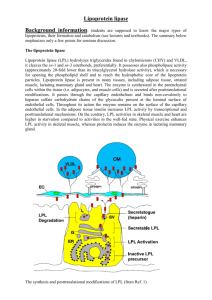
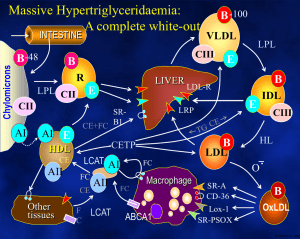
Molecular Pathobiology of the Human Lipoprotein Lipase Gene
advertisement

ISSN 0163-7258/96 $32.00 PI1 SOl63-7258(96)00005-S Pharmacol. Ther. Vol. 70, No. 2, pp. 101-135, 1996 Copynght 0 1996 Elsev~er Science Inc. ELSEVIEK Editor: D. Shugur Associate Molecular Pathobiology of the Human Lipoprotein Lipase Gene Ven Murthy,*t Pierre Julien,+ and Claude Gag& *MOLECULAR BIOLOGY LABORATORY ON HUMAN DISEASES. DEPARTMENT OF BIOCHEMISTRY, FACULTY OF MEDICINE, LAVAL UNIVERSITY, STE-FOY, QUEBEC GIK 7P4, CANADA +DEPARTMENT OF MEDICINE AND CENTRE DE RECHERCHE MALADIES SUR LES Lll'lDlQUES.LAVALUNIVERSITY MEDICAL CENTRE,STE-FOY,QUEBEC GlV 4G2,CANADA is a key enzyme in the metabolism of lipids. Many ABSTRACT. Lipoprotein lipase (LPL; E.C. 3.1.1.34) diseases, including obesity, coronary heart disease, chylomicronemia (pancreatitis), and atherosclerosis, appear to be directly or indirectly related to abnormalities in LPL function. Human LPL is a member of a superfamily of lipases that includes hepatic lipase and pancreatic lipase. These lipases are characterized by extensive homology, both at the level of the gene and the mature protein, suggesting that they have a common evolutionary origin. A large number of natural mutations have been discovered in the human LPL gene, which are located at different sites in the gene and affect different functions of the mature protein. There is a high prevalence of two of these mutations (207 and 188) in the Province of Quebec, and one of them (207) is almost exclusive to the French-Canadian population. A study of these and other naturally occurring mutant LPL molecules, as well as those created in vitro by sitedirected mutagenesis, indicate that the sequence of LPL is organized into multiple structural and functional units that act in concert in the normal enzyme. In this review, we discuss the interrelationships of LPL structure and its function, the molecular etiology of abnormal LPL in humans, and the clinical and therapeutic aspects of LPL deficiency. PHARMACOL. THER. 70(2): 101-135. 1996. KEY WORDS. Familial lipoprotein lipase deficiency, micronemia syndrome, gene mutations. hepatic lipase, pancreatic lipase, lipase gene family, chylo- CONTENTS 1. INTRODUCTION . . . . . .. . . . . . . . . .. . . 2. THE LIPASE SUPERFAMILY . . . . . . . . .. . 2.1. HUMAN LIPASES AND THEIRFUNCTION . . . . . . . .. . . . . . 2.2. GENEORGANIZATIONOFLIPASES .. 2.3. CONSERVATION ANDEVOLUTION OF LIPASES ANDTHEIR STRUCTURALDOMAINS ......... 3. LIPOPROTEIN LIPASE . . . . . . . . . . . . . . . 3.1. FUNCTIONALANATOMYOFTHE LIPOPROTEIN LIPASE MOLECULE ... 3.2. MOLECULARPHYLOGENYOF LIPOPROTEIN LIPASE . . . . . . . . . . . 3.3. MUTATIONSINVOLVINGTHE NONCODINGSEQUENCESOFTHE HUMAN LIPOPROTEIN LIPASEGENE . . . . . . . . . . .. . . . . . 3.4. MUTATIONS INVOLVING THE CODING SEQUENCES OF THE HUMAN LIPOPROTEIN LIPASE GENE 4. THE CHYLOMICRONEMIA SYNDROME .. 4.1. DESCRIPTION . . . . . . .. . . . . . . . . 4.2. DIAGNOSIS . . . . . . . . . . . . . . . . . . . 4.3. CLINICAL MANIFESTATIONS ...... 4.3.1. SYMPTOMS OF CHYLOMICRONEMIA . . .. .. 102 102 102 103 105 107 107 109 113 114 118 118 118 119 119 4.3.2. SIGNSOFCHYLOMICRONEMIA 4.3.3. ANOMALOUS LABORATORY FINDINGSIN CHYLOMICRONEMIA . . .. . . 4.4. CAUSESOFCHYLOMICRONEMIA SYNDROME . . . . . .. . . . . . . . .. . . . 5. PRIMARY LIPOPROTEIN LIPASE DEFICIENCY . .. . . . . . . . . . .. . . . . . . . . 5.1. HOMOZYGOTESTATEOFLIPOPROTEIN LIPASE DEFICIENCY . . . . . . . . . . . . . 5.2. HETEROZYGOTESTATEOF LIPOPROTEIN LIPASE DEFICIENCY . . 5.3. ORIGIN ANDDISSEMINATION OF LIPOPROTEIN LIPASEGENEDEFECTS INQUeBEC . . . . .. . . . . . . .. . . . . . 6. TREATMENT OF LIPOPROTEIN LIPASE DEFICIENCY . . . . . . . . . . . . . . . . . . . . . . 6.1. IDENTIFICATION AND CORRECTION OFSECONDARYFACTORS . . . . . .. . . 6.2. FAMILY SCREENING AND COUNSELING . . . . . . . . .. . . . . . . . 6.3. DIETARYREGIMEN . . . . . . . . .. . . . 6.4. DRUGTHERAPY . . . . . . . . . . . .. . 6.5. PROSPECTS• FGENETHERAPY . . . . ACKNOWLEDGEMENTS . . . . . . .. . . . . . . . . REFERENCES . . . . . . . . . .. . . . . .. . . . . . . . 120 121 121 122 122 122 124 125 125 125 125 126 127 127 127 ABBREVIATIONS, apo, apolipoprotein; FCH, familial combined hyperlipidemia; HDL, high-density lipoprotein; HL, hepatic lipase; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LPL, lipoprotein lipase; LRP, LDL receptor-related protein; MCT, medium-chain TG; PL, pancreatic lipase; TG, triglyceride; VLDL, very low-density lipoprotein. ~Corresponding author. V. Murthy et al 102 1, INTRODUCTION Lipoprotein lipase (LPL; E.C. 3.1.1.34) is the enzyme responsible for the influx of free fatty acids into peripheral tissues for storage in the form of triglycerides (TGs) or as a source of energy. It is known that the action of LPL on TG-rich lipoproteins promotes the exchanges of lipids between lipoproteins and that LPL is, thus, indirectly involved in the maturation of the majority of plasma lipoproteins (Brunzell, 1989). There is evidence to suggest that LPL has a contributory role in the manifestation of a number of pathologic conditions related to the metabolism ofTG-rich lipoproteins (Bergeron et uI., 1991). Dysfunction and deficiency of LPL activity thus has been associated with the pathogenesis of various dyslipoproteinemias and with the production of atherogenic particles, such as intermediate-density lipopro- teins (IDL) and low-density lipoproteins (LDL) (Brunzell, 1989; Eckel, 1989). Because of its physiological importance and its possible role in lipid-related pathologies, LPL has been intensively examined in many in s~ivoand in vitro studies in both human and animal models. The functions of LPL and its regulation have been high- lighted in a number of excellent and detailed recent articles (Borensztajn, 1987; Brunzell, 1989; Eckel, 1989; Bensadoun, 1991; Auwerx et al., 1992; Braun and Severson, 1992; Dolphin, 1992; Lalouel et al., 1992; Santamarina-Fojo, 1992; Wang et (II., 1992; Olivecrona and Bengtsson-Olivecrona, 1993; Santamarina-Fojo and Dugi, 1994). The objective of the present review is to discuss the molecular basis of various LPL deficiencies known to occur in humans and to provide an assessment of the possible therapeutic approaches that are currently available for the treatment of these pathologic conditions. (PL; EC. 3.1.1.3). All three enzymes hydrolyze lipid emulsions and have similar aqueous-lipid interfacial catalytic activity. The three lipases show several individual characteristics, as well as differences in their properties and physiological functions (Table 1). LPL is synthesized in the parenchymal cells of several tissues, including skeletal muscle, heart, adipose tissue, lung, lactating mammary gland, etc. (Borensztajn, 1987). It is expressed transiently in the embryonic liver, but not in the adult (Vilaro et al., 1988; Gimenez Llort et al., 1991). It is transported from the site of synthesis to the luminal surface of vascular endothelia, where it is anchored by ion interaction with heparin sulfate proteoglycans and/or by glycosyl phosphatidylinositol (Braun and Severson, 1992). It can be released from the bound form into the circulation by the administration of heparin. This property has been widely used to release LPL from cell membranes and to purify LPL by heparin-Sepharose affinity chromatography (Bengtsson and Olivecrona, 1977). In its active form, LPL is a glycosylated noncovalent homodimer (Osborne et al., 1985) chat reversibly dissociates to form inactive monomers under physiological conditions, as well as under the effects of pH, ionic strength, and temperature (Olivecrona and BengtssonOlivecrona, 1987). For enzyme activity, it has an obligatory requirement for apolipoprotein (apo) CII, a small protein of 79 amino acid residues. Each LPL subunit contains a site for heparin binding, as well as a site for interaction with ape CII. The physical and kinetic properties of LPL enzyme have been reviewed in detail by Olivecrona and Bengtsson- Olivecrona (1987). The enzymatic activity is optimal at a pH between 8.0 and 8.5 and it is stabilized by the presence of lipids or by binding to lipid-water interfaces and detergents, such as deoxycholate. Although LPL is activated by its cofactor, apo C-II, it is inhibited by various factors, including fatty acids, apo CIII and, possibly, apo E. It is inhibited 2. THE LIPASE SUPERFAMILY 2.1. Human Lipases and Their Function Apart from LPL, there are two other lipases with important also by high salt concentrations (1 M NaCI), an observation that is used to differentiate between plasma LPL and HL lipolytic activities: hepatic lipase (HL) and pancreatic activities. The later remains unaffected TABLE lipase 1. Properties of the Enzymes of the Human Lipase Family HL LPL Site of synthesis Site of action Subunits Glycosylation Binding to cell surface Released into circulation Activators Inhibitors Aqueous-lipid interfacial activity Lipoprotein-ligand activity Substrates for lypolysis Parenchymal cells Capillary endothelia of several Homodimcr + Glycosaminoglycan links By heparin Apo CII Apo CIII and possibly apo E f + Chylomicrons, VLDL Enzyme TG Specificity under these condi- activity lipasc, Sri-1 and minor phospholipase Sri-3 ester bonds tissues Hepatocytes Liver sinusoids M onomer + Glycosaminoglycan By heparin Possible stimulation PL links Exocrine pancreas Duodenum Monomer + Free by apoE Colipase + + Chylomicron remnants, IDL, HDLL TG lipasc, phospholipase Sri-1 and Sri-3 ester bonds + Alimentary TGs TG lipase, variable phospholipase Sn-1 and Sn-3 ester bonds Human Lipoprotein Lipase Gene 103 tions. In vitro, LPL can hydrolyze a number such as long- and short-chain of substrates, glycerides, phospholipids, and transports PL and procolipase, along with other pancreatic hydrolases, into the duodenum, where the enzyme completes various synthetic substrates, but at much lower rates than TG (see Olivecrona and Bengtsson-Olivecrona, 1987, for a review). Chylomicrons and very-low-density lipoproteins (VLDL) constitute the major substrates for lipolysis by LPL the hydrolysis lipase (Verger, the PL to the lytic rate (van in viva (Wang et al., 1992). In the resulting of apo CII in LPL activity. Like LPL and HL, PL acts preferentially on the sn-1 and sn-3 bonds of TGs (Dolphin, 1992). reaction, LPL shows a high degree of substrate specificity. The sn-1 and sn-3 bonds of lipoprotein TGs are preferentially hydrolyzed, generating sn-2 monoglycerides and free fatty acids (Dolphin, 1992). The sn-2 bond is attacked subsequently after nonenzymatic conversion to the sn-1 isomeric form. In addition to its lipolytic activity, LPL also fulfills other important functions in lipid metabolism. It recently has emerged that lipoproteins may bind to cell surfaces through lipase-mediated binding, a ligand function independent of the enzymatic activities of the lipases (Beisiegel et al., 1994). The heparinbinding capacity of LPL (shared by HL, but not by PL) may facilitate the uptake and internalization of plasma lipoproteins through specific receptors (Beisiegel, 1995). Both transcriptional and posttranscriptional steps of LPL gene expression appear to be regulated by various environmental, dietary, and developmental factors and by hormones such as insulin, thyroid hormone, and glucocorticoids (Pykalisto et al., 1976; Nilsson-Ehle et al., 1980; Cryer, 1981). In contrast to LPL, the synthesis of HL is confined mainly to liver where it is localized on the surface of liver sinusoids through glycosaminoglycan links (Table 1) (Olivecrona and Bengtsson-Olivecrona, 1993). Treatment with heparin, therefore, leads to the release of HL, as well as LPL, into circu- of alimentary TGs begun anteriorly by lingual 1984). The colipase, which helps to anchor lipid-water interface, does not affect its cataTilbeurgh et al., 1992), in contrast to the role It shows variable phospholipase activity, depending on the animal species. The active enzyme is glycosylated like the other two lipases. LPL and HL share two conserved glyco- sylation sites, whereas the glycosylation site of PL is in a different position (Olivecrona and Bengtsson-Olivecrona, 1990; Ben Zeev et al., 1994). Like HL, PL is active in the monomeric form, but unlike the other two lipases, it is not anchored to membrane surfaces, but acts as free molecules. 2.2. Gene Organization of Lipases The exon-intron organization and nucleotide sequences of human LPL and HL genes have been studied by several investigators (Martin et al., 1988; Cai et al., 1989; Deeb and Peng, 1989; Kirchgessner et al., 1989; Ameis et al., 1990). Some of the salient features of their gene structure are shown in Table 2. The two lipases are situated on two different chromosomes (8~22 for LPL and 15q21 for HL). The size of the HL gene is twice that of LPL, due mainly to the longer introns in HL. The LPL gene contains 10 exons separated by 9 as a introns, and the HL gene has 9 exons separated by 8 introns. Intron 9 is present only in LPL and interrupts the termination codon TG/A. The LPL and HL mRNAs code for monomer. Although glycosylation has been shown to critically affect the secretion and the activity of LPL, it does signal peptides of 27 and 22 amino acid residues and mature proteins of 448 and 477 amino acid residues, respectively. not seem essential for HL catalytic activity (Stahnke et ul., 1991; Ben Zeev et u1., 1992). HL possesses both TG lipase and phospholipase activities (Deckelbaum et al., 1992). It acts on TGs of chylomicron remnants to further reduce their The greater length of HL is due mainly to the larger number of amino acid residues coded by its exons 2 and 8. The LPL lation. HL is a glycosylated protein and is active size, on the TGs of IDL to produce LDL, and on the TGs and phospholipids of the high-density lipoproteins (HDLs) of HDL: to form HDLI. The conversion of HDLL to HDLi is important in the reverse cholesterol transport process, a mechanism believed to be necessary for the protection of extrahepatic tissues from accumulating excess cholesterol. HL has no requirement for an obligatory activator, unlike LPL, which needs apo CII for its function, but the activity of HL may be stimulated by apo E. In contrast, as noted earlier, apo CIII and, possibly, apo E may act as LPL inhibitors. It has been suggested that the opposite effects of apo E on LPL and HL could serve to direct the action of these two plasma lipases toward specific lipoproteins (Thuren et ul., 1992). Thus, the preferred substrates of LPL are the apo CII-rich lipoproteins, chylomicrons, and VLDL, whereas those of HL are the TGs of apo E-containing lipoproteins, i.e., chylomicron remnants, IDL, and HDL2. PL is synthesized by the acinar cells of the exocrine pancreas (Table I), which also synthesize the precursor molecule of its protein activator, colipase. The pancreatic duct mRNA occurs in two isoforms in the human (3.75 kb and 3.35 kb), but only a single isoform of HL mRNA (1.7 kb) is observed. This is due to the existence of two alternate polyadenylation sites in LPL mRNA and only one in HL mRNA. Although the LPL gene is only half the size of the HL gene, each of the two LPL mRNAs is twice as long as the HL mRNA. This difference in mRNA size is primarily due to the very large noncoding exon 10 that is present in LPL, but absent in HL. Apart from these size differences, the other 9 exons of LPL and the 9 exons of HL are very similar in size and are organized in a very similar fashion. Except for introns 1, 4, and 9, all LPL introns are located at sites identical to HL (Cai et al., 1989). There are multiple TATA Box and CAAT Box consensus sequences in the HL gene, but there is one major transcription initiation site for LPL, as well as for HL. Both genes possess potential 5’-elements responsive to glucocorticoids, cyclic AMP, calcium ions, and adipocyte specific enhancer motifs. The gene organization of human PL is not known. However, such data are available for canine PL, whose transcriptional unit is 10 times larger than the mature mRNA and is organized into 13 exons (Mickle et al., 1989). Exon 104 TABLE 2. Organization of Human LPL and HL Genesa Chromosome LPL HL 8p22 15q21 (kb) 30 60 Number of exons 10 9 Number of introns 9 8 9 37.5 Gene length Intron/exon Major ratio mRNA Exon size (kb) 3.75; 1 3.35 1.7 130 nt 275 nt peptideh Uncleaved 43 nt 188 nt Untranslated Signal portion 81 nt: 27 aa Met-!’ - Ala-’ 66 nt: 22 aa Met-22 - Ala-I 6 nt: 2 aa Ala’ - Asp? 21 nt: 7 aa Leu’ - Glu’ Exon 2’ 162 nt: 54 aa Gln’(c/!A) - Thri” 186 nt: 62 aa G~u”(G/AG)- Se@ Exon 3 180 nt: 60 aa Va15’ - GIu”~ 183 nt: 61 aa Val’(’ - Glu”@ Exon 4 111 nt: 37 aa 117 nt: 39 aa Glu”’ - Thr’h” Exon 5 Glulli - Thrli' 234 nt: 78 aa Asn?” 234 nt:78 aa Glyli’ _ Gly”l G~Y”“(G/GG) - Exon 6 243 nt: 81 aa As~?‘?(c/AT) - Lvs”: 243 nt: 81 aa A~~?‘~(G/cc) - Lys jjh Exon 7 120 nt: 40 aa V~~“)(G/TC) - Thr’sl Vd'*"(G/TT) - Exon 8 Exon 9 - Lcu'~"(cT/G) - LyS’” I05 nt: 35 aa L~s~‘~(AA/G)- Gly++” Termination codon Noncoding sequence Exon 219 nt: 73 aa Gln’-‘l’ 183 nt: 61 aa LeUii3(CT/G) 3 nt (TGA) 3 nt (TG/A) sequence Noncoding Number sequence None 49 nt None None 1948 nt None 4’75 499 of aa coded by mRNA Total Signal peptide Mature protein 5’-flanking Start 27 23 448 476 elementsc -43 -188 nt of transcription TATA Box 215 to -210 nt CAAT Box 253 to -249 nt Nuclear factor-A Glucocorticoid Cal+ 111 nt: 37 aa Arg4+‘(AG/A) - Arg”;’ IO” Coding Cyclic 117 nt: 39 aa Thr”‘; AMP -424 to -419 nt -512 to -508 nt - 1332 to ~ 1327 nt -234 -768 to -227 nt to -761 nt responsive -832 to -827 nt ~ 1022 to 1008 nt -560 to -554 nt -577 responsive -242 to -235 nt _ 550 to 543 nt Adipocyte specific i’-flanking elements? t 2952 to + 2957 nt signals Polyadenylation nt to -66 nt to -111 nt binding responsive Poly-A -70 -116 sites +3348 to +3353 nt t2981 nt; +3376 nt to -571 113 to -91 +1525 nt nt to t1531 +1545 a Compiled from Deeh and Peng (1989) and Kirchgessner et ui. (1989) for LPL, and Marnn et ui. (I%?), Cai et al. (1989) and Am& et ul. (1990) for HL. ’ The ammo acids are numbered starting with the first residue (+ 1) nf the mature protein. ’ Codons that are split between two succcss~vc exons and the correspondmg armn~ acids arc shown in parentheses. d The number of nucleotides in exon 10 of LPL ~ncludcs the third nuclrotidc of the i’-terminal sr<q~ codon (TG/A) and up to nr 3376 (the second polyadenylatlon site). ’ The position of the 5’- and 3’-flank’mg eIements areidentltied m the s<ienrlfic ltteraturcusinga wmetv of startmg sites. ~g., the first nucieotide of a given cDNA clone (Wion et ul., 1987) or the transcnpnon initiation site (Deb and Peng, 1989; A meis et al., 1990). Because these reference points are not unique and may be subject to change, we have numbered the nuclcotidea in this xwew using the first nucleotlde of the Initiator codon (ATG) as + I, becaux there occurs only one such codon 111both LPL and HL. nt, nucleotides; aa, amino acids. Human Lipoprotein Lipase Gene 105 1 codes for all of the untranslated mRNA sequence, and Exon 2 starts with the initiator codon ATG and encodes 1990; Winkler et al., 1990). The sequences of these three lipases are aligned in Fig. 1 so as to obtain maximum homol- the major part of the signal peptide. The stop codon, ogy with the minimum number of gaps. The putative struc- polyadenylation signal, and the 3’ untranslated the sequence are contained in exon 13. The presence of an intron immediately upstream of the initiator codon may afford a mechanism for the use of alternate promoters and differential splicing to achieve tissue-specific expression of PL. From a comparison of the intron-exon organization of PL, HL, and LPL, it has been proposed that these three major lipolytic enzymes may belong to a superfamily of lipase genes and may have descended from a single ancestral gene (Kirchgessner et al., 1989). On the basis of amino acid sequence homology, the membership of this superfamily is occasionally extended to include certain conserved regions of Drosophila yolk proteins YPl, YP2, and YP3 (Kirchgessner et al., tural and functional also indicated. The sequence domains common similarity between to these lipases are LPL and HL (53%) is much greater than that between either LPL and PL (35%) or HL and PL (36%) (Table 3). It is reasonable to assume that amino acid sequences that are associated with critical functions in an enzyme protein may be much better con- served than others. This, indeed, appears to be the case for certain functional and structural domains of the lipase family (Table 4). For example, the catalytic triad consisting of serine, aspartate, and histidine is identical and even the regions surrounding the triad residues are well conserved between the members of the lipase family (Fig. I) (Kirch- 1987; Komaromy and Schotz, 1987; Datta et al., 1988; Kirchgessner et al., 1989). The Drosophila yolk proteins, however, show no lipolytic or other activities in common with the gessner et al., 1989). A second series of conserved regions are those identified as the potential lipid binding sites. Two putative N-linked glycosylation sites are present in HL and three lipases in the group. Two other lipase-like enzymes, lecithin-cholesterol acyltransferase and lingual lipase are found to have no significant homology with LPL, HL, and PL and, presumably, do not belong to the same family (Komaromy and Schotz, 1987). LPL, which are conserved 2.3. Conservation of the lid and its structural relation to other parts of the enzyme may be stabilized by the disulfide bridge that links the two conserved cysteines flanking the lid. Other Lipases and their Structural Domains Of the three human lipases, only the three-dimensional (A2 and A4), and only one such consensus sequence is identified in PL (A3) (Hide et al., 1992). The sequence of the lid covering the active site is not well conserved, although the conformation and Evolution of and in the same positions and A5 in Fig. I). However, HL appears to have two additional potential sites where N-glycosylation can occur (Al struc- ture of PL has been established by X-ray crystallography so far (Winkler et al., 1990). The results show the presence of two distinct domains in the PL molecule. The N-terminal domain (residues l-336) consists of a central P-sheet surrounded by ol-helices (the cr/P-hydrolase fold) and encloses the three amino acid residues of the catalytic triad: Ser153, Asp”‘, HisZh4 (in the porcine enzyme, the catalytic triad is numbered Ser152, Asp *7h, HisZh4, because the amino acid residue Serj’ of human PL is deleted in this species). The C-terminal domain (residues 337-449) has a &sandwich fold and contains the main colipase binding site. The active site in the N-terminal domain is shielded by a flap or a lid consisting of an amphipathic helix. On binding to the substrate, the helical lid is supposed to roll back upon the body of the molecule, thus enhancing the hydrophobicity around the active site. This movement of the lid is related to interfacial activation, a phenomenon by which the lipase activity is increased in the presence of lipid-water interface (Ollis