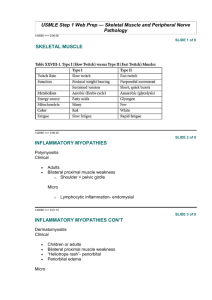
Gene_trherapy_update..
advertisement

GENE THERAPY IN MUSCULAR DYSTROPHY - ABSTRACTS FROM 10TH ANUAL MEETING OF AMERICAN SOCIETY OF GENE THERAPY - MAY, 30 JUNE 3, 2007 SEATTLE 1) Antisense Oligonucleotide Induced Exon Skipping as a Therapy for Duchenne Muscular Dystrophy Steve D. Wilton, Abbie A. Fall, Penny M. Harding, Patrick Iversen, Susan Fletcher Centre for Neuromuscular and Neurological Disorders, University of Western Australia, Perth, Western Australia, Australia; Research and Development, AVI Biopharma, Corvallis, OR Antisense oligonucleotides (AOs) can be used to re-direct normal splicing patterns of a pre-mRNA in order to excise selected exons from the mature mRNA. We have been developing AO induced targeted exon skipping as a potential therapy for Duchenne muscular dystrophy (DMD), the most common severe form of childhood muscle wasting. The huge dystrophin gene (79 exons spanning 2.4 Mb) encodes a protein that links the actin cytoskeleton and a complex of proteins embedded in the sarcolemma. Protein-truncating mutations in the dystrophin gene result in a nonfunctional protein, with subsequent loss of structural integrity of the muscle fibres. An X-linked disease, affected males show signs of muscle weakness from 3-5 years of age and are restricted to a wheelchair by age 12 years. Becker muscular dystrophy (BMD) also arises from dystrophin mutations, but these are typically in-frame, and allow synthesis of an internally deleted protein, which can be of near normal function. Some BMD patients are asymptomatic and diagnosis may only be made later in life, despite missing substantial regions of the dystrophin gene. We have developed a panel of AOs that can induce skipping of each exon of the predominant muscle specific isoform. Forty one of the 77 exons targeted for excision could be removed with high efficiency with single AOs, although no obvious pre-mRNA motif was consistently targeted. The remaining exons were more stubborn and were excised at lower efficiencies, despite many AOs being designed and evaluated. One approach found to induce very efficient skipping of these recalcitrant exons was the use of select AO combinations. Consisting of AOs that were often inactive when used individually, some AO cocktails could induce efficient exon skipping after in vitro transfection in the nanomolar range. AO design and delivery are both crucial aspects of developing a viable therapy. We have observed that trends in exon skipping efficiency induced by AOs composed of 2 -O-methyl modified bases on phosphorothioate backbone (2OMeAOs) were also seen when the corresponding sequences were applied as phosphorodiamidate morpholino oligomers (PMOs). That is, the design of 2OMeAOs can be extrapolated to the morpholino chemistry. However, the more robust PMOs seem much better suited to in vivo applications as shown by substantial systemic dystrophin expression being induced in a mouse model of muscular dystrophy after repeated weekly intraperitoneal administration of a PMO coupled to a cell penetrating peptide tag at a dose of 5 mg/kg. PMOs have already entered clinical trials for other conditions. A clinical trial is scheduled to commence in the UK in 2007 using a PMO directed at dystrophin exon 51. This compound should restore the reading frame in DMD cases arising from responsive dystrophin mutations occurring in the major deletion hotspot. The PMO will be delivered by an intramuscular injection, and if safety concerns are met and proof-of- principle demonstrated in human dystrophic muscle, systemic administration will follow as soon as possible. 2) Prolonged AAV-Mediated Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy with a Brief Course of Immunosuppression Zejing Wang, Christian S. Kuhr, James M. Allen, Eric Finn, Paul Gregorevic, Jeffrey S. Chamberlain, Stephen J. Tapscott, Rainer F. Storb Transplantation Biology, Fred Hutchinson Cancer Research Center, Seattle, WA; Neurology, University of Washington, Seattle, WA; Human Biology, Fred Hutchinson Cancer Research Center, Seattle, WA Introduction: Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene. Studies in the mdx mouse model of DMD have shown that muscle membrane integrity and function can be improved by AAV-mediated delivery of a functional dystrophin protein. Our previous studies have demonstrated that direct intramuscular injection of AAV2 or AAV6 in wild-type random bred dogs resulted in a robust immune response to capsid, or capsid-associated proteins. Recent evidence of immune-mediated loss of AAV vector persistence in human trials also suggests that immune modulation might be necessary to achieve successful long-term transgene expression. In this study, we assessed the potential of various immunosuppression regimens in prolonging AAV-mediated transgene expression in both random-bred normal dogs and in dogs with muscular dystrophy caused by a dystrophin mutation (cxmd dogs). Methods: AAV serotype 6 carrying different promoter-transgene cassettes were produced as previously described for murine and canine studies. Direct intramuscular injections of vectors at 1x1011 vector genomes per site in a total volume of 250 l were performed. Different immunosuppression regimens were utilized between 4 to 18 weeks. The injection sites were biopsied under anesthesia between 4 and 30 weeks after injection for histology, immune responses and transgene expression analysis. Results: Daily immunosuppression with cyclosporine (CSP) and mycophenolate mofetil (MMF) largely prevented the immune response to AAV6-CMV-LacZ in a normal dog for up to four weeks and permitted robust transgene expression. The same immunosuppressive regimen did not as completely prevent an immune response to AAV6-CMV-cFIX or AAV6-CMVhuman-micro-dystrophin (h- -dys) in a cxmd dog, suggesting that a more aggressive immunosuppressive regimen might be necessary. A brief course of five days antithymocyte globulin (ATG) treatment was given to cxmd dogs injected with either AAV6-CMV- h- -dys or AAV6-CMV-canine micro-dystrophin (c- -dys) to deplete T lymphocyte, and followed by daily use of both CSP and MMF. This regimen successfully prevented the immune responses and allowed long-term expression of transgenes in dystrophic muscles. Conclusions: Taken together, our results suggest that the combination of CSP and MMF effectively prevents the immune responses in a normal dog, while a more potent regimen with the combination of ATG, CSP and MMF is required in cxmd dogs, possibly due to the pre-existing inflammatory nature of the DMD muscle disease. The latter immunosuppression regimen is sufficient to permit long-term and robust expression of a c- -dys transgene in the skeletal muscles of a cxmd dog, and its expression restored localization of components of the dystrophin-glycoprotein complex at the muscle membrane. This protocol has potential applications to human clinical trials to enhance AAV-mediated gene therapies for various human diseases. 3) A Clinically Relevant Gene Therapy Approach for Duchenne Muscular Dystrophy by Vascular Delivery of Micro-Dystrophin Louise R. Rodino-Klapac, Paul M. Janssen, Chyrstal L. Montgomery, Ryan Jensen, Louis G. Chicoine, K. Reed Clark, Jerry R. Mendell Center for Gene Therapy, Columbus Children s Research Institute, Columbus, OH; Physiology and Cell Biology, The Ohio State University, Columbus, OH Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder with monogenic mutations setting the stage for successful gene therapy treatment. Longterm therapeutic goals include systemic delivery of a small dystrophin transgene, micro-dystrophin, delivered by adeno-associated virus (AAV) via the vasculature. Current and past clinical trials have limited gene delivery to direct intramuscular injection. As we proceed toward our goal of widespread muscle transduction, we anticipate progress in a stepwise fashion, where the transgene is delivered by selective catheterization to branches of the femoral artery. This approach of isolated lower limb perfusion (ILP) permits a clinical trial for muscular dystrophy assessing both safety and efficacy as for the following reasons: 1) selective delivery of vector to lower limb muscles can produce clinically meaningful results; 2) the lower limb can be compartmentalized to prevent spread of virus to other organ systems providing an important measure of safety; 3) delivery of virus in a compartmentalized system provides safe passage for the virus since pre-existing immunity to AAV may preclude muscle transduction. We first tested ILP of micro-dystrophin in the mdx mouse with the goal of comparing the efficiency of AAV serotypes [AAV1, 6, or AAV8] in crossing the vascular barrier leading to widespread gene expression in muscle in a manner different from previous studies, not relying on excessive pressure, volume, or pharmacologic agents to combat vascular resistance. A micro-dystrophin construct was used with features previously described by Harper et al (2002), deleting the untranslated regions and C-terminus, retaining spectrin repeats 1-3 and 24, and hinges 1, 2, and 4 of full-length dystrophin, under control of a muscle specific promoter, and the addition of an intron to enhance gene expression. Comparative studies conclude that AAV6 and AAV8 deliver and transduce micro-dystrophin by ILP more efficiently than AAV1, with micro-dystrophin levels > 85% for both serotypes. Functional significance was established in extensor digitorum longus (EDL) by demonstrating increased maximum force generation and protection against eccentric contractions (P< 0.05). Extending these studies to non-human primate; we have successfully translated ILP vascular delivery using AAV8 to cross the endothelial barrier of muscle vasculature by specifically targeting the lower limb muscles with a fluoroscopy guided catheter to deliver AAV8.GFP (green fluorescent protein). These findings and ongoing studies set the stage for a future clinical trial in DMD patients with vascular delivery of the micro-dystrophin transgene. 4) Independent Canine Models of Duchenne Muscular Dystrophy Due to Intronic Insertions of Repetitive DNA Bruce F. Smith, Joe N. Kornegay, Dongsheng Duan Scott-Ritchey Research Center, Auburn University, Auburn, AL; School of Medicine, University of North Carolina Chapel Hill, Chapel Hill, NC; School of Medicine, University of Missouri, Columbia, MO Duchenne muscular dystrophy (DMD) is the most common X-linked disease and inherited myopathy of humans. As such, development of effective gene therapy for DMD has been and continues to be a high priority. A number of animal models have allowed the testing of novel approaches in order to determine their validity prior to application to human patients. Of these, the canine model most closely recapitulates the clinical presentation, immune system responses and body mass of human patients. Several canine mutations have been identified, although the Golden Retriever remains the best characterized and therefore most commonly employed model. Additional canine models would further improve the utility of the dog as a model system as these models would mimic the variety of challenges seen with human patients, including variable transcription, the presence or absence of epitopes and the effect of residual mutant or revertant protein. We have characterized two additional canine models of DMD at the level of histology, morphology and molecular basis. These models were identified in the Labrador Retriever and Welsh Corgi breeds. In both cases, affected animals can be identified at birth by elevated creatine kinase levels. Both Welsh Corgis and Labrador Retrievers have a relentlessly progressive and ultimately fatal course of disease . Both models are dystrophin deficient except for rare revertant fibers. They display prominent skeletal muscle pathology identical to these found in human patients such as variable fiber size, central nucleation, fiber splitting, fatty infiltration, macrophage infiltration, fibrosis and calcification. We have identified the molecular basis of each model as the inclusion of repetitive sequence elements creating novel exons in the cDNA. In both cases these elements have been inserted into introns (intron 13 for the Welsh Corgi and intron 19 for the Labrador Retriever), activating splice acceptor sites already present in the normal intron sequence. In both cases, the inserted sequences contain in-frame stop codons leading to early termination of translation. The characterization of the mutations and morphologic features of each of these new models provides the information required to employ these models in gene therapy studies. These models provide important alternatives to the available models and they will broaden our understanding of how relevant approaches will function in the face of different mutations and clinical manifestations in the human population. 