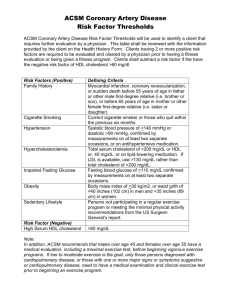
chapter 39 - Department of Medical Genetics
advertisement

Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Human Biochemical Genetics 177-575 From: Chapter Braunwald’s Heart Disease CHAPTER 42 Lipoprotein Disorders and Cardiovascular Disease Jacques Genest • Peter Libby NOTE: For the Human Biochemical Genetics 177-575 course, the required material for the course will be on the first two headings: Lipoprotein Transport System and Lipoprotein Disorders. The remaining text is for additional knowledge for those interested. Please note that tables 42.3 and 42.4 as well as Figure 42.4 is very relevant for this course. 1 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Chapter outline Introduction Lipoprotein Transport System Biochemistry of Lipids Lipoproteins, Apolipoproteins, Receptors, and Processing Enzymes Lipoprotein Metabolism and Transport Lipoprotein Disorders Definitions Genetic Lipoprotein Disorders Low-Density Lipoproteins (Type II Hyperlipidemia) Triglyceride-Rich Lipoproteins High-Density Lipoproteins Secondary Causes of Hyperlipidemia Drugs that Affect Lipid Metabolism Bile acid binding resins HMG-CoA Reductase Inhibitors (Statins) Cholesterol Absorption Inhibitors Fibric Acid Derivatives (Fibrates) Nicotinic Acid (Niacin) Fish Oils Phytosterols CETP inhibitors Other Medications, combination therapy Monitoring of Lipid Therapy Clinical Trials of Drugs Affecting Lipid Metabolism Treating High-Risk (primary prevention) Patients Secondary Prevention and Intensive LDL-C lowering Acute Coronary Syndromes Medical therapy versus revascularization; Regression studies Approach to the Treatment of Lipid Disorders Target Levels Lifestyle Changes Treatment of Combined Lipoprotein Disorders Novel Approaches Drug Development and Future Directions Specific sub groups (diabetics and the elderly) Gene Therapy 2 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Abbreviations ABCA1 ATP-binding cassette transporter A1 ABCG1 ATP-binding cassette transporter G1 ACAT acetyl-CoA acetyltransferase Apo apolipoprotein ApoBec Apo B editing complex C cholesterol CE cholesteryl ester CETP cholesteryl ester transfer protein CoA Coenzyme A EL Endothelial lipase FFA Free fatty acids HDL High-density lipoprotein HL Hepatic lipase HMG CoA Red hydroxymethylglutaryl coenzyme A reductase HSL hormone-sensitive lipase IDL intermediate-density lipoprotein IDL Intermediate density lipoprotein LCAT lecithin cholesterol acyltransferase LDL low-density lipoprotein LDL-R low-density lipoprotein receptor Lp(a) Lipoprotein (a) LRP low-density lipoprotein receptor– related peptide NPC1L1 Niemann-Pick disease type C protein (NPC) like 1 PLTP phospholipid transfer protein PLTP Phospholipid transfer protein sER smooth endoplasmic reticulum SR- B1 scavenger receptor B1 TG triglycerides TIA Transient ischemic attack TRL Triglyceride-rich lipoproteins 3 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease VLDL very-low-density lipoprotein VLDL-R very-low-density lipoprotein receptor. Jan 2009 4 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 INTRODUCTION Serum levels of lipids and lipoprotein lipids have proven among the most potent and best substantiated risk factors for atherosclerosis in general and coronary heart disease (CHD) in particular. Chapter 39 discusses the biological basis of atherosclerosis. Chapter 39 presents the observational data on lipids as a key component of the palette of cardiovascular risk factors. The present chapter deals with the fundamentals of lipid metabolism, the therapeutic approaches to treatment of lipid disorders, and the evidence base regarding their clinical use. Although the term hyperlipidemia has long been used in clinical practice, the term dyslipoproteinemia more appropriately reflects the disorders of the lipid and lipoprotein transport pathways associated with arterial diseases. Dyslipidemia encompasses disorders often encountered in clinical practice such as low high-density lipoprotein (HDL) cholesterol level and elevated triglyceride level but an average total plasma cholesterol level. Certain rare lipoprotein disorders can cause overt clinical manifestations, but most common dyslipoproteinemias themselves only rarely cause symptoms or produce clinical signs that are evident on physical examination. Rather, they require laboratory tests for detection. Dyslipoproteinemias constitute a major risk factor for atherosclerosis and coronary artery disease, and their proper recognition and management can reduce cardiovascular and total mortality rates. Thus the fundamentals of lipidology presented here have importance for the daily practice of cardiovascular medicine. LIPOPROTEIN TRANSPORT SYSTEM Biochemistry of Lipids The lipid transport system has evolved to carry hydrophobic molecules (fat) from sites of origin to sites of utilization through the aqueous environment of plasma. The proteins (apolipoproteins) that mediate this process are conserved throughout evolution in organisms with a circulatory system. Most apolipoproteins derive from an ancestral gene and contain both hydrophilic and hydrophobic domains. This amphipathic structure enables these proteins to bridge the interface between the aqueous environment of plasma and the phospholipid constituents of the lipoprotein [1]. The major types of lipids that circulate in plasma include cholesterol and cholesteryl esters, phospholipids, and triglycerides (Fig. 42–1). Cholesterol constitutes an essential component of mammalian cell membranes and furnishes substrate for steroid hormones and bile acids. Many cell functions depend critically upon membrane cholesterol, and cells tightly regulate cholesterol content. Most of the cholesterol in plasma circulates in the form of cholesteryl esters, in the core of lipoprotein particles. The enzyme lecithin:cholesterol acyltransferase (LCAT) forms cholesteryl esters in the blood compartment by transferring a fatty acyl chain from phosphatidyl choline to cholesterol. Triglycerides consist of a three-carbon glycerol backbone covalently linked to three fatty acids. The fatty acid composition varies in terms of chain length and presence of double bonds (degree of saturation). Triglyceride molecules are nonpolar and hydrophobic; they are transported in the core of the lipoprotein. Hydrolysis of triglycerides by lipases generates free fatty acids (FFA) used for energy. Phospholipids constituents of all cellular membranes consist of a glycerol molecule linked to two fatty acids. The fatty acids differ in length and in the presence of a single (monounsaturated) or multiple (polyunsaturated) double bonds. The third carbon of the glycerol moiety carries a phosphate group to which one of four molecules is linked: choline (phosphatidyl choline—or lecithin), ethanolamine (phosphatidylethanolamine), serine (phosphatidyl serine), or inositol (phosphatidylinositol). A related 5 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 phospholipid, sphingomyelin, has special functions in the plasma membrane in the formation of membrane microdomains, such as rafts and caveolae. The structure of sphingomyelin resembles that of phosphatidylcholine. The backbone of sphingolipids uses the amino acid serine rather than glycerol. Phospholipids are polar molecules, more soluble than triglycerides or cholesterol or its esters. Phospholipids participate in signal transduction pathways: hydrolysis by membrane-associated phospholipases generates second messengers such as diacyl glycerols, lysophospholipids, phosphatidic acids, and free fatty acids such as arachidonate that can regulate many cell functions. Lipoproteins, Apolipoproteins, Receptors, and Processing Enzymes Lipoproteins are complex macromolecular structures, composed of an envelope of phospholipids and free cholesterol, a core of cholesteryl esters and triglycerides. The apolipoproteins comprise the protein moiety of lipoproteins (Fig. 42–2). Lipoproteins vary in size, density in the aqueous environment of plasma, and lipid and apolipoprotein content (Fig. 42–3, Table 42–1). The classification of lipoproteins reflects their density in plasma (1.006 gm/mLl) as gauged by flotation in the ultracentrifuge. The triglyceride-rich lipoproteins consisting of chylomicrons and very-low-density lipoprotein (VLDL) have a density less than 1.006 gm/ml. The rest of the ultracentrifuged plasma consists of low-density lipoprotein (LDL), HDL, and lipoprotein (a) [(Lp(a)]. Apolipoproteins have four major roles: (1) assembly and secretion of the lipoprotein (apo B100 and B48); (2) structural integrity of the lipoprotein (apo B, apo E, apo AI, apo AII); (3) co activators or inhibitors of enzymes (apo AI, CI, CII, CIII); and (4) binding or docking to specific receptors and proteins for cellular uptake of the entire particle or selective uptake of a lipid component (apo AI, B100, E) (Table 42–2). The role of several apolipoproteins (AIV, AV, D, and J) remain incompletely understood. Many proteins regulate the synthesis, secretion, and metabolic fate of lipoproteins; their characterization has provided insight in molecular cellular physiology and provided targets for drug development (Table 42–3). The discovery of the LDL receptor furnished a landmark in understanding cholesterol metabolism and receptor-mediated endocytosis. The LDL receptor regulates the entry of cholesterol into cells, as tight control mechanisms alter its expression on the cell surface, depending on need. Other receptors for lipoproteins include several that bind VLDL but not LDL. The LDL receptor– related peptide, which mediates the uptake of chylomicron remnants and VLDL, preferentially recognizes apolipoprotein E (apo E) [2]. The LDL receptor–related peptide interacts with hepatic lipase. A specific VLDL receptor exists [3]. The interaction between hepatocytes and the various lipoproteins containing apo E is complex and involves cell surface proteoglycans that provide a scaffolding for lipolytic enzymes (lipoprotein lipase and hepatic lipase) involved in remnant lipoprotein recognition [4-6]. Macrophages express receptors that bind modified (especially oxidized) lipoproteins. These scavenger lipoprotein receptors mediate the uptake of oxidized LDL into macrophages. In contrast to the exquisitely regulated LDL receptor, high cellular cholesterol content does not suppress scavenger receptors, enabling the intimal macrophages to accumulate abundant cholesterol, become foam cells, and form fatty streaks. Endothelial cells can also take up modified lipoproteins through a specific receptor, such as Lox-1 [7]. At least two physiologically relevant receptors bind HDL particles: the scavenger receptor class B (SRB1; also named CLA-1 in humans) [8] and the adenosine triphosphate–binding cassette transporter A1 (ABCA1) [9]. SR-B1 is a receptor for HDL (also for LDL and VLDL, but with less affinity). SR-B1 mediates the selective uptake of HDL cholesteryl esters in steroidogenic tissues, hepatocytes, and 6 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 endothelium. The ABCA1 mediates cellular phospholipid (and possibly cholesterol) efflux and is necessary and essential for HDL biogenesis [9]. Lipoprotein Metabolism and Transport The lipoprotein transport system has two major roles: the efficient transport of triglycerides from the intestine and the liver to sites of utilization (fat tissue or muscle) and the transport of cholesterol to peripheral tissues, for membrane synthesis and for steroid hormone production or to the liver for bile acid synthesis. Intestinal pathway (chylomicrons to chylomicron remnants). Life requires fats. The human body derives essential fatty acids that it cannot make from the diet. Fat typically furnishes 20 to 40 percent of daily calories. Triglycerides comprise the major portion of ingested fats. For an individual consuming 2000 kcal/day, with 30 percent in the form of fat, this represents approximately 66 gm of triglycerides per day and approximately 250mg (0.250 gm) of cholesterol. Upon ingestion, pancreatic lipases hydrolyze triglycerides into free fatty acids and mono- or diglycerides. Emulsification by bile salts leads to the formation of intestinal micelles. Micelles resemble lipoproteins in that they consist of phospholipids, free cholesterol, bile acids, di- and monoglycerides, free fatty acids, and glycerol. The mechanism of micelle uptake by the intestinal brush border cells still engenders debate. The Niemann-Pick C1-like 1 (NPC1-L1) protein is part of an intestinal cholesterol transporter complex, and the target for the selective cholesterol absorption inhibitor ezetimibe [10] (see below). The advent of inhibitors of cholesterol uptake has rekindled interest in the mechanisms of intestinal fat absorption. After uptake into intestinal cells, fatty acids undergo re-esterification to form triglycerides and packaging into chylomicrons inside the intestinal cell and enter the portal circulation (Fig. 42–4, part 1). Chylomicrons contain apo B48, the amino-terminal component of apo B100. In the intestine, the apo B gene is modified during transcription into mRNA with a substitution of a uracil for a cytosine by an apo B48 editing enzyme complex (ApoBec). This mechanism involves a cytosine deaminase and leads to a termination codon at residue 2153 and a truncated form of apo B. Only intestinal cells express ApoBec. Chylomicrons rapidly enter the plasma compartment after meals. In capillaries of adipose tissue or muscle cells in the peripheral circulation, chylomicrons encounter lipoprotein lipase (LPL), an enzyme attached to heparan sulfate and present on the luminal side of endothelial cells (Fig. 42–4, part 2). LPL activity is modulated by apo CII (an activator) and by apo CIII (an inhibitor). Lipoprotein lipase has broad specificity for triglycerides; it cleaves all fatty acyl residues attached to glycerol, generating three molecules of free fatty acid for each molecule of glycerol. Muscle cells rapidly take up fatty acids. Adipose cells can store triglycerides made from fatty acids for energy utilization, a process that requires insulin. Fatty acids can also bind to fatty acid–binding proteins and travel to the liver, where they are repackaged in VLDL. Peripheral resistance to insulin can thus increase the delivery of free fatty acids to the liver with a consequent increase in VLDL secretion and increased apo B particles in plasma. As discussed later, this is one of the consequences of the metabolic syndrome (Chap. 43). The remnant particles, derived from chylomicrons following LPL action, contain apo E and enter the liver for degradation and reutilization of their core constituents (Fig. 42–4, part 3). Hepatic pathway (very-low-density lipoprotein to intermediate-density lipoprotein). Food is not always available, and dietary fat content varies. The body must ensure readily available triglyceride to meet energy demands. Hepatic secretion of VLDL particles serves this function (Fig. 42–4, part 4). VLDLs are triglyceride-rich lipoproteins smaller than chylomicrons (Table 42–1 and Fig. 42–3). They contain apo B100 as their main lipoprotein. As opposed to apo B48, apo B100 contains a domain recognized by the LDL receptor (the apo B/E receptor). VLDL particles follow the same catabolic pathway through lipoprotein lipase as chylomicrons (Fig. 42–4, part 2). During hydrolysis of triglyceride-rich lipoproteins by LPL, an exchange of proteins and lipids takes place: VLDL particles (and chylomicrons) acquire apo Cs and apo E, in part from HDL particles. VLDLs also exchange triglycerides for cholesteryl esters from HDL (mediated by cholesteryl ester transfer protein [CETP]) (Fig. 42–4, part 9). Such bidirectional transfer of constituents between lipoproteins serves several purposes, allowing lipoproteins to acquire specific apolipoproteins that will dictate their metabolic fate; transfer of phospholipids onto nascent HDL particles mediated by phospholipid transfer protein (PLTP) (during the loss of core triglycerides, the phospholipid envelope becomes redundant and is shed off to apo AI to form new HDL particles); and transfer of cholesterol from HDL to VLDL remnants so it can be metabolized in the liver. This exchange constitutes a major part of the “reverse cholesterol transport pathway”. After hydrolysis of triglycerides partly depletes VLDL of triglycerides, VLDL particles have relatively more cholesterol, shed several apolipoproteins (especially the C apolipoproteins), and acquire apo E. The VLDL remnant lipoprotein, called intermediate-density lipoprotein (IDL), is taken up by the liver via its apo E moiety (Fig. 42–4, part 3) or further delipidated by hepatic lipase to form an LDL particle (Fig. 42–4, part 6). There are at least four receptors for triglyceride-rich lipoprotein (TRL), TRL remnants, and apo B–containing