et al., 1992; van Tilbeurgh et al., 1992, 1993). Colipase, the protein activator of PL, helps to bind the enzyme to the interface in the presence of bile salts in the intestine that might otherwise interfere with this binding. Similar lids are also presumably present in all three enzymes of the lipase family. The amino acid sequences of human LPL, HL, and PL have all been established either by direct sequencing of selected polypeptide regions or by deciphering from the corresponding cDNA sequences (Wion et al., 1987; Ameis et ui., putative functional structures that do not show marked conservation are the p-5 loop (or hydrophobic flanking wings) and the oxyanion hole, presumed to be involved in controlling access of the substrate to the active site. Proline is an amino acid that can have profound effects on protein structure. The &-tram isomerism of this amino acid generally influences the conformation of proteins. LPL and HL have a striking resemblance in regard to the frequency and distribution of proline residues, but are different from PL in this regard (Table 5). The four proline residues conserved in all three lipases (Pro’60, I’ro1T3, ProZ07, and Pro214 of LPL) are all clustered within a region that contains the lipid-binding domains B3 and B4 and the lid (Fig. 1). There are also other proline residues that are not conserved, but that are present within various lipid-binding domains of LPL: e.g., Pro95, Pro157, and Prolgo. Cysteine residues contribute to protein conformation and stability by their capacity to form covalent disulfide linkages that bring distant regions of the protein molecule into closer proximity. Not all cysteines, however, have such a role (Fig. 1, Table 5). The eight cysteines conserved in all the three lipases participate in the formation of disulfide bridges. One of these, involving CY~*‘~ and Cyszj9 of LPL, encloses the cr-helical flap or lid region of the lipases (Fig. 1). The two additional cysteines that LPL shares with HL, but not with PL (Cys*7 and Cys40 of LPL), are not linked by an S-S bond, but are highly conserved in LPL and HL molecules of different species. Of the six cysteines unique to PL, four are linked by S-S bridges not found in LPL or HL. Of the other two, 106 ++ hLPL:----ADQRRD hHL: -------LGQ hPL: KEVCYERLGC Al + + ++ ---------RTPEDT---AQAVETmL HEMK----TR TE--RPLHIL PWSPKDVNTR ++ FIDIESKFAL SLKPEPFGRR FSDDSPWSGI +++ ++ ___------ + A FLLGETNQ-FLLY-TNENP EDTCHLIPGV --GCQIRINH NNFQEVAA-D 33 45 56 O------o A2 + ++ ++** + + *+ hLPL:AESVATCHFNSKTFMVI H SSLPLVMII Jd hHL: PDTLQECGFN TNRKTRFIIH hPL: SSSISGSNFK D *+ + * *+ GWTVTGMYES GWSVDGVLEN GFIDKGE-EN A Bl +*+ + +++ +** +** hLPL:mVSAGYTK LVGQDVARFI hHL: =IAVRNTR LVGKEVAALL hPL: UQASQNIR IVGAEVAYFV B2 A3 ++**++*+* ****++* ** t ++++ + ++ + + NWMEEEFNYP LD -GYS LGAHAAGIAG S----LTNKK 148 RWLEESVQLS RS SAHVSGFAG SSIGG--THK 163 EFLQSAFGYS PS HVHVIGHS J&AHAAGEAG RRTNG----T 169 AA B3 ++******+* +* *++ hLPL:VNRITGLDJ?A GPNFEYAEAP hHL: IGRITGLDAALFEGSAPS hPL: IGRITGLDPA EPCFQGTPEL A **+*+** * **++**+++ SRLSPDDADF VDVLHTFTBS; NRLSPDDANF VDAIHTFTPF, VRLDPSDAKF VDVIHTDGAP C *** ++*** hLPL:NGGTFQPGm hHL: NGGSFQPGhPL: NGGVEMPGLDF 0 + * ++*+ ++ * ++ RrJaGDVD-OL HGFMIT-OT + ++ ICEAIRVIAF. +* +++*+ * +++* FIVPKLVAALY WIWQMVAALK WLANVCKNLF 0 + *+ + + *++ KREPDS-NV1 SQPAQPVNVG KVE--SVNCI B4 +++ *+ SP--Q HM-GJSVGLK IVPNJaaGMS B5 +*+*+** + +++**+++ VKCSHERSIH LFIDSUNEE ESHERSVH LF-HAG SNHLRSYKNPD .A +++* * ++ ++t +++**+*++* KPVGHVDIYP QPIGHYDFYP QWGHLDFFP + +++* 207 222 229 + NPSKAYRCSS TQSMAYPCGD GF-AGFPCAS +*++++ ++ A5 ** *++ * *++ +++* +*+++ +** +*+* +++++ + + + IGELLMLKLK AFEISLYGTV AESENIPFTL P--EVSTm YSFLIYTEVD IGELIMIKFK TFTMSLLGTK EKMQKIPITL GKGIA-Sm YSFLITLDVD VGDLQMVKFI ---VSLFGNK GNSKQYEIFK GTLKPDS--T HSNEFDSDVD + +++* hLPL:APAVFVKCHD hHL: QEKIFVKCEI hPL: VLLTLTPC-0 ----DSYFSW WDTVQTIIPW --INPTL--- ++ + + + KSLNKKSG-KSKTSKRKIR -----_____ ++ SDWWSSPGFA STGPRHSGLV ------PRVG 448 476 449 +*+*++++ IQKIRVKAGE LKTIRVKAGE ASKIIVET-N +++ +** TQKKVIFCSR TQQRMTPCSE VGKQFNFCSP 266 281 28 8 l- A4 + ++++ hLPL:GTESETHTNQ hHL: ImETPIQT hPL: GKKVTGHIL- + KTRSQMPYKV VTRAQSPFKV DTGDASNFAR 92 105 113 SCRKNRCNNL SCKKGRCNTL PCPSGGCPQM o--a * R---SSKMYL K---SKRLFL TNDVGQKFYL *** VVDWLSRim LVDWITLAm CVDWKGGSm 0 hLPL:KEAFEKGLCL hHL: MNSFSQGLCL hPL: YNVFTANKCF d hLPL:WKS------hHL: WENSAVWANV hPL: WYNNV----- GYEINKVRAK GYHVRQEPRS GHYADRYPGK + + FHYQVKIHFS YHYQLKIQFWRYKVSVTLS +++ 323 337 348 381 396 402 + + EKVSHLQKGK NTDDLLLRPT ETV----REE 430 456 441 Human Lipoprotein Lipase Gene 107 TABLE 3. Amino Acid Sequence Homology in Human Lipases Homology Lipases (“4 LPL and HL LPL and PL HL and PL ,’ From Hdc 53 35 36 and Divergence Dayhoff distancea (W 72 132 130 er ui. (1992). tions of the lipases. On the basis of these data, a number of structural domains with possible specific functions have been identified in the LPL molecule (Table 4). Thus, by analogy with PL, LPL is considered to be organized into two large and distinct amino terminal (N: residues 1-312) and carboxy terminal (C: residues 313-448) structural domains. The N-domain is the seat of many important activities of LPL, including catalysis, and the C-domain appears to be implicated in such functions as the initial interaction with lipoproteins one (Cys’02 of LPL) is situated within the sequence bordered by one of the unique S-S bridges of PL, and the other (Cys’HJ of LPL) is part of one of the presumed lipid binding domains (B3, Fig. 1). LPL and HL are much closer to each other than they and the LPL-mediated uptake of lipoproteins by cell surface receptors. The active site of LPL can be considered to be made up of (a) the catalytic triad, (b) the oxyanion hole, (c) the lipid binding site, (d) the lid, and (e) the p-5 loop. Except for the catalytic triad, most residues in the catalytic sites of LPL are to PL in both their amino acid sequence homology and their evolutionary divergence (Table 3). In the course of evo- and PL are hydrophobic and their main chains are not readily accessible for hydrogen bonding. This has the effect of diminishing the affinity of the catalytic site for the phos- lution of the lipase gene gene branched off earlier PL gene has the highest imately the same number phoryl groups of the phospholipids (van Tilbeurgh et al., 1994), thus explaining the lower phospholipase activities of these enzymes, as compared with their TG lipase activities. The catalytic triad of LPL is made up of the three amino family, it is probable that the PL than the other two. Although the number of introns, it has approxof codons as the other two lipases (e.g., 448, 477, and 449 amino acid residues in mature pro- acid residues Ser’jz, Asp’j6, and Hi@‘, teins of hLPL, hHL, and hPL). As seen in Fig. 1, the functional domains of the lipases often coincide with specific in all the three members of the human lipase family (LPL, HL, and PL) (Fig. 1) and in all other species so far studied (Hide et al., 1992). The oxyanion hole in LPL is probably axons. Based on these observations, it has been suggested that the molecular evolution of the lipases could have occurred through gene duplication events, selective loss of introns, and fusion of exons (Kirchgessner et al., 1989; Hide that has also been applied to explain the evolution of other multidomain proteins, such as the LDL receptor (Sudof et al., 1985) and serine proteases et ul., 1992), a mechanism (Rogers, 3. 3.1. LIPOPROTEIN may also show hydrophobic interactions with the lipid a mobile surface loop in the putative three-dimensional structure of LPL that covers the catalytic site and is rearranged to permit the substrate to have access LII’ASE Functional Anatomy the Lipoprotein formed by the main chain nitrogens of Trpis and Leu”‘, which are next to Ser”: of the catalytic triad (van Tilbeurgh et al., 1994). The lipid-binding site constitutes a very hydrophobic groove that probably binds the aliphatic chain of the acyl-enzyme intermediate. Tyry4, ProiS’, AlalSH, and Ile’“’ substrates. The lid represents 1985). which are identical of Lipase Molecule to the catalytic domain. The lid sequence is contained between two conserved cysteines (Cys*‘h to Cysz”), which The structural and functional domains of LPL and HL have mostly been extrapolated from the available physical information on PL. Some of these inferences have been further corroborated by (a) nucleotide and amino acid homologies form one of the four disulfide bridges in the LPL molecule and could have the effect of stabilizing the lid within the protein during its action. Dugi et al. (1992) have identified two highly amphiphilic a-helical structures (residues Asn?ii to Arg2ls and Asp?‘? to Lys?‘” connected by a p-turn) in the existing among the three lipases, (b) analysis of degrees of conservation in different regions of the enzyme molecules, (c) identification of naturally occurring human mutations and characterization of the biochemical lesions involved, and (d) production of in vitro site-directed mutations and lid with opposite polar and hydrophobic faces. In addition to its role in the hydrolysis of TGs and phospholipids, the lid may be essential for specifying the lipase substrate. For example, there are found to be differences in the open (active) conformation of LPL and PL, which may account for their examination different substrate specificities (van Tilbeurgh of their effect on the catalytic or other func- et al., 1994). FIGURE 1. Amino acid sequences of human LI’L (hLPL), human HL (hHL), and human PL (hPL). The amino acid sequences are shown in single letter notation and are aligned according to Hide et al. (1992). Amino acid identity between all three sequences is indicated by an asterisk (*) and identity between any two of the three sequences is indicated by a plus sign (+). The following putative functional sites are underlined: (Al-AS) N-linked glycosylation, (Bl-BS) lipid-binding domain, (C) o-helical lid, and (d) p-5 loop. The three amino acids representing the catalytic triad (serine, aspartate, and histidine) are indicated by solid triangles (A). Amino acids of the oxyanion hole are indicated by open triangles (A). The four cysteine pairs involved in disulfide linkages in all three lipases (0) and the two pairs unique to PL (0) are each connected by continuous lines. 108 V. Murthy TABLE 4. Functional and Structural Anatomy Functional Catalytic domain Amino triad Oxyanion Trp5j, site Lid p-loop site,’ Heparin-binding clusters Cys?‘h up to cysl’q interaction Dimerization van Tilbeurgh YS Lys’WArg?W, 148 2110, A$82 C-terminal residues 56 amino et al., 1994 Wong et ul., 1991; Dichek et al., Bengtsson Olivecrona, 1993 acid van uptake of by cell surface et al., 1992; et ul., 1993 Semenkovich et al., 1990; Ben Zecv et al., 1994; Busca et ul., 1995 site LPL-mediated lipoproteins receptors al., 1992; Lys4”‘, Lys”l) Asn-“-His44-Ser-‘5 Asn~jY_Lys’h@_Thr’hl with lipoproteins 1994 1990; Hide et et al., 1994 Davis et al., 1992; Berryman and Bensadoun, 1993; Dichek et al., 1993; Hata et al., 1993; Ma et al., 1994a; van Tilbeurgh et al., 1994 LYs’O” Arg151 Lys’“‘, Arg4”;, Lys+“-Lys4’+ sitesh et al., et al., 1991a,b; et al., 1992 Yang et al., 1989; Davis et al., 1992; Dichek Arg""_L "i-Lys Tilbeurgh Dugi et al., 1992; Tashiro Henderson et al., 1993 up to Trp”4 &I"', et nl., 1989; Faustinella et al., 1992; Faustinella Winkler et al., van Tilbeurgh 14'_Lys14" LYS LYS Kirchgessner Emmerich van up to ProYi up to Prolh” up to Ile”” up to Gly”q up to Leulj’ Arg:“‘, Initial Reference Leu”’ Glu9’ Pro’57 ArglR; Vallzb Ser*“” Glyj’ Apo CII binding Glycosylation LPL Gene acid residues implicated Ser’32, AsP’~~, His*4’ hole Lipid-binding of the Human et al. Tilbeurgh et ul., 1993; Lookene and 1994 Intact carboxy terminal folding domain Williams et al., 1992; Al-Haideri et nl., 1993; Nykjaer et ul., 1993; Zhang et al., 1994a Prolines Residues: 19, 31, 122, 157, 160, 190, 199, 207, 310, 350, 354, Bruin Cysteines Residues: 27, 40, 216, 239, 264, 275, 278, 283, 418, 438 66, 168, 214, 397, 77, 95, 173, 258, 432 et al., 1994a :’ LyslqY of LPL in the human is changed to Thr m the gutnea pig. h Ser” 1s changed to Thr m chicken, and Lys “” is changed to Asn m the guinea pig. Asn”‘-Pro”‘~Srr”‘, (Won et al., 1987), may not be eflic~ent because of the presence of proline (Marshall, 1974). Using deletion Henderson of charge ments and tion. Ile225Thr tion that The periodicity in the ogy with the &loop rendering occupies Tilbeurgh hole its protein upon into segthe to this func- human LPL muta- region. in the LPL protein Hisi to Trph”. In anal- on opening of the LPL lid, the core of the protein, accessible a catalytically necessary for believed activator, The thus and bringing competent position sequence functional catalysis. One to be responsible apo CII. The domains of them the N-terminal sites other The in the endothelial These heparin-binding presence consensus proteins, of two S-S bridges Cys?“‘) in LPL may confer heparin binding. Additional heparin-binding (in the N-terminal region) Lysj’j, His”” is the site, tetrapeptide of region). Lys”‘q, A noncharged the consensus sulfated and sequence proteoglycans, including and X is a small are found on these and CysL;“regions charged are: Lys’q’, and Lys”“, clusters Lys”“, with Arg’j’ C-terminal Trp’“P up to Trp’“j Trp-Set--Asp-Trp, is also upon LYS~“‘, Arg”Oi, LyP, up to GIYJ”~ (in the tetrapeptide, in apo E and apo B. (Cy~?~‘-Cys?‘~ positively two heparin- and X-B-B-B-X-X- residue sequences activity in addition and for the wall. The hypothetical stability in medi- for the interaction X-B-B-X-B-X charged site LPL. vessel to the sequences, B is a positively residue. and sites are essential correspond consensus neutral implicated apo CII cell wall glycosaminoglycans of LPL where as a pClsn1ble glycosylatlorl has been between of activities for the binding C-terminal with localization B-X, Glu-Glu) interaction heparin-binding of LPL possible in a complex multiple the binding loop site even more is a participant LYS ‘t7-Lys’qH, minimally region that distal and that of apo CII (Lys-Gly ating et al., 1994). and has, therefore, to those the may fold back the active this and catalysis occurring of the lid, the maintenance proximal mobile PL, it is supposed oxyanion LPL within is another parts that for normal is a naturally is located that different shown of the loop contribute p-5 loop structure (van affecting have of the lid is crucial apical residues the mutants et al. (1993) proposed present capable in the with of binding C-terminal Human region. Lipoprotein Lipase Gene The relative importance 109 of these various sites in hep- arin binding is not clear, although it may be presumed that they may have differential activities, depending upon different reaction conditions. As compared with LPL, PL, which has no significant interaction with heparin, shows a more symmetrical charge distribution (van Tilbeurgh et al., 1994). Two putative glycosylation sites have been identified in human LPL. In the absence of glycosylation at the N-terminal domain (Asn” up to Serq5), LPL is completely inactive and the enzyme is not secreted (Ben Zeev et al., 1992). In contrast, glycosylation at the C-terminal domain (Asn3jy up to Thr36’) does not appear to affect either enzyme activity or secretion (Semenkovich Busca et al., 1995). The initial interaction et al., 1990; Ben Zeev et al., 1994; of lipoprotein substrates with LPL, which is a necessary prerequisite for their subsequent change in the enzyme mole- cule, leading to the opening of the lid that normally masks the catalytic triad. This opening exposes the hydrophobic residues of the lid in a process called interfacial activation (Tashiro et aI., 1992) and forms a cleft lined by the hydrophobic amino acid side chains of the LPL backbone or the amphiphilic helices of the lid (Dugi et al., 1992). The fatty acid chains of TGs are thought to bind to this hydrophobic cleft, with the glycerol moiety occupying the oxyanion hole. The hydrolysis of the TG is then brought about with the participation of the catalytic triad. The active form of LPL is a homodimer (Osborne 5. Frequency and Distribution line Residues in Human Lipasesa et al., 1985). Both head-to-head and head-to-tail dimeric forms have been considered. Based on structural analysis of LPL and pancreatic lipase, van Tilbeurgh et al. (1994) have proposed a head-to-tail dimeric form in which the N-terminal domain of each monomer is in contact with the C-terminal domain of the other in such a manner that the heparin binding sites are available for reaction and both lids are free to open upon interfacial activation. The specific amino acid residues in the two monomers that may participate in dimer formation have not been identified. In addition to its action on TGs, LPL may also play a role as an intermediary in the uptake and degradation of lipoproteins by cells. Although LPL, by itself, may mediate the binding of lipoproteins to cell surfaces by the interaction of the free heparin binding sites of the LPL-lipoprotein complexes with the membrane proteoglycans, it appears that cell surface receptors, such as the LDL receptor or the LDL receptor-related protein (LRP), may also have a role in this phenomenon (Williams et al., 1992; Nykjaer et al., 1993). Using various deletions, spanning positively charged amino acids, in the C-terminal region of LPL, Zhang et al. (1994a) found that the binding of lipoproteins to LRP was independent of catalytic function and heparin binding properties of LPL, but was dependent on the region Ile404 up to Lys4’4, which includes the very highly conserved residue Glyq”“. of Cysteine Cysteines Total number LPL HL PL Unique LPL HL PL Common to: LPL and HL LPL and PL HL and PL LPL, HL and PL ;’ Derlvcd 3.2. and ProProlines 10 (4 SS) 10 (4 SS) 20 21 14 (6 SS) 26 0 0 6 (2 SS) 11 10 16 to: hydro- lysis, is believed to be mediated by the carboxy terminal domain of LPL, particularly the region containing the last 56 amino acids (Lookene and Bengtsson Olivecrona, 1993). The interaction of LPL with the lipoproteins is supposed to result in a conformational TABLE 10 8 8 8 (4 (4 (4 (4 SS) SS) SS) SS) 7 6 8 4 from Fig. 1. SS, disulfide linkages. Molecular Phylogeny of Lipoprotein Lipase The amino acid sequences of chicken, guinea-pig, mouse, rat, bovine, and human LPL are shown in Fig. 2. There are two extra amino acid residues at the N-terminus of the mature LPL of bovine, sheep, and chicken. In chicken, there are also 15 additional amino acids following the stop codon of the human LPL. In rat and mouse, there is deletion of the residue corresponding to Asn444 in the human. The sheep, rat, mouse, guinea-pig, and chicken LPL mRNAs code for 28, 27, 27, 17, and 23 amino acids of the signal peptide as compared with 27 in the human LPL. The bovine signal peptide has not yet been determined. The bovine and sheep LPL, both of which have 450 amino acid residues in the mature protein, differ from each other by only one residue in exon 1 (Glys in the ox replacing Arg5 in the sheep). The rodents, rat and mouse, differ from each other in 11 of their 447 residues. In spite of these differences, there exists a very high proportion of identical amino acids (92-94%) and high degree of sequence similarity (96-97%) in the LPLs of the rodents and the ruminants (sheep and ox) as compared with the human (Table 6). The identity and similarity are less in the guinea-pig (87% and 92%) and even less in the chicken (74% and 82%). These differences between the species are consistent with their evolutionary divergence, as determined by the Dayhoff distance (Hide et al., 1992). The proportions of amino acid residues in the various LPL species, identical or similar to the human enzyme, are the highest in the middle exons and they tend to be lower towards the N-terminal or C-terminal exons. The similarity is even greater if only those regions of the LPL molecule are considered that are presumed to subserve specific functions of the enzyme. There are also specific amino acids, such as proline and cysteine, that may have a disproportionate influence on protein conformation and remain unchanged even within certain regions of the LPL molecule that are not generally well conserved. For example, Proi60, Proiij, ProZoi, and Pro214 of LPL are not only conserved in all species of LPL, but also in HL and PL (Figs. 1 and 2). Prolines in positions 95, 157, Exon 1 hLPL: - ------ N- _--__-_fJ-- "--__--___ _-sy_----- TlhLPL: ADQRRDFIDI I bLPL:DRITGGK--R-sLPL:DRITRGK--R---GG---S-rLPL: --AG-m-S-mLPL: gLPL: -NCQK-YT-cLPL:SDPEAEMN-EG- ESKFALRTPE DTAEDTCHLI ----------------se- ------------------- ____----___-------- PGVAESVATC HFNXSKTFM _--T----N---T-_--N- __-----___-----_--- --L-j-j--SN- --L-j-,--SN- -----_---v ----- ------------------ V -----R-m-----S----A N-"------EPD--V-Y-V --QMD-L-Q- N---T----V TKLVGQDVAR _--T----N- +--- -----i---____-,--------A---- hLPL: hLPL: YEVPKLVA ALYKREPDSN VIVVDWLSRA QEHYPVSAGY bLPL: sLPL: rLPL: mLPL: gLPL: cLPL: _--------________-_ _____----------------------- --------__ _____----__------__ --------__ _-------__ -------_-------------____y-_------y__ -Q-------- ----m----K -M---AD_-- -Q----___- ---------K +,--_f(D--- -Q____---- -----N__-_ -a----____ ----L_,____ -----N_--- ----_+__K -------R__ -H---E--D_ -----E___- _--------D -----___-- __A_-e-V-- -Q------A- -----K---M Exon hLPL: hLPL: bLPL: sLPL: rLPL: mLPL: gLPL: cLPL: ---------- -----R--S- 82 YPLDNVHLLG YSLGAHAAGI A A --- G -------------_--- G --------------- 4 -E-------_ AGSLTNKKVN RI ------------------- --D------- -C---T---G NFEYAEAPSR LSPDDADFVD ------------------- ----_____----___--- ------------------- --------._v ---------- ---------- ---------- ---------v ---------- ---------- ---------- -sv-----_- -------m-V ---R--T--S -------T-s ------Q--- ---N-_-__- ---------- -----K-_-- T----D--I- ----______ __f_______ Exon hLPL: R _-Q-_--E_- __-TE---_- -a-ES-L-@- -----S---- 0---T----C EM AIRVIAERGL 5 - -_--__ aI GDVDQLVKCS hLPL: VLHTFTRGSP GRSIGIQKPV GHVDIYPNGG TFQPGCNIGE bLPL: sLPL: rLPL: mLPL: gLPL: cLPL: ---------_ -_--__---_ ---------_ ------------------- ____-----____--------------------------------- ___----____-----_----------------------------- ------------------------------------- ---------------- S-------QD -L---SQK-F _ M------- ----y____- D--------- --I-----__ G_____-L__ -L-L---K-F S _------- -L-__----- -+-------- -I-------- -L___----- K -- -t-------- -------- f i3 hLPL: hLPL: bLPL: sLPL: rLPL: mLPL: gLPL: cLPL: --HT-----N B5 HERSIHLFID A P ---- CA------R_ TH---P---- ______---- ----- SLLNEENPSK AYRCSSKEAF 0 EKGLCLSCRK 0.4 NRCNNLGYEI -_--V----_ ---------- ----N---_- ------____ ----- -_--V----_ _-------__ ----N-m--- ----__---- -----M---- ---------- ---------- ----N--___ ------____ ----- ---------------------------- --------__ ------------Y--K--M ----N-m-----_N-------_NT_--- -_____--------_--------_---_ M ---- V -----___---------V__---------KV S__------NKVRAKRSSK ---------___---------------- -__----_----_------R--T--NT- Human Lipoprotein Exon hLPL: 111 Lipase Gene 6 --------em -- 7 Exon I _____-- I ----__---- ---T-____- ____------ 1 _I@------ I hLPL : MYLKTRSQMP YKVFHYQVKI HFSGTESETH TNQAFEISLY GTVAESENIP FTLPEVSTNK bLPL : ---______- -- ------- _______N-y -____----- ----_----- -- SLPL : ---------- -- ----_-- -------N-y _____----_ ____------ -- ------- rLPL : -________- -- ------- ------NDKQ N------m-m ____------ -- ------- mLPL gLPL : : CLPL : i A2 -----__ -----____- -- ------- ------NGKQ H------s-m ____------ -- ------- ---------- -- ----e-s y----_TT-y ---------- ---------- -- ----A-N --____A__- -- ----_-- --F-KTNV-K VD-p-L---- --LD------ --+___s-- ------__-- l-3 -fi-----___ ---------_ Exon --------- DIGELLMLKL KWKSDSYFSW SDWWSSPGFA IQKIRVKAGE TQKKVIFCSR _N--_----D _G__------ --- -N-------D -G-- t a --- hLPL: hLPL : bLPL : V----- TYSFLIYTEV sLPL: ___--L---_ ---------- --I---__-- -----L---_ ---------- -- ---------- ------- M -- ---------- -----_-s-v _E__------ M -- I ------- : mLPL : ---------- ------- --M_------ p----_-s-v _ER_------ gLPL : ---------- ---------- --ITE-____ -S--GR-T-T -E-_------ CLPL : _F-------_ --- Q-EK-TF--- -N--TPFA-T hLPL : ---------- ---------- hLPL : EKVSHLQKGK bLPL : --M-Y----- SLPL : : : _--------_ DR-------- ---X-__ _____----_ DS-------- ---X--- ----K--__- EAP--___-- : DGS-R-G--E EA-I---- rLPL mLPL -1 ------ ----------- --RV---S-- 9 Exon ------ R --M-Y--__- gLPL: CLPL --- ______ rLPL D ------ 8 V FIGURE 2. Amino acid replacements in LPL sequences of various species and in human mutant LPL. Note that hLPL, bLPL, sLPL, rLPL, mLPL, gLPL, and cLPL refer to mature LPL sequences from human (Wion et al., 1987), bovine (Senda et al., 1987), sheep (Edwards et al., 1993), rat (Bra& et al., 1992), mouse (Kirchgessner et al., 1987), guinea-pig (Enerback et al., 1987), and chicken (Cooper etal., 1989), respectively. The human mutant LPL molecule is indicated by hLPL (in italics). The LPL sequences are aligned according to Hide et al. (1992). The amino acid sequence of the hLPL is shown in full; for others, only those amino acids that differ from the human sequence are indicated at the appropriate sites. The nine coding exons of human LPL are identified by the corresponding numbers at their C-termini and a vertical line following each number. When a vertical line passes through the letter symbol of an amino acid, it indicates that the codon for that amino acid is split between two successive exons by an intronic sequence within the gene. Mutations in the human LPL gene involving amino acid substitutions are indicated by amino acid letter symbols, stop codons by p, frame shift mutations by 0, and silent mutations by a. Specific amino acid or polypeptide regions of the human LPL protein suspected of being involved in the spatial structure and functions of the active enzyme (Table 10) are overlined with the following identifications: (4) N-linked glycosylation; (g) lipid-binding domain; (c) lid; (B) the p-5 loop. The th ree amino acids of the catalytic triad (Ser13Z, As~‘~~, and HisZG1) are denoted by solid triangles (A). Amino acids of the oxyanion hole are indicated by open triangles (A). The four cysteine pairs involved in disulfide linkages are denoted by solid circles (o), and the corresponding pairs are connected by continuous lines. and 190 of LPL are conserved necessarily lines in other in positions LPL by alanine plete 160, to have 168, effect by another tion) or replacement of another similar 397, of pro- in almost com- substitutions and 432 et al., 1994a). amino acid (Type of were found (Bruin amino but not and 310 of human to result but 199, 350, of LPL, substitution 214, 258, reported activity, 173, no significant of proline in all species Site-directed 157, 190, 207, has been loss of catalytic prolines ment lipases. Replace- A substitu- acid by proline (Type B substitution) may both LPL Two naturally activity. mutations are Prol57Arg activate enzyme present in human replaced in the animals with B mutations are Ser266Pro activity have undesirable occurring no evident in humans on LPL and Pro207Leu, (Table LPL (Pro’??, natural consequences Type A human LPL Pro’@, that Proj9i molecules adverse and Leu286Pro. both 7). Certain effect. of which other and Pro432) are of other Examples lead to the inactivation This in- prolines loss of enzyme species of of Type of LPL activity V. Murthv et al. 112 TABLE 6. Amino Acid Replacements in the LPL of Different Species as Compared with the Human Enzymea Exon 1 LPL (species) Exon Signal Uncleaved 2 3 4 5 6 7 8 9 Mature protein 27 2 54 60 37 78 81 40 61 35 448 UK? 4-o-4 0 0 54-2-5 87 91 60-3-l 93 98 37-1-1 95 81-3-O 96 100 40-O-2 95 95 61-2-3 92 95 35-2-4 83 89 450-14-20 92 96 115-3-O 97 78-1-O 99 100 Human Number of amino acids Bovine T-C-NJ % identity % homologyi Functional regionsb 96 100 Sheep T-C-N % identity % homology 28-l-5 79 82 4-o-4 0 0 54-2-5 87 91 60-3-l 93 98 37-1-1 95 Y7 78-1-O 99 100 81-3-O 96 100 40-O-2 95 95 61-2-3 92 95 35-2-4 83 89 450-14-20 92 96 115-3-O Rat T-C-N % identity % homology 27-3-2 81 93 2-o-o 100 100 54-3-5 85 91 60-3- 1 93 98 37-1-o 97 100 78-1-O 99 100 81-2-O 98 100 40-3-2 88 95 61-3-l 93 98 34-o-4 88 88 447-16-13 94 97 115-2-l 97 99 Mouse T-C-N % identity % homology 27-3-2 81 93 2-o-o 100 100 54-3-5 85 91 60-2-I 95 98 37-1-l 95 97 78-0-O 81-I-O 40-I-4 88 90 61-4-3 89 95 34-o-4 88 88 447-12-18 93 96 115-2-O Guinea-pig T-C-N % identity B homology 17-l-16 0 6 2-1-o 50 100 54-4-6 81 89 60-l-4 92 93 37-4-4 78 89 78-4-6 87 92 81-2-I 96 09 40-O-4 90 90 61-2-Y 82 85 34-3-3 82 91 448-21-37 Chicken T-C-N % identity o/ohomology 23-2-21 0 9 4-I-3 0 25 54-6- 16 59 60- 3-4 88 78-8-5 83 70 93 37-1-2 92 95 8 l-4-8 85 YO 40-4-8 70 80 61-7-10 72 84 50-7-26 34 48 465-4 l-82 74 82 Y9 100 100 100 94 97 100 98 100 115-4-10 88 Yl 87 92 I 15-5-h YO 95 ,’ Derived from Fig. 2. ” Onlv those regions of LPL presumed to participate in specific funcm)ns (If rhc CIIZWWare taken into ionslderation (Table 4. Fig. 2). LWhen an amino aud m human LPL 1s coded bv a codon split between two successive axons, due to an ~nrervcn~ng sequrnce (intron), it IS nrbitrarlly assigned to the following cxon in calculating the number of amino acids III each coon. The same rule is applied to other LPL sequences, in the absence of specific mformation on their gene organization. J T 3C I and N refer to the total number of eron ammo acids and the number of ronscrvat1ve or nonconscr~x~v~ replacements in the var~~b LPL m&c&s. cThe bovine signal peptide has nor been idenuficd (Senda er ui., 1957). f Homology is calculated by adding the number of ammo acids that are &ntlcnl and those that represent conservative suhstlturlons. The amino xld changes are classified as conscrx~ative or nonconservative from the Venn diagram of ammo acid subsets &rived by Taylor (lY86), hxxd on Dayhoff’s amino acid changes are arhltrarily defined as those involving not more than a single property. mutation odds matrix (Davhoff et al., 1978). C onservative Substitutions of one amino acld by another differing m more than one propertv are rlarsed as nonconscrwtlw. Extra amino RCI~\ or deletions, compared with the normal human LPL, are also considered nonconservative seems line to be due to the appearance and not because due substitutions amino acids species other of LPL On the other line with at certain present in Ala”’ sites species the effect by proline in the chicken to inactivation LPL of the (Table 7). acid by proappears to Ser’““, Alaq”, and of substitution may not always also on the nature For example, effect its replacement human In view of the importance replacement because of it occurs by threonine leads and stability proteins, residues in the for- of the three-dimensional it is to be expected by amino acid Replacement occurring (Table inactivation SerliZCys, highly S-S in conserved affect Pro’“. (Table 8), results interference with cases of human the introduction of other amino Arg”’ or other to loss of cata- from that Ser’;’ is in close encloses to His?“’ of the catalytic by cysteine of a naturally leading and SerZ5ICys. amino triad. acids, in enzyme of new acids, proximity the e.g., is next to the to an lid sequence, Substitution such and/or of as histidine inactivation, the S-S bridge as such as LPL. is the cause LPL, of of cystcines the structure, enzyme are also known place (Cy~~‘~-Cys!~‘) and is close ing that 8). There Arg243Cys, bridge A$” in human resulting residues cysteine would by serine of Cys”” structures loss or gain of a multidomain mutation lytic activity that substitutions well as the function, or leucine LPL. of cysteine mation LPL are naturally but may depend involved. but by in other molecule (Ser’“‘, proacids, Leu?“” activity substitutions has no apparent LPL, and of an amino in the such amino naturally no loss of catalytic LPL residue S@h occur substitution due to proline, of the other original same proline because However, be entirely of the other some of a new additional loss of the than hand, be well tolerated Se+?). to the indicatthe His”’ 113 Human Lipoprotein Lipase Gene TABLE Various 7. Amino Acid Substitutions Involving Proline LPLs as Compared with the Human Species Site Ser Ax Thr LW Phe Arg Ser Ala LPL species guinea-pig human mutant guinea-pig human mutant chicken rat mouse guinea-pig, chicken Site Ala’ Ala’ Thr12 Se+ Se+” LeuLsh LeuL8” Led”6 Ala”+ Aias3’ Serj”’ Ser”h Ser396 Ala”’ Ala”’ Serqq2 Replacing amino acid k-0 Ile Pro Pro Thr Pro Met Val Pro Thr Pro Pro A rg Pro Val Pro LPL species chicken bovine, sheep chicken human mutant chicken human mutant bovine, sheep rat, guinea-pig chicken human mutant mouse chicken guinea-pig guinea-pig bovine, sheep chicken All amino acid residues are numbered as in hLPL. Conserved proline sites in LPL are 19, 31, 66, 77, 95, 160, 173, 190, 199, 214, 258, 310, 350, and 354. of the catalytic triad may be involved in this action. SerZ5’ forms a part of one of the lipid-binding domains (B5 in Fig. 2). Among the LPL species so far investigated, the guineapig LPL is the only natural molecule that contains 11 cysteines instead of 10. This extra cysteine, which replaces Glu’ of human LPL, occurs very near the extreme N-terminus of exon 1 and, therefore, may be expected to have little effect on enzyme activity. 3.3. Mutations Involving the Noncoding Sequences of the Human Lipoprotein Lipase Gene Although intron nucleotides do not code for amino acids, at least some parts of their sequence, particularly those near the intron-exon junctions, play a critical role in the processing of the mRNA precursor and in the correct splicing of the coding exons. Mutations in the introns, therefore, may affect the maturation and turnover of the mRNA, as well as its size, its translatability, and the nature and number of the protein products TABLE Various formed. The 5’ and 3’ noncoding sequences also contain various regulatory elements, including the transcription initiation site, polyadenylation site, etc. (see Table 2 for a description of the elements present in the human LPL gene), whose activity may be influenced positively or negatively by a given polymorphism. Several mutations have been discovered in the human LPL gene involving transitions or transversions in the nucleotide sequence of introns or in the flanking regions (Table 9). Some of these occur in, or near, the splice acceptor or splice donor sites and are found to interfere with gene expression by creating 8. Amino Acid Substitutions Involving Cysteine LPLs as Comoared with the Human Soecies Amino acid to cysteine other amino acids Cysteine to other amino acids Amino acid to proline or other amino acids Proline to other amino acids Replacing amino acid in Site Cys216 in or Replacing amino acid LPL soecies Site Replacing amino acid Ser human mutant Glu3 Ser’7L Cys guinea-pig CYS human human human human mutant mutant mutant mutant human mutant ArgZ4j Cys Arg2q3 His Arg2q7 Leu Serz5’ CYS LPL soecies All armn~ acid residues are numbered as in human LPL. Conserved cyst&e sites in LPL are 27, 40, 239, 264, 275, 278, 283, 418, and 438. new splice sites and leading to the formation of aberrantly spliced mRNAs. In a recent report, Yang et al. (1995) have described a naturally occurring mutation in the LPL gene, which occurs in the binding site of the transcription Ott-I and results in a highly reduced promoter Although mutations of the coding sequences factor activity. are often revealed by their effects on enzyme mass and/or function, mutations of the noncoding sequences may have more subtle effects and may require indirect approaches for detection and investigation. The occurrence of alternate types of nucleotides in the same position in the nucleic acid sequence, with no concomitant apparent phenotypic differences, is generally referred to as polymorphism. Such polymorphisms in nucleic acids can be detected easily if they lead to alterations in restriction sites. In fact, many of the polymorphisms in the noncoding sequences of the human LPL gene, which have now been characterized in terms of the actual base change, were originally detected by restriction fragment analysis and, even now, continue to be identified in the same manner. A number of studies have been published supporting or refuting claims that certain restriction polymorphisms of the LPL gene may be associated with various lipid-related pathologies in humans. The most extensively investigated of these are the PvuII and Hind111 sites. Variable results have been reported on the possible association between the PvuII polymorphism and plasma TG levels (Chamberlain et al., 1989; Ahn et al., 1993a), which may be due to ethnic differences in test subjects. The Hind111 site is reported to be associated with hypertriglyceridemia (Chamberlain et al., 1989; Ahn et aI., 1993b), levels of total and HDL cholesterol (Heizmann et at., 1991; Mitchell et al., 1994), coronary heart disease (Chamberlain et al., 1989; Thorn et al., 1990; Mattu et al., 1994), and insulin resistance (Ahn et al., 1993a; Cole et al., 1993). Even though the association between a particular LPL polymorphic site and a pathology is confirmed, it remains to be clarified whether this association is due to the LPL gene itself or due to its metabolic or genetic linkage to a separate unidentified gene that may be primarily responsible for the observed abnormality. 114 V. Murthy TABLE 9. Mutations in the Noncoding Location of the Human Alternate Type 1 (SD site) Intron Sequences 1 bp SB ACCgta - LPL Gene Restriction site forms Reference Chimienti et al., 1992; Pepe and Chimienti, 1993 ACCcta - Gotoda 2 (SD site)” 1 bp SB GACGgt Intron 2-Exon 3 (SA site)h 1 bp SB agGTAAC - aaGTAAC Hata et al., 1990a Intron 3 (20 bp from SD site) GAGACT - GAGCCT Gotoda 3 (SA site) 3 (6 bp from SA site) Intron 4 (SD site) 1 bp SB G - Intron 6 (SA site)’ 1 bp SB C-A Intron 6 (1.57 kb from SA site)” cttcagGT Intron 8 (495 bp from SD site) Intron 9’ - tttcagGT (TTTA)n, AAGCTT - TAGCTG Fisher rt al., 1987; Li er al., 1988a; Gotoda et ul., 1989; Oka et al., 1989; Gotoda er al., 199213 PVUII Zuliani and Hobbs, Ahn et al., 1992 n = 9- 13 - AAGCGT 101: element” et al., 1992 Holzl et ul., 1994 1990; Hind111 Heinzmann et al., 1987; Oka et al., 1990, 1991; Gotoda et al., 1992b XbaI Heizmann BstNI Funke BamHI et al., 1991 et cd., 1988 Repeats (CA)n Fisher et al., 1987; Chamberlain et u1., 1989 Li et al., 1988b Hegele et al., 1989a Hegele et al., 198913 Hegele et al., 198913 Narcisi er al., 1993 1 bp SB T-C Yang et ul., 1995 BstI BgIII PstI TaqI Promoter et ul., 1994 Wiebusch Inn-on 9 3’ flank of exon et al., 1992 Nakamura Mb011 A CAGCTG Repeats rt ul., 1992b Wiebusch C -T 1 bp SB Intron Intron Intron 6, at the 3’-end of an alu sequence’ GACGat et cd., 1990, 1991a,b 2-Intron Exon et al. L’This mutatwn, involwng the first nucleotide of intron 2 at the junction of exon 2 and intron 2, creates multiple cryptic splice sites, givq rise to several aberrantly sphced mRNAs. h This represents a transition mutation of the last nucieotlde of mtron 2 in rhe splice acceptor we at the lunction of intron 2 and exon 3. LThis mutation causes aberrant splicing, resulting in the deletion of c‘xons 6 through 9 in the mRNA. d The region containmg the PvuII site resembles the splicing sire in its homology towards the consensus sequence required for 3’-splicing and formation of lariat structure, suggesting that the C - T change may interfere with the correct splicing of mRNA. v A total of five different alleles were found containing this tetranucleotide repeat. ‘Occurs only among Blacks, not in Caucasians, in the populations surveyed. c Two IOCI of CA repeats were found with a total of 10 d&rent alleles. ‘>This mutation occurs at the blnding site of rhe transcriptlon factor Ocr-I (nt-39 starting from the major transcriptional start site or nt-227 from the ATG initiator codon). It is reported to inhibit promotor activity by 85%. SA, splice acceptor; SB, substitution; SD, sphce donor. 3.4. Mutations Involving the Coding the deletions Sequences of the Human Lipoprotein Mutations merous Table in the and thev coding continue 10 lists 71 different that affect LPL protein a majority amino acid of 46, to be detected mutations structure gene at a rapid in the human and function. pace. These 9 missense mutations involving of various of various mutations in which the amino codon acid codon sizes (from codons acid. stops the are altered Although polypeptide the with creation and growth 4 5 silent no change in of a termiimmediately, of interesting presented deductions in Table LPL mutations is higher in exon number many of can be made from The exons and C-terminal 5 (Fig. 3). The of mutations the distributed. in the middle with the N-terminal it is highest contain and even- 10. are not uniformly of mutations the highest 1 bp to 6 kb), 1 bp to 2 kb) and data lead to frame-shifts of translation. compared by a termination sizes (from A number the density include substitutions, and insertions termination The iPL^gene involving of an amino tual are nu- mutations insertions nation LPL are missense 7 deletions coded of the which the substitution codon, regions Lipase Gene functional as exons, exons with are also those that sites of the LPL molecule. Some regions, to be more example, and even some susceptible 3 of the specific to mutations 4 amino acid codons, than residues appear others; for situated be- Human Lipoprotein TABLE 10. 115 Lipase Gene Mutations in Coding Sequences of the Human Tvue Mutation Location LPL Gene Base or codon change Amino acid change - Reference Rouis et al., 1996; Elbein et ul., 1994; Mailly et al., 1995 Reina et al., 1992 Gag& et al., 1994 Kobayashi et al., 1994 Gag& et al., 1994 Foubert et al., 1994 Gotoda et al., 1991a Sprecher et al., 1992 Bruin et al., 199413 Wilson et al., 1993 Wilson et al., 1993 Ishimura Oka et al., 1992a Henderson et al., 1990 Emi et al., 1990a; Ishimura Oka et al., 1992a Gagne et al., 1994; Nevin et al., 1994 Foubert et al., 1994 Gagne et al., 1994 Emi et al., 1990a Deeb et al., 1991 Bijvoet et al., 1994 Ameis et al., 1991 Elbein et al., 1994 Bruin et al., 1993 Ma et al., 1992a Faustinella et al., 1991a Foubert et al., 1994 Bruin et al., 1992 Gehrisch et al., 1994 Hayden and Ma, 1992; Ma et al., 1993 Beg et al., 1990 Haubenwallner et al., 1993 Tenkanen et al., 1994 Emi et al., 1990b; Monsalve et al., 1990; Bergeron et al., 1992; Henderson et al., 1992 Foubert et al., 1994 Dichek er al., 1991; Henderson et al., 1991; Ma et al., 199413 Hata et nl., 1992 Gehrisch et al., 1994 D9N’ MS Exon 2 GAC - AAC Asp - T18” D2lV’ N43S” H44Yi Exon Exon Exon Exon Exon Exon Exon Exon Exon Exon Exon Exon Exon 2 2 2 2 2 3 3 3 3 3 3 3 3 Multiple GAC AAT CAC _ GTC ACT TAC Multiple Asp - Val Asn - Ser His - Tyr _ Y61Ter W64Ter V69Lh Y73Ter R75S W86R K102’ Q 106Te@ 11 bp DL-FS-Ter MS MS MS 1 bp IN Ter Ter MS Ter MS MS 5 bp IN-FS-Ter Ter TAT - TAA TGG - TGA GTG - CTG TAC - TAG AGA - ACT TCG - CGG Multiple CAG - TAG Vlcw Silent Exon 3 GTG E118E N120-Y1219 H136R G139S’@ G142E” v149v G154P D156N” D156G” D156H” PI 57R” E163D s172clj 2 bp IN Silent 4 bp DL-FS-Ter MS MS MS Silent MS MS MS MS MS MS MS Exon 3 Exon 4 Exon 4 Exon 4 Exon 4 Exon 4 Exon 4 Exon 5 Exon 5 Exon 5 Exon 5 Exon 5 Exon 5 Exon 5 GAG AACTAC CAT GGC CGA GTG GGC CAT CAT CAT CCA GAG TCT - A176T’” D180G” H183QlS G188E’” MS MS MS MS Exon Exon Exon Exon 5 5 5 5 GCA GAC CAC GCG - ACA GAG CAG GAG Ala Asp His Gly G188R’4 MS MS Exon Exon 5 5 GGG ATT - (A/C)GG ACT Gly - Arg Ile - Thr MS Silent MS MS Exon 5 5 5 5 5 CGA CAT - GAA CAC Gly His - GAC ATT - GAG ACT Asp - Glu Ile - Ser Gotoda et al., 1991a Reina et al., 1992 MS Exon Exon Exon Exon CCC - CTG Pro - G2092” C216P 1 bp DL-FS-Ter MS Exon Exon 5 5 CGA TGT - GG ACT Multiple Cys - Ser A221L” 1 bp DL-FS-Ter Exon 5 GCT - CT Multiple 1225T2’ MS Exon 5 ATT - ACT Ile - 6 kb DL C239Ter2” R243HL4 Major Ter MS R243P MS Ma et al., 1991; Normand et al., 1992; Levesque et al., 1994 Wiebusch et al., 1992 Hayden and Ma, 1992; Ma et al., 1992 Deeb ec al., 1991; Goroda et al., 1992a; Takagi et al., 1992 Henderson et al., 1993; Ma et al., 1993 Langlois et al., 1989 Takagi et al., 1994 Dichek et al., 1991; Gotoda et al., 1991a; Ma et al., 1994b Ma et al., 1994b 1194T’@ G195E H202H D204E2’ 1205Sz2 P207L:’ DL - GTA Asn Tyr Trp Val Tyr Arg Trp Multiple Gln - Ter Ter Leu Ter Ser Arg Val Val - Ter _ GAA - AC CGT AGC GAA GTC AGC AAT GGT CAT CGA GA(T/C) TGT Glu - Glu Multiple His - Arg Gly - Ser Gly - Glu Val - Val Gly - Ser Asp - Asn Asp - Gly Asp - His Pro - Arg Glu - Asp Ser - Cys - Thr - Glu - Gln - Glu Glu His Leu Thr Exons 3-5 Exon 6 Exon 6 Multiple TGC CCC - TGA CAC Multiple Multiple Arg - His Exon 6 CCC TGC Arg - - Cys V. Murthy 116 TABLE 10. (continued) Mutation Type Location Base or codon change Amino acid change Exon D250N” MS MS MS 6 Exon 6 Exon 6 CCC - CTC TCC - ACC GAC - AAC Arg - Leu Ser - Thr Asp - Asn S251C L252A’: S259R MS MS MS Exon 6 Exon 6 Exon 6 Ser - Cys Leu - Arg Ser - Arg A26lT’: Y262H” Y262Ter’ ’ S266P’” L286P N291S” MS MS -I-U MS MS MS Exon Exon Exon Exon Exon Exon TCT CTG ACT or CCC TAC TAC TCC CTG AAT 2 kb IN’j Duplication Multiple Multlple A3 34Tlh T3521 L353’7 T36lT MS MS 2 bp DL-FS-ter Silent Exon 6Inn-on 6 Exon 7 Exon 7 Exon 7 Exon 8 GCC - ACC ACT - ATT CTG - G ACC - ACA Ala - Thr Thr - Ile MultIpIe Thr - Thr L365V’” MS Exon 8 CTA - GTA Leu - Val W382Trr’” W382Ter’z,“’ Ter Ter Exon 8 Exon 8 TGG TGG - TAG TGA Trp Trp - Ter Ter E410Vf” MS Exon 8 GAG - GTG Glu - Val S447Ter-” Ter Exon 9 TCA - TGA Ser - 3 kb DL’? Majpr DL Exon 9 Multiple R243LL” S244T’O et al. 6 6 6 6 6 6 - TGT - CGG - CGT AG(A/G) - ACC - CAC - TA(A/G) - CCC - CCG - ACT Ala Tyr Tyr Ser Leu Asn - Multiple Thr His Ter Pro Pro Ser Ter Reference Appelman et al., 1994 Hata et al., 1990a Ishimura Oka et cd., lYY2b; Ma et ul., 199213 Wiebusch et cd., 1992 Ma et al., 1994~ Wiebusch et al., 1992; Foubert et al., 1994 Ma et al., 1994~ Rouis et ul., 1996 Funke et al., 1990 Wiebusch et ul., 1992 Foubert et ul., 1994 Wiebusch et al., 1992; Ma et al., 1994~; Reymer et al., 1995 Langlois et ul., 1989; Devlin er al., 1990 Kobayashi et a!., 1993 Wiebusch et al., 1992 Wiebusch et ul., 1992 Reina et al., 1992; Gagne et al., 1994; Gehrisch et al., 1994 Wlebusch et al., 1992; Pepe et UI., 1994 Ma rr al., 1994~ Gotoda et cd., 1991a; Kozaki er al., 1993 Wiebusch ec al., 1992; Previato et ul., 1994 Hara et ul., 1990b; Kobayashi et al., 1992; Kozaki er ucI., 1993 Be&an et al., 1995 The amino acid residue numbers refer to the mature LPL protein as in Wion et al. (1987). &dons are numbered starting from the imtiator codon (ATG), when mentioned in the following text. DL, deletion; FS, frameshlft; IN, insertion; MS, missense; Ter, translation terminarion. ’ Alters TaqI sate. Appears to contribute to hypertriglyceridemia in some families. In e‘~tr” expression shows the presence of both LPL mass and activity. : Leads to the formation of a truncated protein of only 19 residues. ’ Asp” is highly conserved in all species examined, except guinea-pig and chlcken, where it is replaced by Asn or Glu, both conservative subsritutlons. The rrpiacemenr of Asp hv Val residue leads tu a change in charge and generates a new HaeIII site m the exon. in wtro studies show no effect on catalytic activity. i Asn” is thought to be an N-hnked glycosylation site of the mature LPL proteln. Expression studies show that this mutation affects both enzyme actlvxy and secretion. i HIS+~ is a highly conserved residue. His4iTyr represents a nonconser\‘anve wbstltutlon. Found in two Individuals wth famihal combined hypcrlipidemia, but m rlitro studies show no change m catalytic activity. ’ Located in a conserved hydrophobic region of LPL. Expresa~on of the mutant cDNA prepared by site-directed mutagenesis m COS cells shows 80% reducrion in catalytic activity. (ACC ; Found in an LPL-deficient patient of Malaysian descent, it consists of a 6 hp msertior (TGGGCT) at th e site of n single base d&non AC) at the residue Thr”“ in exon 3, causing a frameshlft. This results in a markedly truncated LPL protein rhat rrrminates prematurely in exon 4 with a random sequence of 44 ammo acid residues in the carhoxy terminal portlo”. of the first base of ’ Found in suhlects of Polish, German, and English descent, this mutation leads to a truncated LPL molecule due to substitution with resultmg suhstirution of the Gln codon (CAG) by T. Th c s a me base is replaced by G and by A m normal guinea-pig and chicken LPL mRNA, Gln by Glu and Lys. In rat and mouse, both rhe first and the third bases of this codon are replaced to give Asn in place of Gin. ’ This deletion causes a frameshIft mutation by removing the last two nucleorides of Asn”” (AAC - A) and the hrst Two nucleotides of Tyr”’ (TAC C), producing a truncated proteln of 142 residues, h>Ith substitution of 23 C-terminal ammo acids. This mutation alters the MSEI restriction site. “I Found in an LPL-d&cent indrvidual of Spanish descent. In tiitro mutagenesis shows that this mutation completely abolishes LPL functmn. normal amounrs ” The region surrounding Gly ‘I’ is highly conserved among lipases from dlffcrent specws. Expression m COS-i cells shows that, although of the proteln are produced, It is deticienr m both catalytic acriviry and secretion. ” This mutation affects the last base of exon 4 and is sxuated within the 5’-consensus sequence for splicing. It cgeates a new BfaI restrlction site. It inactive enzyme when expressed occurs withln a conserved P-sheet region close to Asp”‘, which forms a part of the catalytic site and produces catalytlcally in z’ltr” changes of the first base and the third of ” Three d&rent mutations affect Asp”+ of the catalytic triad, SIX”‘, Asp”“, HIS!