levels of precision among taxa than obtained with either the Hensen-Stempel pipet or the Folsom splitter.3 Normal counting procedures tally organisms on the basis of their abundance in a sample. Therefore, in a sample with a dominant organism making up 50% of total numbers, the tally of the dominant taxon will be large and have a small error. However, error about the subdominants will increase as the tally of each taxon decreases. By accepting one level of precision, the technique3 has been developed to obtain the same error about dominants and subdominants, permitting quantitative comparisons between taxa over successive times or between stations. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. Enumeration Using a compound microscope and a magnification of 100×, enumerate small zooplankton (protozoa, rotifers, and nauplii) in a 1- to 5-mL clear acrylic plastic counting cell fitted with a glass cover slip. For larger, mature microcrustacea use a counting chamber holding 5 to 10 mL. A Sedgwick-Rafter cell is not suitable because of size. An open counting chamber 80 by 50 mm and 2 mm deep is desirable; however, an open chamber is difficult to move without jarring and disrupting the count. A mild detergent solution placed on the chamber before counting reduces organism movements or special counting trays with parallel or circular grooves or partitions4,5 can be used. Count microcrustacea with a binocular dissecting microscope at 20× to 40× magnification. If identification is questionable, remove organisms with a microbiological transfer loop and examine at a higher magnification under a compound microscope. Report smaller zooplankton as number per liter and larger forms as number per cubic meter: where: C= V′ = V′′ = V′′′ = number of organisms counted, volume of the concentrated sample, mL, volume counted, mL, and volume of the grab sample, m3. To obtain organisms per liter divide by 1000. 3. References 1. LONGHURST, A.R. & D.L.R. SEIBERT. 1967. Skill in the use of Folsom’s plankton sample splitter. Limnol. Oceanogr. 12:334. 2. MCEWEN, G.F., M.W. JOHNSON & T.R. FOLSOM. 1954. A statistical analysis of the Folsom sample splitter based upon test observations. Arch. Meteorol. Geophys. Bioklimatol., Ser. A, 6:502. 3. ALDEN, R.W., III, R.C. DAHIYA & R.J. YOUNG, JR. 1982. A method for the enumeration of zooplankton samples. J. Exp. Mar. Biol. Ecol. 59:185. 4. GANNON, J.E. 1971. Two counting cells for the enumeration of zooplankton micro-crustacea. Trans. Amer. Microsc. Soc. 90:486. 5. DODSON, A.N. & W.H. THOMAS. 1964. Concentrating plankton in gentle fashion. Limnol. Oceanogr. 9:455. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 10200 H. Chlorophyll The concentration of photosynthetic pigments is used extensively to estimate phytoplankton biomass.1,2 All green plants contain chlorophyll a, which constitutes approximately 1 to 2% of the dry weight of planktonic algae. Other pigments that occur in phytoplankton include chlorophylls b and c, xanthophylls, phycobilins, and carotenes. The important chlorophyll degradation products found in the aquatic environment are the chlorophyllides, pheophorbides, and pheophytins. The presence or absence of the various photosynthetic pigments is used, among other features, to separate the major algal groups. The three methods for determining chlorophyll a in phytoplankton are the spectrophotometric,3-5 the fluorometric,6-8 and the high-performance liquid chromatographic (HPLC) techniques.9 Fluorometry is more sensitive than spectrophotometry, requires less sample, and can be used for in-vivo measurements.10 These optical methods can significantly under- or overestimate chlorophyll a concentrations,11-18 in part because of the overlap of the absorption and fluorescence bands of co-occurring accessory pigments and chlorophyll degradation products. Pheophorbide a and pheophytin a, two common degradation products of chlorophyll a, can interfere with the determination of chlorophyll a because they absorb light and fluoresce in the same region of the spectrum as does chlorophyll a. If these pheopigments are present, significant errors in chlorophyll a values will result. Pheopigments can be measured either by spectrophotometry or fluorometry, but in marine and freshwater environments the fluorometric method is unreliable when chlorophyll b co-occurs. Upon acidification of chlorophyll b, the resulting fluorescence emission of pheophytin b is coincident with that of pheophytin a, thus producing underestimation and overestimation of chlorophyll a and pheopigments, respectively. HPLC is a useful method for quantifying photosynthetic pigments9,13,15,16,19-21 including chlorophyll a, accessory pigments (e.g., chlorophylls b and c), and chlorophyll degradation products (chlorophyllides, pheophorbides, and pheophytins). Pigment distribution is useful for quantitative assessment of phytoplankton community composition and zooplankton grazing activity.22 1. Pigment Extraction Conduct work with chlorophyll extracts in subdued light to avoid degradation. Use opaque containers or wrap with aluminum foil. The pigments are extracted from the plankton concentrate with aqueous acetone and the optical density (absorbance) of the extract is determined with a spectrophotometer. The ease with which the chlorophylls are removed from the cells varies considerably with different algae. To achieve consistent complete extraction of the pigments, disrupt the cells mechanically with a tissue grinder. Glass fiber filters are preferred for removing algae from water. The glass fibers assist in breaking the cells during grinding, larger volumes of water can be filtered, and no precipitate © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater forms after acidification. Inert membrane filters such as polyester filters may be used where these factors are irrelevant. a. Equipment and reagents: 1) Tissue grinder:*#(118) Successfully macerating glass fiber filters in tissue grinders with grinding tube and pestle of conical design may be difficult. Preferably use round-bottom grinding tubes with a matching pestle having grooves in the TFE tip. 2) Clinical centrifuge. 3) Centrifuge tubes, 15-mL graduated, screw-cap. 4) Filtration equipment, filters, glass fiber†#(119) or membrane (0.45-µm porosity, 47-mm diam); vacuum pump; solvent-resistant disposable filter assembly, 1.0-µm pore size;‡#(120) 10-mL solvent-resistant syringe. 5) Saturated magnesium carbonate solution: Add 1.0 g finely powdered MgCO3 to 100 mL distilled water. 6) Aqueous acetone solution: Mix 90 parts acetone (reagent-grade BP 56°C) with 10 parts saturated magnesium carbonate solution. For HPLC pigment analysis, mix 90 parts HPLC-grade acetone with 10 parts distilled water. b. Extraction procedure: 1) Concentrate sample by centrifuging or filtering as soon as possible after collection. If processing must be delayed, hold samples on ice or at 4°C and protect from exposure to light. Use opaque bottles because even brief exposure to light during storage will alter chlorophyll values. Samples on filters taken from water having pH 7 or higher may be placed in airtight plastic bags and stored frozen for 3 weeks. Process samples from acidic water promptly after filtration to prevent possible chlorophyll degradation from residual acidic water on filter. Use glassware and cuvettes that are clean and acid-free. 2) Place sample in a tissue grinder, cover with the 2 to 3 mL 90% aqueous acetone solution, and macerate at 500 rpm for 1 min. Use TFE/glass grinder for a glass-fiber filter and glass/glass grinder for a membrane filter. 3) Transfer sample to a screw-cap centrifuge tube, rinse grinder with a few milliliters 90% aqueous acetone, and add the rinse to the extraction slurry. Adjust total volume to 10 mL, with 90% aqueous acetone. Use solvent sparingly and avoid excessive dilution of pigments. Steep samples at least 2 h at 4°C in the dark. Glass fiber filters of 25- and 47-mm diam§#(121) have dry displacement volumes of 0.03 and 0.10 mL, respectively, and introduce errors of about 0.3 and 1.0% if a 10-mL extraction volume is used. 4) Clarify by filtering through a solvent-resistant disposable filter (to minimize retention of extract in filter and filter holder, force 1 to 2 mL air through the filter after the extract), or by centrifuging in closed tubes for 20 min at 500 g. Decant clarified extract into a clean, calibrated, 15-mL, screw-cap centrifuge tube and measure total volume. Proceed as in 2, 3, 4, or 5 below. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. Spectrophotometric Determination of Chlorophyll a. Equipment and reagents: 1) Spectrophotometer, with a narrow band (pass) width (0.5 to 2.0 nm) because the chlorophyll absorption peak is relatively narrow. At a spectral band width of 20 nm the chlorophyll a concentration may be underestimated by as much as 40%. 2) Cuvettes, with 1-, 4-, and 10-cm path lengths. 3) Pipets, 0.1- and 5.0-mL. 4) Hydrochloric acid, HCl, 0.1N. b. Determination of chlorophyll a in the presence of pheophytin a: Chlorophyll a may be overestimated by including pheopigments that absorb near the same wavelength as chlorophyll a. Addition of acid to chlorophyll a results in loss of the magnesium atom, converting it to pheophytin a. Acidify carefully to a final molarity of not more than 3 × 10−3M to prevent certain accessory pigments from changing to absorb at the same wavelength as pheophytin a.13 When a solution of pure chlorophyll a is converted to pheophytin a by acidification, the absorption-peak-ratio (OD664/OD665) of 1.70 is used in correcting the apparent chlorophyll a concentration for pheophytin a. Samples with an OD664 before/OD665 after acidification ratio (664b/665a) of 1.70 are considered to contain no pheophytin a and to be in excellent physiological condition. Solutions of pure pheophytin show no reduction in OD665 upon acidification and have a 664b/665a ratio of 1.0. Thus, mixtures of chlorophyll a and pheophytin a have absorption peak ratios ranging between 1.0 and 1.7. These ratios are based on the use of 90% acetone as solvent. Using 100% acetone as solvent results in a chlorophyll a before-to-after acidification ratio of about 2.0.3 Spectrophotometric procedure—Transfer 3 mL clarified extract to a 1-cm cuvette and read optical density (OD) at 750 and 664 nm. Acidify extract in the cuvette with 0.1 mL 0.1N HCl. Gently agitate the acidified extract and read OD at 750 and at 665 nm, 90 s after acidification. The volumes of extract and acid and the time after acidification are critical for accurate, consistent results. The OD664 before acidification should be between 0.1 and 1.0. For very dilute extracts use cuvettes having a longer path length. If a larger cell is used, add a proportionately larger volume of acid. Correct OD obtained with larger cuvettes to 1 cm before making calculations. Subtract the 750-nm OD value from the readings before (OD 664 nm) and after acidification (OD 665 nm). Using the corrected values calculate chlorophyll a and pheophytin a per cubic meter as follows: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: V1 = volume of extract, L, V2 = volume of sample, m3, L = light path length or width of cuvette, cm, and 664b, 665a = optical densities of 90% acetone extract before and after acidification, respectively. The value 26.7 is the absorbance correction and equals A × K where: A = absorbance coefficient for chlorophyll a at 664 nm = 11.0, and K = ratio expressing correction for acidification. c. Determination of chlorophyll a, b, and c (trichromatic method): Spectrophotometric procedure—Transfer extract to a 1-cm cuvette and measure optical density (OD) at 750, 664, 647, and 630 nm. Choose a cell path length or dilution to give OD664 between 0.1 and 1.0. Use the optical density readings at 664, 647, and 630 nm to determine chlorophyll a, b, and c, respectively. The OD reading at 750 nm is a correction for turbidity. Subtract this reading from each of the pigment OD values of the other wavelengths before using them in the equations below. Because the OD of the extract at 750 nm is very sensitive to changes in the acetone-to-water proportions, adhere closely to the 90 parts acetone:10 parts water (v/v) formula © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater for pigment extraction. Turbidity can be removed easily by filtration through a disposable, solvent-resistant filter attached to a syringe or by centrifuging for 20 min at 500 g. Calculate the concentrations of chlorophyll a, b, and c in the extract by inserting the corrected optical densities in following equations:5 a) Ca = 11.85(OD664) − 1.54(OD647) − 0.08(OD630) b) Cb = 21.03(OD647) − 5.43(OD664) − 2.66(OD630) c) Cc = 24.52(OD630) − 7.60(OD647) − 1.67(OD664) where: Ca, Cb, and Cc = concentrations of chlorophyll a, b, and c, respectively, mg/L, and OD664, OD647, and OD630 = corrected optical densities (with a 1-cm light path) at the respective wavelengths. After determining the concentration of pigment in the extract, calculate the amount of pigment per unit volume as follows: 3. Fluorometric Determination of Chlorophyll a The fluorometric method for chlorophyll a is more sensitive than the spectrophotometric method and thus smaller samples can be used. Calibrate the fluorometer spectrophotometrically with a sample from the same source to achieve acceptable results. Optimum sensitivity for chlorophyll a extract measurements is obtained at an excitation wavelength of 430 nm and an emission wavelength of 663 nm. A method for continuous measurement of chlorophyll a in vivo is available, but is reported to be less efficient than the in-vitro method given here, yielding about one-tenth as much fluorescence per unit weight as the same amount in solution. Pheophytin a also can be determined fluorometrically.24 a. Equipment and reagents: In addition to those listed under 1a and 2a above: Fluorometer,i#(122) equipped with a high-intensity F4T.5 blue lamp, photomultiplier tube R-446 (red-sensitive), sliding window orifices 1×, 3×, 10×, and 30×, and filters for light emission (CS-2-64) and excitation (CS-5-60). A high-sensitivity door is preferable. b. Extraction procedure: Prepare sample as directed in 1b above. 1) Calibrate fluorometer with a chlorophyll solution of known concentration as follows: Prepare chlorophyll extract and analyze spectrophotometrically. Prepare serial dilutions of the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater extract to provide concentrations of approximately 2, 6, 20, and 60 µg chlorophyll a/L. Make fluorometric readings for each solution at each sensitivity setting (sliding window orifice): 1×, 3×, 10×, and 30×. Using the values obtained, derive calibration factors to convert fluorometric readings in each sensitivity level to concentrations of chlorophyll a, as follows: where: Fs = calibration factor for sensitivity setting S, Rs = fluorometer reading for sensitivity setting S, and, C′a = concentration of chlorophyll a determined spectrophotometrically, µg/L. 2) Measure sample fluorescence at sensitivity settings that will provide a midscale reading. (Avoid using the 1× window because of quenching effects.) Convert fluorescence readings to concentrations of chlorophyll a by multiplying the readings by the appropriate calibration factor. c. Determination of chlorophyll a in the presence of pheophytin a: This method normally is not applicable to freshwater samples. See discussion under Section 10200H and ¶ 2b above. 1) Equipment and reagents—In addition to those listed under ¶ 1a and ¶ 2a above, pure chlorophyll a##(123) (or a plankton chlorophyll extract with a spectrophotometric before-and-after acidification ratio of 1.70 containing no chlorophyll b). 2) Fluorometric procedure—Calibrate fluorometer as directed in ¶ 3b1). Determine extract fluorescence at each sensitivity setting before and after acifidication. Calculate calibration factors (Fs) and before-and-after acidification fluorescence ratio by dividing fluorescence reading obtained before acidification by the reading obtained after acidification. Avoid readings on the 1× scale and those outside the range of 20 to 80 fluorometric units. 3) Calculations—Determine the ‘‘corrected’’ chlorophyll a and pheophytin a in sample extracts with the following equations:8,24 where: Fs = conversion factor for sensitivity setting S (see ¶ 2b, above), © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Rb = fluorescence of extract before acidification, Ra = fluorescence of extract after acidification, r = Rb/Ra, as determined with pure chlorophyll a for the instrument (redetermine r and Fs if filters or light source are changed), Ve = volume of extract, and Vs = volume of sample. d. Extraction of whole water, nonfiltered samples: Alternatively, to prevent cell lysis during filtration, extract whole water sample. 1) Equipment and reagents—Fluorometer equipped with a high-sensitivity R928 phototube**#(124) with output impedance of 36 ma/W at 675 nm and a high-sensitivity door. Place neutral density filter (40–60N) in the rear light path,††#(125) selected to permit reagent blanking on the highest sensitivity scale. 2) Extraction procedure—Decant 1.5 mL sample into screw-cap test tube and add 8.5 mL 100% acetone. Mix with vortex mixer and hold in the dark for 6 h at room temperature. Filter through glass fiber filter‡‡#(126) or centrifuge. Measure fluorescence as described in Section 10200H.3 and estimate concentrations as in ¶ 3c. Because humic substances interfere, if they are present filter a sample portion (see Section 10200H.1b) and process filtrate with sample. Subtract filtrate (blank) fluorescence from that of sample. 4. High-Performance Liquid Chromatographic Determination of Algal Chlorophylls and Their Degradation Products a. Equipment and reagents: In addition to those listed for pigment extraction, ¶ 1a above: 1) High-pressure liquid chromatograph capable of a flow rate of 2.0 mL/m. 2) High-pressure injector valve equipped with a 100-µL sample loop. 3) Guard column (4.0 × 0.5 cm, C18 packing material, 3-µm particle size, or equivalent protection system) for extending life of primary column. 4) Reverse-phase HPLC column.§§#(127) 5) Fluorescence detector capable of excitation at 430 ± 30 nm and measuring emission at wavelengths greater than 600 nm. 6) Data recorder device: Strip chart recorder or, preferably, an electronic integrator. 7) Syringe, glass, 250-µL. 8) HPLC eluents: System A (80:15:5; methanol:reagent water: ion-pairing solution) and System B (80:20; methanol:acetone). Use HPLC-grade solvents; measure volumes before mixing. Filter eluents through a solvent-resistant 0.4-µm filter before use and degas with helium. Prepare the ion-pairing (IP) solution from 15 g tetrabutylammonium acetatei i#(128) and 77 g ammonium acetate###(129) made up to 1 L with reagent water.15 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 9) Calibration standards: Individually dissolve 1 mg each pure chlorophyll a and bi i#(130) in 100 mL 90% acetone. Determine the exact concentrations spectrophotometrically (ε664 for chlorophyll a in 90% acetone = 87.67 L g−1 cm−1; ∈647 for chlorophyll b in 90% acetone = 51.36 L g−1 cm−1).5 Prepare pheophytin a + a′ and b + b′ standards from the primary chlorophyll a and b standards by acidification with hydrochloric acid; correct respective concentrations for Mg2+ loss. Extract chlorophyll c with 90% acetone from diatoms, purify by thin-layer chromatography (TLC)25 and calibrate spectrophotometrically (ε631 for a mixture containing equal amounts of chlorophylls c1 and c2 in 90% acetone containing 1% pyridine = 42.6 L g−1 cm−1; the absence of this small amount of pyridine is presumed to cause only small differences in the absorption properties of chlorophyll c.26 Alternatively, determine the chlorophyll c content of a 90% acetone extract made from diatoms, spectrophotometrically (chlorophyll c1 + c2, µg/mL = 24.36E630 − 3.73E664)5 and use as standard. Prepare chlorophyllide a from diatoms,27 purify by TLC25 and calibrate spectrophotometrically in 90% acetone (ε664 for chlorophyllide a = 128 L g−1 cm−1).28 Prepare pheophorbide a by acidification of chlorophyllide a, purify by TLC,25 and calibrate spectrophotometrically in 90% acetone (∈665 for pheophorbide a = 69.8 L g−1 cm−1).28 Standards stored under nitrogen in the dark at −20°C are stable for about 1 month. b. Procedure: 1) Set up and equilibrate the HPLC system with solvent System A at a flow rate of 2 mL/min. Adjust fluorometer sensitivity to provide full-scale reading with the most concentrated chlorophyll a standard. 