Metabolism 1: An Introduction to Protein Structure
advertisement

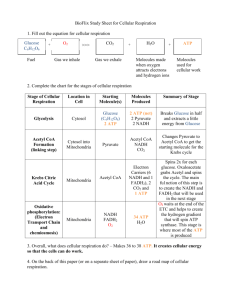
Metabolism 1: An Introduction to Protein Structure Outline the reaction by which amino acids are joined together; sketch a trimeric peptide, illustrating the amino terminus, carboxyl terminus and side chains Protein: any of a group of organic compounds composed of one or more chains of amino acids and forming an essential part of all living organisms Human body: 20% protein Protein mutations are responsible for a variety of diseases (e.g. sickle cell anaemia) Structure of an amino acid: Substitutions of the side chain (R group) give rise to the 20 different amino acids Backbone: the whole of the amino acid minus the side chain Amino acids with hydrophobic side chains Amino acids with hydrophilic side chains Glycine: the simplest amino acid (R group = H) Asparagine Alanine Glutamine Valine Cysteine Leucine Histidine Isoleucine Serine Proline Threonine Methionine Tyrosine Phenylalanine Tryptophan Aspartate Glutamate Lysine Arginine Amino acids with charged side chains Arginine and Lysine Protonated at physiological pH; therefore basic Histidine Often protonated Aspartic acid and Glutamic acid Deprotonated at physiological pH; therefore acidic Amino acids with charged side chains: The ionisation state of an amino acid provides vital biological properties to many proteins Therefore cells cannot generally tolerate wide changes in pH If the ionisation state of key amino acids within a protein is altered: a loss of biological activity often results The ability to take up and release protons gives amino acids some buffering capacity to resist some changes in pH Chirality: The central Cα carbon atom is a chiral centre: i.e. it has 4 different substituents bound to it This produces optical isomers (enantiomers) of each amino acid, which are non-superimposable mirror images of each other Glycine (Gly) has no side chain; therefore it is the only non-chiral amino acid All amino acids found in proteins are L-enantiomers Peptides are formed by a condensation reaction between 2 amino acids: Structure of a peptide: Protein structure: The polypeptide chain of a protein rarely forms a disordered structure Proteins have functions which rely upon specificity Functionality requires a definite 3D structure (conformation) of the polypeptide chain Proteins generally possess a degree of flexibility which is necessary for function (e.g. muscle fibres) Characteristics of the peptide bond: There is no free rotation about the peptide bond The C=O and N–H groups are in the same plane of the molecule The other 2 bonds in the backbone of the polypeptide chain are able to rotate Understand the concepts of primary structure, secondary structure, tertiary structure & quaternary structure with respect to proteins Folding of proteins: Proteins generally fold into a single conformation of lowest energy Folding of proteins may occur spontaneously or it may involve chaperones Chaperones: molecules which bind to a partially folded polypeptide chain and ensure that folding continues along the most energetically favourable pathway By breaking the bonds which hold a protein together, a protein is denatured into the original flexible polypeptide Common laboratory denaturants include: o Urea: breaks hydrogen bonds o 2-mercaptoethanol: breaks disulphide bonds Structural levels of proteins: Primary structure: the linear sequence of amino acids which constitute the protein Nomenclature: the protein sequence is written from the amino terminus to the carboxyl terminus Secondary structure: local structural motifs within a protein (e.g. α-helices and ß-pleated sheets) Tertiary structure: arrangement of motifs of the secondary structure into domains (compact globular structures) Quaternary structure: the 3D structure of a multimeric protein, composed of several subunits Distinguish between an α-helix and a ß-pleated sheet and appreciate the bonds that stabilise their formation α-helices: Hydrogen bonds between the C=O group of 1 residue and the N–H group of another residue stabilise the helix Side chains of individual amino acids project out from within the helix Right-handed helices are more common since L-enantiomers of amino acids are used in proteins Proline: when proline is joined to a polypeptide chain, it loses its N–H group This prevents the N atom from hydrogen bonding with the C=O group of another residue within the helix The helical conformation is distorted and kinked ß-pleated sheets: Hydrogen bonds between the C=O and N–H groups of 2 or more ß-strands stabilise the sheet C=O and N–H groups project out perpendicular to the line of the backbone Parallel ß-pleated sheet: alternate ß-strands run in the same direction Anti-parallel ß-pleated sheet: alternate ß-strands run in opposite directions Pleating allows for the best alignment of the hydrogen-bonded C=O and N–H groups Appreciate the different types of bond that combine to stabilise a particular protein conformation Covalent bonds: 2 atoms share electrons The strongest bonds within a protein Exist in the primary structure May exist as disulphide bridges: Disulphide bridges occur when cysteine side chains within a protein are oxidised This results in a covalent link between the 2 amino acids –CH2-SH + HS-CH2– –CH2-S-S-CH2– Hydrogen bonds: 2 atoms with partial negative charges share a partially positively charged hydrogen atom May occur either between atoms on different Side chains and the backbone of the protein or between water molecules Ionic interactions: electrostatic attractions between charged side chains Relatively strong bonds, particularly when the Side chains are within the interior of the protein and excluded from water The majority of charged Side chains are at the surface of a folded protein, where they can be neutralised by counter-ions (e.g. salts) Van der Waals forces: electrostatic attractions between 2 atoms due to the fluctuating electron cloud surrounding each atom, which has a temporary electric dipole Weak and transient forces Due to the sheer number of Van der Waals interactions within a protein, they play a large part in the overall conformation of a protein Van der Waals radius: the appropriate distance required for Van der Waals attractions This varies for different atoms, based on the size of the electron cloud If the 2 electron clouds of adjacent atoms are quite close: the transient dipole in 1 atom can induce a complementary dipole in another atom, with weak attractive properties If the 2 electron clouds of adjacent atoms are too close: there are repulsive forces between the 2 atoms due to the electrons Hydrophobic interactions: Pack hydrophobic side chains into the interior of the protein This creates a hydrophobic core and a hydrophilic surface Give examples of the post translational modifications of amino acids, with reference to glycosylation, hydroxylation and carboxylation Amino acids may be modified following protein synthesis Hydroxylation: Proline hydroxyproline This requires: prolyl hydroxylase and vitamin C Hydroxyproline is present in collagen fibres: additional hydroxyl groups help to stabilise the fibres Scurvy: caused by vitamin C deficiency Glycosylation: N-linked glycosylation of asparagine: addition of sugar residues This increases the solubility of proteins and protects them from enzymatic degradation Primary structure motif: N-X-S/T (asparagine-any amino acid-serine/threonine) Carbohydrate-deficient glycoprotein (CDG): associated with N-linked sugar chain transfer deficiency Carboxylation: Glutamate γ-carboxyglutamate This requires vitamin K-dependent carboxylase Formation of γ-carboxyglutamate residues within several proteins of the blood clotting cascade is critical for their normal function: it increases their calcium binding capability Warfarin (anticoagulant): inhibits the above carboxylation reaction Lecture summary: Proteins are chains of amino acids linked by peptide bonds which have evolved to fulfil specific functions within the cell Such functions rely upon the conformation (3D structure) of the protein which is determined by a variety of forces The α-helix and ß-pleated sheet are the two staple motifs that define the conformation of a protein The nature of the amino acid side chain dictates its position within the conformation of the protein Post-translational modifications of proteins add more diversity to protein structure Metabolism 2: Energetics and Enzymes Define the 1st and 2nd Laws of thermodynamics 1st Law of Thermodynamics: energy can neither be created nor destroyed; it is simply converted from one form to another 2nd Law of Thermodynamics: in any isolated system, such as a single cell or the universe, the degree of disorder can only increase Entropy: the amount of disorder in a particular system Reactions proceed spontaneously towards products with greater entropy Biological systems are very well ordered: energy is taken from the environment and invested into chemical reactions within the cell which maintain order Explain the concept of free energy and how we can use changes in free energy to predict the outcome of a reaction Free energy (G) [kJ/mol]: the amount of energy within a molecule that can perform useful work at a constant temperature ∆G: the amount of disorder that results from a particular reaction For the reaction: A + B C + D ∆G = G (C + D) − G(A + B) A reaction can only occur spontaneously if ∆G is negative (energetically favourable reactions) A reaction