lipoproteins: the VLDL receptor, the remnant receptor, the LDL receptor (also called the apo B/E receptor), and the LDL receptor–related peptide. Most hepatic receptors share in their ability 7 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 to recognize apo E, an engagement which mediates uptake of several classes of lipoproteins, including VLDL and intermediate-density lipoprotein. The interaction between apo E and its ligand is complex and involves the “docking” of TRL on heparan sulfate proteoglycans before presentation of the ligand to its receptor. Low-density lipoproteins. LDL particles contain predominantly cholesteryl esters packaged with the protein moiety apo B 100. Normally, triglycerides constitute only 4 - 8 percent of the LDL mass (Table 42.1). In the presence of elevated plasma triglyceride levels, LDL particles can become enriched in triglycerides and depleted in core cholesteryl esters. LDL particle size variation results from changes in core constituents, with an increase in triglycerides and a relative decrease in cholesteryl esters leading to smaller, denser LDL particles. The LDL particles in most higher mammals, including humans and nonhuman primates, serve as the main carriers of cholesterol. In other mammals, such as rodents or rabbits, VLDL and HDL particles transport most of the cholesterol. Cells can either make cholesterol from acetate through enzymatic reactions requiring at least 33 steps or obtain it as cholesteryl esters from LDL particles. Cells internalize LDL via the LDL receptor (LDL-R) (Fig. 42–5A). LDL particles contain one molecule of apo B. While several domains of apo B are highly lipophilic and associate with phospholipids, a region surrounding residue 3500 binds saturabily and with high affinity to the LDL-R. The LDL-R localizes in a region of the plasma membrane rich in the protein clathrin (Figs. 42–4, part 7 and 42–5A). Once bound to the receptor, clathrin polymerizes and forms an endosome that contains LDL bound to its receptor, a portion of the plasma membrane, and clathrin. This internalized particle then fuse with lysosomes whose catalytic enzymes (cholesteryl ester hydrolase, cathepsins) release free cholesterol and degrade apo B. The LDL-R will detach itself from its ligand and recycle to the plasma membrane. Cells tightly regulate cholesterol content by (1) cholesterol synthesis in the smooth endoplasmic reticulum (via the ratelimiting step hydroxymethylglutaryl coenzyme A [HMG-CoA] reductase); (2) receptor-mediated endocytosis of LDL (two mechanisms under the control of the steroid-responsive element binding protein [SREBP]); (3) cholesterol efflux from plasma membrane to cholesterol acceptor particles (predominantly apo AI and HDL) via the ABCA1 transporter; and (4) intracellular cholesterol esterification via the enzyme acyl-CoA: cholesteryl acyltransferase (ACAT) (see Fig. 42–5A, B). The SREBP coordinately regulates the first two pathways at the level of gene transcription. Cellular cholesterol binds to a protein called SCAP (SREPB cholesterol-activated protein), which is located on the endoplasmic reticulum. Cholesterol inhibits the interaction of SCAP with SREPB. In the absence of cholesterol, SCAP will mediate the cleavage of SREBP at two sites by specific proteases and release an amino (NH2) fragment of SREBP. The SREBP NH2 fragment will migrate to the nucleus and increase the transcriptional activity of genes involved in cellular cholesterol and fatty acid homeostasis. Cleavage of SREBP depends on a proprotein convertase related to the subtilisin/kexin family of convertases. Another member of the convertase superfamily, proprotein convertase subtilisin/kexin 9 (PCSK9) may participate in the cellular processing of the LDL-R; gain of function mutations in this gene cause dominant familial hypercholesterolemia, while loss of function increases LDL-R and lowers LDL-C significantly [11]. The ACAT pathway regulates cholesterol content in membranes [12]. Humans express two separate forms of ACAT. ACAT1 and ACAT2 derive from different genes and mediate cholesterol esterification in cytoplasm and in the endoplasmic reticulum lumen for lipoprotein assembly and secretion. Regulation of cholesterol efflux depends in part on the ABCA1 pathway, controlled in turn by hydroxysterols (especially 24- and 27-OH cholesterol, which act as ligands for the liver-specific receptor [LXR] family of transcriptional regulatory factors). In conditions of cholesterol sufficiency, the cell can decrease its input of cholesterol by decreasing the de novo synthesis of cholesterol. The cell can also decrease the amount of cholesterol that enters the cell via the LDL-R, increase the amount stored as cholesteryl esters, and promote the removal of cholesterol by increasing its movement to the plasma membrane for efflux. High-density lipoprotein and reverse cholesterol transport. Epidemiological studies consistently have shown an inverse relationship between plasma levels of HDL cholesterol and the presence of coronary artery disease. HDL promotes reverse cholesterol transport and can prevent lipoprotein oxidation and exert anti-inflammatory actions in vitro. The metabolism of HDL is complex and incompletely understood. The complexity arises because HDL particles acquire their components from several sources while these components also are metabolized at different sites. Apolipoprotein AI, the main protein of HDL, is synthesized in the intestine and the liver. Approximately 80% of HDL originates from the liver [13] and 20% from the intestine [14] (Fig 42-4 part 5). Lipid-free apo AI acquires phospholipids from cell membranes and from redundant phospholipids shed during hydrolysis of triglyceride-rich lipoproteins. Lipid-free apo AI binds to ABCA1 and promotes its phosphorylation via cyclic adenosine monophosphate, which increases the net efflux of phospholipids and cholesterol onto apo AI to form a nascent HDL particle (Fig. 42–4, part 10) [15,16]. This particle, containing apo AI and phospholipids (and little cholesterol) resembles a flattened disk in which the phospholipids form a bi-layer surrounded by two molecules of apo AI arranged in a circular fashion at the periphery of the disk (Fig. 42–5B). These nascent HDL particles will mediate further cellular cholesterol efflux. Currently, standard laboratory tests do not measure these HDL precursors because 8 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 they contain little or no cholesterol. Upon reaching a cell membrane, the nascent HDL particles will capture membraneassociated cholesterol and promote the efflux of free cholesterol onto other HDL particles (Fig. 42–4, part 10). Conceptually, the formation of HDL particles appears to involve two-steps , the first step ABCA1-dependent and the second probably does not require ABCA1 [17]. The efflux of cellular cholesterol from peripheral cells, such as macrophages does not contribute importantly to overall HDL-C mass but may have an important effect on export of cholesterol from atheromata. Macrophage can efflux cholesterol onto apo AI and apoE, onto nascent HDL particles via the ABCA1 transporter or onto spherical HDL particles, via the ABCG1 transporter (Fig 42-5E). ABCG1 transporter does not promote cellular cholesterol efflux to lipidfree or lipid-poor apo AI but to mature HDL particles. The plasma enzyme LCAT, an enzyme activated by apo AI, then esterifies the free cholesterol (Figs. 42–4, part 8, and 42–5B). LCAT transfers an acyl chain (a fatty acid) from the R2 position of a phospholipid to the 3’-OH residue of cholesterol, resulting in the formation of a cholesteryl ester (Fig. 42–1). In a process called selective uptake of cholesterol, HDL also provides cholesterol to steroid hormone–producing tissues and the liver through the scavenger SR-B1 receptor (Fig. 42–5C) [18]. Because of their hydrophobicity, cholesteryl esters move to the core of the lipoprotein and the HDL particle now assumes a spherical configuration (a particle denoted HDL3). With further cholesterol esterification, the HDL particle increases in size to become the more buoyant HDL22. Cholesterol within HDL particles can exchange with triglyceride-rich lipoproteins via cholesteryl ester transfer protein (CETP), which mediates an equimolar exchange of cholesterol from HDL to triglyceride-rich lipoprotein and triglyceride movement from triglyceride-rich lipoprotein onto HDL (Fig. 42–4, part 9). Inhibition of CETP increases HDL-C in the blood and represents a therapeutic target for cardiovascular disease prevention. Phospholipid transfer protein (PLTP) mediates the transfer of phospholipids between triglyceride-rich lipoprotein and HDL particles. Triglycerideenriched HDL are denoted HDL2b. Hepatic lipase can hydrolyze triglycerides and endothelial lipase can hydrolyze phospholipids within these particles, converting them back to HDL3 particles (Fig 42-6, part 6). One mechanism of reverse cholesterol transport includes the uptake of cellular cholesterol from extra-hepatic tissues, such as lipid-laden macrophages, and its esterification by LCAT, transport by large HDL particles, and exchange for one triglyceride molecule by CETP. Originally on an HDL particle, the cholesterol molecule can now be taken up by hepatic receptors on a triglyceride-rich lipoprotein or LDL particle. HDL particles, therefore, act as shuttles between tissue cholesterol, triglyceride-rich lipoprotein, and the liver. Most HDL originates in the liver [13] and intestines [14]. Reverse cholesterol transport by HDL constitute a small but potentially important portion of the plasma HDL mass. Indeed, selective inactivation of macrophage ABCA1 does not change HDL-C levels in mice but there is an increase in atherosclerosis [19]. The catabolism of HDL particles has engendered debate among lipoprotein researchers. The protein component of HDL particles is exchangeable with lipoproteins of other classes. The kidneys appear to be a route of elimination of apolipoprotein AI and other HDL apolipoproteins. The lipid component of HDL particles also follow a different metabolic route (Fig. 42–5A, B, and C). 9 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 LIPOPROTEIN DISORDERS Definitions Time and new knowledge have stimulated changes to the classification of lipoprotein disorders. The original classification of lipoprotein disorders by Fredrickson, Lees, and Levy was based on the measurement of total plasma cholesterol and triglycerides and analyzed lipoproteins patterns after separation by electrophoresis. This classification recognized elevations of chylomicrons (type I), VLDL or pre-beta lipoproteins (type IV), “broad beta” disease (or type III hyperlipoproteinemia), beta lipoproteins (LDL) (type II), and elevations of both chylomicrons and VLDL (type V). In addition, the combined elevations of pre-beta (VLDL) and beta (LDL) lipoproteins was recognized as type IIb hyperlipoproteinemia. Though providing a useful conceptual framework, this classification has some drawbacks: it does not include HDL cholesterol, and it does not differentiate severe monogenic lipoprotein disorders from the more common polygenic disorders. Subsequently, the World Health Organization, the European Atherosclerosis Society and, more recently, the National Cholesterol Education Program have classified lipoprotein disorders on the basis of arbitrary cut-points. A practical approach describes the lipoprotein disorder by the absolute plasma levels of lipids (cholesterol and triglycerides) and lipoprotein cholesterol levels (LDL and HDL cholesterol) and considers clinical manifestations of hyperlipoproteinemia in the context of biochemical characterization. For example, a young patient presenting with eruptive xanthomas and a plasma triglyceride level of 11.3 mmol/L (1000 mg/dL) likely has familial hyperchylomicronemia. An obese, hypertensive middle-aged man with a cholesterol level of 6.4 mmol/L(247 mg/dL), a triglyceride level of 3.1mmol/L (274 mg/dL), a HDL cholesterol level of 0.8 mmol/L (31 mg/dL), and a calculated LDL cholesterol level of 4.2 mmol/L (162 mg/dL) likely has the metabolic syndrome, and should spur identification of its components including hypertension and hyperglycemia, should be sought. The clinical usefulness of apolipoprotein levels has stirred debate. Although a useful research tool in general, the measurement of apolipoproteins AI and B practically may add little substantial information to that provided by the conventional lipid profile. Taken as a single measurement, the apo B level provides information on the number of potentially atherogenic particles and can be used as a goal of lipid-lowering therapy [20]. Similarly, LDL particle size correlates highly with plasma HDL cholesterol and triglyceride levels, and most studies do not show it to be an independent cardiovascular risk factor. Small, dense LDL particles tend to track with features of the metabolic syndrome, which usually involves dyslipoproteinemia with elevated plasma triglycerides and reduced HDL cholesterol levels [21]. It remains uncertain whether in addition to LDL particle number reduction, a change in LDL particle size will bring further clinical benefit. Genetic Lipoprotein Disorders Understanding of the genetics of lipoprotein metabolism has expanded rapidly. Classification of genetic lipoprotein disorders usually requires a biochemical phenotype in addition to a clinical phenotype. With the exception of familial hypercholesterolemia, monogenic disorders tend to be infrequent or very rare. Disorders considered heritable on careful family study may be difficult to characterize unambiguously because of age, gender, penetrance, and gene-gene and environmental interactions. Most common lipoprotein disorders encountered clinically result from the interaction of increasing age, lack of physical exercise, weight gain, and a suboptimal diet with individual genetic make-up. 10 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Genetic lipoprotein disorders can affect LDL, lipoprotein (a) [Lp(a)], remnant lipoproteins, triglyceride-rich lipoproteins (chylomicrons and VLDL), or HDL (Table 42–4). Within each of these, genetic disorders can cause an excess or a deficiency of a specific class of lipoprotein. Low-Density Lipoproteins (Type II Hyperlipidemia) Familial hypercholesterolemia. Familial hypercholesterolemia is the most thoroughly studied lipoprotein disorder. The elucidation of the pathway by which complex molecules enter the cell by receptor-mediated endocytosis and the discovery of the LDL receptor represent landmarks in cell biology and clinical investigation. Affected subjects have an elevated LDL cholesterol level greater than the 95th percentile for age and gender. In adulthood, clinical manifestations include corneal arcus, tendinous xanthomas over the extensor tendons (metacarpophalangeal joints, patellar, triceps and Achilles tendons), and xanthelasmas. Transmission is autosomal codominant. Familial hypercholesterolemia affects approximately approximately 1:500, although this prevalence is higher in populations with founder effects. Patients with familial hypercholesterolemia have a high risk of developing coronary artery disease (CAD) by the third to fourth decade in men and approximately 8 to 10 years later in women. Diagnosis is based on elevated plasma LDL cholesterol level, family history of premature CAD, and the presence of xanthomas. A molecular diagnosis is sometimes required. Defects of the LDL-R gene cause an accumulation of LDL particles in plasma and thus alter the function of the LDL-R protein and cause familial hypercholesterolemia (Fig. 42–4, part 7). To date, there are well over 600 identified mutations of the LDL-R gene (see http://www.umd.necker.fr) [22]. Familial defective apo B. Mutations within the apo B gene that lead to an abnormal ligand-receptor interaction can cause a form of familial hypercholesterolemia clinically indistinguishable from the primary form. Several mutations at the postulated binding site to the LDL-R cause familial defective apo B100 (Fig. 42–4, part 7). These consist of apo BArg3500Gln, apo BArg3500Trp, and apo BArg3531Cys [23]. The apo BArg3500Gln results from a GA substitution at nucleotide 3500 within exon 26 of the apo B gene. The defective apo B has a reduced affinity (20 to 30 percent of control) for the LDL-R. LDL particles with defective apo B have a plasma half-life three- to fourfold greater than the half-life of normal LDL. Because of their increased persistence, these LDL particles can more readily undergo oxidative modifications that can enhance their atherogenicity. Affected subjects usually have elevated LDL cholesterol levels up to 400 mg/dL (10.4 mmol/L) but may also have normal levels. Familial defective apo B 100 has a prevalence similar to that of familial