2) Calibrate HPLC system by preparing working standards from the primary standards (on day of use). Once retention times of the standards are determined for a particular system, simplify standardization by preparing serial dilutions from mixed standards. Prepare separately mixed standards for the chlorophylls and chlorophyllide a and for the pheophytins and pheophorbide a. Mix 1-mL portions of standards with 300 µL ion-pairing solutions and equilibrate for 5 min before injection (use of ion-pairing agents greatly enhances separation of dephytolated pigments, chlorophyllide a, chlorophyll c, and pheophorbide a). Prepare blanks by mixing 1 mL 90% acetone with 300 µL IP solution. Rinse syringe twice with 150 µL standard and draw about 250 µL standard into syringe for injection. Place syringe in injector valve, overfilling the 100-µL sample loop. Construct calibration curves by plotting fluorescence peak areas (or heights) against standard pigment concentrations. 3) Prepare samples for injection by mixing a 1-mL portion of the 90% acetone pigment extract with 300 µL IP solution. 4) Use a two-step solvent program to optimize separation of the chorophylls from their degradation products.15 After injection, change from solvent System A to System B over 5 min © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater and follow with System B for 15 min at a flow rate of 2 mL/min. Re-equilibrate the column with System A for 5 min before the next injection for a total analysis time of approximately 25 min. Degas the solvent systems with helium during analysis. Increase lifetime of HPLC column by storing it in 100% methanol between runs. Periodically flush the HPLC system with reagent water to avoid buildup of ion pairing agents. 5) Calculate individual pigment concentrations using the following formula: where: Ci = As = Fi = peak area). VI = VE = VS = individual pigment concentration, mg/L, area of individual pigment peak from sample injection, standard response factor (mg pigment/0.1 mL standard divided by corresponding injection volume (0.1 mL), extraction volume, mL, and sample volume, L. 6) This method is designed only for quantification of chlorophylls and their degradation products. Detect carotenoid pigments, which also are present in 90% acetone extracts but do not fluoresce, by absorbance spectroscopy (at about 440 nm).21 7) The elution order and approximate retention times for the major chlorophyll pigments and their degradation products are shown in Figure 10200:11. The detection limits (s/n = 2) vary with fluorometer configuration and flow rate; however, they range from 10 to 100 pg per injection for most chlorophylls and their degradation products.15,21,29 The accuracy of the HPLC method depends primarily on purity of pigment standards. Preferably measure absorption spectra (350 to 750 nm) of the standards and compare with published data. Pigment purity also can be assessed by HPLC analysis, providing there are no co-eluting contaminants with absorption and fluorescence bands overlapping those of the standards. HPLC and spectrophotometrically derived pigment concentrations for available EPA standards agree reasonably well (± 20%) if spectrophotometric results are corrected for the presence of pheopigments and the HPLC results are expressed as pigment equivalents (e.g., chlorophyll a equivalents = chlorophyllide a + chlorophyll a + chlorophyll a′, provided that the proper molecular weight corrections are applied).30 Thus, if significant amounts of chlorophyll derivatives are present, pigment concentrations determined spectrophotometrically will be overestimated. The agreement between HPLC and fluorometrically derived results depends on the presence of accessory chlorophylls b, c, and their derivatives. Triplicate injections of a fivefold dilution of an EPA © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater sample gave coefficients of variation of 7.5% (chlorophyllide a), 9.1% (chlorophyll c), 13.4% (pheophorbide a), 9.6% (chlorophyll b), 0.5% (chlorophyll a), 6.2% (pheophytin a), and 22.9% (pheophytin a′), with an average value of 10% for the seven pigments analyzed. 5. High-Performance Liquid Chromatographic Determination of Algal Chlorophyll and Carotenoid Pigments (PROPOSED) a. Equipment and reagents: In addition to those listed for pigment extraction, ¶ 1a above: 1) High-performance liquid chromatographic pump capable of gradient delivery of three different solvents at a flow rate of 1 mL/min. 2) High-pressure injector valve equipped with a 200-µL sample loop. 3) Guard column (50 × 4.6 mm, C18 packing material,***#(131) 5-µm particle size) for extending life of primary column. 4) Reverse-phase HPLC column with endcapping (250 × 4.6 mm, 5-µm particle size, C18 column***#(132)). 5) Variable wavelength or filter absorbance detector with low-volume flowthrough cell. Detection wavelength is 436 nm. 6) Data recording device: Strip chart recorder or, preferably, an electronic integrator or computer equipped with hardware and software for chromatographic data analysis. 7) Syringe, glass, 500-µL. 8) HPLC eluents: Eluent A (80:20, v:v; methanol:0.5M ammonium acetate, pH 7.2); Eluent B (90:10, v:v; acetonitrile:water), and Eluent C, ethyl acetate. Use HPLC-grade solvents. Measure volumes before mixing. Filter eluents through a solvent-resistant 0.4-µm filter before use and degas with helium. 9) Calibration standards: Chlorophylls a and b, and β,β-carotene can be purchased†††#(133) as can zeaxanthin and lutein.‡‡‡#(134) Other pigment standards can be purified from plant extracts by thin-layer chromatography (TLC)25 or preparative-scale HPLC. Determine concentration of all standards using a monochromator-based spectrophotometer in the appropriate solvents before calibration of the HPLC system. The recommended extinction coefficients for most common algal pigments found in freshwater systems are given in Table 10200:III. Measure absorbance in a 1-cm cuvette at the appropriate wavelength (usually at λmax) and 750 nm (to correct for light scattering). Calculate concentrations of standards as follows: where: Ci = individual pigment concentration, mg/L, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater A = absorbance at specific wavelength, E1cm = weight-specific absorption coefficient, L g−1 cm−1, b = pathlength of cuvette, cm, and 1000 = conversion factor, g to mg. Standards stored under nitrogen in the dark at −20°C are stable for about 1 month. b. Procedure: 1) Set up and equilibrate the HPLC system with Eluent A at a flow rate of 1 mL/min. 2) Calibrate the HPLC using working standards (about 0 to 1000 ng/mL) prepared from primary standards on day of use. Mix 1 mL standard with 300 µL distilled water, shake, and equilibrate for 5 min before injection (diluting standards and sample extracts with water increases affinity of pigments for the column in the loading step, resulting in an improved separation of more polar pigments). Rinse syringe twice with 300 µL standard and draw 500 µL standard into syringe for injection. Place syringe in injector valve, overfilling the 200 µL sample loop 2.5-fold. To check for possible interferences in the extraction solvent and/or filter, prepare a blank by extracting a glass fiber filter in 90% acetone; mixing 1 mL 90% acetone filter extract and 300 µL distilled water; and injecting into the HPLC system. Plot absorbance peak areas (or heights) against standard pigment concentrations. Calculate response factors as the slope of the regression between the weights of the injected standards (ng) and the areas of the parent pigment (plus areas of structurally-related isomers if present). These isomers contribute to the absorption signal of the standards; disregarding them results in over-estimation of pigments in sample extracts.31 3) Prepare samples for injection by mixing a 1 mL portion of the 90% acetone pigment extract and 300 µL distilled water, shake, and equilibrate for 5 min before injection. 4) Following sample injection, use a gradient program to optimize separation of chlorophyll and carotenoid pigments. The system described in Table 10200:IV has been developed from the original method32 to insure elution of most hydrophobic pigments. Degas solvent system with helium during analysis. Periodically flush HPLC system with distilled water to avoid accumulation of ion-pairing reagents. 5) Routinely determine peak identities by comparing retention times of sample peaks with those of pure standards. Confirm peak identities spectrophotometrically by collecting eluting peaks from the column outlet (or directly with an on-line diode array spectrophotometer). Absorption maxima for most common pigments found in freshwater systems are given in Table 10200:III. 6) Calculate individual pigment concentrations using the formula given in ¶ 4b5) preceding. 7) This method is designed for separation of chlorophyll and carotenoid pigments (Figure 10200:12), however, it also separates major chlorophyll breakdown products. 8) Method precision was assessed by making triplicate injections of a mixture of phytoplankton and plant extracts. Coefficients of variation ranged from 0.6 to 6.0% (Table © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 10200:III). Using an appropriate internal standard increases precision. 6. References 1. ROTT, E. Spectrophotometric and chromatographic chlorophyll analysis: comparison of results and discussion of the trichromatic method. Ergebn. Limnol. (Suppl. to Arch. Hydrobiol.) 14:37. 2. MARKER, A.F.H., E.A. NUSCH, H. RAI & B. RIEMANN. 1980. The measurement of photosynthetc pigments in freshwaters and standardization of methods: Conclusions and recommendations. Ergebn. Limnol. (Suppl. to Arch. Hydrobiol.) 14:91. 3. LORENZEN, C.J. 1967. Determination of chlorophyll and pheo-pigments: spectrophotometric equations. Limnol. Oceanogr. 12:343. 4. FITZGERALD, G.P. & S.L. FAUST. 1967. A spectrophotometric method for the estimation of percentage degradation of chlorophylls to pheopigments in extracts of algae. Limnol. Oceanogr. 12:335. 5. JEFFREY, S.W. & G.F. HUMPHREY. 1975. New spectrophotometric equations for determining chlorphylls a, b, and c, in higher plants, algae and natural phytoplankton. Biochem. Physiol. Pflanzen 167: 191. 6. YENTSCH, C.S. & D.W. MENZEL. 1963. A method for the determination of phytoplankton chlorophyll and phaeophytin by fluorescence. Deep Sea Res. 10:221. 7. LOFTUS, M.E. & J.H. CARPENTER. 1971. A fluorometric method for determining chlorophylls a, b, and c. J. Mar. Res. 29:319. 8. HOLM-HANSEN, O., C.J. LORENZEN, R.W. HOLMES & J.D.H. STRICKLAND. 1965. Fluorometric determination of chlorophyll. J. Cons. Cons. Perma. Int. Explor. Mer 30:3. 9. ABAYCHI, J.K. & J.P. RILEY. 1979. The determination of phytoplankton pigments by high-performance liquid chromatography. Anal. Chim. Acta 107:1. 10. LORENZEN, C.J. 1966. A method for the continous measurement of in vivo chlorophyll concentration. Deep Sea Res. 13:223. 11. JACOBSEN, T.R. 1978. A quantitative method for the separation of chlorophylls a and b from phytoplankton pigments by high-pressure liquid chromatography. Mar. Sci. Comm. 4:33. 12. BROWN, L.M., B.T. HARGRAVE & M.D. MACKINNON. 1981. Analysis of chlorophyll a in sediments by high-performance liquid chromatography. Can. J. Fish. Aquat. Sci. 38:205. 13. GIESKES, W.W. & G.W. KRAAY. 1983. Unknown chlorophyll a derivatives in the North Sea and the tropical Atlantic Ocean revealed by HPLC analysis. Limnol. Oceanogr. 28:757. 14. GOWEN, R.J., P. TETT & B.J.B. WOOD. 1983. Changes in the major dihydroporphyrin plankton pigments during the spring bloom of phytoplankton in two Scoottish © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. sea-lochs. J. Mar. Biol. Assoc. U.K. 63:27. MANTOURA, R.F.C. & C.A. LLWEWLLYN. 1983. The rapid determination of algal chlorophyll and caroteniod pigments and their breakdown products in natural waters by reverse-phase high-performance liquid chromatography. Anal. Chim. Acta 151:297. GIESKES, W.W.C. & G.W. KRAAY. 1984. Phytoplankton, its pigments, and primary production at a central North Sea station in May, July and September 1981. Neth. J. Sea Res. 18.71. HALLEGRAEFF, G.M. & S.E. JEFFREY. 1985. Description of new chlorophyll a alteration products in marine phytoplankton. Deep Sea Res. 32:697. TREES, C.C., M.C. KENNICUTT II & J.M. BROOKS. 1985. Errors associated with the standard fluorometric determination of chlorophylls and phaeopigments. Mar. Chem. 17:1. ESKINS, K., C.R. SCHOFIELD & H.J. DUTTON. 1977. High-performance liquid chromatography of plant pigments. J. Chromatogr. 135:217. WRIGHT, S.W. & J.D. SHEARER. 1984. Rapid extraction and high performance liquid chromatography of chlorophylls and carotenoids from marine phytoplankton. J. Chromatogr. 294:281. BIDIGARE, R.R., M.C. KENNICUTT II & J.M. BROOKS. 1985. Rapid determination of chlorophylls and their degradation products by high-performance liquid chromatography. Limnol. Oceanogr. 30:432. JEFFRY, S.W. 1974. Profiles of photosynthetic pigments in the ocean using thin-layer chromatography. Mar. Biol. 26:101. PHINNEY, D.A. & C.S. YENTSCH. 1985. A novel phytoplankton chlorophyll technique: toward automated analysis. J. Plankton Res. 7: 633. STRICKLAND, J.D.H. & T.R. PARSONS. 1968. A Practical Manual of Sea Water Analysis. Fish. Res. Board Can. Bull. No. 167. Queen’s Printer, Ottawa, Ont. JEFFREY, S.W. 1981. An improved thin-layer chromatographic technique for marine phytoplankton pigments. Limnol. Oceanogr. 26:191. JEFFREY, S.W. 1972. Preparation and some properties of crystalline chlorophyll c1 and c2 from marine algae. Biochim. Biophys. Acta 279: 15. 27. BARRETT, J. & S.W. JEFFREY. 1971. A note on the occurrence of chlorophyllase in marine algae. J. Exp. Mar. Biol. Ecol. 7:255. 28. LORENZEN, C.J. & J. NEWTON DOWNS. 1986. Specific absorption coefficients of chlorophyllide a and pheophorbide a in 90 percent acetone, and comments on the fluorometric determination of chlorophyll and pheopigments. Limnol. Oceanogr. 31:449. 29. SARTORY, D.P. 1985. The determination of algal chlorophyllous pigments by high performance liquid chromatography and spectrophotometry. Water Res. 19:605. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 30. MURRAY, A.P., C.F. GIBBS & A.R. LONGMORE. 1986. Determination of chlorophyll in marine waters: Intercomparison of a rapid HPLC method with full HPLC, spectrophotometric and fluorometric methods. Mar. Chem. 19:211. 31. BIDIGARE, R.R. 1991. Analysis of algal chlorophylls and carotenoids. In D.C. Hurd & D.W. Spencer, eds., Marine Particles: Analysis and Characterization. America Geophysical Union, Washington, D.C. 32. WRIGHT, S.W., S.W. JEFFREY, R.F.C. MANTOURA, C.A. LLEWELLYN, T. BJORNLAND, D. REPETA & N. WELSCHMEYER. 1991. Improved HPLC method for the analysis of chlorophylls and carotenoids from marine phytoplankton. Mar. Ecol. Prog. Ser. 77:183. 33. JEFFREY, S.W. & F.T. HAXO. 1968. Photosynthetic pigments of symbiotic dinoflagellates (zooxanthallae) from corals and clams. Biol. Bull. 135:149. 34. JENSEN, A. 1978. Chlorophylls and carotenoids. In J.A. Helleburst & J.S. Craige, eds., Handbook of Phycological Methods: Physiological and Biochemical Methods. Cambridge University Press, Cambridge, England. 35. DAVIS, B.H. 1976. Carotenoids. In T.W. Goodwin, ed., Chemistry and Biochemistry of Plant Pigments. Academic Press, New York, N.Y. 36. JOHANSEN, J.E., W.A. SVEC & S. LIAAEN-JENSEN. 1974. Carotenoids of the Dinophyceae. Phytochem. 13:2261. 10200 I. Determination of Biomass (Standing Crop) Biomass is a quantitative estimate of the total mass of living organisms within a given area or volume. It may include the mass of a population (species biomass) or of a community (community biomass) but gives no information on community structure or function. The most accurate methods for estimation of biomass are dry weight, ash-free dry weight, and volume of living organisms. Indirect methods include estimates of total carbon, caloric content, nitrogen, lipids, carbohydrates, silica (diatoms), and chlorophyll (algae). Adenosine triphosphate1 (ATP) and deoxyribonucleic acid2,3 (DNA) also have been used as indirect estimates. All estimates of biomass can be affected by the presence of organic and inorganic detritus; ATP and DNA analyses include contributions from the bacterial flora.4 1. Chlorophyll a Chlorophyll a is used as an algal biomass indicator.5 Assuming that chlorophyll a constitutes, on the average, 1.5% of the dry weight of organic matter (ash-free weight) of algae, estimate the algal biomass by multiplying the chlorophyll a content by a factor of 67. 2. Biovolume (Cell Volume) Plankton data derived on a volume-per-volume basis often are more useful than numbers per © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater milliliter.6 Determine cell volume by using the simplest geometric configuration that best fits the shape of the cell being measured (such as sphere, cone, cylinder).7 Cell sizes of an organism can differ substantially in different waters and from the same waters at different times during the year; therefore, average measurements from 20 individuals of each species for each sampling period. Calculate the total biovolume of any species by multiplying the average cell volume in cubic micrometers by the number per milliliter. Compute total wet algal volume as: where: Vt = total plankton cell volume, mm3/L, Ni = number of organisms of the ith species/L, and Vi = average volume of cells of ith species, µm3. 3. Cell Surface Area An estimation of cell surface area is valuable in analyzing interactions between the cell and surrounding waters. Compute average surface area in square micrometers and multiply by the number per milliliter of the species being considered. 4. Displacement Volume This method8 measures an equivalent volume of liquid that is displaced by the sample. Displacement volume may be determined by several methods; for simple, direct measurement proceed as follows: Place sample in sieve of mesh size equal to or smaller than net used in capture; let sample drain and transfer to a measured volume of water in a graduated cylinder; measure the new volume containing sample plus known volume. The displacement volume equals the new volume minus original measured volume of water. 5. Gravimetric Methods The biomass of the plankton community can be estimated from gravimetric determinations, although silt and organic detritus interfere. Determine dry weight by placing 100 mg wet concentrated sample in a clean, ignited, and tared porcelain crucible and dry at 105°C for 24 h. Alternatively, filter a known volume of sample through 0.45-µm-pore-diam membrane or a prerinsed, dried, and preweighed glass-fiber filter. (Note that the small sample used in direct filtration may lead to error if not handled properly.) Cool sample in a desiccator and weigh. Obtain ash weight by igniting the dried sample at 500°C for 1 h. Cool, rewet ash with distilled water, and bring to constant weight at 105°C. The ash is rewetted to restore water of hydration of clays and other minerals; this may amount to as much as 10% of weight lost during © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater incineration.9 The ash-free dry weight is the difference between the dry weight and the weight of the ash residue after ashing. The ash-free dry weight is preferred to dry weight to compare mixed assemblages. The ash content may constitute 50% or more of the dry weight in phytoplankton having inorganic structures, such as the diatoms. In other forms the ash content is only about 5% of dry weight. 6. Adenosine Triphosphate (ATP) Methods of measuring adenosine triphosphate (ATP) in plankton provide the only means of determining the total viable plankton biomass. ATP occurs in all plants and animals, but only in living cells; it is not associated with nonliving particulate material. The ratio of ATP to biomass varies from species to species, but appears to be constant enough to permit reliable estimates of biomass from ATP measurements.10 The method is simple and relatively inexpensive and the instrumentation is stable and reliable. The method also has many potential applications in entrainment and bioassay work, especially plankton mortality studies. a. Equipment and reagents: 1) Glassware: clean, sterile, dry borosilicate glass flasks, beakers, and pipets. 2) Filters: 47-mm-diam, 0.45-µm-porosity membrane filters. 3) Filtration equipment. 4) Freezer (−20°C). 5) Boiling water bath. 6) Detection instruments designed specifically for measuring ATP.*#(135) 7) Microsyringes: 10-, 25-, 50-, 100-, and 250-µL. 8) Reaction cuvettes and vials. 