cannot occur spontaneously if ∆G is positive (energetically unfavourable reactions) Draw the chemical structure of ATP and explain how it acts as a carrier of free energy and is used to couple energetically unfavourable reactions ATP: ATP structure: Phosphoanhydride bonds have a large negative ∆G of hydrolysis ATP ADP + Pi ∆G = −31 kJ/mol Coupled reactions: Biosynthetic pathways are generally energetically unfavourable (e.g. peptide synthesis) They take place because they are coupled to an energetically favourable reaction A reaction will proceed if the sum of ∆G for the overall reaction is positive Most energetically unfavourable biochemical reactions are coupled to the hydrolysis of high-energy phosphoanhydride bonds (e.g. ATP hydrolysis) Example 1: glucose + fructose sucrose ∆G = +23 kJ/mol This reaction is coupled to ATP hydrolysis: Glucose + ATP glucose-1-phosphate + ADP Glucose-1-phosphate + fructose sucrose ∑∆G = −31 + (+23) = −8 kJ/mol; therefore it is energetically favourable Example 2: glucose-6-phosphate + H2O glucose + Pi ∆G = −13.8 kJ/mol This energetically favourable reaction will not occur at a useful rate unless it is catalysed by an enzyme N.B. enzymes do not change the value of ∆G Example 3: glucose + oxygen carbon dioxide + water ∆G = −2872 kJ/mol This energetically favourable reaction results in an increase in the entropy (disorder) of the universe It does not occur spontaneously since energy must be supplied to overcome the activation energy barrier Describe how enzymes act as catalysts of reactions with reference to the reaction catalysed by lysozyme Enzyme: a protein that acts as a catalyst to induce chemical changes in other substances itself remaining apparently unchanged by the process Mode of action: enzymes lower the activation energy barriers that impede chemical reactions from taking place 1 or more substrates bind to the enzyme tightly at the active site The enzyme arranges the substrate(s) in such a way that certain bonds are strained Key residues within the enzyme participate in either the making or breaking of bonds by altering the arrangement of electrons within the substrate(s) This can either take the form of oxidation or reduction reactions Transition state: the particular conformation of the substrate in which atoms of the molecule are rearranged both geometrically and electronically so that the reaction can proceed Enzymes work by bending substrates in such a way that the bonds to be broken are stressed: the substrate molecule resembles the transition state Lysozyme: a component of tears and nasal secretions, involved in defence against bacteria Lysozyme catalyses the hydrolysis of sugar molecules within bacterial cell walls: this results in lysis of bacteria Lysozyme hydrolyses alternating polysaccharide copolymers of N-acetyl glucosamine (NAG) and Nacetyl muramic acid (NAM) Lysozyme hydrolyses the ß-1,4 glycosidic bond which links C1 of NAM to C4 of NAG Mechanism of action: 1. Glu35 protonates the oxygen in the glycosidic bond: this breaks the glycosidic bond 2. Glu35 deprotonates a water molecule: this forms a hydroxide ion 3. Asp52 stabilises the positive charge on the transition state 4. The hydroxide ion attacks the transition state and adds an –OH group 5. Glu35 and Asp52 are in their original state to continue catalysis Optimum pH for lysozyme: 5.0 At pH 5.0: Asp52 is deprotonated (–COO−) Glu35 is protonated (–COOH) This is essential for lysozyme function Describe how oxidation and reduction involve the transfer of electrons Oxidation: loss of electrons Reduction: gain of electrons Since the cellular environment is generally aqueous, often when a molecule gains an electron, it simultaneously gains a proton Outline the reaction catalysed by glucose-6-phosphatase and explain what clinical symptoms are linked to inherited deficiencies of this enzyme In the liver: glycogen glucose-6-phosphate glucose Glucose-6-phosphate + H2O glucose + Pi This reaction is catalysed by glucose-6-phosphatase Glucose-6-phosphatase is predominantly a liver enzyme It catalyses the above reaction when blood glucose levels are low and releases glucose from the liver into the bloodstream Glucose-6-phosphatase deficiency (Von Gierke’s disease) Symptoms: Low blood sugar levels Slow growth Large livers Short stature Outline the differences between lock and key and induced fit models of substrate-enzyme interactions Lock and key model: the shape of the substrate is complementary to that of the active site of the enzyme This model explains the specificity of most enzymes for a single substrate Induced fit model: the substrate induces a change in the conformation of the enzyme which results in the formation of the active site; upon release of the products the enzyme reverts back to its original conformation This is the correct model since proteins generally possess a degree of flexibility necessary for function Crystallographic analysis of enzymes supports this model Describe graphically, the effects of a) substrate concentration, b) temperature and c) pH on enzyme catalysed reactions Effect of substrate concentration: Enzyme kinetics: Michaelis constant (KM): the concentration of substrate at which a given enzyme works at half its maximal velocity (vmax) KM is useful to compare the strength of enzyme-substrate complexes: Low KM value: tight binding within the enzyme-substrate complex High KM value: weak binding within the enzyme-substrate complex At vmax the rate of product formation depends on the turnover number (i.e. how rapidly the substrate can be processed) Lineweaver-Burk Plot: a double reciprocal plot of 1/V against 1/[S] Effect of temperature: Catalysis increases as temperature is increased Each enzyme has an optimum temperature; above this the enzyme’s conformation is denatured Effect of pH: The above graph is typical of most enzymes: they have an optimum pH at which the catalytic side chains are in the correct ionisation state Illustrate the role of the coenzyme NAD in the reactions catalysed by glyceraldehyde 3-phosphate dehydrogenase, lactate dehydrogenase and malate dehydrogenase, referring to the biochemical changes involved in its reduction to NADH NAD+: a cofactor for dehydrogenation reactions It is a coenzyme which only functions after binding to a protein Dehydrogenases catalyse dehydrogenation reactions: RCH(OH)R’ + NAD+ RCOR’ + NADH + H+ NAD+ catalyses dehydrogenation of substrates by accepting a hydrogen atom and 2 electrons: NAD+ + H+ + 2e- NADH Glyceraldehyde 3-phosphate dehydrogenase catalyses… Glyceraldehyde 3-phosphate + NAD+ + Pi 1,3-bisphosphoglycerate + NADH + H+ The substrate is oxidatively phosphorylated Lactate dehydrogenase catalyses… Pyruvate + NADH + H+ lactate + NAD+ Pyruvate is converted into lactate by anaerobic respiration during intense exercise This generates free NAD+ which is needed by muscle for other reactions Lactate is transported from muscles to the liver by the bloodstream The liver has high NAD+ levels which can be used by lactate dehydrogenase to regenerate pyruvate Malate dehydrogenase catalyses… Malate + NAD+ oxaloacetate + NADH + H+ Lecture summary: Energetically unfavourable reactions can occur by coupling them to energetically favourable reactions, generally involving the hydrolysis of high-energy phosphoanhydride bonds Enzymes are specific biological catalysts which increase the rate of biochemical reactions by lowering the activation energy barriers that impede chemical reactions Enzymes are sensitive to extremes of temperature and pH, as they are proteins Metabolism 3: Metabolic Pathways and ATP Production I Sketch a cartoon of the three stages of cellular metabolism that convert food to waste products in higher organisms, illustrating the cellular location of each stage Cellular metabolism: energy is liberated from food molecules to provide energy 3 food molecules are used by cells: 1. Polysaccharides simple sugars 2. Proteins amino acids 3. Fats fatty acids and glycerol 3 stages of metabolism: 1. Digestion: enzymes liberate small molecules in the small intestines 2. Cellular metabolism I: small molecules are oxidised to generate ATP and NADH in the cell cytosol 3. Cellular metabolism II: small molecules are oxidised to generate ATP within the mitochondria Glucose combustion: single-step reaction The relatively large activation energy is overcome by a heat source Free energy is released as heat Glucose metabolism: multi-step reaction The relatively small activation energies of each step are overcome by enzymes and body temperature Free energy liberated is invested into carrier molecules, such as ATP This reaction is ~50% efficient Outline the metabolism of glucose by the process of glycolysis, listing the key reactions, in particular those reactions that consume ATP and those that generate ATP Glycolysis: an anaerobic process which occurs in the cell cytoplasm 1. Glucose + ATP glucose 6-phosphate + ADP + H+ Hexokinase Glucose is committed to further reactions and it is trapped inside the cell (due to negative charge) 2. Glucose 6-phosphate fructose 6-phosphate Phosphoglucose isomerase 3. Fructose 6-phosphate + ATP fructose 1,6-bisphosphate + ADP Phosphofructokinase Fructose 1,6-bisphosphate is a highly symmetrical, high energy compound 4. Fructose 1,6-bisphosphate glyceraldehyde 3-phosphate + dihydroxyacetone phosphate Aldolase 5. Dihydroxyacetone phosphate glyceraldehyde 3-phosphate Triose phosphate isomerase 6. Glyceraldehyde 3-phosphate + NAD+ + Pi 1,3-bisphosphoglycerate + NADH Glyceraldehyde 3-phophate dehydrogenase 7. 