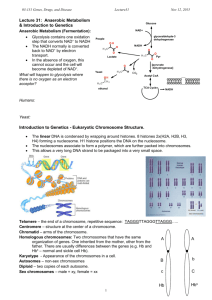
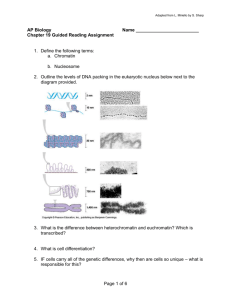
etd-plt-035-part 2v2
advertisement

INTRODUCTION 1 DNA hypermethylation-associated gene silencing Epigenetics defines all heritable changes in gene expression that are not the result of alterations in the primary DNA sequence. It is increasingly apparent that epigenetics, heritable changes in gene expression that are not caused by changes in DNA sequence, plays an important role in tumorigenesis (Rountree et al., 2001). DNA can be methylated at the 5-carbon position of cytosines in the context of a CpG dinucleotide. In mammals, DNA methylation patterns are established by at least three DNA methyltransferases: Dnmt1, Dnmt3a, and Dnmt3b. Dnmt3a and Dnmt3b are characterized mainly as de novo methyltransferases and are essential in establishing methylation patterns during development (Okano et al., 1999). Dnmt1 appears to be the main enzyme required for the maintenance of these methylation patterns (Bestor et al., 1988). In addition, Dnmt1 may also play a role in de novo methylation (Jair et al., 2006) and may cooperate with Dnmt3b to maintain DNA methylation in cancer cells (Rhee et al., 2002). Clusters of CpG dinucleotides, called CpG islands, are present near or within the promoter regions of about 40% of mammalian genes (Robertson and Wolffe, 2000). Normally, CpG islands are protected from methylation while those CpG dinucleotides outside the island are methylated (Graff et al., 1997). The mechanism of this protection is unknown. Methylation of CpG dinucleotides is a way to silence noncoding regions of the genome, such as pericentric heterochromatin and transposons (Baylin, 2005; Bird, 2002; Wolffe and Bird; 1999). Hypermethylation of CpG islands in the promoter regions in normal cells is a means to silence non-expressed genes, such as imprinted genes and those on the inactive X chromosome (Baylin, 2005; Bird, 2002). In the cancer cell, however, promoter CpG islands somehow lose the protection and become 2 aberrantly hypermethylated. In various tumor types, different gene promoter have been identified and shown to have aberrant hypermethylation of CpG islands that are normally unmethylated (Baylin and Herman, 2000a). Aberrant promoter DNA hypermethylation and associated epigenetic gene silencing frequently provide for a loss of tumor suppressor gene function in cancer (Baylin and Herman, 2000b; Jones and Laird, 1999). Two models have been proposed for the mechanism of DNA hypermethylation associated silencing. The first model suggests that DNA hypermethylation directly prevents sequence specific transcription factors from binding to the DNA (Robertson and Jones, 2000). The second model proposes that DNA hypermethylation is indirectly involved in silencing. Many studies have shown that silencing may be mediated through methyl-CpG binding proteins (MBDs; Wade, 2001; Bird, 2002). The MBDs do not bind to DNA in a sequence specific manner but preferentially bind to methylated CpGs. Some of the MBDs have been shown to be an integral part of, or associated with, repressive protein complexes containing histone deacetylases (HDACs) and chromatin remodeling proteins (Ballestar and Wolffe, 2001; Bird, 2002; Wolffe and Bird; 1999). These repressive complexes alter the chromatin structure surrounding hypermethylated DNA into a transcriptionally inactive conformation (Varga-Weisz, 2001). Histone modification The N-terminal tails of histones can be modified by covalent post-translational modifications. Specific residues on these histone tails can be phosphorylated, acetylated, and methylated (Zhang and Reinberg, 2001; Jenuwein and Allis, 2001; Litt et 3 al., 2001; Noma et al., 2001; Peters et al., 2002). The existence of these histone modifications has been known for many years, but for some of these modifications, their functional significance is just being discovered. The idea that combinations of modifications on the histone tails can determine what protein, or protein complexes, may bind to the chromatin to determine biological activity is referred to as the “histone code” hypothesis (Strahl and Allis, 2000). Specifically phosphorylated serine and threonine residues are critical in ensuring proper mitotic chromosomal condensation and segregation (Nowak and Corces, 2004). Histone acetylation is the most studied histone modification. The tails of H3 and H4 can be acetylated at particular lysine residues and these histones are found at euchromatic regions of the genome and at actively transcribed genes (Hake et al., 2004). Methylation can occur on certain lysine and arginine residues. Currently, many studies are focusing on lysine methylation. Methylation of some lysines is associated with inactive genes, while other methylated lysines correspond to active genes. Different modifications, or combinations of modifications, may alter the functional properties of chromatin. Studies of histone tail modifications at genes in different organisms reveal that histone methylation, in addition to histone acetylation, is associated with different chromatin conformational states and transcriptional status. Methylation of lysine 4 on the N-terminal tail of histone H3 (methyl-H3-K4) is associated with euchromatic chromosomal domains (Noma et al., 2001) and transcriptionally active genes (Heard et al., 2001; Xin et al., 2001; Boggs et al. 2002). Areas enriched with methyl-H3-K4 also correlate with acetylation of H3 (Litt et al., 2001). In contrast, methylation of lysine 9 on the N-terminal tail of H3 (methyl-H3-K9) 4 is localized to heterochromatic domains (Nakayama et al., 2001; Noma et al., 2001; Litt et al., 2001) and transcriptionally inactive genes (Noma et al., 2001; Litt et al. 2001; Heard et al., 2001; Peters et al., 2002). The chromatin at unmethylated CpG islands has characteristics of transcriptionally active chromatin. It is enriched in hyperacetylated histones H3 and H4 and has a more open conformation when compared to bulk chromatin (Tazi and Bird, 1990; Litt et al., 2001). The chromatin of in vitro methylated, CpG-rich DNA is associated with hypoacetylated histones H3 and H4 and is resistant to nuclease and restriction enzyme digestion (Schubeler et al., 2000; Davey et al., 1997; Kass et al., 1997). It is speculated that MBDs recruit chromatin-remodeling complexes to hypermethylated CpG islands and these complexes facilitate the access of HDACs to the histones and, in turn, alter the chromatin into a closed and transcriptionally inactive conformation. Histone variants and histone replacement Studies of Neurospora crassa and Arabidopsis thaliana reveal that patterns of DNA methylation depend on histone methylation (Tamaru and Selker, 2001; Johnson et al., 2002). Additionally in Arabidopsis, loss of DNA methylation leads to loss of methyl-H3-K9 only when coupled with transcription (Johnson et al., 2002). A model has been suggested in which the histone variant H3.3 replaces the core histone H3 during transcription and this replacement leads to the loss of methyl-H3-K9 (Ahmad and Henikoff, 2002). This concept of histone replacement was postulated from studies done in Drosophila melanogaster. H3.3 is highly conserved across multiple species and 5 differs from H3 at only four amino acids positions (Albig et al., 1995). In Drosophila, H3.3 is deposited at transcriptionally active loci in a replication-independent manner, while histone H3 is deposited in a replication-coupled manner (Ahmad and Henikoff, 2002). More recently, it was shown that induction of a heat shock gene displaces both H3.3 and H3, followed by selective deposition of H3.3 (Wirbelauer et al., 2005). When comparing histone tail modification profiles of H3 and H3.3, although none of the modifications examined were exclusive to either histone, H3.3 was shown to be enriched with modifications that are associated with transcriptionally active genes whereas H3 was shown to be enriched with methyl-H3-K9, the modification associated with silent genes (McKittrick et al., 2004). There are, as yet, no extensive data as to how H3.3 functions in mammalian cells in comparison with how it does in Drosophila. In HeLa nuclear extracts, H3 and H3.3 are in distinct complexes and mediate nucleosome assembly in a DNA synthesis dependent and independent manner, respectively (Tagami et al., 2005). In human cancer cells, upon induction of a stably integrated gene, H3.3 is deposited at the active locus (Janicki et al., 2004). This may be where the known functional similarities end. In Drosophila, H3.3 is enriched throughout the body of active genes, while enrichment of active histone modifications is greatest at the promoter region and decreases gradually through the coding region (Wirbelauer et al., 2005). In one study examining mouse NIH3T3 cells, H3.3 was enriched at the promoter and the coding regionof genes (Daury et al., 2006). However in another study of mouse cells, both H3.3 and active histone modifications are enriched at the promoter region and depleted in the body of the gene 6 (Chow et al., 2005). The consequence of histone replacement may be different in different organisms. In summary, replacement of core histones with histone variants is another way in which chromatin can be altered. Histone replacement may be a mechanism by which inactive genes that are silenced through repressive histone tail modifications can be reactivated by replacing them with histones carrying active modifications. The involvement of histone variants in the re-activation of genes introduces another way in which chromatin can regulate gene transcription. Goals The goal of this thesis is to elucidate the interaction of histone tail modifications and histone replacement with DNA hypermethylation in re-activating genes silenced with DNA hypermethylation in cancer. We will determine how covalent posttranslational modifications on histone tails differ at the chromatin around a hypermethylated and unmethylated promoter and asses if there is a pattern of histone modifications that determines the status of gene expression. Then, we will ascertain if these histone modifications influence patterns of DNA methylation, or vice versa, by examining changes in the histone modifications along a hypermethylated promoter after treatment with drugs inhibiting DNA methylation or histone deacetylation. We will also investigate whether histone replacement is involved in the re-expression of a gene silenced with DNA hypermethylation by determining if the histone variant H3.3 is enriched at a hypermethylated, unmethylated, or demethylated gene promoter. 