5) A Myoblast Expansion System for Improving the Efficiency of Autologous Stem Cell Therapy To Treat Muscular Dystrophy Sheng Li, Brent Fall, Miki Haraguchi, Jeffrey S. Chamberlain Senator Paul D Wellstone Muscular Dystrophy Co-operative Research Center, Department of Neurology, The University of Washington School of Medicine, Seattle, WA Autologous stem cell-based transplantation is a promising approach to treating inherited muscle disorders. A successful strategy would be facilitated by regimes that allow for expansion both in vitro and in vivo of cells with myogenic potential. A previous study demonstrated that a chemical inducible dimerizer (CID), AP20187, could maintain in vitro proliferation of myoblasts expressing F36VFGFR-1, a chimeric protein composed of the cytoplasmic phosphorylation domain of fibroblast growth factor receptor 1 (FGFR-1) and a mutated dimerization domain of FK506 binding protein (F36V). Here, we generated a lentiviral vector carrying a bicistronic expression cassette composed of a microdystrophin/GFP fusion gene and the F36VFGFR-1 gene under the control of a synthetic muscle-specific promoter. The mdx myoblasts transduced with this vector were prevented from differentiating and were expanded in culture medium containing AP20187, but lacking FGF-2. When intramuscularly transplanted into mdx tibialis anterior muscles, these AP20187expanded cells formed large clusters of myofibers expressing microdystrophin/GFP and the developmental isoform of myosin heavy chain. Although CID-expanded myoblasts differentiated to form myotubes and myofibers in skeletal muscle, we found that in culture, residual AP20187 retained in those CID-expanded cells dramatically delayed myotube formation. These observations imply that exogenous control of FGFR-1 activation in myoblasts can be used to prevent terminal differentiation and enable large-scale expansion of myogenic precursors, potentially increasing the efficiency of myoblast engraftment in muscle disorders. 6) Gene Therapy for Duchenne Muscular Dystrophy by the Helper-Dependent Adenovirus Vector (HDAdv) Mediated Full-Length Dystrophin Expression Masatoshi Ishizaki, Ryoko Kawano, Yuji Uchida, En Kimura, Makoto Uchino, Yasushi Maeda Neurology, Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan Backgrouud Duchenne muscular dystrophy (DMD) is a progressive muscle wasting disorder caused by the absense of dystrophin. Among various obstacles against the DMD gene therapy, the huge cDNA size, 14kb, limits the use of many kinds of virusbased vectors. We have generated a helper-dependent adenovirus vector, which has a cloning capacity of up to 37kb, that carried myc-tagged murine full-length dystrophin cDNA. In this study we evaluated the therapeutic effect of the HDAdmediated full-length dystrophin gene transfer into mdx mice and utrophin/dystrophin double knockout mice (dko mice), which are severely dystrophic mice. Method and Results [We have constructed HDAd vector contained the murine full-length dystrophin expression cassette and myc-tag,integral protein (HDAdv-mFLmyc-dys). (1) Gene delivery by multiple intramuscular injection into neonatal dko mice Each 7day-old dko mice were injected with the HDAdv-mFLmyc-dys into the following muscle groups: lower limbs, upper limbs and lattissimus dorsi. The transgene was widely expressed and prevented the dystrophic changes pathologically and physiologically in injected dko mice. We observed the restoration of dystrophin and dystrophin associated proteins, and nNOS. Furthermore, motor performance in injected dko mice could be improved and their lifespan became longer. (2) Systemic gene delivery intravenously in young adult mdx mice Young adult mdx mice were injected with the HDAdv-mFLmyc-dys via the tail vein. Body-wide expression of fulllength dystrophin expression was detected in the skeletal muscle and diaphragm. Conclusion These results offer a hopeful prospect for DMD therapy. Therapeutic gene transfer with HDAd may ameliorate DMD patients. 7) Inhibition of Myostatin by Gene Therapy Increases Muscle Mass and Strength in a Mouse Model of Muscular Dystrophy Amanda M. Haidet, Liza Rizo, Chalonda R. Handy, Chris J. Shilling, Zarife Sahenk, Jerry R. Mendell, Brian K. Kaspar Integrated Biomedical Science Graduate Program, The Ohio State University, Columbus, OH; Center for Gene Therapy, Columbus Children s Research Institute, Columbus, OH; Department of Pediatrics, The Ohio State University, Columbus, OH Increasing the size and strength of muscles represents a promising therapeutic strategy for musculoskeletal disorders. Significant interest has focused on myostatin, a negative regulatory factor of muscle growth. Inhibition of myostatin significantly increases muscle mass. Several proteins including follistatin, follistatin-related gene (FLRG) and growth and differentiation factor-associated serum protein 1 (GASP-1) inhibit myostatin. We have cloned the genes expressing these proteins into Adenoassociated viral (AAV) vectors. Administering AAV1 at a single time point by intramuscular injection provides a long-lasting therapeutic benefit. Wild-type mice injected in the hindlimbs with 1 x 1011 viral particles of AAV1-Follistatin, FLRG, or GASP-1 have shown increased overall body mass, with a subsequent 30% increase in hindlimb and forelimb grip strength compared to AAV1-GFP treated controls. We have evaluated these animals over 1.5 years following gene injection and have found no untoward effects due to post-natal gene delivery of these muscle-enhancing factors. The overall strength increase was greatest for animals receiving AAV1Follistatin followed by AAV1-FLRG and then AAV1-GASP-1. We next tested the potential of AAV1-Follistatin to increase muscle mass and strength and delay muscle deterioration in the mdx mouse model of Duchenne muscular dystrophy (DMD). DMD is an X-linked recessive disease resulting in the wasting of skeletal muscles and cardiac function, ultimately resulting in death. Mdx animals were injected bilaterally in the hindlimbs with AAV1-Follistatin at 3 weeks of age and followed for 6 months before sacrifice. These animals showed a 30-50% increase in skeletal muscle mass, enhanced muscle grip strength, and decreased serum creatine kinase levels compared to AAV1-GFP controls. These findings indicate that follistatin slows or delays muscle damage. Histological analysis of AAV1-Follistatin treated muscles demonstrated myofiber hypertrophy at the local site of injection and at remote sites, including paraspinal and upper limb muscles reinforcing conclusions that circulating follistatin had biological efficacy. No adverse organ pathology was detected and animals were capable of normal reproduction in preliminary studies. Furthermore, mdx animals treated with AAV1-Follistatin at 6.5 months of age also showed increased muscle strength, demonstrating the ability of follistatin to improve strength in aged animals. These results suggest that inhibition of myostatin by myostatin inhibitory proteins, in particular follistatin, delivered by viral mediated gene therapy represents a promising therapeutic strategy that warrants consideration for clinical trials in human muscle diseases. 8) In Vitro Identification of Optimal Exon-Skip Targets for Treatment of Duchenne Muscular Dystrophy Using a Human Muscle Cell Line Nathaniel A. Walton, Clifford J. Beall, Jerry R. Mendell, K. Reed Clark Center for Gene Therapy, Columbus Children s Research Institute, Columbus, OH; Department of Pediatrics, The Ohio State University, Columbus, OH Introduction: DMD is a chronic, debilitating disease of childhood caused by the absence of dystrophin. Muscle biopsies from at least half of dystrophin deficient patients show revertant myofibers expressing the mutant protein. This is likely related to second-site mutations that restore the reading frame or possibly create alternative or cryptic splice-sites. Clinical trials are underway to exploit similar mechanisms to skip exons and restore the dystrophin reading frame in DMD patients using antisense oligonucleotides (AONs) and morpholinos. While this method holds promise, the potential need for re-administration and excessive production costs keeps enthusiasm in check. A potential option to achieve the same goal is through the use of modified snRNA genes with anti-sense exon/intron sequences delivered by adenoassociated virus (AAV) to disrupt spliceosomal recognition of the targeted exon during pre-mRNA processing. In making such a system clinically applicable to as many patients as possible, we have developed a system to identify potential antisense targets through recognition of exon splice enhancer (ESE) motifs. To achieve this goal, we have established a myocyte cell line derived from a patient with an exon 33 stop codon mutation. We describe a matrix assay to screen potential anti-sense targets in tandem that results in enhanced dual-target snRNA vectors. Methods: AONs (2 -O-methyl phosphothioate) were designed to target multiple ESE motifs using the ESE finder program. Myotube cultures were differentiated using serum starvation and incubation at 37o. AONs (10 nM - 200 nM) were introduced by transfection using Lipofectin and total RNA isolated 24 hr post-transfection. RT-PCR (100 ng of template) was performed using dystrophin specific primers located in exons 31 and 35. The full-length amplicon was 626 bp, while mRNA with exon 33 skipped yielded a 470 bp amplicon. Results: Six AONs were synthesized that were predicted to block recognition of 2 or more contiguous ESE binding motifs. AONs were transfected alone or in tandem at several final concentrations (12.5, 25 and 50 nM) into mature (6 days post-differentiation) myotube cultures. AON matrix analysis using RT-PCR revealed that individual AONs were not as efficient (based on minimal titration end-point) in producing skipped transcripts as several dual AON combinations. Currently, we are evaluating the optimal AONs combinations in dualtarget snRNA AAV vectors in this culture system. Discussion: Specific targeting with AONs to regions within exons should increase specificity by avoiding canonical splice donor and acceptor sites. We identified several active anti-sense target regions that when combined in tandem allowed activity down in the low nanomolar range (12.5 nM). Our data suggest that targeting two regions within an exon facilitates disruption of spliceosomal recognition that can be incorporated into AAV vectors for sustained delivery of anti-sense sequences at doses within the expectation of a clinical trial needed to achieve exon exclusion and possible therapeutic efficacy. 9) Gait in BioTO-2 and Bio14.6 Dystrophic Hamsters Thomas G. Hampton, Ivo Amende, Ajit Kale, Scott Mccue, Hemmi N. Bhagavan, Anton H. M. Terpstra, Case VanDongen R D, Biobreeders Inc., WaterTown, MA; R D, MouseSpecifics Inc., Boston, MA The delta-sarcoglycan-deficient hamster strains BIO 14.6 and BIO TO2 are excellent models to study muscular dystrophy and the efficacy of gene therapy. Gait disturbances, important clinically, have not yet been described in these hamster models. Accordingly, we compared the gait of BIO 14.6 (n=12) and BIO TO2 (n=12) dystrophic hamsters to healthy BIO F1B (n=12) control hamsters. We used ventral plane videography to determine gait indices in 3-mo and 9-mo old male BIO 14.6, BIO TO2, and BIO F1B hamsters walking on a transparent treadmill belt at 16 cm/s. Gait indices were based on 10 consecutive strides for each of the 4 limbs. We also studied 1-mo old BIO TO2 (n=4) and F1B (n=4) hamsters and found kinematic and postural changes in both BIO 14.6 and BIO TO2 hamsters, including significantly shorter swing, stride, and stance durations. Stride length was 13% shorter (P<0.05) in BIO 14.6 and BIO TO2 dystrophic hamsters at 3-mo and 9-mo of age compared to BIO F1B hamsters. Propulsion duration of the hind limbs, an indicator of muscle strength, was shorter in 9-months BIO 14.6 (236 14 ms) and BIO TO2 hamsters (244 8 ms) compared to BIO F1B (303 11 ms; P<0.05). Braking duration, reflecting generation of ground reaction forces, was delayed in 9-mo old BIO14.6 (142 7 ms) and BIO TO2 hamsters (185 9 ms) compared to BIO F1B (112 14 ms; P<0.05). Hind paw eversion, evidence of muscle weakness, was greater in 9-mo old BIO TO2 than in BIO F1B hamsters (20.1 1.1 vs. 6.7 1.7 ; P<0.05). The propulsive deficit was apparent in BIO 14.6 and BIO TO2 at 3-mo and in BIO TO2 animals at 1-mo of age. Our quantitative analysis of gait demonstrates gait disturbances in hamsters with muscular dystrophy that commence as early as 1-mo of age. Our findings show that these dystrophic BIO hamsters recapitulate the functional features of human muscular dystrophy. Early detection and quantitative measures of these gait abnormalities will accelerate the development of gene therapy for treating muscular dystrophy. 