1,3-bisphosphoglycerate + ADP 3-phosphoglycerate + ATP Phosphoglycerate kinase 8. 3-phosphoglycerate 2-phosphoglycerate Phosphoglycerate mutase 9. 2-phosphoglycerate phosphoenolpyruvate + H2O Enolase 10. Phosphoenolpyruvate + ADP pyruvate + ATP Pyruvate kinase Net output of glycolysis per glucose molecule: 2 pyruvate; 2 ATP; 2 NADH + H+ Substrate-level phosphorylation: ATP is produced by the direct transfer of a high-energy phosphate group from an intermediate substrate in a biochemical pathway to ADP, such as occurs in glycolysis Oxidative phosphorylation: ATP is produced using energy derived from the transfer of electrons in an electron transport system Distinguish between the aerobic and anaerobic metabolism of glucose with reference to the enzymes involved and the comparative efficiencies of each pathway with respect to ATP generation Anaerobic metabolism of glucose: Alcoholic fermentation: characteristic of yeasts 1. Pyruvate + H+ acetaldehyde + CO2 Pyruvate decarboxylase 2. Acetaldehyde + NADH + H+ ethanol + NAD+ Alcohol dehydrogenase Lactate production: characteristic of mammalian muscle during intense activity when oxygen is a limiting factor Pyruvate + NADH + H+ lactate + NAD+ Lactate dehydrogenase Anaerobic metabolism of glucose involves a dehydrogenase: this allows NAD+ to be regenerated to allow glycolysis to continue in conditions of oxygen deprivation NAD+: required for the dehydrogenation of glyceraldehyde 3-phosphate Oxygen deprivation: conditions in which the rate of NADH formation by glycolysis is greater than the rate of oxidation of NADH by the respiratory chain Aerobic metabolism of glucose: Acetyl CoA production: Pyruvate + HS-CoA + NAD+ acetyl CoA + CO2 + NADH Pyruvate dehydrogenase complex This series of reactions occur in the mitochondrial matrix This series of reactions proceeds through the pyruvate dehydrogenase complex Acetyl CoA is committed to enter the TCA cycle Aerobic metabolism of glucose yields considerably more ATP than anaerobic metabolism Anaerobic metabolism yields 2 ATPs; whereas complete aerobic metabolism can yield 38 ATPs Describe the reactions catalysed by lactate dehydrogenase and creatine kinase and explain the diagnostic relevance of their appearance in plasma Lactate dehydrogenase (LDH): catalyses the interconversion of pyruvate and lactate LDH is present in the heart, liver, kidneys, skeletal muscle, brain and lungs Elevated LDH levels can be used to diagnose several disorders including: Stroke Myocardial infarction Liver disease (e.g. hepatitis) Muscle injury Muscular dystrophy Pulmonary infarction Creatine kinase: Creatine kinase catalyses the following reaction: Creatine phosphate + ADP + H+ creatine + ATP ∆G = −43.1 kJ/mol (c.f. −31 kJ/mol for ATP) A large reservoir of creatine phosphate is present in muscle: it buffers the demand for phosphate (which is required for ATP synthesis) When a muscle is damaged, creatine kinase leaks into the bloodstream Either total creatine kinase levels (70% accurate) or the level of the tissue specific isoform (90% accurate) can be measured to determine which tissue has been damaged Elevated creatine kinase levels can be used to: Diagnose myocardial infarction Determine the extent of muscular disease Evaluate the cause of chest pain Help to identify carriers of Duchene muscular dystrophy Outline the oxidative decarboxylation reaction catalysed by pyruvate dehydrogenase, with reference to the product and the five co-enzymes required by this enzyme complex Pyruvate forms acetyl CoA via the pyruvate dehydrogenase complex The pyruvate dehydrogenase complex is gigantic: it consists of 3 enzymes and 5 cofactors (60 polypeptides) Enzyme Prosthetic group Reaction catalysed E1 Pyruvate decarboxylase TPP Oxidative decarboxylation of pyruvate E2 Lipoamide reductase-transacetylase Lipoamide Transfer of acetyl group to CoA E3 Dihydrolipoyl dehydrogenase FAD Regeneration of oxidised lipoamide Cofactors: thiamine pyrophosphate (TPP), lipoamide, FAD, CoA and NAD+ Prosthetic group: a cofactor which forms a permanent part of the complex with an enzyme Coenzyme: a cofactor which binds reversibly to its corresponding enzyme o CoA: a coenzyme for E2 o NAD+: a coenzyme for E3 Thiamine pyrophosphate (TPP): Derivative of thiamine (vitamin B1) It readily loses a proton: this forms a carbanion which attacks that of pyruvate to form hydroxyethylTPP Thiamine deficiency causes Beriberi Symptoms include: damage to the peripheral nervous system, muscle weakness and reduced cardiac output Lipoamide: The dithiol group undergoes redox reactions The long flexible arm allows the dithiol group to swing between active sites within the complex Arsenite (AsO33−) and mercury inhibit the pyruvate dehydrogenase complex since they have a high affinity for sulphydryl groups which are found in reduced lipoamide Flavine adenine dinucleotide (FAD): FAD + 2e- + 2H+ FADH2 Mechanism of the pyruvate dehydrogenase complex: 1. E1: decarboxylation of pyruvate Pyruvate + carbanion of TPP hydroxyethyl TPP + CO2 2. E1: oxidation of hydroxyethyl TPP Hydroxyethyl TPP + lipoamide acetyllipoamide + carbanion of TPP 3. E2: transfer of acetyl group from acetyllipoamide to CoA Acetyllipoamide + CoA acetyl CoA + dihydrolipoamide 4. E3: regeneration of oxidised lipoamide (dehydrogenation) Dihydrolipoamide + FAD lipoamide + FADH2 5. Regeneration of FAD: FADH2 + NAD+ FAD + NADH + H+ Overall reaction: pyruvate + HS-CoA + NAD+ acetyl CoA + CO2 + NADH Describe the different processes by which the fatty acid palmitate and the amino acid alanine are converted into acetyl-CoA Fatty acid metabolism: occurs in the mitochondria Acetyl CoA is produced from both polysaccharides and fats within the mitochondria: 1. Polysaccharides sugars glucose pyruvate acetyl CoA 2. Fats fatty acids acetyl CoA Fatty acids are the most compact fuel since they are fully reduced (i.e. the carbon skeleton is saturated with hydrogen) Fatty acid metabolism yields several times the useful energy compared with carbohydrates Mechanism of fatty acid metabolism: 1. The fatty acid is converted into an acyl CoA species: Fatty acid + ATP + HS-CoA acyl CoA + AMP + PPi Acyl CoA synthetase 2. The acyl CoA species undergoes ß-oxidation: Cn acyl CoA + NAD+ + FAD + H2O + CoA Cn−2 acyl CoA + acetyl CoA + NADH + FADH2 E.g. palmitoyl CoA + NAD+ + FAD + H2O + CoA myristyl CoA + acetyl CoA + NADH + FADH2 ß-oxidation: involves a sequence of dehydration, hydration, oxidation and thiolysis reactions Each ß-oxidation reaction removes 2 carbons from the acyl CoA and produces 1 acetyl CoA Metabolism of palmitic acid [C16]: involves 7 ß-oxidation reactions 1. Palmitic acid + ATP + HS-CoA palmitoyl CoA + AMP + PPi 2. Palmitoyl CoA + 7 NAD+ + 7 FAD + 7 H2O + 7 CoA 8 acetyl CoA + 7 NADH + 7 FADH2 Amino acid metabolism: occurs in the liver The pathway of amino acid metabolism depends upon the number of carbon atoms in the amino acid C3 amino acids pyruvate C4 amino acids oxaloacetate C5 amino acids α-ketoglutarate Transamination: alanine [C3] undergoes transamination Alanine + α-ketoglutarate pyruvate + glutamate Alanine aminotransferase Pyruvate can enter the TCA cycle Glutamate is converted to α-ketoglutarate by glutamate dehydrogenase This generates NH4+ ions which are ultimately converted to urea Persistently elevated levels of alanine aminotransferase are indicative of hepatic disorders (e.g. Hepatitis C) Metabolism 4: Metabolic Pathways and ATP Production II Outline the Krebs or TCA (tricarboxylic acid cycle) with particular reference to the steps involved in the oxidation of acetyl Co-A and the formation of NADH and FADH2 and the cellular location of these reactions Acetyl CoA: possesses a high-energy thioester bond (C-S) between the acetyl group and CoA The thioester bond is readily hydrolysed; therefore acetyl CoA can donate its acetyl group to other molecules (i.e. oxaloacetate in the Krebs cycle) Krebs cycle: an aerobic process which occurs in the mitochondrial matrix 1. Oxaloacetate + acetyl-CoA citrate + HS-CoA + H+ Citrate synthase 2. Citrate isocitrate Aconitase 3. Isocitrate + NAD+ α-ketoglutarate + NADH + H+ + CO2 Isocitrate dehydrogenase 4. α-ketoglutarate + NAD+ + HS-CoA succinyl CoA + NADH + H+ + CO2 α-ketoglutarate dehydrogenase complex 5. Succinyl CoA + GDP + Pi + H2O succinate + GTP* + HS-CoA Succinyl CoA synthetase 6. Succinate + FAD fumarate + FADH2 Succinate dehydrogenase 7. Fumarate + H2O malate Fumerase 8. Malate + NAD+ oxaloacetate + NADH + H+ Malate dehydrogenase Net output per turn of the cycle: 1 GTP, 3 NADH, 1 FADH2 and 2 CO2 *GTP (guanosine triphosphate): GTP can act as a phosphoryl donor in protein synthesis or in signal transduction processes (e.g. G protein-coupled receptors) The γ-phosphate group of GTP can be transferred to ADP to generate ATP: GTP + ADP GDP + ATP Nucleoside diphosphokinase Cellular location of the Krebs cycle: Krebs cycle enzymes: soluble proteins which are located in the mitochondrial matrix, except for… Succinate dehydrogenase: an integral membrane protein which is embedded in the inner mitochondrial membrane N.B. The Krebs cycle only occurs under aerobic conditions since reduced cofactors are required: these must be reoxidised via oxidative phosphorylation Reoxidation of reduced cofactors via oxidative phosphorylation generates ATP: Re-oxidation of NADH 3 ATP Re-oxidation of FADH2 2 ATP Therefore overall the Krebs cycle generates: 3 NADH + 1 FADH2 + 1 GTP = 12 ATP per acetyl CoA Calculate