7 There is evidence for a relationship between DNA hypermethylation and histone tail modification in Neurospora, Arabidopsis, and Drosophila. However, there are only a few studies examining this relationship in humans, much less in human cancer. Our lab has previously shown that DNA hypermethylation appears to be dominant over at least histone deacetylation in maintaining gene silencing in cancer cells (Cameron et al., 1999; Suzuki et al., 2002). A panel of genes typically hypermethylated in cancer were re-expressed following treatment with the DNA demethylating agent 5-aza-2'deoxycytidine (5-Aza-dC), but not with the HDAC inhibitor Trichostatin A (TSA). TSA was able to enhance gene re-expression only if the cells were first treated with 5-AzadC. Therefore, it seems DNA hypermethylation is at least dominant over histone deacetylation in silencing genes. Further study of the relationship between chromatin changes and DNA hypermethylation may elucidate the mechanism of re-expressing genes silenced with DNA hypermethylation in human cancer. 8 DEPENDENCE OF HISTONE MODIFICATIONS AND GENE EXPRESSION ON DNA HYPERMETHYLATION IN CANCER 9 Introduction Aberrant promoter DNA hypermethylation and associated epigenetic gene silencing frequently provide for loss of tumor suppressor gene function in cancer (Baylin and Herman, 2000b; Jones and Laird, 1999). DNA methylation-mediated gene silencing is closely linked to the deacetylation of histones (Baylin and Herman, 2000b; Jones and Laird, 1999). More recently, methylation of histones at key lysine residues has been shown to work in concert with acetylation and other modifications to provide a “histone code” that may determine heritable transcriptional states (Jenuwein and Allis, 2001). The data for this have come predominantly from studies examining broad domains in chickens (Litt et al., 2001), yeast (Noma et al., 2001), and the mammalian inactive X chromosome (Heard et al., 2001; Peters et al., 2002; Boggs et al., 2001), as well as individual promoter sites known to be expressed or silenced by epigenetic mechanisms such as X inactivation (Boggs et al., 2002) and genomic imprinting (Xin et al., 2001). These studies reveal that acetylated histone H3 and methyl-H3-K4 are enriched in euchromatic domains and correlate with active gene expression, while methyl-H3-K9 is enriched at deacetylated, transcriptionally silent heterochromatic regions (Litt et al., 2001; Noma et al., 2001; Heard et al., 2001; Peters et al., 2002; Boggs et al., 2001; Xin et al., 2001). The above histone modifications have been widely hypothesized to determine active versus inactive gene expression status (Litt et al., 2001; Noma et al., 2001; Heard et al., 2001; Peters et al., 2002; Boggs et al., 2001; Xin et al., 2001). Moreover, presence of methyl-H3-K9 has recently been shown to be essential for all or a subset of DNA methylation in Neurospora crassa (Tamaru and Selker, 2001) and Arabidopsis thaliana 10 (Jackson et al., 2002), respectively. However, for hypermethylated tumor suppressor genes in human cancer, DNA hypermethylation appears to be dominant over at least the histone deacetylation part of the histone code for maintaining a silenced state (Cameron et al., 1999). In this regard, we have shown previously that the DNA demethylating agent 5-Aza-2’deoxycytidine (5-Aza-dC), but not the histone deacetylase (HDAC) inhibitor Trichostatin A (TSA), reactivates the expression of such genes (Cameron et al., 1999; Suzuki et al., 2002). We now provide evidence that promoter DNA hypermethylation can control transcriptional silencing and, either directly or indirectly, the state of key elements of the histone code. At hMLH1, a mismatch repair gene often silenced with aberrant CpG island hypermethylation in colorectal cancers (Herman et al., 1998), a zone of deacetylated H3 (deacetylated histone H3-K9 and -K14) plus methylH3-K9 (dimethyl-H3-K9) surrounds the hypermethylated, silenced promoter. This same promoter when unmethylated and active is embedded in methyl-H3-K4 (dimethyl-H3K4) and acetylated H3 (acetylated histone H3-K9 and -K14). Treatment with TSA fails to reactivate the hypermethylated gene or dramatically alter the histone modifications examined. However, 5-Aza-dC treatment leads to initiation of demethylation by 12 hours, appearance of transcription by 24 hours, and full reversal of key elements of the histone code by 48 hours. Thus, DNA hypermethylation, either directly or indirectly through suppressing transcription, appears to specify for repressive histone modifications at a tumor suppressor gene promoter. An important element of the findings is that the demethylating drug 5-Aza-dC appears to be a potent tool for dissecting the components of this DNA methylation-mediated transcriptional control and for potentially reversing their interaction for therapeutic purposes in cancer. 11 Materials and methods Cell culture. SW480 cells were maintained in McCoy’s 5A modified medium. RKO cells were maintained in MEM. All media (Invitrogen Corporation) were supplemented with 10% fetal bovine serum (Gemini Bio-Products) and 1% penicillin/streptomycin (Invitrogen Corporation) and grown at 37C in 5% CO2 atmosphere. 5-Aza-dC and TSA treatments. Cells were treated with mock or 1 µM 5-Aza-dC (Sigma-Aldrich) for 12 hr, 24 hr, 48 hr, or 5 days, or with 300 nM TSA (Wako) for 24 hr, as previously described (Cameron et al., 1999; Herman et al., 1998). Chromatin immunoprecipitation. We used the ChIP Assay Kit from Upstate USA Inc. and followed the manufacturer’s protocol with some modifications. Briefly, proteins were cross-linked to DNA by addition of formaldehyde directly to the culture medium to a final concentration of 1% for 10 min at room temperature. The crosslinking reaction was quenched by adding glycine to a final concentration of 0.125 M for 5 min at room temperature. The medium was then removed and cells were washed with 1X phosphate buffered saline (PBS) containing a combination of protease inhibitors (1mM Pefabloc and 1X Complete protease inhibitor cocktail; Roche Molecular Biochemicals). The PBS was removed and 0.2X trypsin (Mediatech) was added to the cells. After a 5 min incubation at 37C, ice-cold 1X PBS containing 10% FBS was added to stop trypsinization. The cells were scraped off the culture flask, pelleted, and washed twice with 1X PBS plus protease inhibitors as above. For each ChIP assay 12 approximately 106 cells were used. The sonicated samples were pre-cleared with 80 µl of Protein A and Protein G agarose beads (Upstate USA, Inc.; 3 parts Protein A and 1 part Protein G with 200 µg/ml salmon sperm DNA and 0.5 mg/ml bovine serum albumin in TE buffer as a 50% gel slurry) for 1 hr at 4°C with agitation. The soluble chromatin fraction was collected, and 5 µl of either anti-acetyl-Histone H3 (Lys 9 and Lys 14), anti-dimethyl-Histone H3 (Lys 4), anti-dimethyl-Histone H3 (Lys 9), or no antibody was added and incubated overnight with rotation (all antibodies from Upstate Biotechnology). Immune complexes were collected with 60 µl of the 3:1 salmon sperm DNA/Protein A and Protein G agarose beads. The beads were washed as recommended, but were transferred to a new tube before each wash. After elution, the cross-links were reversed and the samples were digested with proteinase K. DNA was recovered by phenol extraction, ethanol precipitated, and resuspended in 1X TE buffer. PCR amplification and analysis. Primer sets for PCR were designed to amplify overlapping fragments of approximately 200 bp along the hMLH1 promoter. One primer set for GAPDH was designed to amplify a 128 bp fragment of the genomic sequence to serve as an internal control. All primers were purchased from Invitrogen Corporation or IDT. All PCR reactions were performed with JumpStart REDTaq DNA Polymerase (Sigma-Aldrich) in a total volume of 25 µl, using 1-2 µl of either immunoprecipitated (bound) DNA, a 1:10 dilution of non-immunoprecipitated (input) DNA, or a no antibody control. All reactions were optimized with input DNA to ensure that PCR products for both hMLH1 and GAPDH were in the linear range of amplification. Ten µl of PCR product were size fractionated by PAGE and were 13 quantified using Kodak Digital ScienceTM 1D Image Analysis software. Enrichment was calculated by taking the ratio between the net intensity of the hMLH1 PCR product from each primer set and the net intensity of the GAPDH PCR product for the bound sample and dividing this by the same ratio calculated for the input sample. Values for enrichment were calculated as the average from at least two independent ChIP experiments and multiple independent PCR analyses of each. ChIP primer sequences are listed in Table 2. Methylation-specific PCR (MSP). Genomic DNA was isolated using the Wizard Genomic DNA Purification Kit (Promega). The genomic DNA was modified by bisulfite treatment, as previously described (Magdinier and Wolffe, 2001). The primers used for MSP have been previously described (Herman et al., 1998) and were purchased from Invitrogen Corporation. MSP primer sequences are listed in Table 3. RT-PCR. We isolated total RNA with Trizol (Invitrogen Corporation), according to the manufacturer’s instructions. RNA was reverse-transcribed using SuperscriptTM II Rnase H Reverse Transcriptase (Invitrogen Corporation). PCR was performed using 1 µl of cDNA and primers unambiguous for GAPDH or hMLH1 (Invitrogen Corporation). All PCR reactions were performed with JumpStart REDTaq DNA Polymerase (SigmaAldrich) in a total volume of 25 µl. RT-PCR primer sequences are listed in Table 3. 