10) Retrograde Axonal Transport of AAV Vectors Allows Extended Restoration of Dystrophin Expression by Exon Skipping in mdx Mice Brain Elise Peltekian, Ros Carine, Carole Gruszczynski, Cyrille Vaillend, Serge Laroche, Luis Garcia, Olivier Danos Duchenne Muscular Dystrophy, Genethon/CNRS UMR 8115, Evry, France; Duchenne Muscular Dystrophy, Genethon/CNRS UMR 8115, Evry, France; NAMC, CNRS UMR 8620, Orsay, France; NAMC, CNRS UMR 8620, Orsay, France; Duchenne Muscular Dystrophy, Genethon/CNRS UMR 8115, Evry, France; Hopital Necker, Inserm U781, Paris, France; Duchenne Muscular Dystrophy, Genethon/CNRS UMR 8115, Evry, France Dystrophin is selectively localized in cortical, hippocampal and cerebellar neurons. In addition to the progressive muscle wasting, one third of Duchenne patients suffer a moderate to severe form of mental retardation. Behavioural studies also suggest that the loss of dystrophin expression in mdx mice brain results in a mild cognitive deficit and memory impairment. We have recently developed an original approach to obtain a complete, functional and enduring restoration of dystrophin in the skeletal muscle of mdx mice [Goyenvalle et al., 2004]. The muscle rescue was achieved by means of vectorized antisense sequences that are potentially able to eliminate the mutated exon 23 of the dystrophin gene in this mutant, thereby allowing a local restoration of dystrophin expression. AAV vector considerably enhanced the proper sub cellular localization of antisense sequences and their inclusion into the spliceosome. This vector has been shown to be particularly efficient for gene transfer into pyramidal neurons of the hippocampus. We show here that dystrophin expression in the mdx mice dorsal hippocampus was achieved by a bilateral intracerebral stereotaxic administration of an AAV-vector expressing antisense sequences linked to a modified U7 small nuclear RNA (snRNA). mRNA analyses and protein detection by immunohistochemistry and Western blotting were carried out on serial brain tissue sections. The DNA sequence confirmed that the 688 bp band resulting from RT-PCR corresponds to the exon 23-skipped mRNA. Moreover, this exon 23-skipped mRNA and the corresponding protein were selectively detected in a remote cerebral strucuture, the entorhinal cortex, directly connected to the hippocampus. These findings suggest a retrograde axonal transport of AAV particles to this afferent neuroanatomic area, such allowing the rescue of dystrophin expression in this neuronal netwotk. These results already suggest that mRNA manipulation may allow the reversion of genetic alterations in the mature brain. One main interest of this approach is the restoration of the lost protein not only in the target structure of the brain but also in the target cells, within the complex cell population of the brain tissue. Potentially, exon skipping may be used to selectively inactivate the expression of genes involved in cognitive processes and synaptic plasticity. It may open new opportunities for therapeutic strategies targeted to a functional neuroanatomical structure. 11) Restoration of Dystrophin Expression in the Diaphragm of the Dystrophic mdx Mice by Polymer-Tagged Morpholino Antisense Oligonucleotides Bo Wu, Pei Juan Lu, Jignya Ashar, Ehsan Benrashid, Elizabeth Keramaris, Yong Fu Li, Paul A. Morcos, Qi Long Lu McColl-Lockwood Laboratory for Muscular Dystrophy Research, Cannon Research Center, Carolinas Healthcare System, Charlotte, NC; Gene Tools, LLC, Philomath, OR Duchenne and Becker muscular dystrophies are allelic disorders arising from mutations in the dystrophin gene. Duchenne muscular dystrophy (DMD) is characterised by nonsense or frame-shifting mutations in the dystrophin gene that result in an absence of functional protein, dystrophin, while Becker muscular dystrophy is usually caused by in-frame deletions resulting in shortened yet partially functional dystrophin. The application of antisense oligonucleotides (AOs) is a promising strategy for the treatment of DMD. This treatment removes specific exons from the dystrophin mRNA transcript with AOs, thus altering pre-mRNA processing to restore the mRNA reading frame and to express shortened but functional Becker-like protein. Local and systemic deliveries of phosphodiamidate morpholino oligonucleotides (PMOs) have been demonstrated to effectively induce exon skipping and dystrophin expression in the Golden retrieval muscular dystrophic (GRMD) dog and the dystrophic mdx mouse . However, systemic delivery of PMOs in both models also demonstrated that expression of dystrophin can not be achieved homogeneously in all muscle of the body with especially poor induction in the cardiac muscle and diaphragm. Efforts have been made to search for and achieve improved delivery and improved antisense oligonucleotide efficiency. In this study, we examine the effect of PMOs tagged at the 3 prime with an arginine-based polymer (Vivoporter, supplied by GeneTools) on skipping of E23 of the dystrophin gene in the mdx mice in vivo. We found that just 10 g morpholino- vivoporter improved local efficiency of PMO induced exon skipping with almost 100% TA muscle fibres expressing dystrophin. Intraperitoneal injection of the morpholino- vivoporter achieved full induction of dystrophin in the diaphragm and abdomen muscles, but limited dystrophin expression in other skeletal and cardiac muscles. Since DMD severely affects respiratory muscles specifically the diaphragm, resulting in progressive deteriorations and failure of respiratory system, full induction of dystrophin in these muscles by i.p. injection may provide a realistic alternative for the rescue of the respiratory functions in DMD. 12) Screening of Antisense Oligonucleotides for Human Dystrophin Exon Skipping in a GFP Reporter Culture System with High Sensitivity and Specificity Ehsan Benrashid, Saafan Malik, Allen Zillmer, Randy J. Thresher, Jeffrey Rosenfeld, Qi Long Lu McColl-Lockwood Laboratory for Muscular Dystrophy Research, Carolinas Medical Center, Charlotte, NC; Animal Models Core Facility, University of North Carolina, Chapel Hill, NC Frameshift mutations in the dystrophin gene, located on the X chromosome (Xp21), can result in the progressive muscle wasting disease Duchenne s Muscular Dystrophy (DMD), which affects 1 in 3500 live male births. Antisense oligonucleotide (AON) therapy has previously been shown to selectively induce the skipping of particular exons both in vitro and in vivo, by targeting exon splicing enhancer (ESE) regions, 3 -, and 5 -splice sites. The DMD phenotype can thus be alleviated to the milder Becker s Muscular Dystrophy (BMD) form. Due to limitations in the quantification of AON efficacy via RT-PCR and nested PCR assay systems, as well as practical limitations of primary cultures for AON screening, this study employed a GFP reporter culture system to provide for efficient and high throughput screening of numerous AON compounds for human dystrophin exon 50 skipping, which could rescue approximately 8% of DMD mutations. AON screening was performed using both 2 -O-methyl phosphorothioate (2OMe) and morpholino (PMO) chemistry via fluorescence microscopy, RT-PCR/nested-PCR analysis, as well as flow cytommetry. After initial selection of AONs, the effect of overall AON length was also determined on the culture system. The efficient AONs selected were further tested with normal human myoblasts and relevant patient fibroblasts (with hEx51 deletion; further skipping of E50 can restore the reading frame). The results showed that AONs selected with the reporter system are highly effective for specific exon skipping in all cells. Our results demonstrate that the in vitro cell culture-based system is a valuable tool for the screening of effective AONs with high specificity and reliability, both of which are essential for clinical trials involving antisense therapy in DMD 13) rAAV Type 8-Mediated Extensive Therapeutic Gene Delivery into Skeltal Muscle of -Sarcoglycan Deficient Mice Akiyo Nishiyama, Beryl N. Ampong, Jin-Hong Shin, Hiroyuki Nakai, Takashi Okada, Shin'ichi Takeda Molecular Therapy, National Institute of Neuroscience, NCNP, Kodaira, Tokyo, Japan; Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine, Pittsburgh, PA Backgrounds: Autosomal recessive limb-girdle muscular dystrophy type 2D (LGMD2D) is caused by the mutations in the -sarcoglycan gene ( -SG) and the absence of -SG results in a reduction of whole SG complex, composed of -, -, -, SG, from the sarcolemma. The recombinant adeno-associated virus (rAAV) is expected to be useful to transduce -SG cDNA. However, the transduction by the rAAV type 2 (rAAV2) encoding -SG cDNA showed cytotoxicity with immune response in the -SG-deficient mice. The rAAV2 only allows transduction of the gene through local injection, but the rAAV type 8 (rAAV8) enables systemic deliverly of the gene through intravenous injection. We examined the therapeutic effects of the rAAV8 encoding the -SG cDNA on the -SG-deficient mice. Methods: We generated the rAAV2 or rAAV8 encoding the -SG cDNA driven by a CMV promoter (rAAV2- SG or rAAV8- -SG). We injected the rAAVs into the anterior tibial (TA) muscle of the -SG-deficient mice. The neonatal or adult -SG-deficient mice were injected with the 1 x 1011 vg or 5 x 1011 vg of rAAVs, respectively. Moreover, we injected the rAAV8- - SG (5 x 1012 vg) into the tail vein of the 5-week-old -SG-deficient mice for systemic delivery. Results and Discussion: At 4 weeks after the injection of the rAAV8- -SG into -SG-deficient TA muscle, expression of -SG was widely distributed in the posterior leg muscles, including the TA, extensor digitorum longus, gastrocnemius, soleus, and tibialis posterior muscles. In particular, the rAAV8- -SG effectively transduced the posterior leg muscles as well as the heart of the adult mice. In contrast, the rAAV2- -SG merely achieved local transduction of muscle fibers in the TA muscle. Together with wide expression of -SG, expression of other SGs was also detected in the posterior leg after the local injection of the rAAV8- -SG. The rAAV8- -SG-transduced muscle did not induce the severe immune response or cytotoxicity after the low dose injection of the rAAV8- -SG. In association with the rAAV8- -SG transduction, the -SG-deficient muscle significantly decreased the degeneration-regeneration cycle. Furthermore, the contractile force of the rAAV8- SG-injected muscle has been largely improved compared to the non-injected muscles. By systemic delivery of the rAAV8- -SG into the adult -SG-deficient mice, we observed the -SG expression in the various tissues including the skeletal muscle, cardiac muscle, and liver at 4 weeks. Conclusion: The rAAV8 is an effective tool to deliver therapeutic genes into the dystrophic skeletal muscle and enables wide distribution of the gene. This extensive rAAV8-mediated -SG transduction in the limb-girdle muscular dystrophy type 2D model mice would pave the way for the future clinical application. 