G(x) O or x 0 1 AG
advertisement
 O or x 0 1 AG")
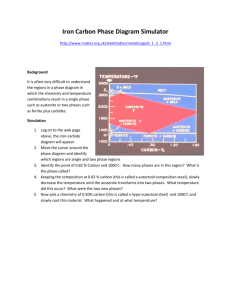
Chapter 8. Phase transitions
When chemical reactions occur, the system makes transition among multiple minima at
the molecular level. The figure below illustrates the molecular connection between free
energy G, its derivative Δ G, and the free energy as a function of reaction coordinate,
G(x).
When an infinitesimal amount of A
reacts to form A', ΔG kJ/mole are
released, and G changes by a corresponding amount.
G(x)
A
A'
ΔG
0
1
O or x
Figure 8.1: The free energy G(x) as a function of coordinate (e.g. bond
distance) has two local minima, A’ being lower in free energy. When A
converts to A’, Δ G kJ/mole are released per infinitesimal amount of
reaction dξ. Note that both x and ξ can vary between 0 and 1, but the
meaning is very different: when all of the substance is in state A, x=ξ=0,
and when it is all in state A, x=ξ=1. However, while the substance can
have ξ=0.5 when the reaction is half completed, almost none of it will
ever be at x=0.5. Rather, half will be at x=1 and half at x=1.
So far, we have treated macroscopic pure substances and mixtures as though they had a
single minimum in the free energy as a function of reaction coordinate. However,
thermodynamics does not forbid multiple minima even at the macroscopic level, and can
be used to make comparative statements about the minima.
Definition: a phase is a local minimum in the free energy surface.
Unlike ordinary chemical reactions, transition between phases can occur even when only
one pure substance is present:
A(1) A(2)
For phase transitions, we call the reaction coordinate “order parameter.” The superscripts
refer to the phases.
Definition: an order parameter is a thermodynamic variable scaled to zero in one phase,
nonzero in an(other) phase.
Example: a gas-liquid transition order parameter
O = ρ − ρgas ; or O =
ρ − ρgas
ρliq − ρgas
; in general
X − X (2)
X (2) − X (1)
O = X − X (2) ; or O =
Thermodynamics cannot make statements about the details of the barrier (e.g. its height),
or how fast a transition can occur. The transition itself is a rather delicate matter – it
violates P2 since temporarily ΔG > 0 if the transition occurs at constant T,P.
The solution to this dilemma: if climbing the barrier were required of the entire
macroscopic system, phase transitions could indeed never occur. Rather, a small portion
of phase (1), called a nucleus, fluctuates to look like phase (2). The nucleus is at the
barrier top in fig. 8.1. From this nucleus, phase (2) grows downhill in chemical potential
if it is at lower free energy (P2). Thus the transition itself relies on microscopic
fluctuations, and microscopic information is required to determine the barrier height,
which is rather small. We need statistical mechanics to compute rates.
If we are interested only in equilibrium, not how we get there, we can treat the phase
transition like any other chemical reaction: A(1) and A(2) interconvert to yield mole
(1)
(2)
& neq
numbers neq
or concentrations or pressures that minimize G:
A(1) A(2) ⇒ G(T , P, n (i ) ) = µ(1)n (1) + µ(2)n (2) = µ(1)n (1) + µ(2) (n − n (1) ) where n = const.
dG = 0 ⇒ µ(1)dn (1) + µ(2)dn (2) = (µ(1) − µ(2) )dn (2) = 0 ⇒ µ(1) = µ(2) at equilibrium
µT,P
µ(2)
µ(1)
T
P
Fig. 8.2: Chemical potential of two phases as a function of
temperature and pressure (Gibbs ensemble). At high T, phase 1 is
more stable, at (3)-(5) both phases coexist, at low T phase 2 is
more stable. In this diagram at high T and P, the two chemical
potentials become degenerate and only one phase exists at (6).
Definition: a 1st order phase transition occurs when the chemical potential difference ΔG
between two phases separated by a barrier vanishes.
Definition: a critical phase transition occurs when the chemical potential barrier between
two phases just vanishes.
The figure below show diagrams of the chemical potentials at the local minima, and of
the resulting phase diagram:
µ
P
0
1 O
µ
0
triple
point
A(1)
1
O
1 O
0
0
µ
1 O
A(2)
A(3)
µ
µ
0
1
O
µ
0
1 O
T
Fig. 8.3 Phase diagram showing the nature of the chemical potential
at various points. It is a double well for first order phase transitions,
and merges into a single quartic well at the critical point (6).