14 Results Mapping the histone code at hMLH1. We compiled a detailed map of histone acetylation and histone methylation across a 2 kb region of the promoter for hMLH1. We performed chromatin immunoprecipitation (ChIP) for two human colorectal cancer cell lines, RKO and SW480, in which the hMLH1 promoter is hypermethylated and transcriptionally silenced or unmethylated and transcriptionally active, respectively (Herman et al., 1998). The histone-associated DNA regions were analyzed using a multiplex PCR approach with overlapping primer sets spanning the promoter (Fig. 1A). Overall, acetylated histone H3 was enriched throughout the unmethylated hMLH1 promoter; however, there was essentially no acetylation of these same sites along the hypermethylated promoter (Figures 1B and 1C). Virtually identical results were observed for methyl-H3-K4 at the two promoters (Figures 1D and 1E). In stark contrast, methyl-H3-K9 was enriched along the entire hypermethylated, silenced hMLH1 promoter, especially over a region where it was depleted to virtually undetectable levels along the unmethylated, transcriptionally active promoter (Figures 1F and 1G). Thus, it seems that key elements of the histone code surrounding an aberrantly hypermethylated tumor suppressor gene in human cancer resemble the histone modifications that may be necessary for proper genomic imprinting and gene expression along the X chromosome in other mammalian cells (Heard et al., 2001; Peters et al., 2002; Boggs et al., 2001; Xin et al., 2001) and for defining large heterochromatic and euchromatic domains in chickens (Litt et al., 2001) and yeast (Noma et al., 2001). 15 Inhibition of HDACs with TSA. Although little is known about the relationship between DNA methylation and the methylation component of the histone code in mammals, it has been known for some time that DNA methylation and histone deacetylation work in concert to silence genes in cancer (Baylin and Herman, 2000b; Jones and Laird, 1999; Magdinier and Wolffe, 2001; Nguyen et al., 2001). In fact, we have shown previously that DNA methylation is dominant over histone deacetylation in maintaining a silent state at hypermethylated promoters, as 5-Aza-dC can reactivate genes silenced with aberrant promoter hypermethylation but TSA alone cannot reactivate these same genes (Cameron et al., 1999). We explored these findings further at the level of the histone code. We treated RKO cells with TSA, which alone did not reactivate hMLH1, but which was able to reactivate FABP4, a control gene silenced in these cells by a mechanism distinct from one involving DNA hypermethylation (Suzuki et al., 2002; data not shown). Subsequent ChIP and PCR analysis revealed only a slight increase in acetylated H3 at the hMLH1 promoter (Figures 2A and 2B) and essentially no change in methyl-H3-K4 (Figures 2C and 2D) and methyl-H3-K9 (Figures 2E and 2F). These data show that, in addition to being unable to reactivate expression of a hypermethylated, silenced hMLH1 gene, TSA alone is unable to evoke obvious changes in key parameters of the histone code at this promoter. Inhibition of DNMTs with 5-Aza-dC. Recent genetic studies in Arabidopsis (Jackson et al., 2002) and Neurospora (Tamaru and Selker, 2001) suggest that methyl-H3-K9 may determine sites of DNA 16 methylation, although evidence for the existence of this relationship in higher eukaryotes has not been explored. We therefore set out to probe this relationship between key elements of the histone code and DNA methylation. We addressed this question by using ChIP and PCR analysis to examine the fate of histone modifications at the hMLH1 promoter upon inhibition of DNMTs by a dose of 5-Aza-dC, which is sufficient to cause demethylation of the promoter region (Herman et al., 1998) and to reactivate the expression of the hypermethylated, silenced hMLH1 gene in RKO cells (Herman et al., 1998; data not shown). Surprisingly, after drug treatment, we observed a complete reversal of the histone code components examined at the hMLH1 promoter in RKO cells. Acetylated H3 and methyl-H3-K4 levels became markedly enriched (Figures 3A, 3B, 3C, and 3D), while methyl-H3-K9 levels were severely depleted (Figures 3E and 3F). Thus with 5-Aza-dC, we recapitulated in RKO cells the state of the unmethylated, expressed promoter originally observed in SW480 cells (compare Figures 3A, 3C, and 3E to Figures 1B, 1D, and 1F). This transformation of key parameters of the histone code upon inhibition of the DNMTs suggests that, in human colorectal cancer cells, DNA hypermethylation, or another activity mediated by DNMTs, may be essential for maintaining a particular combination of histone modifications at gene promoters silenced with aberrant DNA hypermethylation. Furthermore, the observation that 5Aza-dC, but not TSA, can both reactivate expression of the silenced hMLH1 gene and completely reverse key histone modifications surrounding the gene promoter strengthens the idea that there exists some interdependence between reversal of important histone code components and reactivation of a gene silenced with aberrant DNA hypermethylation. 17 Time course analysis after 5-Aza-dC treatment. The observation that inhibition of the DNMTs leads to both steady state reactivation of hMLH1 expression and complete reversal of key histone code parameters surrounding the gene promoter, invited delineating the sequence of events to help dissect the operative mechanisms. We performed time course studies in which RKO cells were treated with 5-Aza-dC and monitored over 5 days for the states of key elements of the histone code, DNA methylation, and gene expression using ChIP and PCR analysis, methylation-specific PCR (MSP), and reverse transcriptase-polymerase chain reaction (RT-PCR), respectively. For the ChIP and PCR analysis, we used four of the original thirteen primer sets (Figure 1A), which cover the region of greatest difference in histone modification observed between RKO and SW480 cells and also between 5-Aza-dCtreated and untreated RKO cells at hMLH1 (Figures 1A, 1B, 1D, and 1F; Figures 3A, 3C, and 3E). At 12 and 24 hours after the start of 5-Aza-dC treatment, there was no dramatic change in the histone code components examined (Figure 4). By 48 hours, acetylated H3 and methyl-H3-K4 showed dramatic enrichment in 5-Aza-dC-treated samples compared to mock-treated samples; at the same time, methyl-H3-K9 became severely depleted (Figure 4). We next used MSP to examine the methylation status of the hMLH1 promoter following treatment with 5-Aza-dC. The region examined covers the area of greatest CpG density in the promoter and overlaps with the region examined by ChIP and PCR analysis in these time course studies (Figure 1A). By 12 hours, we observed onset of demethylation of the promoter, which was maximal by 24 hours and sustained until 5 days after the start of drug treatment (Figure 5A). Finally, we examined re-expression of hMLH1 by 5-Aza-dC using RT-PCR. Transcriptional 18 reactivation became apparent by 24 hours after the start of 5-Aza-dC treatment, and gene expression continued throughout the time course (Figure 5B). The observed sequence of events, then, is demethylation of the hMLH1 promoter by 12 hours, appearance of hMLH1 transcript by 24 hours, and complete reversal of all examined histone code components along the gene promoter by 48 hours (Table 1). Although in these experiments it appears that demethylation distinctly occurred first, as it was detectable by 12 hours and maximal by 24 hours, we are less certain about the order of events with respect to reactivation of transcription and reversal of the histone code parameters examined, due to the different sensitivities of the techniques used. To help sort this out, we examined data from several immunofluorescence experiments in which we stained for hMLH1 protein after treatment of RKO cells for 24 hours with the same dose of 5-Aza-dC used in the present studies. No nuclear staining was visible in mock-treated cells, but distinct re-expression of hMLH1 protein was present in 33-50% of RKO cells by 24 hours (data not shown). These data suggest that a substantial percentage of cells were transcribing hMLH1 by 24 hours in our present time course experiments and that the ChIP procedures would likely have detected a distinct change in the histone code parameters examined if these changes had preceded transcription. Our data, then, suggest a sequence of events in which 5-Aza-dC produces demethylation first, transcriptional reactivation second, and reversal of important histone code components third. 19 Discussion Our data provide the first detailed map of H3 acetylation and H3 methylation for a hypermethylated versus an unmethylated gene promoter in cancer cells. We also demonstrate here that inhibiting the DNMTs, but not the HDACs, essentially recapitulates at a hypermethylated, silenced promoter a combination of histone modifications similar to that at an unmethylated, active promoter. Finally, our results show unequivocally that DNA demethylation precedes both the reactivation of the silenced gene and, somewhat surprisingly, the reversal of key elements of the repressive histone code. These findings are consistent with the idea that DNA demethylation, either directly, or indirectly by reactivating transcription of the hMLH1 gene, reverses important components of the repressive histone code surrounding the hypermethylated promoter. These results favor the idea that DNA hypermethylation, not a particular combination of histone modifications containing elevated methyl-H3-K9, is the dominant epigenetic mechanism involved in maintaining silencing of the hMLH1 gene. In considering the mechanisms which underlie our observation that upon treatment with 5-Aza-dC, demethylation precedes reactivation of transcription, which precedes reversal of key histone code parameters, at least two scenarios may be considered. The first potential mechanism is one in which DNA methylation plays a direct role in both gene silencing and maintaining a repressive histone code at a hypermethylated gene promoter in cancer. We could speculate that the DNA modification itself, or components of the DNA methylating machinery such as the DNMTs or methyl-CpG binding proteins, could directly interact with histone methyltransferases or proteins that target them, directing them to regions containing 20 DNA methylation and allowing them to set up a repressive histone code (Jones and Baylin, 2002). If this turns out to be the case, it would suggest a new paradigm, seeing that data from Neurospora (Tamaru and Selker, 2001) and Arabidopsis (Jackson et al., 2002) suggest the opposite and point to a role for methyl-H3-K9 in targeting and maintaining DNA methylation. Our data stress the importance of identifying the enzymes responsible for modifying the histones in the setting of mammalian gene promoters and developing histone methyltransferase inhibitors to formally test relationships between histone modifications and DNA methylation in mammalian cells. A second and more indirect mechanism may better fit the changes we have observed and relate to an important new view of relationships between histone code parameters and gene transcription (Goll and Bestor, 