14) Induction of Dystrophin Expression by Exon Skipping in mdx Mice by PNA and PNA-Peptide Conjugates HaiFang Yin, QiLong Lu, Matthew J. A. Wood Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, Oxfordshire, United Kingdom; McCollLockwood Laboratory, Neuromuscular/Amyotrophic Lateral Sclerosis Center, Carolinas Medical Center, North Carolina Duchenne muscular dystrophy (DMD) is the most common, serious form of muscular dystrophy, arising from mutations in the dystrophin gene that preclude the synthesis of functional protein. Antisense oligonucleotides (AOs) have been used to manipulate splicing of the pre-mRNA to induce specific exon skipping and thereby restore the reading frame and expression of functional dystrophin. The efficacy of this approach has now been established in animal models and in DMD patients. In this report, we describe in vitro and in vivo studies examining the effects of peptide nucleic acid oligonucleotides (PNA) and PNA with peptide conjugates including TAT, musclespecific peptide (MSP), AAV6 functional domain (AAV6), AAV8 functional domain (AAV8) on exon skipping efficiency. In cultured mdx mouse cells treated with unmodified neutral PNA or PNA-peptide conjugates, RT-PCR showed the efficient skipping of exon 23. In vivo, single intramuscular injections of PNA or PNA-peptide conjugates into TA muscles of mdx mice resulted in substantial numbers of dystrophin-positive fibres at 2 weeks after injection, with no apparent cytotoxicity. Mice of different ages treated with PNA and PNA-peptide conjugates all showed significant improvement in dystrophin expression compared with untreated mdx mice. Western analysis confirmed dystrophin expression in these muscles. Dose-response experiments demonstrated that PNA and PNA-AAV6, PNA-AAV8 conjugates showed dose-dependent characteristics. Surprisingly, there was no significant difference between the PNA and PNA-peptides conjugates in both delivery and exon skipping efficiency. Together, our results to date suggest that PNA and PNA-peptide conjugates have potential for the treatment of DMD. 15) Complementary Dystrophin and Utrophin Expression Prevents Severe Dilated Cardiomyopathy in 2-Year-Old Mdx Mice Brian P. Bostick, Yongping Yue, Chun Long, Dongsheng Duan Molecular Microbiology and Immunology, School of Medicine, University of Missouri, Columbia, MO Background. A cure for dystrophin-deficient muscular dystrophy requires concomitant correction in both skeletal muscle and the heart. Recent advance in gene transfer technique has made it possible to deliver a therapeutic dystrophin gene to more than half cardiomyocytes. We have previously shown that dystrophin expression in 50% of cardiomyocytes ameliorated stress-induced cardiomyopathy in young dystrophin-null mdx mice. However, it is not clear whether this level of expression can halt end-stage heart disease. Here we evaluated the protective effect of partial dystrophin expression in aged mdx mice that displayed severe dilated cardiomyopathy. Methods and Results. To mimic 50% transduction of the myocardium, heterozygous female mice were generated by crossing normal and affected animals. We confirmed that half of the heart cells in these mice expressed dystrophin. Heart function and pathology were evaluated using a comprehensive panel of anatomical, histopathological and in vivo physiological assays in 2-year-old mice. Heart weight/body weight ratio was completely normalized. Morphology study revealed an absence of cardiac fibrosis and calcification. Importantly, all the ECG and hemodynamic parameters were corrected. Further studies suggest that the protection was not due to expansion of dystrophin positive cells in the aged heart. Interestingly, a double immunostaining for dystrophin and utrophin revealed complementary expression in the heart of heterozygous mice. Conclusions. We demonstrated that dystrophin expression in half cardiomyocytes is sufficient to eliminate severe heart disease in aged mdx mice. We propose a model where the interspersing dystrophin-positive cells form a latticework that functions as a reinforcing steel grid to strengthen the mechanic stability of the whole heart. Since utrophin is usually up-regulated in DMD patients, our results raise the hope of ameliorating dystrophic cardiomyopathy by partial gene transfer. (Supported by grants from the National Institutes of Health and the Muscular Dystrophy Association). 16) In Utero Delivery of Adeno-Associated Virus Vector-Encoding Human MiniDystrophin Leads to Functional Correction in Dystrophin Deficient Mice Anthony Y. Tsai, Sasha Bogdanovich, Christina F. Hughes, Masayuki Endo, Jesse Vrecenak, Jeremy Traas, Philip Zoltick, Tejvir S. Khurana, Tim Brazelton, Alan W. Flake Surgery, Children s Hospital of Philadelphia, Philadelphia, PA; Physiology, University of Pennsylvania, Philadelphia, PA Introduction: Duchenne muscular dystrophy (DMD) is the most common disabling and lethal congenital muscle disorder. Patients with DMD experience progressive muscle degeneration and weakness until they succumb to respiratory or cardiac failure. Currently there is no cure for DMD. Human mini-dystrophin with truncation of the repeating rod domain has been shown to ameliorate dystrophic histopathology and restores membrane integrity. Adeno-associated virus serotype 9 (AAV2/9) has also been shown to have high muscle specificity. Here, we investigated the possibility of using in utero delivery of AAV2/9 vector-mediated mini-dystophin to achieve longterm functional correction of dystrophin deficient mdx mice. Methods: Fetuses from pregnant mdx mice underwent vitelline vein injection at E14.5 with AAV2/9.CMV.minidystrophin. Specific skeletal muscles, cardiac muscle, and brain and liver tissue from treated and age-matched mdx control mice were harvested on week 5, 10 and 20. Human mini-dystrophin expression was detected by immunohistochemistry (IHC) of frozen sections and quantified by real-time PCR (Q-PCR). Extensor digitorum longus (EDL) muscles of the treated and control mice were used in physiological studies to measure force of maximal twitch, tetanus and eccentric force drop between first and fifth contractions (ECC). Results: Expression of human mini-dystrophin was detected in week 5, 10, and 20 samples using IHC with human specific antibody with no significant difference in the proportion of transduction among the 3 time points. QPCR of heart, TA and soleus at the 20-week time point using the human dystrophin probe (hDys) with the ribosomal 18S probe as the endogenous control detected a normalized hDys/18S RNA ratio of 4.46 3.04, 2.29 1.12, and 2.76 3.86, respectively. Functional studies showed that ECCs demonstrated significantly (P<0.01) less reduction in treated vs. control animals (4.30 2.02% vs.14.01 6.54%) respectively. There was no difference in force observed during twitch (93.00 26.94mN vs. 103.15 31.15mN) or tetanus (459.17 98.81mN vs. 528.24 141.77mN) between treated and untreated animals respectively. Conclusion: These data demonstrate that systemic in utero administration of AAV 2/9 vector encoding for human mini-dystrophin can 1) attain significant transgene expression in skeletal muscles and the heart corroborated by Q-PCR results 2) achieve sustained expression of the transgene without evidence of immune response and 3) result in functional improvement in the mdx model of muscular dystrophy. In utero gene transfer remains an attractive strategy for the treatment of the muscular dystrophies. 17) Inhibiting Myostatin in Dystrophic Mice with Follistatin Improves the Success of Myoblast Transplantation Basma F. Benabdallah, Manaf Bouchentouf, Joel Rousseau, Pascal Bigey, Annick Michaud, Pierre Chapdelaine, Daniel Scherman, Jacques P. Tremblay Genetique Humaine, Centre de Recherche du CHUL, Quebec, QC, Canada; Genetique Humaine, Centre de Recherche du CHUL, Quebec, QC, Canada; Genetique Humaine, Centre de Recherche du CHUL, Quebec, QC, Canada; U640, Inserm, Paris, France; Genetique Humaine, Centre de Recherche du CHUL, Quebec, Canada; Ontogenie et Reproduction, Centre de Recherche du CHUL, Quebec, QC, Canada; U640, Inserm, Parid, France; Genetique Humaine, Centre de Recherche du CHUL, Quebec, France Duchenne muscular dystrophy is a recessive disease due to a mutation in the dystrophin gene. Myoblasts transfer permits to introduce dystrophin gene in dystrophic muscle fibers. However, this approach success is reduced by the short duration regeneration post-transplantation; wich reduces the number of hybrid fibers. In this manuscript, the aim was to verify whether the success of the myoblast transplantation is enhanced by the blockade of the myostatin signal with an antagonist, follistatin. Three different approaches were studied to over-express the follistatin in mdx transplanted muscles. First, transgenic follistatin/mdx mice were generated; second, a follistatin plasmid was electroporated in mdx muscles, and finally, follistatin was induced in mdx mice muscles by a histone deacetylase inhibitor treatment. All the three approaches induced an improvement of the success of the myoblast transplantation. Moreover, a fiber hypertrophy was also observed in all muscles, proving that the inhibition of the myostatin by the follistatin may represent a good method to improve myoblast transplantation and increase muscle function. The inhibition of the myostatin by follistatin could be a promising novel therapeutic approach toward the treatment of muscle wasting in muscle diseases such as Duchenne muscular dystrophy in combination with myoblast transplantation. 18) Distinct Transduction Profiles in the Dystrophic Dogs with rAAV Serotype 8 Sachiko Ohshima, Jin-Hong Shin, Akiyo Nishiyama, Katsutoshi Yuasa, Hiroyuki Nakai, Takashi Okada, Shin'ichi Takeda Molecular Therapy, National Institute of Neuroscience, NCNP, Kodaira, Tokyo, Japan; Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine, Pittsburgh, PA Background: Duchenne muscular dystrophy (DMD) is an X-linked, lethal disorder of the striated muscle caused by mutations in the dystrophin gene, which encodes a large sub-sarcolemmal cytoskeletal protein. The absence of dystrophin associated with the loss of dystrophin-glycoprotein complex (DGC) from the sarcolemma results in progressive muscle weakness, cardiomyopathy, and early death. Several treatment modalities have been attempted to correct the dystrophic phenotypes, but effective therapy still awaits to be developed. A recombinant adeno-associated virus (rAAV) serotype 2 (rAAV2) have been utilized in the various preclinical and clinical studies, but the rAAV serotype 8 (rAAV8) demonstrated the better gene transfer efficiency in the skeletal and cardiac muscle. Here we investigated the transduction profiles with the rAAV2 and rAAV8 in the muscles of canine X-linked muscular dystrophy in Japan (cxmdJ). Methods: The rAAV2 or rAAV8 encoding the lacZ gene driven by a CMV promoter was injected into the tibialis anterior and extensor carpi ulnaris of the normal beagles at 6-10 weeks old (1x1011 - 2x1013 vg). We did biopsy of the transduced muscle 2 weeks after the injection and analysed histologically to examine -gal expression and immune response. We also injected the rAAV8 encoding the microdystrophin (M3, c CS2) gene driven by the CMV promoter into the tibialis anterior and extensor carpi ulnaris of cxmdJ at 6-8 weeks old (1x1012 - 2x1013 vg) and examined the efficiency of rAAV8. The transduced muscle has been sampled 4 weeks after the injection to perform histochemical and immunohistochemical analysis, and Western blotting. Results: We detected the greater number of the gal-positive fibers in the rAAV8-transduced canine skeletal muscles than those in the rAAV2-transduced muscles 2 weeks after the injection. Moreover, we can detect much less inflammatory cell infiltration in the rAAV8-transduced muscle than the rAAV2-transduced muscles. Immunohistochemistory of CD4, CD8, CD11b, and MHC class I, revealed less CTL-mediated immune response with the rAAV8 than those with the rAAV2. However, we can detect a certain degree of immune response with a high dose of rAAV8. Consequently, extensive microdystrophin expression in the rAAV8-injected skeletal muscle of cxmdJ was achieved. Discussion: rAAV8mediated gene transfer showed effective transgene expression with less immune responses than those in the rAAV2-mediated transduction. Even though rAAV8mediated gene transfer is less immunogenic, it is indispensable to clarify the appropriate condition of virus titer and dose according to the encoding gene. We are currently attempting either the limb perfusion or systemic delivery through saphenous vein in cxmdJ to examine the rAAV8-mediated transduction efficiency of microdystrophin. Conclusion: The rAAV8 is the efficient tool for the therapeutic gene delivery into the dystrophic canine skeletal muscle. The systemic microdystrophin transduction with the rAAV8 would be promising for the future DMD gene therapy. 