“, two of them involving the second base of wdon 181 and resultmg in substxution of aspartare by asparagine, histldme, or glyc~ne. All three mutations alter the TaQl restriction site and give rise ta inactlve LPL when expressed m vitro. proline. It alters a PvuII site. ” This muratmn 1s situated next to Asp”” and results in the loss of a conserved Ii Leads ro only partial loss of LPL catalytic activity, compared with several other m~ssensc mutations. ” Also termed LPL-Bethesda, this mutant LPL shows loss of catalytic activity and alrered affinity for heparln. Also alters a SfaNl site. (contmued) Human Lipoprotein 117 Lipase Gene TABLE 10. (continued) ‘7 Although this amino acid substitution is highly conservative, is no effect on secretion or heparm binding. Ii: His’“’ hes close to a putanve lipld-blnding domain and outside the proposed of the LPL molecule active site of LPL, the catalytic (B4, Fig. 2), and KS replacement by glutamine activity is abolished; there leads to charge alteration and production of lnacuve LPL. The mutant gene is thought to be of Russian or Swiss origm. ” Glyl88Glu, highly prevalent in the Province of Quebec, is believed to be a ubiquitous and probably ancient mutation. The frequency of occurrence and distribuuon of Glyl8HArg is not known. Both mutations alter Sau961 and AvaII sites. x Ilc”’ is located wthm a highly conserved, putative lipid-binding domain of LPL (84, Fig. 2). By analogy with pancreatic lipase, it is in the proximity of both the catalync triad and the lid that covers the hydrolytic pocket of the enzyme. Introduction of the hydroxyl group of threonine provides a potential site for phosphorylation or glycosylation that could induce conformational changes. The mutation results in loss of enzyme activity, but retains normal heparin affinity. The occurrence of this mutanon in two different DNA haplotypes is suggested to indicate a multicentric origin. ” Alters a Hincl site in cxon 5. 1LIk? 1s a highly conserved ammo acid and is situated in strand 10 of the proposed three-dimensional structure of LPL (as do Pro’@’ and Asp”+). Substitutions of these residues with other amino acids could disrupt hydrophobic interactions between strands 10 and 11, thus changing the conformation of the central catalytic domain. li This mutation, which appears to have originated in the Northwest region of France, is now found almost exclusively in the French-Canadian population of Quebec. It alters the BslI restriction site. after 223 residues. ” This involves deletion of the thnd nucleotide of the codon for Gly”“, resulting in termination :’ This mutatlon destroys a conserved dlsulfide bridge that may be crmcal for LPL structure and function. “~The frameshift caused by the single bp d&non leads to termination of translation three residues downstream. The deletion of G (GCT - CT) abolishes an AluI we (AGCT). I; Ile’!i is located in the proximal xctwn of the amphipathic surface loop shielding the active site of LPL. L oss of LPL activity by this mutation suggests the rnportance of maintaining charge and periodicity in this region of the molecule. !’ This mutation, which results in total loss of LPL activity or mass, abolishes an HgiAl site and creates an Mbol site. :” Three mutations have been reported that change Arg”’ (which is close to the catalytic triad: Set”‘, Asp’jh , Hi@‘) to histidine, cyst&e, ot leucine. One of them affects the first base of codon 270 and the other two the second base of the same codon. Al1 three mutations alter HhaI and Eco47111 restriction sites. Arg243His was found m Caucasian, Chinese, and Japanese subjects, and Arg243Cy s was found in those of French and German descent. Haplotype analysis indicates two separate origins for the ArgZ43Cys mutation. This, together with ArgZ43His, suggests high mutability of this CpG dinucleotide in the LPL gene. ‘j’ Also occurs near H&l’ of the catalytic wad. ” The location of Asp”” in the proposed three-dimenslonal structure of LPL suggests that it may be involved in a charge interaction wth an cY-helix III the armno terminal region of LPL. The mutation alters a TaqI site. ” L~u’~‘, like the neighboring AspziO and SC?‘, is a highly conserved amino acid and is replacedonly by residues wth similar size and hydrophobicity XI other wld-type lipases. Substwutmn with the positively charged arginine may lead to disruption of spatial structure and results in the observed partial loss of LPL actwty. Pregnancy-induced chylomicronemia is reported to be associated with this mutation or one of the other three mutatmns (Ala261Thr, AsnZUlSer, Trp382Ter) that result in parnal LPL deficiency. ” Two dlffcrent types of substitutions are reported, involving the third base of the codon of the amino acid Ty?, one leading to replacement of tyrosine by hlsndine and the other to termination of translation. The Tyr262His mutant LPL has reduced heparin-binding activity and a destabihzmg effect on dimcr formation. It can be detected by alteranon in an Awl site in exon 6. ” Ser266Pr0, a mutation that inactivates human LPL, is caused by T - C change m the serine codon. T - A change in the same codon results in the suhstltutlon serine - threonine in the normal chicken LPL. ” This mutatwn 1s produced by the juxtaposition of intron 6 to a partially duplicated exon 6 and involves exon-Alu interchange. It is found in subjects of different ancestries and possibly represents an ancient mutation. “’ In contrast to Ala334Thr, Ala334Pro apparently has no effect on LPL activny, because this substitution is found in normal chicken LPL. ‘- The codon involved m this mutation is split between exons 7 and 8, but both the deleted bases of the codon are in exon 7. The frameshift leads to termination after 355 residues. ii It is suggested that this mutation interferes with the correct folding and assembly of active LPL homodimers. “I Two different mutatwns of this codon have been reported, involvmg the second or the thrd base. Both lead to immediate translation termination. ” Glu”“ 1s conserved in LPL and in analogous regions of pancreatic hpase in different species. Replacement of this residue by mutatcon may affect formanon of homodlmers. The A to T base change in the codon generates a Mae111 restnction site. ‘! This represents the longest prematurely terminated natural LPL molecule, with only two residues less than the normal protein at the C-terminus. Less, equal, or higher catalytic activities than normal have been reported for the truncated protein. The mutation generates a MnII restriction site. ” This deletion (2.116 kb) includes the end of intron 8, the whole of exon 9, and about two-thirds of mtron 9. Of the three AIu sequences present in the normal lntron 9, the mutant LPL contains only the third Alu sequence plus the right arm of the second, suggesting that a stem-loop structure hetwccn the end of lntron 8 and an Alu sequence m intron 9 may have been involved in this mutation. tween Gly’j’ and Pro ‘57, 5 of the 8 situated between HiP2 and Gly?@“, and all the 3 successive residues in Asp”5”-Serzi’-Leu?iZ undergo mutations. As~‘~~, a member of the catalytic triad, and Arg2”, which is a close neighbor of another member of the triad, His!“, each suffer three missense mutations resulting in three different amino acid substitutions. Two missense mutations affect each of the following amino acids: Gly’ss, Tyr262, and Trp3sz. One of these, Gly188Glu, is very prevalent in the Qukbec population (Bergeron et al., 1992). 3. The presence of an amino acid residue with unique properties at a given position in the protein sequence may be critical for enzyme activity, and replacement by any other residue may not be tolerated. Such appears to be the case for most of the human LPL mutations. However, in some cases, LPL from other species are found to contain normal variants of the amino acid residue whose replacement leads to mutation in the human LPL. Thus, for example, although Asp9Asn, AspZlVal, Leu286Pro, and Ala334Thr are mutational substitutions in the human LPL, Asp9Gly, AspZlAsn (Giu), Leu286Met(Val), and Ala334Pro substitutions are normal variants in other LPL species (Fig. 2). 118 mucosa and response to the consumption cleared appear from (Cohen, the types lipoproteins, and I is defined the in which lipoprotein the presence from FIGURE 3. Distribution of mutations in different exons of the human LPL gene. The mutation density is defined as the number of mutations in a specific exon divided by the total number of amino acid residues coded by that exon. 4. All LPL mutations the same manner. arin binding 0: these tural domains are also degrees found dimensional A search involving the amino acid alternate acid and, therefore, ceivable the depending the new codon exons and disorders the nucleotide atic studies effects shows three- at least are silent protein specify degree (Table the does not same result the consequence the turnover have been conducted of the rate rate of synthesis of such mutations However, to examine polymorphisms func- preference rate of mRNA action. in the but it is con- may influence possible 10). of acceptability change hence, to amino The protein of codon and, five in regard to the rules of endonuclease of codon acid resi- overall to be affected, on the of mRNA ing the sites that by alter- no system- the quantitative on LPL mRNA CHYLOMICRONEMIA 4.1. Description The chylomicronemia or protein. syndrome Brunzell, presence (>15 is characterized hypertriglyceridemia Chylomicrons are formed are the predomiis marked as various but secondary the causes has been in the plasma more defects. by resulting classification of chylomicrons from Type activity recently, it is The chylomicro- clinical consequences and Bierman, 1982; of Chait and 1992). 4.2. Diagnosis Diagnosis of chylomicronemia ing the appearance (lactescent, white). in the fasting state in the intestinal After for 24 hr, chylomicrons plasma (Type in large amounts, V). With tion and in specialized laboratory 4.4, of heparin into the plasma assay U/kg body circulation i.v. holus which the LPL in S.C. biopsies mutations 1991, 1992b). of adipose syndrome in are by apo of apo E and Vernier, (Table of LPL tissue useful activation In the newborn assay enzyme LPL activ- techniques of chylomicronemia of apo CII, LPL et al., 1987; Huff et ul., 1990), analysis (Hixson gene LPL et ul., 1992). laboratory diagnosis releases mass by immuno- defective (Brunzell Other using injection et cd., 1989). Postheparin ity can also be determined 1992). cause In this procedure, (Peeva et al., such by measurement TG substrate weight), if a catalytically into the plasma of evalua- are available is one after (Brun causes As will be discussed activity. is also used to measure to determine is released usually deficiency 10 min (Type to formulate exact by clinical with a synthetic obtained (50-100 blood The that lipolytic is measured samples LPL plasma accuracy laboratories. layer may frequently and can be confirmed postheparin LPL activity levels can be pinpointed techniques in Section of plasma sufficient surface are also present lactescent TG approach. lipid of chylomicronemia plasma with then, only in detail plasma therapeutic chylomicronemia, VLDL or plasma left in the re- form a creamy or frankly practice, visually, an immediate has been I) or, when a turbid some by observ- (“cream of tomato”) the plasma on clear CII (Connelly by the is easily performed of the blood frigerator genotypes of marked mmol/L). in LPL and VLDL, past, on of plasma manifestations. Type V pattern of molecular (Brunzell into based profiles resulting metabolism, arises chylomicronemia measurements SYNDROME syndrome In the based on the detection the differential THE as well 1973). I-V), clinical chylomicrons in lipid be estimated mutation in the human, in changing the acid by another. according Another to variable the site of a given of the mature of translation may consist struc- mutations of the amino literature is not expected that of protein. functions and of the codon of one amino Different and of LPL. LPL therefore, may be of LPL to different upon local published sequence forms replacement the structure mutations tion, to of the The these probably, and the contribution affected nature the molecule. to affect depending, mutation due within 10). This functions in and hep- causes, et ul., comFredrick- disorders (Types chylomicrons The hr from and in certain to Table of the multifunctional assignment function secretion, differentially (see footnotes a reflection the activity, are affected combinations LPL do not influence Catalytic fasting species. 8-10 results in Types I and V disorders. made based on the appearance nemia lipoprotein disorder, of elevated genetic (Brunzell within in the plasma. associated as a genetic deficiency EXON classified in are, then, large macromolecular presence is present stream syndrome and the electrophoretic Chylomicronemia nant of these have blood fat. They compartment of dyslipoproteinemias the concentrations 0 the chylomicronemia continued et al. (1967) several in of dietary vascular in the clearance plexes and their son transiently 1989). The defects 4. ct ul. V. Murthv 1990), and detection 10) (Monsalve with primary et al., of LPL 1990; Ma et ul., LPL deficiency, chylomicro- Human Lipoprotein 119 Lipase Gene TABLE 11. Symptoms, Signs, and Laboratory is present infants very often after early the first after milk, plications of the disease children, diffuse reported (Black patients have and expected medical It is presumed during the of LPL and/or have a less severe clinic, first diagnosis year of life and “Cream trauma, high (Fig. been make The nal pain, vious are more TGs common complication with or without levels eating be elicited. patients do not with comparatively show when clinical plasma signs, TGs and are very 4). major plasma the 1995). However, of plasma of TGs habits, of chylomicronemia pancreatitis, (Gagne et al., unusually The precise rich reasons is abdomi- and is related 1989). History in fatty foods, for repetitive to the of pre- can often colicky abdomi- tooth abscess, the Two grandmothers diagnosis hyperbilirubinemia conditions leading icterus, for other and in our was unusually g-o- newborn 330- had led to suspicions because than - and often : n 2O4 L;: lo- of the aspect 0 - et al., 1989). are diverse purposes in investiga- (Gagne suspected * that have deficiency which to diagnosis to LPL deficiency, drawn asthma, the clinical of LPL deficiency 60- of LPL blood conditions grandchild in E 5 Physical of the a chylomicronemic without and, later, confirmation pains. LPL ages and, led to diagnosis initiated that their case, the 1 ao- 2 was made screening mononucleosis, of primary they reported In another between appearance 100 disease. of patients the diagnosis have rarely 1983). to avoid of the in 25% in 50% adults et al., learned form are some of the coincidental of the blood to com- childhood and of abdominal of tomato” allowed unrelated signs in young or family evaluation patients. Thus, early have 70%, findings signs of chylomicronemia tion when such experience, (Hoeg was made In almost following “pale? our deficiency fortuitously individuals coincidental pregnancy In levels from have 1994a) such through epistaxis, lower 1993). In young since et al., that 20 years. deficiency. related hemorrhages 1993). These findings TG > 15 mmol/L Fasting chylomicronemia Pseudohyponatremia Pseudohypocalcemia Pseudo-increased hemoglobinemia Normal amylasemia with pancreatitis Pseudohyperbilirubinemia 1989; Brunzell, may 1977). may refrain problems observation is made of 1 and others, pains, and Sprecher, (I’erron diagnosis In our referral et al., Syndrome progression. patients foods growth Sprecher, growth Sometimes, fatty colicky (Black and diagnosis (Sadan gastrointestinal school and older have and under normal feeding, birth have repetitive drinking Laboratory Lipemia retinalis Eruptive xanthomas Hepatomegaly Splenomegaly Abdominal pains with or without pancreatitis Dyspnea Paresthesias Flushing with alcohol Recent memory loss Peripheral neuropathy nemia of Chylomicronemia Signs Symptoms be made Findings measurement 5. I I I I of lipids. 4.3. Clinical Manifestations 4.3.1. Symptoms plasma triglyceridemia the chylomicronemia presence of one or more in Table 11. However, tions is unpredictable equal lence 1995). of each Thus, of these to or greater syndrome the occurrence for each than patient, and/or of clinical and by is no consensus clinical manifestations the signs listed manifesta- some may have no symptoms there with 15 mmol/L, is characterized of the symptoms with severe chylomicronemia (Brunzell, In patients of chylomicronemia. patients or signs on the preva(Gagne et al., FIGURE 4. Plasma TG concentrations and frequency of physical signs of chylomicronemia syndrome in patients with primary LPL deficiency. The frequency was evaluated on the basis of the first clinical evaluation of 56 patients, carried out at the Quebec Lipid Research Clinic (Gagne et al., 1989). The asterisk (*) indicates a significant difference (I’< 0.05) for the plasma TG concentration compared with that of patients with no clinical signs. (Mean * SD). V. Murthy FIGURE 5. Retinal photographs of normolipidemic subject (A) and chylomicronemic ciency (B) showing lipaemia retinalis. Eruptive xanrhomas in female (C) and male deficiency. nal pains and pancreatitis presumably et al., related 1967). Pregnancy ciency (Ma et al., the ceptives, production 1992). Rarely, leads to chronic and diabetes of mellitus pancreatitis (Searles (Howard with fasting, plasma 1992). The 1977). by which is still unknown. to determine of irritation microns in the blood by the of pancreatitis done after the acute rapidly is generally (Chait event; acids (Chait from but the leads to pancreatitis could and lysolecithins PL from capillaries and Brun- resulting of the pancreas the circulating and Brunzell, This possibility elevated patient homozygote for familial LPL defi(D) homozygote patients with familial LPL is supported concentrations in chylomicronemic Other mono- (Chait and Brunzell, Patients of flushing symptoms and 1992). only loss subjects, often However, on treatment with chylomicronemia are unex- of hypertriglycoccasionally consumption of alcohol, present, as com- mechanisms manifestations are rare and, when elicited are memory of dyslipidemia, of the extremities. upon syndrome Chylomicronemic forms as yet and disappear eridemia. 