hypercholesterolemia (1/500). In subjects with the classic presentation of familial hypercholesterolemia, the prevalence of familial defective apo B 100 is reported to be 1 in 50 to 1 in 20. The reasons for the variability of plasma LDL cholesterol levels remain unexplained. An autosomal dominant form of hypercholesterolemia that maps to chromosome 1p34.1 involves a mutation within the proprotein convertase, subtilisin/kexin type 9 gene (PCSK9). PCSK9 codes for a protein identified as neural apoptosisregulated convertase 1 (NARC1), a novel proprotein convertase belonging to the subtilase family of convertases. It is related to subtilisin/kexin isoenzyme-1 (site-1 protease) required for cleavage of SREBP [11]. Subjects with loss of function mutation of PCSK9 have a markedly lower LDL-C than subjects without the mutation. Black Amricans have a higher prevalence of this protective mutation than whites In the Atherosclerosis Risk in Communities study, subjects with life-long low LDL-C because of a mutation at the PCSK9 gene locus had a marked reduction in coronary events [24], confirming that genetic low LDL-C states confer cardio protective advantage. An autosomal recessive form of familial hypercholesterolemia has been identified in kindred from Sardinia and is caused by mutations in the ARH gene that encodes a protein involved in the recycling of the LDL-receptor [25]. Hypobetalipoproteinemia and abetalipoproteinemia Mutations within the apo B gene can lead to truncations of the mature apo B 100 peptide. Many such mutations cause a syndrome characterized by reduced LDL and VLDL cholesterol but little or no clinical manifestations and no known risk of cardiovascular disease, a condition referred to as hypobetalipoproteinemia. Apo B truncated close to its amino terminus loses the ability to bind lipids, producing a syndrome similar to abetalipoproteinemia, a rare recessive lipoprotein disorder of infancy that causes mental retardation and growth abnormalities. Abetalipoproteinemia is caused by a mutation in gene coding for the microsomal triglyceride transfer protein (MTP) required for assembly of apo B–containing lipoproteins in the liver and the intestine. The resulting lack of apo B–containing lipoproteins in plasma causes a marked deficiency of fat- 11 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 soluble vitamins (A, D, E, and K) that circulate in lipoproteins. In turn, this results in mental and developmental retardation in affected children. Sitosterolemia. A rare condition of increased intestinal absorption and decreased excretion of plant sterols (sitosterol and campesterol) can mimic severe familial hypercholesterolemia, with extensive xanthoma formation. Premature atherosclerosis, often apparent clinically well before adulthood, occurs frequently in patients with sitosterolemia. Diagnosis requires specialized analysis of plasma sterols demonstrating an elevation in sitosterol, campesterol, cholestanol, sitostanol, and campestanol. Interestingly, plasma cholesterol is normal or reduced, and triglycerides are normal. Positional cloning techniques have localized the defect to chromosome 2p21. Mutations in the adenosine triphosphate binding cassette G5 and G8 genes (ABCG5 and ABCG8) have been found in patients with sitosterolemia. The gene products of ABCG5 and ABCG8 are half ABC transporters and are thought to form a heterodimer characteristic of the full ABC transporters. The complex is located in the villous border of intestinal cells and actively pumps plant sterols back into the intestinal lumen. A defect in either of the genes renders the complex inactive, and absorption of plant sterols (rather than their elimination) ensues. ABCG5 and ABCG8 mutations leading to sitosterolemia are very rare [26]. Lipoprotein (a) (see Chap. 36). Lipoprotein(a) (pronounced “lipoprotein little a”)consists of an LDL particle linked covalently with one molecule of apo (a). The apo (a) moiety consists of a protein with a high degree of homology with plasminogen. The apo (a) gene appears to have arisen from the plasminogen gene by non-homologous recombination. The apo (a) gene has multiple repeats of one of the kringle motifs (kringle IV), varying in number from 12 to more than 40 in each individual. Plasma lipoprotein(a) levels depend almost entirely on genetics and correlate inversely with the number of kringle repeats and, therefore, with the molecular weight of apo (a) [27]. Few environmental factors or medications modulate plasma lipoprotein(a) levels. The pathogenesis of lipoprotein(a) may result from an antifibrinolytic potential and/or ability to bind oxidized lipoproteins. Some prospective epidemiological studies have shown a positive (albeit weak) association between lipoprotein(a) and coronary artery disease (see Chap. 39) [28]. Triglyceride-Rich Lipoproteins (TRL) In subjects with the metabolic syndrome and in diabetic patients, elevation of plasma triglyceride level occurs most often in the presence of visceral (abdominal) obesity and a diet rich in calories, carbohydrates, and saturated fats. Severe elevation of plasma triglycerides can result from genetic disorders of the processing enzymes or apolipoproteins and poorly controlled diabetes. Familial hypertriglyceridemia (type IV hyperlipoproteinemia). Familial hypertriglyceridemia is not associated with clinical signs such as corneal arcus, xanthoma, and xanthelasmas. Plasma triglycerides, VLDL cholesterol, and VLDL triglycerides are moderately to markedly elevated; LDL cholesterol level is usually low as is and HDL cholesterol. Total cholesterol is normal or elevated, depending on VLDL cholesterol levels. Fasting plasma concentrations of triglycerides are in the range of 2.3 to 5.7 mmol/L (200 to 500 mg/dL). After a meal, plasma triglycerides may exceed 11.3 mmol/L (1,000 mg/dL). The disorder is found in first-degree relatives, but phenotypic variability is related to gender, age, hormone use (especially estrogens), and diet. Alcohol intake potently stimulates hypertriglyceridemia in these subjects, as does caloric or carbohydrate intake. The relationship with coronary artery disease is not as strong as with familial combined hyperlipidemia and has not been seen in all studies. Depending on criteria used, the prevalence of familial hypertriglyceridemia ranges from 1 in 100 to 1 in 50. The disorder is highly heterogeneous and likely results from several genes, with a strong environmental influence. An unrelated disorder, familial glycerolemia, a chromosome X-linked genetic disorder, may mimic familial hypertriglyceridemia because most measurement techniques for triglycerides use the measurement of glycerol after enzymatic hydrolysis of triglycerides [29]. The diagnosis of familial hyperglycerolemia requires ultracentrifugation of plasma and analysis of glycerol. Hepatic overproduction of VLDL causes familial hypertriglyceridemia is (Fig. 42–4, part 4); the catabolism (uptake) of VLDL particles can be normal or reduced. Lipolysis by LPL appears not to be a limiting factor, although the triglyceride load, especially in the postprandial state, may limit processing of VLDL particles. The genetic basis of familial hypertriglyceridemia is unknown, and the candidate approach to find the gene or genes involved (apo B, LDL, apo CIII) has not yielded fruit thus far. Treatment is based first on lifestyle modifications, including withdrawal of hormones (estrogens and progesterone), limiting alcohol intake, reducing caloric intake, and increasing exercise. The decision to treat this disorder with medications (see below) depends on global cardiovascular risk. An infrequent disorder characterized by severe elevation in plasma triglyceride levels (both VLDL and chylomicrons) is associated with a fat-rich diet, obesity, and poorly controlled diabetes. Recognized as type V hyperlipidemia, the pathogenesis is multifactorial and results from overproduction of both VLDL and chylomicrons and from decreased catabolism of these particles. 12 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Familial hyperchylomicronemia (type I hyperlipidemia). This rare disorder of severe hypertriglyceridemia associates with elevations in fasting plasma triglycerides greater than 11.3 mmol/L (>1,000 mg/dL). These patients have recurrent bouts of pancreatitis and eruptive xanthomas. Interestingly, severe hypertriglyceridemia can also be associated with xerostomia, xerophthalmia, and behavioral abnormalities. The hypertriglyceridemia results from a markedly reduced or absent LPL activity or, more rarely, the absence of its activator, apo CII (Fig. 42–4, part 2) [30]. These defects lead to a lack of hydrolysis of chylomicrons and VLDL and their accumulation in plasma, especially after meals. Extreme elevations of plasma triglycerides (>113 mmol/L; >10,000 mg/dLl) can result. Plasma from a patient with very high triglycerides is milky white, and a clear band of chylomicrons can be seen on top of the plasma after it stands overnight in a refrigerator. Populations with a founder effect can have high prevalence of LPL mutations. At least 60 LPL mutations can cause LPL deficiency. LPL188, LPLasn291ser, and LPL207 are frequently associated hyperchylomicronemia. Heterozygotes for the disorder tend to have an increase in fasting plasma triglycerides and smaller, denser LDL particles. Many patients with complete LPL deficiency present in childhood fail to thrive and have recurrent bouts of pancreatitis. To underscore the importance of LPL’s role, the LPL deficient mouse leads to a perinatal lethal phenotype [31]. The treatment of acute pancreatitis includes intravenous hydration and avoidance of fat in the diet (including in parenteral nutrition). Plasma filtration is required only rarely. Chronic treatment includes avoidance of alcohol and dietary fats. To make the diet more palatable, short-chain fatty acids (which are not incorporated in chylomicrons) can be used to supplement the diet. Type III hyperlipoproteinemia. Type III hyperlipoproteinemia, also referred to as dysbetalipoproteinemia or broad beta disease, is a rare genetic lipoprotein disorder characterized by an accumulation in plasma of remnant lipoprotein particles. On lipoprotein agarose gel electrophoresis, a typical pattern of a broad band between the pre-beta (VLDL) and beta (LDL) lipoproteins is observed, hence the name “broad beta disease.” Patients with this disease clearly have increased cardiovascular risk. The clinical presentation consists of pathognomonic tuberous xanthomas and palmar striated xanthomas. The lipoprotein profile shows increased cholesterol and triglyceride levels and reduced HDL cholesterol. Remnant lipoproteins (partly catabolized chylomicrons and VLDL) accumulate in plasma and accumulate cholesterol esters. The defect results from abnormal apo E, which does not bind to hepatic receptors that recognize apo E as a ligand (Fig. 42–4, part 3) [32]. The ratio of VLDL cholesterol to triglycerides, normally less than 0.7 mmol/L (<0.30 mg/dL), is elevated in patients with type III hyperlipoproteinemia, owing to cholesteryl ester enrichment of remnant particles. The diagnosis includes plasma ultracentrifugation for lipoprotein separation, lipoprotein electrophoresis, and apo E phenotyping or genotyping. Patients with type III hyperlipoproteinemia have the apo E 2/2 phenotype or genotype. There are three common alleles for apo E: apo E2, E3, and E4. The apo E2 allele has markedly decreased binding to the apo B/E receptor. In a normal population, the prevalence of the apo E2/2 phenotype is approximately 0.7 to 1.0 percent. Type III hyperlipoproteinemia occurs in approximately 1 percent of subjects bearing the apo E2/2 phenotype. The reasons for the relative rarity of type III dyslipoproteinemia are not fully understood. As discussed previously, a second “hit” is thought to impart the full expression of the disorder. Other rare mutations of the apo E gene can cause type III hyperlipoproteinemia [32]. In general, type III dyslipoproteinemia responds well to dietary therapy, correction of other metabolic abnormalities (diabetes, obesity), and, in cases requiring drug therapy, fibric acid derivatives or statins. The importance of the apo E gene and protein is underscored by the widespread use of the apo E deficient mouse which develop experimental atherosclerosis [33]. Familial combined hyperlipidemia. One of the most common familial lipoprotein disorders is familial combined hyperlipoproteinemia (FCH). Described initially in survivors of myocardial infarction, the definition of familial combined hyperlipoproteinemia has undergone several refinements. It is characterized by the presence of elevated total cholesterol and/or triglyceride levels based on arbitrary cut-points in several members of the same family. Advances in analytical techniques have added the measurement of LDL cholesterol and, in some cases, apo B levels. Because of the lack of a clearcut clinical or biochemical marker, considerable overlap exists between familial combined hyperlipoproteinemia, familial dyslipidemic hypertension, the metabolic syndrome, and hyperapobetalipoproteinemia. Genetic heterogeneity probably underlies familial combined hyperlipoproteinemia, which has a prevalence of approximately 1 in 50 and accounts for 10 to 20 percent of patients with premature CAD. The condition has few clinical signs; corneal arcus, xanthomas, and xanthelasmas occur infrequently. The biochemical abnormalities include elevation of plasma total and LDL cholesterol levels (>90th to 95th percentile) and/or an elevation of plasma triglycerides (>90th to 95th percentile)—a type IIb lipoprotein phenotype, often in correlation with low HDL cholesterol and elevated apo B levels; small, dense LDL particles are seen frequently. For a diagnosis of familial combined hyperlipoproteinemia, the disorder must be identified in at least one first-degree relative. The underlying metabolic disorder appears to be hepatic overproduction of apo B–containing lipoproteins, delayed postprandial triglyceride-rich lipoprotein clearance, and increased flux of free fatty acids (FFA) to the liver. Experimental data have shown that substrate levels drive hepatic apo B secretion, the most important substrates being FFA and cholesteryl esters. Increased delivery of FFA to the liver, as occurs in states of insulin resistance, leads to increased hepatic apo B secretion (Chap. 40). Familial combined hyperlipoproteinemia has complex genetics. It was initially 13 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 considered an autosomal codominant trait; modifying factors include gender, age of onset, and co morbid states such as obesity, lack of exercise, and diet. Initial reports of linkage with the apo AI-CIII-AIV and LPL genes remain unsubstantiated. A novel locus on chromosome 1 and 16 in Finnish families currently appears to be a promising candidate gene related to familial combined hyperlipoproteinemia [34]. Recent reports of the acylation-stimulating protein (ASP), also known as complement C3desARG pathway, suggests that abnormal peripheral uptake of FFA may underlie some cases of familial combined hyperlipoproteinemia and the insulinresistance metabolic syndrome [35]. A putative receptor for ASP has been identified recently as the orphan receptor C5L2, the complement C5 receptor that also binds complement C3desARG. Abnormal binding of ASP to peripheral cells has been reported in subjects with familial combined hyperlipoproteinemia. Abnormal ASP binding causes decreased uptake of FFAs into adipocytes and subsequent increased flux of FFAs to the liver (Fig. 42–5D). FFAs are a major substrate for hepatic apo B-containing lipoprotein assembly and secretion. High-Density Lipoproteins Reduced plasma levels of HDL cholesterol consistently correlate with the development or presence of CAD (see Chap. 36). Most cases of reduced HDL cholesterol result from elevated plasma triglycerides or apo B levels and often keep company with other features of the metabolic syndrome. Primary forms of reduced HDL cholesterol which occur in cases of premature CAD and helped shed light on the complex metabolism of HDL particles. Genetic disorders of HDL can result from decreased production or abnormal maturation and increased catabolism [36]. Genetic lipoprotein disorders leading to moderate to severe elevations in plasma triglycerides cause a reduction in HDL cholesterol levels. Familial hyperchylomicronemia, familial hypertriglyceridemia, and familial combined hyperlipoproteinemia are all associated with reduced HDL cholesterol levels. In complex disorders of lipoprotein metabolism such as familial combined hyperlipidemia, the metabolic syndrome, and common forms of hypertriglyceridemia, several factors most likely correlate to low HDL cholesterol level. Plasma triglycerides and