19) Intravascular Delivery of AAV-9 Leads to Efficient Transduction of Multiple Muscles in Dog Yongping Yue, Janet Bogan, Arka Ghosh, Dan Bogan, Chun Long, Bruce F. Smith, Joe N. Kornegay, Dongsheng Duan Molecular Microbiology and Immunology, University of Missouri, Columbia, MO; Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO; Scott-Ritchey Research Center, Auburn University, Auburn, AL; Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, NC Duchenne muscular dystrophy (DMD) is the most common childhood fatal muscle disease. Since nearly every muscle in the body is affected, an effective DMD gene therapy will require efficient transduction of all body muscles. Proof-of-concept bodywide transduction has been demonstrated in mice with adeno-associated viruses (AAV). However, the size of the human body is several hundreds fold larger than that of a mouse. Translating results in the mouse to human patients remains a titanic challenge. The size of a dog is only several fold different from that of a human patient. The dog model therefore represents an ideal system to test whether intravascular delivery can target multiple muscles prior to human clinical trials. Previous attempts to deliver AAV directly to canine muscle have largely failed to achieve efficient transduction. This is mainly due to a strong immune response. In this study, we evaluated whether systemic gene delivery in newborn dogs could avoid inducing immune responses and lead to high level expression. An AAV-9 vector carrying the human placental alkaline phosphatase reporter gene was injected into neonatal dogs via the jugular vein in the absence of immuno-suppressive drugs. Transgene expression was examined by histochemical staining at 2, 3, 4, and 6 weeks after AAV infection in the cranial sartorius, vastus lateralis, long digital extensor and the lateral head of the gastrocnemius muscles. In contrast to the previous reports of low expression and strong immune reaction in dog muscle, we observed high-level persistent expression in multiple muscles. In some sections, up to 95% of myofibers were transduced. Taken together, our results provide the first evidence that systemic AAV delivery can reach multiple muscles in a large animal and that body size is not a barrier to intravascular AAV gene transfer. Our results raise the hope of whole body correction for many systemic diseases (such as DMD) in human patients. (Supported by grants from the National Institutes of Health and the Muscular Dystrophy Association). 20) Ameliorating Dystrophic Pathology Via AAV-Mediated Gene Delivery of Myostatin Propeptide Chunping Qiao, Jianbin Li, Bing Wang, Juan Li, Xiao Xiao Molecuar Pharmaceutics, UNC Shool of Pharmacy, Chapel Hill, NC; Department of Orthopaedic Surgery, University of Pittsburgh, Pittsburgh, PA Duchenne muscular dystrophy (DMD), caused by mutations in the dystrophin gene, is the most common, disabling and lethal muscle disease. Myostatin has been extensively documented as a negative regulator of muscle growth. Myostatin blockades therefore offers an effective strategy for treating a number of muscle degenerative diseases, including sarcopenia and muscular dystrophy. In this study, we investigated whether gene delivery of myostatin inhibitors, specifically, the propeptide, could improve muscle growth and ameliorate the pathologies of DMD in mouse model. The serotype 8 AAV-MPRO76AFc vector was delivered into 3-monthold mdx mice by simple tail vein injection. The treated mdx mice started to gain weight two weeks after vector injection (p<0.01), and the difference became highly significant after one and half months of treatment (p<0.001). The average serum CK level of treated mice (610 261 IU/L) was slightly lower than mdx mice (906 289) (n=5 for each group, p=0.06), although it was still far above the wild type mouse (6.7 2.3 IU/L). We then did cryo-thin-sectioning on TA muscle, and performed histology examination. The treated muscle showed improved muscle pathology with more uniformed muscle fiber sizes and less mononuclear cells. The treated muscle also displayed less fibrosis. Because of the limited number of studied animals, at present time, we are still in the progress to evaluate whether AAV-MPRO76AFc gene delivery into mdx mice can increase their muscle strength. In conclusion, our study indicated that delivering myostatin propeptide gene offered therapeutic benefit in Duchenne Muscular Dystrophic mice. 21) Antisense Oligonucleotide Selection for Exon Skipping by a Readout Reporter System with High Capacity and Specificity Allen E. Zillmer, Qi L. Lu, Jeffrey Rosenfeld McColl-Lockwood Laboratory for Muscular Dystrophy Research, Carolinas Medical Center, Charlotte, NC Duchenne muscular dystrophy (DMD) presents a unique opportunity to apply antisense technology (AON). Since the majority of DMD mutations occur in a region non-critical to the gene function, exons or groups of exons can be skipped by AONs resulting in truncated but functional protein. The critical factor determining the efficiency of exon skipping is the AON selection. RT-PCR detection of mRNA with target exon skipped in human myoblast culture is highly variable. In this study, we explore the use of an expression vector-based readout reporter system for the selection of AONs. This system utilizes EGFP reporter to demonstrate the efficiency of specific AON on exon skipping. The sequence of a human dystrophin intron was inserted into a specific site within the EGFP coding region. The insertion however did not disrupt the reading frame of the gene as the intron sequence was recognized by the transcription machinery and reliably spliced out of the transcripts. The insertion of E23 of the mouse dystrophin together with 900bp of its two flanking introns produced a chimeric transcript with the insertion of E23 in the EGFP transcript, leading to the disruption of EGFP reading frame. Targeting the transcripts with specific AONs to E23 removed the exon and restored the expression of EGFP. The levels of EGFP expression reliably indicated the efficiency of AON both in vitro and in vivo. The high levels of EGFP expression and consistency in culturing the transfectant C2C12 myoblast provide highly sensitive and reliable measurement for the selection of AON with highest potency in skipping specific human dystrophin exon. We conclude that this system will be complementary to the human myoblast cell culture systems, providing high capacity to distinguish the differential antisense effect and to select most potent AON for skipping many dystrophin exons required for treating the majority of DMD. 22) The Evaluation of Novel Photoreceptor Specific Promoters in an EIAV Lentiviral Vector for Targeted Gene Expression in the Photoreceptors of the Eye On Kan, Marjorie Nicoud, Sharifah Iqball, Stuart Sims, Jian Kong, Peter Gouras, Rando Allikmets, Stuart Naylor, Katie Binley Biological Systems, Oxford BioMedica (UK) Ltd, Oxford, United Kingdom; Ophthalmology and Pathology Cell Biology, Columbia University, New York Many human diseases resulting in blindness such as Stargardt disease (STGD) primarily affect the photoreceptors. STGD is a macular dystrophy caused by a mutation in the photoreceptor ATP binding transporter gene (ABCR) resulting in the accumulation of the toxic lipofuscin pigment A2E. This leads to degeneration of RPE and photoreceptor cells. We wanted to develop an EIAV based lentiviral vector that displays photoreceptor specific gene expression and evaluate these vectors in the murine ABCR-/- k/o model of Stargardt disease. We investigated the ability of several truncated human photoreceptor specific promoters: rhodopsin (Rho), phosphodiesterase (PDE) and retinitis pigmentosa 1 (RP1) to drive reporter gene expression in vitro, by means of transient plasmid transfections in various cell lines, including the human retinoblastoma Y-79. Subsequently, the promoters were transferred to the EIAV lentivector platform for further evaluation in vitro and in vivo. These promoters were either evaluated alone or in conjunction with an enhancer element derived from the human interphotoreceptor retinoid binding protein gene (IRBP). We discovered that the PDE promoter coupled with three copies of the IRBP enhancer element showed significant expression in Y-79 cells, comparable to the CMV promoter, but showed little or no activity in non-photoreceptor cell types in vitro. Subretinal delivery of these EIAV vectors into mice demonstrated that reporter gene expression was restricted to the photoreceptor cell layer. The PDE promoter coupled with a single IRBP enhancer element displayed the strongest photoreceptor specific expression in vivo. We will present data to show that these novel photoreceptor promoters can be used to drive ABCR expression in the photoreceptors of ABCR-/k/o mice resulting in a significant reduction in A2E accumulation and thus show promise for addressing this disease in the clinical setting. 23) Viral Mediated Transfer of the ELOVL4 Gene Can Result in High Level ELOVL4 Protein Expression in Human RPE Cells, Increasing the Major AntiInflammatory Retinal Fatty Acid DHA Sergey S. Seregin, Maria O. Tikhonenko, Julia V. Busik, Andrea Amalfitano Departments of Microbiology and Molecular Genetics, Pediatrics, Physiology, Michigan State University, East Lansing, MI The highly conserved Elongation of very long chain fatty acids-like 4 protein (ELOVL4) belongs to a family of proteins functioning in elongation of long chain fatty acids. ELOVL4 is expressed in retina, skin, brain and testis suggesting important roles for ELOVL4 in these tissues. Inherited mutations in the ELOVL4 gene are known to account for autosomal dominant Stargardt-like macular degeneration, and autosomal dominant macular dystrophy (ADMD), possibly due to significant decreases in retinal production of very long chain fatty acids (VLCFA). All of these abnormalities are associated with the loss of functional ELOVL4 protein, thus gene therapy approaches based upon transduction of the ELOVL4 gene, and expression of ELOVL4 protein in retinal tissues, may be a viable strategy for several retinal diseases. To establish the proof of concept for this goal, we investigated ELOVL4 gene delivery via the use of an Adenovirus (Ad) based gene transfer vector. Ad based vectors are known to transduce a wide variety of cell types, are the most widely utilized gene transfer vector currently utilized in human clinical gene therapy trials, and most importantly, are currently being safely utilized in advanced human clinical trials for treatment of other retinal diseases. A recombinant rat or human ELOVL4 cDNA was generated, and each was placed under transcriptional control of a CMV enhancer/promoter element, within an Ad shuttling plasmid. Recombination with an E1-,E3- Ad plasmid, resulted in the successful generation of an [E1-,E3]Ad+(rat or human)ELOVL4 vector. These recombinant Ads were successfully amplified on 293 cells, and utilized to infect human Retinal Pigmented Epithelial (hRPE) cells. The hRPE cells infected with either human, or rat ELOVL4 recombinant Ads expressed up to 25 fold higher protein levels of ELOVL4 relative to hRPE cells infected with a control (CMV-LacZ) Ad vector, as determined by Western blotting of proteins derived from the infected cells. More importantly, the increased levels of ELOVL4 expression were also associated with significantly elevated levels of docosahexaenoic acid (DHA) in the hRPE cells, as determined by RP-HPLC followed by UV and evaporative light scatter detection methods. These data provide proof of concept that efficient transfer of the ELOVL4 gene can be accomplished in hRPE cells, and that over-expression of ELOVL4 protein can increase the levels of the major retinal anti-inflammatory fatty acid DHA. Future studies will determine how useful this approach will be in relevant animal models of retinal disease. 24) Effect of Insulin-Like Growth Factor-1 (IGF-1) on Global Gene Expression Profiles in Murine Skeletal Muscle C. Ramana Bhasker, Shaochun Song, Theodore Friedmann Center for Molecular Genetics, Dept. of Pediatrics, University of California San Diego, La Jolla, CA The peptide hormone IGF1 is a powerful mitogen that plays vital roles in the action of growth hormone and is a central participant in growth and function of skeletal muscle. It has been shown to bring about important therapeutic effects in the mdx mouse model of muscular dystrophy and other forms of muscular and neuromuscular disease such as a mouse model of ALS. There is little detailed understanding of the global effects of IGF-1 on global patterns of gene expression. To develop a more thorough understanding of the mechanisms of action of IGF-1, we have recently undertaken microarray-based and proteomic studies of IGF-1 on murine myoblasts in culture and in skeletal muscle in vivo. We report here a description of the acute in vivo effects of IGF-1 on global patterns of gene expression in skeletal muscle of the mouse, using an Affymetrix (mouse genome 430 2.0 array) platform and data analysis with the Vampire Software (SDSC and UCSD) and the Genespring Software (Agilent/Silicon Genetics). Our results indicate that more than 200 genes representing many functional families are down-regulated while far fewer genes are up-regulated by IGF1 at 1, 2, 4 and 6 hours after subcutaneous administration of IGF-1. In contrast, IGF-1 produces a far greater degree up-regulation of many gene families in cultured myoblasts. Interestingly, after the in vivo exposure to IGF-1, some important muscle-specific genes are strongly down-regulated while a number of small and large ribosomal subunit proteins are up-regulated, consistent with stimulated protein synthesis These kinds of detailed global gene expression and proteomic studies represent powerful approaches to understanding the mechanisms of action of IGF-1 and related growth factors, both in the development of disease phenotypes and in their treatment. 