1995). et al., and recent in psychoneuropathic plained plain (Cantin of chylomicronemia with other of numbness involved of abnormally lysophosphatidylcholine or polyparesthesias, well as patients plain patients likely symptoms dyspnea, by the presence of plasma com- but are most often such subtle by a questionnaire. evaluaof pan- understood, chylomicronemia by fatty quantities if hyper- because of pancreatitis Inflammation in large relation overlooked is part of the treatment pathogenesis mechanism The 1973), TG levels decrease as cholelithiasis even sub- et al., is often such causes result et al., 1986; Chait is often It is difficult which contra- and alcohol with fat malabsorption and Levy, 1964; Cameron trimesters, pancreatitis, is the cause or the consequence er ul., creatitis, (Kraus LPL defi- Oral types, (Stuyt repetitive are at increased is increased. estrogenic pancreatitis 1992). incipient and third chylomicronemia of hyperlipidemia indeed, zell, to and Ooi, triglyceridemia tion of VLDL the more they (Fredrickson patients the second the risk of pancreatitis and Brunzell, clinical, may also unmask during especially may increase although levels 1993) and put these risk of pancreatitis when are unknown, to TG-chylomicron et al. be the released 4.3.2. Signs of chylomicronemia. tive xanthomas, nemia that are observed Lipemia retinalis nal vessels, due as revealed are the which result chylo- macrophages 1992). They have intermittently (Table chylomicrons of the accumulation a yellowish of the reti- examination, (Fig. in groups in the skin (Parker erup- 11 and Fig. 4). appearance by funduscopic may cluster retinalis, are signs of chylomicro- refers to the whitish to circulating thomas, Lipcmia and hepatomegaly 5B). Eruptive and become appearance 1970; Brunzell, with xan- confluent, of chylomicrons et al., and is a reddish by the 1995). ring at Human Lipoprotein Lipase 121 Gene the base, and may become pruritic (Fig. 5C and D). Eruptive xanthomas are found mainly on the extensor surfaces of the arms, the back, the buttocks, and the thighs. Lipemia retinalis, eruptive xanthomas, and hepatomegaly should be considered signs of very elevated plasma TG levels, as well as high risk factors for pancreatitis. These signs may also be used to evaluate the efficacy of therapeutic treatment. In contrast to these, splenomegaly often persists in LPL-deficient patients, despite adequate control of plasma TG levels (Gagne et al., 1989; Bertrand et al., 1990*). It is of interest to note that 28% of the patients with splenomegaly had no other clinical sign and their plasma TGs were only 16.7 f 3.7 mmol/L, whereas other patients with TABLE 12. Causes Primary apo CII deficiency Familial LPL inhibitor Autoimmune chylomicronemia Combination of primary hypertriglyceridemia, and/or heterozygous state for LPL deficiency Primq h~pertriglyceridemia Familial hypertriglyceridemia Dysbetalipoproteinemia Familial combined hyperlipidemia 4.3.3. Anomalous laboratory findings in chylomicronemia. In chylomicronemic patients, laboratory anomalies are encountered that are caused either directly by high levels of blood chylomicrons or indirectly due to chylomicron interference in the analytical procedures (Table 11). In vitro hemolysis is often observed when blood is collected from chylomicronemic patients for analysis (Cantin et al., 1995). This is possibly due to an increased fragility of the erythrocyte clinical consequences, but can become misleading in terms of diagnosis. There are other instances where chylomicronemia gives rise to false blood analysis results. Pseudohyponatremia is a well-known anomaly in chylomicronemia. Thus, a plasma level of 15 mmol/L of TGs may decrease natremia by about 1 mmol/L in the presence of normal plasma osmality (Steffes and Frier, 1976). Similarly, a concentration of 12 mmol/L of chylomicron TGs increases hemoglobin measurement by 10 g/L, as determined by the Coulter counter method (Gagne et al., 1977a). This false increase in hemoglobin concentration may be corrected by replacing the plasma with an isotonic solution. Ordinarily, pancreatitis is associated with increases in plasma amylase concentrations. Syndrome Primary LPL deficiency splenomegaly combined with one or more extra clinical signs had plasma TG levels of 50.7 f 16.4 mmol/L (mean ? SEM). membrane resulting from exchange of lipoprotein material between the membrane and the ambient plasma (Cantin et ul., 1992). This hemolysis does not appear to have any of the Chylomicronemia 4.4. factors Common secondan factors Secondary hypertriglyceridemia Diabetes mellitus Hvuothvroidism Nephrotic syndrome Chronic renal failure Alcohol Obesity Drugs raising the level of plasma TGs Estrogens Oral contraceptives Corticosteroids Tamoxifen Isotretinoids Diuretics P-Blockers Pregnancy I. Causes of the Chylomicronemia The most common secondary Syndrome genetic and nongenetic causes leading to chylomicronemia are listed in Table 12. The catabolism of chylomicrons and VLDL is dependent on the catalytic activity of LPL and the availability of its protein activator, the apo CII. Defects in either LPL or apo CII may lead to chylomicronemia. Familial LPL deficiency and apo CII deficiency are autosomal recessive disorders, each of which can cause massive chylomicronemia due to complete or partial loss of LPL activity. Primary LPL deficiency is discussed in Section 5. Readers are referred to detailed recent reviews and articles on familial apo CII deficiency (Breckenridge et al., 1978; Breckenridge, 1987; Connelly et al., 1987; Dolphin, 1992; Tuzgol et al., 1994; Brunzell, 1995). An inherited LPL However, amylasemia has often been found to be normal in pancreatitis due to chylomicronemia. This paradoxical finding has been attributed to an interference factor (Fallat inhibitor leading to chylomicronemia and very low postheparin LPL activity has also been reported in a family et ul., 1973) or to the presence of an enzyme inhibitor (Lesser and Warshaw, 1975; Warshaw et al., 1975). Real values are obtained if amylase activity is determined using diluted disorders (autoantibodies against LPL) has been reported in a patient with idiopathic thrombocytopenic purpura and Grave’s disease (Kihara et al., 1989), and heparin resistance plasma (Fallat er al., 1973). Pseudohyperbilurinemia has also been observed in chylomicronemia. These uncertainties in laboratory analyses may make the diagnosis and the follow- was noted in a case of disseminated up of chylomicronemic patients somewhat difficult. All laboratory results, therefore, should be interpreted with caution in the presence of a lactescent plasma. *Bertrand. M., Gagne, C.. Bun, L. D., J&en, P., Pineault, S., White, J., hlurthy, M. R. V. and Lupien, P. J. (1990) Familial hyperchylomicronemia and splenomegaly. In: International Symposium on Triglycerides: The Role in Diabetes and Atherosclerosis, May 23-26, 1990, p. 130, Vienna, Austna. (Brunzell et al., 1983). Chylomicronemia due to autoimmune lupus erythematosus (Glueck et al., 1969a,b). Abnormalities in the LPL and apo CII genes do not represent the most frequent causes of the chylomicronemia syndrome in the general population. Familial forms of hypertriglyceridemia, such as familial hypertriglyceridemia, dysbetalipoproteinemia, and familial combined hyperlipidemia (FCH), are often associated with one or more other genetic and/or nongenetic factors that tend to increase the plasma TG levels (Table 12). The genetic predisposition to hypertriglyceridemia is exacerbated by secondary factor(s), lead- V. Murthy 122 ing to the chylomicronemia syndrome 1992). The same individual may possess ent genetic hypertriglyceridcmic triglyceridemia, type, heterozygocity Gaudet rt al., Uncontrolled nemia 1983; Wilson fasting (Bierman 1973; et ul., the Wilson catalytic activity and Brunzell, (especially Gleeson and 1987; Iverius (Molitch (Chait mild to moderate uals, may familial forms secondary (Stuyt et u1., 1988), 1992), which oral Plasma TGs Cholesterol 1.31 t 0.55 4.92 + 0.92 22.35 t 5.44 t VLDL TG:: Cholesterol Cholesterol/npo concentrations mmol/L, 36 mmol/L of these eliminate the When LPL activity LPL gene, VLDL is impaired (Brunzell, of these particles. of chylomicrons is unclear, in the but it could of HL or the direct endothelial The and do plasma, that not but there complete absence be the result continue reach exists mechanism occurs a process of amount of ingested siderable variations The with between different viduals, over long-chain patients, long periods of LPL (Table no postheparin TGs. as well as within 10) may Patients plasma acids. The due LPL produce heterogeneity have been activity, and/or reported but show profiles significant of cholesterol and HDL The (Table vascular apos ably found in B and apo AI), that as in both these particles reduced level, which relative atherosclerotic in the homozygotes attacks and the presence the HDL but also drastically are rarely cholesterol explain been were reduced indicating in recurrent complications ratio, (apo of chylomicronemia mainly low LDL for with females, 13). morbidity is expressed have ratios, were not only poor in cholesterol, in number Except 13. occa- the sexes (GagnC et al., 1989). and fractions, and in Table level are observed differences between well as the cholesterol-to-apo the LDL for females are shown chylomicronemia. profiles TG 6 and 234 1989). is lower in males as compared such as chylomicrons, of pancreatitis; observed cardio- (Nikkila, 1983). the low LDL-to-HDL cho- of large plasma lipoproteins, are nonatherogenic, freedom could prob- of homozygote complications (Gagne patients et ul., 1989). to the con- observed indi- et ul., 1989). Variations the secretion levels to vary between et ul., (Brun- the plasma of 26 mmol/L (Gagne to severe which activity the same value in another patients, total cholesterol lesterol However, activity found lipoprotein in plasma activity is related and normal a mean of LPL TG levels have been (Gagne that may influence levels of plasma fatty 0.0042 0.0001 0.0001 for males plasma Increases from in plasma 0.36 * 0.10 0.33 * 0.08 0.45 + 0.06 The by the reticulo- of hypcrchylomicronemia AI 0.27 t 0.04 1.25 ? 0.33 0.78 * 0.10 for the turnover of the catalytic of chylomicrons to an even- system. severity in factors also The uptake lesions chylomicron hypertriglyceridt-mia in the indicating of genetic of both Chylomicrons indefinitely equilibrium, turnover port DEFICIENCY as a result and massive 1995). accumulate tual is defective the catabolism 0.021 0.0001 0.0001 have been the lipoprotein Homozygote State of Lipoprotein Lipase Deficiency in the 0.36 k 0.25 0.49 k 0.26 1.19 + 0.35 in one tissue individ- Correction LII’ASE B O.lY k 0.11 3.14 t 0.78 3.M k 0.71 TGs :lllii cholesterol values xc expressed as mmol/L. Mean * SD (n = 16). I,‘, mu slgnificnnt. Datn from CantIn et 01. ClYO?). sionally LIPOPROTEIN B ns ns ns zell, 1989). In the French-Canadian condition. PRIMARY 12.84 3.91 4.33 2 7.41 1.64 rt 3.00 17.80 t 25.12 HDL T(;s Cholcstcrol Cholcs~crol/apo no statistically 5.1. 0.0001 ns 0.72 + 0.48 0.38 + 0.19 6.79 -’ 8.33 LDL I-G Cholesterol Cholestert-rl/apo cholesterol, 5. P 27.39 k 18.23 7.91 k 6.42 Chylomicrons TGS Cholesterol carrying in normal to rapidly Homozygotes Controls enzyme induce in patients found 13. Plasma Lipoprotein Concentrations in Control and Homozygotes for Primary LPL Deficiency (Bag- rt cd, 1986), isotretinoids et al., 1987), diuretics, of hypertriglyceridemia. chylomicronemic (Ma et ul., 199513). Flynn Brunzell, has been et condition corticosteroids chylomicronemia factors with (Pykalisto (julien hypertriglyceridemia produce failure, combined and Brunzell, (Brun and as hypo- the third trimester) 1974), 1980; TV (IL., renal as estrogens et ul., and Connolly, P-blockers such physiological during such particles 1992). Chylomicronemia by obesity dade rt u1., 1970), tamoxifen (Dicken diseases, when drugs, et al., produc- (Brunzcll and chronic by a transient lipid-raising 1986; of LPL 1983, et al., 1993), and exacerbated contraceptives may induce to hypertriglyceridemia may also be induced Several and Brunzell, the hepatic chylomicronemia such as pregnancy use are the with chylomicron syndrome, predisposition d., 1976; Chait et ul., in the chylomicro- and alcohol et al., 1994). V arious induce alcohol by increasing nephrotic also E: geno- (Julien 1966; Chait TGs that compete thyroidism, familial and involved et al., 1993). Diabetes saturate may mellitus factors chylomicronemia tion of VLDL and due to apo TABLE Subjects hyper- 1995). secondary syndrome of differ- such as familial for LPL deficiency diabetes most frequent and Brunzell, an amalgam traits, hypertriglyceridemia and/or 1994; (Chait et al. the transin the 5.2. Heterozygote State of Lipoprotein Lipase Deficiency The heterozygote ically and zygotes are considered who have chylomicronemia abnormal ease, and state for LPL biochemically they is still not characterized. The to be asymptomatic in regard and other escape deficiency well usual manifestations unambiguous clinical clin- heteroto of the dis- identification. Human Lipoprotein TABLE 14. Lipase Gene Phenotypic 123 Expression Subjects LPL gene mutation Number of subjects Male/Female Reduction in LPL activity Total TG Total cholesterol VLDL cholesterol LDL cholesterol HDL cholesterol Apo R Ape AI Denser LDL of the Heterozygote State for LPL Deficiency Minnicha Maillyb Babirakc Wilsond Emi’ Het 291 19 nd slight - or nd nd Het (Obligate Het 9 25 25/o 2Om30% nd 14 8/6 52% Het 188 29 II/18 50% Het 188 11 5/6 - 50% t 1 - or I t nd t I nd nd nd nd or t nd nd nd Julieng Het 188 8 4/4 62% Het 207 48 23/25 40-50% t - or 1 t - Miesenback’ nd nd nd I nd nd 1 1 HDLL 1 HDLL presence presence 1 Het, heterozygote; nd, not determined; -, unchanged; 1, increased; I, decreased. ‘Minmch et al. (1995): hMa~llv et al. (1995): <Babirak et al. (1989): dWilson et ai. (19901; ‘E mi et ui. (1990b); ‘Mxsenbock (1994). (1995~4 (unpubil;hed results) and Sniderman et al. (1995). et ui. (1993); EJulien et al. Babirak et al. (1989) have reported that carriers of LPL defi- dense in subjects carrying LPL gene mutations ciency could be identified on the basis of reduced postheparin (Miesenbbck et al., 1993; Julien et al., 1994, 1995a). Heterozygotes for LPL gene deficiency show impaired TG plasma LPL activity and mass. However, in our own study (Gagne et al., 1977b), even though adipose tissue LPL activity in heterozygotes was half the normal level, we could not clearly distinguish individual carriers from individual noncarriers based on postheparin plasma LPL activity because of significant overlapping in LPL activity between these two groups of subjects. Wilson et al. (1990) also reported that adipose tissue LPL activity was reduced by 50% in carriers, but this reduction did not allow reliable identification of the carriers from the noncarriers. tainties in biochemical In view of these uncer- and clinical data, the only reliable way to identify the carriers of LPL deficiency of the gene defects. is by analysis tolerance and postprandial lipemia (Miesenbbck et al., 1993). Postprandial hyperlipidemia is known to be a predisposing condition for atherosclerosis French-Canadian heterozygotes is consistent with impaired clearance of plasma TGs, due to a catalytically defective LPL enzyme. This probably leads hyperapo profiles in most by higher than normal levels of plasma TG. Hypertriglyceridemia is found not only in individuals with a marked decrease in LPL activity (40-60% of normal), but also in those with only moderate reduction in LPL activity (<30%). is significant heterogeneity However, there in hypertriglyceridemia (normal do not exhibit increases in either total apo B or LDL apo B (Sniderman et al., 1995). The phenotypic expression of the heterozygote state, thus, LPL activity (Table 14). Plasma lipoprotein characterized 1979). However, deficiency could represent a subgroup of FCH, a dyslipidemia characterized by an overproduction of apo B. However, to mild hypcrtriglyceridemia, are abnormal, (Zilversmith, precocious atherogenesis has not been investigated in these carriers. Babirak et al. (1989, 1992) have suggested that LPL Phenotypic expression of LPL mutations in heterozygotes is found to result in a 20-62% decrease in postheparin plasma of these individuals 188 and 207 the presence hypoalphalipoproteinemia, of cholesterol-poor LDL particles and without B. The heterogeneity of triglyceridemia in the heterozygote carriers of LPL gene mutations indicates that factors other than LPL deficiency may also play a role in the phenotypic expression of this familial dyslipoproteinemia. This possibility is supported by the observation that as high as 13-20% of French-Canadian patients with Type IV and V hyperlipo- to severe), even in subjects carrying the same LPL gene mutation. The major feature of this hypertriglyceridemia is the presence of larger than normal VLDL particles (TG- and proteinemia are carriers of an LPL gene defect (Julien et al., 1994; Minnich et al., 1995). Age, obesity, hyperinsulinemia, and lipid-raising drugs have been shown to contribute to cholesterol-rich particles) (Julien et al., 1995a). As shown in Table 14, an investigation of a group of Quebec heterozygotes, the largest of any group studied so far, shows that the expression of hypertriglyceridemia in heterozygotes (Wilson et al., 1990, 1993; Julien et al., 1995b). Thus, severe hypertriglyceridemia observed in some heterozygotes could these heterozygote subjects had reduced HDL cholesterol, as well as reduced apo AI, indicating that these HDL particles are less numerous and poor in cholesterol. But this reduction, was not as pronounced as in the homozygotcs. In two other studies, only the particles in the HDLI subfraction were found to be altered. Generally, no significant changes were found in total plasma apo B and LDL cholesterol. However, LDL particles have been reported to be more be the result of a second independently inherited defect in lipoprotein metabolism (Chait and Brunzell, 1983; Wilson et al., 1983). Similarly, it has been shown that homozygosity for LPL deficiency protects familial hypercholesterolemic patients against increased LDL cholesterol. This indicates the importance of gene-gene interactions in the phenotypic expression of dyslipoproteinemia (Zambon et u1., 1993). Furthermore, a variable lipid phenotype has also been dem- V. Murthy et ul. 124 TABLE Activity 15. Frequency of LPL Gene Mutations Among 82 Homozygous French-Canadian Number of alleles Exon Mutation 5 pro’“; j 6 6 9 Gly’s” - Glu A,$” - Asn ASTP - Ser Se+ - Ter - W) 113 37 4 1 Le” (1992) and unpublished (69) (22) (2) (1) 0 0 0 0 0 0 data. onstrated in a family affected by combined and LPL activities Plasma postheparin LPL activity 1 (1) 8 (5) Unidentified (’Data from J&n and LPL Probands= deficiency of HL (Auwerx et al., 1990). The interaction of genetic, dietary, and other factors in the phenotypic expression of dyslipidemia (in LPL heterozygote subjects) has not tury from the Northwestern are homozygous for a specific LPL gene haplotype named H2 (Hind111 and PvuII: + / +), indicating a common origin and a founder effect for this mutation (Ma et al., 1991). After arrival in Charlevoix, some of the settlers migrated towards the Northeast along the St. Lawrence River and then along the Saguenay River towards Lac St-Jean (Fig. 7B) (Dionne et al., 1992). These are the regions now populated by the descendants of the original immigrants tion 207 in their LPL gene (Fig. 6). has led to identification of 4 founders originating from different regions of France, one of them possibly of Scottish oriMutation occurrence As mentioned in Sections 3.3 and 3.4, gene analysis of individuals with documented enzyme deficiency has led to the of a large number of LPL gene variants. These variants have been found in subjects from different ethnic origins, including Caucasians, Japanese, Malaysian, and Black Americans (Lalouel et al., 1992). Detailed studies have been carried out in two large populations affected by LPL deficiency, one North European and the other French- Canadian (Wilson et al., 1990; Julien et al., 1994). Genealogical reconstructions of affected families have been attempted in the French-Canadian The incidence of homozygosity Northeastern population of Quebec. for LPL deficiency in the region of the Province of Quebec et al., 1992; Dionne et al., 1993). 188 has been shown to be specifically associated with haplotype Hl (Hind111 and PvuII: +/ -), suggesting a common origin for this mutation (Ma et al., 1991). The 5.3. Origin and Dissemination of Lipoprotein Lipase Gene Defects in Qukbec identification who carried muta- The second most common mutation, 188, is present mainly in Western Quebec (Fig. 6) (Bergeron et al., 1992). Genealogical reconstruction of 14 families carrying mutation 188 gin (Fig. 7A) (Bergeron yet been investigated. part of France, especially from Perche (Fig. 7A) (Dionne et al., 1993). DNA haplotype analysis has revealed that LPL alleles containing mutation 207 has been calculated to be at least one in 10,000, and the incidence in the general population is as low as 1 in 1 million (Julien rt ul., 1994). Five LPL gene defects have been identified to date in the Province of Quebec, and they all lead to complete loss of plasma postheparin LPL activity (Table 15). Of the 82 French-Canadian patients identified, a majority are of this mutation in different parts of the world and in various ethnic groups suggests that it may be an ancient mutation predating the spread of European and East Indian populations. The European immigrants to Canada carrying mutation 188 arrived at the site presently occupied by the city of Quebec. Their descendants migrated towards the Mauricie region, where they settled. Some of them, however, also moved along the fertile St. Lawrence Valley towards Montrkal (Fig. 7B). We find most of the current carriers of mutation 188 in these areas (Fig. 6). The relative isolation of different regions of the Province of Quebec and the limited demographic movements during the past centuries have served to confine the carriers of mutations 207 and 188, over a long period of history, to the regions where these immigrants originally settled (Dionne et al., 1993). Haplotype analysis of other French-Canadian patients clinically homozygous for LPL deficiency, and carrying still unidentified mutation(s) of the LPL gene, suggests that there may exist at least 3 additional mutations, besides 188, 207, and 250, underlying LPL deficiency in the French-Canadian homozygotes or compound heterozygotes for mutations 207 and 188. Most of the French-Canadians living in the Province of population (Julien et al., 1995a). Recently, 2 new mutations at positions 291 and 447 of the LPL protein were identified Quebec are descendants nich et ul., 1995). The total heterozygote carrier rate for all of these LPL gene mutations has been estimated, based on the HardyWeinberg equilibrium (Table 16) (Julien et al., 1994). The Saguenay-Lac-St-Jean (Northeastern Qukbec) and Mauricie (Western Quebec) regions are found to have the highest carrier rate, with frequencies of l/48 and l/107, respectively. of approximately 8500 settlers who migrated from France between 1608 and 1759 (Charbonneau and Robert, 1987). We previously have hypothesized that the high frequency of familial LPL deficiency in this population was the result of a founder effect (De Braekeleer et al., 1991). Genealogical reconstructions of affected families show two different sets of founders for mutation 188 and 207. Analyses of geographic distributions indicate that mutation 207, which is almost exclusively French-Canadian, is more prevalent in the Northeastern region of the Province (Fig. 6) (Normand et al., 1992). Genealogical reconstruction of families carrying mutation 207 has enabled us to identify 16 founders who migrated to Quebec in the early 17th cen- in 2 French-Canadian homozygote patients (Table 15) (Min- However, within the Charlevoix area, a subdivision of the Quebec region, the incidence is estimated to be l/33. On the basis of the number of homozygote patients referred to the Lipid Clinics, the total number of carriers for these two major mutations is believed to be at least 45,000 in the Province of Quebec (J&en et ul., 1994). 