the theoretical maximum yield of ATP per glucose molecule oxidized by aerobic respiration and compare this to the theoretical maximum yield of ATP per molecule of palmitic acid Theoretical maximum yield of ATP… Per molecule of glucose oxidised by aerobic respiration: Glycolysis: Glucose 2 pyruvate + 2 ATP + 2 NADH Direct production of 2 ATP Oxidative phosphorylation: 2 NADH 6 ATP Total: 8 ATP Pyruvate dehydrogenase complex: 2 pyruvate + 2 NAD+ + CoA 2 acetyl CoA + 2 NADH + 2 CO2 Oxidative phosphorylation: 2 NADH 6 ATP Total: 6 ATP Krebs cycle: 2 acetyl CoA 6 NADH + 6 H+ + 2 FADH2 + 2 GTP Direct production of 2 GTP ≡ 2 ATP Oxidative phosphorylation: 6 NADH 18 ATP Oxidative phosphorylation: 2 FADH2 4 ATP Total: 24 ATP Grand total: 8 + 6 + 24 = 38 ATP Per molecule of palmitic acid: 1st step of ß-oxidation: conversion of fatty acid acyl CoA species Palmitic acid + ATP + HS-CoA palmitoyl CoA + AMP + PPi Total: − 2 ATP (2 high-energy phosphoanhydride bonds are used and this is the equivalent of 2 ATPs) Subsequent steps of ß-oxidation: Palmitoyl CoA + 7 NAD+ + 7 FAD + 7 H2O + 7 CoA 8 acetyl CoA + 7 NADH + 7 FADH2 7 NADH 21 ATP 7 FADH2 14 ATP 8 acetyl CoA 96 ATP Total: 131 ATP Grand total: 21 + 14 + 96 − 2 = 129 ATP Outline the glycerol phosphate shuttle and the malate-aspartate shuttle, in particular stating why these mechanisms are required NAD+ needs to be regenerated by oxidative phosphorylation in order for glycolysis to continue since NAD+ is required for dehydrogenation reactions NADH cannot diffuse directly from the cytosol into the mitochondrial matrix since the inner mitochondrial membrane is impermeable to NADH and NAD+ There are 2 mechanisms by which high-energy electrons can be transferred from cytosolic NADH across the mitochondrial membrane 1) Glycerol phosphate shuttle: found in skeletal muscle and in the brain In the cytosol: Dihydroxyacetone phosphate + NADH glycerol 3-phosphate + NAD+ Cytosolic glycerol 3-phosphate dehydrogenase (GPD) Cytosolic GPD transfers electrons from NADH to form glycerol 3-phosphate Glycerol 3-phosphate diffuses from the cytosol into the mitochondria In the mitochondria: Glycerol 3-phosphate + FAD dihydroxyacetone phosphate + FADH2 Mitochondrial glycerol 3-phosphate dehydrogenase (membrane-bound GPD) Mitochondrial GPD transfers electrons from glycerol 3-phosphate to FAD 2) Malate-aspartate shuttle: found in the heart, liver and kidneys The malate-aspartate shuttle is mediated by 2 membrane carriers and 4 enzymes Net reaction: cytosolic NADH + mitochondrial NAD+ cytosolic NAD+ + mitochondrial NADH In the cytosol: Oxaloacetate + NADH malate + NAD+ Cytosolic malate dehydrogenase (MDH) Cytosolic MDH transfers electrons from NADH to oxaloacetate to form malate Malate is transported into the mitochondrial matrix by an α-ketoglutarate transporter In the mitochondria: Malate + NAD+ oxaloacetate + NADH Mitochondrial malate dehydrogenase (MDH) Mitochondrial MDH catalyses reoxidation of malate by NAD+ to form oxaloacetate Understand the concept of transamination with reference to the malate-aspartate shuttle Oxaloacetate cannot readily cross the inner mitochondrial membrane: It accumulates in the mitochondrial matrix It is depleted from the cytosol A transamination reaction is necessary to convert oxaloacetate to aspartate which can be transported across the mitochondrial membrane by a glutamate/aspartate transporter Transamination: a reaction in which an amino group is transferred from an amino acid to a keto acid, thereby forming a new pair of amino and keto acids One of the pairs is almost always glutamate (an amino acid) and α-ketoglutarate (a keto acid) In the mitochondria: Oxaloacetate + glutamate aspartate + α-ketoglutarate Mitochondrial aspartate aminotransferase Transamination: glutamate transfers an amino group to oxaloacetate to form aspartate Aspartate is transported into the cytosol by a glutamate/aspartate transporter In the cytosol: the same transamination reaction occurs but in reverse Aspartate + α-ketoglutarate oxaloacetate + glutamate Cytosolic aspartate aminotransferase Explain in general terms the relationship between TCA intermediates and those pathways involved in amino acids synthesis and breakdown Amino acid breakdown: the amino group is removed and the carbon skeleton is used to produce Krebs cycle intermediates or glucose Degradation of all 20 amino acids produces 7 Krebs cycle intermediates: pyruvate, acetyl CoA, acetoacetyl CoA, α-ketoglutarate, succinyl CoA, fumarate and oxaloacetate Give two examples of the use of NADPH in reductive biosynthesis Biosynthesis: the synthesis of macromolecules from smaller, simple molecules Glycolysis and the Krebs cycle provide intermediates for many biosynthetic reactions Krebs cycle intermediates which are drawn off for biosynthesis must be replenished E.g. if oxaloacetate is removed, acetyl CoA cannot enter the Krebs cycle; therefore glycolysis stops Anaplerotic reactions: enzyme-catalysed reactions which regenerate Krebs cycle intermediates Pyruvate + CO2 + ATP + H2O oxaloacetate + ADP + Pi + 2 H+ Pyruvate carboxylase NADP+: nicotinamide adenine dinucleotide phosphate Similar structure to NAD+: it differs by a single phosphate group attached to one of the ribose rings The additional phosphate group gives NADP+ a different conformation to NAD+; therefore NADP+ binds to different enzymes than NAD+ NADP+ can accept 2 electrons and 1 proton (i.e. a hydride ion) like NAD+: NADP+ + H+ + 2e− NADPH The hydride ion is held by a high-energy linkage; therefore it can be easily transferred NADPH participates in anabolic reactions; whereas NADH participates in catabolic reactions This allows electron transport in catabolism to be kept separate from that of anabolism NADPH is a cofactor for reduction in: o Biosynthesis of RNA o Biosynthesis of cholesterol: NADPH catalyses the final reaction of cholesterol synthesis The C=C bond in 7-dehydrocholesterol is reduced by the transfer of a hydride ion from NADPH Metabolism 5: Mitochondria and Oxidative Phosphorylation Outline the proposed evolutionary origins of mitochondria Endosymbiotic theory: mitochondria are believed to be the evolutionary descendents of a prokaryote which formed a symbiotic relationship with the ancestors of eukaryotic cells Following this many genes needed for mitochondrial function were translocated to the eukaryotic genome Evidence for the theory: Mitochondria can only arise from pre-existing mitochondria Mitochondria have their own genome: a single circular DNA molecule with no associated histones This resembles the genome of prokaryotes The mitochondrial genome is similar to that of Rickettsia prowazekii Mitochondria have their own protein-synthesising machinery: 70S ribosomes This resembles the protein-synthesising machinery of prokaryotes The first amino acid of the transcript is fMet (as with prokaryotes), not Met (as with eukaryotes) Antibiotics (e.g. streptomycin) block protein synthesis in both prokaryotes and mitochondria, and do not interfere with protein synthesis in the cytoplasm of eukaryotes Draw a cross sectional representation of a mitochondrion, and label its component parts Mitochondrial structure: a rod-shaped organelle with 2 membranes Outer membrane: limits the size of the mitochondrion Intermembrane space: the space between the inner and outer mitochondrial membranes Inner membrane: has many cristae (infoldings that project into the mitochondrial matrix) Cristae: invaginations of the inner membrane which increase the surface area upon which oxidative phosphorylation can occur and enhance ATP production Matrix: the space enclosed by the inner membrane; it contains the Krebs cycle enzymes In some cells mitochondria are mainly distributed where ATP is rapidly consumed E.g. between the myofibrils of muscle cells; wrapped around the flagellum of sperm Outline the chemiosmotic theory Oxidative phosphorylation: ATP synthesis is driven by the transfer of high-energy electrons from reduced coenzyme to molecular O2 via the ETC Within mitochondria reduced coenzyme is reoxidised by molecular oxygen: NADH + H+ + ½ O2 NAD+ + H2O ∆G = −220 kJ/mol FADH2 + ½ O2 FAD + H2O ∆G = −167 kJ/mol Energy released by the reoxidation of reduced coenzyme can be used to generate several phosphoanhydride bonds (∆G ATP hydrolysis = −31 kJ/mol) Some of the energy released by the reoxidation of reduced coenzyme is recovered by components of the ETC and used to synthesise ATP Chemiosmotic hypothesis of oxidative phosphorylation: 1. Protons are translocated from the mitochondrial matrix into the intermembrane space The ETC provides the energy for active transport of protons 2. Protons diffuse back into the mitochondrial matrix through a specific proton channel which is coupled to ATP synthase: flow of protons back into the matrix is coupled to ATP synthesis The proton motive force which drives protons back into the mitochondrial matrix consists of a pH gradient and a transmembrane electric potential Describe the electron transport chain in mitochondria with reference to the functions of coenzyme Q (ubiquinone) and cytochrome c Electron transport chain (ETC): a chain of 3 membrane complexes and 2 mobile carriers Membrane complexes: integral membrane proteins 1. NADH dehydrogenase complex 2. Cytochrome b-c1 complex 3. Cytochrome oxidase complex Mobile carriers: fixed in the membrane 1. Ubiquinone (coenzyme Q) 2. Cytochrome c Ubiquinone: a mobile electron carrier Ubiquinone can accept either 1 or 2 electrons, together with