Kedar & Nupe's Cardio notes Part I
advertisement

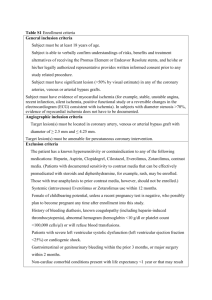
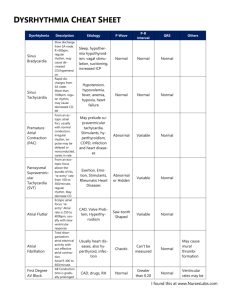
Cardiac Pathophysiology Electrocardiogram (Lilly, Chapter 4) I. II. III. IV. V. Basic Principles a. Depolarization – shows an upward deflection on the EKG indicating that the current of positive charges is going towards the reference electrode b. Repolarization – shows a downward deflection in the EKG indicating that the current of positive charge is flowing away from the reference electrode c. Note: in the normal human heart, the deflection on repolarization is upright, indicating the direction of repolarization is actually opposite that of depolarization. I.e. if the depol happens from base to apex, then the repolarization, which would normally show a downward deflection on the EKG, must happen from apex to base. This makes sense, because AP’s in endocardial cells (the first to become stimulated) are much longer than epicardial cells (the last to become stimulated), thus reversing the expected pattern of depol/repol Lead reference system a. Limb Leads – I, II, III (use the L mnemonic) b. Bipolar Leads – aVR, aVL, aVF (right, left, femur) c. Precordial Leads – measure the heart’s impulses in the direction perpendicular to the frontal plane – V1-V6 Reading the EKG – a. Voltage Calibration – 1mV signing should be 10 large boxes. Sometimes that is changed, such that the 1mV tracing is 5 boxes (if hypertrophy is so large that the tracing goes off the page) b. Rhythm – sinus if: i. Each QRS preceeded by a P, each P is followed by a QRS, P waves are upright in I, II, and III, and PR interval (see below) is greater than 3 small boxes ii. Keep in mind sinus tachy and bradycardias c. Rate – Use the mnemonic of 300, 150, 100, 75, 60, 50 for each large box. Otherwise, each large box is 0.2 s, each small box is 0.4s and five boxes is 1s d. Intervals i. PR – one large block (increased in 1st degree AV block) ii. QRS – ½ large block (BBB, ectopic beats, toxicity, hyperkalemia) iii. QT – difficult b/c it varies w/ HR e. Axis – Should be upright in leads I and II (-30 to 90 degrees). Can also use I and aVF. Use the other leads to determine whether it is right axis or left axis deviation P wave abnormalities – best visualized in lead II (parallel to flow from SA to AV) a. P wave is normally biphasic in lead V1. RA shows upward deflection in the anterior direction, followed by LA showing downward deflection posteriorly b. LA enlargement shows up as a largely biphasic V1 with a large LA downstroke following the upward deflection of the RA c. RA enlargement is just the opposite QRS abnormalities a. Ventricular Hypertrophy – i. big QRS in corresponding leads ii. axis deviations toward the larger ventricle b. BBB’s i. RBBB – initial depolarization of the septum, and left ventricle is normal so you see small R wave in V1, and small Q wave in V6. However, as the RV finally receives a signal, it transmits a widened, asynchronous impulse showing up as a second R (R’) wave over V1 and an S wave to V6 ii. LBBB – septal depolarization doesn’t happen, so NO R wave in V1 (you get Q instead) or Q wave in V6. You actually see the opposite because the right bundle takes precedence and you get signal toward the L instead of the R. Again, as LV catches up, you get a widened and late impulse that shows up as a second R wave over V5/V6 c. Myocardial Infarction i. Pathologic Q wave (small ones are normal in V6 and aVL) – deep and wide, precordial leads designate location of infarct. Pathophysiologically, the reason for the Q wave is that the lead over the infarcted tissue is detecting viable tissue on the other side of the heart ii. In the case of a posterior wall MI, you get large V1 and V2’s. This can be differentiated from RVH by the lack of right axis deviation iii. ST segment elevation – occurs because infarcted tissue directly below the lead changes the baseline so that the ST segments appear elevated or depressed due to diastolic leak of potassium either away or towards the lead depending on the depth of the infarct. I.e. in the case of an MI, K+ leak occurs away from the lead, which shifts the baseline lower (positive charge moving away from the lead) and