125 Human Lipoprotein Lipase Gene 08 Lac St-Jean Administrative Regions 01: Bas-St-Laurent 02: Saguenay - Lac St-Jean 03: Quebec 04: Mauricie - Trois-Rivikres 05: Estrie 06: Montreal 07: Outaouais 08: Abitibi - Tkmiscaminque 09: C&e-Nord 10: Nouveau-QuGbec - 207-m 188-m 250-m Unknown - 0 Compound Heterozygote - H FIGURE 6. Geographic distribution of LPL-deficient patients homozygous for different LPL gene mutations in the Province of Quebec. The distribution is based on the place of birth of each patient. The size of each box is proportional to the number of patients identified in each administrative region. Reprinted from Julien et al. (1994), with permission of the copyright holder, Canadian Cardiology Publications, Inc., Mississauga, Ontario, Canada. 6. TREATMENT LIPOPROTEIN 6.2. OF LIPASE DEFICIENCY Family Screening and Counseling Identification and Correction of Secondary Factors Statistically, the siblings of a patient with primary LPL deficiency have a relative risk of 25% of having the disease and As mentioned in Section 4.4, primary LPL deficiency is not the most common cause of chylomicronemia. Understanding the multiple etiology underlying chronic and transient 50% of being heterozygote. Diagnosis of chylomicronemia in one individual, therefore, implies family screening. In families where primary LPL deficiency is detected, the only way chylomicronemia is necessary for the proper management of this syndrome, as well as to avoid recurrent clinical complications. The long-term goal of the treatment must be directed towards the prevention of recurrences. This can be achieved by maintaining plasma TG levels below 10 to predict the risk of having a child with the disease is to identify the mutation at the molecular level in both prospective parents. As discussed in Section 5.2, the measurement 6.1. mmol/L. Factors that may aggravate hypertriglyceridemia, such as alcohol consumption, use of hypertriglyceridemic drugs, oral contraceptive agents, and estrogens, should be avoided. Careful attention should be given to detection and treatment of secondary factors, such as diabetes mellitus and hypothyroidism. If a woman desires pregnancy, the followup should be planned in advance, as pregnancy may be complicated and has the potential for high risk (Watts et al., 1992; Ma et al., 1993). of postheparin LPL activity is not useful for identification of heterozygotes, because enzyme levels change over time and overlap with those of normal individuals, although the mean is lower in heterozygotes (Gag& et al., 197713). 6.3. (about 50% of normal levels) Dietary Regimen Fat restriction applies to patients with familial LPL deficiency, as well as to patients with secondary hypertriglyceridemia, when secondary causes cannot be treated or treat- V. Murthy 126 familial LPL deficiency. limiting the intake it by complex should caloric and minerals. cess depends, The low-fat increased by using micronemia, administration a milk of MCT formula amounts PROVINCE Formulation in the nutrients, daily diet. in women be limited in iron. 1993), The milk reduction reduce signs of triglyccridemia. triglyceridemia of chylomicronemia pancreatitis TGs can be achieved at lo-20 Fat restriction The to a level are aim of the where the eliminated. by maintaining therapy is to symptoms and Prevention fasting plasma Perron et zell, 1992). emulsions TG is progressively are identified by other nutrition 1992; introduced, and Brun- be given, but lipidic After while and possible also be desirable to consider drugs, necessary. in the same (Chait lipids are metabolized be monitored and corrected. et al., as their et u1., 1985). as chylomicrons. should Ma not nurse, causes as these should and Sprecher, is treated should be avoided, if judged et al., may adapted attention should pancreatitis to iron, the diet (Black in fats (Steiner mechanisms levels be given Special (Watts of are present for whom children by a quantities conditions. produced should must children women Parenteral be made be periodically poor as that should adequate the acute alimentation secondary For long-term factors therapy, it may the use of hypotriglyceridemic mmol/L. is the only dietary treatment available TABLE 16. Estimation of the Heterozygote Carrier LPL Gene Mutations in the Province of Quebeca Administrative of available and minerals with LPL deficiency is extremely phase, for adequate the adequate 1990; diet should Chylomicron-induced manner diet attention growing and pregnant by the same is not sufficient and er al., that vitamins, and to infants, 1993). Women causes (Schluter Special especially be given secondary MCT with For infants, is commercially to ensure tc age and physiological of these vein. Chylo- relieved et u1., 1992). mostly of an adequate dietitian all necessary ment and MCT 199413). professional FIGURE 7. A: Spread of mutations 188 (solid line) and 207 (dashed line) from France to Quebec in the 17th century. B: Founding regions for mutations 188 and 207 in the Province of Quebec and movement of settlers along the St. Lawrence River and the Saguenay. via the portal acids Mead-Johnson) may be oils for cook- into chylomicrons oil (Shirai fatty with the of the diet significantly containing of essential (Portagen@, ill., be to Suc- for their catabolism. transported can compliance TG (MCT) on LPL activity thus, of fat is limited palatability fats are not incorporated fatty acids are directly the diet with the usual 40%. medium-chain are not dependent However, consumption The by and replacing as well as essential on the patient’s diet. accomplished acids intake, compared however, extremely fatty and proteins. adequate 10-l 5% of total calories, ing. MCT is efficiently carbohydrates provide vitamins This of long-chain et al. region 01 Rx St Laurent 02 Saquenay Lac-St-Jean” 03 Quebec (Charlcvolx)” 04 Maurlcie’ 06 Montr~ul 00 core Nod Eastern Quebec” Western Quebec’ Whole Quebec Estimated Rate for carrier l/245 l/48 l/l16 l/107 l/24’) l/166 l/M l/220 l/l43 for 6.4. Drug Therapy Drug therapy is used to lower plasma triglyceridemia, such as familial and dysbetalipoproteinemia, rate LPL deficiency glyceridemia. effective action Fibric lipolysis develop moderate acid derivatives is on plasma plasma in adipocytes, and activation LDL in HDL of fibrates and high-carbohydrate when heterozygotes. are known VLDL However, hyperFCH, inhibition principal through of VLDL cholesterol produc- Lowering with a reduction (or nicotinic with are elevated, the use of fibrates of in denser (Shepherd, acid) diet is useful in lowering levels as the most The 1994). for hypertri- metabolism of LPL (Davignon, and an increase especially to severe TG levels. TG plasma TG levels is often associated A combination in primary as well as in heterozygotes drugs for lowering of fibrates reduced tion, who TGs hypertriglyceridemia, 1993). a low-fat plasma TGs, as in the case of to treat homo- Human Lipoprotein zygote patients is not indicated 127 Lipase Gene for primary because LPL- they and apo respond Two approaches (X-deficiencies only to low-fat oped. diet. from the patient, Prospects of Gene Therapy 6.5. As described in Sections of LPL deficiency restrictions or lipidemia. consists administration Both methods the part of medical of the patient. and, 6.3 and 6.4, the current in humans professionals Chronic and compliance use of drugs in the techniques genes normal cells of direct could means a cure of continuous Several that are directly mouse (Field, 1993; Liu et al., et al., 1994), its activator and HL (Busch et al., 1994). of an optimum The model chylomicrons, thomas, and deficiency (Jones inherited in the LPL et al., LPL gene, zinger er al., bined lipase-deficient recessive 1994). The glycosylated that but remains within cytes above pathway, two models directly by defects l-3 1990). The appears because could help by lesions in other genes. and for an secretory if the target can sophisticated to migrate and techniques more work is needed can technical the gene expressed. to develop loss, to in a form Although is receiving in clinical direct to the target or qualitative and to deliver be tested of the a highly where (Wilson method, cells in viuo. This, the vector of attention, they disease consists to the target integrated use of such in the treat- An alternative quantitative cells, be properly the possible a great deal them to a point situations. Ackno~,lrdgements-The authors are deeply indebted co Professor David Shugar for his interest and careful correction of the drafts of this article. We wish to express our thanks to Professor Alan Sniderman for reading the final manuscript and for useful suggestions. We thank Gervais Lapointe, Merck Frosst Canada, and Car& Bra&, CHUL Research Centre, for assistance in complling the references. Research from the authors’ laboratories discussed in this review was supported by grants from the Heart and Stroke Foundation of Canada, Parke-Davis Canada, the Canadian Diabetes Association, the Natural Scirnces and Engineering Research Council of Canada and the Medical Research Council of Canada. Dr. I? Julien was a Career Scwnt~st of the Fends de la Recherche en Sante: du Quebec. et for the to the lipase glyco- in cld adipo- characteristics the LPL deficiency of the efficacy of is caused itself or indirectly Ahn, Y. I., Kamboh, M. I. and Ferrell, R. E. (1992) Tm,o new alleles in the tetranucleotide repeat polymorphism lipase (LPL) locus. Hum. Genet. Ahn, Y. I., Ferrell, R. E., Hamman, of lipoprotein iological components at the lipoprotein 90: 184. R. E and Kamboh, lipase gene variation of the insulin-resistance of the San Luis Valley, Colorado. M. I. (1993a) with the physsyndrome Diabetes in the Care 16: 1502-1506. Ahn, Y. I., Kamboh, M. I., Hamman, R. F., Cole, S. A. and Ferrell, R. E. (1993b) Two DNA polymorphisms gene and their associations in the lipoprotein lipase with factors related to cardiovascu- lar disease. J. Lipid Res. 34: 421-428. Al-Haideri, M., Granot, E., Schwiegelshoh, I. J. and Deckelbaum, B., Vogel, T., Gorecki, R. J. (1993) Apoprotein E simulates non receptor triglyceride-rich Circulation terization particle cellular uptake. 88: I-321. M., Schotz, protein (Davis References Ameis, D., Stahnke, abnormally N-linked normally no significant recognize M., Goldberg, LPL gene gene responsible in the LPL gene related of appears inactive in evaluating when and reticulum are processed in situations LPL The to be specific other a single, chylomicronemia it produces permit tech- hypercholesterol- lipid-related 1994). less invasive, require site with population com- homozygote days. would that would Association for T/t complex catalytically endoplasmic is (Gin- is the has in utero I erent 1990). Th e d’ff et al., LPL gene therapy either et al., such as adipsin (Davis but the of Arg of both massive mannose), glycosylation processing proteins exhibits normally, (il., 1990; Masuno defective normally and dies within (high LPL base change model et ul., 1983). The develops to suckle, animal a deficiency cld mutation is transcribed second causes the xan- deficiency substitution the (Paterniti show 412 of the LPL protein within 17 that at birth, LPL mutation activities that to human by a single in the of LPL transgene subcutaneous feline be much success familial gene This for the devel- of cats which chromosome allowed models mouse, HL normal attempted two animal similar The (cld/cld) autosomal of complica- is eventually for human to be caused acid residue are useful of the LPL transgene activity resulting Gly at the amino studies the nature retinalis, 1983). and is found and LPL itself et al., Animal prevalent functioning patient. partial from et al., the devel- are harvested that 199413; is a colony lipemia reduced Zhang serve as test systems protocol first genes deficiency et al., also are available that could con- apo CII (Shachter when such a procedure There expression. fasting 1994; how the expression LPL deficiency opment of human in LPL in viuo and to evaluate in humans. a some suffering highly into of the gene of interest however, that treatthan have been 1983), including et al., tions to expect this rather recently a number Zsigmond in understanding up found are being cells by the normal back Grossman would design genes by their disease, implicated lipid diseases is regulated which ad- opened has already transfer inva- of foreign accomplished, for the (Shimada 1994), 1992; on Recent have of defective models overexpress or indirectly the attendant another hyper- management. transgenic structed and in animals If properly represent emia, et al., and expression replacement counterparts. ment of a patient on the part side effects. of transfer in mammalian possibilities ment is physiologically sive and may lead to unpredictable vances nique supervision gene therapy the target transfected transplanted of dietary to control long-term then, treatment of a regimen of drugs require to somatic In the ex viva strategy, G., Kobayashi, J., McLean, J., Lee, G., Buscher, M. C. and Will, H. (1990) Isolation and charac- of the human hepatic lipase gene. J. Biol. Chem. 265: 6552-6555. Ameis, D., Kobayashi, Kane, J. I?, Lee, (1991) Familial due to a single J. Clin. Appelman, LPL 87: T, mutation (type mutation R. J. and Schotz, M. J., M. C. I hyperlipoproteinemia) in the lipoprotein lipase gene. 1165-1170. E. E. G., Bijvoet, deficiency. H., Havel, chylomicronemia missense Invest. P W., Bruin, de novo J., Davis, R. C., Ben Zeev, O., Malloy, G., Wong, Hayden, S. M., Wiebusch, M. R. and in the lipoprotein Atherosclerosis H., Ma, Y., Reymer, Castelein, lipase 109: 63. (LPL) J. J. (1994) gene A causing 128 V. Murthv et al. Auwerx, J. H., Babirak, S. P., Hokanson, J. E., Stahnke, G., Will, H., Deeb, S. S. and Brunzell, J. D. (1990) Coexistence malities of hepatic lipase and lipoprotein Am. J. Hum. Genet. lipase in a large family. contributions K. (1992) Lipoprotein from molecular biology. Crit. lipase: Rev. Clin. S. P., Iverius, I? H., Fujimoro, (1989) Detection and state for lipoprotein W. Y. and Brunzell, characterization lipase deficiency. of the J. D. heterozygotr Arteriosclerosis 9: 326- S. I’., Brown, B. G. and Brunzell, J. D. (1992) Familial com- bined hyperlipidemia scler. Thromb. and abnormal lipoprotein lipemia. A complication Arch. Intern. lipase. Arterio- Med. U., Meng, Bethesda: of high-dosage corticosteroid therapy. Tuzgol, M. S., Skarlatos, heparin binding S. I., Previato, L., Brunzell, Beisiegel, lipase (Ala176-Thr) leads and loss of enzymic activity. Proc. 87: 1474-3478. Natl. Acad. Sci. USA U. (1995) Receptors for triglyceride-rich role in Ilpoprotein metahollsm. lipoproteins Curr. Opin. and Lipidol. 6: U., Krapp, A., Weber, W. and Olivecrona, role of alpha 2M receptor/LRP olism. Ann. Bengtsson, in chylomicron T. (19i7) lipase with heparin-Sephnrose. affinity binding. Benlian, Biochem. P., itienne, Raisonnier, remnant metab- lipase gene (Gly13Y-Ser) in a boy of Spanish Black, D. M. and Sprecher, exon 9 causes hpoprotein recombination. Evaluation J., de Gennes, A., Arnault, of lipoprotein of conditions for F., Hamelin, L., Brault, J., Fouhert, E (1995) Homozygous lipase deficiency; D.. L., Chuat, dcletlon of possible intron-Alu lipase. Annu. Rec. Nutr. 11: M. H., Davis, R. C., Elovson, J. and Schotz, M. C. (1992) Maturation activity of lipoprotein requires tion to rhe cis-Golgi Ilpase. Expression glucose trimmlng compartment. of full hut not transloca- J. Viol. Chem. Xi: asparagine-linked G., Liu, G., Da\%, R. C. and Doolittle, lipase and hepatic lipase: the role of glycosylation in the expression of a functional J., Julien, la lipoprotcln I? and Murthy, lipase humaine: M. R. V. (1991) Expression mutations de Rergeron, J., Normand, processing A., Murthy, M. R. V., Jullcn, I’., Gagne, C., Dianne, h(. R. and Lupien, C., De Braekcleer, M., Brun, D., Hayden, P. J. (1992) P revalence, geographical distri- and genealogical investigations of mutation 188 of lipo- prorein lipase gene in the French Canadian population of Quebec. Genet. Berryman, 41: 206-210. D. E. and Bensadoun, genesis of a putative protein lipasc. J. Biol. Chem. heparin A. (1993) Site-directed binding domain muta- of avian Ilpo- 268: 3272-3276. E. L., Bagdade, J. D. and Porte, D. J. (1966) Concept pathogenesis 79: 148. of diabetic 147: 60-62. J., Hamelin, J., Raisonnicr, Boulange, D. L. (1992) Kegulation and translocation of lipoprotein \X<C. (1987) D e fi clencies Breckenridge, Inc., A., Souli, A., Lavau, M. of the synthesis, lipase. Biochem. J. of lipemia. Trans. Assoc. Am. Physicians of plasma lipolytic activ- ities. Am. Heart J. 113: 567-573. Breckenridge, \x: C., (lY78) Little, Poast, bl. ciency of apolipoprotein J. A., Steiner, Hypertriglyceridemia C-II. G., Cho\v, A. and associated N. Engl. J. Med. with defi- 298: 1265- 1273. Rruln, T., Knsteleln, J. J., Van Diermen, P. M., Stalenhoef, D. E., Ma, Y., Henderson, A. E, Sturk, A., Brunzell, J. D. and Hayden, M. R. (1992) A missense mutation Ilpoprotein Bruin, llpase (LPL,,,,,,,,,,,) rcsu x. Itin Linden, N., Brunzell, Recurrent extended D. E., Hoogerbrugge J. D., Hayden, pancreatitis Dutch Pro157 Arg in g In I055 .. 0 f i,lta ” Iytic activ- 208: 267~272. T., Tuzgol, S., van Diermcn, van der M. R. and Kastelein, and chylomicronemia tion in lipoprotein J. J. in kindred is caused by a Gly154-Ser Bruin, T., Appelman, an substitu- lipase. J, Lipid Res. 34: 2109-2119. E. E., Blanchard, J. J. P. and Derewenda, H., Groat, N. R., Knstelein, Z. S. (1994a) Th c role of proline residues and function of lipoprotein lipase. Atherosclero- his 109 hi. W. J., van den En&, H., Hayden, M. R. and Kastelein, heterozygote for lipoprotein GlyI88-Glu: correlation A. E., Jnnsen, J. J. (1994h) A compound lipase deficiency, Val69-LLeu and between in vitro LPL activity and clim- J. Lipid Res. 35: 438~445. L. D., Gagne, C., Rousseau, I’. J. (1986) Se\wc C., Moorlani, lipemla Induced S. and Luplen, by tamoxifen. Cancer 57: A., Moorjani, S., 7123-2126. Rrun, L. D., Gagne, C., Julien, I’., Tremhlay, C. and Lupien, I’. J. (1989) Familial lipoprotein lipnse- activity deficiency: study ofrotal body fatness and subcutaneous Brunzrll,J. 1, Bharucha, and restricted 121: 237-246. fat tissue distrihution. et physiopathologic. Med. Sci. 7: 1061~1068. bution Gene Rouchard, enzyme. J, Lipid Res. 35: 1511-1523. Bergeron, treatment severely and Galibert, E (1992) Sequence of rat lipoprotein lipase-encoding Brun, M. H. (1994) Lipoprotein 93: 339- Lipase. Evener Publishers, A., Chuat, J. C., Dugail, I., Quignard cal expression. 6219- 6227. Ben Zeev, O., Stahnke, Genet. Chicago. Bruin, T., Tuzgol, S., Mulder, 217-237. cntalytlc Hum. children J. (1987) Lipoprotein in the structure J. Lipid Res. 36: 356-366. Ben Zwv, O., Doolittle, for a mutation causes chylomicro- D. L. (1993) Dietary in dietary far. Am. J. DIS. Child. Borensztajn, (1993) J. L., No& A. (1991) Lipoprotein descent. of hypcrchylomicronemic ity. Eur. J, Biochem. Interaction J. 167: 109-119. J. C., Tsc, C. and Gallbert, Bensadoun, G. (1994) The NY Acad. Sci. 737: 53-69. G. and Olivecrona, H. D., Hayden, 343. H. E., Stuyt, 117-122. Beisiegcl, S., Bakker, J. J. (1994) Homozygosity 287: 3 37-347. 125: 12Y-134. a single amino acid substitution to abnormal Bicrman, 1, Braun, J. E. and Severson, E. L. (lY70) Steroid-induced J. D., Brewer, H. B., Jr. and Fojo, S. S. (1990) Lipoprotein Clin. in the lipoprotein cDNA. 12: 1176-1183. Bagdade, J. D., Porte, D. J. and Bicrman, their Bruin, Brault, D., Noe, L., Etienne, 334. Babirak, Beg, 0. S. M., M. R. and Kastelein, growth Lab. Sci. 29: 243-268. Babirak, Bijvoet, naemia 46: 470-477. Auwerx, J., Leroy, l? and Schoonjans, recent of abnor- Metabolism cause5 of chylolnicronemia of Inherited A. 38: 1005-1009. D. (1989) Familial lipoprotein Disease, syndrome. and other In: The Mctaholic pp. 1165%1180, Striver, S.. an d Valle, L., Sly, W lipascdeficicncy D. (cds.) Rasi\ C. R., Beaudet, Mc(;ra\v Hill, Ne\\ York. Rrunzell, J, D. (lYY5) Familial lipoprotein iipnse deficiency and other causes of the chylomi~ronemia syndrome. and Molecular Discasc, pp. lY11~1Y32, Striver, C. R., Beau&t, Basis ofInherited Metabolic A. L., Sly, W. S. and Vallc, D. (eds.) McGraw- Hill lx, New Yoak. Brunzell, J. D. and Bierman, drome: internctlon Med. Clin. In: The North E. L. (1982) Chylornicronemia syn- of genetlc and acquired hypertrlglyceridemia. Am. 66: 455-468. Human Lipoprotein Brunzell, 129 Lipase Gene J. D., Hazzard, W. R., Porte, D. and Bierman, Evidence for a common, saturable, anism for chylomicrons man. J. Clin. Brunzell, Invest. triglyceride and very low density D. L., Bloom, chylomicronemia lipase activity. lipoproteins R. J., Wang, S. R. and Lewis, B. (1983) Familial due to a circulating inhibitor of the Ninth International 271-273, Stein, Creative Communications Busca, pp. Y. (eds.) R 6, L J., Deeb, S. S., of N-glycosylation at lipase induces its accumu- reticulum and alters this cel- R. L., Martin, M. T, Mao, S. J., Thomas, hepatic triglyceride G. A., Fitzgerald, M. C., Yates, C. E. and Jackson, lipase expression protein and aortic cholesterol 269: R. L. (1994) Human reduces high density lipo- in cholesterol-fed transgenic mice. S. H. and Ghan, of the human hepatic triglyceride L. (1989) Structure lipase gene. Biochemistry 28: 89668971. Cameron, lipid abnormalities Cantin, B., Brun, I? J. and J&en, with hyperlipidemia: in acute pancreatitis. L. D., Gagne, the incidence lipid composition and fluidity in primary lipoprotein Biophys. Cantin, B., Boudriau, Hemolysis membrane lipase defi- M., Brun, L. D., Gagne, C., lipase deficiency. Metabolism A. and Brunzell, disorders. Metab. Clin. Exp. 32: A. and Brunzell, Adv. Intern. Chamberlain, Med. associations in Atherosclerosis Charbonneau, population Canada J. D. (1992) Chylomicronemia J. A., Oka, K., Galton, normal and D. J. and Stocks, at the lipoprotein lipase gene: hypertriglyceridaemic subjects. H. and Robert, N. (1987) Origines 1608-1759. In: Atlas a 1800, pp. 118119, francaises de la Historique du Harris, R. C. and L. (eds.) Les Presses de I’Universite de Montreal, Montreal. G., Capurso, change at the donor lipase deficiency Res. Commun. Cohen, splice site of intron in a southern-Italian 187: 620-627. Association issues and recent D. and Doolittle, M. H. (1990) in the mouse. Evidence of impaired lipase processing and secretion. J. Biol. Chem. 265: 17960-17966. Davis, R. C., Wong, H., Nikazy, J., Wang, K., Han, Q and Schotz, M. C. (1992) Chimeras of hepatic lipase and lipoprotein Domain localization of enzyme-specific Dayhoff, lipase. properties. J. Biol. Chem. M. O., Schwartz, R. M. and Orcott, B. C. (1978) Model of evolutionary change in proteins. In: Atlas of Protein Sequence and Structure, pp. 345-358, Research Dayhoff, Foundation, M., Dionne, M. 0. (ed.) National Washington, C., Gagne, Bio- DC. C., Julien, P., Brun, D., Murthy, M. R. V. and Lupien, P. J. (1991) Founder effect in familial hyperchylomicronemia among French Canadians of Quebec. Hum. Hered. 