protons (from solution) It passes electrons to cytochrome b-c1 complex It has a hydrophobic tail: this confines it to the lipid bilayer of the membrane Succinate dehydrogenase: an integral membrane protein Succinate dehydrogenase is embedded in the inner surface of the inner mitochondrial membrane: this allows it to pass its electrons directly to ubiquinone via FAD Ubiquinone is the entry point for electrons donated by FADH2 Therefore fewer protons are pumped into the intermembrane space compared with NADH and less ATP is produced Cytochrome oxidase: involved in the final step of electron transfer Cytochrome oxidase receives 4 electrons and 4 protons from cytochrome c 4e− + 4H+ + O2 2H2O It pumps 4 additional protons into the intermembrane space: this enhances the proton gradient Molecular O2 is an ideal terminal electron acceptor since it has a high affinity for electrons: this provides a driving force for oxidative phosphorylation Proteins of the ETC accept electrons and protons from the aqueous solution Each unit of the ETC has a higher affinity for electrons than the previous unit Electron transfer along the ETC is energetically favourable: as electrons progress along the chain, they lose energy As electrons pass through each membrane complex, protons are pumped into the intermembrane space Redox reactions: Redox reaction: an electron transfer reaction involving a reduced substrate and an oxidised substrate Redox couple: a substrate that can exist in both oxidised and reduced forms E.g. NAD+/NADH + H+ and FAD/FADH2 Redox potential: the ability of a redox couple to either accept or donate electrons Standard redox potential (E’o): determined experimentally by allowing a reaction to reach equilibrium and measuring the resultant e.m.f. E’o conditions for biological systems: 25°C; pH 7.0; reactant concentrations of 1.0M Standard hydrogen electrode: used as a reference electrode Describe how ATP synthase is able to generate and utilise ATP respectively, with reference to its structure ATP synthase: a multimeric enzyme complex that catalyses the synthesis of ATP from ADP and Pi Structure of ATP synthase: 2 parts: F0 and F1 F0 unit: membrane-bound F1 unit: projects into the matrix Structure of the F0 unit: a, b and c subunits Structure of the F1 unit: α, ß and γ subunits The c subunits are arranged into a disc which is fixed to the γ subunit The α and ß subunits cannot rotate since they are locked in a fixed position by the b subunit The b subunit is anchored to the a subunit in the membrane Mechanism of ATP synthesis: 1. Protons flow across the inner mitochondrial membrane via a pore 2. This causes the disc of c subunits to rotate 3. This in turn causes the attached γ subunit to rotate 4. The catalytic portions of the ß subunits undergo conformational changes: this changes their affinities for ADP and ATP 5. Torsional energy flows from the catalytic portion to the bound ADP and Pi: this forms ATP Binding change mechanism: the active site of a ß subunit cycles between 3 states Open state: ADP and Pi bind to the active site Loose binding state: the enzyme closes up around the molecules and binds them loosely Tight binding state: the enzyme changes shape and forces the molecules together to form ATP ATP synthesis is a reversible reaction: ADP + Pi ATP Position of equilibrium depends on the direction of proton flow through ATP synthase: Protons flowing into the matrix ATP synthesis Protons flowing into the intermembrane space ATP hydrolysis Explain why carbon monoxide, cyanide, malonate and oligomycin are poisonous in terms of their effects on specific components of the electron transport chain If oxidative phosphorylation is disrupted, cells become rapidly depleted of ATP and may die Lack of oxygen causes failure of oxidative phosphorylation: hypoxia (diminished oxygen) and anoxia (complete lack of oxygen) Drug/Toxin Effect on the ETC Cyanide (CN−) Bind to the ferric (Fe3+) form of the haem group in cytochrome oxidase with high affinity Azide (N3−) Cyanide is supertoxic: i.e. ingestion of a few drops can be lethal Carbon monoxide Binds to the ferrous (Fe2+) form of the haem group in cytochrome oxidase with high affinity Malonate Acts as a competitive inhibitor of succinate dehydrogenase: it competes with succinate Oligomycin Inhibits oxidative phosphorylation: it binds to the stalk of ATP synthase and blocking the flow of protons through it Dinitrophenol Causes metabolic uncoupling: it transports protons across the mitochondrial membrane Describe how oxidative phosphorylation can be measured experimentally Oxygen electrode: measures the oxygen concentration of a solution 2 electrodes: platinum cathode and silver anode The reaction chamber is isolated from the electrode compartment by a Teflon membrane Teflon membrane: permeable to oxygen A small voltage is applied between the electrodes (0.6 V) Oxygen is reduced at the platinum cathode: O2 + 4H+ + 4e− 2H2O The circuit is completed at the silver anode, which is slowly corroded by the KCl electrolyte: Ag + Cl− AgCl + e− The resulting current is proportional to the oxygen concentration in the reaction chamber The oxygen electrode can be used to measure oxidative phosphorylation: Incubate a suspension of mitochondria from homogenised tissue with an isotonic medium containing substrate (e.g. succinate and Pi) Add ADP: this causes a sudden burst of oxygen uptake since ADP is converted into ATP Understand the difference between oxidative phosphorylation and substrate level phosphorylation Substrate level phosphorylation: the synthesis of ATP by transfer of a high-energy Pi from an intermediate substrate in a biochemical pathway to ADP Oxidative phosphorylation: the synthesis of ATP from ADP and Pi using energy from the transfer of highenergy electrons from reduced coenzyme to O2 via the ETC Metabolism 6: Lipids and Membranes Describe the structure of: fatty acids, triglycerides, phospholipids, cholesterol and sphingomyelin Fatty acids: the simplest lipids Structure: hydrocarbon chain + carboxyl group Fatty acids are amphipathic: i.e. they possess both hydrophobic and hydrophilic regions Hydrophilic head: the carboxyl group (undergoes esterification reactions) Hydrophobic tail: the hydrocarbon chain (may be either saturated or unsaturated) Function: fatty acids are constituents of more complex lipids (phospholipids and sphingolipids) Triacylglycerols (triglycerides): Structure: 3 fatty acid chains + glycerol linked by ester bonds Ester bonds: the carboxyl group on each fatty acid is esterified to a hydroxyl group on glycerol Function: triacylglycerols are metabolic energy stores Membrane structure: 2 membrane types: 1. Plasma membranes: define the outer limit of the cell 2. Intracellular (organelle) membranes: separate and limit the size of organelles Fluid mosaic model of the membrane: Membranes are 2D solutions of oriented proteins and lipids which are held non-covalently The membrane is a continuous bilayer ~5 nm thick Proteins are either (a) integral or (b) peripheral o Integral proteins: span the entire bilayer o Peripheral proteins: present on either side of the membrane Lipid rafts: Membranes are not homogeneous and are dynamic Lipid rafts are specialised domains of the plasma membrane which are enriched in certain lipids (glycolipids and cholesterol) and proteins Structure of the lipid bilayer: The lipid bilayer is composed of phospholipids, sphingolipids, glycolipids and steroids Phospholipids: Structure: glycerol backbone + tail (2 fatty acid chains) + head group (contains phosphate) Fatty acid chain variables: (a) length and (b) saturation Common phospholipids include: Phosphatidylcholine (PC): choline head group Phosphatidylserine (PS): serine head group Phosphatidylethanolamine (PE): ethanolamine head group Phospholipids in aqueous solution shield the hydrophobic core by: Bilayer formation: in the case of amphipathic lipids (e.g. phospholipids and glycolipids) Micelle formation: in the case of non-amphipathic lipids (e.g. triglycerides) Sphingolipids and glycosphingolipids: Derived from sphingosine (an amino alcohol with a long, unsaturated hydrocarbon chain) Sphingolipid structure: a long hydrocarbon chain is linked to the amino group on sphingosine by an amide bond Sphingosine + fatty acid ceramide Ceramide + sugar glycosphingolipid Glycosphingolipids include cerebrosides and gangliosides Function: structural role; recognition of molecules Sphingomyelin: phosphocholine + ceramide Sphingomyelin is a common membrane lipid Give examples of how the lipid composition can differ for different cellular membranes, and indicate the significance of this Membrane flexibility: the ability of the membrane to bend Membrane fluidity: the ability of lipid molecules to move in the plane of the bilayer (the monolayer) Individual lipid molecules very rarely move from one monolayer to the other (flip-flop) Factors affecting fluidity: Length of the hydrocarbon tail: o Longer carbon chains have a greater surface area over which dispersion forces can act o Therefore long hydrocarbon chains have a greater tendency to aggregate lower fluidity Degree of saturation of the hydrocarbon tail: o Unsaturated fatty acids have kinked chains and the dispersion forces between the hydrocarbon chains are less stable o Therefore hydrocarbon chains with a greater degree of unsaturation have a lower tendency to aggregate greater fluidity Cholesterol: decreases membrane fluidity (i.e. increases membrane rigidity) Asymmetry of membrane lipids: Within cell membranes: lipid composition of cell membranes varies according to the cell and they type of cell membrane (i.e. plasma membrane or intracellular membrane) E.g. the plasma membrane of human erythrocytes has a much higher percentage of glycolipids, phosphatidylcholine and cholesterol compared to the mitochondrial membrane of beef heart Within bilayers: Phospholipids and glycolipids are unevenly distributed in the extracellular and cytosolic monolayers o Extracellular monolayer: composed of glycolipids, sphingomyelin and phosphatidylcholine o Cytosolic monolayer: composed of phosphatidylserine, phosphatidylethanolamine and phosphatidylinositol Certain lipids are organised into microdomains within monolayers of the plasma membrane Lipid rafts: microdomains which are enriched in sphingomyelin, glycosphingolipid, phospholipid, cholesterol, cell-surface receptor proteins and signalling proteins Lipid rafts act as signalling sites for signalling across the plasma membrane Outline the pathway for synthesis of fatty acids Fat digestion, absorption and storage: 1. Bile salts emulsify fats 2. Hydrolytic enzymes (secreted by the pancreas) hydrolyse fats in the small intestine 3. Triacylglycerols are transported from the small intestine to the liver in the bloodstream via chylomicrons 4. Triacylglycerols are transported from the liver to muscles and the heart in the bloodstream via VLDLs 5. Triacylglycerols are stored in adipose tissue Fatty acid synthesis (aka lipogenesis): Acetyl CoA: the key intermediate between fat and carbohydrate metabolism Acetyl CoA + oxaloacetate citrate Acetyl CoA fatty acids If there is too much acetyl CoA: it is stored as fatty acids If there is too little acetyl CoA: sources of acetyl CoA are mobilised o Carbohydrates are short-term stores of acetyl CoA o Triacylglycerols are long-term stores of acetyl CoA Fatty acid biosynthesis: acetyl CoA palmitate [C16] 1. Production of malonyl CoA: acetyl CoA [C2] malonyl CoA [C3] Enzyme: acetyl CoA carboxylase Cofactor: biotin 2. Activation of acetyl CoA/malonyl CoA by acyl carrier protein (ACP): acyl CoA acyl ACP Cofactor: phosphopantetheine 3. Elongation of acetyl ACP/malonyl ACP by successive addition of 2 carbons Enzyme: fatty acid synthase (FA synthase) This stage is very similar to fatty acid degradation by ß-oxidation: each cycle adds 2 carbons 2 enzymes are involved in fatty acid synthesis: 1. Acetyl CoA carboxylase: this catalyses the first step 2. FA synthase: this contains multiple activities which catalyse the elongation reactions Overall reaction: Acetyl CoA + 7 malonyl CoA + 14 NADPH + 14 H+ palmitate + 7 CO2 + 6 H2O + 8 HS-CoA + 14 NADP+ Further metabolism of palmitate: Esterification: this forms triacylglycerols Formation of other fatty acids by: o Desaturation: this forms unsaturated fatty acids, including monounsaturated fatty acids o Elongation: this forms longer chain fatty acids Regulation of fatty acid synthesis: Feedback inhibition of palmitoyl CoA to: o Acetyl CoA carboxylase o FA synthase o Pentose phosphate pathway: this is a source of NADPH Glucose 6-phosphate pentose phosphate + NADPH Hormonal regulation of acetyl CoA carboxylase Transcriptional regulation of acetyl CoA carboxylase and FA synthase: o Activated by insulin (and citrate) o Inhibited by glucagon (and adrenaline and palmitoyl CoA) Distinguish between the pathways for synthesis and metabolism of fatty acids in terms of: substrates and products, coenzymes used, carrier molecules, cellular location Cellular location Substrates Products Fatty acid synthesis Fatty acid degradation Cytosol Mitochondrial matrix Acetyl CoA + malonyl CoA + NADPH + H+ Fatty acid + CO2 + H2O + HS-CoA + NADP+ 1. Fatty acid + ATP + HS-CoA 2. Acyl CoA + NAD+ + FAD + H2O + CoA 1. Acyl CoA + AMP + PPi 2. Acetyl CoA + NADH + FADH2 Carrier molecules ACP HS-CoA Redox cofactor NADPH (reducing agent) NAD+ and FAD (oxidising agents) Metabolism 7: Cholesterol Explain the physiological functions of cholesterol in membrane stability Structure of cholesterol: Derivative of a saturated tetracyclic hydrocarbon Planar, rigid structure: all cyclohexane rings are in the chair conformation The storage form of cholesterol is acylated at position 3 (i.e. where the–OH group is usually present) Physiological functions of cholesterol: Regulates membrane fluidity: high cholesterol leads to reduced membrane fluidity o It reduces phase transitions of lipids o It reduces the lateral mobility of polar lipids Precursor of steroid hormones: cortisol (a glucocorticoid), aldosterone (a mineralocorticoid), progesterone (a progestin), oestradiol (an oestrogen) and testosterone (an androgen) Precursor of bile salts Signalling molecule: cholesterol is involved in lipid raft formation at the plasma membrane Outline the synthesis of cholesterol from acetate Cholesterol biosynthesis: occurs in the liver 1. Acetyl CoA [C2] + acetoacetyl CoA [C4] HMG CoA [C6] 2. HMG CoA [C6] + 2NADPH + 2H+ mevalonate [C6] + 2NADP+ + HS-CoA HMG CoA reductase (3-hydroxy-3-methylglutaryl CoA reductase) This is the regulated step in cholesterol synthesis: it is inhibited by statins, mevalonate, bile acid and cholesterol 3. Isoprenoid metabolism (head-to-tail condensations of isoprene units): mevalonate squalene a. Mevalonate [C6] isoprene units [C5] b. C5 + C5 C10 c. C10 + C5 C15 (farnesyl pyrophosphate) There is a branched pathway at farnesyl pyrophosphate d. C15 + C15 C30 (squalene) 4. Cyclisation reaction: squalene cholesterol a. Squalene [C30] lanosterol [C30] b. Lanosterol [C30] cholesterol [C27] Outline the synthesis of bile acids and steroid hormones from cholesterol Synthesis of steroid hormones: Site of synthesis: gonads and adrenal glands 5 classes: progestins, glucocorticoids, mineralocorticoids, androgens and oestrogens Pregnenolone is the precursor of all steroid hormones: Cholesterol [C27] pregnenolone [C21] Cholesterol desmolase: catalyses side-chain cleavage of cholesterol Regulation: the level of steroid is controlled by their rate of synthesis Synthesis of bile acids: Function of bile salts: facilitate digestion via emulsification of dietary fats Site of synthesis: liver Enterohepatic circulation: circulation of bile between the liver (site of synthesis) and the small intestine (site of action) Regulated step of bile acid synthesis: Cholesterol 7-α-hydroxycholesterol Cholesterol 7-α-hydroxylase: this is a cytochrome P450 monooxygenase Describe the mechanism of transport of cholesterol around the body and its uptake into cells Fat digestion in the GI tract: 1. Large fat globules are emulsified by bile salts in the duodenum 2. Lipase (a pancreatic enzyme) digests fat fatty acids + monoglycerides 3. Fatty acids and monoglycerides form micelles, along with bile salts 4. Fatty acids and monoglycerides leave the micelles and diffuse into epithelial cells 5. Chylomicrons containing fatty substances are transported out of epithelial cells into lacteals 6. Fatty substances are carried away from lacteals by lymph Lipoproteins and cholesterol transport: Cholesterol is insoluble in blood; it is transported in the circulatory system within lipoproteins Structure of a plasma lipoprotein: a core of cholesterol esters and/or triacylglycerols surrounded by a shell of cholesterol, phospholipids and proteins (apoproteins) Apoproteins: (i) emulsify lipids and (ii) contain cell-targeting signals (determine which cells cholesterol is transported to) Lipoproteins (in order of increasing density/decreasing size): chylomicrons; VLDL; LDL; IDL; HDL Lipoproteins can be separated either by ultracentrifugation or by electrophoresis Chylomicrons, VLDL and LDL: transport cholesterol to peripheral tissues Mechanism of cholesterol transport: 1. Chylomicrons transport triacylglycerols and cholesterol from the intestine to the liver Triacylglycerols (in chylomicrons) are hydrolysed by lipoprotein lipase in the capillaries 2. Chylomicron remnants and cholesterol are taken up by the liver, where they are repackaged as VLDLs 3. VLDLs transport excess triacylglycerols and cholesterol from the liver in the bloodstream Triacylglycerols (in VLDLs) are hydrolysed by lipoprotein lipase in the capillaries: VLDLs IDLs 4. IDLs are either converted into LDLs by lipoprotein lipase or they are taken up by the liver 5. LDLs transport cholesterol to peripheral tissues or they are taken up by the liver 6. HDLs transport cholesterol from peripheral tissues to the liver or to endocrine glands for steroid hormone synthesis Draw a diagram of low density lipoprotein (LDL) LDL structure: Core: cholesterol in the form of cholesteryl esters Shell: unesterified cholesterol, phospholipids and a single apolipoprotein B-100 molecule Apolipoprotein B-100 (Apo B-100): Maintains the solubility of LDLs in the aqueous environment of the blood Acts as a recognition site and binds with the receptor of target cells Explain why disturbances in cholesterol homeostasis cause disease Cholesterol homeostasis: Dietary cholesterol reduces the activity of HMG CoA reductase Hepatocytes (and enterocytes): synthesise cholesterol de novo All cells except those of the liver and intestine obtain cholesterol from the plasma LDLs are the primary source of cholesterol Receptor-mediated endocytosis: LDL uptake o LDLs bind to LDL receptors on the surface of cells and are taken up by endocytosis o LDL cholesterol is removed from the blood o The mevalonate pathway (and hence cholesterol biosynthesis) is suppressed When cholesterol is abundant inside the cell, new