25) Spliceosome-Mediated RNA Trans-Splicing for Muscular Dystrophies Chunping Qiao, Jianbin Li, Bing Wang, Juan Li, Xiao Xiao Department of Molecular Pharmaceutics, UNC School of Pharmacy, Chapel Hill, NC; Department of Orthopaedic Surgery, University of Pittsburgh, Pittsburgh, PA Spliceosome-mediated RNA trans-splicing (SMaRT) is a novel RNA-reprogramming strategy. It offers several potential advantages over traditional gene replacement therapy, such as reduced transgene size and elimination of ectopic expression of the repaired product by retaining endogenous regulation. In this study, we explored the potential application of SMaRT technology for muscular dystrophies. We use the Duchenne muscular dystrophy mouse, the mdx, as a model system, in which the mutated gene dystrophin was shown to carry a premature stop codon in exon 23. We intended to use pre-trans-splicing molecules (PTM) containing mouse dystrophin intron 22 sequence linked the C-terminal coding region of dystrophin to intercept the RNA splicing before exon 23. The resultant is a trans-spliced mRNA encoding a functional dystrophin. To examine the trans-splicing efficiency in cultured cells, we first created a target plasmid by inserting the dystrophin intron 22 into mini-dystrophin gene 3858, between Rod 2 (R2) and Rod 22 (R22) regions, generating plasmid pAAV-cmv-3858-spolyA-in22. We then constructed three PTMs containg different binding domains of intron 22 and the C-half of the mini-dystrophin 3990 with a myc tag. The PTM plasmids were respectively co-transfected with the target plasmid for trans-splicing into 293 cells. Trans-splicing events were monitored by both RT-PCR and western blot. Sequencing results of RT-PCR fragment confirmed the expected trans-splicing products, while western blot showed expected myc-tagged minidystrophin. We then packaged those three PTMs into adeno-associated viral vector and injected them into the leg muscle of three-month-old mdx mice. Two months after vector injection, the mice were sacrificed and the injected muscles were carefully dissected. The trans-splicing events were analyzed by both immunofluorescent (IF) staining and RT-PCR. IF staining indicated low percentage of mini-dystrophin positive cells, while RT-PCR failed to detect trans-splicing product. Our results indicated that SMaRT technology offers some potential for the treatment of muscular dystrophies; however, its efficiency has to be improved to render therapeutic effect. 26) Isolation, Characterization, and Myogenesis of Satellite Cells Derived from Skeletal Muscle Nicholas Ieronimakis, Gayathri Balasundaram, Jeffrey S. Chamberlain, Morayma Reyes Pathology, University of Washington, Seattle, WA; Neurology, University of Washington, Seattle, WA Satellite cells constitute the natural stem cell reservoir for regeneration in adult skeletal muscle. The regenerative role and capability of satellite cells (SC) in skeletal muscle makes them a primed candidate for treatment of degenerative diseases such as muscular dystrophy. However, the challenge of isolation and expansion of pure satellite cell populations has encumbered their use in clinical applications. We report here the direct isolation of SC from skeletal muscles by fluorescence-activated cell sorting based on the expression of Sca-1, CD34, CD31 and CD45 cell surface antigens. Sca-1 is predominantly a marker of hematopoietic stem cells and as we have reported a marker of endothelial cells in the skeletal muscle. Conveniently SC are Sca-1 negative. CD34 is also a marker of hematopoietic stem cells and some endothelial cells, but in the skeletal muscles CD34 is highly expressed by SC. By utilizing a host of antibodies we have isolated a very distinct and homogenous population recognized as Sca-1-, CD31-, CD34+ and CD45- from both wt and mdx (dystrophyn -/-) mice, ages ranging from newborn to 25 months. Because some hematopoietic and endothelial cells also express these markers we excluded all CD45+ cells (hematopoietic) and CD31+ (endothelial) cells. Furthermore, the forward and size scatter pattern of this population corresponds with the size morphology of SC as a homogenously small mononuclear population. Depending on age, this population represents 0.5-2% of all mononuclear cells derived from skeletal muscles. The abundance of SC in the skeletal muscle declines with age though more severely in mdx mice to almost undetectable levels by 25 month. RT-PCR analysis and immunohistochemistry of freshly sorted cells confirms this population expresses many satellite cell markers such as Pax7, NCAM, MyoD, Myf5, Syndecan 3, CD34, and c-met while lacking expression of endothelial markers TEK, vWF, and Flt. Nearly 100% of these cells express Pax7 and Myf5 signifying this population to be very pure and homogenous. In addition, staining of freshly FACS-sorted SC for NCAM and cmet are polarized which correlates with polarization of these markers in muscle tissue sections towards the basal lamina. Interestingly, SC derived from mdx mice express lower levels of NCAM, MyoD, Myf5 and syndecan 3, perhaps due to impaired myogenesis. We have culture-expanded these cells in vitro in F10C with 15% horse serum and 10 ng/ml bFGF, obtaining clones of more than 1000 SC (>10 cell doublings) for seven days. These cells can differentiate into robust myotubes when bFGF is withdrawn and horse serum is reduced to 1.5%. To demonstrate the myogenic potential of these cells, Sca-1-, CD31-, CD45-, CD34+ cells were FACSsorted from GFP mice and directly injected intramuscularly in the tibialis anterior muscles of mdx mice. Two weeks after transplantation multiple green myofibers were seen throughout the TA muscle, demonstrating the regenerative capability of these cells. This new approach using flow cytometry to directly isolate SC will be extremely useful in studying their biology for the development of regenerative treatments for muscular diseases. 27) Global Force Improvement by Systemic AAV Delivery of -Sarcoglycan Transgene in Deficient Mice Marc Bartoli, Francoise Fougerousse, Jérôme Poupiot, Olivier Danos, Isabelle Richard LGMD, Genethon, Evry, France -sarcoglycanopathy (Limb Girdle Muscular Dystrophy type 2D, LGMD2D) is a recessive muscular disorder caused by deficiency in -sarcoglycan, a transmembrane protein part of the dystrophin-associated complex. We constructed recombinant adeno-associated virus (rAAV) vectors expressing the human - sarcoglycan cDNA under the control of a muscle specific promoter. Efficient and sustained transgene expression with correct sarcolemmal localization and without evident toxicity was obtained after intra-arterial injection into the both hindlimbs of a LGMD2D murine model. Transgene expression resulted in restoration of the sarcoglycan complex, histological improvement, membrane stabilization with full rescue of the contractile force deficits and stretch sensibility that led to an increase of the global activity of the animals. We will also present the analyses carried-out to monitor the immune response against the transgene. This poster establishes the feasibility for whole body AAV-mediated -sarcoglycan gene transfer as a therapeutic approach 28) Viral Based Therapeutic for LGMD Type 2D Deficiencies Demonstrates Long-Term Results in Alpha-Sarcoglycan Deficient Animals Louise R. Rodino-Klapac, Christopher J. Shilling, Zarife Sahenk, K. Reed Clark, Jerry R. Mendell Center for Gene Therapy, Columbus Children s Research Institute, Columbus, OH Limb-girdle muscular dystrophy (LGMD) type 2D is characterized by skeletal muscle weakness and results from mutations occurring in the -sarcoglycan gene. Localized in the sarcolemma, the sarcoglycans ( , , , and ) are a subcomplex of the dystrophin-associated proteins (DAP). Alpha-sarcoglycan ( -SG) deficiency is the most common form of sarcoglycan-LGMD and no therapeutic treatments are currently available. The -SG mouse model provides an opportunity to test translational treatment approaches. Prior studies have suggested that AAV-mediated -SG gene transfer could not sustain expression because of transgene toxicity, potentially precluding clinical gene transfer for LGMD2D. Herein, we describe our in vivo work comparing -SG gene expression from either the ubiquitously expressed cytomegalovirus (CMV) promoter, or muscle specific promoters, muscle creatine kinase (MCK) and desmin (DES) in the -SG KO mouse in the context of rAAV gene delivery. We injected the tibialis anterior (TA) muscle of 4-6 week old -SG KO mice with rAAV1. -SG (CMV, MCK, DES promoter) at low (3 x 109 vg) and high (3 x 1010 vg) doses. Sustained gene expression was observed irrespective of promoters at six and twelve weeks post gene transfer. Quantitation of -SG gene expression by fiber counts yielded similar levels of myofiber transduction for CMV and MCK at the 6 week high dose, 61.4% versus 64.4% respectively. However, -SG expression using the DES promoter was significantly lower at the high dose with 34% of the myofibers transduced. Similar levels of expression were seen at the 12 week time point for MCK and DES, while CMV exhibited a 25% reduction in expression. Our studies show sustained and robust gene expression well beyond the time at which transgene cytotoxic effects were previously reported using rAAV2. CMV. -SG. Mononuclear cell analysis in our studies showed no evidence of infiltrating B or T cell subsets or macrophages. Differences in our findings compared to previous work could relate to AAV serotype. In summary, our data demonstrate robust -SG gene expression as long as 3 months using AAV1, with no reduction in expression using the muscle specific MCK promoter. These findings enhance the possibility for gene therapy as a potential treatment option for LGMD2D and support our current efforts for launching a gene therapy study for this disease. 29) Functional Analysis of alpha-Dystrobrevin-3 in Striated Muscle with rAAV6 Gene Transfer Guy Odom, Glen Banks, James Allen, Marv Adams, Leonard Meuse, Miki Haraguchi, Stan Froehner, Jeffrey Chamberlain Neurology, University of Washington, Seattle, WA; Physiology and Biophysics, University of Washington, Seattle, WA Alpha Dystrobrevin ( Db) is a member of the dystrophin subfamily of proteins, and is an integral component of the dystrophin-glycoprotein complex (DGC) at the sarcolemma of striated muscles. Decreasing amounts of dystrobrevin from the sarcolemma contributes to the severity of disease in several muscular dystrophies including DMD and LGMD. Mice lacking the three isoforms of Db ( Db-/-) found in skeletal muscle present with several phenotypic changes consistent with dystrophy including a mild cardiomyopathy, myopathy, fragmented neuromuscular synapses and shallow folds within the muscle-tendon junction. Both DB1 and DB2 transgenes restore the skeletal muscle abnormalities in the DB knockout to varying degrees because of subtle differences in their location within the muscles membrane and varying functional domains contained in each protein. Because DB3 lacks many of these functional domains it has previously not been considered a relevant isoform of Db. In the present study we tested whether Db-3 has any functional significance in skeletal muscle integrity, myotendonous junction folding, and maintenance of the neuromuscular synapses. We intravenously administered recombinant adenoassociated viral (rAAV6) vectors containing GFP-tagged Db-2 or Db-3 transgenes in order to transduce the striated musculature of -Db knockout mice. The mice were injected at 4 weeks of age and analyzed at 8 and 12 weeks after administration. Db expression was viewed using immunohistochemistry of frozen muscle sections. Muscle integrity was analyzed in frozen sections stained with hematoxylin and eosin. Neuromuscular synapses were analyzed in wholemount teased muscle fibers stained with rhodamine conjugated alpha-bungarotoxin. The lengths of myotendonus junctions of gastrocnemius muscle were viewed using transmission electron microscopy. We also tested how DB3 might interact with the DGC in vitro and in vivo utilizing coimmunoprecipitation assays. Muscles throughout DB-/- mice were transduced with rAAV6-DB2 and rAAV6-DB3. DB2 and DB3 both localized to the sarcolemma and were concentrated within the folds of the myotendinous and neuromuscular junctions. Db3 and Db-2 were equally effective at preventing central nucleation in most skeletal muscle fibers, preventing fragmentation of neuromuscular synapses and restoring folds at the myotendinous junction. DB2 was more effective than DB3 in preventing fingerlike projections from protruding out of the synaptic borders. In coimmunoprecipitation studies, Db-3 was demonstrated to interact with the sarcoglycan subcomplex through an individual sarcoglycan interaction in vitro. In vivo coimmunoprecipitation of DB3 from whole muscle lysates resulted in the majority of the DGC components being detected. Db-3 is a significant component of the DGC that functions to prevent muscle degeneration, synapse destabilization and myotenindous junction defects within skeletal muscle. These results suggest the reduction of DB3 from the sarcolemma in various muscular dystrophies likely contributes to their pathogenesis in a similar manner to DB1 and DB2. 