41: 168-173. Deckelbaum, pholipid R. J., Ramakrishnan, Olivecrona, hydrolysis protein R., Eisenberg, S., Olivecrona, G. (1992) Triacylglycerol and phos- in human plasma lipoproteins: and hepatic lipase. Biochemistry lipase gene. Biochemistry role of lipo- 31: 8544-8551. Deeb, S. S. and Peng, R. L. (1989) Structure of the human 28: 4131-4135. Erratum, lipoBio- 28: 6786 (1989). Deeb, S. S., Reina, M., Petersen, J., Takata, J. D. (1991) Gene mutations tein lipase deficiency. tial gene duplication lipoprotein Dichek, Arterioscler. involving lipase deficiency. K., Kajiama, in patients Thromb. G. and with lipopro- 11: 1418. exon-Alu interchange Am. J. Hum. Genet. H. L., Fojo, S. S., Beg, 0. U., Skarlatos, results in 46: 112-119. S. I., Brunzell, of two separate allelic mutations levels in Hispanic Biophys. clearance: compari- Am. J. Clin. Nutr. 49: 306-313. R. F. and Ferrell, R. E. (1993) men. Genet. lipase locus with Epidemiol. 10: in the lipoprotein lipase gene of a patient with the familial hyperchylomicronemia J. Biol. Chem. Dichek, syndrome. 266: 473477. H. L., Parrott, C., Ronan, R., Brunzell, J. D., Brewer, H. B., Fojo, S. (1993) F unctional lipase genetically engineered characterization from human of lipopro- tein lipase and human hepatic lipase. J. Lipid Res. 34: 139331340. Dicken, C. H. and Connolly, associated of a PvuII RFLP at the lipoprotein fasting insulin 1777188. C. E., Hamman, 1 causes lipoprotein family. Biochem. triglyceride assessment methods. Cole, S. A., Aston, lipase and pancreatic 10: 61B-71B. lipase deficiency a chimeric A., Resta, F. and Pepe, G. (1992) A G-C J. C. (1989) Chylomicron son ofthree frag- and evo- 263: 1107-1110. Davis, R. C., Ben Zeev, O., Martin, Jr. and Santamarina Chimienti, restriction localization, J. D., Cutler, G. B., Jr. and Brewer, H. B., Jr. (1991) Identification 79: 85-91. canadienne, with lipoprotein D. H., L. (1988) Devlin, R. H., Deeb, S., Brunzell, J. and Hayden, M. R. (1990) Par- polymorphisms 1. Des Origines Dechene, syndrome. 37: 249-273. J. C., Thorn, J. (1989) DNA I?, Ledbetter, chromosomal Can. J. Cardiol. Brunzell, 209-213. Chait, relationships chemistry J. D. (1983) S evere hypertriglyceridemia: role of familial and acquired and its action 13: 525-541. J. (1994) Fibrates: a review of important protein 44: 652-658. Chait, lipase activity Int. J. Biochem. S. H., Liu, S. W. and Ghan, lipase. J. Biol. Chem. T and Bengtsson M. R. V., Lupien, I? J. and Julien, P. (1995) in primary lipoprotein lipoprotein metabolism. ment length polymorphisms, findings. A. and levels in adipose and heart. Bio- Human hepatic lipase. Cloned cDNA sequence, Davignon, sequence 1008: 92-101. M. A., Chen, De Braekeleer, 1139: 25-31. S., Bertrand, Rogers, I? A., Murthy, P. J. and Bensadoun, lipase: cDNA S., Luo, C. C., Li, W. H., VanTuinen, Brown, of M. R. V., Lupien, in erythrocyte ciency. Acta Datta, of mRNA Acta A. (1981) Tissue in lipoprotein medical Ann. Surg. 177: 483-489. C., Murthy, I? (1992) Alterations Biochim. regulation Biophys. S. J. L., Capuzzi, D. M., Zuidema, G. D. and Margolis, (1973) Acute pancreatitis adipose lipoprotein 267: 21499-21504. 16376-16382. Cai, S. J., Wang, D. M., Chen, reciprocal Combined J. Lipid Res. 36: 939-951. Busch, S. J., Barnhart, D. A., Stein, J. C., Strieleman, (1989) Avian lutionary P., Auwerx, S. (1995) Absence in the rough endoplasmic J. Biol. Chem. on Atherosclerosis, Ltd., Tel Aviv. 43 in human lipoprotein lular compartment. IX. Proceedings S. and Stein, M. A., Pognonee, Reina, M. and Vilaro, lation Symposium O., Eisenberg, R., Pujina, asparagine In: Atherosclerosis G. F. and Little, J. A. (1987) Apolipopro- tein CIIS, ~,~h~.+Familial apolipoprotein CII deficiency associated with premature vascular disease. J. Clin. Invest. 80: 1597-1606. Cryer, S., Julien, H. E. and Hayden, M. R. (1992) Familial lipase deficiency. P. W., Maguire, chim. of lipoprotein J. Lipid Res. 241: 12-19. I?, Ma, Y., Henderson, Connelly, Cooper, P., St-Hilaire, Brunzell, J. D., Peterson, J., Deeb, S. S., Santamarina-Fojo, lipoprotein in 52: 1578-1585. J. D., Miller, N. E., Alaupovic, C. S., Sarson, E. L. (1973) removal mech- with isotretinoin S. M. (1980) Eruptive (13~cis-retinoic xanthomas acid). Arch. Derma- tol. 116: 951-952. Dianne, C., Gagne, Roederer, de Braekeleer, Genet. J., Hayden, M. (1992) Genetic lipase deficiency Ann. C., Julien, P., Murthy, G., Davignon, M. R. V., Lambert, M. R., Lupien, epidemiology in Saguenay-Lac-St-Jean 35: 89-92. M., I? J. and of lipoprotein (Quebec, Canada). 130 Dianne, C., Gagne, Davignon, C., Julien, I’., Murthy, J., Lambert, H., Lupien, and regional distribution Function CRC enzymes in the regulation lipase defi- Hum. Biol. 65: 29-39. and the role of apolipo- of their activity. of Apolipoproteins, M. (1993) of lipoprotein of Quebec. P. J. (1992) Lipolytic proteins D., Ma, R., Henderson, M. R. and de Braekeleer, ciency in French-Canadians Dolphin, M. R. V., Roederer, G., M., Chitayat, I? J., Hayden, Genealogy Dugi, et al. V. Murthy In: Structure pp. 295-362, Rosseneu, and M. (ed.) Santamarina covering H. L., Talley, G. D., Brewer, H. B., Jr. and Fojo, S. (1992) Human lipoprotein the catalytic substrates. site is essential J. Biol. Chem. to common lipase: the loop for interaction \vith lipid lipase: a multifunctional metabolic enzyme diseases. N. Engl. J. Med. 320: 1060~1068. S. E., Page, R. A., Volpe, C. I’., Kille, I’., G. E. and Cryer, A. (1993) Cloning of a full length cDNA chim. Biophys. encoding and sequencing ovine lipoprotein lipase. Bio- 1172: lhi-170. Acta J. M. and Wilson, D. E. (1994) Molecular screening protein lipase gene in hypertriglyceridemic iloninsulin-depcndcnt Metah. mutation lipase deficiency resulting from lipase gene. 47: 107~111. R. R. and Lalouel, J. M. (1990h) Missense of human lipoprotein tional deficiency. J. Biol. Chcm. lipase imparting mutafunc- J., Beg, 0. U., Peterson, J., Previato, L., Brunzell, J. D., Fojo, S. (1992) Human lipo- protein lipasc. Analysis ofthe catalytic triad by site-directed mutagenesis of Scr-132, Asp-156, and His-241. J. Biol. Chem. 267: Hermansson, H., Bengtsson-Olivecrona, M. L., Olivecrona, G., Carlsson, I’., T. and Bjursell, G. (1987) Molec- Fredrickson, orders. an integrated Funke, H., Reckwerth, Janscn, tification C. J. (1973) Suppression F., Chang, Vinaimont, Catalytic A., N., Smith, mutation L. C., Chen, Faustinclla, (1991h) Structural J. Aiol. Chem. and functional causing familwith a nonsense 266: C. F. and Ghan, L. that serine J. Biol. Chem. L. C. and Ghan, I32 is essen- L. (1992) Functional topol- ogy of a surface loop shielding the catalytic center in lipoprotein Iipase. Biochemistry Field, L. J. (1983) Transgenic Rev. Physiol. morphisms mice in cardiovascular researc-h. Annu. G. A. and La\vn, R. M. (1987) Tivo poly- in the human Acids Res. 15: 7657. lipoprotein S., Brun, lipase (LPL) gene. Nucl. L. D. and Lupien, Effect of hyperchylomicronemia ment of hemoglobin. chylomicronemie Am. J. Clin. familiale: 68: 584-586. S. and Lupien, I? J. (1977b) Hyperlipolytique dans 106: 333-338. C., Arun, L. D., Julien, P., Moorjani, (1989) Primary lipoprotein-lipase-activity of a French on the measure- Pathol. etude de l’activite une famille. Un Med. Can. Canadian S. and Lupien, P. J. deficiency: clinical inves- population. Can. Med. Assoc. J. 140: 405-411. Gagne, E., Genest, J., Jr., Zhang, H., Clarke, M. R. (1994) Analysis ofDNA L. A. and Hayden, changes in the LPL gene in patients hyperlipidemia. Arterioscler. Thromb. 14: 1250-1257. Gaudet, D., Moorjani, I’., Despres, disorders S., Gagne, C., Julien, I?, Tremblay, G., Perron, J. P. and Lupien, P. J. (1995) Premature Gehrisch, province. coronary of monogenic traits for lipid Canadians in a North Eastern region Atherosclerosis 109: 203. among French of Quebec S., Steinke, M., Kosta, H., Hoche, I., Julius, U. and Jaross, W. (1994) Point mutations Gimene: in the lipoprotein (N) and combined Atherosclerosis lipase (LPL) gene hyperlipidemic (CH) subjects. 109: 61. Llort, L., Vilanova, J., Skottova, N., Bengtsson Olivecrona, M. and Robert, Am. J. Physiol. Ginzinger, M. Q. (1991) Lipoprotein hydrolysis by perfused newborn colony M. R. (1994) Molecular of cats with lipoprotein a mutation lipase rat liver. 261: G641-G647. D. G., Krapp, A., Zhang, H. F., Gagne, U. and Hayden, S. E., Beisiegel, characterization lipase deficiency that impairs catalytic activity Gleeson, of a (LPL) reveals and interaction the LDL receptor related protein (LRP). Atherosclerosis with 109: 10. J. M., Dukes, C. S., Elstad, N. L., Ghan, I. F. and Wilson, of estrogen/progestin retinoids and chylomicron agents remnant metabolism. on plasma Contraception 35: 69-78. Glueck, C. J., Kaplan, A. P., Levy, R. I., Greten, Ann. Intern. C. J., Levy, R. I., Glueck, Med. Furuichi, familial H. R., Greten, H. D. S. (1969h) Acquired type I hyperlipoproteinM., Murase, lipoprotein lipase deficiency. 164: 1391-1396. Am. J. Med. 47: 318-324. T., Yamada, N., Takaku, Y. (1989) Gene polymorphism Commun. H. of exogenous 71: 1051l1062. H. I., Gralnick, cmia with systemic lupus erythematosus. T., Senda, H., Gralnick, D. S. (1969a) A new mechanism hypertriglyceridemia. Glueck, Gotoda, 55: 977114. Fisher, K. L., FitzGerald, 10: 830. C., Auger, I’. L., Moorjani, and Fredrickson, 31: 7219-‘7223. M., defect in a patient with Type 1 hyper- Arteriosclerosis and Fredrickson, 266: 9481-9485. D., Schulze Beiering, H., Paulwehe, B. and Assman, G. (1990) Iden- D. E. (1987) Effects roles of highly conserved serines lipase. Evidence tial for enzyme catalysis. F., Smith, L. (1991a) 267: 7194 (1992). L. C., Semenkovich, lipoprotein M., in a Turkish family. J. Biol. Chcm. Erratum, E, Smith, in human S. H. and Ghan, (Aspl56-Gly) of 225: 1331l1334. J. P, Rosseneu, lipase deficiency. Co-inheritance (Sert47-Ter) 14418~14424. Fnustinella, Van Biervliet, triad residue mutation ial lipoprotein JAMA and dis- G. (1988) Bst NI (Eco RR) RFLP in of the molecular I? J. (197ia) Gngne, to mechanisms lipase gene (LPL). Nucl. Acids Res. 16: 2741. enables triacylglycerol Faustinella, approach A., Stapenhorst, M. and Assmann, Funke, H., Wiehusch, Gagne, lipo- 109: 67. N. Engl. J, Med. 276: 34-44. Iipase of guinea pig. Gene 58: 1-12. I’. and underlying D. S., Levy, R. I. and Lees, R. S. (1967) Fat transport G., Llobera, amylase activity by hypertriglyceridemia. I., Liu, J. I’., Benlian, protein lipasc (LPL) deficiency in France. Atherosclerosis ular cloning and sequence analysis of cDNA encoding lipoprotein Fallat, R. W., Nestor, J. W. and Glueck, J. L., Ma, Y., Forsythe, F., Lagarde, Hayden, M. R. (1994) An analysis of mutations in normal 4161-4165. S., Semh, Ann. artery disease and high prevalence 265: 5910-5916. Bre\ver, H. B., Jr. and Santamarina E., De Gennes, H., Dairou, \vith familial combined D. E., Iverius, P. H., Wu, L., Hata, A., Hegele, tion (Gly-Glu188) Enerhack, L., Gagne, M. S., Zhang, tigation M., Iverius, P. H., Hegele, R. and in exon 3 of the lipoprotein Am. J. Hum. Genet. Emi, M., Wilson, R., Williams, members of familial 79: 1450-1456. Lalouel, J. M. (lY9Oa) Lipoprotein a nonsense of the lipo- diabetes mellitus families. J. Clin. Endo- Emi, M., Hata, A., Robertson, Emmerich, Fouhert, L. F. (1987) Pancreatitis hypertriglyceridemia. 107: 63. Gagne, C., Brun, L. D., Moorjani, Elhein, S. C., Yeager, C., Kwong, L. K., Lingam, A., Inoue, I., Lalouel, crinol. Med. lipidemin. Edwards, W. D., Daniels, Sweeney, Intern. the lipoprotein 267: 25086-25091. Eckel, R. H. (1989) Lipoprotein relevant associated with isotretinoin-induced in lipoproteins: Press, Inc, Boca Raton. K. A., Dichek, Flynn, W. J., Freeman, P. G. and Wickboldt, identified Biochem. F. and by PvuIl in Biophys. Res. Human Lipoprotein Lipase Gene Gotoda, T, Murase, T., Ishibashi, Yamada, N. (1990) Splicing, in familial lipoprotein Gotoda, T, Yamada, Ishibashi, 131 S., Shimario, nonsense N., Kawamura, Acids Res. 17: 10146. Kozaki, 10: 833. K., Mori, mutations Y. in the human lipoprotein lipase gene in patients with familial lipoprotein deficiency. J. Clin. Invest. N., E, Yazaki, Y., Furuichi, and Murase, T (1991a) Heterogeneous lipase Gotoda, T, Yamada, N., Murase, T, Inaba, T, Ishibashi, S., Shimano, rence of multiple aberrantly site mutation Gotoda, spliced mRNAs upon a donor splice lipase deficiency. T, Miyake, M., Kozaki, K., Mori, S., Murakami, N., Shimano, M. and Yazaki, Y. (1992a) A newly identified tion in the human lipoprotein hetcrozygote Acta Gotoda, with familial R., H., Shimada, null allelic muta- lipase (LPL) gene of a compound LPL deficiency. Biochim. Biophys. T., Yamada, N., Murase, Harada, K., Kawamura, T, Shimano, M., Kozaki, H., Shimada, nuclease digestion. M., K. and Yazaki, Y. (1992b) of three separate DNA polymorphisms lipoprotein lipase gene by gene amplification Grossman, in the human and restriction endo- J. F., Muller, D., Lupien, I? J. and Wilson, J. M. (1994) Successful ex viva gene therapy directed to liver in a patient hypercholesterolaemia. Nature Genet. lipase deficiency: Ser-Thr244 Am. J. Hum. Genet. Hata, A., Robertson, detection nonsense in 3’ lipase gene. sequencing of individual identification in exon 9 of the human alleles after of a common lipoprotein lipase Acids Res. 18: 540775411. Ham, A., Ridinger, L. K., Shuhua, Iverius, I’. H., Wilson, S. D., Emi, M., Kwong, A., Guy Grand, B., Basdevant, A., D. E. and Lalouel, J. M. (1992) Missense mutations in exon 5 of the human activation correlates lipoprotein with loss of dimerization. lipase gene. InJ. Biol. Chem. 267: 20132220139. Hata, A., Ridinger, D. N., Sutherland, S., Emi, M., Shuhua, I? H. and Lalouel, J. M. (1993) Binding segments Identification Z., of five critical of the amino-terminal of lipoprotein residues domain. lipase to in two distinct J. Biol. Chem. 268: S., Horl, G., Shachter, S. K., Hofler. G., Kostner, N. S., Presta, in the gene for lipoprotein lipase resulting in a highly conservative amino acid substitution causes familial chylomicronemia lipoproteinemia). Hayden, Genomics lipase deficiency. (type I hyper- Mol. Cell. genetics of human Biochem. 113: 171- 176. Hegele, R. A., Nakamura, R. (1989a) A BglII RFLP Acids Res. 17: 8899. with high density lipase gene: pos- lipoprotein levels. Hum. Henderson, H. E., Devlin, R., Peterson, protein lipase gene causes a premature tein lipase deficiency. Henderson, J., Brunzell, mutation stop codon and lipopro- Mol. Biol. Med. 7: 511-517. H. E., Ma, Y., Hassan, M. E, Monsalve, A. D., Winkler, J. D. and in exon 3 of the lipo- F., Gubernator, K., Peterson, M. V., Marais, J., Brunzell, and Hayden, M. R. (1991) Amino acid substitution J. D. (Ile194-Thr) lipase gene causes lipoprotein lipase deficiency in three unrelated probands. Support for a multicentric origin. J. Clin. Henderson, Invest. 87: 2005-2011. H. E., Hassan, (1992) The lipoprotein of Indian descent: gins and an increased Henderson, F., Berger, G. M. and Hayden, lipase Gly188-Glu evidence frequency. mutation suggesting J. J., Brunzell, ori- 29: 119-122. Lewis, I., Maeder, J. D. and Hayden, relationships analysis and mutagenesis common J. Med. Genet. H. E., Ma, Y., Liu, M. S., Clark D. L., Kastelein, M. R. in South of lipoprotein M. R. (1993) lipase: mutation of the loop region. J. Lipid Res. 34: 1593-1602. Hide, W. A., Chan, L. and Li, W. H. (1992) Structure tion of the lipase superfamily. apolipoprotein and evolu- J. Lipid Res. 33: 167-178. E by gene amplii?cation isotyping of human and cleavage with HhaI. J. Lipid Res. 31: 545-548. Hoeg, J. M., Osborne, of lipoprotein J. C. and Gregg, R. E. (1983) Initial diagnosis lipase deficiency in a 75 year-old man. Am. J. Med. Holzl, B., Huber, R., Paulweber, F. (1994) Lipoprotein tion in intron Y., Emi, M., Lalouel, J. M. and White, at the lipoprotein B., Patsch, J. R. and Sandhofer, lipase deficiency 6 of the lipoprotein due to a 3’splice site muta- lipase gene. J. Lipid Res. 35: 2161-2169. Howard, J. M., Ehrlich, E., Spitzer, J. J. and Singh, in patients L. M. (1964) with acute pancreatitis. Ann. Surg. 160: 210-214. Huff, M. W., Evans, A. J., Wolfe, B. M., Connelly, G. E and Strong, W. L. (1990) Identification acteristics of an apolipoprotein triglyceridemic subject. Ishimura E, Kihara, L. (1992a) A missense in exon 3 of the lipoprotein micronemia. S., Smith, mutation L. C., Oka, (Trp86-Arg) lipase gene: a cause of familial chylo- Am. J. Hum. Genet. Oka, K., Semenkovich, I. J., Shachter, char- C-II variant isolated from a hyper- J. Lipid Res. 31: 385-396. Oka, K., Faustinella, K. and Chan, P. W., Maguire, and metabolic 50: 1275-1280. C. E, Faustinella, N., Smith, L. C., Coleman, E, Goldberg, T, Hide, W. A., Brown, W. V., Oka, K. and Chan, L. (199213) A missense (Asp250-Asn) 18: 392396. M. R. and Ma, Y. (1992) Molecular lipoprotein E., Fried, G. M., Breslow, J. L. and Zechner, R. (1993) A novel missense mutation (Aspl80-Glu) I’. O., Ladias, J. A., 86: 578-584. Ishimura 8447-8457. Hauhenwallner, Genet. Hyperlipidemia Myers, R. L., Ren, K., Cheng, T, Inoue, I., Wilson, D. E., Iverius, heparin. T., Kwiterovich, morphism haplotypes of the human lipoprotein sible association lipase 75: 889-892. D. N., Sutherland, J., Lubbers, T, Schotz, S. E. and Lusis, A. J. (1991) DNA poly- Hixson, J. E. and Vernier, D. T (1990) Restriction 47: 721-726. strand separation: mutation gene. Nucl. and transition in the lipoprotein for M., Emi, M. and Lalouel, J. M. (1990b) Direct and automated electrophoretic I?, Iverius, heterozygote S., Kirchgessner, Nucl. Acids Res. 15: 6763. C., Kirchgessner, Structure-function A., Gambert, J. M. (1990a) Compound splice site of intron 2 (AG-AA) with familial 6: 335-341. Hata, A., Emi, M., Luc, G., Basdevant, lipoprotein Heizmann, Africans J. Lipid Res. 33: 106771072. M., Raper, S. E., Kozarsky, K., Stein, E. A., Engelhardt, P H. and Lalouel, (LPL) gene: HindIII. in exon 5 of the lipoprotein 1138: 353-356. Detection C., Ladias, J., Antonarakis, lipase (LPL) gene. Nucl. M. and Lusis, A. J. (1987) RFLP for the human lipoprotein Hayden, M. R. (1990) Frameshift 266: 24757-24762. T, Yamada, N., Murase, Kawamura, Y. and Takaku, F. (1991b) Occur- that causes familial lipoprotein J. Biol. Chem. Heinzmann, Derby, C., Antonarakis, 88: 1856-1864. H., Koga, S., Yazaki, Y., Furuichi, Y., Emi, M., Lalouel, J. M. and White, Arteriosclerosis M., H., Takaku, Hegele, R. A., Nakamura, R. (198913) Two RFLPs at the lipoprotein lipase deficiency. S., Shimano, H., Harada, K. and and missense mutations lipase gene. Nucl. mutation with in the lipoprotein familial lipoprotein lipase gene in two unrelated families lipase deficiency. J. Lipid 745-754. Iverius, I? H. and Brunzell, J. D. (1988) Relationship protein lipase activity women. J. Clin. and plasma sex steroid Invest. 82: 1106-1112. Res. 33: between lipolevel in obese 132 V. Murthy Jones, B., Wallace, A., Harding, D. R., Hancock, bell, C. H. (1983) Occurence micronemia Julien, of idiopathic in a cat. Vet. Rec. 112: 543-547. I’. (1992) High frequency the Quebec Julien, population. P., Gagne, Moorjani, C., Murthy, tein lipase gene as Julien, Sniderman, pop- S. P., Murthy, C. and Lupien, and dyslipopro- Woodford, Symposium F. I?, Davignon, on J. and G., Despres, J. P., Murthv, M. R. V., Gagne, C., Gaudet, D., Brun, D., Cadelis, F., Moorjani, S. and Lupien, I? J. (1995b) Effect of obesity on plasma triglyc- eride levels in familial lipoprotein Yamashita, Y., Kubo, M., Nozaki, S., Funahashi, S., Sho, N. and Tarui, S. (1989) Autoimmune chylomicronemia. Kirchgessner, N. Engl. J. Med. T G., Svenson, (1987) The sequence Kirchgessner, hyper- encoding lipoprotein T. G., Chuat, M. C. lipase. A lihi, A., Schotz, J. C., Heinzmann, M. C., Galibcrt, of the human C., Etienne, J., lipase F. and Lusis, A. J. (1989) Orga- lipoprotein gene family. lipase gene and evolution Proc. Natl. Acad. Sci. USA 86: J., Nishida, Hashimoto, T., Ameis, D., Stahnke, H., Fukamachi, S. (1992) A heterozygous stop codon) in lipoprotein interface them. recognition Biophys. Kohayashi, Levesque, G., Julien, G., Schotz, M. C., I., Shirai, K., Saito, Y. and Yoshida, mutation (the codon lipase contributes for Mutat. Res. Commun. J., Sasaki, K., Galton, I? J. and Murthy, site screening D. and Stocks, method to a defect in lipid Bio- J. (1988a) Pvu-II RFLP D. and Stocks, J. (1988h) Bst-I RFLP J. Biol. Chem. R. C., Henderson, H., Castellani, I. J., Zhang, H., Kirk, E., Lookenc, A. and Bengtsson functional J., Inadera, H., Saito, (Ala334-Thr) H. E., Murthy, salve, M. V., Clarke, Lamhert, Res. Commun. Hayden, tein Iipnse: in viva evidence nemia in French Canadians. Biophys. 43 lipopro- 43 is essential Res. Commun. for 205: J., Kastelein, bases of codon M. C. and Schotz, M. C. (1987) Cloning of rat hepatic lipase cDNA: evidence for a lipase gene family. Proc. Natl. Acad. 84: 1526-1530. Y., Orimo, T., Kawamura, M., Shimano, H., Yazaki, Y., H. and Yamada, N. (1993) Mutational ysis of human lipoprotein lipase by carhoxy-terminal anal- truncation. (1977) I hyperlipoproteinemia. Subclinical chronic pancreatitis Am. J. Med. 62: 144-149. Lalouel, J. h4., Wilson, D. E. and Iverius, P (1992) Lipoprotein and hepatic triglyceride Curr. Lipidol. Opin. lipase: molecular 3: 86-95. B. I., Roederer, G., Liu, J. J., Brunzell, J. D. and Hayden, occurring aspartic acid I56 is essential B. I., Bijvoet, G., Murthy, mutations at the first acid 156 in the proposed lipase. In viva evidence that for catalysis. J. Biol. Chem. 267: mutation lipoprotein different and genetic lipase aspects. S., Henderson, H. E., Cramb, I’., Bakker, H. D., Kas- J. D. and Hayden, M. R. (19921~) A mis- (Asp250-Asn) in exon 6 of the lipase gene causes chylomicronemia ancestries. Genomics Ma, Y., Liu, M. S., Ginzinger, version of a mild-to-severe D., Frohlich, phenotype mutation human in patients of 13: 64’1-653. Hayden, M. R. (1993) Gene-environment for a SerI72-Cys E., M. R. V., Julien, J., Brunzcll, interaction J. D. and in the con- in a patienr homozygous in the lipoprotein lipase gene. J. Invest. 91: 1953-1958. Ma, Y., Henderson, H. E., Liu, M. S., Zhang, H., Forsythe, 1. J., Clarke-Lewis, I., Hayden, M. R. and Brunzell, J. D. (1994a) Mutagenesis in four candidate 279-282, 291-304, 390-393 heparin binding and 439-448) regions (residues and identitication residues affecting heparin binding of human lipoprorein of lipase. J, Lipid Rcs. 35: 2049-2059. Ma, Y., Liu, M. S., Chitnyat, J. Lipid Res. 34: 176551772. Kraus, R. M. and Levy, R. I. lipoprotein cause of familial chylomicro- catalytic triad of human lipoprotein Clin. 5066515. Kozaki, K., Gotoda, in the human C., J. D. and N. Engl. J. Med. 324: 1761-1766. M. R. (1992n) Two naturally aspartic P. J., Brunzell, T, Tuzgol, S., Wilson, M. S., Davignon, G., Mon- T, Julien, I?, Gagne, J., Lupien, M. R. (1991) A mutation Roederer, 191: 1046~1054. site of human that asparagine Biochcm. catalysis and secretion. sites and Eur. J. Biochem. M. R. V., Roederer, lipase gene as the most common Ma, Y., Wilson, at the second base of codon asparagine glycosylation ofcleavage molecule. L. A., Normand, M., Davignon, Y. and S., Saito, Y., Fojo, S. S. and Brewer, H. B., Jr. (1994) A naturally N-linked G. (1993) Chymotryptic 213: 1855194. in exon Kobayashi, J., Inadera, H., Fujita, Y., Talley, G., Morisaki, N., Yoshida, mutation Olivecrona, lipase. Identification studies of the truncated telein, J. J., Rrunzell, in the proposed lipase. 269: 11417-11424. sense Biochem. at lipase (LPL) gene locus. Nucl. Acids Res. 1918-1923. 182: 70-77. N., Tashiro, for lipase gene. Hum. lipase (LPL) gene locus. Nucl. Acids L. W., Lusis, A. J., Ma, Y., Forsythe, lipasc gene in a case with type I hyper- in type 82: 795- 16: 11856. Biophys. Ouchi, Y., Lupien, in the lipoprotein Liu, M. S., Jirik, F. R., LeBoeuf, 7 of the lipoprotein Sci. USA of pancreatiMed. Res. 16: 2358. Li, S., Oka, Iipidcmia. Komaromy, Intern. 