HDL cholesterol levels vary inversely. For several reasons, patients with elevated apo B levels also have reduced HDL cholesterol levels. First, decreased lipolysis of triglyceride-rich lipoproteins (each VLDL contains one molecule of apo B) decrease the substrate (phospholipids) available for HDL maturation. Second, triglyceride enrichment of HDL increases their catabolic rate and hence reduces their plasma concentration. Third, exchange of lipids between HDL and triglyceride-rich lipoprotein is reduced, leading to a more rapid disappearance of HDL from plasma [37]. The inverse relationship between HDL cholesterol levels and plasma triglycerides reflects the interdependency of the metabolism of triglyceride-rich lipoproteins and HDL particles. Apo AI gene defects. Primary defects affecting production of HDL particles consist predominantly of apo AI-CIII-AIV gene defects. More than 46 mutations affect the structure of apo AI [38], leading to a marked reduction in HDL cholesterol levels. Not all of these defects are associated with premature cardiovascular disease. Clinical presentations can vary from extensive atypical xanthomatosis and corneal infiltration of lipids to no manifestations at all. Treatment of these apo AI gene defects generally fails to raise HDL cholesterol levels. Other mutations of apo AI lead to increased catabolic rate of apo AI and may not be associated with cardiovascular disease. One such mutation, apo AI Milano (apo AIArg173Cys), appear to confer longevity despite very low HDL levels [38]. LCAT, CETP deficiency. Genetic defects in the HDL-processing enzymes give rise to interesting phenotypes. Deficiencies of LCAT, the enzyme that catalyzes the formation of cholesteryl esters in plasma, cause corneal infiltration of neutral lipids and hematological abnormalities due to abnormal constitution of red blood cell membranes. LCAT deficiency can lead to an entity called “fish eye disease” because of the characteristic pattern of corneal infiltration observed in affected individuals [39]. Patients without CETP have very elevated HDL cholesterol levels, enriched in cholesteryl esters. Because CETP facilitates the transfer of HDL cholesteryl esters into triglyceride-rich lipoproteins, a deficiency of this enzyme causes accumulation of cholesteryl esters within HDL particles. CETP deficiency is not associated with premature CAD but may not afford protection against CAD [40]. Tangier disease and familial HDL deficiency. A rare disorder of HDL deficiency was identified in a proband from the Chesapeake Bay island of Tangier in the United States. The proband, whose sister was also affected, had markedly enlarged yellow tonsils and nearly absent HDL cholesterol levels, an entity now called Tangier disease. The cellular defect in Tangier disease consists of a reduced cellular cholesterol efflux in skin fibroblasts and macrophages from affected subjects [41]. A more common entity, familial HDL deficiency, was also found to result from decreased cellular cholesterol. The genetic defect in Tangier disease and in familial HDL deficiency results from mutations at the ATP binding cassette A1 gene (ABCA1) that encodes the ABCA1 transporter (Fig. 42–5B) [42]. At least 100 mutations have been reported within ABCA1, causing Tangier disease (homozygous or compound heterozygous mutations) or familial HDL deficiency (heterozygous mutations). Although subjects with Tangier disease and familial HDL deficiency are at increased risk for CAD, their very low levels of LDL cholesterol appear to have a protective effect. ABCA1 appears to shuttle from the late endosomal compartment to the plasma membrane and act as a membrane-bound transporter of phospholipids (and cholesterol) onto acceptor proteins 14 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 such as apo AI and apo E. Hydroxysterols regulate ABCA1 via the LxR/RxR nuclear receptor pathway. ABCA1 undergoes phosphorylation via protein kinase A and acts as a receptor for apo AI. Other cholesterol transport defects. Niemann-Pick type C disease is a disorder of lysosomal cholesterol transport. In patients with Niemann-Pick type C disease, mental retardation and neurological manifestations occur frequently. The cellular phenotype involves markedly decreased cholesterol esterification and cellular cholesterol transport defect to the Golgi apparatus. Unlike Tangier disease/familial HDL deficiency, the cellular defect in Niemann-Pick type C disease appears proximal to the transport of cholesterol to the plasma membrane. The gene for Niemann-Pick type C disease (NPC1) has been mapped to 18q21 and the gene codes for a 1278–amino acid protein, the role of which appears to be involved in cholesterol shuttling between the late endosomal pathway and the plasma membrane. The NPC1 gene product shares homology with the morphogen receptor patched and the SREBP cleavage activating protein (SCAP) [43]. NiemannPick type C cells lack NPC1 protein and cholesterol sequestration within the late endosome compartment prevents upregulation of ABCA1 and these patients thus have impaired cellular cholesterol efflux and HDL assembly [44]. NiemannPick type I disease (subtypes A and B), caused by mutations at the sphingomyelin phosphodiesterase-1 (SMPD-1) gene, associates with a low HDL cholesterol level. The SMPD-1 gene codes for a lysosomal (acidic) and secretory sphingomyelinase. The low HDL cholesterol level in Niemann-Pick A and B patients appears to result from a decrease in LCAT reaction because of abnormal HDL constituents [45]. Secondary Causes of Hyperlipidemia and the Metabolic Syndrome Several clinical disorders lead to alterations in lipoprotein status (Table 42–5). Hormonal causes. Hypothyroidism, a not infrequent cause of secondary lipoprotein disorders, often manifests with elevated LDL cholesterol, triglycerides, or both. An elevated level of thyroid-stimulating hormone is key to the diagnosis, and the lipoprotein abnormalities often revert to normal after correction of thyroid status. Rarely, hypothyroidism may uncover a genetic lipoprotein disorder such as type III hyperlipidemia. Estrogens can elevate plasma triglycerides and HDL cholesterol levels, probably because of increases in both hepatic VLDL and apo AI production. In postmenopausal women, estrogens may reduce LDL cholesterol by up to 15 percent. The use of estrogens for the treatment of lipoprotein disorders is no longer recommended because of the slight increase in cardiovascular risk with prolonged use of estrogens in the postmenopausal period (see Chaps. 42 and 73) [46]. Rarely, pregnancy causes severe increases in plasma triglycerides, on a background of lipoprotein lipase deficiency. Such cases present a serious threat to mother and child and require referral to specialized centers. Male sex hormones and anabolic steroids can increase hepatic lipase activity and have been used in the treatment of hypertriglyceridemia in men. Growth hormone can reduce LDL cholesterol and augment HDL cholesterol but is not recommended in the treatment of lipoprotein disorders. Metabolic causes. The most frequent secondary cause of dyslipoproteinemia is probably the constellation of metabolic abnormalities seen in patients with the metabolic syndrome (see Chaps. 39 and 43). The finding of increased visceral fat (abdominal obesity), elevated blood pressure, and impaired glucose tolerance often clusters with increased plasma triglycerides and a reduced HDL cholesterol level and comprise the major components of the metabolic syndrome [21,47]. The lack of internationally recognized uniform criteria for the metabolic syndrome among other issues cast doubt on the usefulness of this syndrome as a diagnostic entity [48]. Overt diabetes, especially type 2 diabetes, frequently elevates plasma triglycerides and reduces HDL cholesterol. These abnormalities have prognostic implications in patients with type 2 diabetes. Poor control of diabetes, obesity, and moderate to severe hyperglycemia can yield severe hypertriglyceridemia with chylomicronemia and increased VLDL cholesterol levels. Subjects with type I diabetes can also have severe hypertriglyceridemia when poorly controlled. Familial lipodystrophy (complete or partial) may be associated with increased VLDL secretion. Dunnigan lipodystrophy, a genetic disorder with features of the metabolic syndrome, is caused by mutations within the Lamin A/C gene and is associated with limb-girdle fat atrophy. Excess plasma triglycerides often accompany glycogen storage disorders. 15 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Renal disorders. In subjects with glomerulonephritis and protein-losing nephropathies, a marked increase in secretion of hepatic lipoproteins can raise LDL cholesterol levels, which may approach the levels seen in subjects with familial hypercholesterolemia. By contrast, patients with chronic renal failure have a pattern of hypertriglyceridemia with reduced HDL cholesterol. Patients with end-stage renal disease, including those on hemodialysis or chronic ambulatory peritoneal dialysis, have a poor prognosis and accelerated atherosclerosis and should undergo aggressive treatment of lipoprotein disorders. This approach, however has been recently challenged when a recent trial of statins in diabetic patients on dialysis showed no reduction in cardiovascular end-points. After organ transplantation, the immunosuppressive regimen (glucocorticoids and cyclosporine) typically elevates triglycerides and reduces HDL cholesterol levels. Because transplant patients generally have an increase in cardiovascular risk, a secondary hyperlipidemia may warrant treatment. Patients receiving the combination of statin plus cyclosporine merit careful dose titrations and monitoring for myopathy. Liver disease. Obstructive liver disease, especially primary biliary cirrhosis may lead to the formation of an abnormal lipoprotein termed lipoprotein-x. This type of lipoprotein is found in cases of LCAT deficiency and consists of an LDL-like particle but with a marked reduction in cholesteryl esters. Extensive xanthoma formation on the face and palmar areas can be the result from accumulation of lipoprotein-x. Lifestyle. Factors contributing to obesity, such as an imbalance between caloric intake and energy expenditure, lack of physical activity, and a diet rich in saturated fats and refined sugars, contribute in large part to the lipid and lipoprotein lipid levels within a population (see Chaps. 36 and 41). Medication. Several medications can alter lipoproteins. Thiazide diuretics can increase plasma triglyceride levels. Beta blockers, especially non-beta-1 selective, increase triglycerides and lower HDL cholesterol levels. Retinoic acid and estrogens can increase triglyceride levels, sometimes dramatically. Corticosteroids and immunosuppressive agents can increase plasma triglyceride levels and lower HDL cholesterol levels. Estrogens can increase plasma HDL cholesterol significantly and often increase triglyceride concentrations. In clinical practice, many dyslipoproteinemias, other than the genetic forms mentioned earlier, share an important environmental cause. Lifestyle changes (diet, exercise, reduction of abdominal obesity) should be the foundation for the treatment of most dyslipidemias. The effects of marked alterations in lifestyle, reduction in dietary fats, especially saturated fats, and exercise can improve cardiovascular prognosis. Translating these findings into practice, however, has been more difficult. For example, dietary manipulations as performed in a physician’s office lead to relatively small reductions in plasma lipid and lipoprotein cholesterol levels (see Chaps. 36, 41, and 42). 16 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Drugs that Affect Lipid Metabolism (Table 42–6) Bile acid binding resins The bile acid–binding resins interrupt the enterohepatic circulation of bile acids by inhibiting their reabsorption in the intestine (more than 90 percent of bile acids reabsorbed). Currently, their main use is adjunctive therapy in patients with severe hypercholesterolemia due to increased LDL cholesterol. Since bile acid–binding resins are not absorbed systemically (they remain in the intestine and are eliminated in the stool), they are considered safe in children. Cholestyramine (Questran) is used in 4-gm unit doses as powder, and colestipol (Colestid) is used in 5-gm unit doses; a 1-gm tablet of colestipol is available. Effective doses range from 2 to 6 unit doses/day, always taken with meals. The most important side effects are predominantly gastrointestinal, with constipation, a sensation of fullness, and gastrointestinal discomfort. Hypertriglyceridemia can result from the use of these drugs. Decreased drug absorption dictates careful scheduling of medications 1 hour before or 3 hours after the patient takes bile acid– binding resins. Bile acid–binding resins can be used in combination with statins and/or cholesterol absorption inhibitors in cases of severe hypercholesterolemia. Hydroxymethylglutaryl–Coenzyme A Reductase Inhibitors (Statins) Statins inhibit HMG-CoA reductase and prevent the formation of mevalonate, the rate-limiting step of sterol synthesis. To maintain cellular cholesterol homeostasis, expression of the LDL-R increases and the rate of cholesteryl ester formation declines. These homeostatic adjustments to HMG-CoA reductase inhibition increase LDL cholesterol clearance from plasma and decrease hepatic production of VLDL and LDL. In addition to blocking the synthesis of cholesterol, statins also interfere with the synthesis of lipid intermediates with important biological effects. In the cholesterol synthetic pathway, intermediate molecules of dimethylallyl pyrophosphate are metabolized by prenyl transferase into geranyl pyrophosphate and subsequently into farnesyl pyrophosphate. This step occurs before the formation of squalenes [49]. These intermediates, geranylgeranyl and farnesyl, are used for protein prenylation, a mechanism by which a lipid moiety attaches covalently to a protein, allowing anchoring into cell membranes and enhancing its biological activity. This is the case for the GTP-binding proteins Rho A, Rac, and Ras. Indeed, statins may increase HDL cholesterol in part by preventing the geranylgeranylation of Rho A and phosphorylation of peroxysome proliferator–activated receptor alpha (PPAR), a factor that regulates apo AI transcription [49]. Altered protein prenylation may also mediate some of the putative effects of statins not related to a reduction in LDL cholesterol levels. Atherosclerosis is an inflammatory disease. Statins decrease C-reactive protein [50], induce apoptosis in smooth muscle cells, alter collagen content of atherosclerotic plaques, alter endothelial function, and decrease the inflammatory component of plaques [51,52] (See also chapters 38 & 39). Some investigators argue that statins possess effects independent of their inhibition of HMG CoA reductase. The clinical importance of these possible LDL-independent actions, and differences in efficacy between statins for a given percentage reduction in LDL cholesterol remain speculative. Statins are generally well tolerated; side effects include reversible elevation in transaminases and myositis, which causes discontinuation of the drug in less than 1 percent of patients. The currently available drugs are fluvastatin (Lescol), 20 to 80 mg/day; lovastatin (Mevacor), 20 to 80 mg/day; pravastatin (Pravachol), 20 to 40 mg/day; simvastatin (Zocor), 10 to 80mg/day; atorvastatin (Lipitor), 10 to 80 mg/day; and rosuvastatin (Crestor), 5 to 40 mg/day. Concomitant drugs that interfere with the metabolism of statins by inhibiting the cytochrome P450 3A4 and 2C9 systems can increase plasma 17 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 concentrations of statins. These include antibiotics, antifungal medications, certain antiviral drugs, grapefruit juice, cyclosporine, amiodarone, and several others. Cholesterol Absorption Inhibitors The development of selective inhibitors of intestinal sterol absorption has significantly advanced the treatment of lipoprotein disorders. Ezetimibe is the first such compound. Ezetimibe appears to limit selective uptake of cholesterol and other sterols by intestinal epithelial cells, by interfering with the Niemann-Pick C1-like 1 protein 1 (NPC1L1) [10]. It is particularly indicated for patients with LDL cholesterol levels above target on maximally tolerated statin dose. Ezetimibe lowers LDL cholesterol by about 18 percent and adds to the effect of statins [53]. Because ezetimibe also prevents the intestinal absorption of sitosterol, it might be the drug of choice in cases of sitosterolemia. The current dose of ezetimibe is 10mg/day. Fibric Acid Derivatives (Fibrates) Two derivatives of fibric acid are currently