Important properties of 1st order O phase transitions illustrated by points in figure 6:
i) Cooperativity
(2), (3), (4)
Although 1st order phase transitions can be thought of as ‘chemical reactions’ they are
far more cooperative than chemical reactions. The reason is that the “order parameter”
reaction coordinate corresponds to many degrees of freedom changing in concert, not just
a few bonds. Once a nucleus has formed and the top of the barrier is reached, all the
molecules in the less stable phase ‘react’ by adding to the nucleus. Although Δµ(24 ) per
degree of freedom may be very small, N Δµ(24 ) where N ~ O(1020) may be very large, so
equilibrium switches rapidly.
ii) Coexistence curve
(3), (5), (6)
Everywhere along the coexistence curve, µ(1) = µ(2) ⇒ dµ(1) = dµ(2) as shown in
chapter 4 and again above. From the Gibbs-Duhem equation
s (1) − s (2) Δs (12)
Δh (12)
⎛ dP ⎞
⇒ −s dT + v dP = −s dT + v dP or ⎜ ⎟ = (1)
=
=
⎝ dT ⎠ µ v − v(2) Δv(12) T Δv(12)
iii) Discontinuity of thermodynamic variables in 1st order phase transitions (2),(3),(4)
From the plot of µs, derivatives of the chemical potentials are clearly discontinuous
across the coexistence curve, according to ii).
(1)
⎛ ∂µ ⎞
⎜⎝
⎟
∂T ⎠ P
(1)
(2)
⎛ ∂µ ⎞
−⎜
⎝ ∂T ⎟⎠ P
side 1
(2)
side 2
⎛ ∂µ ⎞
⎛ ∂µ ⎞
= Δ⎜
= Δs (12) and Δ ⎜ ⎟ = Δv(12)
⎟
⎝ ∂T ⎠ P
⎝ ∂P ⎠ T
If Δs = Δh / T is discontinuous, so is Δh (12) . To account for the latent heat
Δh (12) released during the phase transition, c p must have a δ − fucntion type
(12)
(12)
discontinuity. In reality any sample of substance is always finite in size and not perfectly
pure, so cP just has a very narrow sharp spike:
T
∫ c dT = H − H
P
0
T0
cP (T)
T
T0
T trans
H(T)
Δh(12)
{
T
T0
T trans
Fig. 8.4: relationship between heat capacity and enthalpy. At the
transition temperature where µ 1=µ2, heat is released while T
remains constant. Thus the heat capacity has a sharp spike at the
transition temperature.
iv) Number of coexisting phases
(1) vs. (3)
Usually, only two phases coexist. However, more phases can coexist (have the same
chemical potential at the same T, P). There is however a limit. The maximum number of
coexisting phases is given by Gibbs’ phase rule:
f =r−m+2
f = number of independent variables, r = number of components=substances, m = number
of phases
Proof: f = total number of variables – number of constraints
T , P, and r of the χ
r
(1)
q
µi(1)=1r = µi(2) = ...µi(m ) , and ∑ χ q(i ) = 1
in
q =1
each of m phases = 2 + rm
− r(m − 1)
−
m
Example: for one component, the maximum value of m is achieved when f = 0:
0 = 1 − m + 2 ⇒ m = 3 ; at most three phases can coexist, and it occurs at a single point in
the phase diagram, since f=0. Hence the name ‘triple point’ point for point (1) in figure
8.3. Two phases (m = 2) leave f = 1 and coexist on a line, for example P(T) in figure 8.3.
One phase leaves f = 2 independent variables, so phases of single component systems
generally occupy two-dimensional patches on the P(T) phase diagram.
Example: for r = 2 substances and m = 2 phases, f = 2 − 2 + 2 = 2 . The phase transition
reaction would be
A1(1) + A2(1) A1(2) + A2(2)
where subscripts refer to different substances (e.g. A1 = methanol, A2 = water) and
superscripts refer to different phases (e.g. (1)=liquid, (2)=gas). There are now two
degrees of freedom, so instead of a coexistence line P(T), there is a coexistence surface.
Picking T , P, χ1 as variables, P = P(T , χ1 ). (χ2 = 1-χ1 is not linearly independent of χ1.)
A sketch of the coexistence surface and a 2-D cut through it is shown below.