3: 416-417. Li, S. R., Oka, K., Galton, and second Ser447-n in a case with type I hyperlipidemia. Yoahida, S. (1993) A missense mutation occurring Ann. I’., Deshaies, the Pro207 - Leu mutation Ma, Y. H., Bruin, 9647-9651. Kohayashi, 86: 948-952. 798. Ma, Y., Henderson, 262: 8463-8466. S., Svenson, K., Amens, D., Pilon, C., d’Auriol, L., Anda- of the by hyperlipidemia. cleavage of lipoprotein K. L., Lusis, A. J. and Schotz, of cDNA T., 320: 1255-1259. member of a lipase gene family. J. Biol. Chem. nization tis masqued pro- lipase defi- in plasma of transgenic mice expressing human lipoprotein S., Matsuzawa, Guilhot, ciency. Proc. Natl. Acad. Sci. USA for a significant Brunzell, J. D. and Hayden, M. R. (1994) Alteration of lipid profiles lipase deficiency. Atherosclero- sis 115: S8. Kihara, human lipoprotein the human lipoprotein Amsterdam. Levesque, portion of mutations underlying al. J. J. and Hayden, accounts at the human lipoprotein cohort. In: Atherosclero- 10th International A. (eds.) Elsevier, C., of the lipopro- F., Gagne, of the M. E, lipase gene haplotypes pp. 254-257, I?, Vohl, B., Cadelis, in the Quebec Halappanavar, study of a French-Canadian sis X. Proceedings S., Deeh, S., Brunzell, J. D., Kastelein, M. R. (1989) A major insertion M. R. V. (1994) A rapid restriction G., Cadelis, I? J. (1995a) Lipoprotein Julien, J., Langlois, Lesser, I? B. and Warshaw, A. L. (1975) Diagnosis 10: 54-60. C. M. R. V., Levesque, in 6’75-676. M. R. V., Cantin, cause of dyslipidemias a I?, Savanurmath, Atherosclerosis, lipase deficiency 8: I? J. (1994) Mutations Can. J. Cardiol. teinemias: of lipoprotein Can. J. Cardiol. S. and Lupien, ulation. W. S. and Camp- familial hyperchylo- et D., Bruin, T, Beisiegel, U., Benlian, I?, Fouhert, L., De Genncs, J. L., Funke, H., Forsythe, I., Blaichmnn, S., Papanikolnou, and Hayden, M., Erkelens, D. W., Kastelein, J., Brunrell, J. D. M. R. (1994b) Recurrent missense mutations at the first and second base of codon Arg243 in human lipoprotein lipase in patients of different ancestries. Hum. Mutat. 3: 52-58. Human Lipoprotein Lipase Gene 133 Ma, Y., Ooi, T C., Liu, M. S., Zhang, H., McPherson, A. L., Forsythe, I. J., Frohlich, J., Brunzell, M. R. (1994~) High frequency of mutations protein lipase gene in pregnancy-induced sible association R., Edwards, J. D. and Hayden, in the human lipo- chylomicronemia: with apolipoprotein E? isoform. pos- J. Lipid Res. Mailly, F., Tugrul, Y., Reymer, P W. A., Bruin, T, Seed, M., Groenemeyer, 8. F., Asplund-Carlson, G. J., Kastelein, Humphries, S. E. and Talmud, and prevalance Arterioscler. Marshall, A., Valiance, J. J. I?, Hamsten, in the gene for lipoprotein cations D. The carbohydrate-peptide G., variant Functional and hyperlipidemic impli- subjects. and metabolism of glycoprotein. of Biochem. the Sot. G. A., Busch, Blankenship, S. J., Meredith, 263: H., Blanchette and cDNA S. S. and Scow, nonsecretable brown high mannose- adipocytes mice. J. Biol. Chem. cld/cld disorders of chylomicron of Inherited variants E. W., Morgan, J., Elwood, of com- 265: 1628- R., Rees, A., Hackshaw, I? C. and Galton, at the LPL gene locus associate defined severity of atherosclerosis in a Welsh population. F. S., Weidenbach, Scheele, Miesenbock, with angiographically Thromb. E, Swarovsky, G. A. (1989) Structure J. Biol. Chem. D. J. (1994) DNA and serum lipoprotein Arterioscler. 14: 1090~1097. B., LaForge, of canine pancreatic McGraw-Hill, Nilsson-Ehle, K. S. and lipase gene. E., Paulweber, F. and Patsch, J. R. (1993) Heterozygous due to a missense mutation lipopro- as the cause of impaired triglyceride tolerance with multiple lipoprotein malities. Minnich, J. Clin. encoding glyceridemic S. and Davignon, defective lipoprotein J. (1995) Prevalence lipase patients of French Canadian G., in hypertri- descent. J. Lipid Res. 36: 117-124. Mitchell, DNA R. J., Earl, L., Bray, P., Fripp, Y. J. and Williams, Molitch, variation and triacylglycerides. M. E., Oill, P. and Ode& lipidemia Monsalve, at the lipoprotein with quantitative lipoproteins during estrogen S., Kastelein, and Hayden, W. D. (1974) Massive H., Roederer, C., Davignon, Normand, hyper- Genet. G., Julien, M., Kumon, Y. and Hashimoto, Atherosclerosis mutation T., Nakauchi, T, Margallo, Nykjaer, in intron of mutation A., Bengtsson Annu. Rev. 1, Bharucha, Olivecrona, A., 188 cause of lipo- Y., Yamamoto, of lipo- 3 with hypertri- of Quebec. Hum. G., Lookene, J. (1993) The alpha 2-macroglobulin protein receptor-related receptor/low migrating very low density lipoprotein J. Biol. Chem. density lipo- lipase and beta- associated with the lipase. 268: 15048-15055. K., Tkalcevic, G. T., Stocks, W. V. (1989) Nucleotide the human A., Moestrup, U. and Gliemann, protein binds lipoprotein lipoprotein J., Galton, sequence D. J. and Brown, of PvuII polymorphic site at lipase gene locus. Nucl. Acids Res. 17: 6752. Oka, K., Tkalcevic, G. T., Nakano, K. and Brown, of human lipoprotein 21-26. T., Tucker, H., Ishimura W. V. (1990) Structure Erratum, Biochim. Biophys. G., Nakano, map Biophys. Acta 1049: Acta. 1090: 357 (1991). T., Tucker, H., Ishimura-Oka, and Virgil, W. (1991) Corrigendum: map of human lipoprotein Oka, and polymorphic lipase gene. Biochim. structure K. and polymorphic lipase gene. Biochim. Biophys. Acta 1090: 357. Olivecrona, T. and Bengtsson-Olivecrona, from research. Evener milk-The model In: Lipoprotein Publishers, Olivecrona, enzyme in lipoprotein lipase J. (ed.) Inc., Chicago. T and Bengtsson-Olivecrona, in lipoprotein G. (1987) Lipoprotein Lipase, pp. 15-58, Borensztajn, metabolism. Curr. G. (1990) Lipases involved Opin. T. and Bengtsson-Olivecrona, lipase and hepatic Ollis, D. L., Cheah, S. M., Harel, lipase. Curr. Opin. E., Cygler, M., Dijkstva, M., Remington, J. L., Verschueren, The o//3 hydrolase Lipidol. 1: 116-121. G. (1993) Lipoprotein Lipidol. 4: 187-196. B., Frolow, E, Franken, S. J., Silman, I., Schrag, K. H. G. and Goldman, fold. Protein G., A. (1992) Eng. 5: 197-211. J. C., Jr., Bengtsson-Olivecrona, role of the dimer to monomer J. D. population C. M., Weber, W., Beisiegel, Murthy, at codon K. (1994) Association 109: 62. M. C. (1980) Lipo- metabolism. P., Gagne, C., Dionne, C., De Braekeleer, and genealogy G., T (1985) Studies on inactivation Lee, N. S. and of lipoprotein dissociation. lipase: Biochemistry 24: 5606-5611. Parker, F., Bagdade, J. D., Odland, Evidence diabetic for the chylomicron eruptive histochemical lipase gene polymorphism glyceridemia. J., Fernandez Olivecrona, Invest. 86: 728-734. protein A. S. and Schotz, P., Deeb, P. J., Brunzell, lipase gene is a frequent T, Yasuoka, N., Suehiro, J. L. and Brown, M. S. (eds.) 89: 671-675. Osborne, protein lipase deficiency in persons of different ancestries. J. Clin. Nakamura, T., Bergeron, Sussman, 227: 522-525. R., Bruin, J., Lupien, M. R. (1990) A missense of the human lipoprotein in plasma high-density Hum. Biol. 66: 3833397. J. J., Peritz, L., Devlin, M. R. V., Gagne, J. (1994) lipase gene and their therapy. JAMA M. V., Henderson, Basis J. B., Wyngaarden, 49: 667-693. Olivecrona, polymorphisms association D. S., Goldstein, I?, Garfinkel, Biochem. lipase A., Roy, M., Giry, C., DeLangavant, Lavigne, J., Lussier-Cacan, of alleles abnor- Invest. 91: 448-455. A., Kessling, In: The Metabolic Stanbury, New York. Oka, K., Tkalcevic, G.. Holzl, B., Foger, B., Brandstatter, B., Sandhofer, levels 264: 12895512901. tein lipase deficiency metabolism. Disease, pp. 604-642, J. B., Fredrickson, Oka, R. K., Needham, A. K., Stocks, Mickle, Nikkila, E. A. (1983) Familial lipoprotein lipase deficiency and related S. K., Petersen, lipase by cultured lipase-deficient and 14: 869-873. 207 of the lipoprotein E. J., Chernick, of inactive hyperlipidemia Thromb. lipase gene in the French Canadian plasma hepatic triglyceride lipase. Mackie, combined Arterioscler. M. C., Burke, D. M., 1638. Mattu, familial P. J. (1992) Geographic A. D., 10907710914. R. 0. (1990) Synthesis type lipoprotein with LPL activity. M., Ma, R., Hayden, M. R. and Lupien, R. L. (1988) Isolation sequence of human postheparin bined 92: 312-313. J. D. and Deeb, S. S. (1994) The LPL gene distribution M. P., Fitzgerald, M. A. and Jackson, J. Biol. Chem. Masuno, in individuals C. C. (1993) at the lipoprotein lipase (LPL) A. E., Woods, C. W., G. D., Cardin, D. T., Mao, S. J., Rechtin, Racke, M. M., Schafer, Flanagan, locus. Hum. Genet. Nevin, D. N., Brunzell, Murthy, M. R. V., J&en, Symp. 40: 17-26. Martin, M. C., Scott, J. and Shoulders, repeat polymorphisms lytic enzymes and plasma lipoprotein 15: 468-478. nature linkages A. F., A., Olivecrona, I’. J. (1995) A common Vast. Biol. (1974) D., Winder, lipase (Asp9-Asn). in normal Thromb. R. Dinucleotide decreased 35: 1066-1075. Miller, Narcisi, T. M., Schotz, xanthomas: and electron G. F. and Bierman, E. L. (1970) origin in lipids accumulating a correlative microscopic in lipid biochemical, study. J. Clin. Invest. 49: 2172-2187. Paterniti, J. R., Brown, bined lipase deficiency W. V. and Ginsberg, H. N. (1983) Com- (cld): a lethal mutation 17 of the mouse. Science 221: 1677169. on chromosome 134 V. Murthy Peeva, E., Brun, T., Gagne, L. D., Mu&y, C., Lupien, slzc and distribution J. Obcs. Relat. in familial Metah. splice site and deficient Pep’, Lovecchlo, Mol. mun. in loss of enzymr activity. I?, Gaudet, G. and Lupien, interfere with infant! Perron, D., Gagne, 1 lipasc- in the very Atherosclerosis 109: 203. D., Moorjani, I? J. (19%) level in very low birth protein lipase Prcwato, C., A. (1994) A new Biophys. [Glu410-Val) Pykalisto, change Res. Com- leads to enzyme Rcs. Goldgerg, of decreased adipose after treatment 43: 591~600. M., Brunzell, and J. D. and apolipoprotein Reymer, I., Jansen, Hayden, (Asn291Ser) M. R. (1995) genes. M., Lohse, (LPL) gene Sadan, I!, Dugl, Shapiro, infant. J. Clin. EndoMolecular basks in the lipoprotein lipase H., Forsyth, llpase mutation cholesterol levels in serine pro- P., Beg, 0. U., Ronan, with familial J. Lipid M. M., Arher, Fojo, S. (1996) in the lipoprotein LPL lipase deficiency: LPL I., Joseph, D., France, M. S. (1977) Type I hyperllpoproteinemia __* _^_ 90: t i>-, , /. S. (1992) G cnetic S. and in a R-day-old C-II. Curr. role of Opin. Lipidol. Dug~, K. A. (1994) Structure, and role of lipoprotein Opin. Llpidol. lipase in lipoprotein function metabolism. Curr. W. (1990) [Primary hyper- lipoproteinemia E. and Andlcr, Type I in the neonatal period]. Klin. Padiatr. of chyloSearles, G. E. and Ooi, T. C. (1992) U n d errccognition micronemla as a cause of acute pancreatitis. Can. Med. Assoc. C. F., Luo, C. C., Nakanishi, Smith, L. C. and specific mutagenesis Ghan, M. K., Chen, L. (1990) I n eltm expression of the cloned human lipoprotein H. N. of apolipoprotein in transgenic J. (1993) Mechanism Shimada, of action M., Shimano, H., Gotoda, CII mice. J. Clin. offibrates. Invest. Postgrad. T., Yamamoto, M., Inaba, T., Yazaki, Y. and Yamada, of human Ilpoprotein Med. lipase K., Kawamura, N. (1993) Overexpression in transgenlc mice. Resistance to and hypercholesterolemia. 17924-17929. Erratum, J., Inadera, H., Ohkubo, J. J. Biol. Chem. 269: 11673 (1994). K., Kobayashi, Y. and Yoshida, relieved ollsm Y., Mori, S., Saito, S. (1992) Type I hyperlipoproteinemia hy lipoprotein lipase defect by administration in lipid-interface caused recognition of medium-chain was triglyceride. Metah- 31: 1161~1164. A. D., Julien, triglyceride I? and clearance, pp. xix-sxxvii, Bagdade, Inc., Sprecher, W. G. and IV 1995, K., L., Molitch, W. D., Pochlman, E. 1, Rogol, A. D. (cds.) Mosby-Year St. Louis. B. V., Bellet, D. M., Stein, Trp64- type L. E., Cianflone, C. R., Landsberg, Sniderman, D. L., Kohayashi, Harris, and of Endocrinology, J. D., Braverman, P., Kannan, D. M., Odell, A. D., Ryan, K. (1995) Peripheral pathway, In: Year Book E. S., Julicn, M. E., Nathan, Cianflone, the adipsin-ASP hyperlipoproteinemia. J., Rymaszewski, I’. S., Ameis, E. A., Scholtz, nonsense Stahnkc, mutation C., Davis, M., Goldberg, D., Yunker, I. J., R. L., Black, M. C. and Wiginton, in the lipoprotein hepatic Steffes, and S. H., sltc’- lipase gene. R. C., Doolittle, D. A. (1992) lipasc gene. J. Lipid lipase activity. J. Lipid of N-linked tions true sodium, in hypcrllpidemia. G., Mvher, potassium J. Lab. Clin. J. J. and Kuksis, lipid composition in a lactating proteinemia. J. Clin. Am. I? M., Demackcr, by oestrogen proteinaemia. Br. Med. R. and A., Ikeda, deficiency. transcript. protein lipase teinemia. Z., Shoji, M. S., Sanchez- Science T. and shared by 228: 893-895. Yamamoto, lipoprotein Iipase A. (LPL) of LPL mRNA 89: 581-591. A., Tsutsumi, A. (1994) A newly J. Lipid I hyperlipo- of 8 exons due to the absence Invest. Y., Mori, gene type (G916) in cxon 5 of LPL gene causes LPL protein mutation LPL i,h,,,,,,,) in a patient A. E (1986) Pancreawith J. L., Brown, on primary One base deletion A., Ikeda, plasma I hyperlipo- 41: 121-128. and EGF precursor. studies J. Clin. and type J. 293: 734. Y., Tsutsumi, Molecular method 88: 683-688. with Bell, G. (1985) Cassette genes for LDL receptor (1992) Med. in a patient T C., Russel, D. W., Goldstcin, Pescador, on concentra- A. (1985) Milk patient Nutr. and precise and chloride P. N. and Stalenhoef, titis induced H., Schotz, glycosylation Res. 32: 477-484. M. W. and Frier, E. F. (1976) A simple of determlning Takagi, M. H., Wang, Will, H. (1991) Effect and Yamamoto, 1806-1808. Semenkowch, D. W., I. J., Ginsberg, J. L. (1994) 0 bcrexpression no detectable 202: 355- 360. J. 1-t;: J. D., Rosenberg, R., Goldberg, hypertriglyceridcmia Takagi, 5: lli-125. B., Tro\vltzsch, 84: J. 69: S34-S41. Sudof, 3: 186-195. S. and coding Sci. USA Y 3: 168im 1690. Stuyt, dyslipoproteinemias: lipase and apolipoprotcin Santamarina-Fojo, Schluter, causes Steiner, Res. 37: 651~661. J. Pediat. lipoprotein Breslow, M. C. and mutations Acad. Res. 33: 859-866. IO: 28-33. insertion J. D. and Santamarina Tyr262-His). Santamarlna-Folo, and Book Res. 33: 1823-1832. Genetics K. A., Lohsc, in a patient N., Brucker, lipase and hypcr- HDL and intron for two point (Aap9-Asn, J. D. (1976) Reversal Furuichi, of a cDNA Natl. T., Leff, T., Smith, A., Ramakrishnan, Sniderman, 315: 458-459. R., Talley, G. D., Brunzell, Homozygoilty chylo- A lipoprotein Nature shuffling Nature R., Talley, D., Lie, K. E., Kastelein, lvith reduced atherosclerosis. J. (1985) Exon tease N. S., Hayek, Walsh, Shirai, and famlllal B. E., Zhang, J. C., Kromhout, is associated in premature Rogers, J. Lipid E., Grocnmeyer, H., Seidell, lipo- lipase S. S. (1992) mutations C-II genes. P. W, Gagne, J. and Deeh, Proc. 268: with P. K. and sequence for 265: 5429-5433. 4369-4373. Horton, chylomicronemia: lipase. Biol. Chcm. of lipoprotein of hypothyroidism. and triglyceride H. B., Jr. (1994) A novel inactivation lipoprotein 43 is important J. Biol. Chem. W. V., Qasha, cloning hypertriglyccridemia horn K. A., Ronan, Brunzell, K., Brown, diet-induced 109: 142. tissue lipoprotein triglyceridemia of familial twins site asparagine and secretion. G., Gagne, 35: 1552-1560. crinol. Metnh. premature on plasma domain A. l? and human deficiency weight P., Trcmblay, premature O., Dugi, P., Tremhlay, lipase birth Atherosclerosis in the C-terminal J. Lipid 0, loa Fo~o, S. and Brewer, mutation microncmia. lipoprotein S., J&en, weight deficiency. G. D., Santamarina S., lullen, Effect of feeding L., Guardamagna, missensc for hovinc Shachter, V., Tarricone, a Leu 365-Val C., Moorlanl, I? J. (lYY4a) Does P., Gaudet, M., Oka, Shepherd, growth C. and Lupien, Rouis, G + 2: 1455-1459. Riochcm. glycosylation activity Y. (lY87) Molecular F., Di Perna, lipase deficiency: N-linked both enzyme Senda, 199: 570-576. Perron, Reina, Int. A. M. and Capurso, Italian cabc of lipoprotein resulting lipase deficiency. in a lipoprotein Gcnet. G., Resta, M., Colacicco, cell usage of a mutated quantitation Hum. G., Chimienti, I’. (1992) Adipose 16: 737-‘744. G. (1993) C orrect transcript patient. J&en, lipoprotein Disord. Pepe, G. and Chimienti, Potential M. R. V., Despr@s, J. I’., Normand, P. J. and et ul. wth Z., Oida, identified (Cys2 39primary Res. 35: 2008-2018. K., Nakai, heterozygous stop/TGC972 type IV T. lipo--GA; hyperlipopro- Human Lipoprotein Lipase Gene Tashiro, J., Kobayashi, 135 J., Shirai, K., Saito, Y., Nakamura, moto, Y. and Yoshida, S. (1992) Trypsin the interfacial Tokyo activation treatment action of lipoprotein Wilson, H., Morimay impair protein lipase. J. Biochem. Wilson, 111: 509-514. Taylor, W. R. (1986) The classification J. Theor. Biol. Tenkancn, H., Taskinen, stitution (His183-Gln) M., Ulmanen, in exon 5 of the lipoprotein results in loss of catalytic I., C. (1994) A novel amino acid subactivity: phenotypic gene in a heterozygous lipase gene expression of the state. J. Lipid Res. 35: 220- Thorn, J. A., Chamberlain, L., Stocks, J. C., Alcolado, J. and Galton, pedigree atherosclerosis. Wilson, homozygous pholipid E in hepatic lipase catalyzed hydrolysis of phoslipoproteins. Biochemistry D. E., Hata, A., Kwong, L. K., Lingam, Ridinger, D. N., Yeager, C., Kaltenborn, and Lalouel, J. M. (1993) Mutations M. R. (1994) Apolipoprotein of chylomicronaemia Med. Genet. J. J. and Hayden, CII-Padova (Tyr37-stop) in an Italian kindred as a cause from Siculiana. J. H., Sarda, L., Verger, R. and Camhillau, of the pancreatic lipase-procolipase C. (1992) complex. Nature van Tilbeurgh, H., Egloff, M. I’., Martinez, R. and Cambillau, C. (1993) Interfacial C., Rugani, N., Verger, activation of the lipase- prolipase complex by mixed miscelles revealed by X-ray c*ystal- lography. 362: 814-820. Nature van Tilbeurgh, H., Roussel, (1994) Lipoprotein and catalysis. A., Lalouel, J. M. and Cambillau, R. (1984) Borgstrom, Pancreatic C. model based on the pan- consequences J. Biol. Chem. for heparin binding 269: 4626-4633. lipases. B. and Brockman, In: Lipases, pp. 82-150, H. (eds.) Elsevier Science Puhlish- ers B.V., Amsterdam. Vilaro, S., Llobera, M., Bengtsson-Olivecrona, T (1988) Synthesis of lipoprotein rats and localization them. Wilson, J. M., Grossman, and functional phys. Acta. J. and McConathy, properties of lipoprotein W. J. (1992) Structure lipase. Biochim. Ann. Surg. DNA Wang, sequence. H., Davis, in pancreatitis with hyper- I? and Lewis, B. (1992) Man- with severe hypertriglyceridaemia during Br. J. Obstet. D., Armstrong, Gynaecol. H., Kastelein, V. and Assman, lipase G. (1992) Mutations T., Pometta, in the lipowith type 1 T L. (1992) Mechanisms lipase alters cellular density lipoprotein, metabolism and heparan 13284-13292. by which lipo- of lipoprotein(a), and nascent lipoproteins. receptors 267: 235: 1638-1641. exchange: K. E. and Schotz, characterization of a chimeric lipase. Proc. Natl. Acad. 88: 11290-11294. lipase. J. Biol. Chem. Yang, W. S., Nevin, M. E, Gotto, 264: 16822-16827. D. N., Peng, R., Brunzell, in the promoter (LPL) gene in a patient with familial combined and low LPL activity. Proc. Natl. Acad. J. D. and Deeb, of the lipoprotein lipoprotein cholesterol Sci. USA in a patient and lipoprotein lipase hyperlipidemia 4466. Zambon, A., Torres, A., Bijvoet, S., Gagne, C., Moorjani, hypercholesterolaemia A. M., of bovine milk lipoprotein S. S. (1995) A mutation 92: 4462S., Lupien, of raised with familial lipase deficiency. H., Krapp, A., Ma, Y., Ginzinger, low Roles for low den- sulfate proteoglycans. J. D., Beisiegel, Hayden, M. R. (1994a) In e:itro mutagenesis sclerosis lipase critical Lancer Brunzell, for mediation of the binding protein (LRP). Athero- R. C., Liu, M., Jirik, F. R., Henderson, L. W., Lusis, A. J., Forsythe, J. D. (1994b) Lipoprotein expressing Zilversmith, human lipoprotein profiles in transgenic mice 109: 66. a postprandial phenome- 60: 473-485. E., Scheffler, E., Forte, T. M., Potenz, R., Wu, W. and L. (1994) Transgenic mice expressing lipase driven by the mouse metallothionein type associated H., I., Ma, Y., Kirk, E. and lipase on a regular and high fat D. B. (1979) Atherogenesis: non. Circulation Zsigmond, U. and studies defining resi- 109: 66. H., Lehoeuf, Castellani, Chan, K. J., Fless, G. M., Petrie, K. A., Snyder, M. L., Brocia, sity lipoprotein M. C. and lipase complementary R. C., Nikazy, J., Seebart, diet. Atherosclerosis 99: 163-166. J. J. P, Bruin, protein lipase gene are not restricted to patients hyperlipidemia. Circulation 86: I-609. R. W. and Swenson, lipoprotein Yang, C. Y., Gu, Z. W., Yang, H. X., Rohde, Zhang, deficiency. H., Funke, T. G., Lusis, A. J., Schotz, Science of 343: 771-774. lipase of hepatic lipase and lipoprotein Sci. USA 3: 179-222. W. (1990) Structure of lipoproteins to the LDL receptor-related 182: 72-75. K., Jackson, of patients Biol. Chem. lipase. Nature dues of lipoprotein report of two cases with familial lipoprotein protein pancreatic gene therapy of familial Ther. A. and Hunziker, K. L., Kirchgessner, Zhang, Bio- C. A. and Lesser, I? B. (1975) Inhibition pregnancy: Williams, Bio- 1123: l-17. Watts, G. E, Morton, Wichusch, Wion, Hum. Gene 341: 1119-1121. of serum and urine amylase activity agement human the end 736: 53-61. M., Raper, S. E., Baker, J. R., Newton, J. G. (1992) Ex ho E K., D’Arcy, low-density lipase in the liver of newborn of the enzyme by immunofluorescence. Warshaw, A. L., Bcllini, lipidemia. Winkler, in diabetes: Med. Suppl. I? J., Hayden, M. R. and Brunzell, J. D. (1993) Prevention G. and Olivecrona, J. 249: 549-556. Wang, C. S., Hartsuck, diabetes. J. Clin. Invest. Jr. and Pownall, H. J. (1989) Structure lipase. Molecular creatic lipase X-ray structure: in exon 3 of the lipoprotein to hyperlipidaemia J. Intern. M. C. (1991) Domain 359: 159-162. Verger, predisposition of the beginning? Lawn, R. M. (1987) Human 31: 622-626. van Tilheurgh, Structure T, Kastelein, J., P. H. in a family with hypertriglyceridemia, hypercholesterolemia. S. M., Bruin, A., Shuhua, K. C., Iverius, and noninsulin-dependent R. S. and Thoene, 31: 2332- 2338. Tuzgol, S., Bijvoet, mutation. Wilson, D. E., Kwong, L. K., Elbein, S. C. and Lalouel, J. M. (1994) Atherosclerosis K. H., Sisson, I’. and Waite, M. (1992) Role in high-density for a missense Invest. 86: 7355750. Genetic of apolipoprotein expres- lipase deficiency in the extended 92: 203-211. and hepatic 85: 55-60. T., Weisgraber, with lipo- 32: 1107-1114. R. R. and Lalouel, J. M. (1990) Phenotypic of a prohand J. Clin. pancreatitis, J. C., Oka, K., Ghan, D. J. (1990) Lipoprotein lipase gene variants in coronary Thuren, Metabolism D. E., Emi, M., Iverius, P. H., Hata, A., Wu, L. L., Hillas, lipase gene segregating 228. I. F. (1983) Phenotypic pedigree of a proband sion of heterozygous lipoprotein M. R., Antikainen, K. and Ehnholm, in the extended lipase deficiency. E., Williams, of amino acid conservation. 119: 205-218. Kontula, mutant D. E., Edwards, C. Q and Ghan, heterogeneity with increased perinatal plasma very low density lipoprotein human lipoprotein promoter. A pheno- mortality and reduced of normal size. J. Biol. Chem. 269: 18757718766. Zuliani, G. and Hobbs, morphism H. H. (1990) Tetranucleotide repeat poly- in the LPL gene. Nucl. Acids Res. 18: 4958.