shifts up the ST segment in relative terms. In the case of a subendocardial ischemic event, K+ leak goes from endocardium towards the lead, leading to baseline shift and therefore a relative drop in the ST segment Myocardial Ischemia/Stable Angina Ischemia is defined as the state of the heart in which myocardial oxygen demand exceeds the body’s ability to supply the heart with oxygen I. Myocardial Oxygen Supply – coronary perfusion occurs during diastole, so perfusion pressure can be measured by aortic end-diastolic pressure a. Oxygen carrying capacity b. Coronary Blood flow – depends on the perfusion pressure and the resistance of coronary vasculature to blood i. External compression – during cardiac cycle, the epicardial and subendocardial vessels are typically compressed during systole ii. Unlike most tissues, the heart cannot increase O2 extraction, and so must rely on increasing blood flow. This is accomplished by dilation of major resistance vessels iii. Mediators such as Adenosine, NO, prostacyclin, endothelium-derived hyperpolarizing factor, endothelin-1 (a constrictor) iv. Neural control – sympathetic receptors (alpha and beta-2) both promote vasodilation. Balance between alphas and betas and above factors that mediate total tone II. Myocardial Oxygen Demand a. Wall Stress – causes an increase in O2 demand because increased force pulling myocardial cells apart requires energy to withstand. Increased diameter in the case of MR or AI causes increased wall stress. Increased ventricular pressures also increase wall stress. Finally, compensatory hypertrophy works to reduce wall stress because force is spread out over a larger mass b. Heart Rate – a higher rate means more energy utilized c. Contractility – increasing the force of contraction increases the demand for O 2 III. Pathophysiology of Ischemia – myocardial ischemia is a result of fixed vessel narrowing and abnormal vascular tone from the damaged endothelium a. Fixed Vessel Narrowing i. Usually, the distal, smaller vessels are the first to become stenotic and occluded. The larger resistance vessels are usually plaque free and are able to adapt when necessary. Thus the significance of vessel narrowing depends on the extent to which distal arteries are occluded ii. Any greater than 60% occlusion may cause progressively worse downstream ischemia – first on exertion, then at rest b. Endothelial Cell Dysfunction i. Inappropriate Vasoconstriction – because endothelial derived factors (NO, prostacyclin, etc) are not released, the catecholamines are unopposed and vasoconstriction occurs IV. V. VI. VII. VIII. IX. X. ii. Platelet Aggregation – platelet derived anti-thrombotic factors (prostacyclin) is not released and local hypercoagulability occurs. Furthermore, because of the impaired function of the endothelium, the factors secreted by platelets (thromboxane, ADP, serotonin) go unopposed Non CAD causes of Myocardial Ischemia a. Decreased aortic perfusion pressure – hypotension or aortic regurgitation b. Decrease in O2 carrying capacity from anemia c. Extreme myocardial demand from aortic stenosis Consequences of Ischemia a. Immediately, there is decreased contraction, resulting in flow backup and pulmonary congestion (dyspnea) b. Stunned myocardium – in times of prolonged ischemia, there is a reversible change in which the heart will still lack full contractile function, but will eventually recover c. Hibernating myocardium – chronic ventricular dysfunction even though irreversible damage has not yet occurred Stable Angina a. Fixed obstructive plaques in one or more coronary arteries i. Symptoms directly correlated to degree of occlusion ii. When myocardial demand goes up, not enough reserve to compensate, leading to transient ischemia iii. With endothelial dysfunction, vessels may paradoxically constrict in response to stress due to catecholamine alpha adrenergic stimulation unopposed by endothelial derived factors iv. Stable threshold vs. Variable threshold angina – i.e. does the angina occur after the same activity always or only sometimes? Variant Angina and Silent Ischemia a. Variant or Prinzmetal’s angina refers to vasospasm as the source of angina which occurs at rest often due to early atherosclerotic