2002; Ahmad and Henikoff, 2002; Johnson et al., 2002). In this scenario, DNA demethylation leads to gene reactivation, which in turn, leads to reversal of key elements of the histone code. This possibility is supported by our temporal data and by recent exciting findings in Arabidopsis (Johnson et al., 2002) and Drosophila (Ahmad and Henikoff, 2002) by others. Johnson et al. report that loss of DNA methylation itself does not lead to a decrease in methyl-H3-K9; rather, only at loci where reactivation of transcription occurs due to loss of DNA methylation does methyl-H3-K9 decrease (Johnson et al., 2002). They postulate that methyl-H3-K9 may be replaced by replication-independent deposition of new nucleosomes containing variant histone H3.3 once transcription occurs (Johnson et al., 2002), a concept suggested by studies from Ahmad and Henikoff in Drosophila (Ahmad and Henikoff, 2002). In light of these findings, our data could be interpreted as showing that 5-Aza-dC leads to demethylation of the DNA, which causes reactivation of hMLH1 21 gene transcription and, possibly, subsequent deposition of H3.3. The newly deposited variant histones would lack methyl-H3-K9 and could undergo post-translational modification, including methylation at K4 or acetylation, resulting in a heritable histone code that supports active transcription at the hMLH1 promoter. This type of mechanism could also help to explain our previous findings that TSA alone cannot reactivate hypermethylated genes in cancer but can synergize with low doses of 5-Aza-dC to reactivate such genes (Cameron et al., 1999; Suzuki et al., 2002). In this model, TSA may be working by facilitating the acetylation of the newly deposited histones thus helping to augment newly initiated transcription. Although further studies must continue to verify the above proposed sequence of events, our new findings are important to multiple aspects of abnormal, epigenetically mediated gene silencing in cancer. Pooling all the available data, including ours and those from studies in Neurospora (Tamaru and Selker, 2001) and Arabidopsis (Jackson et al,. 2002; Johnson et al., 2002), the following sequence of events is a plausible model for DNA methylation-mediated silencing of tumor suppressor genes in cancer. Our extensive histone code map along the hMLH1 promoter in SW480 cells suggests that enrichment of acetylated H3 and methyl-H3-K4 within and upstream of promoter CpG islands could protect the islands at normally expressed mammalian genes from DNA hypermethylation, similar to the postulated methyl-H3-K4- and acetylation-mediated protection from transcriptional repression that has been suggested to occur in chickens (Litt et al., 2001) and yeast (Noma et al., 2001). Such protection may be lost in some cancers at selected sites because these key components of the histone code break down, allowing histone deacetylation to occur, methyl-H3-K9 to spread into the promoter, and 22 aberrant DNA hypermethylation of the CpG island and silencing to result. Our data suggest that DNA hypermethylation firmly maintains this new heritable silenced state by repressing transcription and, directly or indirectly, sustaining these key elements of a repressive histone code. Importantly, 5-Aza-dC is able to disrupt this established heritable state of the histones. These findings stress the usefulness of this drug for dissecting the basic relationships between DNA methylation and histone modifications for their contribution to gene expression patterns in normal and disease states, as well as the possibilities for reversing DNA hypermethylation and repressive components of the histone code for prevention and treatment of cancer. 23 Figure 1. Map of histone H3 modifications along a hypermethylated versus an unmethylated hMLH1 promoter. (A) Schematic of the hMLH1 promoter. The vertical lines represent the location of CpG dinucleotides, and the arrow indicates the approximate position of the transcription start site. The CpG island extends 3’ from approximately –800 (relative to the transcription start site) into exon 1. The doubled horizontal line denotes the region examined by MSP. In SW480 cells the promoter is unmethylated, and the gene is expressed. However in RKO cells, the promoter is hypermethylated, and the gene is silenced. The horizontal bars below the schematic indicate the location of the DNA fragments amplified by PCR done on the DNA recovered from ChIP experiments. The broken bars denote the primer sets used in the time course experiments. (B), (D), and (F) Enrichment of hMLH1 promoter DNA immunoprecipitated with antibodies specific for acetylated histone H3 (K9 and K14), dimethyl-H3-K4, and dimethyl-H3-K9, respectively. Points on the graphs represent data from the corresponding DNA fragment amplified by PCR, as shown at the bottom of panel (A). The value of each point was calculated as the average from two independent ChIP experiments and a total of four independent PCR analyses. Each error bar indicates the standard deviation from the mean. Open squares represent data from SW480. Closed squares represent data from RKO. (C), (E), and (G) Representative PCR analyses of ChIP on RKO and SW480 from areas typical of enrichment for acetylated H3, methyl-H3-K4, and methyl-H3-K9, respectively. Multiplex PCR was performed on bound (B) immunoprecipitated DNA and input (I) non-immunoprecipitated DNA with each hMLH1 primer set. 24 Figure 2. Inhibition of histone deacetylation by TSA fails to dramatically alter key components of the histone code map along the hypermethylated hMLH1 promoter. ChIP was done on RKO cells after treatment with 300 nM TSA for 24 hours. (A), (C), and (E) Enrichment of acetylated histone H3 (K9 and K14), dimethyl-H3-K4, and dimethyl-H3-K9, respectively, at the hMLH1 promoter. Open circles represent enrichment in RKO cells treated with TSA. Filled circles represent data from untreated RKO cells. Points on each graph represent data from the corresponding DNA fragment amplified by PCR, as illustrated in Figure 1A. The value of each point was calculated as the average from two independent ChIP experiments and a total of four independent PCR analyses. Each error bar indicates the standard deviation from the mean. (B), (D), and (F) Representative PCR analyses of ChIP performed on RKO cells, before and after treatment with TSA, from areas typical of enrichment for acetylated H3, methyl-H3-K4, and methyl-H3-K9, respectively. Bound DNA (B) and input DNA (I) were coamplified with primers for hMLH1 and GAPDH. 26 Figure 3. Inhibition of DNA methylation by 5-Aza-dC completely reverses all examined components of the histone code map along the hypermethylated hMLH1 promoter. ChIP was performed on RKO cells after treatment with 1 µM 5-Aza-dC for five days. (A), (C), and (E) Enrichment of hMLH1 promoter DNA precipitated by antibodies specific for acetylated histone H3 (K9 and K14), dimethyl-H3-K4, and dimethyl-H3-K9, respectively. Open circles represent enrichment in RKO cells treated with 5-Aza-dC. Filled circles represent data from untreated RKO cells. The value of each point was calculated as the average from two (untreated) or three (drug-treated) independent ChIP experiments and four independent PCR analyses from each untreated or drug-treated experiment. Each error bar indicates the standard deviation from the mean. Points on each graph correspond to the overlapping DNA fragments amplified by PCR as depicted in Figure 1A. (B), (D), and (F), Representative PCR analyses of ChIP done on RKO cells, with or without treatment with 5-Aza-dC, from areas typical of enrichment for acetylated H3, methyl-H3-K4, and methyl-H3-K9, respectively. DNA from bound (B) and input (I) fractions were coamplified with primers for hMLH1 and GAPDH. 28 Figure 4. Treatment with 5-Aza-dC completely reverses all components of the histone code examined at a hypermethylated hMLH1 promoter by 48 hours. The data represent two independent time course experiments in which RKO cells were treated with 1 M 5-Aza-dC (or mock-treated) and harvested at each time point shown for ChIP analysis. A total of four to seven PCR analyses were performed on the immunoprecipitated DNA from each time point. Each point on the graphs represents the average value of enrichment, and each error bar indicates the standard deviation from the mean. Open circles represent data from RKO cells treated with 5-Aza-dC. Filled circles represent data from mock-treated RKO cells. Data from a five day time point served as positive controls to ensure that drug treatment was effective. The broken horizontal bars under the hMLH1 promoter schematic in Figure 1A indicate the location of the primer sets used in this ChIP and PCR analysis. 30 Figure 5. Treatment with 5-Aza-dC initiates demethylation by 12 hours and transcription by 24 hours at a hypermethylated hMLH1 promoter. RKO cells were treated with 1 M 5-Aza-dC and harvested at the indicated time points. (A) MSP analysis of hMLH1. A doubled line above the hMLH1 promoter schematic in Figure 1A indicates the region examined by MSP. Methylation was detected by the presence of a PCR product amplified by methylation-specific primers in the “M” lanes. Demethylation was detected by PCR products amplified by unmethylated-specific primers in the “U” lanes. Bisulfite dH2O denotes bisulfite-treated dH2O, which served as a negative control for the treatment. RKO and SW480 served as positive controls for the methylated and unmethylated PCR reactions, respectively. (B) RT-PCR analysis of hMLH1 expression. GAPDH expression served as a loading control. Five day mock- and five day 5-Aza-dC-treated RKO cells served as positive and negative controls, respectively, for hMLH1 expression. 