T
T
gas
(a)
liq+gas
(b)
liq
P
χ1
Fig. 8.5 Liquid-gas coexistence surface (shaded) when there are
two components and two phases. (a) is the boiling point of pure
substance “A2”, (b) is the boiling point of pure substance “A1”,
whose mole fraction is given by χ1. An important question we
would like to answer is what the composition of the liquid and of
the gas is when we are at the point • . This can be answered by the
lever rules.
v) Unstable equations of state and lever rules
In a diagram like the above, it would be nice to have a rule for the composition
(l )
X A & X A(g) in each phase. Such ‘lever rules’ exist and are best derived by analyzing the
equations of state.
The prototype such equation is the Van der Waals equation of sate, a modification of the
ideal gas law:
2
nRT
⎛ n⎞
P=
− a⎜ ⎟
⎝V⎠
V − nb
We already saw in a homework set how additional terms to the ideal gas equation can
arise. By solving the full W = N!/[(N-M)! M!] without the assumption of small number
of particles compared to volume, the lowest order correction increased the pressure and
was of the type +a’(n/V)2. Expansion to all orders actually yields the 1/(V-nb) term,
whereas the net -a(n/V)2 in the van der Waals equation comes from intramolecular
attractions and has a (-) sign. We will later derive the van der Waals equation of state
and related equations using statistical mechanics. In thermodynamics, we must take it as
a given.
P
T2
T1
µ
spinodal
point
0
1 O
F
C
µ
B
E
D
spinodal
point
0
1
O
A
µ
0
1 O
V
Fig. 8.6 Plot of the van der Waals equation phase diagram. It contains a
region where κ<0. which is unphysical by P2 and the proof in chapter 7.
Instead, the sample begins to coexist in two phases, one corresponding
to the dense branch of P(T) on the left, one to the low density branch.
At high T, this region eventually disappears at the critical temperature Tc.
Consider the figure above. In the region CD,
⎛ ∂P ⎞
⎜⎝
⎟ > 0 ⇒κ < 0.
∂V ⎠
This violates the stability criterion entirely and cannot be realized as a metastable state.
Points C and D are known as spinodal points: it is where the barrier in the free energy just
vanishes and two stable phases cannot be maintained separately.
Even regions BC and DE are not usually realized, as seen in figure 8.7, which is a 1D projection of figure 8.2: µ(1) = µ(2) at B and E, and the gas (1) is converted to a liquid
(2) while the pressure is unchanged and the volume gradually decreases (line BE): µ(gas )
first increases; at B and E µ1 = µ2 and µ remains unchanged; finally µ increases with a
smaller slope in the liquid state.
⎛ ∂µ(1) ⎞
⎛ ∂µ(2) ⎞
or v(1) > v(2) (liquid is always denser than gas).
⇒⎜
>
⎟
⎜
⎟
⎝ ∂P ⎠ T ⎝ ∂P ⎠ T
By Le Chatelier’s principle, you must go from the larger slope ∂P/∂V to the smaller slope
when pressure increases.
µ = ∫vdP + f(T)
C
F
µ(2) = µ(liq)
O
D
B,E
V
B
area 1
µ(1) = µ (gas)
C
A
O
D
area 2
E
P
P
Fig. 8.7 The chemical potentials of gas and liquid intersect: as P is
increased, both chemical potentials rise, but the one of the gas rises
faster because its volume decreases more rapidly with pressure
than the liquid volume: gases are more compressible than liquids.
The surfaces µ(ι)(P) intersect at points B and E, shown here in the
µ-P diagram but also shown in the P-V diagram in fig. 8.6. Inset:
lever rule integral, like fig. 8.6 rotated by 90° angle.
Note that κ > 0 from B to C: the gas is stable in the sense that d 2 µ(1) > 0 , even though
the liquid chemical potential µ(2) minimum is lower. To get over the barrier, a nucleation
site (liquid droplet) must first form by fluctuation. Then the whole sample can liquefy by
condensing on the nucleus. Nucleation is the result of density fluctuations, which
increase from B to C until Δρ ~ ρliq and nucleation occurs without barrier. The
likelihood of density fluctuations cannot be computed in the framework of postulates P0
– P4; we will need statistical mechanics. Between B and C we call the state
“metastable:” it is a local free energy minimum, but not the global free energy minimum.
Only the lack of formation of a nucleus prevents part or all of the substance in going from
B to E.