available in the United States and two more are available in Canada and Europe. Gemfibrozil (Lopid) is used at a dose of 600mg twice a day and is indicated in cases of hypertriglyceridemia and in the secondary prevention of cardiovascular diseases in patients with a low HDL cholesterol levels. These latter recommendations are based on the Veterans Administration HDL Intervention Trial (VA-HIT). Fenofibrate (Tricor; Lipidil Micro, EZ) is used to treat hypertriglyceridemia and combined hyperlipoproteinemia. The dose is 200 mg/day and a new formulation is available to vary the dose from 67 mg (especially in cases of renal failure) to 267 mg/day. Clofibrate (Atromid) is still available in some centers, although its use has declined since the introduction of newer molecules. Ciprofibrate (Lypanthyl, Lipanor) and bezafibrate (Bezalip) are more widely used in Europe. The main indications for the use of fibrates is the treatment of hypertriglyceridemia when diet and lifestyle changes are not sufficient. Another potential indication is in the prevention of cardiovascular diseases in patients with elevated plasma triglycerides and low HDL cholesterol, although the data supporting their use is weaker than that for statins. The mechanism of action of fibrates involves interaction with the nuclear transcription factor PPAR that regulates the transcription of the LPL, apo CII, and apo AI genes. The side effects of fibrates include cutaneous manifestations, gastrointestinal effects (abdominal discomfort, increased bile lithogenicity), erectile dysfunction, elevated transaminases, interaction with oral anticoagulants, and elevated plasma homocysteine, especially with fenofibrate and, to a lesser extent, with bezafibrate [54]. Because fibrates increase LPL activity, LDL cholesterol levels may rise in patients with hypertriglyceridemia treated with this class of medications. Fibrates, especially gemfibrozil, can inhibit the glucuronidation of statins, and thus retard their elimination. For this reason, combination of gemfibrozil with statins may increase the risk of myotoxicity. Nicotinic Acid (Niacin) Niacin has been used for decades for the treatment of dyslipidemias and is particularly effective in increasing HDL cholesterol and lowering triglyceride levels. The effect of niacin on LDL cholesterol is more modest. Effective doses of niacin are in the range of 3,000 mg/day, in three separate doses. It is preferable to use an escalating dose schedule to reach the full dose in 2 to 3 weeks rather than starting with the full dose. Slow-release forms of niacin, including Niaspan (1-2 g/day) decrease the side effect profile of the drug. Skin flushing can be attenuated by taking a daily aspirin. Niacin decreases the hepatic secretion of VLDL from the liver and decreased FFA mobilization for the periphery. Although niacin has been shown in the long-term follow-up of the Coronary Drug Project to decrease mortality at 15 years. 18 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Significant and common minor side effects and much less frequent serious adverse actions, and the development of statins hamper its use. Side effects of niacin include flushing, hyperuricemia, hyperglycemia, hepatotoxicity, acanthosis nigricans, and gastritis. Close laboratory monitoring of side effects is warranted. Long-acting niacin has the advantage of a once- or twice-daily dosing schedule, but older preparations of slow-release niacin were potentially more hepatotoxic. Niacin effectively raises HDL cholesterol levels and, in combination with low-dose statin, can retard the angiographic progression of CAD and decrease adverse cardiac events [55]. Recent work has identified cell surface receptors for nicotinic acid that belong to the G-protein–coupled heptahelical superfamily. This discovery may speed the elucidation of the molecular mechanism of nicotinic acid’s effects on lipid metabolism [56]. CETP inhibitors The inhibition of cholesteryl ester transfer protein (CETP) by pharmacological agents mimics the genetic heterozygous CETP deficiency state (Fig. 42–4, part 9). Of several agents tested in man, Torcetrapib is currently in phase III studies. At doses of 120 mg / day, Torcetrapib increases HDL-C levels by 40-50% and is generally well tolerated [57]. A marked increase in larger, more buoyant HDL particles is observed in patients on Torcetrapib; these particles appear to promote cellular cholesterol efflux efficiently. Reported side effects include elevation in hepatic transaminases, an increase in blood pressure and abdominal symptoms. Fish Oils Fish oils are rich in polyunsaturated fatty acids such as eicosapentaenoic acid or docosahexaenoic acid, with the first double on the omega-3 (-3) position. These fatty acids lower plasma triglyceride levels and have antithrombotic properties. Although employed in the treatment of hypertriglyceridemia, their use is reserved in cases of severe hyper-triglyceridemia refractory to conventional therapy. Fish oils decrease VLDL synthesis and decrease VLDL apo B. The response to fish oils depends on dose, requiring a daily intake of up 10 gm of eicosapentaenoic acid or docosahexaenoic acid for a significant benefit on plasma triglyceride levels. A prescription form of fatty acids has become available in the US for use in extreme hypertriglyceridemia (>500 mg/L or 5.6 mmol/L) Phytosterols Phytosterols are derivatives of cholesterol from plants and trees. They interfere with the formation of micelles in the intestine and prevent intestinal cholesterol absorption. They can be obtained as “neutraceuticals” or can be incorporated in soft margarines. The sterols may prove useful for the adjunctive management of lipoprotein disorders and are part of the therapeutic lifestyle change regimen in the current guidelines (see later). Other Medications Probucol was used as an antioxidant and had modest effects on plasma lipoprotein levels. The lack of conclusive evidence that probucol has beneficial effects and profound reduction in HDL levels and prolongation of QT interval led to its withdrawal. A study indicates that it may have a role in the prevention of restenosis after coronary angioplasty if used before the procedure [58]. Thyroxine is no longer used as a lipid modulator unless hypothyroidism has been documented. Monitoring of Lipid Therapy After initiation of medical therapy, the response should be checked within the first 3 months, along with transaminases and creatinine kinase. Thereafter, clinical judgment should dictate the interval between 19 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 follow-up visits. Although frequent visits are probably not useful in the detection of serious side effects, they serve to encourage compliance and adherence to diet and lifestyle changes. Clinical Trials of Drugs Affecting Lipid Metabolism Numerous pathological, epidemiological, genetic, and interventional trials have validated the central tenet of the “lipid hypothesis,” which proposes a causal relationship between dyslipidemia and atherogenesis and identifies lipid modification as a risk-reducing strategy for CHD (see Chap. 38). A number of small trials using dietary or drug therapies have demonstrated the angiographic benefit of managing elevated total cholesterol and LDL cholesterol. Early clinical trials using bile-acid sequestrants, fibrates, or nicotinic acid reported modest reductions in coronary risk with modest reductions in LDL cholesterol. The advent of the HMG-CoA reductase inhibitors, or statins, in the mid-1980s made possible more aggressive reduction of LDL cholesterol. By the late 1990s, several large-scale, prospective randomized trials with these drugs had reported robust reductions in relative cardiovascular risks, compared with placebo [59-68]. This section discusses the more recent trials in this area, specifically in high-risk primary prevention, intensive LDL-C lowering in secondary prevention, the benefits of intensive therapy in acute coronary syndromes, regression studies, and specific sub-group analysis (the elderly and diabetic patient). The focus on statin trials reflects the widespread clinical success of this category of drugs. Ample reviews have considered the results of the older trials. We refer readers interested in these earlier trials to previous editions of this textbook. Treating High-Risk (primary prevention) Patients (Table 42.7) As the interaction of multiple coronary risk factors has received greater clinical importance, recommendations for clinical practice have increasingly embraced the concept of global risk management. This global risk is calculated using an algorithm that considers not only total cholesterol but also HDL cholesterol, smoking, age, hypertension, and gender (see Chap. 39). Adult diabetic subjects are considered to be at high cardiovascular risk, with few exceptions. Cardiovascular risk is determined as cardiovascular death or non-fatal myocardial infarction in the next 10 years, based on the Framingham Heart Study risk score and determines the intensity of lipid intervention. Based on the recent evidence from clinical trials, the 2004 U.S. Adult Treatment Panel (ATP III) modified its guidelines, recommending a more aggressive LDL-C target in very high-risk subjects to an LDL-C <70 mg/dL (1.8 mmol/L) [69]. Similar risk stratification and targets are proposed in Europe [70]. Because global risk assessment shifts emphasis away from abnormal lipids alone to the patient’s overall risk profile, it raises an important issue for the future of lipid management. That is, should the decision to initiate lipid modification for cardiovascular risk reduction be based on high risk or high cholesterol? Recent trials have evaluated this question. The Medical Research Council/British Heart Foundation Heart Protection Study (HPS) evaluated the role of statin therapy in at-risk patients for whom guidelines at the time would not have recommended drug intervention [64]. In HPS, however, over 65% of patients had evidence of atherosclerosis (coronary of peripheral) and the remainder had diabetes or multiple risk factors. The broad inclusion criteria of cardiovascular risk and age (40 to 80 years), simple entry criteria (total cholesterol >135 mg/dL (3.5 mmol/L)), and number randomized (20,536) makes this trial a seminal contribution in the field. The twoby-two factorial design of the HPS intended to analyze the effect of simvastatin, 40 mg/day, versus placebo with or without a combination of antioxidant vitamins (600 mg alpha tocopherol, 250 mg ascorbic acid, 20 mg beta carotene) on the risk of major vascular events. The combination of antioxidant vitamins did not affect morbidity or mortality. Simvastatin treatment, on the other hand, reduced the risk for any major vascular event by 24 percent (p=0.0001), with an absolute risk reduction of 5.4 percent (28.3 % in the placebo group minus 19.4 % in the simvastatin group). The all-cause mortality rate fell by 13 percent (p=0.0003), 20 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 with a significant 17 percent reduction (p=0.0001) in deaths attributed to any vascular cause. There was no increase in non cardiac causes of mortality, such as neoplasia, respiratory disease, or other nonvascular deaths. Subgroup analysis in HPS had sufficient power to examine women (n=5,072), diabetics (n=4,282), CHD patients (n=7,150) and subjects >75 years of age (n=1,263). All of these subgroups benefited from the statin therapy. The Prospective Evaluation of Pravastatin in the Elderly (PROSPER) assessed the impact of treatment with pravastatin 40mg/day versus placebo in 5804 men and women aged 70 to 82 years of age with a history of vascular disease (coronary, cerebrovascular, or peripheral vascular) or a risk factor profile consistent with high risk (smoking, hypertension, or diabetes) [65]. Participants in this study had plasma total cholesterol levels between 155 and 350 mg/dL (4.0 to 9.0 mmol/L) and triglyceride concentrations less than 530 mg/dL (6.0 mmol/L). Women accounted for more than half of the individuals in PROSPER. The mean follow-up was 3.2 years and the composite primary endpoint included coronary death, nonfatal myocardial infarction, and fatal or nonfatal stroke. Pravastatin treatment reduced the relative risk for this endpoint significantly by 15 percent (p=0.014) and for CHD death by 24 percent (p=0.043). The treatment showed similar benefit across many subgroups: those with versus those without prior vascular disease; men versus women; tertiles of baseline LDL cholesterol; current smokers versus nonsmokers; and those with and those without a history of hypertension. Participants in the lowest tertile of baseline HDL cholesterol (<1.11 mmol/L or 43 mg/dL) experienced greater benefit than those in the higher tertiles. Lipid-modifying treatment had no effect on the risk for stroke (see also Chapt 58) or on cognitive function, both of which are important endpoints in an older population. Hypertensive patients The large Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) sought to determine whether cholesterol lowering with open-label pravastatin 20 to 40mg/day (plus resin, if needed) would reduce total mortality in 10,355 moderately hypercholesterolemic, hypertensive men and women, aged 55 years or older, with at least one other CHD risk factor, as compared with usual care [66]. The ALLHAT patients met lipid criteria of a LDL cholesterol level of 120 to 189 mg/dl (3.1 to 5 mmol/liter) (or 100 to 129 mg/dL [2.6 to 3.3 mmol/L] in those with CHD) and triglycerides less than 350mg/dL (<3.9 mmol/L). In contrast with other large statin studies, ALLHAT reported no benefit and no harm of statin treatment compared with usual care on any study endpoint. The study may have been underpowered because of difficulties related to patient recruitment. The trial was open-labeled, and there was declining adherence in the pravastatin group and a high crossover rate to statin therapy in the usual-care group (by 6 years, more than 25 percent of these patients were receiving a statin). Indeed, the absolute difference in total cholesterol between the pravastatin and the usual-care group by the end of the trial was only 9.6 percent, which was approximately half of the reduction achieved in older statin trials. The lipid-lowering arm of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) assessed the clinical effect of atorvastatin, 10 mg/day, versus placebo in 10,305 hypertensive patients with a total cholesterol level of less than 250 mg/dL (6.5 mmol/L) and a high-risk profile [67]. Although the trial excluded individuals with previous myocardial infarction, current angina, or cerebrovascular disease within 3 months before randomization, the randomized patients had evidence of other vascular disease or CHD risk equivalents (left ventricular hypertrophy, other electrocardiographic abnormalities, peripheral arterial disease, previous cerebrovascular disease, diabetes), or several other CHD risk factors. Originally planned to have a follow-up period of 5 years, the ASCOT ended early after a median follow-up of 3.3 years, after 100 primary endpoint events had occurred in the atorvastatin group compared with 154 in the placebo group. The relative risk reduction was 36 percent (p=0.0005), and the benefit became apparent within a year of the study’s initiation. Benefit was similar across pre-specified subgroups. Atorvastatin reduced the relative risk for stroke by 27 percent (p=0.024) and for total cardiovascular events by 21 percent (p=0.0005). Total mortality and adverse events did not differ between the treatment groups. Diabetic patients Because of their high risk for developing CHD, diabetic patients warrant aggressive lipid modification, according to U.S. guidelines (see Chap. 43 & 45). However, few trials have evaluated the effects of lipid modification on clinical endpoints in this population, and the evidence of benefit derives largely from subgroup analyses. The Collaborative Atorvastatin Diabetes Study (CARDS) studied patients with type 2 diabetes without manifest CAD [68]. Entry criteria included not only diabetes but also at least one other CHD risk factor: current smoking, hypertension, retinopathy, or micro- or macroalbuminuria. The trial enrolled individuals with LDL cholesterol levels less than 160 mg/dL (4.14 mmol/L) and triglycerides less than 600 mg/dL (6.78 mmol/L). The study included 2,838 men and women, 40 to 75 years of age. The trial utilized a fixed dosage of atorvastatin of 10 mg/day compared with placebo. The primary efficacy parameter is the time from randomization to the occurrence of a first primary endpoint event, which may include major coronary events, revascularizations, stroke, unstable angina, or resuscitated cardiac arrest. The study was stopped prematurely for benefit. There was a 36% reduction (p=0.001) in primary end-points (CHD death, non-fatal MI, unstable angina, revascularization, fatal and non-fatal stroke). 