LDL receptors are not synthesised; therefore uptake of additional cholesterol from plasma LDLs is blocked Cholesterol homeostasis dysfunction: LDLs transport cholesterol via the arteries LDLs may be retained in the arteries and form atherosclerotic plaques Increased LDL cholesterol levels are associated with: atherosclerosis and cardiovascular disease Hypercholesterolemia: Caused by high serum LDL cholesterol levels (high LDL to HDL ratio) Hypercholesterolemia induces the formation of atherosclerotic plaques in arteries (as above) Familial hypercholesterolemia: hereditary form of hypercholesterolemia Cause: there is an absence/deficiency of functional LDL receptors due to a mutation in the LDL receptor gene Cholesterol is deposited in various tissues due to the high serum level of LDL cholesterol Serum cholesterol level in homozygotes ~ 680 mg/dl Serum cholesterol level in heterozygote ~ 300 mg/dl Desirable serum cholesterol level < 200 mg/dl Give an example of how a selective enzyme inhibitor can be used as a pharmacological agent controlling cholesterol metabolism HMG CoA reductase inhibitors (statins): inhibit the de novo synthesis of cholesterol in hepatocytes HMG CoA reductase catalyses the formation of mevalonate: this is the committed step in cholesterol synthesis Mode of action: statins act as competitive inhibitors of HMG CoA reductase; therefore cholesterol synthesis in hepatocytes is reduced The decrease in cholesterol synthesis is detected and LDL receptor production is increased Therefore more LDL cholesterol is taken up into hepatocytes by endocytosis Bile acid sequestrants (e.g. cholestyramine): inhibit the intestinal reabsorption of bile salts Bile salts promote the absorption of dietary cholesterol and dietary fats Mode of action: bile acid sequestrants bind negatively charged bile salts and prevent their reabsorption Control of hypercholesterolaemia: Dietary restriction of fats and cholesterol Increased secretion of bile acid sequestrants HMG CoA reductase inhibitors inhibit cholesterol synthesis Metabolism 8: Membrane Trafficking Explain the terms "endocytosis" and "exocytosis" Endocytosis: uptake of extracellular material into a cell via invagination of the plasma membrane to form vesicles Phagocytosis: uptake of solid particles A large internal phagosome is formed around the extracellular material by the plasma membrane Pinocytosis: uptake of extracellular fluid and dissolved solutes Small vesicles are formed around the extracellular material by the plasma membrane Receptor-mediated endocytosis: molecule-specific endocytosis A specific receptor on the plasma membrane binds to an extracellular molecule which it recognises Exocytosis: discharge of intracellular material from a cell via fusion of secretory vesicles with the plasma membrane Describe the pathway and cellular locations for synthesis, post-translational modification and exocytosis of a secreted protein 3 types of intracellular transport: 1. Gated transport: e.g. nuclear import 2. Transmembrane transport: e.g. import of newly synthesised proteins into the ER 3. Vesicular transport: e.g. transport between organelles Protein targeting: newly synthesised proteins are delivered to the appropriate cellular destinations Following protein synthesis proteins may be directed to: A cell membrane: either the plasma membrane or an intracellular (organelle) membrane The aqueous interior of an organelle The cell exterior (via secretion) Secretory pathway: a sequence in which proteins are moved out of a cell Ribosomes ER Golgi apparatus secretory vesicles plasma membrane cell exterior Protein synthesis: occurs on the ribosomes in the cytosol A common pool of ribosomes in the cytosol synthesises all proteins: i.e. both proteins which remain in the cytosol and those which are transported into the ER The ER signal peptide directs an engaged ribosome to the ER membrane Signal peptide: the amino acid sequence within a protein which directs its post-translational transport Polyribosome: the mRNA molecule may remain permanently bound to the ER membrane Ribosomes move along the mRNA and are recycled At the end of protein synthesis, ribosomes are released from mRNA and rejoin the common pool of ribosomes in the cytosol Translocation of secretory proteins across the ER membrane: 1. The ER signal peptide at the N-terminus of a protein directs it to the ER membrane 2. The protein is guided to the ribosome by a signal recognition particle (SRP) which binds to the signal peptide 3. The SRP in turn binds to an SRP receptor on the ER membrane: this targets the protein to the ER 4. Cotranslational translocation: the protein is fed into the ER lumen via a translocation channel while it is still being synthesised on the ribosome 5. Post-translational translocation: the protein is fed into the ER lumen after being fully synthesised by the ribosome 6. Following translocation of the protein into the ER lumen, a signal peptidase cleaves the ER signal peptide from the protein Post-translational modification and quality control: Post-translational modification: occurs in the ER, Golgi and secretory vesicles Glycosylation: addition of carbohydrate chains to specific amino acids (ER) and processing (Golgi) Formation of disulphide bonds (ER): occurs between cysteine residues Folding of proteins (ER): facilitated by chaperones and ER proteins Assembly of multimeric proteins (ER): facilitated by chaperones and ER proteins Specific proteolytic cleavages (ER, Golgi and secretory vesicles) Quality control: unfolded or misfolded proteins are retained in the ER and transported back into the cytosol for degradation Exocytosis: Soluble and membrane proteins move to their final destinations via the secretory pathway This is mediated by transport vesicles which transport cargo proteins between organelles The transport vesicles bud from one organelle and fuse with another organelle Mechanism of the secretory pathway: 1. Secretory proteins are packaged into anterograde transport vesicles by budding from the ER 2. Anterograde ER transport vesicles fuse together to form new cis-Golgi cisternae 3. ER-resident proteins are retrieved from the cis-Golgi via retrograde transport vesicles 4. Cisternal progression: cis-Golgi cisternae successively move from the cis to the trans face of the Golgi complex Cis-Golgi cisternae medial-Golgi cisternae trans-Golgi cisternae 5. Golgi-resident proteins are retrieved from later Golgi cisternae (i.e. trans-Golgi cisternae) via retrograde transport vesicles 6. Vesicles eventually reach the trans-Golgi network: this is a major branchpoint in the secretory pathway and results in… a. Constitutive secretion b. Regulated secretion c. Lysosome formation: soluble proteins within vesicles bud from the trans-Golgi network and move to the late endosome and finally to the lysosome Distinguish "constitutive" and "regulated" secretion Constitutive secretion: soluble proteins are continuously secreted from the cell No external signals are required for secretion Secretion is not affected by environmental factors The Golgi apparatus is scattered throughout the cytoplasm E.g. fibroblasts (secrete collagen) and activated B lymphocytes (secrete antibodies) Regulated secretion: soluble proteins are stored inside the cell A specific signal is necessary for secretion (neural or hormonal stimulation) Cells are usually apical or polarised The Golgi apparatus is between the nucleus and the secretory surface E.g. goblet cells (secrete mucus) and beta cells of the pancreas (secrete insulin) Describe the process of receptor-mediated endocytosis and the roles played by endocytic vesicles, early endosomes, late endosomes, and lysosomes Receptor-mediated endocytosis: the process whereby cells internalise extracellular material LDL is taken up into cells via receptor-mediated endocytosis: LDL receptors are localised in clathrincoated pits Mechanism of endocytosis for internalising LDL: LDL receptors on the cell surface bind to Apo B-100 on the surface of LDLs Clathrin-coated pits containing LDL receptor-LDL complexes bud off from the plasma membrane enclosed in endocytic vesicles (early endosomes) The vesicle coat is shed and the uncoated early endosome fuses with a late endosome The late endosome fuses with a lysosome: the constituent lipids and proteins of LDL are hydrolysed by lysosomal enzymes; cholesteryl esters are hydrolysed by a lysosomal lipase LDL receptors are recycled to the cell surface Give a general description of the molecular mechanisms of vesicular transport within cells Give examples of diseases resulting from defects in the secretory and endocytic pathways I-cell disease (inclusion cell disease): lysosomal storage defect Mannose is phosphorylated to mannose 6-phosphate (M6P) by a phosphotransferase at the cis-Golgi network The M6P residue acts as a lysosomal sorting signal at the trans-Golgi network I-cell disease: there is a mutation in the phosphotransferase which catalyses the formation of M6P; therefore affected individuals lack the M6P sorting signal Consequences: o The lysosomal enzymes are secreted rather than being sorted into lysosomes o Undigested glycolipids and extracellular components accumulate in lysosomes (they would usually be degraded by lysosomal enzymes) Familial hypercholesterolemia: endocytic pathway defect The affected individual produces a mutant form of the LDL receptor This causes impaired endocytosis of LDL which results in high serum LDL cholesterol levels Metabolism 9: Integration