Now to the famous lever rule. The line BE has a simple geometrical interpretation:
µE = µB ⇒ ∫ v(P)dP = 0
at constant temperature. This is because µ is a state function, and because the GibbsDuhem relation relates µ to the integral over VdP. It follows that
D
0
C
B
E
D
0
C
∫ vdP + ∫ vdP + ∫ vdP + ∫ vdP = 0
or
0
E
C
C
D
D
B
0
∫ vdP − ∫ vdP = ∫ vdP − ∫ vdP
or area 1 = area 2 in the V-P plot inset in figure 8.7. Thus the onset of the phase
transition assuming the barrier can be crossed the moment µ1 – µ 2 is when the areas of
the equation of state below and above the line segment EB in figure 8.6 are equal.
Once we know where the line of phase coexistence lies, it is easy to derive the lever
rule. At some volume V during condensation (x-axis in figure 8.6, y-axis of inset in fig.
8.7)
v = χ1v1 + χ 2 v2
⇒ ( χ1 + χ 2 )v = χ1v1 + χ 2 v2
χ (1) v − v(2) v − v(liq)
=
=
χ (2) v(1) − v v(gas ) − v
In words: from A to B, the gas compresses as we decrease V. Finally, the volume of the
gas reaches vB=v(gas). At B, a liquid phase begins to appear, with smaller molar volume
vE=v(liq). As we reduce volume further, more liquid appears and gas disappears.
Therefore the pressure remains constant because vE < vB as we move along line segment
EB. The mole fraction of liquid to gas increases as V (or v = V/ntot) decreases further.
The ratio of mole fractions is given by the ratio of the lever arms from v to B or v to E,
respectively. Finally, we reach point E, and all gas has been converted to liquid. The gas
lever χ (1) = χ (gas ) ~ v − v(liq) = v(liq) − v(liq) reaches 0 at point E, so all gas has disappeared.
(OK, almost all, as an infinitesimal amount given by a Boltzmann factor remains.)
⇒
The mole fractions are in inverse ratio of the lever arms v(1) − v & v − v(2) . Similar lever
rules apply in the multi-component system we discussed, or if parameters other than P are
held constant:
T
gas
χ1(gas)
liq
χ 1(liq) start
χ1
Fig. 8.8 Lever rule for the phase diagram coexistence surface in
fig. 8.5. The solid horizontal lever arms give the mole fraction of
substance A1 in the liquid and gas. The starting point for the
distillation was “start.”
vi) Critical phrase transitions:
Last, we briefly discuss critical phase transitions. Here we quickly get into a realm where
statistical mechanics is needed because microscopic phenomena are amplified straight to
the macroscopic level.
So far, we avoided point (6), where the phases A(1) and A(2) fuse such that their local
minima become one minimum. At that point, the system switches from having three
places where dG(x)=0, to having only one, and also 2 positive+1 negative d2G to having
d2G=0 and only d4G≠0. The figure below shows what happens when one approaches (6)
along an extension of the coexistence curve, and what the order parameter looks like.
P
O
µ
ε
O~(Tc-T)
µ
liq
µ
0
0
1
1
O
O
Tc
µ
gas
0
0
T
1 O
1 O
T
Fig. 8.9 d G(x)=0 at the critical point; above Tc, the system has one
quadratic minimum, below Tc, it has two quadratic minima and a
quadratic barrier. Right: two values of the order parameter (e.g. 0
and 1) merge to a single value at the critical point. Near the
merger the order parameter (e.g. density) obeys a power law.
2
At (6):
-
a single minimum in free energy splits into two minima: only one local
minimum µi (T , P) exists above Tc, two µ(1) and µ(2) below Tc.
d 2G = 0 (i.e. G is a function of the type aO 4 − b(T )O 2 ; b switches from
positive to negative at Tc, and exactly equals 0 at Tc.
the order parameter is zero above Tc, and can take on one of two equally
likely values below Tc (as long as we stay on the coexistence curve).
Because d 2G = 0 , thermodynamic stability analysis fails at Tc. The macroscopicity
assumption of thermodynamics fails: N N usually implies small fluctuations so
δ S 2 S, δU 2 δU, δV 2 δV, etc. Because G is flat to 4th order, fluctuations
become comparable to the difference in the order parameter between phases.
δρ 2 ~ ρliq at the critical point: bubbles of fluid of any density from gas to
Example:
liquid coexist.
As a result, quantities such as c p diverge not just at a point, but over a broad range of T.
Thermodynamics is a theory of averages and breaks down when δ 2 x ~ x . It cannot
predict critical points or the values of exponents for diverging parameters or order
parameters. It cannot predict certain constants in equations of sate. We need
STATISTICAL MECHANICS