32 Table 1 Summary of time course data Changes observed DNA demethylation 0h no 12 h 24 h 48 h 5d yes yes yes yes Gene re-expression no no yes yes yes Acetylated H3 Methyl-H3-K4 Methyl-H3-K9 , depletion; , enrichment 34 Table 2 Sequences of ChIP primers Primer name Sequence GAPDH-AS1 5’ GTCCACCACCCTGTTGCTGTA 3’ GAPDH-S1 5’ CAGAGACTGGCTCTTAAAAAGTGC 3’ MLH1pro 1799R 5’ CACGAACGACATTTTGGCGCC 3’ MLH1pro 1601F 5’ GCAACCCACAGAGTTGAGAAATTTG 3’ MLH1pro 1669R 5’ CACCCTTCAGCGGCAGCTATTG 3’ MLH1pro 1471F 5’ GGATATTCCGTATTCCCCGAGCTCC 3’ MLH1pro 1565R 5’ CCGCTACCTAGAAGGATATGCG 3’ MLH1pro 1344F 5’ CAACGTTAGAAAGGCCGCAAGG 3’ MLH1pro 1432R 5’ GCCTCTGCTGAGGTGATCTGG 3’ MLH1pro 1226F 5’ GGCTCCACCACTAAATAACGCTG 3’ MLH1pro 1294R 5’ CAAGATGGAAGTCGACGAGGC 3’ MLH1pro 1105F 5’ GTCCGCCACATACCGCTCGTA 3’ MLH1pro 1194R 5’ TGTCGCCGCCTCATCGTAGCT 3’ MLH1pro 934F 5’ CAACACCTCCATGCACTGGTATAC 3’ 35 Table 2 Sequences of ChIP primers (continued) MLH1pro 1037R 5’ AAGAGAGAGCTGCTCGTGCAG 3’ MLH1pro 829F 5’ GGTTGCGTAGATTCCGTCAATGC 3’ MLH1pro 900R 5’ CTGCAAGGCGTTGACTTATCTCC 3’ MLH1pro 702F 5’ TCTTGCACCTCCAACTCAGGG 3’ MLH1pro 766R 5’ GTGGCCTATGAGAACTACCTCC 3’ MLH1pro 566F 5’ CCTCAAAGTATGGGTCGTGGTC 3’ MLH1pro 642R 5’ CAATCCTAGAGTCCCTGCAGAC 3’ MLH1pro 433F 5’ GATTAACATCTACATCATAGGAGCTC 3’ MLH1pro 481R 5’ GGATTTCTTCACTTGGAACTGTTGAG 3’ MLH1pro 284F 5’ CCCTCTCCTAAGCCAATTGTTCAG 3’ MLH1pro 398R 5’ GATTAAGACCAGAGGCGTTAGGC 3’ MLH1pro 188F 5’ CCATTGTTTGTCTGAGAAGTGGAC 3’ MLH1pro 240R 5’ CGTTCTTGGTTTCAGTAGGGGC 3’ MLH1pro 4F 5’ CTCTGAGGGCAGGAAAGTCTG 3’ 36 Table 3 Sequences of MSP and RT-PCR primers Primer name Sequence MSP primers MLH1 U F 5’ TTTTGATGTAGATGTTTTATTAGGGTTGT 3’ MLH1 U R 5’ ACCACCTCATCATAACTACCCACA 3’ MLH1 M F 5’ ACGTAGACGTTTTATTAGGGTCGC 3’ MLH1 M R 5’ CCTCATCGTAACTACCCGCG 3’ RT-PCR primers MLH1RT F 5’ GAATGCGCTATGTTCTATTCCATCC 3’ MLH1RT R 5’ ATAGATCAGGCAGGTTAGCAAGCTG 3’ 37 A HISTONE VARIANT ASSOCIATED WITH ACTIVE TRANSCRIPTION IS TARGETED TO AN UNMETHYLATED GENE PROMOTER IN CANCER CELLS 38 Introduction DNA in human cells is packaged as a nucleoprotein structure known as chromatin. It is becoming increasingly apparent that chromatin-associated proteins, such as histones, are extensively involved in the regulation of gene transcription. Recently there has been great interest in chromatin modification and how these modifications may define the transcriptional status of a gene. The N-terminal tails of histones can be phosphorylated, acetylated, and methylated at specific residues (Zhang and Reinberg, 2001). The association of acetylated histones H3 and H4 with active gene transcription has been shown at many genes (Peterson and Laniel, 2004). Methylation of certain residues is also associated with active genes, while methylation at other residues is linked with silent genes (Peterson and Laniel, 2004). It is thought that different combinations of these modifications can serve as binding sites for specific protein complexes and can define or modify the functional properties of chromatin (Strahl and Allis, 2000). Chromatin can also be altered by histone replacement, in which core histones are interchanged with replacement-subtype variant histones (Hake et al., 2004). All of the core histones, except histone H4, have several variants (Albig et al., 1995). The core histones are only expressed at the beginning of S-phase of the cell cycle (Frank et al., 2003; Malik and Henikoff, 2003). This is because the core histones are only incorporated into nucleosomes during DNA replication (Osley, 1991). In contrast, variant histones are expressed in a replication-independent manner, and therefore are constitutively expressed (Osley, 1991). H3.3, one the most studied histone variants, is highly conserved across many species and differs from H3 at only four amino acid 39 positions (Albig et al., 1995). Recent studies have provided evidence that H3.3 associates with actively transcribed genes and is involved in the reactivation of genes. H3.3 was shown in to be deposited at actively transcribed genes in a replicationindependent manner (Ahmad and Henikoff, 2002; Daury et al., 2006). This is in contrast to H3, which was not enriched at actively transcribed genes and was deposited in a replication-coupled manner (Ahmad and Henikoff, 2002). Detailed studies of H3.3 from plants and animals demonstrated that H3.3 is enriched with histone tail modifications that are associated with active genes (McKittrick et al., 2004; Waterborg, 1990), while H3 is enriched with modifications associated with silent genes (McKittrick et al., 2004; Johnson et al., 2004). When a gene was reactivated, both H3.3 and H3 were displaced, followed by deposition of H3.3 (Janicki et al., 2004; Schwartz and Ahmad, 2005; Wirbelauer et al., 2005). H3.3 deposition was also shown to be tightly coupled with transcription (Schwartz and Ahmad, 2005; Daury et al., 2006). Most importantly, H3.3 was found to combine with active histone modifications to form a stable, heritable mark during mitosis (Chow et al., 2005). Aberrant promoter DNA hypermethylation is process that is associated with gene silencing. In many tumor types, important genes, such as tumor suppressor genes, have been shown to be silenced with DNA hypermethylation. We previously showed that a silent histone modification was enriched at a hypermethylated, silent gene and that active histone modifications were enriched at an unmethylated, expressed gene (Fahrner et al., 2002). In this study we attempted to define the localization of H3.3 in relation to DNA hypermethylation. 40 Materials and methods Construction of plasmids. The H3.3 constructs were made from the plasmid HS-H3.3YFP (a generous gift from K. Ahmad). This plasmid contains the Drosophila H3.3A gene, with an 18 bp linker sequence at the 3’ end of the gene before the YFP ORF. The H3.3 gene and the linker sequence were PCR amplified from this plasmid using Platinum Pfx DNA polymerase (Invitrogen Corporation) and cloned into pBluescript SK+ (Stratagene) and the sequence was verified. Primers containing a triple HA sequence were annealed, then cloned into the pBluescript SK+ plasmid containing H3.3, 3’ of the linker sequence. The entire sequence of H3.3-linker sequence-triple HA was cut from the plasmid and sub-cloned into pIRESneo3 (Clontech) and pEFIRES-P (a generous gift from C.A. Zahnow). Both plasmids contain an internal ribosome entry site (IRES) between the multiple cloning site and the antibiotic resistance gene. In pIRESneo H3.3-HA, H3.3-HA is under the control of the cytomegalovirus (CMV) promoter and the plasmid contains a neomycin resistance gene. The sequence and orientation of H3.3-HA in the final constructs were verified by sequencing. Expression in the pEFIRES-P plasmid was driven by the human polypeptide chain elongation factor 1α (EF1α) promoter and the selectable marker was puromycin. Stable transfection of cells and cell culture. Twenty four hr before transfection, RKO and SW480 colorectal cancer cells were plated so that they were 80% confluent on the day of transfection. pIRES H3.3-HA was transfected into RKO and SW480 with Lipofectamine (Invitrogen Corporation). The transfection complex was diluted in OptiMEM I Reduced Serum Medium (Invitrogen Corporation). The media were removed 41 from the cells and the cells were washed once with 1X PBS (Invitrogen Corporation). Serum- and antibiotic-free media were added to the cells. The cells were incubated in this media for 15 min. The transfection complex was then added to the cells and incubated for 4 hr. The media were then replaced with media containing serum and antibiotics. Forty eight hr after the addition of the transfection complex, the cells were split 1:8 and placed in media with the selective antibiotic, Geneticin (Invitrogen Corporation). RKO and SW480 clones were selected in 0.8 mg/ml and 0.6 mg/ml Geneticin, respectively. After 3 wk, Geneticin was reduced to 0.6 mg/ml for RKO clones and 0.4 mg/ml for SW480 clones. pEFIRES-P H3.3-HA was transfected into RKO and SW480 with Lipofectamine 2000 (Invitrogen Corporation), according to the manufacturer’s instructions. Twenty four hr after transfection, RKO and SW480 cells were split 1:8 and placed in media with 2 and 4 µg/µl puromycin (Sigma-Aldrich), respectively. RKO and SW480 were maintained in Eagle MEM (Mediatech) and McCoy’s 5A modified medium (Mediatech), respectively. The media were supplemented with 10% bovine calf serum (HyClone) and 1% penicillin/streptomycin (Mediatech). All cells were maintained at 37C in 5% CO2 atmosphere. Western blot analysis. Whole cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (1x PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing 1 mM Pefabloc SC and 1X Complete Protease Inhibitor Cocktail (Roche Molecular Biochemicals). Nuclear lysates were prepared using the NEPER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology, Inc.). Whole cell 42 lysates (15 µg) or nuclear lysates (10 µg) were prepared in NuPAGE LDS Sample Buffer and run on a NuPAGE Novex 4-12% Bis-Tris Gel in NuPAGE MES SDS Running Buffer in the XCell SureLock Mini-cell Electrophoresis Apparatus (Invitrogen Corporation). Proteins were transferred to a PVDF membrane (Millipore Corporation) using the XCell SureLock Mini-cell Blot Module (Invitrogen Corporation). Rabbit antiHA (Y-11; Santa Cruz Biotechnology, Inc.) was used at 1:1000, mouse anti-β-Actin (clone AC-15; Sigma-Aldrich) was used at 1:10000, and mouse anti-HA (F-7; Santa Cruz Biotechnology, Inc.) was used at 1:10000. Immunoprecipitation. Protein lysates were prepared as described above. Five hundred µg of whole cell or 100 µg of nuclear lysate were precleared using 80 µl of Protein A and Protein G agarose beads (Upstate USA, Inc.; 3 parts Protein A and 1 part Protein G with 200 µg/ml salmon sperm DNA and 0.5 mg/ml bovine serum albumin in TE buffer as a 50% gel slurry) for 1hr at 4°C with rotation. The precleared samples were incubated with 5 µg of rabbit anti- HA or normal rabbit IgG (Santa Cruz Biotechnology) overnight at 4°C with rotation. The immune complexes were collected with 60 µl of the above Protein A/G slurry for 1 hr at 4°C with rotation. The beads were washed three times with RIPA buffer and the immunoprecipitated samples were separated by gel electrophoresis as described above. Immunofluorescence and microscopy. Cells were prepared for immunofluorescence as previously described (Reese et al., 2003). The cells were incubated with rabbit anti-HA 43 (Santa Cruz Biotechnology, Inc.), diluted 1:500, overnight at 4ºC in a humidifying chamber. A donkey fluorescein isothiocyanate (FITC)-conjugated anti-rabbit antibody (1:100; Jackson ImmunoResearch Laboratories, Inc.) was used for secondary detection and incubated with the cells for 1 h at room temperature. Cells were also stained with 100 µg/ml of 4'-6-Diamidino-2-phenylindole (DAPI, Roche Molecular Biochemicals) for 1 min. Coverslips were mounted onto slides with ProLong Gold antifade reagent (Invitrogen Corporation). Images (60X) were captured with the Nikon Eclipse E800 microscope and Nikon DXM1200F digital camera. The images were analyzed using MetaMorph TE200 (Universal Imaging Corporation). Chromatin immunoprecipitation (ChIP). ChIP was performed as previously described (Fahrner et al., 2002) with some modifications. For sonication, a Branson Ultrasonics Sonifier (model S-450A) with a 3 mm tapered microtip was used at an output of 2 and 40% duty cycle. The samples were sonicated with 20 sets of 10 second pulses, with 30 seconds of rest in between each pulse. The samples were pre-cleared with 80 µl of Protein A and Protein G agarose beads (3 parts Protein A and 1 part Protein G with 200 µg/ml salmon sperm DNA and 0.5 mg/ml bovine serum albumin in TE buffer as a 50% gel slurry) for 1hr at 4°C with rotation. Approximately 5 µg of either rabbit anti-dimethyl-Histone H3 (Lys 4), rabbit anti-HA, or no antibody was added and incubated overnight at 4°C with rotation. DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen), with modifications. Briefly, 5 volumes of PB buffer were added to each sample. The DNA was passed through the spin column and 44 washed once with PE buffer. DNA was eluted twice with 50 µl of EB buffer. Unless specified otherwise, all ChIP reagents were from Upstate USA, Inc. PCR amplification and analysis. Primer sets for PCR were designed as previously described (Fahrner et al., 2002) and purchased from Integrated DNA Technologies. All PCR reactions and analysis were also performed as previously described (Fahrner et al., 2002), except that PCR products were size fractionated on a 3% agarose gel and enrichment was calculated by taking the net intensity of the hMLH1 PCR product from the bound sample and dividing by the net intensity of the product from the input sample. Results Stable expression of tagged H3.3 As there is no commercially available antibody for H3.3, we created an epitope tagged fusion protein of H3.3 in order to follow the expression of H3.3 in our cells. H3.3 was PCR amplified from a Drosophila H3.3 cDNA sequence, which is identical to the human cDNA sequence (Albig et al., 1995) and a triple HA sequence was placed at the 3’ end of H3.3 (Figure 6A). The CMV-driven pIRESneo H3.3-HA construct was transfected into the human colorectal cancer cell lines RKO and SW480. Clones that were stably expressing the fusion protein were selected by resistance to neomycin. Western blot analysis revealed that almost all of the clones selected in RKO cells expressed H3.3-HA (Figure 6B). Three of the 24 clones picked did not survive selection and, of the remaining clones, the only clone not expressing H3.3-HA was clone 1. 45 Random clones were also analyzed by immunofluorescent staining for HA, which revealed that H3.3-HA is localized to the nucleus (Figure 6C). Western blot analysis of clones from SW480 cells showed no expression of H3.3-HA (Figure 6D). Only 11 of the 20 clones picked survived selection, and none of them expressed H3.3-HA. In the hope of obtaining clones from both SW480 and RKO, we sub-cloned H3.3-HA into another plasmid. The EF1α-driven pEFIRES-P H3.3-HA was transfected into RKO and SW480, and clones were selected by resistance to puromycin. Tweleve RKO clones were selected and survived selection. However, none of the RKO clones expressed H3.3-HA by Western blot analysis (Figure 7A). Of the 20 SW480 clones that were selected and survived selection, all expressed H3.3-HA but only clone 20 did not show a degradation product (Figure 7B). In order to define the relationship between H3.3 and DNA methylation, we elected to examine a RKO clone created with CMV-driven construct with SW480 clone 20, created with the EF1α-driven construct. We compared expression of H3.3-HA in the RKO clones against expression in the SW480 clone. From whole cell lysates, the SW480 clone expressed H3.3-HA more than the RKO clones did (Figure 7C). We established that the triple HA tag on our fusion protein was recognizable by an antibody against the tag. Whole cell and nuclear lysates from selected RKO clones were immunoprecipitated with an anti-HA antibody. Immunoprecipitation of whole cell lysates was much weaker than that of nuclear lysates (Figure 8). The antibody was able to recognize the tag in all of the nuclear lysates from the RKO clones, except for clone 1. This was the only RKO clone that did not show any H3.3-HA protein expression (Figure 6B). H3.3-HA was immunoprecipitated from whole cell lysates from three of the four 46 RKO clones expressing H3.3-HA (Figure 8). In the SW480 clone, the HA tag was identifiable in both whole cell and nuclear lysates. Exogenously expressed H3.3-HA is enriched at an unmethylated, and not a hypermethylated, promoter There is one study that has shown that H3.3 is selectively deposited at an induced gene in cancer cells (Janicki et al., 2004). This observation was made at a stably integrated transgene. We wondered if H3.3 would be found at an active, unmethylated gene in an endogenous setting, and how its localization might differ at a silent, hypermethylated gene. Arbitrarily, one RKO clone was chosen to compare against the SW480 clone. We examined the promoter of hMLH1, a mismatch repair gene whose promoter is hypermethylated and transcriptionally silenced in RKO cells, and is unmethylated and transcriptionally active in SW480 cells (Herman et al., 1998). ChIP was used to identify enrichment of H3.3-HA at either of these promoter settings in the respective clones. We analyzed a region of the promoter that we have previously shown to be the region of greatest difference in acetyl-H3 (K9 and K14), dimethyl-H3-K4, and dimethyl-H3-K9 observed between RKO and SW480 cells (Fahrner et al., 2002). H3.3HA was enriched across the unmethylated promoter in the SW480 clone, when compared to its localization at the hypermethylated promoter in the RKO clone (Figure 9A). Enrichment of dimethyl-H3-K4 was also examined and, as expected, there was general enrichment of this modification across the unmethylated promoter, and less enrichment at the methylated promoter (Figure 9B). 47 Discussion H3.3 is associated with active transcription. Recent studies show that H3.3 is deposited at active genes (Ahmad and Henikoff, 2002; Chow et al., 2005) and at induced genes (Janicki et al., 2004; Schwartz and Ahmad, 2005; Wirbelauer et al., 2005). We wondered if H3.3 could also distinguish between a hypermethylated, silent gene and an unmethylated, active gene. We show very preliminary data suggesting that H3.3 may be targeted to an unmethylated, active promoter, and not to a hypermethylated, silent promoter. H3.3-HA in the SW480 clone, where the hMLH1 promoter is unmethylated, is enriched across three of the four points analyzed at the promoter, but not in the RKO clone, where the promoter is hypermethylated. These data are compiled from one PCR analysis of the DNA isolated from one ChIP experiment. Although this is very limited data, we feel confident that the trend we see is valid based on the data that we see of dimethyl-H3-K4 enrichment. Our previous study showed that at these same points on the promoter, dimethyl-H3 K4 is enriched at the unmethylated hMLH1 promoter and depleted at the hypermethylated hMLH1 promoter (Fahrner et al., 2002). In our current study, we see enrichment of dimethyl-H3-K4 in SW480 wild type cells, compared to RKO wild type, at two of the four points. In addition, the SW480 clone shows enrichment at three of the four points, in comparison to the RKO clone. Nevertheless, data from at least two PCR analyses from multiple, independent ChIP experiments must be obtained to confirm our findings. Another challenge to our preliminary result is the fact that H3.3-HA in the RKO clones and the SW480 clone are expressed from different promoters. In the RKO clones, the CMV promoter is driving expression, while the EF1α promoter is driving 48 expression in the SW480 clone. When we evaluate the expression of H3.3-HA in the RKO clones and the SW480 clone, we see that the SW480 clone expresses much more H3.3-HA than any of the RKO clones even though the level of β-Actin appears to be similar in all clones (Figure 7C). In addition, when we compare expression in RKO clones from lysates harvested at an early passage (Figure 6B) to lysates harvested from clones growing in cell culture for two months (Figure 7C), we see a dramatic decrease in H3.3-HA expression at the later passage. However, expression from the EF1α promoter appears to be unchanged after two months in cell culture (compare Figure 7B to 7C). Although both promoters are constitutively active and are used often in expression vectors, their functional properties are not the same. In certain cell types, the CMV and EF1α promoters have different expression levels. In rat brain cells, CMV is most active in non-neuronal cell types and shows some activity in neurons, but EF1α is active only in neurons (Tsuchiya et al., 2002). In undifferentiated mouse embryonic stem (ES) cells, EF1α has high activity while CMV has moderate activity (Zeng et al., 2003). Activity of these promoters can also be affected by the number of cell passages. After three months in cell culture, EF1α activity in the undifferentiated ES cells is unchanged, but CMV activity is dramatically decreased (Zeng et al., 2003). In order to overcome this difference in expression between cell lines and cell passages, we are making new RKO and SW480 clones with the EF1α-driven construct. This promoter gives higher expression of H3.3-HA than the CMV promoter does in our cell lines. Furthermore, we do not see a difference in expression from the EF1α promoter after two months in cell culture. Once clones are selected and verified for 49 expression of H3.3-HA, we will repeat ChIP experiments comparing the targeting of H3.3-HA to a hypermethylated versus an unmethylated promoter. The final experiment is to examine the involvement of H3.3 in the reactivation of a gene silenced with DNA hypermethylation. We plan to treat RKO cells with 5-AzadC in a time course, as was done in our previous study. Upon treatment with 5-Aza-dC we saw DNA demethylation by 12 hours, gene re-expression by 24 hours, and finally reversal of the histone modifications by 48 hours (Fahrner et al., 2002). We will investigate cells harvested at different time points to determine when H3.3 appears at the hMLH1 promoter in our time line for re-expression. Replication-independent histone replacement may be a means to switch a pattern of histone modifications seen at a silent gene to a pattern found at an active gene (Ahmad and Henikoff, 2002). Studies of Drosophila H3 and H3.3 show that while none of the histone tail modifications are exclusive to either histone, H3 is