30) Dp116 Expression in Dystrophin/Utrophin Double Knockout (mdx:utrn-/-) Mice Restores Muscle Mass and Viability but Does Not Correct Dystrophic Pathology Andrea L. H. Arnett, Luke M. Judge, Jeffrey S. Chamberlain Molecular and Cellular Biology, Medical Scientist Training Program, University of Washington, Seattle; Neurology, University of Washington, Seattle Mice deficient in both dystrophin and utrophin (mdx:utrn-/-) exhibit a phenotype similar to that seen in DMD patients, including severe muscle wasting, skeletal deformities, joint contractures, and premature death. In these mice, the absence of both dystrophin and utrophin prevents assembly of the dystrophin-glycoprotein complex (DGC). We previously generated transgenic mice with skeletal muscle expression of Dp116 and have introduced this transgene onto the mdx:utrn-/- background. Dp116 is a non-muscle isoform of dystrophin that lacks the actin-binding domain and the majority of the rod domain found in the full length dystrophin isoform. Thus, it is postulated that Dp116 has no mechanical link to the actin cytoskeleton, but can still assemble and stabilize the DGC at the sarcolemma. We now report that Dp116 expression can dramatically increase both muscle mass and lifespan in mdx:utrn-/and can delay both formation and progression of kyphosis and joint contractures. However, histological examination of muscle from these transgenic mice reveals signs of muscular dystrophy similar to that seen in muscles of dystrophin-deficient (mdx) mice. Our results provide compelling evidence that dystrophin and the DGC participate in signaling mechanisms that are critical for maintenance of muscle mass. Alternatively, Dp116 may contribute to the structural integrity of myofibers via noncanonical binding of Dp116 to the actin cytoskeleton. 31) Targeting Adenovirus Vector to Neural Cell Adhesion Molecule (NCAM) for Gene Delivery to Developing Muscle Bhanu Munil Koppanati, Christoph Volpers, Joel Kaar, Florian Kreppel, Alan J. Russell, Stefan Kochanek, Carl Lagenaur, Paula R. Clemens Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, PA; Center for Molecular Medicine (ZMMK) and Institute for Genetics, University of Cologne, Cologne, Germany; McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA; Division of Gene Therapy, University of Ulm, Helmholtzstrasse, Ulm, Germany; Department of Neurobiology, University of Pittsburgh School of Medicine, Pittsburgh, PA; Neurology Service, Department of Veterans Affairs Medical Center, Pittsburgh, PA Duchenne muscular dystrophy (DMD) is a debilitating disorder affecting approximately 1 in 3500 males. Most primary muscle disorders, such as DMD, affect multiple widespread muscle groups. One significant concern hindering successful gene transfer to muscle is inefficient gene delivery to muscle cells. Gene delivery in utero and vector targeting to a receptor highly expressed and relatively specific for developing muscles are strategies with the potential to overcome this difficulty. Several strategies have been attempted to achieve vector targeting of various cells. In this study genetic modification of adenoviral fiber protein to contain the Immunoglobulin G-binding domain of Staphylococcus aureus protein A combined with post-assembly vector modification with a targeting antibody was pursued. We tested whether a genetically modified adenoviral vector containing a synthetic Immunoglobulin G-binding domain in the capsid conjugated to anti-NCAM antibody can target NCAM, a cell surface receptor on embryonic muscle, to increase muscle gene delivery in vitro. In order to ablate native tropism and thus detarget the modified vector, the vector was conjugated with increasing concentrations of polyethylene glycol (PEG). We demonstrated that PEG ablated native tropism in a dosedependent fashion. Furthermore, in order to retarget the vector to NCAM-expressing myoblasts, the vector was incubated with increasing concentrations of anti-NCAM antibody, prior to conjugation with PEG. We observed an antibody-dependent increase in gene transfer in primary myoblasts, but not in control HepG2 cells. Further increases in antibody concentration resulted in a reduction of gene transfer. In addition, when primary myoblasts preincubated with an RGD peptide were infected with the PEG-treated vector-targeting ligand complex, we observed a reduction in gene delivery suggesting that the modified vector could be using the same pathway for internalization as the unmodified vector. Further studies are underway to study intracellular trafficking of the vector in muscle cells. Our results encourage us to pursue studies of the modified vector in utero, with a hope for synergistic benefits of combined fetal gene delivery and cell-specific targeting. 32) Effect of Animal Age and the Route of Administration on Systemic AAV-9 Transduction Brian P. Bostick, Arkasubhra Ghosh, Yongping Yue, Chun Long, Donsheng Duan Molecular Microbiology and Immunology, School of Medicine, University of Missouri, Columbia, MO Recombinant adeno-associated virus serotype-9 (rAAV-9) has recently been identified as an exceptional vector for systemic gene transfer. Most studies have focused on the transduction profile following intravenous delivery. It is not well appreciated how the route of administration (intra-venous vs. intra-arterial) and the age of the animal at the time of gene transfer (neonatal vs. adult) influence the transduction profile. To examine the effect of the administration route, we injected newborn C57Bl/10 mice with AV.RSV.AP (an rAAV-9 vector carrying the alkaline phosphatase (AP) reporter gene; 12 x 1010 vg/g body weight) either intravenously via the facial vein, or intra-arterially via the left ventricle. To determine the effect of age, we compared 40-hr-old neonatal mice (12 x 1010 vg/g body weight, 3.5 x 1011 vg/mouse) and 7-wk-old adult mice (7 x 1010 vg/g body weight, 1 x 1012 vg/mouse) injected intravenously. Transgene expression was evaluated at 12 weeks postinjection. In general, regardless of the age or the route of administration, we observed broad transduction in all body skeletal muscle with the highest expression in the diaphragm and tongue muscles. Interestingly, muscles composed predominantly of slow twitch fibers (such as the soleus muscle) showed less expression. Alkaline phosphatase activity in the soleus muscle was significantly lower than that in the tibialis anterior muscle (27.97 7.97 units/ g protein vs. 144.57 35.81 units/ g protein, respectively; p < 0.05). We also observed high transduction in cardiac muscle in all groups. In addition to the strong striated muscle expression, we found consistent lung transduction (including small conducting airways and alveolar cells) in all groups. The route of administration seemed to have minimal effect on the overall transduction profile. However, the age at the time of injection seemed to play a role in several tissues/organs. Both the liver and the kidney showed higher expression when AAV was delivered to adult mice. Adult mice also showed relatively high transduction in vascular endothelial cells and vascular smooth muscle. The most striking difference was seen in the retina. The ganglion cell layer was strongly transduced following neonatal administration but not after adult delivery. Taking together, our results provide additional support for the use of rAAV-9 in systemic gene therapy. Efficient transduction via both venous and arterial routes allows maximal flexibility in a clinical setting. However, the age-associated differences should be considered when designing gene therapy protocols with rAAV-9. (Supported by grants from the National Institutes of Health, the Cystic Fibrosis Foundation and the Muscular Dystrophy Association). 33) The AAV9 Capsid Preferentially Transduces Cardiac Tissue and Demonstrates Unique Behavior In Vivo Christina A. Pacak, Cathryn S. Mah, Bijoy Thattaliyath, Melissa A. Lewis, Sean Germain, Glenn A. Walter, Barry J. Byrne Department of Molecular Genetics and Microbiology, University of Florida, Gainesville, FL; Powell Gene Therapy Center, University of Florida, Gainesville, FL; Department of Pediatric Cardiology, University of Florida, Gainesville, FL; Department of Cellular and Molecular Therapy, University of Florida, Gainesville, FL; Department of Physiology and Functional Genomics, University of Florida, Gainesville, FL Pompe disease is a form of muscular dystrophy and metabolic myopathy caused by a deficiency in the enzyme acid alpha glucosidase (GAA). A lack of GAA results in the accumulation of glycogen in lysosomes and consequent dysfunction in cells throughout the body that can result in cardiorespiratory failure. Previously, we have demonstrated that intravenous administration of AAV2/9 to adult and newborn mice and non-human primates can result in efficient transduction of cardiac tissue. Here we demonstrate through MRI, ECG, biochemical, and histological analysis that intravenous administration of AAV2/9 carrying a therapeutic transgene in newborn (prior to disease presentation) or adult (where cardiac abnormalities were present) mice can ameliorate the cardiac phenotype in a mouse model of Pompe disease (Gaa-/-). AAV2/9 treatment normalizes the shortened PR interval in Gaa-/- mice by an average of 5 ms (from 22% shorter than wild-type to only 11% shorter). Cardiac MR demonstrated a return of the EF% to wild-type control levels following therapeutic gene delivery at both ages. Furthermore, NMR, EM and PAS staining revealed clearance of glycogen and healthy sub-cellular morphology in the hearts of treated mice. Ex vivo force mechanics on excised diaphragm sections demonstrated an improvement in the contractile force of treated mice and CT scans show a distinct decrease in spinal kyphosis. In conclusion, these data show that IV administration of rAAV2/9-CMV-Gaa can both prevent and correct the cardiac phenotype and provide global improvement in a mouse model of Pompe disease warranting application of this approach to the clinic setting to assess its efficacy in humans. 34) Scalable Method for Purification of Recombinant Adeno-Associated Viral Vector (rAAV) Type 6 Jingmin Zhou, Bern Hauck, J. Fraser Wright, Katherine A. High, Guang Qu Center for Cellular and Molecular Therapeutics, The Children s Hospital of Philadelphia, Philadelphia, PA Recombinant adeno-associated viral vector serotype 6 (rAAV-6) has been shown to be highly efficient for muscle gene delivery, including delivery to skeletal and cardiac muscle. This unique property makes rAAV-6 an especially attractive vector to deliver therapeutic genes for diseases such as Duchenne muscular dystrophy. However, systemic gene delivery will likely require production and purification of large quantities of this vector, and therefore readily scalable purification processes are critical for clinical development. We previously reported a scalable purification process for rAAV type 2 and successfully implemented the process into cGMP manufacturing to purify clinical vectors (Qu et al, J. Virol. Meth. 2007 Mar.140 18392). Here we described a fully scalable, column chromatography-based method for rAAV-6 purification. The method developed included cation exchange chromatography followed by an anion exchange column chromatography to remove cell culture process impurities (HEK293 cell and cell culture medium derived), and a final single CsCl gradient centrifugation step to remove AAV-6 empty capsids, a product related impurity. In contrast to results obtained during process development for AAV-2 vector purification by ion exchange chromatography and AAV-6 vector purification by heparin resin chromatography, the rAAV-6 particles were found to be very sensitive (ie. easily detached from the ion exchange resins) to the washes using buffers containing ionic detergents such as N-lauroyl sarcosine or sodium cholate. These detergent washes could not be used in rAAV-6 cation exchange column chromatography to remove cellular proteins. Highly efficient vector purification was achieved by optimizing washing and elution conditions, including pH, salt concentration and volume of the washing and elution buffers. Typically 80% of the starting / feed-stream vector genomes were recovered through the two-column process, and highly purified, empty capsid-free rAAV-6 vector was obtained in the final preps. The vector preps have been shown to be effective by transduction in vitro. 