21 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 The FIELD trial [71] was carried out in Australia, New Zealand and Finland and examined the effect of fenofibrate, 200 mg/day on the development of CHD in patients with diabetes for 5 years. The investigators examined 9795 patients, age 5075 years with type 2 diabetes diagnosed after age 35 years, and no clear indication for cholesterol-lowering therapy at baseline at the time of study design. Lipid entry criteria were (total cholesterol 116-251 mg/dL (3.0 – 6.5 mmol/L), plus either total cholesterol to HDL ratio ≥4.0 or triglyceride >88.6 mg/dL (1.0 mmol/L). Patients had stable diabetes for at least 5 years. The primary composite endpoint of CHD death or non-fatal MI was not significantly lower in the fenofibrate group compared to the placebo group (5.2% vs. 5.8%, p=0.16. Sub-group analysis showed a non significant increase in CHD death (2.2% on fenofibrate vs. 1.9 % on placebo, p=0.22), but a significant reduction in non-fatal myocardial infarction on fenofibrate (3.2% vs. 4.2%, p=0.01). The secondary composite endpoint of total CV events was significantly lower in the fenofibrate group compared to the placebo group (12.5 % vs. 13.9%, p=0.035). Cardiovascular Mortality (2.9% fenofibrate vs. 2.6% placebo, p=0.41), total mortality (7.3% fenofibrate vs. 6.9% placebo, p=0.18) , and stroke (3.2% fenofibrate vs. 3.6% placebo, p=0.36) were not significantly different between the fenofibrate and placebo groups. The rate of statin drop-in in the fenofibrate group averaged 8% whereas it averaged 17% in the placebo group during the course of the trial. The CARDS and FIELD trials were similar in several respects, including inclusion criteria and baseline lipid levels. On the basis of these trials, prevention of cardiovascular disease in diabetic subjects should include a statin. Other fibrate trials The Veteran’s Affairs Cooperative Studies Program High Density Lipoprotein Cholesterol Intervention Trial (VA-HIT) [71], a multicenter, randomized study that assessed the effects of gemfibrozil at a dose of 1200 mg/day versus dietary therapy on the incidence of cardiovascular events in 2,531 men with known CHD and low baseline HDL cholesterol levels [73]. Enrollment criteria for VA-HIT required a HDL cholesterol level of 40 mg/dL (1.03 mmol/L) or less or LDL cholesterol level of less than 140 mg/dL (3.6mmol/L) and triglycerides at 300 mg/dL (3.4 mmol/L) or less. The mean age of subjects in the VA-HIT was 64 years, with more than 75 percent of the patients older than 60 years of age. Baseline lipids in the VA-HIT revealed a total cholesterol level of 175 mg/dL, HDL cholesterol of 32 mg/dL, LDL cholesterol of 111 mg/dLl, and triglycerides of 161 mg/dL. Gemfibrozil therapy resulted in essentially minimal alterations in total cholesterol and LDL levels. Total cholesterol was decreased by 4 % as compared with placebo, and no significant change was demonstrable with LDL cholesterol concentrations. Triglycerides, on the other hand, fell by 31 % and HDL cholesterol increased by 6 %. The primary endpoint of the VA HIT was the combination of CHD death and nonfatal myocardial infarction. Patients randomized to receive placebo accounted for 275 events, compared with 219 events in the gemfibrozil group. The decline in coronary events represented a 22 percent risk reduction that was statistically significant (p=0.006). The event curves began to diverge at approximately 2 years after the beginning of the trial. The Bezafibrate Infarction Prevention (BIP) [73] study reported no reduction in fatal and nonfatal myocardial infarction and CHD death in a cohort of 3,090 men and women with CHD, total cholesterol of 180 to 250 mg/dL, HDL cholesterol less than 45 mg/dL (1.2 mmol/L), triglycerides less than 300 mg/dL (3.4 mmol/L), and LDL cholesterol less than 180 mg/dl (4.7 mmol/L), who were treated with either bezafibrate, 400 mg/day, or placebo. Despite producing an increase in HDL cholesterol of 14 % and reduction in triglycerides of 25 % compared with placebo, fibrate treatment did not reduce CHD risk. After 6.2 years, the reduction in the cumulative probability of the primary endpoint was 7.3 percent (p=0.24). However, a substantial risk reduction with bezafibrate (5 percent; p=0.02) was observed post hoc in the small subgroup of patients with elevated triglycerides at baseline (>200 mg/dL [2.3 mmol/L]). The Action to Control Cardiovascular Risk in Diabetes (ACCORD) study, sponsored by the US National Heart, Lung, and Blood Institute, anticipates enrolling 10,000 patients with type 2 diabetes mellitus (from www.accordtrial.org). The trial will compare effects on cardiovascular disease events of three strategies: intensive glycemic control; increasing HDL cholesterol and lowering triglycerides (with strict LDL cholesterol and glycemic control); and intensive blood pressure control (in the context of tight glycemic control). The lipid-modification arm of ACCORD will evaluate a fibrate intended to lower triglycerides and increase HDL cholesterol, combined with a statin to lower the LDL cholesterol level. The ACCORD study, therefore, will assess the effect of multiple risk factor intervention in diabetic patients and will also provide valuable information about the use of the statin-fibrate combination in preventing clinical events. In January 2003, the ACCORD main trial began a 30-month recruiting period. Follow-up will continue until 2009. Secondary Prevention Trials Table 42.8, Figure 42.6 In the past decade and a half, secondary prevention trials have shown the power of large, well designed trials examining the effect of statins on cardiovascular events, cardiovascular mortality and total mortality. The results of the 4S [63], CARE [61], LIPID [62] and HPS [64] have been discussed previously. A recent meta-analysis involving 90,056 subjects in 14 trials of cardiovascular disease prevention with statins, carried by the Cholesterol Treatment Trialists [74] showed a linear relationship between the absolute decrease in LDL-C and reduction in cardiovascular events (CHD death or non-fatal MI). A 1.0 mmol/L (38.67 mg/dL) reduction in LDL-C was associated with a 22% reduction in cardiovascular events. Importantly, this magnitude of effect was preserved when the LDL-C was reduced even further. (Figure 42.6). National guidelines recommended a therapeutic target of 2.6 mmol/L for LDL-C (100 mg/dL); the results of recent clinical trials have stimulated a 22 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 revision of this recommendation to a LDL-C <1.8 mmol/L (<70 mg/dL). Two recent trials have examined the effect of lowering LDL-C below recommended targets; the Treating to New Targets (TNT) trial [75] and the Incremental Decrease in End points through Aggressive Lipid lowering (IDEAL) trial [76]. The TNT trial examined patients with stable CHD and an LDL-C 130-250 mg/dL (3.4-6.5 mmol/L) and Triglycerides 600 mg/dL (6.8 mmol/L). From a screening population of over 18,468 subjects, 15,464 were randomized to atorvastatin 10 mg/day. Patients who did not reach the LDL-C target of <130 mg/dL (3.4 mmol/L) were excluded; 10,001 patients were then randomized to atorvastatin 10 mg/day, to reach an average LDL-C of 100 mg/dl (2.6 mmol/L) or to atorvastatin 80 mg/day, to reach a mean LDL-C of 80 mg/dL (2.0 mmol/L). The primary outcome measure was CHD death, non fatal MI, resuscitated cardiac arrest and fatal or non-fatal stroke. After a mean follow-up of 4.9 years, the primary outcome measure was decreased by 22% (HR = 0.78 (95% CI 0.69, 0.89), p=0.0002). The absolute risk decreased from 8.3% to 6.7% events / 4.9 years. Cardiovascular mortality was decreased by 20% (non significant), from 127 deaths (2.5%) on atorvastatin 10 mg vs. 101 (2.0%) on atorvastatin 80 mg (HR 0.80, p=0.09) Total mortality was not changed (282 deaths (5.6%) on atorvastatin 10 mg vs. 284 deaths (5.7%) (HR 1.01, p=0.92). Overall, TNT confirmed that in patients with stable CHD, intensive LDL-C lowering decreased cardiovascular events. The IDEAL trial evaluated 8,888 patients with stable CHD aged 80 years or younger to simvastatin 20 mg/day or atorvastatin 80 mg /day. Patients were followed for an average of 4.8 years. The primary outcome measure was CHD death, non fatal MI, or resuscitated cardiac arrest. On treatment LDL-C levels were 104 mg/dL (2.6 mmol/L) in the simvastatin 20 mg group, vs. 81 mg/dL (2.6 mmol/L) in the atorvastatin 80 mg group. A major cardiac event occurred in 463 patients on simvastatin (10.4%) compared to 411 patients (9.3%) in the atorvastatin group (HR 0.89, 95% CI 0.78 – 1.01, p=0.07). Non fatal MI occurred in 321 (7.2%) patients on simvastatin and in 267 (6.0%) patients on atorvastatin (HR 0.87, 95% CI 0.77 – 0.98, p=0.02). There were no changes in total mortality (374 (8.4%) in the simvastatin group vs. 366 (8.2%) in the atorvastatin group ((HR 0.98, p=0.81). Both the TNT and IDEAL studies show that intensive LDL-C lowering decreases cardiovascular events; neither study was sufficiently powered to evaluate total mortality. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) [77] will determine, in 12,000 patients, whether intensive treatment with simvastatin 80 mg, compared to 20 mg will result in further CHD events. In SEARCH, and additional arm of the trial will investigate the effects of B vitamins to reduce homocysteine on cardiovascular events. Acute Coronary Syndromes Earlier secondary prevention statin studies generally selected patients who were at least 3 to 6 months post coronary event and stabilized. Substantial interest has turned to the question of statin treatment in the period immediately following an acute coronary syndrome. The Myocardial Ischemia Reduction and Aggressive Cholesterol Lowering (MIRACL) trial [78] examined the premise that early and intensive treatment with high-dose atorvastatin therapy begun immediately after the onset of an acute coronary event might produce beneficial clinical effects in a much larger cohort (n=3,086). The composite primary endpoint of MIRACL included nonfatal acute myocardial infarction, cardiac arrest, or symptomatic myocardial ischemia. Atorvastatin resulted in a modest albeit statistically significant 16 percent improvement in relative risk for the primary endpoint (p=0.048). The major effect of early atorvastatin was a reduction in recurrent myocardial ischemia, which was decreased by 26 % (p=0.02). While statistically significant, the clinical benefits observed in MIRACL were not as robust as those seen in the larger and longer secondary prevention trials (Table 42.8). The Aggrastat-to-Zocor (A-to-Z) study [79] randomized patients with acute coronary syndromes (randomized to tirofiban or heparin) to receive either simvastatin, 40 to 80mg/day, versus diet plus simvastatin, 20mg/day. Overall, the results of this trial were neutral. The Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) trial [80] compared pravastatin, 40 mg/day, with atorvastatin, 80 mg/day, in 4,162 post acute coronary event patients. This study also had an antibiotic treatment arm. The lipid-lowering arm of PROVE-IT showed that in patients who have recently survived an acute coronary syndrome, an intensive statin regimen that yielded a median LDL cholesterol of 62 mg/dL (1.60 mmol/L) provided greater protection against death or major cardiovascular events than less aggressive therapy that lowered LDL cholesterol to a median of 95 mg/dL (2.5 mmol/L), a level below the upper limit recommended by ATP-III as a target for this category of patients. Medical Therapy Versus Revascularization; Regression studies The Atorvastatin versus Revascularization Treatment (AVERT) trial evaluated the potential benefits of aggressive lipid lowering with open-label atorvastatin, 80mg/day, versus usual care on ischemic events in a cohort of 341 patients with stable atherosclerosis who were scheduled to undergo an elective percutaneous revascularization procedure [81]. Follow-up was 18 months. Qualification requirements included at least one native coronary vessel with 50 percent or greater stenosis and an 23 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 LDL cholesterol level in excess of 115 mg/dL. Exclusion criteria included triglycerides in excess of 500 mg/dL, ejection fraction less than 40%, inability to complete 4 minutes of exercise on a standard Bruce protocol, left main or triple vessel coronary atherosclerosis, or recent unstable angina or myocardial infarction. The composite primary endpoint in the AVERT trial encompassed the incidence of ischemic events, defined as cardiac death, resuscitation after cardiac arrest, nonfatal myocardial infarction, stroke, worsening angina, or revascularization (coronary artery bypass graft or repeat angioplasty). The patients randomized to usual care received percutaneous coronary interventions, and approximately one-third of the patients with treated lesions underwent placement of a coronary stent. The patients treated with angioplasty could receive hypolipidemic therapy as part of usual care. Lipid-lowering drugs were administered in 73 % of patients in this group at some time during the follow-up (71 % of the total group received statins). Despite ”drop-in” of lipid-lowering therapy, the atorvastatin group had lower total cholesterol, LDL cholesterol, and triglycerides relative to usual care. A total of 22 ischemic events occurred in the group randomized to receive aggressive lipid-lowering therapy, compared with 37 events in the usual-care group (36 percent reduction; p=0.048). The difference in the treatment arms tended toward statistical significance, since the significance level was adjusted to 0.045 because of the performance of two interim analyses. Atorvastatin treatment significantly delayed the time to the first event (p=0.027) compared with usual care, but 54 percent of angioplasty patients compared with 41 percent of atorvastatin patients had improvement of Canadian Cardiovascular Society (CCS) classification of angina symptoms (p=0.009). Seventeen serious adverse events were reported in the atorvastatin group, although none were attributed to atorvastatin. Twenty-eight of the patients in the angioplasty group had serious adverse events; six of these patients had events attributable to the angioplasty. These results support a strategy of aggressive lipid-lowering therapy to complement revascularization in patients with stable angina. The multicenter, randomized Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation (COURAGE) trial may elucidate the issue further through its investigation of whether combining percutaneous coronary intervention with maximal statin intervention will provide greater benefits than statin intervention alone in more than 3,000 patients [82]. The COURAGE protocol targets global risk reduction, emphasizing (1) lifestyle modification; (2) maximal use of drugs to lower blood pressure to Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC) VI goals; (3) maximal use of simvastatin, 80mg/day, to lower cholesterol to below current US goals for secondary prevention; and (4) maximal use of drug to alleviate anginal symptoms with or without the best interventional devices to conduct percutaneous coronary intervention. Patients will be treated to a target LDL cholesterol level of 60 to 85 mg/dl and will be followed for a minimum of 3 years. The main outcome will be all-cause mortality or nonfatal myocardial infarction. Regression studies The Reversal of Atherosclerosis with Lipitor (REVERSAL) trial [83] used intravascular ultrasonography to examine the effect of differing degrees of lipid-lowering on plaque volume. Over 18 months, patients treated with pravastatin (40mg/day) had a 25 percent drop in LDL cholesterol, and those randomized to atorvastatin (80 mg/day) had a 46 percent decrease, to an average LDL cholesterol level of 79 mg/dL (2.6 mmol/L). The more aggressive lipid-lowering regimen reduced lesion volume. The primary end-point was a reduction in the total atheroma volume which increased by 0.4% on pravastatin but decreased by 2.7% (p=0.02) on atorvastatin. The ASTEROID study [84] was designed to evaluate whether 24 months treatment with rosuvastatin 40 mg will result in regression of coronary atherosclerosis as measured by IVUS in 507 patients. During treatment with rosuvastatin 40 mg. LDLC decreased from 130 mg/dL (3.4 mmol/L) to 61 mg/dL (1.6 mmol/L); HDL-C increased by 14.7%. 