enriched with the silent modification, dimethyl-H3-K9, and H3.3 is enriched with active modifications, including dimethyl-H3-K4, acetyl-H3-K9, and acetyl-H3-K14 (McKittrick et al., 2004). Upon re-activation of a gene, histone modifications could be switched by replacing a silent region having only H3-containing nucleosomes, enriched with a silent modification and depleted of active modifications, with H3.3-containing nucleosomes, enriched with active modifications and depleted of a silent modification. Additionally, histone modifications may be switched directly. The existence of histone acetyltransferases and deacetylases has been known for some time. A recently discovered lysine-specific histone demethylase, LSD1, can demethylate H3-K4 (Shi et al., 2004) or H3-K9 (Metzger et al., 2005). The substrate for LSD1 may differ 50 depending on its protein binding partners. In the BRAF-HDAC complex, the transcriptional co-repressor, CoREST, promotes the demethylation of H3-K4 by LSD1 (Lee et al., 2005). LSD1 can also co-localize with androgen receptor to stimulate androgen receptor-dependent transcription by demethylating H3-K9 (Metzger et al., 2005). It will be critical to determine if switching of histone modifications is occurring indirectly by histone replacement, directly by histone modifying enzymes, or a combination of both. 51 Figure 6. CMV-driven H3.3-HA is expressed in RKO, but not SW480 cells, and is targeted to the nucleus. (A) Schematic representation of the H3.3-HA fusion gene. (B) Expression of pIRESneo H3.3-HA in stably transfected RKO clones. Whole cell lysates from clones were analyzed by Western blot analysis. The top band is the loading control, β-Actin, and bottom band is H3.3-HA. Numbers across the top indicate clone number. WT is wild type, untransfected RKO. All clones, except clone 1, are expressing H3.3-HA. (C) H3.3-HA is localized to the nucleus. Two RKO clones were randomly selected for detection of H3.3-HA by immunofluorescence and compared with wild type. H3.3-HA was detected by an anti-HA antibody (left). DNA was counterstained with DAPI (center). The expression of H3.3-HA in both clones was confined to the nucleus. (D) pIRESneo H3.3-HA is not expressed in stably transfected SW480 clones. Whole cell lysates from clones were analyzed by Western blot analysis. The top band is the loading control, β-Actin, and bottom band is H3.3-HA. Numbers across the top indicate clone number. Clone number 2 from RKO (RKO 2) was used as a positive control for H3.3-HA (right). 52 Figure 7. EF1α-driven H3.3-HA is expressed in SW480 but not RKO. Whole cell lysates from clones were analyzed by Western blot analysis. The top band in all panels is the loading control, β-Actin. Numbers across the top indicate clone number. (A) pEFIRES-P H3.3-HA is not expressed in stably transfected RKO clones. Clone number 2 from RKO (RKO 2) was used as a positive control for H3.3-HA. The bottome band is H3.3-HA. WT is wild type, untransfected RKO. None of the clones show expression of H3.3-HA. (B) Expression of pEFIRES-P H3.3-HA in stably transfected SW480 clones. All clones, except clone 20, showed a doublet. The top band in the doublet is H3.3-HA, as confirmed by clone number 2 from RKO (RKO 2). WT is wild type, untransfected SW480. (C) A comparison of H3.3-HA expression among clones. Protein expression of H3.3HA from selected RKO clones were compared with the SW480 clone. The SW480 clone expresses more H3.3-HA than the RKO clones. 54 Figure 9. H3.3-HA is enriched at the unmethylated, expressed hMLH1 promoter. (A) Enrichment of H3.3-HA. (B) Enrichment of dimethyl-H3-K4. The data from ChIP were quantified and enrichment was calculated at four overlapping primer sets at the hMLH1 promoter. The points on the graph are as follows: the SW480 clone is represented by open squares, SW480 wild type by filled squares, RKO clone by open triangles, and RKO wild by filled triangles. 57 CONCLUSIONS 59 We set out to further our understanding of how reversal of DNA hypermethylation-associated gene silencing in cancer cells may be aided by chromatin changes. First, we established a relationship between promoter DNA hypermethylation and histone tail modifications. Through the examination of the active marks acetylated H3 (K9 and K14) and dimethyl-H3-K4, and the silent mark dimethyl-H3-K9, we discovered that distinct patterns of histone modifications are associated with the status of gene expression along a DNA hypermethylated versus a DNA unmethylated gene promoter. The two active marks are enriched and the silent mark is depleted at an unmethylated promoter. The reverse pattern was observed at a hypermethylated promoter. Second, we determined how DNA methylation influences patterns of histone modifications. Treatment of cells containing the hypermethylated gene with a histone deacetylase inhibitor did not alter the pattern of histone modifications. Therefore, DNA hypermethylation is dominant over histone deacetylation in silencing a gene. Treatment with a DNA demethylating agent, on the other hand, reversed the pattern of modifications at the hypermethylated promoter to one that was identified at the unmethylated promoter. A time course experiment of drug-induced DNA demethylation revealed that DNA demethylation precedes gene re-expression, which precedes reversal of histone modifications. Finally, we examined the relationship between DNA hypermethylation and a histone variant associated with active transcription. Preliminarily, we found the variant H3.3 to be enriched along an unmethylated promoter and depleted along a hypermethylated promoter. We need to explore this association further by assessing the role of H3.3 in DNA demethylation-induced gene re-activation. Replacement of H3 with H3.3 may be a way to switch the pattern of histone 60 modifications that accompanies the gene re-expression induced by treatment with 5Aza-dC, specifically the decrease in dimethyl-H3-K9 and increases in acetylated H3 and dimethyl-H3-K4. Understanding the mechanism of gene re-expression initiated by DNA demethylation provides an avenue to examine the interactions between promoter DNA hypermethylation and chromatin changes, which mediate heritable patterns of gene silencing. Defining this relationship could be critical in finding therapies that will reverse the gene silencing that is associated with DNA hypermethylation in cancer. 61 REFERENCES 62 Ahmad, K. and Henikoff, S. (2002). The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9, 1191-1200. Albig, W., Bramlage, B., Gruber,,K., Klobeck,,H.G., Kunz, J., and Doenecke, D. (1995). The human replacement histone H3.3B gene (H3F3B). Genomics 30, 264-272. Ballestar, E. and Wolffe, A.P. (2001). Methyl-CpG-binding proteins. Targeting specific gene repression. Eur. J. Biochem. 268, 1-6. Baylin, S.B. and Herman, J.G. (2000a). In DNA Alterations In Cancer: Genetic And Epigenetic Changes (ed. Ehrlich, M., Eaton Publishing, Natick, MA) 293-310. Baylin, S.B. and Herman, J.G. (2000b). DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 16, 168-174. Baylin, S.B. (2005). DNA methylation and gene silencing in cancer. Nat.Clin.Pract.Oncol. 2, Suppl. 1 S4-S11. Bestor, T., Laudano, A., Mattaliano, R., and Ingram, V. (1988). Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal 63 domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 203, 971-983. Bird, A.P. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 621. Boggs, B.A., Cheung, P., Heard, E., Spector, D.L., Chinault, A.C., and Allis, C.D. (2002). Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat. Genet 30, 73-76. Cameron, E.E., Bachman, K.E., Myohanen, S., Herman, J.G., and Baylin, S.B. (1999). Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 21, 103-107. Chow, C.M., Georgiou, A., Szutorisz, H., Maia e Silva, A., Pombo, A., Barahona, I., Dargelos, E., Canzonetta, C., and Dillon, N. (2005). Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 6, 354-360. Daury, L., Chailleux, C., Bonvallet, J., and Trouche, D. (2006). Histone H3.3 deposition at E2F-regulated genes is linked to transcription. EMBO Rep. 7, 66-71. 64 Davey, C., Pennings, S., and Allan, J. (1997). CpG methylation remodels chromatin structure in vitro. J. Mol. Biol. 267, 276-288. Fahrner, J.A., Eguchi, S., Herman, J.G., and Baylin, S.B. (2002). Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 62, 7213-7218. Frank, D. Doenecke, D., and Albig, W. (2003). Differential expression of human replacement and cell cycle dependent H3 histone genes. Gene 312, 135-143. Gabrielli, F., Aden, D.P., Carrel, S.C., von Bahr, C., Rane, A., Angeletti, C.A., Hancock, R. (1984). Histone complements of human tissues, carcinomas, and carcinoma-derived cell lines. Mol. Cell. Biochem. 65, 57-66. Goll, M.G. and Bestor, T.H. (2002). Histone modification and replacement in chromatin activation. Genes Dev. 16, 1739-1742. Graff, J.R., Herman, J.G., Myohanen, S., Baylin, S.B., and Vertino, P.M. (1997). Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J. Biol. Chem. 272, 22322-22329. 65 Hake, S.B., Xiao, A., and Allis, C.D. (2004). Linking the epigenetic “language” of covalent histone modifications to cancer. Brit. J. Cancer 90, 761-769. Heard, E., Rougeulle, C., Arnaud, D., Avner, P., Allis, C.D., and Spector, D.L. (2001). Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107, 727-738. Herman, J.G., Graff, J.R., Myohanen, S., Nelkin, B.D., and Baylin, S.B. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U S A 93, 9821-9826. Herman, J.G., Umar, A., Polyak, K., Graff, J.R., Ahuja, N., Issa, J.P., Markowitz, S., Willson, J.K., Hamilton, S.R., Kinzler, K.W., Kane, M.F., Kolodner, R.D., Vogelstein, B., Kunkel, T.A., and Baylin, S.B. (1998). Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. U S A 95, 6870-6875. Jackson, J.P., Lindroth, A.M., Cao, X., and Jacobsen, S.E. (2002). Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416, 556-560. 66 Jair, K.-W., Bachman, K.E., Suzuki, H., Ting, A.H., Rhee, I., Yen, R.W., Baylin, S.B., and Schuebel, K.E. (2006). De novo CpG island methylation in human cancer cells. Cancer Res. 66, 682-692. Janicki, S.M., Tsukamoto, T., Salghetti, S.E., Tansey, W.P., Sachidanandam, R., Prasanth, K.V., Ried, T., Shav-Tal, Y., Bertrand, E., Singer, R.H., and Spector, D.L. (2004). From silencing to gene expression: real-time analysis in single cells. Cell 116, 683-698. Jenuwein, T. and Allis, C.D. (2001). Translating the histone code. Science 293, 10741080. Johnson, L., Cao, X., and Jacobsen, S. (2002). Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr. Biol. 12, 1360-1367. Johnson, L., Mollah, S., Garcia, B.A., Muratore, T.L., Shabanowitz, J., Hunt, D.F., and Jacobsen, S.E. (2004). Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res. 32, 65116518. Jones, P.A. and Laird, P.W. (1999). Cancer epigenetics comes of age. Nat. Genet. 21, 163-167. 67 Jones, P.A. and Baylin, S.B. (2002). The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3, 415-428. Kass, S.U., Pruss, D., and Wolffe, A.P. (1997). How does DNA methylation repress transcription? Trends Genet. 13, 444-449. Lee, M.G., Wynder, C., Cooch, N., and Shiekhattar, R. (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 415, 432-435. Litt, M.D., Simpson, M., Gaszner, M., Allis, C.D., and Felsenfeld, G. (2001). Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 293, 2453-2455. Magdinier, F. and Wolffe, A.P. (2001). Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc. Natl. Acad. Sci. U S A 98, 4990-4995. Malik, H.S. and Henikoff, S. (2003). Phylogenomics of the nucleosome. Nat Struct Biol. 10, 882-891. McKittrick, E., Gafken, P.R., Ahmad, K., and Henikoff, S. (2004). Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. U S A 101, 1525-1530. 68 Metzger, E., Wissmann, M., Yin, N., Muller, J.M., Schneider, R., Peters, A.H., Gunther, T., Buettner, R., and Schule, R. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436-439. Nakayama, J., Rice, J.C., Strahl, B.D., Allis, C.D., and Grewal, S.I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110-113. Nguyen, C.T., Gonzales, F.A., and Jones, P.A. (2001). Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res. 29, 4598-4606. Noma, K., Allis, C.D., and Grewal, S.I. (2001). Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293, 11501155. Nowak, S.J. and Corces, V.G. (2004). Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 20, 214-220. 69 Okano, M., Takebayashi, S., Okumura, K., and Li, E. (1999). Assignment of cytosine-5 DNA methyltransferases Dnmt3a and Dnmt3b to mouse chromosome bands 12A2-A3 and 2H1 by in situ hybridization. Cytogenet. Cell Genet. 86, 333-334. Orphanides, G. and Reinberg, D. (2002). A unified theory of gene expression. Cell 108, 439-451. Osley, M.A. (1991). The regulation of histone synthesis in the cell cycle. Annu. Rev. Biochem. 60, 827-861. Peters, A.H., Mermoud, J.E., O'Carroll, D., Pagani, M., Schweizer, D., Brockdorff, N., and Jenuwein, T. (2002). Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30, 77-80. Peterson, C.L. and Laniel, M.A. (2004). Histones and histone modifications. Curr Biol. 14, R546-R551. Rhee, I., Bachman, K.E., Park, B.H., Jair, K.-W., Yen, R.W., Schuebel, K.E., Cui, H., Feinberg, A.P., Lengauer, C., Kinzler, K.W., Baylin, S.B., and Vogelstein, B. (2002). DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552-556. 70 Robertson, K.D. and Jones, P.A. (2000). DNA methylation: past, present and future directions. Carcinogenesis 21, 461-467. Robertson, K.D. and Wolffe, A.P. (2000). DNA methylation in health and disease. Nat. Rev. Genet. 1, 11-19. Rountree, M.R., Bachman, K.E., Herman, J.G., and Baylin, S.B. (2001). DNA methylation, chromatin inheritance, and cancer. Oncogene 20, 3156-3165. Schubeler, D., Lorincz, M.C., Cimbora, D.M., Telling, A., Feng, Y.Q., Bouhassira, E.E., and Groudine, M. (2000). Genomic targeting of methylated DNA: influence of methylation on transcription, replication, chromatin structure, and histone acetylation. Mol. Cell Biol. 20, 9103-9112. Schwartz, B.E. and Ahmad, K. (2005). Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 19, 804-814. Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J.R., Cole, P.A., Casero, R.A., and Shi, Y. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941-953. Strahl, B.D. and Allis, C.D. (2000). The language of covalent histone modifications. Nature 403, 41-45. 71 Suzuki, H., Gabrielson, E., Chen, W., Anbazhagan, R., van Engeland, M., Weijenberg, M.P., Herman, J.G., and Baylin, S.B. (2002). A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 31, 141-149. Tagami, H., Ray-Gallet, D., Almouzni, G., and Nakatani, Y. (2004). Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51-61. Tamaru, H. and Selker, E.U. (2001). A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 414, 277-283. Tazi, J. and Bird, A. (1990). Alternative chromatin structure at CpG islands. Cell 60, 909-920. Tsuchiya, R., Yoshiki, F., Kudo, Y., and Morita, M. (2002). Cell type-selective expression of green fluorescent protein and the calcium indicating protein, yellow cameleon, in rat cortical primary cultures. Brain Res. 956, 221-229. Varga-Weisz, P. (2001). ATP-dependent chromatin remodeling factors: nucleosome shufflers with many missions. Oncogene 20, 3076-3085. 72 Wade, P.A. (2001). Methyl CpG-binding proteins and transcriptional repression. Bioessays 23, 1131-1137. Waterborg, J.H. (1990). Sequence analysis of acetylation and methylation in two histone H3 variants of alfalfa. J Biol Chem. 265, 17157-17161. Wirbelauer, C., Bell, O., and Schubeler, D. (2005). Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 19, 1761-1766. Wolffe, A.P. and Bird, A.P. (1999). Methylation-induced repression--belts, braces, and chromatin. Cell 99, 451-454. Xin, Z., Allis, C.D., and Wagstaff, J. (2001). Parent-specific complementary patterns of histone H3 lysine 9 and H3 lysine 4 methylation at the Prader-Willi syndrome imprinting center. Am. J. Hum. Genet. 69, 1389-1394. Zeng, X., Chen, J., Sanchez, J.F., Coggiano, M., Dillon-Carter, O., Petersen, J., and Freed, W.J. (2003). Stable expression of hrGFP by mouse embryonic stem cells: promoter activity in the undifferentiated state and during dopaminergic neural differentiation. Stem Cells 21, 647-653. 73 Zhang, Y. and Reinberg, D. (2001). Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343-2360. 74 CURRICULUM VITA 75 Sayaka Eguchi 1111 Park Avenue, Apartment 1509 (410) 462-2443 Baltimore, Maryland 21201 eguchis@ureach.com EDUCATION 1999 – Present Johns Hopkins University – Baltimore, Maryland Ph.D. candidate, completion in March 2006 1994 – 1997 Rensselaer Polytechnic Institute – Troy, New York B.S., Biology and Psychology (dual major), Magna Cum Laude RESEARCH TRAINING 1999 – Present Johns Hopkins University – Baltimore, Maryland Graduate Student, Program in Cellular and Molecular Medicine Project: The role of histone modifications in the reversal of abnormal gene silencing in cancer. Advisor: Stephen B. Baylin, M.D. 1997 – 1999 Memorial Sloan Kettering Cancer Center – New York, New York Research Technician Performed experiments contributing to research projects investigating chromatin remodelling and transcriptional regulation of yeast histone genes. Managed and organized laboratory. Advisor: Mary Ann Osley, Ph.D. 76 RESEARCH TRAINING (continued) 1997 Genzyme Genetics – Yonkers, New York Laboratory Technician Developed pictures and cut karyotypes of normal and abnormal chromosomes from human amniotic, blood, and bone marrow samples. 1996 – 1997 Rensselaer Polytechnic Institute – Troy, New York Undergraduate Researcher Participated in research project studying life cycle of Zebra Mussels in Lake George. Advisor: Sandra Nierzwicki-Bauer, Ph.D. SUMMARY OF TECHNICAL EXPERTISE Molecular Biology: Transformation of yeast and bacteria Alkaline lysis plasmid purification DNA isolation by gel extraction Nucleic acid purification from cultured cells Polymerase chain reaction (PCR) Quantitative real-time PCR Bisulfite treatment of DNA Methylation-specific PCR (MSP) Reverse transcriptase PCR (RT-PCR) DNA cloning and sequencing 77 Molecular Biology (continued): Chromatin immunoprecipitation (ChIP) 5’ rapid amplification of cDNA ends (RACE) Northern blot analysis S1 ribonuclease protection assay Protein Analysis: Construction and purification of fusion proteins β-galactosidase enzyme activity assay Whole cell and nuclear extract isolation from cultured cells Western blot analysis Immunoprecipitation Cell Biology: Mammalian cell culture and maintenance Drug treatment of cultured cells Transient and stable transfection Immunohistochemistry Immunofluorescence Fluorescence in situ hybridization (FISH) and fiber FISH Confocal, epifluorescence, and light microscopy Software: Microsoft Word, Powerpoint, and Excel Scala InfoChannel Designer 78 Software (continued): Thomson Endnote and Reference Manager Adobe Pagemaker and Photoshop WORK EXPERIENCE 2005 – Present Johns Hopkins University – Baltimore, Maryland Editor, Restriction Digest Edit and organize layout of articles and images for graduate student newsletter. 2003 – Present Johns Hopkins University – Baltimore, Maryland Editor, Peer Editing Service Format and edit biomedical research papers and grants for postdoctoral fellows and graduate students. 2003 – Present Maryland Science Center – Baltimore, Maryland Educator, BodyLink Exhibit Plan and demonstrate human health and biology activities, assist visitors with lab experiments, organize special events, and supervise staff and exhibit area. AWARDS AND PROFESSIONAL ACTIVITIES Associate Member, American Association for Cancer Research (2006) Intern, Maryland Science Center (2003) 79 AWARDS AND PROFESSIONAL ACTIVITIES (continued) Member, American Association for the Advancement of Science (1998 – present) Inducted Member, White Key Society of the Phalanx Honors Society at Rensselaer Polytechnic Institute (1997) Secretary, Society of Biological Sciences at Rensselaer Polytechnic Institute (1996 – 1997) President, Society of Biological Sciences (1995 – 1996) Charter Member, Society of Biological Sciences (1994 – 1997) PUBLICATIONS Hellebrekers, D. M. E. I., Jair, K.-W., Vire, E., Eguchi, S., Hoebers, N. T. H., Fraga, M. F., Esteller, M., Fuks, F., Baylin, S. B., van Engeland, M., and Griffioen, A. W. (2006). Angiostatic activity of DNA methyltransferase inhibitors. Molecular Cancer Therapeutics 5: 467-475. Fahrner, J. A., Eguchi, S., Herman, J. G., and Baylin, S. B. (2002). Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Research 62, 7213-7218. *First two authors contributed equally Dimova, D., Nackerdien, Z., Furgeson, S., Eguchi, S., and Osley, M. A. (1999). A role for transcriptional repressors in targeting the yeast Swi/Snf complex. Molecular Cell 4, 75-83. POSTERS Annual graduate program retreat (2002, 2004, and 2005) 80 PERSONAL INFORMATION Place of birth: Flushing, New York Date of birth: June 27, 1976 Fluent in English Conversational in Japanese 81