35) Phase I-IIa Gene Therapy Protocol for LGMD 2C: Clinical Strategy and Implications Hafedh Haddad, Didier Caizergues, Aude Rigolet, Carole Masurier, Anne-Marie Douar, Bernard Gjata, Patricia Noguiez-Hellin, Muriel Audit, Loic Millot, Otto Merten, Pascal Laforet, France Leturcq, Thomas Voit, Bruno Eymard, Pierre Carlier, Olivier Benvéniste, François Lemoine, Norma Romero, David Klatzmann, Jean-Yves Hogrel, Marie Rosier-Montus, Patrice Denèfle, Serge Herson, Anne-Marie Masquelier Labs, Genethon, Evry, France; H pital, La Piti -Salp tri re, Paris, France; Institut de Myologie, (IDM), Paris, France; H pital, Cochin, Paris, France Limb girdle muscular dystrophy type 2C (LGMD 2C) is a rare autosomal recessive muscular disorder caused by mutations in the gamma sarcoglycan ( -SG) gene, the most frequently occurring one being the 525delT mutation. Patients with LGMD 2C commonly present with proximal and progressive muscular weakness before the age of 10 and become wheelchair bound by age 12 on average. Cardiomyopathy and respiratory insufficiency may develop during the course of the disease and lead to poor prognosis and premature death. After completion of the pre-clinical work in collaboration with Harvard Gene Therapy Initiative and University of Pennsylvania School of Medicine, GENETHON has obtained an Orphan Drug Designation by EMEA in October 2004 and approval from the French Agencies in November2006 to initiate a phase I/IIa clinical gene therapy trial using a serotype 1 adeno-associated virus (AAV1) vector harboring the human -sarcoglycan gene under the control of a desmin promoter. The current investigation center is the Department of Internal Medicine and the Institute of Myology at La Piti -Salp tri re Hospital in Paris. The investigational product is administered by a single intramuscular injection into the radial muscle.The primary objective of this phase I-IIa is to evaluate the clinical safety of local intramuscular injection of the gene therapy product and tolerance with a 6 months follow-up. Secondary objectives are to monitor local and systemic immune responses, assess histological modifications and gene transfer into injected muscles, including sarcoglycan protein complex expression and therapeutic gene distribution. A total of 9 patients, aged above 15 and harboring the 525delT mutation, will be enrolled sequentially in the study and assigned to 3 cohorts with a single doseescalation. A 6 month follow-up is planned for each patient. Evaluation will address clinical histological, morphological, biological, immunological and functional parameters as well as MRI. First patient has been treated in Paris in December 2006. The phaseI-IIa is planned to last 18 months. In view of the phase IIb of this clinical trial in which systemic administration will be performed, important preclinical and clinical points to consider are being tackled and will be presented. 36)Transplantation of Autologous Genetically Modified Myoblasts: A Possible Treatment for Recessive Muscular Dystrophies Jacques P. Tremblay, Simon Quenneville, Pierre Chapdelaine, Zoé Coulombe, Christophe Pichavant Human Genetic, CRCHUL, Quebec, Canada Autologous myoblasts are possible vectors to deliver genes into muscle fibers of patients affected by various recessive muscular dystrophies. These genes may be incorporated in the myoblasts in culture by transfection or nucleofection with a plasmid using the Phi C31 integrase system. The gene of interest may also be transduced with a retrovirus, a lentivirus or a dual high-capacity (hc) adenovirus (Ad)adeno-associated virus (AAV) hybrid vector (HV) that can deliver two full-length dystrophin-encoding modules into target cells. These various methods have been used successfully by our research team to introduce successfully the microdystrophin gene and the full length dystrophin gene in myoblasts of animal models or of patients affected by Duchenne Muscular Dystrophy (DMD). These genetically modified cells were than transplanted with success in muscles leading to the expression of the transgene in thousands of muscle fibers, even in non-human primates. We have also used lentiviral vector to introduce a normal UDP-GlcNAc 2epimerase / N-acetylmannosamine kinase (GNE) gene into myoblasts of patients affected by Hereditary Inclusion Body Myopathy (HIBM). This genetic correction permitted to increase the synthesis of sialic acid in the genetically corrected cells and thus increase the sialylation of the surface glycoproteins of these cells. To avoid the potential toxicity of the transgene expression in myoblasts the transgene may be place under the control of a muscle specific promoter so that it is expressed only following fusion with the host muscle fibers. The genetic modification of myoblasts is also an excellent way to introduce U7snRNA to induce exon skipping. This approach has permitted the expression of a quasi-dystrophin both in vitro and in vivo. These results demonstrate the feasibility of ex vivo gene for many recessive muscular dystrophies. 37) Vascular Endothelial Growth Factor Overexpression Improved Survival and Engraftment of Human Myoblasts into SCID Mouse Muscles Manaf Bouchentouf, Basma F. Benabdallah, Pascal Bigey, Jacques P. Tremblay Human Genetics, CHUL, Quebec, Canada; Laboratoire de Pharmacologie Chimique et G n tique, Inserm U640 CNRS, Paris, France Background: Transplantation of muscle precursor cells (MPC) or myoblasts is considered as a potential approach to repair damaged skeletal muscles in a number of recessive muscular dystrophies, in particular Duchenne muscular dystrophy (DMD). This approach is however limited by the death of the majority of the cells within four days following their injection. The purposes of this study were to evaluate whether ischemia was implicated in the early death of myoblasts transplanted into SCID mouse TA muscles and to evaluate the effect of VEGF overexpression on their survival, proliferation and graft success. Methods: In vitro apoptosis was induced by culturing myoblast in 6 well plates at 75% of confluence with serum free medium 24 hours in hypoxic environment. To evaluate effect of VEGF165 on transplanted myoblasts, intramuscular injections were performed into TA muscles of 18 SCID mice using a 30-gauge needle insulin syringe. Nine mice were injected with 40 g of pCEP4-VEGF plasmid ressuspended in 40 l of 0,9% sterile NaCl and nine other mice were injected with 40 g of pCEP4 empty plasmid ressuspended in 40 l of 0,9% sterile NaCl. Electroporation was performed using two stainless steel plate electrodes placed on the injected muscles and eight square electric pulses of 20 ms length with 200 V/cm voltage. Hypoxia within transplanted human myogenic cells was detected using the Hypoxyprobe-1 kit. To quantify the in vivo cell mortality, myoblasts were radio-labeled with [methyl-14C] thymidine (50 mCi/mmol). Radio-labeled cells (1x106) were injected into 8 sites of the TA muscle using a glass micro-pipette.The amount of radio-label within each TA muscle was measured on DNA extracts using liquid scintillation counter Results: VEGF165 overexpression on human myoblast enhanced their survival in vitro but had no effect on their proliferation. Electroporation of SCID mouse TA muscles with pBabe-VEGF-Puro vector promoted VEGF165 production. Electroporation of SCID mouse TA muscles with vector coding for the VEGF165 reduced transplanted myoblast death but did not improve their proliferation. VEGF165 overexpression enhanced myoblast graft success. Conclusion: Our results suggest a role of hypoxia in the death of transplanted myoblast and that VEGF overproduction could be an alternative to improve their survival. 38) AAV Vector-Mediated Expression of Follistatin Stimulates Skeletal Muscle Hypertrophy and Can Enhance Functional Performance in Models of Muscle Disease Paul Gregorevic, Norman A. Meznarich, James M. Allen, Miki Haraguchi, Leonard Meuse, Eric Finn, Jeffrey S. Chamberlain Paul D Wellstone Muscular Dystrophy Cooperative Research Center, Department of Neurology, The University of Washington, Seattle, WA Life threatening loss of skeletal muscle strength is associated with a host of conditions including muscular dystrophies, aging, cancer, and chronic obstructive pulmonary disease. Interventions that enhance muscle function in patients offer the potential for considerably improving quality of life and extending lifespan. We demonstrate that an intervention utilizing rAAV6 vectors carrying an expression cassette encoding the short form of human follistatin can stimulate muscle fiber hypertrophy to increase murine skeletal muscle mass by 100% and force producing capacity by >50% within 6 weeks of intramuscular injection. The hypertrophic effects mediated by this intervention are constrained to the region of transduction, but intravascular administration of this vector to mice facilitates body-wide skeletal muscle hypertrophy and increases contractile capacity on a similar time scale without loss of effect for over 16 months. This potent and rapidly acting intervention represents a powerful model system to study the biology of skeletal muscle adaptation, but may also prove valuable as a timely intervention where rapid loss of muscle strength is a serious health concern. We have ascertained that interventions incorporating rAAV6-Fst administration can restore muscle mass and function in murine models of muscle disease including aging-related muscle wasting, where the muscles of treated animals approach the functional capability of muscles from young adult animals. Ongoing work is aimed at utilizing rAAV6-mediated expression of follistatin to study the mechanisms governing muscle adaptation, and to establish the therapeutic potential for treatment of conditions caused or complicated by loss of skeletal muscle strength. 39) Intravenous Injection Cofactors of Adeno-Associated Virus Produce Varying Tissue-Specific Transduction Levels in Adult Mice Brian R. Schultz, Paul Gregorevic, Eric Finn, Caitlin Doremus, Leonard Meuse, Miki Haraguchi, James M. Allen, Jeffrey S. Chamberlain Molecular and Cellular Biology, University of Washington, Seattle, WA; Medical Scientist Training Program, University of Washington School of Medicine, Seattle, WA; Department of Neurology, University of Washington School of Medicine, Seattle, WA Adeno-associated virus (AAV) is a promising vector for gene replacement therapy in many genetic diseases. In particular, recombinant AAV serotype 6 (rAAV6) has been utilized to administer a therapeutic gene with beneficial results in mouse models of Duchenne muscular dystrophy (DMD). In genetic diseases affecting tissues throughout the body, such as DMD, effective methods of systemic transduction are essential. Manipulating intervention conditions, such as vector dose and co-delivery of transduction-facilitating cofactors, may be important for optimizing the efficacy of viral-vector gene therapy. At increasing doses of intravenously delivered rAAV6CMV-hPLAP to adult C57BL/6 mice, cardiac muscle and diaphragm display a progressive dose-response; however, soleus and tibialis anterior muscles exhibit measurable transduction only after receiving a threshold level of vector genomes above that at which heart and diaphragm begin to show transduction. Administering vector in conjunction with VEGF, heparin sodium, VEGF + heparin sodium, or mouse serum albumin, or under conditions of anesthesia or exercise, results in varying degrees of transduction increase or decrease depending on the muscle group. Another factor to consider is the viral capsid itself. In the preparation of rAAV6, intact virions lacking DNA ( empty capsids ) are produced in excess of those virions that contain vector genomes ( full capsids ). During vector preparation, full capsids are routinely separated from empty capsids by density gradient ultracentrifugation. Adding an excess of empty capsids into the intravenous injection solution increases transduction to varying degrees, again depending on muscle group. These results suggest that the vector genome dose and/or cofactors involved with intravenous injection of rAAV6 can be tailored to target tissue groups and/or levels of transduction.