9) Tris buffer (0.02M, pH 7.75): Dissolve 7.5 g trishydroxymethylaminomethane in 3 L distilled water and adjust to pH 7.75 with 20% HCl. Autoclave 150-mL portions at 115°C for 15 min. 10) Luciferin-luciferase enzyme preparation:†#(136) Rehydrate frozen (−20°C) lyophilized extracts of firefly lanterns with Tris buffer as directed by the supplier; let stand at room temperature 2 to 3 h, then centrifuge at 300 × g for 1 min and decant the supernatant into a clean, dry test tube; let stand at room temperature for 1 h. 11) Purified ATP standard: Dissolve 12.3 mg disodium ATP in 1 L distilled water and dilute 1.0 mL to 100 mL with Tris buffer; 0.2 mL = 20 ng ATP. b. Procedure: 1) Calibration—To determine the calibration factor (F), prepare a series of dilutions of purified ATP standard and record the light emission from several portions of each concentration of standard. Correct mean area of standards by subtracting peak reading or mean area of several blanks using 0.2 mL Tris buffer. Calculate calibration factor FS as: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: FS = calibration factor at sensitivity S, AS = peak reading or mean area under standard ATP curve corrected for blank, and C = concentration of ATP in standard solution, ng/mL. 2) Sample analysis—Collect a 1- to 2-L sample in a clean, sterile sampler. Pass through a 250-µm net to remove large zooplankton10 and filter through a 47-mm 0.45-µm-porosity filter by applying a vacuum of about 30 kPa. (IMPORTANT: Break the suction before the last film of water is pulled through the filter.) Quickly place filter in a small beaker. Immediately cover filter with 3 mL boiling Tris buffer, using an automatic pipet. Place beaker in boiling water bath for 5 min and, with a Pasteur pipet, transfer extract to a clean, dry, calibrated test tube. Rinse filter and beaker with 2 mL boiling Tris buffer; combine extracts, record volume, bring volume up to 5 mL with Tris buffer, cover tubes with parafilm and, if samples cannot be analyzed immediately, freeze at −25°C. Extracts may be stored for many months in a freezer. Prepare at least triplicate extracts of each sample. The analytical procedure depends on detection equipment used. If a scintillation counter is used, pipet 0.2 mL enzyme preparation into a glass vial. Measure the light emission of the enzyme preparation (blank) for 2 to 3 min at sensitivity settings near that anticipated for the sample. Add 0.2 mL sample extract to the vial, record the time, and swirl. Start recording light output 10 s after combining ATP extract and enzyme preparation; record output for 2 to 3 min, using the same time period for all samples. Determine the mean of areas under the curves obtained and correct by subtracting mean of areas under the curves obtained from blanks prepared as directed in Strickland and Parsons.11 c. Calculations: Calculate concentration of ATP as: Ac = Ve = Vs = Fs = mean corrected area under extract curves, extract volume, mL, volume of sample, L, and calibration factor. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater If an ATP content of 2.4 µg ATP/mg dry weight organic matter is assumed,12 total living plankton biomass (B), as dry weight organic matter, is given as: 7. References 1. HOLM-HANSEN, O. & C.R. BOOTH. 1966. The measurement of adenosine triphosphate in the ocean and its ecological significance. Limnol. Oceanogr. 11:510. 2. HOLM-HANSEN, O., W.H. SUTCLIFFE, JR. & J. SHARP. 1968. Measurement of deoxyribonucleic acid in the ocean and its ecological significance. Limnol. Oceanogr. 13:507. 3. HOLM-HANSEN, O. 1969. Determination of microbial biomass in ocean profiles. Limnol. Oceanogr. 14:740. 4. PAERL, H.W., M.M. TILZER & C.R. GOLDMAN. 1976. Chlorophyll a vs. ATP as algal biomass indicators in lakes. J. Phycol. 12:242. 5. CREITZ, G.I. & F.A. Richards. 1955. The estimation and characterization of plankton populations by pigment analysis. J. Mar. Res. 14: 211. 6. KUTKUHN, J.H. 1958. Notes on the precision of numerical and volumetric plankton estimates from small sample concentrations. Limnol. Oceanogr. 3:69. 7. VOLLENWEIDER, R.A. 1969. A Manual on Methods for Measuring Primary Production in Aquatic Environments. IBP Handbook No. 12. Blackwell Scientific Publ., Oxford, England. 8. JACOBS, F. & G.C. GRANT. 1978. Guidelines for zooplankton sampling in quantitative baseline and monitoring programs. EPA-600/3-78-026, U.S. Environmental Protection Agency. 9. NELSON, D.J. & D.C. SCOTT. 1962. Role of detritus in the productivity of a rock-outcrop community in a Piedmont stream. Limnol. Oceanogr. 7:396. 10. RUDD, J.W.M. & R.D. HAMILTON. 1973. Measurement of adenosine triphosphate (ATP) in two precambrian shield lakes of northwestern Ontario. J. Fish. Res. Board Can. 30:1537. 11. STRICKLAND, J.D.H. & T.R. PARSONS. 1968. A Practical Manual of Sea Water Analysts. Fish. Res. Board Can. Bull. No. 167. Queen’s Printer, Ottawa, Ont. 12. WEBER, C.I. 1973. Recent developments in the measurement of the response of plankton and periphyton to changes in their environment. In G. Glass, ed. Bioassay Techniques and Environmental Chemistry. Ann Arbor Science Publ., Ann Arbor, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Mich. 10200 J. Metabolic Rate Measurements The physiological condition and the spectrum of biological interactions of the aquatic community must be considered for evaluation of the state of natural waters. Earlier, numbers, species composition, and biomass were the prime considerations. Recognition of the limitations of this approach led to the measurement of rates of metabolic processes such as photosynthesis (productivity), nitrogen fixation, respiration, and electron transport. These provide a better understanding of the complex nature of the aquatic ecosystem. An indication of photosynthetic efficiency can be determined by the productivity index (mg C fixed/unit chlorophyll a).1 1. Nitrogen Fixation The ability of an organism to fix nitrogen is a great competitive advantage and plays a major role in population dynamics. Two reliable methods for estimating nitrogen fixation rates in the laboratory are the 15N isotope tracer method2,3 and the acetylene reduction method.4 Because the rate of nitrogen fixation varies greatly with different organisms and with the concentration of combined nitrogen, nitrogen fixation rates cannot be used to estimate biomass of nitrogen-fixing organisms. However, the acetylene reduction method is useful in measuring nitrogen budgets and in algal assay work.5 2. Productivity, Oxygen Method Productivity is defined as the rate at which inorganic carbon is converted to an organic form. Chlorophyll-bearing organisms (phytoplankton, periphyton, macrophytes) serve as primary producers in the aquatic food chain. Photosynthesis ultimately results in the formation of a wide range of organic compounds, release of oxygen, and reduction of carbon dioxide (CO2) in the surrounding waters. Primary productivity6 can be determined by measuring the changes in oxygen and CO2 concentrations.7 In poorly buffered waters, pH can be a sensitive property for detecting variations in the system. As CO2 is removed during photosynthesis, the pH rises. This shift can be used to estimate both photosynthesis and respiration.8 The sea and many fresh waters are too highly buffered to make this useful, but it has been applied successfully to productivity studies in some lake waters. Two methods of measuring the rate of carbon uptake and net photosynthesis in situ are the oxygen method9 and the carbon 14 method.10 In both methods, clear (light) and darkened (dark) bottles are filled with water samples and suspended at regular depth intervals for an incubation period of several hours or samples are incubated under controlled conditions in environmental growth chambers in the laboratory. The basic reactions in algal photosynthesis involve uptake of inorganic carbon and release of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater oxygen, summarized by the relationship: CO2 + H2O → (CH2O) + O2 The chief advantages of the oxygen method are that it provides estimates of gross and net productivity and respiration and that analyses can be performed with inexpensive laboratory equipment and common reagents. The dissolved oxygen (DO) concentration is determined at the beginning and end of the incubation period. Productivity is calculated on the assumption that one atom of carbon is assimilated for each molecule of oxygen released. a. Equipment: 1) BOD bottles, numbered, 300-mL, clear and opaque borosilicate glass, with ground glass stopper and flared mouth, for sample incubation. Acid-clean the bottles, rinse thoroughly with distilled water, and just before use, rinse with water being tested. Do not use phosphorus-containing detergents. If suitable opaque bottles are not available, make clear BOD bottles opaque by painting them black and wrapping with black waterproof tape. As a further precaution, wrap entire bottle in aluminum foil or place in light-excluding container during incubation. 2) Supporting line or rack that does not shade suspended bottles. 3) Nonmetallic opaque acrylic Van Dorn sampler or equivalent, of 3- to 5-L capacity. 4) Equipment and reagents for dissolved oxygen determinations: See Section 4500-O. 5) Pyrheliometer. 6) Submarine photometer. 7) Thermometer. b. Procedure: 1) Obtain a profile of the input of solar radiation for the photoperiod with a pyrheliometer. 2) Determine depth of euphotic zone (the region that receives 1% or more of surface illumination) with a submarine photometer. Select depth intervals for bottle placement. The photosynthesis-depth curve will be approximated closely by placing samples at intervals equal to one-tenth the depth of the euphotic zone. Estimate productivity in relatively shallow water with fewer depth intervals. 3) Measure oxygen concentration with probe or by titration and temperature and salinity to determine whether water is supersaturated with respect to oxygen (see Table 4500-O:I). If water is supersaturated, bubble nitrogen gas through sample to lower initial oxygen concentration to less than 80% saturation. 4) Keep samples out of direct sunlight during handling. Introduce samples taken from each preselected depth into duplicate clear, darkened, and initial-analysis bottles. Insert delivery tube of sampler to bottom of sample bottle and fill so that three volumes of water are allowed to overflow. Remove tube slowly and close bottle. Use water from the same grab sample to fill a © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater ‘‘set’’ (one light, one dark, and one initial bottle). 5) Immediately treat (fix) samples taken for the chemical determination of initial dissolved oxygen (see Section 4500-O) with manganous sulfate (MnSO4), alkaline iodide, and sulfuric acid (H2SO4) or check with an oxygen probe. Analyses may be delayed several hours if necessary, if samples are fixed or iced and stored in the dark. 6) Suspend duplicate paired clear and darkened bottles at the depth from which the samples were taken and incubate for at least 2 h, but never longer than it takes for oxygen-gas bubbles to form in the clear bottles or DO to be depleted in the dark bottles. 7) At the end of the exposure period, immediately determine DO as described above. c. Calculations: The increase in oxygen concentration in the light bottle during incubation is a measure of net production which, because of the concurrent use of oxygen in respiration, is somewhat less than the total (or gross) production. The loss of oxygen in the dark bottle is used as an estimate of total plankton respiration. Thus: Net photosynthesis = light bottle DO − initial DO Respiration = initial DO − dark bottle DO Gross photosynthesis = light bottle DO − dark bottle DO Average results from duplicates. 1) Calculate the gross or net production for each incubation depth and plot: mg carbon fixed/m3 = mg oxygen released/L × 12/32 × 1000 L/m3 × K where K is the photosynthetic quotient (PQ), ranging from 1 to 2, depending on the nitrogen supply.11,12 Use the factor 12/32 to convert oxygen to carbon; under ideal conditions 1 mole of O2 (32 g) is released for each mole of carbon (12 g) fixed. 2) Productivity is defined as the rate of production and generally is reported in grams carbon fixed per square meter per day. Determine the productivity of a vertical column of water 1 m square by plotting productivity for each exposure depth and graphically integrating the area under the curve. 3) Using the solar radiation profile and photosynthesis rate during incubation adjust the data to represent phytoplankton productivity for the entire photoperiod. Because photosynthetic rates vary widely during the daily cycle,13,14 do not attempt to convert data to other test circumstances. 3. Productivity, Carbon 14 Method © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater A solution of radioactive carbonate (14CO32–) is added to light and dark bottles that have been filled with sample as described for the oxygen method. After incubation in situ, collect the plankton on a membrane filter, treat with hydrochloric acid (HCl) fumes to remove inorganic carbon 14, and assay for radioactivity. The quantity of carbon fixed is proportional to the fraction of radioactive carbon assimilated. This procedure differs from the oxygen method in that it affords a direct measurement of carbon uptake and measures only net photosynthesis.15 It is basically more sensitive than the oxygen method, but fails to account for organic materials that leach from cells16,17 during incubation. a. Equipment and reagents: 1) Pyrheliometer. 2) Submarine photometer. 3) BOD bottles and supporting apparatus: See ¶ 2a1) and 2), above. 4) Membrane-filtering device and 25-mm filters with pore diameters of 0.22, 0.30, 0.45, 0.80, and 1.2 µm. 5) Counting equipment for measuring radioactivity: Scaler with end-window tube, gas flow meter, or liquid scintillation counter (see Section 7030B). The thin-window tube is the least expensive detector and, when used with a small scaler, provides acceptable data at modest cost. 6) Fuming chamber: Use a glass desiccator with a depth of about 1.4 cm conc HCl in desiccant chamber. The fuming chamber is recommended for filter decontamination.18,19 7) Syringe or pipet, nonmetallic. 8) Chemical reagents: See Section 4500-CO2 (Carbon Dioxide) and Section 2320 (Alkalinity). 9) Radioactive carbonate solutions: a) Sodium chloride dilution solution, 5% NaCl (w/v): Add 0.3 g sodium carbonate (Na2CO3) and one pellet sodium hydroxide (NaOH) per liter. Use for marine studies only. b) Carrier-free radioactive carbonate solution, commercially available in sealed vials having approximately 5 µCi 14C/mL. Confirm absence of suspended and dissolved toxic metals20 or filter and pass through an ion-exchange column.*#(137) c) Working solutions with activities of 1, 5, and 25 µCi 14C/2 mL. For studies of fresh water use carrier-free radioactive carbonate and for studies of marine water prepare by diluting carrier-free radioactive carbonate solution with NaCl dilution solution. d) Stock ampules: Prepare ampules containing 2 mL of required working solution. Fill ampules and autoclave sealed ampules at 121°C for 20 min.21 b. Procedure: 1) Obtain a record of incident solar radiation for the photoperiod with a pyrheliometer. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2) Determine depth intervals for sampling and incubation as described above. 3) Use duplicate light and dark bottles at each depth. Also use dark bottles or bottles harvested at time zero. Fill bottles with sample, add 2 mL radioactive carbonate solution (using a nonmetallic pipet) to the bottom of each bottle, and mix thoroughly by repeated inversion. The concentration of carbon 14 should be approximately 10 µCi/L in relatively productive waters, to 100 µCi/L, or higher, in oliogotrophic (open ocean) waters. To obtain statistical significance, have at least 1000 cpm in the filtered sample. Take duplicate samples at each depth to determine initial concentration of inorganic carbon (CO2, HCO3–, and CO32–) available for photosynthesis (see Section 4500-CO2). For estuarine and marine samples, estimate total inorganic carbon concentrations with a simple titration procedure22 and make initial temperature, salinity, and pH measurements. 4) Incubate samples for up to 4 h. If measurements are required for the entire photoperiod, overlap 4-h periods from dawn until dusk. A 4-h incubation period may be sufficient provided energy input is used as the basis for integrating incubation period to entire photoperiod. For incubation procedure, see ¶ 2b6) above. 5) After incubating remove sample bottles and immediately place in the dark. Filter unpreserved samples without delay. Avoid sample preservation to avoid lysing cells or determine extracellular products. 6) Filter two portions of each sample through a membrane filter, taking care that the largest pore size is consistent with quantitative retention of plankton. Although the 0.45-µm pore filter usually is adequate, determine the efficiency of sample retention immediately before analysis, with a wide range of pore sizes.23,24 Apply approximately 30 kPa of vacuum during filtration. Excess vacuum may cause extensive cell rupture and loss of radioactivity through the membrane.25 Use maximum sample volume consistent with rapid filtration (1 to 2 min), but do not clog filter. 7) Place membranes in HCl fumes for 20 min. Count filters as soon as possible, although extended storage in a desiccator is acceptable. 8) Determine radioactivity by counting with an end-window tube, windowless gas flow detector, or liquid scintillation counter. 9) Determine counting geometry of thin-window and windowless gas flow detectors.26 Using three ampules of carbon 14, prepare a series of barium carbonate (BaCO3) precipitates on tared 0.45-µm membrane filters as directed below. The precipitates will contain the same amount of carbon 14 activity but will have different thicknesses ranging from 0.5 to 6.0 mg/cm2. Dilute each ampule to 500 mL with a solution of 1.36 g Na2CO3/L CO2-free distilled water. Pipet 0.5-mL portions into each of seven conical flasks containing 0, 0.5, 1.5, 2.5, 3.5, 4.5, and 5.5 mL, respectively, of a solution of 1.36 g Na2CO3 /L CO2-free distilled water. Add, respectively, 0.3, 0.6, 1.2, 1.8, 2.4, 3.0, and 3.6 mL 1.04% barium chloride (BaCl2) solution. Let BaCO3 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater precipitate stand 2 h with gentle swirling every half hour. Collect each precipitate on a filter (using an apparatus with a filtration area comparable to that of the samples). With suction, dry filters without washing; place in a desiccator for 24 h, weigh, and count. The counting rate increases exponentially with decreasing precipitate thickness. Extrapolate graphically (or mathematically) to zero precipitate thickness and multiply the zero-thickness counting rate by 1000 to correct for ampule dilution. This represents the amount of activity added to each sample bottle used to determine fraction of carbon 14 taken up in light and dark bottles. c. Calculations: 1) Subtract the mean dark-bottle or time-zero sample count from the mean light-bottle counts for each replicate pair. 2) Determine the total dissolved inorganic carbon available for photosynthesis (carbonate, bicarbonate, and free CO2) from pH and alkalinity measurements; make direct measurement of total CO2 according to Section 4500-CO2 or the methods described in the literature.27-30 3) Determine quantity of carbon fixed by using the following relationship: 4) Integrate productivity for the entire depth of euphotic zone and express as grams carbon fixed per square meter per day [see ¶ 2c2) above]. 5) Using the solar radiation records and photosynthesis rates during incubation, adjust data to represent phytoplankton productivity for the entire photoperiod. If samples were incubated for less than the full photoperiod, apply a correction factor. 4. References 1. GUNDERSEN, K. 1973. In-situ determination of primary production by means of the new incubator, ISIS. Helgolander wiss. Meeresunters. 24:465. 2. BURRIS, R.H., F.J. EPPLING, H.B. WAHLIN & P.W. WILSON. 1942. Studies of biological nitrogen fixation with isotopic nitrogen. Proc. Soil Sci. Soc. Amer. 7:258. 3. NEESS, J.C., R.C. DUGDALE, V.A. DUGDALE & J.J. GOERING. 1962. Nitrogen metabolism in lakes. I. Measurement of nitrogen fixation with N15. Limnol. Oceanogr. 7:163. 4. STEWART, W.D.P., G.P. FITZGERALD & R.H. BURRIS. 1967. In situ studies on N2 fixation using the acetylene reduction technique. Proc. Nat. Acad. Sci. 58:2071. 5. STEWART, W.D.P., G.P. FITZGERALD & R.H. BURRIS. 1970. Acetylene reduction assay for determination of phosphorus availability in Wisconsin lakes. Proc. Nat. Acad. Sci. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 66:1104. 6. GOLDMAN, C.R. 1968. Aquatic primary production. Amer. Zoologist 8:31. 7. ODUM, H.T. 1957. Primary production measurements in eleven Florida springs and a marine turtle-grass community. Limnol. Oceanogr. 2:85. 8. BEYERS, R.J. & H.T. ODUM. 1959. The use of carbon dioxide to construct pH curves for the measurements of productivity. Limnol. Oceanogr. 4:499. 9. GAARDER, T. & H.H. GRAN. 1927. Investigations of the production of plankton in Oslo Fjord. Rapp. Proces-Verbaux. Reunions Cons. Perma. Int. Explor. Mer 42:1. 