of Metabolism Outline general features of metabolic activity in liver, brain, muscle, adipose tissue Metabolism: the sum of all processes in the body Methods of measuring metabolism: O2 uptake CO2 release Heat production Metabolic features of tissues: % Body mass % Resting MR (metabolic rate) Liver 2.5% Muscle Heart Tissue Metabolites Other features 20% Carbohydrates, fats and amino acids Glycogen store 40% Variable Carbohydrates and fats 1% 10% Carbohydrates and fats Brain and nervous 2% 20% Glucose (and ketone bodies during starvation) Adipose tissue 15% Low n/a Major source of blood glucose May have periods of very high ATP requirement during vigorous muscle activity Cannot metabolise fats Continuously high ATP requirement Long term store of fats Further metabolic features of the liver: The liver plays a central role in the coordination of metabolism throughout the body: It receives nutrients absorbed at the intestines immediately (via the hepatic portal vein) Performs a wide repertoire of metabolic processes including: o Glycolysis o Glucose production (gluconeogenesis) o Glucose storage High metabolic activity: 2.5% of body mass vs. 20% of resting metabolic rate It can interconvert nutrient types Plays a central role in the maintenance of blood glucose at 4.0-5.5 mM It performs lipoprotein metabolism (transport of triglycerides and cholesterol) Gluconeogenesis: the process whereby glucose or glycogen is made from oxaloacetate Primarily occurs in the liver (and also in the kidneys to a small extent) Requires ATP hydrolysis: 6 ATPs are used Certain enzymes bypass the irreversible steps of glycolysis Gluconeogenesis and glycolysis are reciprocally regulated (i.e. they inhibit each other) Gluconeogenesis pathway: (only the enzymes for the bypass reactions are shown) 1. Pyruvate oxaloacetate Pyruvate carboxylase (in the mitochondria) 2. Oxaloacetate phosphoenolpyruvate Phosphoenolpyruvate carboxykinase (in the cytosol) 3. (Phosphoenolpyruvate … fructose 1,6-bisphosphate) 4. Fructose 1,6-bisphosphate fructose 6-phosphate Fructose 1,6-bisphosphatase 5. (Fructose 6-phosphate glucose 6-phosphate) 6. Glucose 6-phosphate glucose Glucose 6-phosphatase: specifically found in the liver Muscle Liver Enzymes Hexokinase I Hexokinase IV and glucose 6-phosphatase Reaction Glucose glucose 6-phosphate Glucose glucose 6-phosphate High glucose affinity: the rate is half-maximal at 0.1 mM Low glucose affinity: the rate is half-maximal at 4 mM Highly sensitive to G6P inhibition Less sensitive to G6P inhibition Saturated at low [glucose] Small ∆[glucose] big ∆rate Therefore it is poor for glucose regulation Therefore it is good for glucose regulation Hexokinase Metabolism of ethanol: CH3CH2OH CH3CHO CH3COOH CH3CO-CoA Ethanol metabolism is unregulated; therefore this leads to perturbation of normal metabolism Ethanol + NAD+ acetaldehyde + NADH + H+ Alcohol dehydrogenase Medicinal drugs metabolised by a similar pathway may cause liver damage in alcoholics Unregulated ethanol metabolism leads to the accumulation of NADH Glycolysis is suppressed due to a lack of NAD+ TCA intermediates are reduced to sustain ethanol metabolism: Lactate/malate + NAD+ pyruvate/oxaloacetate + NADH + H+ Lactate accumulates as it cannot be oxidised to pyruvate due to a lack of NAD+ Regulation of glucose metabolism: Glucose metabolism is regulated at a step which is: Strongly dependent on enzyme activity Unique to the pathway Early in the pathway Regulatory mechanisms include: End-product inhibition: i.e. the products of the reaction pathway regulate glucose metabolism Signalling molecules which relay information from other pathways or cells Give four examples of blood-borne hormones which act as metabolic regulators Insulin Glucagon Secreted by the islets of Langerhans of the pancreas Secreted in response to a rise in blood glucose level Secreted in response to a fall in blood glucose level Stimulates uptake and use of glucose Stimulates production of glucose by gluconeogenesis Stimulates storage of glucose as glycogen and fat Stimulates breakdown of glycogen and fat glucose Adrenaline Glucocorticoids Secreted by the adrenal glands Strong and fast metabolic affects to mobilise glucose in response to ‘fight or flight’ stimuli Steroid hormones which increase the synthesis of metabolic enzymes that catalyse formation of glucose Know the effects of eating and fasting on metabolism In a real person, metabolism has to be able to cope with: Different types of food Sporadic food intake The different requirements of different tissues After a meal: blood glucose rises initially and then falls Control mechanisms for the initial rise in blood glucose: Islets: increased insulin secretion (and reduced glucagon secretion) Liver: increased glucose uptake; glucose is used for glycogen synthesis and for glycolysis Acetyl CoA produced by glycolysis is used for fatty acid synthesis Muscles: increased glucose uptake; glucose is used for glycogen synthesis Adipose tissue: increased triglyceride synthesis Increased usage of metabolic intermediates throughout the body Summary of effects due to the rise in blood glucose: 1. High glucose stimulates insulin release from the pancreas 2. High insulin stimulates glucose uptake into the liver and tissues 3. Insulin stimulates anabolic pathways Control mechanisms for the subsequent fall in blood glucose: Islets: increased glucagon secretion (and reduced insulin secretion) Liver: glucose production occurs due to gluconeogenesis and glycogen breakdown Fatty acid breakdown: this provides an alternative metabolite for ATP production This is important for preserving glucose for the brain Glucagon stimulates catabolic pathways Adrenaline: Adrenaline has similar effects to glucagon on the liver It also has additional effects on: Skeletal muscle: adrenaline stimulates glycogen breakdown and glycolysis Adipose tissue: adrenaline stimulates fat lipolysis (hydrolysis) to provide other tissues (i.e. tissues other than the brain) with alternative metabolites to glucose (i.e. fatty acids) After prolonged fasting (i.e. longer than can be covered by glycogen reserves): The glucagon to insulin ratio increases further Adipose tissue: hydrolysis of triglyceride occurs to provide fatty acids for metabolism The quantity of TCA cycle intermediates decreases to provide substrates for gluconeogenesis Protein breakdown occurs: this provides amino acid substrates for gluconeogenesis Liver: ketone bodies are produced from fatty acids and amino acids to partially substitute the glucose requirement of the brain Fatty acid breakdown: 1. ß-oxidation: fatty acids acetoacetyl CoA Branched pathway at acetoacetyl CoA: 2. Acetoacetyl CoA acetyl CoA/acetate Branched pathway at acetate: 3. Acetate acetone/ß-hydroxybutyrate Acetate, acetone and ß-hydroxybutyrate are ketone bodies There is excess accumulation of acetyl CoA: It is used by the heart under normal circumstances It is used by the brain when glucose availability is low Brain: Glucose dependent metabolism: the brain requires a continuous supply of glucose for metabolism o Hypoglycaemia: causes faintness and coma o Hyperglycaemia: may cause irreversible damage The brain cannot metabolise fatty acids directly; it can metabolise ketone bodies Ketone bodies (ß-hydroxybutyrate) can partially substitute for glucose Describe glucose interactions with lipid and amino acid synthesis and breakdown See flow charts Know basic details of contractile metabolism in muscle Skeletal muscle: During light contraction: ATP demand is met by oxidative phosphorylation using O2 and blood-borne glucose and fatty acids During vigorous contraction: ATP demand cannot be met by oxidative phosphorylation since the diffusion of O2 and blood-borne metabolites is a limiting factor Glycogen stores in the muscle are broken down to produce ATP Under anaerobic conditions, pyruvate is converted into lactate and H+ which can leave muscle Aerobic exercise: 1. Actomyosin ATPase, Ca2+ ATPase and Na+–K+ cause muscle contraction 2. Contractions stimulate: 3. o Increased ATP demand o Increased glucose transport from the liver to muscles via the bloodstream Adrenaline stimulates: o Increased muscle glycolysis: this produces ATP o Increased gluconeogenesis o Increased fatty acid production Anaerobic exercise: 1. ATP demand cannot be met by O2 diffusion 2. Transport cannot keep up with the demand for glucose 3. Breakdown of glycogen stores in the muscle increases 4. Lactate production increases: it is transported to the liver via the bloodstream 5. Recovery: the liver uses lactate to form glucose Heart: The heart must beat constantly; therefore it is designed for completely aerobic metabolism as it has a large number of mitochondria It can use TCA cycle substrates for metabolism: e.g. fatty acids and ketone bodies Loss of oxygen is devastating: it leads to cell death and myocardial infarction since energy demand is much greater than energy supply Be able to describe some of the metabolic disturbances that arise in diabetes 2 types of diabetes: 1. Type I diabetics cannot produce insulin 2. Type II diabetics have reduced responsiveness to insulin Metabolism is controlled as if for starvation, regardless of dietary uptake Complications of diabetes: Hyperglycaemia with progressive tissue damage Increase in plasma fatty acids and lipoproteins with possible cardiovascular complications Increase in ketone bodies with possible acidosis Hypoglycaemia with constant coma if the insulin dosage is imperfectly controlled