349 patients underwent follow-up IVUS after 2 years. Several efficacy parameters were used: the median change in percent atheroma volume decreased by 0.79%, and the median change in most diseased sub-segment decreased by 5.6%; both p<0.001. No control or comparator group was used. The ASTEROID showed that rosuvastatin at a dose of 40 mg promotes coronary atherosclerosis regression. The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial examined the effects of high-dose atorvastatin in patients after a stroke or transient ischemic attack (TIA) [85]. The investigators randomized 4731 patients who had a stroke or TIA within one to six months of entry into the study to placebo or atorvastatin 80 mg/day. During a mean follow-up of nearly 5 years, the LDL-C fell to 1.9 mmol/L (73 mg/dL) in the atorvaststin arm, compared with 3.3 mmol/L (129 mg/dL) in the placebo arm. The primary end-point of non-fatal or fatal stroke was decreased by 16% (absolute risk reduction 2.2%). The secondary end-point of major cardiac events was reduces by 20% (absolute reduction 3.5%). The SPARCL study confirms that patients with stroke or TIA should be considered as CHD risk equivalent. Approach to the Treatment of Lipoprotein Disorders 24 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Patients with lipoprotein disorders should undergo comprehensive evaluation and management in the context of a global risk reduction program (see Chap. 45). Most patients with dyslipoproteinemias lack symptoms, except for those with severe hypertriglyceridemia who can present with acute pancreatitis and those with familial lipoprotein disorders who have cutaneous manifestations (xanthomas, xanthelasmas). In the evaluation of patients with dyslipidemia, secondary causes should be sought and treated. The clinical evaluation should include a thorough history, including a complete family history that may reveal clues as to the genetic cause but also to the genetic susceptibility to cardiovascular disease. The physician should seek and address other risk factors (cigarette smoking, obesity, diabetes) and institute a management plan to improve lifestyle, such as diet, physical activity, and alcohol intake. Such interventions should make use of non-physician health professionals (e.g., those with training in diet and nutrition, physical therapy and smoking cessation). The ATP III Therapeutic Lifestyle Change program offers one such approach. Concomitant medication use in addition to lifestyle change will often be needed to achieve current guideline goals. The physical examination should include a search for xanthomas (in extensor tendons, including hands, elbows, knees, Achilles tendons, and palmar xanthomas); the presence of xanthelasmas, corneal arcus, and corneal opacifications. The blood pressure, waist circumference, weight, and height should be recorded and signs of arterial compromise sought and a complete cardiovascular examination must be performed. The evaluation of peripheral pulses and the determination of the ankle-brachial index may reveal important clues for the presence of peripheral vascular disease (see Chap. 57). The diagnosis of lipoprotein disorders depends on laboratory measurements (Table 42–9). The fasting lipid profile generally suffices for most lipoprotein disorders, and specialized laboratories can refine the diagnosis and provide expertise for extreme cases. Additional tests often involve considerable expense and may not increase the predictive value beyond that of the lipid profile, although they can help in refining the diagnosis. To assess baseline risk in individuals on lipid-lowering therapy, the medication should be stopped for 1 month before a lipid profile is measured. Many advanced lipid tests are available in specialized centers (Table 42–9) but seldom add to the clinical assessment specified above. After diagnosis of a lipid disorder (based on at least two lipid profiles), secondary causes should be evaluated by measurement of thyroid stimulating hormone and glucose. Patients who will receive medications should have measurement of baseline liver function (alanine aminotransferase [ALT]) and creatinine kinase. A decision to treat high-risk subjects (for example, patients with an acute coronary syndrome or post-myocardial infarction or coronary revascularization) should be start immediately and should commence concomitantly with lifestyle changes [69]. Target Levels (see Chap. 42) The National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) [69] has made recommendations for the treatment of hypercholesterolemia. Target levels depend on overall risk of cardiovascular death or nonfatal myocardial infarction. Patients with CAD or atherosclerosis of other vascular beds (carotids or peripheral vascular disease), adults with diabetes, and those patients with an estimated 10-year risk of developing CAD of greater than 20 percent fall into a high-risk category and merit aggressive treatment, including medications along with lifestyle modifications, exercise, and diet to achieve a primary target of an LDL cholesterol level less than 2.6 mmol/L (100 mg/dL). In subjects with triglycerides greater than 200 mg/dL, ATP III presents a secondary target of a non-HDL cholesterol level less than 3.4 mmol/L (130 mg/dL). Many of these individuals have the metabolic syndrome. Lifestyle Changes 25 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 TREATMENT. The therapeutic options consist of lifestyle modifications, treatment of secondary causes, and, if possible, diet and medications. DIET (see Chap. 44). Individuals with dyslipoproteinemias should always adopt dietary therapy. High-risk subjects should have medications started concomitantly with a diet because in many cases, diet may not suffice to reach target levels. The diet should have three objectives. First, it should allow the patient to reach and maintain ideal body weight. Second, it should provide a well-balanced diet with fruits, vegetables, and whole grains and third, it should be restricted in saturated fats and refined carbohydrates. Dietary counseling should involve a professional dietitian. Often, the help of dietitians, weight loss programs, or diabetic outpatient centers can aid sustained weight loss. Currently, the ATP III and the American Heart Association recommend a diet in which protein intake represents 15 to 20 % of calories, fats represent less than 35 %, with only 7 % from saturated fats, and the remaining calories derive from carbohydrates. Cholesterol intake should be less than 300 mg/day. Treatment of Combined Lipoprotein Disorders Combined lipoprotein disorders, characterized by an increase in plasma total cholesterol and triglycerides, frequently occur in clinical practice and present difficult challenges. Patients with combined lipoprotein disorders have an increase in LDL cholesterol and LDL particle number (as reflected by an increase in total or LDL apo B), small, dense LDL particles, increased VLDL cholesterol and VLDL triglycerides, and a reduced HDL cholesterol level. Patients with this pattern of combined dyslipidemia often have obesity and the metabolic syndrome. Treatment should begin with lifestyle modifications, with a diet reduced in total calories and saturated fats, weight reduction, and increased physical activity. Drug treatment, when warranted, aims to correct the predominant lipoprotein abnormality. Statins can reduce plasma triglyceride levels, particularly in individuals with high baseline triglyceride levels. Fibrates reduce triglycerides and may change the composition of LDL to larger and less dense particles. The role of fibric acid derivatives in the secondary prevention of cardiovascular diseases in diabetic subjects with low HDL cholesterol levels has been cast into doubt after publication of the FIELD trial [71]. Fibrates can paradoxically increase LDL cholesterol levels (because of increased lipoprotein lipase activity). Plasma total homocysteine levels may increase with fenofibrate and to a lesser extent with bezafibrate. In view of the effects of gemfibrozil on glucuronidation of statins, we advise against gemfibrozil use in combination therapy with statin. The combination of a statin with a fibrate, however has proven highly effective in correcting the combined dyslipoproteinemias; whether this approach prevents recurrent cardiovascular events remains to be shown by appropriate clinical trials.. Patients taking a fibrate plus a statin merit close medical follow-up for evidence of hepatotoxicity or myositis within the first 6 weeks of therapy and every 6 months thereafter. The combine use of a statin and ezetimibe for the treatment of severe hypercholesterolemia or to reach recommended target levels when monotherapy with a statin is either insufficient or causes unwanted side effects has a good rationale, although unsubstantiated by clinical trials. Other combinations, including fibric acid derivatives with bile acid–binding resins and niacin with bile acid–binding resins, have also proven useful in specific cases. The combination of fibrates or statins with niacin requires experience and care because of the risk of hepatotoxicity and myositis. The search for correctable causes (e.g., uncontrolled diabetes, obesity, hypothyroidism, and alcohol use) of combined dyslipidemia and the benefit of lifestyle modifications require reemphasis. Often, the help of dietitians, weight loss programs, or diabetic outpatient centers adds considerably to mamagement. Extracorporeal low-density lipoprotein filtration. Patients with severe hypercholesterolemia, especially those with homozygous familial hypercholesterolemia or severe heterozygous familial hypercholesterolemia may warrant treatment by extracorporeal LDL elimination. These techniques use 26 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 selective filtration, adsorption, or precipitation of LDL (or apo B–containing particles) after plasma separation. Specialized centers have LDL-pheresis available. This approach can dramatically reduce the risk of developing cardiovascular disease and improve survival [86]. Novel Approaches The development of novel pharmaceutical agents for the treatment of lipoprotein disorders will likely continue because cardiovascular disease due to atherosclerosis represents the largest burden of disease for the near future. Better targeting of high-risk individuals will allow optimization of expensive therapies. The finding that subjects who were previously identified as being at relatively low risk of CAD on the basis of their LDL cholesterol levels but who have an elevated C-reactive protein level derive benefit from a statin in the primary prevention of CAD may radically alter the concept of cardiovascular risk stratification [50] (See also Chapts 39 & 45). If markers of inflammation can better identify cardiovascular risk, in addition to conventional risk factors, physicians’ attitudes should embrace these findings. This hypothesis is undergoing rigorous testing in a clinical trial. Novel therapies to raise HDL-C have received considerable support in a proof-of-concept clinical trial where patients with an acute coronary syndrome were given weekly injections of apo AIMilano reconstituted proteoliposomes, in effect nascent HDL particles [87]. Remarkably, a reduction of 4.7% of atheroma volume was documented by IVUS after 5 weeks of treatment. While difficult to apply clinically, this small trial affirmed the benefits of raising HDL-C in patients with CHD. The burgeoning field of pharmacogenetics might, in the near future, allow treatment of patients on the basis of their genetic make-up. Genetic screening may become a useful clinical tool, as technology improves and rapid genotyping for diagnostic and prognostic purposes becomes available for clinicians. Other than cost issues, the ethics of screening for genetic predisposition to disease and access to information represent daunting challenges. The discovery that the apo E4 allele carries the risk of earlyonset Alzheimer disease, one of the familial forms of the disorder, illustrates the ethical complexities of genetic testing in the realm of lipoprotein disorders. Drug Development and Future Directions Novel proteins that regulate the synthesis of lipids have become therapeutic targets for drug development. The development of competitive inhibitors of HMG-CoA reductase leading to statin drugs provides a good example. These drugs have an important impact on cardiovascular morbidity and mortality reduction in high-risk individuals. Inhibitors of pancreatic lipases are being used to treat obesity. Future potential drug targets might include inhibition of apo B secretion by inhibiting the microsomal triglyceride transfer protein, which is crucial in the assembly of apo B–containing lipoproteins. Hepatic steatosis has limited this therapeutic option in humans. Recent work has shown that selective inhibitors of ACAT do not retard atherosclerosis and their anti-atherogenic role remains unproven. Drugs that increase cellular cholesterol efflux to increase HDL cholesterol levels may also prove useful in the treatment of dyslipoproteinemias. The PCSK9 protein associated with genetically low LDL-C levels represents another potential therapeutic target. Other therapeutic modalities in the treatment of atherosclerosis by modulating lipoprotein metabolism include the development of new inhibitors of CETP to increase HDL cholesterol levels or modulation of LCAT, and inhibitors of bile acid transport to decrease intestinal cholesterol uptake. Pharmacological modulation of HDL cholesterol levels, other than by niacin, has not led to results proportional to those achieved for LDL cholesterol. Potential modulators of HDL cholesterol levels include SR-B1, ABCA1 and ABCG1 pathways, apo A1, and its homologues and mimetics. Specific sub groups (diabetics and the elderly) 27 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 Patients with diabetes should receive a statin. Multiple observational studies have documented a markedly increased risk of CHD in adult diabetics over the long-term. Preventive strategies with aspirin, angiotensin convertase inhibitors, tight glycemic control and statins have all shown benefit. Data from CARDS and TNT support intensive LDL-C lowering in diabetics [88]. The elderly represent a unique challenge; most of the attributable risk in patients >75 or 80 years old is due to age and the predictive value of an elevated cholesterol decreases with increasing age [69]. The value of statins and other lipid-lowering drugs in the elderly in primary prevention lacks an evidence base at present. Yet, the data from the PROSPER, HPS, 4S and several other studies, strongly suggest that secondary preventive measures should not be withheld from older patients solely because of age. Gene Therapy Severe, homozygous, monogenic disorders may eventually be treated by gene therapy. The initial trials of gene therapy in cases of homozygous familial hypercholesterolemia have not led to a major improvement and have largely been abandoned. However, the lifelong burden of these rare disorders and the potential for cure makes this approach very appealing. Other diseases, such as abetalipoproteinemia, LPL deficiency, Niemann-Pick type C disease, sitosterolemia, and Tangier disease may become targets for gene therapy. If the approach to correct these disorders is successful, the more widespread applications of gene-based therapies for the purpose of reducing potential cardiovascular risk will become a daunting medical, social, and ethical problem. Acknowledgments: The authors thank Dr. Antonio Gotto who contributed to earlier versions of this chapter in previous editions. 28 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 REFERENCES Lipoprotein transport system 1. Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy HM. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci U S A. 2006 Feb 14;103(7):2126-31 2. Hiltunen TP, Luoma JS, Nikkari T, Yla-Herttuala S: Expression of LDL receptor, VLDL receptor, LDL receptor-related protein, and scavenger receptor in rabbit atherosclerotic lesions: Marked induction of scavenger receptor and VLDL receptor expression during lesion development. Circulation 97:1079, 1998. 3. MacDougall ED, Kramer F, Polinsky P, Barnhart S, Askari B, Johansson F, Varon R, Rosenfeld ME, Oka K, Chan L, Schwartz SM, Bornfeldt KE. Aggressive very low-density lipoprotein (VLDL) and LDL lowering by gene transfer of the VLDL receptor combined with a low-fat diet regimen induces regression and reduces macrophage content in advanced atherosclerotic lesions in LDL receptor-deficient mice. Am J Pathol. 2006 Jun;168(6):2064-73. 4. Mahley RW, Ji ZS: Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res 40:1, 1999. 5. Brown ML, Ramprasad MP, Umeda PK, et al: A macrophage receptor for apolipoprotein B48: Cloning, expression, and atherosclerosis. Proc Natl Acad Sci U S A 97:7488, 2000. 