10. STEEMAN-NEILSEN, E. 1952. The use of radioactive carbon (C-14) for measuring organic production in the sea. J. Cons. Perma. Int. Explor. Mer 18:117. 11. WILLIAMS, P.J. LEB., R.C.T. RAINE & J.R. BRYAN. 1979. Agreement between the 14C and oxygen methods of measuring phytoplankton production: Reassessment of the photosynthetic quotient. Oceanol. Acta 2:411. 12. DAVIES, J.M. & P.J. LEB. WILLIAMS. 1984. Verification of 14C and O2 derived primary organic production using an enclosed system. J. Plankton Res. 6:457. 13. RYTHER, J.H. 1956. Photosynthesis in the ocean as a function of light intensity. Limnol. Oceanogr. 1:61. 14. FEE, E.J. 1969. A numerical model for the estimation of photosynthetic production, integrated over time and depth, in natural waters. Limnol. Oceanogr. 14:906. 15. STEEMAN-NEILSEN, E. 1964. Recent advances in measuring and understanding marine primary production. J. Ecol. 52(Suppl.):119. 16. ALLEN, M.B. 1956. Excretion of organic compounds by Chlamydomonas. Arch. Mikrobiol. 24:163. 17. FOGG, G.E. & W.D. WATT. 1965. The kinetics of release of extracellular products of photosynthesis by phytoplankton. In C.R. Goldman, ed. Primary Productivity in Aquatic Environments. Suppl. 18, Univ. California Press, Berkeley. 18. WETZEL, R.G. 1965. Necessity for decontamination of filters in C14 measured rates of photosynthesis in fresh waters. Ecology 46:540. 19. MCALLISTER, C.D. 1961. Decontamination of filters in the C14 method of measuring marine photosynthesis. Limnol. Oceanogr. 6:447. 20. CARPENTER, E.J. & J.S. LIVELY. 1980. Review of estimates of algal growth using 14C tracer techniques. In P.G. Falkowski, ed. Primary Productivity in the Sea. Brookhaven Symp. Biol. No. 31. Plenum Press, New York, N.Y. 21. STRICKLAND, J.D.H. & T.R. PARSONS. 1968. A Practical Manual of Sea Water Analysis. Fish. Res. Board Can. Bull. No. 167. Queen’s Printer, Ottawa, Ont. 22. PARSONS, T.R., Y. MAITA & C.M. LALLI. 1984. A Manual of Chemical and Biological Methods for Seawater Analysis. Pergamon Press, New York, N.Y. 23. LASKER, R. & R.W. HOLMES. 1957. Variability in retention of marine phytoplankton by © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater membrane filters. Nature 180:1295. 24. HOLMES, R.W. & C.G. ANDERSON. 1963. Size fractionation of C14-labelled natural phytoplankton communities. In C.H. Oppenheimer, ed. Symposium on Marine Microbiology. Charles C. Thomas, Springfield, Ill. 25. ARTHUR, C.R. & F.H. RIGLER. 1967. A possible source of error in the C14 method of measuring primary productivity. Limnol. Oceanogr. 12:121. 26. JITTS, H.R. & B.D. SCOTT. 1961. The determination of zero-thickness activity in Geiger counting of C14 solutions used in marine productivity studies. Limnol. Oceanogr. 6:116. 27. SAUNDERS, G.W., F.B. TRAMA & R.W. BACHMANN. 1962. Publ. No. 8, Great Lakes Research Div., Univ. Michigan, Ann Arbor. 28. DYE, J.F. 1944. The calculation of alkalinities and free carbon dioxide in water by use of nomographs. J. Amer. Water Works Assoc. 36:859. 29. MOORE, E.W. 1939. Graphic determination of carbon dioxide and the three forms of alkalinity. J. Amer. Water Works Assoc. 31:51. 30. PARK, K., D.W. HOOD & H.T. ODUM. 1958. Diurnal pH variation in Texas bays and its application to primary production estimations. Publ. Inst. Mar. Sci. Univ. Tex. 5:47. 10300 PERIPHYTON*#(138) 10300 A. Introduction 1. Definition and Significance Microorganisms growing on stones, sticks, aquatic macrophytes, and other submerged surfaces are useful in assessing the effects of pollutants on lakes, streams, and estuaries. Included in this group of organisms, here designated periphyton,1,2 are the zoogleal and filamentous bacteria, attached protozoa, rotifers, and algae, and the free-living microorganisms that swim, creep, or lodge among the attached forms. Unlike the plankton, which often do not respond fully to the influence of pollution in rivers for a considerable distance downstream, the periphyton show marked responses immediately below pollution sources. Examples are the beds of Sphaerotilus and other ‘‘slime organisms’’ commonly observed in streams below discharges of organic wastes. Because the abundance and composition of the periphyton at a given location are governed by the water quality at that point, observations of their condition generally are useful in evaluating conditions in bodies of water. The use of periphyton in assessing water quality often is hindered by the lack of suitable natural substrates at the desired sampling station. Furthermore, it often is difficult to collect © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater quantitative samples from these surfaces. To circumvent these problems artificial substrates have been used to provide a uniform surface type, area, and orientation.3 2. References 1. ROLL, H. 1939. Zur Terminologie des Periphytons. Arch. Hydrobiol. 35:39. 2. YOUNG, O.W. 1945. A limnological investigation of periphyton in Douglas Lake, Michigan. Trans. Amer. Microsc. Soc. 64:1. 3. SLADECKOVA, A. 1962. Limnological investigation methods for the periphyton (‘‘Aufwuchs’’) community. Bot. Rev. 28:286. 10300 B. Sample Collection 1. Station Selection In rivers, locate stations a short distance upstream and at one or more points downstream from the suspected pollution source or intended study area in the areas of central mixing. In large rivers, sample both sides of the stream in main flow areas. Because the effects of a pollutant depend on the assimilative capacity of the stream and on the nature of the pollutant, progressive changes in water quality downstream from the pollution source may be caused entirely by dilution and cooling—as in the case of nutrients, toxic industrial wastes, and thermal pollution—or by gradual mineralization of degradable organic compounds. Cursory examination of shoreline and bottom periphyton growths on natural substrates downstream from an outfall may indicate conspicuous zones of biological response to water quality that will be useful in determining appropriate sites for sampling stations. When an intensive sampling program is not feasible, a minimum of three sampling stations, one in a reference area upstream from a pollution source and the others in the community downstream from the source, where complete mixing with the receiving water has occurred, will provide minimal data on the periphyton community. In lentic waters (e.g., lakes, reservoirs, ponds) and other standing-water bodies where zones of pollution may be arranged concentrically, locate stations in areas adjacent to a waste outfall and in unaffected areas. Use control stations similar to the affected ones (e.g., similar in water depth and distance from shore). 2. Sample Collection a. Natural substrates: Collect qualitative samples by scraping submerged stones, sticks, pilings, and other available substrates. Many devices have been developed to collect quantitative samples from irregular surfaces. Appropriate techniques for the removal of periphyton from both living and nonliving surfaces have been described.1-4 b. Artificial substrates: The most widely used artificial substrate is the standard, plain, 25- by 75-mm glass microscope slide, but other materials such as clear vinyl plastic also are suitable. Do not change substrate type during a study because colonization varies with substrate. In small, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater shallow streams and in the littoral regions of lakes and reservoirs where light penetrates to the bottom, place slides or other substrates vertically in frames anchored to the bottom. In large, deep streams or standing-water bodies where turbidity varies widely, place slides vertically with the slide face at right angles to the prevailing current. A floating rack, as shown in Figure 10300:1,*#(139) is suitable. Expose several slides (minimally five; three for biomass, one for species, and one backup for each time interval) for each type of analysis to assure collecting sufficient material and to determine variability in results caused by normal differences in colonization of individual slides. In addition to effects of pollutants, length of substrate exposure and seasonal changes in temperature and other natural environmental conditions may have a profound effect on sample composition. No community on an artificial substrate is representative of the natural community. Place, expose, and handle all artificial substrate samplers in conditions as nearly identical as possible, whether they are replicate samplers at a particular sampling location or samplers at different locations. Sampler type and/or construction cause changes in surrounding physical conditions that in turn affect periphyton growth. Variations of 10 to 25% between sample replicates are common. Therefore, to reduce sampling error and increase interpretive power, reduce the magnitude of all possible test variables and use sufficient replication. c. Exposure period: Colonization on clean slides proceeds at an exponential rate for the first 1 or 2 weeks and then slows. Because exposures of less than 2 weeks may result in very sparse collections, and exposures of more than 2 weeks may result in loss of material due to sloughing, sample for 2 weeks during the summer. This exposure period precludes collecting sexually mature thalli of larger, slow-growing filamentous algae such as Cladophora and Stigeoclonium. To obtain optimum growth during the winter, use a longer exposure period. For the most exacting work, determine the optimum exposure period by testing colonization rates over a period of about 6 weeks. Secondary problems associated with macroinvertebrate infestation and grazing may occur, often within 7 to 14 d. To reduce the confounding influence of grazing, increase substrate sampling area and expose for 7 to 10 d. 3. Sample Preservation Preserve samples that are taken for counting and identification in 5% neutralized formalin, Lugol’s iodine, or merthiolate (see Section 10200B.2). Preserve slides intact in bottles of suitable size or scrape into containers in the field. Air-dry slides for dry and ash-free dry weight in the field and store in a 3.0- × 7.7-cm glass bottle. Place slides for chlorophyll analyses in acetone or methanol in the field or collect and freeze with trichlorotrifluoroethane†#(140) (or alternative) or CO2 and hold on dry ice until returned to the laboratory. Store all samples in the dark. 4. References 1. SLADECKOVA, A. 1962. Limnological investigation methods for the periphyton © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater (‘‘Aufwuchs’’) community. Bot. Rev. 28:286. 2. GOUGH, S.B. & W.J. WOELKERLING. 1976. On the removal and quantification of algal aufwuchs from macrophyte hosts. Hydrobiologia 48: 203. 3. BOOTH, W.E. 1981. A method for removal of some epiphytic diatoms. Botanica Marina 24:603. 4. DELBECQUE, E.J.P. 1985. Periphyton on nymphaeids: An evaluation of methods and separation techniques. Hydrobiologia 124:85. 5. Bibliography COOKE, W.B. 1956. Colonization of artificial bare areas by microorganisms. Bot. Rev. 22:613. HOHN, M.H. 1966. Artificial substrate for benthic diatoms—collection, analysis, and interpretation. In K.W. Cummings, C.A. Tryon, Jr., & R.T. Hartman, eds. Organism-Substrate Relationships in Streams. Spec. Publ. No. 4, p. 87. Pymatuning Lab. Ecology, Univ. Pittsburgh, Pittsburgh, Pa. KEVERN, N.R., J.L. WILHM & G.M. VAN DYNE. 1966. Use of artificial substrata to estimate the productivity of periphyton communities. Limnol. Oceanogr. 11:499. ARTHUR, J.W. & W.B. HORNING. 1969. The use of artificial substrates in pollution surveys. Amer. Midland Natur. 82:83. TIPPETT, R. 1970. Artificial surfaces as a method of studying populations of benthic micro-algae in fresh water. Brit. Phycol. J. 5:187. ERTL, M. 1971. A quantitative method of sampling periphyton from rough substrates. Limnol. Oceanogr. 16:576. ANDERSON, M.A. & S.L. PAULSON. 1972. A simple and inexpensive woodfloat periphyton sampler. Progr. Fish-Cult. 34:225. NORTH AMERICAN ENTHOLOGICAL SOCIETY. 1974–1991. (Annual) Current and Select Bibliographies on Benthic Biology. Springfield, Ill. MARKER, A.F.H., C.A. CROWTHER & R.J.M. GUNN. 1980. Methanol and acetone as solvents for estimating chlorophyll a and phaeopigments by spectrophotometry. Arch. Hydrobiol. Ergebn. Limnol. 14:52. NEROZZI, A. & P. SILVER. 1983. Periphytic community analysis in a small oligotropic lake. Proc. Penn. Acad. Sci. 57:138. WETZEL, R., ed. 1983. Periphyton of Freshwater Ecosystems. Developments in Hydrobiology 17. Dr. W. Junk BV Publishers, The Hague, The Netherlands. HAMILTON, P.B. & H.C. DUTHIE. 1984. Periphyton colonization of rock surfaces in a boreal forest stream studied by scanning electron microscopy and track autoradiography. J. Phycol. 20:525. NIELSEN, T.S., W.H. FUNK, H.L. GIBBONS & R.M. DUFFNER. 1984. A comparison of periphyton growth on artificial and natural substrates in the Upper Spokane River, Washington, USA. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Northwest Sci. 58: 243. PIP, E. & G.G.C. ROBINSON. 1984. A comparison of algal periphyton composition on 11 species of submerged macrophytes. Hydrobiol. Bull. 18:109. POULIN, M., L. BERARD-THERRIAULT & A. CARDINAL. 1984. Benthic diatoms from hard substrates of marine and brackish waters of Quebec Canada 3. Fragilarioideae, Fragilariales, Fragilariaceae. Nat. Can. (Que). 111:349. STEVENSON, R.J. 1984. How currents on different sides of substrates in streams affect mechanisms of benthic algal accumulation. Int. Rev. ges. Hydrobiol. 69:241. VYMAZAL, J. 1984. Short-term uptake of heavy metals by periphytic algae. Hydrobiologia 119:171. AUSTIN, A. & J. DENISEGER. 1985. Periphyton community changes along a heavy metals gradient in a long narrow lake. Environ. Exper. Bot. 25:41. FLOWER, R.J. 1985. An improved epilithon sampler and its evaluation in two acid lakes. Brit. Phycol. J. 20:109. LAMBERTI, G.A. & V.H. RESH. 1985. Comparability of introduced tiles and natural substrates for sampling lotic bacteria, algae, and macroinvertebrates. Freshwater Biol. 15:21. PIEKARCZYK, R. & E. MCARDLE. 1985. Pioneer colonization and interaction of photosynthetic and heterotrophic microorganisms on an artificial substrate of polyurethane foam in E.J. Beck Lake, Illinois, USA. Trans. Ill. State Acad. Sci. 78:81. 10300 C. Sample Analysis 1. Sedgwick-Rafter Counts Remove periphyton from slides with a razor blade and rubber policeman. Disperse scrapings in 100 mL or other suitable volume of preservative with vigorous shaking, or use a blender. Transfer a 1-mL portion to a Sedgwick-Rafter cell, and make a strip count as described in Section 10200F.2a. If material in the Sedgwick-Rafter cell is too dense to count directly, discard and replace with a diluted sample. Sedgwick-Rafter cells do not permit examination at magnifications higher than 200×. The Palmer cell1, a thinner version of the S-R cell, permits examination at 400 to 500× with a standard compound microscope. Express counts as cells or filaments per square millimeter of substrate area, calculated as in ¶ C.2. 2. Inverted Microscope Method Counts Using an inverted microscope for periphyton counts permits magnifications higher than those possible with the Sedgwick-Rafter cell. Remove periphyton quantitatively from slides with a razor blade and policeman. Transfer a measured portion, after serial dilution if necessary, into a standardized plankton sedimentation chamber. After a suitable period of settling (see © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Section 10200C.1), count organisms in the settling chamber by counting all organisms within a known number of strips or random fields. Calculate algal density per unit area of substrate as follows: where: N = number of organisms counted, At = total area of chamber bottom, mm2, Vt = total volume of original sample suspension, mL, Ac = area counted (strips or fields), mm2, Vs = sample volume used in chamber, mL, and As = surface area of slide or substrate, mm2. Separation of periphyton from silt and detritus may be enhanced by adding a drop or less of a saturated iodine solution to the counting chamber just before counting. This method is especially useful when Chlorophyta are the predominant organisms because iodine stains starch food reserves blue. Iodine can be added even to preserved samples. 3. Diatom Species Counts Preparation of permanent diatom mounts from periphyton samples differs from preparation of mounts from plankton samples because of the need to remove extracellular organic matter (such as gelatinous materials). If this organic matter is not removed it will produce a thick brown or black carbonaceous deposit on the cover glass when the sample is incinerated. Clear organic matter by incineration by placing a small, known volume of sample (< 1 mL) directly on a cover slip. Let water evaporate and ash at 525°C (not more) for 6 to 10 min. Mount cover slip for direct examination of diatom frustules. Alternatively, decompose organic substances by oxidation with ammonium persulfate or with HNO3 or 30% H2O2 and K2Cr2O7 (see Section 10200D.3) before mounting sample. To oxidize with persulfate place a measured sample of approximately 5 mL in a disposable 10-mL vial. Let stand 24 h, withdraw supernatant liquid by aspiration, replace with a 5% solution of (NH4)2S2O8, and mix thoroughly. Do not exceed a total volume of 8 mL. Heat vial to approximately 90°C for 30 min. Let stand 24 h, withdraw supernatant liquid, and replace with reagent-grade water. After three changes of reagent-grade water, with a disposable pipet transfer a drop of the diatom suspension to a cover glass, evaporate to dryness, and prepare and count a mount as described for plankton (Section 10200). Count as least 500 frustules and express results as relative numbers or percentage of each species per unit area. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 4. Stained Sample Preparation and Counting Staining periphyton samples permits distinguishing algae from detritus and ‘‘live’’ from ‘‘dead’’ diatoms. This distinction is especially important because periphyton often contains many dead diatoms of planktonic as well as periphytic origin. In the first method, cells are exposed to a vital stain to evaluate the percentages of live, senescent, and dead algae, particularly diatoms, by estimating relative metabolic activities. The colorless tetrazolium violet is reduced in the cytochrome system of metabolically active cells to form violet-colored triphenylformazan. When cells are senescent or dead, the reaction fails. Make tetrazolium violet solution by adding 2.0 g tetrazolium violet to 1.0 L water. The solution may be buffered to a pH of 7.5 to 7.7 with tris-hydroxymethyl amine. Add 1 mL tetrazolium violet solution to 9 mL sample and incubate 2 to 4 h at room temperature. Count diatom frustules and other cells (at least 300/sample) and place into the following categories: a) active: violet precipitate observed within the cell or mitochondria; b) senescent: chlorophyll present, but no violet precipitate; c) dead: no chlorophyll or violet precipitate present. In the second method, all algal components of periphyton may be studied in one preparation, without sacrificing detailed diatom taxonomy.2 This method yields permanent slides for reference collections. Thoroughly mix preserved samples in the preservative solution. Prepare acid fuchsin stain by dissolving 1 g acid fuchsin in 100 mL reagent-grade water, adding 2 mL glacial acetic acid, and filtering. Place a measured sample in a centrifuge tube with 10 to 15 mL acid fuchsin stain. Mix sample and stain several times during a 20-min staining period; centrifuge at 1000 g for 20 min. Decant stain, being careful not to disturb sediment, or siphon off supernatant. Add 10 to 15 mL 90% propanol, mix, centrifuge for 20 min, and decant supernatant. Repeat using two washes of 100% propanol and one wash of xylene. Centrifuge, decant xylene, and add fresh xylene. At this stage, store sample in well-sealed vials or prepare slides. Slides for periphyton examinations require random dispersion of a known amount of xylene suspension. Use a microstirrer to break up clumps of algae before removing sample portion from xylene suspension. Count a number of drops of suspended sample into a thin ring of mounting medium*#(141) on a slide. Mix the xylene suspension and medium with a spatula until the xylene has evaporated. Warm the slide on a hot plate at 45°C and cover sample with a cover slip. Count diatoms on the prepared slides using the magnification most appropriate to the desired level of taxonomic identification. Count strips or random fields. Calculate diatom density per unit area of substrate: where the terms are as defined in Section 10300C.2. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 5. Dry and Ash-Free Weight Collect at least three replicate slides for weight determinations.3 Slides air-dried in the field can be stored indefinitely if protected from abrasion, moisture, and dust. Use slides expressly designated for dry and ash-free weight analysis. a. Equipment: 1) Analytical balance, with a sensitivity of 0.1 mg. 2) Drying oven, double-wall, thermostatically controlled to within ±1°C. 3) Electric muffle furnace with automatic temperature control. 4) Crucibles, porcelain, 30-mL capacity. 5) Single-edge razor blades or rubber policeman. b. Procedure: 1) Dry slides to constant weight at 105°C, and ignite for 1 h at 500°C. If weights are to be obtained from field-dried material, re-wet dried material with reagent-grade water and remove from slides with a razor blade or rubber policeman. Place scrapings from each slide in a separate prewashed, prefired, tared crucible; dry to constant weight at 105°C; cool in a desiccator and weigh; and ignite for 1 h at 500°C. 2) Re-wet ash with reagent-grade water and dry to constant weight at 105°C. This reintroduces water of hydration of clay and other minerals, which is not driven off at 105°C but is lost during ashing. If not corrected for, this water loss will be recorded as volatile organic matter.4 c. Calculations: Calculate mean weight from slides and report as dry weight [(crucible + sample weight at 105° C) minus (tare weight of crucible)] per square meter of exposed surface. If 25-by 75-mm slides are used, then Calculate ash weight for sample [(crucible + sample weight at 500°C) minus (tare weight of crucible)]. Subtract ash weight from dry weight to obtain ash-free weight, and report as ash-free weight per square meter of exposed surface. 6. Chlorophyll and Pheophytin The chlorophyll content of attached algal communities is a useful index of the phytoperiphyton biomass. Quantitative chlorophyll determinations require the collection of periphyton from a known surface area. Extract the pigments with aqueous acetone or methanol (see Section 10200G) and use a spectrophotometer or fluorometer for analysis. If immediate pigment extraction is not possible, samples may be stored frozen for as long as 30 d if kept in the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater dark.5 The ease with which chlorophylls are removed from cells varies considerably with different algae; to achieve complete pigment extraction disrupt the cells mechanically with a grinder, blender, or sonic disintegrator, or freeze them. Grinding is the most rigorous and effective of these methods. The Autotrophic Index (AI) is a means of determining the trophic nature of the periphyton community (see Section 10200G). It is calculated as follows: Normal AI values range from 50 to 200; larger values indicate heterotrophic associations or poor water quality. Nonviable organic material affects this index. Depending on the community, its location and growth habit, and method of sample collection, there may be large amounts of nonliving organic material that may inflate the numerator and produce disproportionately high AI values. Nonetheless, the AI is an approximate means of describing changes in periphyton communities between sampling locations. a. Equipment and reagents: See Section 10200G. b. Procedure: In the field, place individual glass microscope slides used as substrates directly into 100 mL of a mixture of 90% acetone (water with 10% saturated MgCO3 solution). Immediately store on dry ice in the dark. (NOTE: Vinyl plastic is soluble in acetone. If vinyl plastic is used as the substrate, scrape periphyton from it before solvent extraction.) If extraction cannot be carried out immediately, freeze samples in the field and keep frozen until processed. Rupture cells by grinding in a tissue homogenizer and steep in acetone for 24 h in the dark at or near 4°C. To determine pigment concentration, follow the procedures given in Section 10200G. c. Calculation: After determining pigment concentration in the extract, calculate amount of pigment per unit surface area of sample as follows: where: Ca is as defined in Section 10200G. 7. References 1. WETZEL, R.G. & G.E. LIKENS. 1991. Limnological Analyses, 2nd ed. Springer-Verlag, New York, N.Y. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. OWEN, B.B., JR. 1977. The effect of increased temperatures on algal communities of artificial stream channels. Ph.D. dissertation, Univ. Alberta, Edmonton. 3. NEWCOMBE, C.L. 1950. A quantitative study of attachment materials in Sodon Lake, Michigan. Ecology 31:204. 4. NELSON, D.J. & D.C. SCOTT. 1962. Role of detritus in the productivity of a rock outcrop community in a piedmont stream. Limnol. Oceanogr. 7:396. 5. GRZENDA, A.R. & M.L. BREHMER. 1960. A quantitative method for the collection and measurement of stream periphyton. Limnol. Oceanogr. 5:190. 8. Bibliography EATON, J.W. & B. MOSS. 1966. The estimation of numbers and pigment content in epipelic algal populations. Limnol. Oceanogr. 11:584. MOSS, B. 1968. The chlorophyll a content of some benthic algal communities. Arch. Hydrobiol. 65:51. CRIPPEN, R.R. & J.L. PERRIER. 1974. The use of neutral red and Evans blue for live-dead determinations of marine plankton. Stain Technol. 49:97. OWEN, B.B., M. AFZAL & W.R. CODY. 1978. Staining preparations for phytoplankton and periphyton. Brit. Phycol. J. 13:155. OWEN, B.B., M. AFZAL & W.R. CODY. 1979. Distinguishing between live and dead diatoms in periphyton communities. In R.L. Weitzel, ed. Methods and Measurements of Periphyton Communities: A Review. STP 690, American Soc. Testing & Materials, Philadelphia, Pa. WETZEL, R.G., ed. 1983. Periphyton of Freshwater Ecosystems. Developments in Hydrobiology 17. Dr. W. Junk BV Publishers, The Hague, The Netherlands. DELBECQUE, E.J.P. 1985. Periphyton on Nymphaeids: An evaluation of methods and separation techniques. Hydrobiologia 124:85. TREES, C.C., M.C. KENNICUTT & J. M. BROOKS. 1985. Errors associated with the standard fluorometric determination of chlorophylls and phaeopigments. Mar. Chem. 17:1. 10300 D. Primary Productivity The productivity of periphyton communities is a function of water quality, substrate, and seasonal patterns in temperature and solar illumination. Productivity may be estimated from temporal changes in biomass (standing crop) or from the rate of oxygen evolution or carbon uptake.1 1. Biomass Accumulation a. Ash-free dry weight: The accumulation rate of organic matter on artificial substrates by attachment, growth, and reproduction of colonizing organisms has been used widely to estimate © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater the productivity of streams and reservoirs.2,3 To use this method, expose several replicate clean substrates for a predetermined period, scrape the accumulated material from the slides, and ash as described previously. where: P = net productivity, mg ash-free weight/m2/d, t = exposure time, d, and A = area of a slide, m2. Obtain estimates of seasonal changes in biomass of established communities by placing many replicate substrates at a sampling point and then retrieving a few at a time at regular intervals. Replace removed slides with new clean slides. The recommended collection interval ranges from 2 to 4 weeks for a year or longer.2 Gain in ash-free weight per unit area from one collection period to the next is a measure of net production. b. ATP estimates: Measurement of adenosine triphosphate (ATP) has been used in recent years to estimate microbial biomass in water. This technique is applicable to periphyton.4 It provides an additional tool for assessing the magnitude and rate of biomass accumulation on substrates in natural waters. At present, the procedure should be limited to communities colonizing artificial substrates. 1) Equipment and reagents—See Section 10200I.6a. 2) Procedure—Either scrape periphyton from an exposed artificial substrate or, if standard glass microscope slides are used, place them in polyethylene slide mailers containing preheated (99°C) Tris buffer. Immerse in a boiling water bath for 10 min to extract ATP. If samples are not assayed immediately, freeze at −25°C; they may be stored in a freezer for up to several months. Complete analysis as directed in Section 10200I.6b. Slides exposed in waters containing high turbidity may collect substantial amounts of particulates including clays. ATP sorbs to these materials; the sorption results in a quenching effect. 3) Calculations—See Section 10200I.6c. 2. Standing Water Productivity Measured by Oxygen Method Hourly and daily rates of oxygen evolution and carbon uptake by periphyton growing in standing water can be studied by confining this community briefly in bottles, bell jars, or other chambers. In contrast, the metabolism of organisms in flowing water is highly dependent on current velocity and cannot be determined with precision under static conditions. Productivity estimates for flowing waters and those for standing waters present different problems; therefore, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater separate procedures are given. Productivity and respiration of epilithic and epipelic periphyton in littoral regions of lakes and ponds can be determined by inserting transparent and opaque bell jars or open-ended plastic chambers into substrata along transects perpendicular to the shoreline.5,6 Chambers are left in place for one-half the daily photoperiod. The DO concentration in a chamber is determined at the beginning and end of the exposure period. Gross productivity is the sum of the net gain in DO in the transparent chamber and the oxygen used in respiration. Values obtained are doubled to estimate productivity for the entire photoperiod. Alternatively, determine the proportion of the incubation period of the total insolation during the photoperiod more accurately by measuring the insolation of the incubation period as a percentage of the total daily insolation. Both these methods assume that photosynthesis is proportional to irradiance (i.e., not light saturated and no photoinhibition). Failure to account for changes in DO in chambers caused by phytoplankton photosynthesis and respiration may cause serious errors in the estimates of periphyton metabolism. It is essential that these values be obtained at the time the periphyton is studied by using the light- and dark-bottle method (see Section 10200I). a. Equipment and reagents: 1) Clear and darkened glass or plastic*#(142) chambers, approximately 20 cm in diameter and 30 cm high, with a median lateral port, sealed with a serum bottle stopper for removal of small water samples for DO analyses or for the insertion of an oxygen probe. Fit the chamber with a small, manually operated, propeller-shaped stirring paddle. 2) Dissolved oxygen probe, or equipment and reagents required for Winkler dissolved oxygen determinations: See Section 4500-O. b. Procedure: At each station place both a transparent and an opaque chamber over the substrate at sunrise or mid-daylight and leave in place for one-half the daily photoperiod. In extremely productive environments or to define the hourly primary productivity changes throughout the day, use incubation periods shorter than one-half the photoperiod. The minimum incubation period giving reliable results is 2 h. Determine DO concentration at the beginning of the incubation period. Include a set of Gaarder-Gran light- and dark-bottle productivity and respiration measurements with each set of chambers to obtain a correction for phytoplankton metabolism. Incubate for the same time period as the chambers. See Section 10200I. At end of exposure period, carefully mix the water in the chambers and determine DO concentration. c. Calculations: When the exposure period is one-half of the photoperiod, calculate gross primary productivity of the periphyton community as: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: PG = gross production, mg O2/m2/d12, Vc = volume of clear chamber, L, C′fc and C′ic = final and initial concentrations, respectively, of DO in the clear chamber, mg/L, corrected for phytoplankton metabolism, Vo = volume of opaque chamber, L, C′io and C′fo = initial and final concentrations, respectively, of DO in the opaque chamber, mg/L, corrected for phytoplankton metabolism, and A = substrate area, m2. Correct for the effects of phytoplankton metabolism in the overall oxygen change in the clear chamber by the following equations: C′fc = Cfc − Cf lb C′ic = Cic − Cilb C′fo = Cfo − Cfdb C′io = Cio − Cidb where: Cfc = Cfl b = Cic = Cilb = Cfo = Cfdb = Cio = Cidb = final DO concentration in clear chamber, mg/L, final DO concentration in light bottle, mg/L, initial DO concentration in clear chamber, mg/L, initial DO concentration in light bottle, mg/L, final DO concentration in opaque chamber, mg/L, final DO concentration in dark bottle, mg/L, initial DO concentration in opaque chamber, mg/L, and initial DO concentration in dark bottle, mg/L. Calculate periphyton community respiration by: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: R = community respiration, mg O2/m2/d24h, and t = length of exposure, h. Determine the net periphyton comm unity productivity (PN) as the difference: PN = PG − R If the incubation time is different from one-half the photoperiod, modify the daily gross production calculation as follows: where: tp = length of the daily photoperiod, h. Community respiration and net production calculations for incubation periods other than one-half the photoperiod are not changed. 3. Standing Water Productivity Measured by Carbon-14 Method The approach is similar to that described above for the oxygen method. Transparent and opaque chambers are placed over the substrate, carbon-14-labeled Na2CO3 is injected into the chamber by syringe, mixed well, and allowed to incubate with the periphyton for one-half the photoperiod. The concentration of dissolved inorganic carbon available for photosynthesis is determined by titration. At the end of the incubation period, the periphyton is removed from the substrate and assayed for carbon-14.5 a. Equipment and reagents: 1) Incubation chamber: See Section 10300D.2a. 2) Special equipment and reagents: See Section 10200I. 3) Carbon-14-labeled solution of sodium carbonate, having a known specific activity of approximately 10 µCi/mL. 4) Other equipment and reagents: See Section 4500-CO2. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater b. Procedure: At each station place a transparent and opaque chamber over the substrate and add approximately 10 µCi carbon-14/L of chamber volume. Mix water in the chambers well, taking care to avoid disturbing the periphyton. Determine concentration of dissolved inorganic carbon as described in Section 2320. At end of exposure period, remove surface centimeter of periphyton and sediment enclosed in the chamber, freeze, and store frozen in a vacuum desiccator. Immediately before analysis, expose sample to fumes of HCl for 10 to 15 min to drive off all inorganic carbon-14 retained in the periphyton. Combust sample (or portion) by the Van Slyke method6 or oxidize by heating in a closed system. Collect all CO2 for radioassay either by flushing CO2 into a two-vial train of ethanolamine (2-aminoethanol) or alternative CO2 absorber, such as methoxyethanol (1:7)7 or flushing CO2 produced by combustion into a gas-flow counter or electrometer. Alternatively, extract known amounts of periphyton biomass with a tissue solubilizer,†#(143) using, for example, 1.0 mL in closed vials at 60°C for 48 h.8 Radioassay subsamples (100-µL) by liquid scintillation. c. Calculations: where: PN = net primary productivity per unit area of substrate per unit time, mg C/m2/d, a = 12C available = dissolved inorganic carbon, mg 12C/L = (total alkalinity − phenolphthalein alkalinity) × 0.2406 = mg 12C/L, b = 14C assimilated = [(radioactivity of sample in light chamber × k1) − (background activity of dark chamber × k2)] × (isotope effect, 1.06). Express radioactivity as disintegrations per second (dps), i.e., counts per second corrected to 100% radioassay counter efficiency. k1 = correction factor to convert individually different light-chamber volumes to 1 L, k2 = correction factor to convert individually different dark-chamber volumes to 1 L, 1.06 = isotope effect to correct for slightly greater mass of 14C than of 12C, which results in a 6% slower assimilation rate, c = 14C available = 14C activity added = (µCi 14C added) × (disintegrations of © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 14C/s/µCi) = 3.7 × 104 µCi 14C added, mL, d = a dimensional factor to convert area of substrate sampled to m2, and e = factor to expand incubation period to the total daylight period. After integration by planimetry or electronic digitizer of the total amount of insolation for the day, determine percentage of total represented by the incubation period. 4. Flowing Water Productivity Measured by Oxygen Method Primary productivity of the periphyton community in a stream or river ecosystem can be related to changes in DO. These changes are the integrated effects of photosynthesis, affected by light levels and turbidity, that occur during the photoperiod by stream phytoplankton, periphyton, and the submerged portions of macrophytes. Respiration results from metabolism of plant communities, aquatic animals, and attached and free-floating microbial heterotrophs. Water depth, turbulence, and water temperature all influence the process of reaeration. Oxygen also can enter by accrual of groundwater and surface waters. Daily fluctuations in photosynthetic production of oxygen are imposed on the relatively steady demand of respiratory activity. However, this latter process may fluctuate greatly in streams receiving a significant load of organic wastes, particularly under intermittent loads such as oxygen demand from urban stormwater runoff. Respiration rates also may vary diurnally under certain conditions, but the factors involved are not well understood. The rate of change in stream DO (q) in grams per cubic meter per hour is represented by the following function of the photosynthetic rate (p), respiration (r), reaeration (d), and accrual from groundwater inflow and surface runoff (a):9 q=p−r+d+a If the equation is multiplied through by depth in meters (z), the resulting values are in terms of grams oxygen per square meter per hour. Figure 10300:2 illustrates this conceptual relationship between q, primary productivity, and respiration of the stream plant community. The procedure measures the time-variable oxygen concentrations in a stream over a 24-h period. Compensations are made for oxygen changes due to physical factors (accrual and reaeration) and the rate of oxygen change due to biological activity that is separated into components due to respiration and primary production. The metabolic rates are the sum of the activity of the entire stream community. Planktonic productivity and respiration can be separated from overall community activity by the use of the light- and dark-bottle oxygen technique (see Section 10200I). However, in most small streams planktonic production is insignificant. The component of production and respiration due to macrophytes is very difficult to separate from periphytic metabolic activity in systems where vascular plants are common. Because periphyton attach to plant surfaces as well as nonliving substrates, radiotracer techniques are required to separate the component of production due to macrophytes from that due to attached algae.10 When vascular plants are present use techniques discussed in Section 10400 to estimate their contribution to net primary productivity. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Respiration by fish and benthic fauna also is difficult to quantitate directly and usually is not separated from periphyton respiration. If compartmentalized animal metabolism is required, calculate this contribution from laboratory respiration rates extrapolated to the field situation based on animal population sizes.11,12 Estimate primary productivity in flowing water by either the free water demand method or the chamber method.13,14 The first does not introduce artificiality to the system; however, it is difficult to separate the components of metabolic activity except for the contribution due to plankton. The chamber method measures periphyton activity alone.15-19 Depending on the hydrologic characteristics of the stream system, accrual and reaeration may be significant. Accrual can be accounted for by simple mixing equations if estimates of the accrued flow and its oxygen concentration are known. In practice, select for study reaches that do not incur significant accrual. Measure reaeration rates either directly15-18 or by estimation from physical and hydrodynamic features of the stream itself.17,18 a. Equipment: 1) BOD bottles, for light- and dark-bottle measurements. See Section 10200I. 2) DO meter and probe for measurement of DO. 3) Bottom chamber, 60 × 20 × 10 cm, with 32-cm lengthwise dividing baffle, rheostat-controlled submersible pump, temperature thermistor, and DO probe.14 Use clear and opaque plastic sleeves for covering chamber and petri dishes or other means of placing periphyton within chambers. 4) Current meter, capable of detecting water current velocities ranging from 0.03 to 3 m/s in water depths as shallow as 0.3 m. 5) Tape measure (30 m) and depth staff, or similar equipment, as required to measure stream cross sections. 6) Fluorometer, capable of detecting fluorescent dye concentration at 0.5 to 100 µg/L (required only if direct measurement of reaeration is made). 7) Liquid scintillation counter, capable of sensitive detection of 85Kr and 3H (required only if direct measurement of reaeration is made). b. Procedure: 1) Light- and dark-chamber method—Grow samples of typical periphyton communities on artificial substrate or collect natural material. Transfer identical portions to both clear and opaque chambers, taking care to use sufficient periphyton to make the ratio of chamber volume to periphyton area equivalent to the ratio of stream volume to periphyton substrate area. Measure current in the stream and match the circulation rate in the clear and opaque chambers to the current. Measure DO concentrations initially in both clear and opaque chambers and after 1 to 3 h to estimate the rate of oxygen increase or decrease. Make concurrent measurements of phytoplankton activity using light- and dark-bottle techniques as described in Section 10200I.2. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Incubate light and dark bottles for the same time interval as the chambers. Make several measurements during the photoperiod to define daily primary productivity. In addition, collect sufficient natural substrate samples of the study reach to estimate periphyton biomass (see Section 10300B). At end of incubation period harvest enclosed periphyton and determine ash-free biomass (see Section 10300B). 2) Free-water diurnal curve methods—Measure, hourly or continuously, DO concentration and water temperature for a 24-h period at one or two stations, depending on stream conditions, precision desired, and availability of equipment. If similar conditions exist for some distance upstream from the reach being studied, diurnal measurements of DO at a single station are sufficient to estimate productivity. Where upstream conditions are significantly different from those in the reach being studied, make measurements at the upstream and downstream limits of the reach. If the single-station method is used, measure depth at several points along the study reach to define average depth. Map and/or make physical surveys to estimate magnitude of possible sources of accrual via effluents or tributary streams and springs. If the two-station method is used, measure the wetted cross-sectional stream area as well as current velocity at several points to define flow (in cubic meters per second) and average cross-sectional area. Correct for phytoplankton activity by light- and dark-bottle measurements (see Section 10200I.2). 3) Direct measurement of reaeration16—Under special circumstances it may be desirable to estimate reaeration directly although the results may not be more accurate than those of the empirical formulations usually used. The tracer gas technique is satisfactory, but is difficult and requires sophisticated equipment not routinely available. Use this method with care and with full recognition of its restrictions. Depending on stream flow, release 10 to 250 µCi 85Kr with 5 to 125 µCi 3H at the upstream end of the reach together with sufficient fluorescent dye to produce a concentration of 10 µg/L when completely mixed across the river cross section. Make fluorometric measurements at the downstream end of the reach until the dye peak appears, then collect water samples to measure the 85Kr/3H ratio by liquid scintillation techniques. Record time of travel for the dye peak from the injection point. c. Calculations: 1) Chamber method—Calculation is analogous to that used for the bell jar technique discussed in Section 10300D.2. where: Pn = hourly rate of net primary production, mg O2/m2/h, Vc = volume of clear chamber, L, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater B = average periphyton biomass estimated for the study reach, mg/m2, t = incubation period, h, W c = total biomass of periphyton contained in clear chamber, mg, C′fc = final oxygen concentration in clear chamber, corrected for phytoplankton metabolism, mg/L: C′fc = Cfc − Cf lb Cfc = final DO in clear chamber, Cf lb = final DO in light bottle, and C′ic = initial oxygen concentration in clear chamber corrected for light-bottle measurement, mg/L: C′ic = Cic − Cilb Cic = initial DO in clear chamber, and Cilb = initial DO in light bottle. where: r = hourly periphyton respiration rate, mg O2/m2/h, Vo = volume of opaque chamber, L, B = average periphyton biomass for the study reach, mg/m2, W o = total biomass of periphyton contained in opaque chamber, mg, C′io = initial oxygen concentration in opaque chamber, corrected for phytoplankton respiration, mg/L: C′io = Cio − Cidb Cio = initial DO in opaque chamber, mg/L, Cidb = initial DO in dark bottle, mg/L, and C′fo = final oxygen concentration in opaque chamber, mg/L: C′fo = Cfo − Cfdb Cfo = final DO in opaque chamber, mg/L, and Cfdb = final DO in dark bottle, mg/L. For each pair of chamber measurements, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Pg = Pn + r where: Pg = hourly gross periphytic primary production, mg O2/m2/h. PG is the area under the curve of primary production per hour through the photoperiod, mg O2/m2/d (see Figure 10300:3). Also, where: R = total periphyton community respiration, mg O2/m2/d, and n = number of observations. Thus, PN = PG − R where: PN = net periphytic production, mg O2/m2/d. 2) Free water methods a) Calculation of reaeration or diffusion for both the single and upstream-downstream methods—Calculate k2 from radiotracer data as follows: and © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: k2 = reaeration coefficient (base e), d−1, KKr = base e transfer coefficient for 85Kr, d−1, t = time of travel, d, (CKr/CH)u = ratio of released radioactivities (µCi/mL) 85Kr to 3H at the upstream station, and (CKr/CH)d = ratio of radioactivities (µCi/mL) 85Kr to 3H at the downstream station. The reaeration coefficient also can be calculated from an equation relating the rate of energy dissipation in a stream to k2.16,17 where: K = escape coefficient, ∆h = change in water surface elevation in a stream reach, and t = time of flow through a stream reach. This can be expressed in terms of hydrodynamic and physical data: where: K′ = 28.3 × 103 s/m ⋅ d for stream flows between 0.028 and 0.28 m3/s; 21.3 × 103 s/m ⋅ d for stream flows between 0.28 and 0.56 m3/s; and 15.3 × 103 s/m ⋅ d for stream flows above 0.56 m3/s, k2 = reaeration coefficient, d−1, at 20°C, 20 ∆H/∆X = slope, m/km, and V = velocity, m/s. Convert k2 to the temperature of the stream by the following equation: 20 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater where: k2t = k2 at ambient water temperature, d−1, and T = ambient water temperature, °C. Convert to D in mg/L/h: where: Cs = oxygen concentration at saturation at ambient stream temperatures, mg/L. b) Single-station method—Calculation of primary productivity and respiration from diurnal oxygen and temperature measure ments at a single station is summarized in Figure 10300:4 and Table 10300:I. Tabulate hourly DO measurements and temperatures. Determine Cs (DO of air-saturated H2O at each temperature from Table 4500-O:I) and compute uncorrected DO consumption, milligrams per liter per hour, for each period: Plot on the half hour, as shown in Figure 10300:4b. Calculate the net primary production and respiration of phytoplankton as shown in Section 10200I. Determine the 24-h average hourly plankton respiration, in milligrams per liter per hour every half hour. Calculate the hourly net phytoplankton production and tabulate for the approximate hours during the photoperiod. Plot as shown on Figure 10300:4c. Calculate and tabulate k2t and substitute D for each Cs, as outlined in ¶ a), above. Plot as shown in Figure 10300:4c. Correct each ∆DO for diffusion and phytoplankton metabolism: © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater ∆DOcorrected, mg/L/H = ∆DOuncorrected − D − Pp − Rp Plot each point as shown in Figure 10300:4d. Gross primary productivity of the benthic and attached algal populations is computed as the area under the curve in Figure 10300:4d from sunrise to sunset. This is primary production in grams per cubic meter per day. Multiply by the average depth for a reach, z meters, to obtain PG in grams per square meter per day. Calculate community respiration: R = 24 z F where: R = community respiration, g/m2/d, z = depth, m, and F = average hourly ∆DO for the dark period (without regard to sign), mg/L/h. Calculate net primary productivity PN as: PN = PG − R c) Upstream-downstream method—Calculation of primary productivity and respiration for a stream reach from upstream and downstream pairs of diurnal curves of oxygen and water temperature is summarized in Figure 10300:5 and Table 10300:II. Alternatively, calculate as below, with oxygen change expressed as the difference between stations rather than as change per hour. The calculations are analogous. Multiply the area under a curve of oxygen change between two stations, corrected for diffusion and plankton metabolism and expressed in milligrams per liter, by the discharge in cubic meters per hour, and divide by the water surface area between the two stations. This, multiplied by 24, yields gross primary productivity in grams per square meter per day. To compute gross primary productivity by this method, tabulate upstream and downstream DO and average water temperature for the reach at each hour. Calculate ∆DO between upstream and downstream stations for each hour as ∆DO = DOdownstream − DOupstream Tabulate Cs and determine the planktonic activity. Correct for planktonic respiration by relating average hourly dark bottle DO change to the time of travel in the stream reach; correct for planktonic production by the hourly change in DO in the light bottle times the time of travel (see Table 10300:II). Calculate or tabulate k2 and convert it to the total oxygen diffusion for the reach. Because diffusion, D, is expressed as milligrams per liter per hour, multiply it by the travel time to obtain © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater the diffusion correction. Correct each hourly upstream-downstream ∆DO as shown in Table 10300:II. Integrate the area under this ∆DO curve from sunrise to sunset to give P as in Figure 10300:5d. where: Q = flow, m3/h, and A = reach area, m2 (average reach width × reach length). and Net production PN = PG − R 5. References 1. VOLLENWEIDER, R.A., ed. 1969. A Manual on Methods for Measuring Primary Production in Aquatic Environments. IBP Handbook No. 12. F.A. Davis Co., Philadelphia, Pa. 2. SLADEEK, V. & A. SLADECKOVA. 1964. Determination of periphyton production by means of the glass slide method. Hydrobiologia 23: 125. 3. KING, D.L. & R.C. BALL. 1966. A qualitative and quantitative measure of aufwuchs production. Trans. Amer. Microsc. Soc. 82:232. 4. CLARK, J.R., D.I. MESSENGER, K.L. DICKSON & J. CAIRNS, JR. 1978. Extraction of ATP from aufwuchs communities. Limnol. Oceanogr. 23:1055. 5. WETZEL, R.G. 1963. Primary productivity of periphyton. Nature 197: 1026. 6. WETZEL, R.G. 1964. A comparative study of the primary production of higher aquatic plants, periphyton, and phytoplankton in a large shallow lake. Int. Rev. ges. Hydrobiol. 49:1. 7. LOEB, S.L. 1981. An in situ method for measuring the primary productivity and standing crop of the epilithic periphyton community in lentic systems. Limnol. Oceanogr. 26:394. 8. BEER, S., A.J. STEWART & R.G. WETZEL. 1982. Measuring chlorophyll a and 14C-labeled photosynthate in aquatic angiosperms by use of a tissue solubilizer. Plant Physiol. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 69:54. 9. ODUM, H.T. 1956. Primary production in flowing waters. Limnol. Oceanogr. 1:102. 10. ALLEN, H.L. 1971. Primary productivity, chemo-organotrophy, and nutritional interactions of epiphytic algae and bacteria or macrophytes in the littoral of a lake. Ecol. Monogr. 41:97. 11. HALL, C.A.S. 1972. Migration and metabolism in a temperate stream ecosystem. Ecology 53:585. 12. NIXON, S.W. & C.A. OVIATT. 1974. Ecology of a New England salt marsh. Ecol. Monogr. 43:463. 13. MCINTIRE, C.D., R.L. GARRISON, H.K. PHINNEY & C.E. WARREN. 1964. Primary production in laboratory streams. Limnol. Oceanogr. 9:92. 14. THOMAS, N.A. & R.L. O’CONNELL. 1966. A method for measuring primary production by stream benthos. Limnol. Oceanogr. 11:386. 15. COPELAND, B.J. & W.R. DUFFER. 1964. Use of a clear plastic dome to measure gaseous diffusion rates in natural waters. Limnol. Oceanogr. 9:494. 16. TSIVOGLOU, E.C. & L.A. NEAL. 1976. Tracer measurement of reaeration. III. Predicting the capacity of inland streams. J. Water Pollut. Control Fed. 48:2669. 17. GRANT, R.S. 1976. Reaeration-coefficient measurements of 10 small streams in Wisconsin. U.S. Geol. Surv. Water Resources Publ. 76-96. 18. ODUM, H.T. & C.M. HOSKIN. 1958. Comparative studies of the metabolism of marine water. Publ. Inst. Mar. Sci. Univ. Tex. 4:115. 19. BOTT, T.L., J.T. BROCK, C.E. CUSHING, S.V. GREGORY, D. KING & R.C. PETERSEN. 1978. A comparison of methods for measuring primary productivity and community respiration in streams. Hydrobiologia 60:3. 6. Bibliography POMEROY, L.R. 1959. Algal productivity in salt marshes. Limnol. Oceanogr. 4:386. CASTENHOLZ, R.W. 1961. An evaluation of a submerged glass method of estimating production of attached algae. Verh. Int. Ver. Limnol. 14:155. WHITFORD, L.A. & G.J. SCHUMACHER. 1964. Effect of a current on respiration and mineral uptake in Spirogyra and Oedogonium. Ecology 45:168. DUFFER, W.R. & T.C. DORRIS. 1966. Primary productivity in a southern Great Plains stream. Limnol. Oceanogr. 11:143. MCINTIRE, C.D. 1966. Some factors affecting respiration of periphyton communities in lotic environments. Ecology 47:918. CUSHING, C.E. 1967. Periphyton productivity and radionuclide accumulation in the Columbia River, Washington, USA. Hydrobiologia 29:125. HANSMANN, E.W., C.B. LANE & J.D. HALL. 1971. A direct method of measuring benthic primary © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater production in streams. Limnol. Oceanogr. 16:822. SCHINDLER, D.W., V.E. FROST & R.V. SCHMIDT. 1973. Production of epilithiphyton in two lakes of the experimental lakes area, northwestern Ontario. J. Fish. Res. Board Can. 30:1511. NORTH AMERICAN BENTHOLOGICAL SOCIETY. 1974–1990 (annual). Current and Select Bibliographics on Benthic Biology. Springfield, Ill. WETZEL, R.G. & G.E. LIKENS. 1991. Limnological Analyses, 2nd ed. Springer-Verlag, New York, N.Y. 10300 E. Interpreting and Reporting Results Although several systems have been developed to organize and interpret periphyton data, no single method is universally accepted. The methods may be qualitative or quantitative. Qualitative methods deal with the taxonomic composition of the communities in zones of pollution, whereas quantitative methods deal with community structure using diversity indices, similarity indices, and numerical indices of saprobity. 1. Qualitative Methods (Indicator Species and Communities) The saprobity system developed by Kolkwitz and Marsson is a widely used method of interpreting periphyton data. This scheme divides polluted stream reaches into polysaprobic, α and mesosaprobic, and oligosaprobic zones, and lists the characteristics of each. The system has been refined1,2 and enlarged by Fjerdingstad3,4 and Sladecek.5,6 Evaluation of the saprobity system requires microscopic evaluation of living indicator biota, particularly for the sensitive sessile protozoans. Glass slides and other transparent substrates are advantageous because they permit direct microscopic examination and identification. Removal of periphyton from slides and preservation for subsequent examination may be acceptable for diatoms, but observation of preserved material is not acceptable for most flagellated organisms. 2. Quantitative Methods These methods use cell counts per unit area of substrate and numerical indices of pollution or water quality. Considerable data on cell densities and species composition of periphyton collected on glass slides in polluted rivers in England are available.7 Other indices include the Shannon-Weaver,8 Simpson’s,9 and Pinkham-Pearson.10 The saprobity system11 also may be used where code numbers assigned for the saprobial value and the abundance of individual species are used to calculate a Mean Saprobial Index. Results also may be expressed by the truncated-log normal distribution of diatom species12,13 as well as the Autotrophic Index (AI).14 3. References 1. KOLKWITZ, R. 1950. Oekologie der saprobien. Ver Wasser-, Boden, Lufthyg. Schriftenreihe (Berlin) 4:1. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2. LIEBMANN, H. 1951. Handbuch der Frischwasser und Abwasserbiologie. Bd. I. Oldenbourg, Munich, Germany. 3. FJERDINGSTAD, E. 1964. Pollution of streams estimated by benthal phytomicroorganisms. I. A saprobic system based on communities of organisms and ecological factors. Int. Rev. ges. Hydrobiol. 49:63. 4. FJERDINGSTAD, E. 1965. Taxonomy and saprobic valency of benthic phytomicroorganisms. Int. Rev. ges. Hydrobiol. 50:475. 5. SLADECEK, V. 1966. Water quality system. Verh. Int. Ver. Limnol. 16:809. 6. SLADECEK, V. 1973. System of water quality from the biological point of view. Arch. Hydrobiol. Ergebn. Limnol. 7:1. 7. BUTCHER, R.W. 1946. Studies in the ecology of rivers. VI. The algal growth in certain highly calcareous streams. J. Ecol. 33:268. 8. SHANNON, C.E. 1948. The Mathematical Theory of Communication. Univ. Illinois Press, Urbana. 9. SIMPSON, E.H. 1949. Measurement of diversity. Nature 163:688. 10. PINKHAM, C.F.A. & J.G. PEARSON. 1976. Applications of a new coefficient of similarity to pollution surveys. J. Water Pollut. Control Fed. 48:717. 11. PANTLE, R. & H. BUCK. 1955. Die biologische uberwachung der Gewasser und der Darstellung der Ergebnisse. Gas-Wasserfach 96:604. 12. PATRICK, R., M.H. HOHN & J.H. WALLACE. 1954. A new method for determining the pattern of the diatom flora. Bull. Philadelphia Acad. Natur. Sci. 259:1. 13. PATRICK, R. 1973. Use of algae, especially diatoms, in the assessment of water quality. In J. Cairns, Jr., ed. Biological Methods for the Assessment of Water Quality. ASTM STP 528, American Soc. Testing & Materials, Philadelphia, Pa. 14. WEBER, C. 1973. Recent developments in the measurement of the response of plankton and periphyton to changes in their environment. In G. Glass, ed. Bioassay Techniques and Environmental Chemistry. Ann Arbor Science Publ., Ann Arbor, Mich. 4. Bibliography FJERDINGSTAD, F. 1950. The microflora of the River Mlleaa, with special reference to the relation of the benthal algae to pollution. Folia Limnol. Scand. 5:1. BLUM, J.L. 1956. The ecology of river algae. Bot. Rev. 22:291. YOUNT, J.L. 1956. Factors that control species numbers in Silver Springs, Florida. Limnol. Oceanogr. 1:286. BUTCHER, R.W. 1959. Biological assessment of river pollution. Proc. Linnean Soc. London 170:159. HOHN, M.H. 1959. The use of diatom populations as a measure of water quality in selected areas of Galveston and Chocolate Bay, Texas. Publ. Inst. Mar. Sci. Univ. Tex. 5:206. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater HOHN, M.H. 1961. Determining the pattern of the diatom flora. J. Water Pollut. Control Fed. 33:48. PATRICK, R. 1963. The structure of diatom communities under varying ecological conditions. Ann. N.Y. Acad. Sci. 108:359. SCHLICHTING, H.E., JR. & R.A. GEARHEART. 1966. Some effects of sewage effluent upon phyco-periphyton in Lake Murray, Oklahoma. Proc. Okla. Acad. Sci. 46:19. SLADECKOVA, A. & V. SLADECEK. 1966. Periphyton as indicator of reservoir water quality. Technol. Water (Czech) 7:507. TAYLOR, M.P. 1967. Thermal effects on the periphyton community in the Green River. Tennessee Valley Authority, Div. Health & Safety, Water Qual. Br., Biol. Sect., Chattanooga, Tenn. PATRICK, R. 1968. The structure of diatom communities in similar ecological conditions. Amer. Natur. 102:173. DICKMAN, M. 1969. A quantitative method for assessing the toxic effects of some water soluble substances, based on changes in periphyton community structure. Water Res. 3:963. BESCH, W.K., M. RICARD & R. CANTIN. 