6. Spillmann D, Lookene A, Olivecrona G. Isolation and characterization of low sulfated heparan sulfate sequences with affinity for lipoprotein lipase. J Biol Chem. 2006 Jun 16; [Epub ahead of print] 7. Sawamura T, Kume N, Aoyama T, et al: An endothelial receptor for oxidized low-density lipoprotein. Nature 386:73, 1997. 8. Acton S, Rigotti A, Landschulz KT, et al: Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271:518, 1996. 9. Krimbou L, Hassan HH, Blain S, et al.. Biogenesis and speciation of nascent apo AI – containing particles in various cell lines. J Lipid Res 2005;46:1668-1677 10. Altmann SW, Davis HR Jr, Zhu LJ, et al: Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science 303:1201, 2004. 11. Abifadel M, Varret M, Rabes JP, et al: Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 34:154, 2003. 12. Willner EL, Tow B, Buhman KK, et al: Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A 100:1262, 2003. 13. Timmins JM, Lee JY, Boudyguina E, et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest. 2005;115(5):1333-42 14. Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116(4):1052-62. 15. Denis M, Haidar B, Marcil M, Krimbou L, Genest J. Molecular and cellular physiology of apolipoprotein A-I lipidation by the ATP biding cassette A1 (ABCA1). J Biol Chem 2004:279:7384-7394 16. Haidar B, Denis M, Marcil M, Krimbou L, Genest J. Apolipoprotein A-I activates cellular camp signaling through the ABCA1 receptor. Evidence for molecular interactions between ABCA1 receptor and G protein. J Biol Chem 2004;279:99639 29 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 17. Chau P, Nakamura Y, Fielding CJ, Fielding PE. Mechanism of prebeta-HDL formation and activation. Biochemistry. 2006;45(12):3981-7 18. Li XA, Titlow WB, Jackson BA, et al: High density lipoprotein binding to scavenger receptor, class B, type I activates endothelial nitric-oxide synthase in a ceramide-dependent manner. J Biol Chem 277:11058, 2002. 19. Van Eck M, Singaraja RR, Ye D, Hildebrand RB, James ER, Hayden MR, Van Berkel TJ. Macrophage ATP-binding cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2006;26(4):929-34 Lipoprotein disorders 20. Barter PJ, Ballantyne CM, Carmena R, et al.. Apo B versus cholesterol in estimating cardiovascular risk and in guiding therapy: report of the thirty-person/ten-country panel. J Intern Med. 2006 Mar;259(3):247-58. 21. Grundy SM, Cleeman JI, Daniels SR, et al.; American Heart Association; National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-52 22. Wilson DJ, Gahan M, Haddad L, et al: A World Wide Web site for low-density lipoprotein receptor gene mutations in familial hypercholesterolemia: Sequence-based, tabular, and direct submission data handling. Am J Cardiol 81:1509, 1998. 23. Whitfield AJ, Barrett PH, van Bockxmeer FM, Burnett JR. Lipid disorders and mutations in the APOB gene. Clin Chem. 2004 Oct;50(10):1725-32 24. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-72. 25. Garuti R, Jones C, Li WP, Michaely P, Herz J, Gerard RD, Cohen JC, Hobbs HH. The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem. 2005;9;280(49):40996-1004 26. Berge KE, Tian H, Graf GA, et al: Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290:1771, 2000. 27. Mooser V, Mancini FP, Bopp S, et al: Sequence polymorphisms in the apo(a) gene associated with specific levels of Lp(a) in plasma. Hum Mol Genet 4:173, 1995. 28. Danesh J, Collins R, Peto R: Lipoprotein(a) and coronary heart disease: Meta-analysis of prospective studies. Circulation 102:1082, 2000. 29. Sjarif DR, Sinke RJ, Duran M, et al: Clinical heterogeneity and novel mutations in the glycerol kinase gene in three families with isolated glycerol kinase deficiency. J Med Genet 35:650, 1998. 30. Santamarina-Fojo S: The familial chylomicronemia syndrome. Endocrinol Metab Clin North Am 27:551, viii, 1998. 31. Weinstock PH, Bisgaier CL, Aalto-Setala K, et al: Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice: Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J Clin Invest 96:2555, 1995. 32. Mahley RW, Huang Y, Rall SC Jr: Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): Questions, quandaries, and paradoxes. J Lipid Res 40:1933, 1999. 33. Zhang SH, Reddick RL, Piedrahita JA, Maeda N: Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258:468, 1992. 30 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 34. Pajukanta P, Lilja HE, Sinsheimer JS, et al. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1). Nat Genet. 2004;36(4):371-6. 35. Kalant D, MacLaren R, Cui W, Samanta R, Monk PN, Laporte SA, Cianflone K. C5L2 is a functional receptor for acylation-stimulating protein. J Biol Chem. 2005;280(25):23936-44. 36. Dastani Z, Engert JC, Genest J, Marcil M. Genetics of High-Density Lipoproteins. Curr Op Cardiol 2006 37. Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96(12):1221-32 38. Sorci-Thomas MG, Thomas MJ: The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med 12:121, 2002. 39. Boekholdt SM, Kuivenhoven JA, Hovingh GK, Jukema JW, Kastelein JJ, van Tol A. CETP gene variation: relation to lipid parameters and cardiovascular risk. Curr Opin Lipidol. 2004;15(4):393-8. 40. Calabresi L, Pisciotta L, Costantin A, et al. The molecular basis of lecithin:cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol. 2005;25(9):1972-8 41. Brooks-Wilson A, Marcil M, Clee SM, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22(4):336-45. 42. Marcil M, Brooks-Wilson A, Clee SM, et al: Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. Lancet 354:1341, 1999. 43. Carstea ED, Morris JA, Coleman KG, et al: Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 277:228, 1997. 44. Choi HY, Karten B, Chan T, Vance JE, Greer WL, Heidenreich RA, Garver WS, Francis GA. Impaired ABCA1dependent lipid efflux and hypoalphalipoproteinemia in human Niemann-Pick type C disease. J Biol Chem. 2003;278(35):32569-7 45. Lee CY, Lesimple A, Denis M, Vincent J, Larsen A, Mamer O, Krimbou L, Genest J, Marcil M. Increased sphingomyelin content impairs HDL biogenesis and maturation in human Niemann-Pick disease type B. J Lipid Res. 2006;47(3):622-32 46. Anderson GL, Limacher M, Assaf AR: Effects of conjugated equine estrogen in post-menopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 291:1701, 2004. 47 Meigs JB, Wilson PW, Fox CS, Vasan RS, Nathan DM, Sullivan L, D'Agostino RB. Body Mass Index, Metabolic Syndrome and Risk of Type 2 Diabetes or Cardiovascular Disease. J Clin Endocrinol Metab. 2006 May 30 48. Kahn R, Buse J, Ferrannini E, Stern M; American Diabetes Association; European Association for the Study of Diabetes. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28(9):2289-304 Drugs that affect lipid metabolism 49. Martin G, Duez H, Blanquart C et al. Statin-induced inhibition of the Rho-singaling pathway activates PPAR alpha and induces apo AI. J Clin Invest 2001;107:1423 50. Ridker PM; JUPITER Study Group. Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: rationale and design of the JUPITER trial. Circulation. 2003;108(19):2292-7 51. Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109(23 Suppl 1):III39-43. 31 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 52. Libby P, Aikawa M: Stabilization of atherosclerotic plaques: New mechanisms and clinical targets. Nat Med 8:1257, 2002. 53. Genest J. Combination of Statin and Ezetimibe for the Treatment of Dyslipidemias and the Prevention of Coronary Artery Disease. Can J Cardiol 2006 54. Sebestjen M, Keber I, Zegura B, Simcic S, Bozic M, Fressart MM, Stegnar M. Statin and fibrate treatment of combined hyperlipidemia: the effects on some novel risk factors. Thromb Haemost. 2004 Nov;92(5):1129-35. 55. Brown BG, Zhao XQ, Chait A, et al: Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 345:1583, 2001. 56. Wise A, Foord SM, Fraser NJ, et al: Molecular identification of high and low affinity receptors for nicotinic acid. J Biol Chem 278:9869, 2003. 57. Brousseau ME, Schaefer EJ, Wolfe ML, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med. 2004 Apr 8;350(15):1505-15. 58. Cote G, Tardif JC, Lesperance J, et al: Effects of probucol on vascular remodeling after coronary angioplasty. Multivitamins and Protocol Study Group. Circulation 99:30, 1999. Clinical trials of drugs affecting lipid metabolism 59. Downs JR, Clearfield M, Weis S, et al: Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: Results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 279:1615, 1998. 60. Shepherd J, Cobbe SM, Ford I, et al: Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 333:1301, 1995. 61. Sacks FM, Pfeffer MA, Moye LA, et al: The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med 335:1001, 1996. 62.Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 339:1349, 1998. 63. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 344:1383, 1994. 64. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 360:7, 2002. 65. Shepherd J, Blauw GJ, Murphy MB, et al: Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 360:1623, 2002. 66. Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). JAMA 288:2998, 2002. 67. Sever PS, Dahlof B, Poulter NR, et al: Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial— Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet 361:1149, 2003. 68. Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with artovastatin in type 2 diabetes in the Collaborative Artovastatin Diabetes Study (CARDS) : mulitcentre randomised placebo-controlled trial. Lancet. 2004;364;685-695 32 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 69. Grundy, S. M., Cleeman, J. I., Merz, C. N. B., Brewer, H. B., Clark, L. T., Hunninghake, D. B., Pasternak, R. C., Smith, S. C., and Stone, N. J. Implications of Recent Clinical Trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Circulation 2004;110(2):227-39. 70. de Backer, G., Ambrosioni, E., Borch-Johnsen, K., et al. European Guidelines on Cardiovascular Disease Prevention in Clinical Practice - Third Joint Task Force of European and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (Constituted by Representatives of Eight Societies and by Invited Experts). Atherosclerosis 2003;171(1):145-55. 71. Keech, A., Simes, R. J., Barter, P.et al.. Effects of Long-Term Fenofibrate Therapy on Cardiovascular Events in 9795 People With Type 2 Diabetes Mellitus (the FIELD Study): Randomised Controlled Trial. Lancet 2005;366(9500):1849-61. 72. Rubins HB, Robins SJ, Collins D, et al: Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 341:410, 1999. 73. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: The Bezafibrate Infarction Prevention (BIP) study. Circulation 102:21, 2000. 74. Baigent, C., Keech, A., Kearney, P. M., Blackwell, L., Buck, G., Pollicino, C., Kirby, A., Sourjina, T., Peto, R., Collins, R., and Simes, J. Efficacy and Safety of Cholesterol-Lowering Treatment: Prospective Meta-Analysis of Data From 90,056 Participants in 14 Randomised Trials of Statins. Lancet 2005;366(9493):1267-78 75 Larosa, J. C., Grundy, S. M., Waters, D. et al.. Intensive Lipid Lowering With Atorvastatin n Patients With Stable Coronary Disease. New Eng J Med 2005;352(14):1425-35. 76 Pedersen, T. R., Faergeman, O., Kastelein, J. J. P.et al. High-Dose Atorvastatin Vs Usual Dose Simvastatin for Secondary Prevention After Myocardial Infarction - The IDEAL Study: A Randomized Controlled Trial. J Am Med Ass 2005;294(19):2437-45. 77 McMahon M, Kirkpatrick C, Cummings CE, et al: A pilot study with simvastatin and folic acid/vitamin B 12 in preparation for the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH). Nutr Metab Cardiovasc Dis 10:195, 2000. 78. Schwartz GG, Olsson AG, Ezekowitz MD, et al: Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The MIRACL study: A randomized controlled trial. JAMA 285:1711, 2001. 79. Wiviott SD, de Lemos JA, Cannon CP, Blazing M, Murphy SA, McCabe CH, Califf R, Braunwald E. A tale of two trials: a comparison of the post-acute coronary syndrome lipid-lowering trials A to Z and PROVE IT-TIMI 22. Circulation. 2006 Mar 21;113(11):1406-14. 80. Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004 Apr 8;350(15):1495-504. 81. Pitt B, Waters D, Brown WV, et al: Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 341:70, 1999. 82. Chiquette E, Chilton R: Aggressive medical management of coronary artery disease versus mechanical revascularization. Curr Atheroscler Rep 5:118, 2003. 83. Nissen SE, Tuzcu EM, Schoenhagen P, et al: Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: A randomized controlled trial. JAMA 291:1071, 2004. 84 Nissen SE, Nicholls SJ, Sipahi I et al. Effect of very high intensity statin therapy on regression of coronart atherosclerosis: the ASTEROID trial JAMA 2006;295(13):1556-65 33 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 85 The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators. High-dose atorvastatin after stroke or transient ischemic attack. New Eng J Med 2006;355:549-559 86 Nishimura S, Sekiguchi M, Kano T, et al: Effects of intensive lipid lowering by low-density lipoprotein apheresis on regression of coronary atherosclerosis in patients with familial hypercholesterolemia: Japan Low-density Lipoprotein Apheresis Coronary Atherosclerosis Prospective Study (L-CAPS). Atherosclerosis 144:409, 1999. 87. Nissen SE, Tsunoda T, Tuzcu EM, et al: Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 290:2292, 2003. 88. Shepherd J, Barter P, Carmena R et al. Effect of LDL cholesterol lowering substantially below currently recommended levels in diabetic patients with coronary artery heart disease and diabetes. The treating to New Targets (TNT) study. Diabetes Care 2006;29:1220-1226 34 Chapter 42 Lipoprotein Disorders and Cardiovascular Disease Jan 2009 TABLES TABLE 42-1 Plasma Lipoprotein Composition TABLE 42-2 Apolipoproteins TABLE 42-3 Lipoprotein Processing Enzymes, Receptors, Modulating Proteins TABLE 42-4 Genetic Lipoprotein Disorders TABLE 42-5 Secondary Causes of Dyslipoproteinemias TABLE 42-6 Current Lipid-Lowering Medications TABLE 42-7 Trials of Statin Therapy in High-Risk Primary Prevention TABLE 42-8 Secondary Prevention Trials of Statin Therapy TABLE 42-9 Laboratory Tests for the Diagnosis of Lipoprotein Disorders FIGURES FIGURE 42-1 Biochemical structure of the major lipid molecules: cholesterol, cholesteryl esters, triglycerides, and phospholipids (phosphatidylcholine and sphingomyelin). FIGURE 42-2 Structure of lipoproteins. Phospholipids are oriented with their polar group toward the aqueous environment of plasma. Free cholesterol is inserted within the phospholipid layer. The core of the lipoprotein is made up of cholesteryl esters and triglycerides. Apolipoproteins are involved in the secretion of the lipoprotein, provide structural integrity, and act as cofactors for enzymes or as ligands for various receptors. FIGURE 42-3 Relative size of plasma lipoproteins according to their hydrated density. HDL: high-density lipoprotein; IDL: intermediate-density lipoprotein; LDL: low-density lipoprotein. FIGURE 42-4 Schematic diagram of the lipid transport system. Numbers in circles refer to explanation in text. Apo: apolipoprotein; CETP: cholesteryl ester transfer protein; EL: endothelial lipase; FFA: free fatty acids; HL: hepatic lipase; HDL: high-density lipoprotein; IDL: intermediate-density lipoprotein; LCAT: lecithin cholesterol acyltransferase; LDL: lowdensity lipoprotein; LPL: lipoprotein lipase; PLTP: phospholipid transfer protein; VLDL: very-low-density lipoprotein. FIGURE 42-5 Cellular cholesterol homeostasis in various tissues. A, Cholesterol homeostasis (hepatocytes). B, Cellular cholesterol efflux (peripheral cells). C, Selective uptake of cholesterol (adrenal cells, hepatocytes, endothelial cells). D, Adipocytes and E, macrophages foam cells. ABCA1: ATP-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; ACAT: acyl–coenzyme A:cholesterol acyltransferase; Apo: apolipoprotein; ASP: acylation-stimulating protein; CE: cholesterol esters; CETP; cholesteryl ester transfer protein; HDL: high-density lipoprotein; HMG CoA Red: hydroxymethylglutaryl coenzyme A reductase; HSL: hormone-sensitive lipase; IDL: intermediate-density lipoprotein; LCAT: lecithin cholesterol acyltransferase; LDL: low-density lipoprotein; LDL-R: low-density lipoprotein receptor; LRP: lowdensity lipoprotein receptor– related peptide; PLTP: phospholipid transfer protein; sER: smooth endoplasmic reticulum; SRB1: scavenger receptor B1; TG: triglycerides; VLDL: very-low-density lipoprotein; VLDL-R: very-low-density lipoprotein receptor. FIGURE 42–6 Clinical trials of statin therapy have demonstrated benefits in patients across the spectrum of coronary disease. From ref. 74: Baigent, C., Keech, A., Kearney, P. M., Blackwell, L., Buck, G., Pollicino, C., Kirby, A., Sourjina, T., Peto, R., Collins, R., and Simes, J. Efficacy and Safety of Cholesterol-Lowering Treatment: Prospective Meta-Analysis of Data From 90,056 Participants in 14 Randomised Trials of Statins. Lancet 2005;366(9493):1267-78 35