1970. Use of benthic diatoms as indicators of mining pollution in the N.W. Miramichi River. Tech. Rep. Fish. Res. Board Can. 202:1. NUSCH, E.A. 1970. Ecological and systematic studies of the Peritricha (Protozoa, Ciliata) in the periphyton community of reservoirs and dammed rivers with different degrees of saprobity. Arch. Hydrobiol. (Suppl.) 37:243. ROSE, F.L. & C.D. MCINTIRE. 1970. Accumulation of dieldrin by benthic algae in laboratory streams. Hydrobiologia 35:481. WHITTON, B.A. 1970. Toxicity of zinc, copper and lead to Chlorophyta from flowing waters. Arch. Mikrobiol. 72:353. BURROWS, E.M. 1971. Assessment of pollution effects by the use of algae. Proc. Roy. Soc. Lond. Ser. B. 177:295. PATRICK, R. 1971. The effects of increasing light and temperature on the structure of diatom communities. Limnol. Oceanogr. 16:405. ARCHIBALD, R.E.M. 1972. Diversity of some South African diatom associations and its relation to water quality. Water Res. 6:1229. CAIRNS, J., JR., B.R. LANZA & B.C. PARKER. 1972. Pollution-related structural and functional changes in aquatic communities with emphasis on freshwater algae and protozoa. Proc. Acad. Natur. Sci. Philadelphia 124:79. OLSON, T.A. & T.O. ODLAUG. 1972. Lake Superior Periphyton in Relation to Water Quality. Water Pollut. Control Res. Ser., 18080 DEM 02/ 72. Univ. Minnesota School Public Health, Minneapolis. HANSMANN, E.W. 1973. Effects of logging on periphyton in coastal streams of Oregon. Ecology 54:194. RUTHVEN, J.A. & J. CAIRNS, JR. 1973. Response of fresh-water protozoan artificial communities to metals. J. Protozool. 20:127. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater MIDWEST BENTHOLOGICAL SOCIETY. 1964–1973 (annual). Current and Select Bibliographies on Benthic Biology. Springfield, Ill. NORTH AMERICAN BENTHOLOGICAL SOCIETY. 1974–1991 (annual). Current and Select Bibliographies on Benthic Biology. Springfield, Ill. BAXTER, R.M. 1977. Environmental effects of dams and impoundments. Annu. Rev. Ecol. Systematics 8:255. WEITZEL, R.L., ed. 1979. Methods of Measurement of Periphyton Communities: A Review. ASTM Spec. Tech. Publ. 690, American Soc. Testing & Materials, Philadelphia, Pa. WETZEL, R.G., ed. 1983. Periphyton of Freshwater Ecosystems. Developments in Hydrobiology 17. Dr. W. Junk B.V. Publ., The Hague, The Netherlands. STEVENSON, R.J. 1984. Epilithic and epipelic diatoms in the Sandusky River USA with emphasis on species diversity and water pollution. Hydrobiologia 114:161. KOSINSKI, R.J. 1984. The effect of terrestrial herbicides on the community structure of stream periphyton. Environ. Pollut. Ser. A, Ecol. Biol. 36:165. LINDSTROM, E.A. & T.S. TRASAN. 1984. Influence of current velocity on periphyton distribution and succession in a Norwegian soft water river. Verh. Int. Ver. Limnol. 22:1965. MCGUIRE, M.J., R.M. JONES, E.G. MEANS, G. IZAGUIRRE & A.E. PRESTON. 1984. Controlling attached blue-green algae with copper sulfate. J. Amer. Water Works Assoc. 76:60. PARKER, B.C., G.J. SCHUMACHER & L.A. WHITFORD. 1984. Some rarely reported algae of the Appalachian Mountains, Eastern North America: Why so rare? Va. J. Sci. 35:197. 10400 MACROPHYTON*#(144) 10400 A. Introduction 1. General Discussion The macrophyton consists principally of aquatic vascular flowering plants, but it also includes the aquatic mosses, liverworts, ferns, and the larger macroalgae. Like other primary producers, these plants respond to the quality of the water in which they grow. The use of biota, including macrophyton, is an increasingly important and recognized technique for assessing aquatic habitats.1 Macrophyton often constitute a dominant factor in the habitat of other aquatic organisms. Freshwater forms range in size from the tiny watermeal (Wolffia spp.), about the size of a pinhead, to plants such as the cattail (Typha spp.), up to 4 m high, and finally to cypress trees (Taxodium spp.), up to 50 m high. Higher aquatic plants often are clustered in large numbers, many in pure stands, covering extensive areas of shallow lakes, reservoirs, marshes, and canals. A few of the larger freshwater algae (Chara spp. and Nitella spp.) resembling higher plants in © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater size, form, and habit sometimes are included in the macrophyton. In marine water, the intertidal rockweeds (Fucus spp. and Ascophyllum spp.) and offshore kelps (Fucus spp. and Macrocystis spp.) are conspicuous. Vascular marine or estuarine plants, such as the eelgrass (Zostera spp.) and the marshgrass (Spartina spp.), are essential to the aquatic ecosystem. Three growth forms of macrophyton generally are recognized: floating, submersed, and emersed. Floating plants may be rooted or free-floating; their principal foliage or crown floats freely on the water surface. All or most of the foliage of submersed plants grows beneath the water surface: nearly all submersed vascular plants have roots. Growing tips of submersed plants may emerge to flower and some species can produce floating leaves. Emersed plants have their principal foliage in the air at or above the water surface; they are attached by roots to the bottom mud. In some cases the same species may grow as a floating or emersed type, or as a submersed or emersed type. Submersed and emersed vascular plants typically are rooted to the bottom but they may be found detached and floating. The distribution and abundance of higher plants is subject to considerable spatial and temporal variation. Among the many factors that determine their presence, density, and morphology are sediment type, water turbidity, water current, nutrient concentrations, water depth, shoreline disturbance, herbivore grazing, and human activities. Zonation in the littoral zone of lakes and shallow, slow-moving streams is common. Emergent macrophytes generally are found in the most shallow portion of the littoral zone. During periods of low water level they may occupy the terrestrial as well as the aquatic habitat. The depth of inhabitation seldom exceeds 1 m. Floating-leaved plants commonly are found in the shallower littoral areas in water depths between 1 and 3 m. Submersed plants may occur from the edge of the shore to the interface of the littoral and profundal zones, but rarely extend beyond a depth of 10 m because of limitation of underwater light. 2. Reference 1. NORRIS, R.H., B.T. HART & M. FIULAYSAR, eds. 1995. Use of biota to assess water quality. Austral. J. Ecol. 20:1. 3. Bibliography BUTCHER, R.W. 1933. Studies on the ecology of rivers. I. On the distribution of macrophytic vegetation in the rivers of Britain. J. Ecol. 21:58. WELCH, P.S. 1948. Limnological Methods. Blakiston Co., Philadelphia, Pa. MILLIGAN, H.F. 1969. Management of aquatic vascular plants and algae. In Eutrophication: Causes, Consequences, Correctives. National Acad. Science, Washington, D.C. BOYD, C.E. 1971. The limnological role of aquatic macrophytes and their relationship to reservoir management. In G.E. Hall, ed. Reservoir Fisheries and Limnology. Spec. Publ. Amer. Fish. Soc. No. 8. HUTCHINSON, G.E. 1975. A Treatise on Limnology. Vol. III. Limnological Botany. John Wiley © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater & Sons, New York, N.Y. WESTLAKE, D.F. 1975. Macrophytes. In B.A. Whitton, ed. River Ecology. Univ. California Press, Berkeley & Los Angeles. WILE, I. 1975. Lake restoration through mechanical harvesting of aquatic vegetation. Verh. Int. Ver. Limnol. 19:660. WOOD, R.D. 1975. Hydrobotanical Methods. University Park Press, Baltimore, Md. RASCHKE, R.L. 1978. Macrophyton. In W.T. Mason, Jr., ed. Methods for the Assessment and Prediction of Mineral Mining Impacts on Aquatic Communities: A Review and Analysis. U.S. Dep. Interior, Harpers Ferry, W. Va. WETZEL, R.G. 1983. Limnology, 2nd ed. Saunders College Publishing, Philadelphia, Pa. DENNIS, W.M. & B.G. ISOM, eds. 1984. Ecological Assessment of Macrophyton: Collection, Use, and Meaning of Data. ASTM STP 843, American Soc. Testing & Materials, Philadelphia, Pa. 10400 B. Preliminary Survey 1. General Considerations A macrophyte survey includes species identification, location, assessment of health, and quantity. More detailed studies may involve functioning of aquatic plants in nutrient and heavy metal uptake and turnover, use of plants as indicator organisms, and effects of plants on water quality conditions. Several sampling protocols are required to meet diverse survey needs. The usefulness of a given study and the appropriate types of statistical analyses are determined and fixed initially. To develop a good sample design, determine what information is desired, prevailing environmental conditions, the life and growth form of the species being sampled, and the methods for obtaining reproducible data that are comparable to other or future studies. In defining reporting requirements, consider matters such as the use of scientific names; the selection of appropriate descriptors such as frequency, density, biomass, cover, diversity, productivity, and outer limit of vegetation growth; and the use of proper statistical techniques. 2. Pre-Field Investigations During pre-field investigations assemble maps, charts, aerial photographs, taxonomic keys, and past reports. Maps, charts, and aerial photographs help determine access routes, project size, plant community distribution, habitat characteristics that may influence plant distribution, and sampling obstacles or hazards. These items also provide a base for doing field work and reporting results. They may be available locally from municipal engineering departments, zoning boards, planning commissions, drainage districts, and land conservation commissions. At the state level information may come from natural resource agencies, natural history survey organizations, universities, and transportation departments. At the federal level, the Geological Survey, Natural Resources Conservation Services, Bureau of Land Management, Forest Service, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Park Service, Fish and Wildlife Service, Tennessee Valley Authority, Bureau of Indian Affairs, Army Corps of Engineers, Environmental Protection Agency, National Biological Service, and the National Oceanographic and Atmospheric Administration have many maps, charts, and aerial photographs available. A final source is private map companies that publish hydrographic maps for fishermen and recreational boaters. Past reports provide historical records useful for planning sampling logistics and interpreting results. Comparable studies taken at different times provide a dynamic look at the vegetation. An often-overlooked resource is a herbarium storing pressed and mounted plant specimens. These generally are located in universities and natural history museums. 3. Field Reconnaissance Sampling efficiency is improved and a sampling scheme can be refined during a field reconnaissance. It provides an opportunity to learn the species of plants present and to sketch their distribution. The species-area curve technique frequently is used to determine the likelihood of finding more species during a preliminary survey. A field reconnaissance allows the investigator to answer logistical questions that are the bane of all sampling efforts. 4. Bibliography ONDOK, J.P. & J. KVET. 1978. Selection of sampling areas in assessment of production. In D. Dykyjova & J. Kvet, eds. Pond Littoral Ecosystems: Structure and Functioning. Ecological Studies 28. Springer-Verlag, Berlin. MACEINA, M.J., J.V. SHIREMAN, K.A. LANGELAND & D.E. CANFIELD, JR. 1984. Prediction of submersed plant biomass by use of a recording fathometer. J. Aquat. Plant Manage. 22:35. Also see Section 10400A.2. 10400 C. Vegetation Mapping Methods Mapping vegetation stands may be necessary. Do this during the preliminary survey.1 1. Baseline Method Vegetation maps constructed using the baseline method or the basepoint-stadia rod-alidade method generally are limited to pure stands of floating or emersed littoral macrophyton in all bodies of water. In clear water, the outline of pure stands of submersed vegetation can be determined by using a viewing box (usually a wooden or plastic box with a watertight glass lens) from the surface or by underwater observation by a diver using a snorkel or SCUBA (self-contained underwater breathing apparatus). The baseline method and the basepoint-stadia rod-alidade method provide accurate maps of vegetation in areas up to 1 × 10 5 m2 where most of the vegetation outline is visible. The baseline method uses intercepting lines from each end of a predetermined base line to closely spaced markers (i.e., chaining pins) around the stand. By presetting the map scale, the ratio between the length of the base line and its reduction on the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater map (drawn on a plane table) can be determined. The basepoint-stadia rod-alidade method is a modification of the baseline method in that the distance between the vegetational outline and the base point is determined with an alidade, range finder, or portable Loran-C unit. One unit on the stadia rod, as viewed between the cross-hairs of the alidade, is equivalent to a distance of 3.05 m between the stadia rod and the alidade. Chaining pins are not required when this method is used. In practice, more readings closely spaced along the vegetational outline usually are taken using the basepoint-stadia rod-alidade method. It is not necessary to measure all distances and angles; some can be determined trigonometrically. After all angles and distances are calculated, fill in irregularities through inspection and use of other maps and photographs. The technology for using global positioning systems (GPS), especially when linked to geographical information systems (GIS), is evolving rapidly.2 These systems probably will have wide applicability to mapping aquatic vegetation, thereby rendering older surveying techniques obsolete or at least time-consuming and tedious. 2. Line Intercept Method The line intercept method is preferable for mapping mixed stands and/or large areas. Select sampling points at equal intervals along a base line. Choose interval length by the degree of accuracy desired: the closer the sampling points, the more accurate the map. Run transects perpendicularly from the base line to the boundary of the plant stand. Use an intercept line (transect line) of plastic-coated wire rope to prevent stretching. If line flotation is a problem, use lead weights applied at regular intervals to sink the line and act as interval markers on the rope to designate sampling units. Use 0.5-m segments (in dense vegetation) to 5-m sampling units (in sparse vegetation) for determining plant species that vertically intercept the line at each segment. During underwater surveys, record data with a wax or soft lead pencil on a writing board constructed of plastic overlays. Construct the vegetation map by placing points where plant species are found within an outline map (or aerial photograph) of the sample area. Determine the area that a single species or total vegetation occupies by planimetry or digitization and computer calculation. Determine frequency from line intercept or quadrat data, or with a set sampler consisting of a 2-cm steel tube, 2 m long, to which five 0.75-cm by 25-cm steel rods are attached on 40-cm centers. Record vegetation touching each of the five points within 2.5 cm of the distal tip. If more than one plant species is touching, record the plant nearest the tip. If no plant is touched, record bare ground. 3. Remote Sensing a. General considerations: Remote sensing techniques are used to detect, assess, and monitor aquatic macrophytes. These techniques include analog aircraft and satellite serial photography, digital aircraft and satellite multispectral scanning in the visible, infrared, and thermal bands; microwave techniques, primarily side-looking airborne radar (SLAR); and shuttle imaging radar (SIR). They provide a synoptic view of large areas, and allow quick surveys to © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater further delineate areas of interest and repeated viewing at relatively low cost. In selecting remote sensing system(s) consider expected results, time for project completion, and available resources. The larger the area, the greater the advantage of remote sensing. Remote sensing also lends itself to studies over time, because each image is a historical record. For determining general associations of a widespread macrophytic growth, ground resolutions of between 30 and 80 m are recommended. Widespread multitemporal coverage is available at scales ranging from 1:100 000 to 1:1 000 000 at a reasonable cost in the form of paper prints, transparencies, and digital formats. Landsat Multispectral Scanner (MSS) and Thematic Mapper (TM) have been used to identify areas with emergent vegetation or topped-out submergent vegetation. Surface roughness is a requisite for an imaging return with SLAR and SIR. Landsat provides limited capability for species discrimination, but availability, cost, and repeat cycles make it useful in determining presence of large populations over time. For a detailed vegetation survey, including discrete species identification, use much larger-scale imagery, (1:10 000 or greater). High-altitude aerial coverage available through the National High Altitude Mapping Program (NHAP) and other sources at original scales of approximately 1:60 000 to 1:120 000 provides both good initial areal coverage and the capacity for enlargement up to five times.3 After determining scale, select film/filter or sensor combinations. These include black and white imagery, color infrared, and black and white-infrared imagery; color infrared film used with a yellow filter is widely applicable, but other combinations also are useful.4-7 For detailed flight planning consider growth stage of plants, water depth and clarity, tidal conditions, cloud cover, and sun angle.3,8,9 Available resources ultimately determine remote sensing activities. b. Aerial photography: 1) Equipment—For all-format aerial photography, use a good-quality 35-mm single-lens reflex (SLR) camera with manual or through-the-lens metering or any good 70-mm camera system, preferably with a motor drive. Intervalometers providing for exact exposure intervals for stereo photography are available for both systems. A 28-mm-focal-length lens with a 35-mm SLR camera gives wide coverage at low altitudes; 40-mm and 80-mm lenses are successful with 70-mm camera systems. When photographing with black-and-white or color film, use a skylight or haze filter. When photographing with color infrared film below 1700 m, use a Wratten 12 filter; at altitudes above 1700 m use a Wratten 15 filter. Time of year and condition of vegetation also may influence the choice of yellow/orange filter. In turbid waters, color film*#(145) provides better water penetration and is more useful in mapping submersed vegetation than is color infrared film.4 Color infrared†#(146) yields more detail, is more useful in mapping emergent vegetation or wetlands, and may provide more detail in clear, nonturbid waters. Preferably mount camera in the belly of a high-wing, single-engine aircraft for low-altitude, small-format photography. Belly mounts require special aircraft modification, but provide a stable platform, protection for the camera, and good access. Alternatively, camera may be mounted on the aircraft door. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 2) Procedures—Because sun angle is critical in obtaining high-quality aerial photography, schedule flight for a time when solar altitude is between 40 and 68 degrees.7 Set camera at designated ISO reading for the film (assume 100 to 125 for color infrared film) and shoot in the automatic exposure mode. At a typical airspeed of about 190 km/h (approximately 120 mph) a shutter speed of 1/250 or 1/500 is adequate. Determine proper f-stop from an aerial exposure computer; in general, f 5.6 to 11 (f 8 is optimum) gives acceptable color exposure. Proper exposure of color infrared film depends on such factors as time of day and year, altitude, humidity, and type of landscape. Process film through the manufacturer, a photo laboratory, or aerial photography service. Development of color infrared film is available solely through the manufacturer. c. Fathometry: Recording fathometers are best applied in water more than 1 m deep where the instruments can determine accurately the height and distribution of subsurface macrophytes. A recording fathometer can be mounted on most boats and can accurately determine one-dimensional (percent cover) and two-dimensional (percent vertical area) profiles of submersed vegetation. Fathometry is especially useful for determining the outer edge of plant growth. Make linear and planimetric measurements on chart tracings that provide permanent records for ready comparison over time. Calculate percent cover by dividing the linear measurement for a macrophyte species or community by the total chart paper length for any given transect. Dividing the area of the tracing with macrophytes by the total water area gives percent vertical area. Use a fathometer accurate to the nearest 0.1 m. Mount the transducer for the recording fathometer with brackets on the boat’s transom. Keep speed and recorder speed constant to produce tracings of similar length and resolution. Only a few minutes are required to replicate transects several kilometers long. Unless gross morphological differences exist, species discrimination on the chart tracings is difficult or impossible. Mark boundaries of monotypic colonies and community types with a fixed line on the chart tracings. Dense vegetation mats that reach the surface may impede boat movement, prevent the transducer signal from reaching the hydrosoil, and merge tracings of macrophytes with the transducer line. Tracing patterns from water less than 1 m deep may be difficult to interpret.10 Location of transects or points along a transect also can be determined using GPS techniques.2 4. References 1. RASCHKE, R.L. 1984. Mapping—Surface or ground surveys. In W.M. Dennis & B.G. Isom, eds. Ecological Assessment of Macrophyton: Collection, Use, and Meaning of Data. Spec. Tech. Publ. 843, American Soc. Testing & Materials, Philadelphia, Pa. 2. KRESS, R. & D. MORGAN. 1995. Application of New Technologies for Aquatic Plant Management. U.S. Army Waterways Experiment Sta., Corps of Engineers, Aquatic Plant Control Research Program. Bulletin, Vol. A-95. 3. AVERY, E.T. & G.L. BERLINE. 1985. Interpretation of Aerial Photographs, 4th ed. Burgess Publishing Co., Minneapolis, Minn. © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater 4. ANDREWS, D.S., D.H. WEBB & A.L. BATES. 1984. The use of aerial remote sensing in quantifying submersed aquatic macrophytes. In W.M. Dennis & B.G. Isom, eds. Ecological Assessment of Macrophyton: Collection, Use, and Meaning of Data. Spec. Tech. Publ. 843, American Soc. Testing & Materials, Philadelphia, Pa. 5. LONG, K.S. 1979. Remote Sensing of Aquatic Plants. Tech. Rep., Waterways Experiment Sta., U.S. Army Corps of Engineers, Vicksburg, Miss., Vol. A-79–2. 6. BREEDLOVE, B.W. & W.M. DENNIS. 1984. The use of all-format aerial photography in aquatic macrophyton sampling. In W.M. Dennis & B.G. Isom, eds. Ecological Assessment of Macrophyton: Collection, Use, and Meaning of Data. Spec. Tech. Publ. 843, American Soc. Testing & Materials, Philadelphia, Pa. 7. BENTON, A.R., JR. 1976. Monitoring aquatic plants in Texas. Tech. Rep. RSC-76, Texas A & M Remote Sensing Center, College Station, Tex. 8. COLWELL, R.H., ed. 1983. Manual of Remote Sensing, 2nd ed. American Soc. Photogrammetry & Remote Sensing, Falls Church, Va. 9. SLAMA, C., ed. 1980. Manual of Photogrammetry, 4th ed. American Soc. Photogrammetry & Remote Sensing, Falls Church, Va. 10. MACEINA, M.J. & J.V. SHIREMAN. 1980. The use of a recording fathometer for determination of distribution and biomass of hydrilla. J. Aquat. Plant Manage. 18:34. 5. Bibliography SCHMID, W.D. 1965. Distribution of aquatic vegetation as measured by line intercept with SCUBA. Ecology 48:816. OSBORNE, J.A. 1977. Ground Truth Measurements of Hydrilla verticillata Royle and Those Factors Influencing Underwater Light Penetration to Coincide with Remote Sensing and Photographic Analysis. Res. Rep. No. 2, Bur. Aquatic Plant Research & Control, Florida Dep. Natural Resources, Tallahassee. 10400 D. Population Estimates 1. Sampling Design The design of a sampling program depends on study aims, collection methods, variation and distribution of vegetation, personnel and funds available, and accuracy expected. Variation in space usually is not random; distribution is determined by water depth, shoreline activity, sediment type, or other factors. The parametric statistic for estimating the true population mean assumes that the population being sampled has a normal distribution and that all sample units have the same probability of being selected. Avoid fixed sampling stations in sampling programs to determine population means, unless they are chosen at random at the beginning of the study. Because normally distributed plant populations may not be a characteristic of contiguous plant © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater communities, use parametric statistics with caution. The simple random sampling design is best applied to homogeneous, noncontiguous plant communities. The number of stations required to obtain an estimate of the true population mean with a predetermined level of confidence and permissible error can be determined by applying the data from a pilot study to the following equation: where: N = number of sampling stations, t = Student’s t at a given probability level; because N is unknown, set t = 2.0; t is approximately equal to 2.0 for N > 30, S = standard deviation, x = estimator of true population mean usually determined by conducting a pilot study; and d = permissible error of the final mean; d = 0.1 is recommended for vegetation studies (± 10%). An estimate of sampling program cost may be obtained as the sum of initial fixed cost (such as cost of equipment purchase) and variable cost (cost per sample multiplied by number of samples). Apply stratified random sampling to populations having many homogeneous stands. This design is best applied to populations with obvious gradients and, in practice, to gain precision by the minimized variance within strata. Determine placement of strata by a pilot study. To maximize precision, place stratum boundaries around homogeneous areas; generally, the fewer strata, the greater precision. Allocate sampling in stratified random sampling design according to: where: Ni = number of samples in stratum I, N = total number of samples, © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater Wi = a weight reflecting the size (number of quadrats, for example) of stratum i, and Si = standard deviation of sampled characteristic within stratum I. Means for population measurement taken along randomly placed stations on a transect line do not represent large areas of lake populations unless the transect line is placed randomly. Arbitrarily placed transect lines within a sampling area may or may not reflect the true variation of the vegetation within. 2. Collection Methods a. Field inventory/reconnaissance: 1) Manual collection—If water depth, clarity, temperature, flow, and other circumstances permit, collect specimens by hand. Under ideal conditions, manual collection by wading, snorkeling, or with SCUBA in deeper water habitats permits a detailed and comprehensive evaluation of the macrophyte community. 2) Drag chains—Construct drag chains by welding sharpened U-shaped hooks to a short length (0.6 to 1.0 m) of medium-weight chain. Attach chain to a rope and pull it through the water. Attach a float to the end of the rope to prevent its loss if the chain is snagged and/or dropped. The drag chain can be used readily from a slow-moving or stationary boat and is most efficient in collection of submersed macrophyte species with tall growth forms. 3) Rakes and tongs—Rakes with various handle lengths and oyster tongs may be useful in collecting macrophytes. A rope may be attached to the rake handle for sampling in deep water or to facilitate sampling over a wider radius. 4) Grab samplers—Devices developed for sampling benthic organisms, such as the Ekman, Ponar, and similar grab samplers (see Section 10500B.3), may be used to collect macrophytes. The light weight of the Ekman grab makes it preferable for the rapid and numerous samplings often required for survey inventories. 5) Recording fathometers—Use to determine height and distribution of subsurface macrophytes. Species with similar morphology usually cannot be distinguished from chart tracing; use supplemental methods to identify species. b. Quantitative sampling: Numerical data collected to describe vegetation commonly include such measures of abundance as density, frequency, cover, and biomass/standing crop.1-3 Collect these data from plots or quadrats or, less frequently, by plotless sampling techniques. The choice of analytical method depends on vegetation density and types, water depth, flow, height of vegetation in the water column, and nature of the sediment. 1) Line intercepts4—This plotless sampling technique entails use of a weighted nylon or lead core line laid along the bottom between two known points or oriented by a compass reading. For dense floating mat vegetation, a floating line may be laid on top of the mat. A surveyor measures the linear distance occupied by various species that underlie the transect line. Express these as a percentage of the total line length for individual species as well as for all species © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater combined. If frequency data are desired, mark the line in increments (e.g., 1 m) and treat species presence/absence in a manner similar to data from quadrat sampling. The line intercept has been used to characterize and map aquatic communities5 and to correlate distribution of macrophytes with selected environmental factors.6 Line transects also are useful for determining patterns of plant distribution.7 In aquatic environments, the line-intercept method is time-consuming and may require a diver equipped with SCUBA. Problems arise in determining whether a plant underlies the transect line.8 2) Belt transects—This technique is similar to the line transect and is useful for biomass or density determinations. Data are collected along a fixed line, but from a two-dimensional plot or belt. The belt can be treated as a series of contiguous quadrats or quadrat location may be selected on the basis of a fixed interval or water depth. Use floating or sinking frames. 3) Quadrats—Quadrats can be used for such population and community estimates as frequency, cover, density, and biomass. Quadrats can be any two-dimensional shape but are typically round or rectangular. The sampling area of quadrat samplers can be of any size, but typically varies from 0.1 to 1 m.2 With the exception of frames, most sampling devices described have been used to obtain estimates of above-ground biomass (standing crop).9 Above-ground biomass generally is used because of the difficulty in collecting underground plant parts, such as rhizome and roots. Without the underground parts, however, the data are of limited value for estimates of primary production. a) Manual samplers—These are relatively simple devices for sampling macrophytes, such as cutting shears. Although they can be used in deep water and manipulated by a diver, they work best in shallow water. They are relatively inexpensive and can be constructed easily or purchased from commercial sources. Frames are suitable for sampling in shallow water. For sampling short, erect plants, use a square sinking frame constructed of metal. For dense or tangled vegetation, a square assembly frame with pins or wing nuts at the corners or a fixed-corner three-sided frame may be useful. Decide whether to include only macrophytes rooted within the frame or also overlapping plants. In deep water, difficulty and bias may occur in sampling tall submersed vegetation. For macrophytes forming a dense floating mat, use a floating frame constructed of wood or PVC pipe. Box samplers are useful for sampling where water is shallow and the bottom consists of unconsolidated sediments. The sampler consists of an open-ended box with a metal cutting flange at the bottom and lateral handles; a sampler constructed of 7-mm plexiglass with dimensions 0.5 m × 0.5 m × 0.6 m and aluminum cutting flange and corner reinforcements weighs about 12 kg. With modifications, a box sampler can be used in deep water.10 Benthic dome (BeD) samplers11 may be used for sampling in deep flowing waters. The sampler consists of a plastic dome with a stainless steel circular collar that can be pushed into the © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater substrate. It weighs approximately 11 kg and has a sampling area of 0.25 m.2 Various samplers12,13 developed for macroinvertebrate sampling also may be used to collect macrophytes. These include the Surber sampler, suitable for shallow rivers with moderate current [see Section 10500B.3b1)]; the stovepipe (cylindrical) sampler, suitable for wadable waters with unconsolidated sediment bottoms [see Section 10500B.3c4)]; and the Ekman grab sampler, best suited for soft sediment bottoms with short, erect vegetation [see Section 10500B.3a6)]. b) Mechanically operated samplers—Mechanical sampling devices are costly and complex, and require a floating platform with winches, cables, and booms. The samplers described below are useful in deep water. They may decrease sample collection time, increase accuracy of above-ground and total biomass estimates, and be subject to less bias than many manual methods. CAUTION: Use extreme caution for safe operation. The Louisiana box sampler is an open-ended 35-cm-high box made of sheet metal or similar material that samples a 61- × 61-cm quadrat (sampling area = 0.37 m2).14 It can be used from a V-hulled or pontoon boat and is hoisted above the water with a cable and boom. A quick-release mechanism lets the sampler fall free through the water column. Aquatic vegetation is trapped against the bottom and severed by cutting edges along the base of sampler. A nylon net sack over the top retains severed plant fragments. A diver inserts a horizontal cutting blade in a slot at the level of the substrate before the sampler is hoisted to the surface. Manual insertion of the cutting plate by a diver makes use of the Louisiana box sampler comparatively efficient. In soft sediments, the sampler may penetrate too deeply and require lifting before the cutting plate can be inserted. Rocks, stumps, roots, and other debris may prevent complete closure of the cutting door. The Osborne sampler15 is a stainless steel box having outside dimensions of 50 cm × 50 cm × 60 cm high and a sampling area of 0.25 m.2 The sampler weighs 110 kg and is operated by winch and cable from a pontoon boat. After hoisting and suspending the sampler alongside the pontoon boat, a quick-release mechanism allows free-fall through the water column. Tempered steel blades along the bottom edge of the sampler cut vegetation during the descent. A wire mesh screen fastened to the top prevents loss of plant fragments. A hinged slotted door is closed with a lift cable and the sampler is winched to the pontoon boat platform for removal of macrophytes and sediments. Because the sampler penetrates and collects sediments, the sample includes roots and rhizomes and can be used to estimate total biomass as well as above-ground biomass. Efficient operation and accurate biomass estimates require an unconsolidated substrate free of rocks and other debris. The Waterways Experiment Station (WES)16 sampler is made of perforated stainless steel and operated from a pontoon boat with an overhead beam that allows it to be hydraulically raised and lowered through a circular opening in the pontoon’s platform. Two types are available: one is cylindrical with a sampling area of 0.28 m2 and the other is square with sampling area of 0.39 © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater m.2 Rotating cutting blades at the base of each sampler sever vegetation as the samplers are lowered. The bottom cutting plate of each is closed hydraulically. A major advantage of the WES sampler is its capability to obtain plant samples from any depth. The Louisiana Box and Osborne samplers, once released, free-fall to the substrate, whereas the hydraulic operation of the WES sampler controls its descent. The size and weight of the trailer and pontoon boat for the WES sampler restrict its use in certain water bodies and require an improved ramp for launching. Although the WES square sampling head is reported to provide a more accurate estimate of above-ground biomass than the circular one, a substantial underestimate of actual above-ground biomass still is reported.16 3. Sample Preparation and Analysis a. Biomass: 1) Fresh weight (wet weight)—Wash samples free of silt and debris, place in a nylon bag (mesh size 0.75 cm) and spin in a garment washer at 560 rpm for 6 to 7 min to remove excess moisture. Weigh sample to nearest 0.1 g. 2) Dry weight—Dry subsample (not less than 10%) in a forced-air oven at 105°C for 48 h or until a constant weight is achieved. The coefficient of variation for a series of subsamples should not exceed 10%. Calculate dry weight by dividing dry weight of subsample by fresh weight of subsample times fresh weight of sample. 3) Ash-free dry weight—Transfer dried subsample to a covered and preweighed crucible. Ignite at 550°C for 6 h. The amount of ash is the weight of material remaining after combustion. Calculate ash-free dry weight by determining the ratio between ash and dry weight times dry weight of sample (see Section 10300C.5). b. Chlorophyll content: Extract fresh plant material with acetone made basic with MgCO3. Grind the plant material and centrifuge at 2500 rpm for 10 to 15 min. Wash residue with acetone and add filtered washings to extract. Dry overnight in a container with anhydrous Na2SO4. Dilute with 90% acetone and water. Determine chlorophyll content (see Section 10200G). c. Carbon content: Most plants (entire) contain 46 to 48% carbon on a dry-weight basis. A factor (46.5%) can be used to calculate carbon content and make comparisons. d. Caloric content: Determine energy content by bomb calorimetry. e. Species identification: 1) Sample preparation—Use fresh specimens for identification wherever possible. Avoid immature plants or plants lacking flowers. Because aquatic plants contain from 80 to 95% water and have less supportive tissue than terrestrial forms, a different procedure is required for drying, preserving, and mounting them. Collect plants during peak growth when flowers and/or fruits are present, if practical. Collect the entire plant (stems, rhizomes, leaves, roots, flowers, and fruits). After collection, either press plants in the field17-19 or wrap specimen in several layers of paper and submerge in water. Label wrapped specimens with date and location of collection on © Copyright 1999 by American Public Health Association, American Water Works Association, Water Environment Federation Standard Methods for the Examination of Water and Wastewater an index card and place sample and card in a plastic bag. Preferably use an ice chest containing crushed ice for storage in the field. Press plants as soon as practical. They can be kept for several days under refrigeration at 4°C. Clean plant of all silt and residue. Prepare a mount by centering the plant on 100% rag herbarium paper. Place emergent plants immediately on paper because they take on a natural posture. Place a limp plant in a shallow pan of water and slide the herbarium paper under it; with a slow motion, raise the paper at a 30° angle while keeping the plant centered. Leaves and stems should lie flat on the paper. Drain off excess water, cover with wax paper to prevent plant from sticking to blotters, and place in a plant press between paper and blotters. Place plant press in a dryer. Dry plants at room temperature, but change blotters at least every other day until the plant is sufficiently dry for permanent mounting. To prepare a wet mount place specimen in an airtight glass vessel filled with 1 part 10% formalin, 3 parts water, and a trace of powdered copper sulfate. Plants will remain lifelike and retain their color for many years in this condition. 2) Identification—A stereomicroscope is needed to identify many plants, especially aquatic grasses and sedges. Observe vegetative and floral structures by dissecting them, under magnification, with forceps and fine needle probes. Preferably identify to species. Numerous references are available to assist in identifying aquatic macrophytes (see Section 10900E.4). 3) Plant label—An important part of the species collection is the label that identifies the plant, the collector, the location of the collection, and the date of the collection.19 Attach label to the sheet with the mounted plant. The mounted plant is a permanent record that is most useful when placed in an herbarium where it can be utilized by others. 4. Data Presentation Express fresh weight (wet weight), dry weight, and ash-free dry weight as grams or kilograms per square meter. Data are best expressed as ash-free dry weight of total biomass. Determine significant digits for dry weight and ash-free dry weight from the accuracy of the scale used to obtain fresh weight: do not use more significant digits than those used for expressing fresh weight. Report pigment as grams chlorophyll per gram dry plant matter and caloric value as gram calories per gram dry plant matte