changes b. Silent Ischemia – occurs often in the presence of symptomatic stable angina, but is unrecognized by the patient. Use a Holter monitor to truly test Differential Diagnosis of intermittent chest pain a. Cardiac – myocardial ischemia, pericarditis b. Gastrointestinal – GERD, ulcers, esophageal spasm, biliary colic c. Musculoskeletal – costochondritis, cervical radiculopathy Diagnosis a. EKG – shows acute ST segment depressions, but may be normal between episodes b. Stress Test – i. considered positive if EKG changes are found or angina is experienced ii. considered very positive if EKG changes occur in 1st three minutes, ST depression is deep, systolic dysfunction causes a drop in systolic BP, ventricular arrhythmias develop, or the patient cannot exercise for more than two minutes c. Nuclear Exercise – nuclear labeling of myocardial perfusion during exercise d. Exercise Echo e. Pharmacologic Stress testing – give dobutamine and see what happens f. Coronary Angiography – the “gold standard” for diagnosing CAD, it does not really tell you how vulnerable the plaques are to thrombose Treatment – increase myocardial O2 supply and decrease demand a. Medical Treatment of Acute Angina – venodilation with nitrates reduces venous return to the heart cause decreased wall stress via preload (lower R) b. Medical treatment of recurrent ischemic episodes i. Nitrates prior to vigorous activity ii. B-Blockers – reduce myocardial oxygen demand by decreasing chronicity and contractility (note: not used in decompensated LV dysfunction – CHF) iii. Ca++ channel blockers – 1. dihydropyridines (nifedipine, amlodipine)– vasodilators that 1) decrease oxygen demand via preload 2) decrease oxygen demand via 2. afterload (arterial dilation) and 3) increase myocardial supply via coronaries Nondihydropyridines (verapamil, diltiazem) are also vasodilators but not as potent. These also have antianginal effects via chronotropic and contractile effects Drug Dihydropyradine (nifedipine) Non-dihydropyradine (verapamil, diltiazem) c. d. Vasodilatory +++ + Contractility + +++ Chronotropy + +++ Treatment to prevent MI and death i. Antiplatelets – Aspirin, clopidogrel ii. Lipid-lowering iii. ACE inhibitors Revascularization – only if patient does not respond to medical management i. PTCA has high ~50% chance of restenosis ii. PTCA w/ stent has much better outcome iii. CABG, which often have poor patency rates -especially veins such as saphenous Valvular Heart Disease (Chapter 8, Lilly) I. II. Rheumatic Fever – inflammatory condition involving heart, skin, and connective tissue a. Pathogenesis of ARF remains unclear, but direct bacterial colonization of the heart does not happen. Mechanism appears to be elaboration of bacterial toxin or autoimmune cross-reactivity with some Ag b. Histology shows “Aschoff body,” an area of focal fibrinoid necrosis surrounded by inflammatory cells (lymphs, plasma, and MO’s) c. While RF can affect all layers of myocardium, the most devastating effects occur in the valvular endothelium Mitral Valve Disease a. Mitral Stenosis – almost always a result of RF i. Pathology – fibrous thickening of the leaflets, with fusion at the commissures and thickening of the chordae ii. Pathophysiology – due to the increased resistance from which the LA must overcome, there is increases in LA pressure and LV stroke volume may be diminished despite normal pressures 1. High LA pressures get passively transmitted to pulmonary capillary pressure causing pulmonary edema 2. Pulmonary edema can result in two types of pulmonary HTN a. Passive – in response to increased backflow b. Reactive – medial hypertrophy and intimal fibrosis of the pulmonary vasculature which 1) prevents pulmonary congestion but 2) causes increases in RV/RA pressures 3. Chronic LA enlargement leads to atrial fibrillation 4. Finally, relative stagnation of blood may predispose to thromboembolus iii. Clinical Manifestations – 1. Dyspnea either on exertion or at rest depending on severity 2. Occasionally, MS may present as the complications, AF, thrombus,etc 3. On PE, palpation may reveal an RV “heave”, increased S1 sound due to slamming shut of non-compliant valves 4. Major feature is the opening snap of MS, thought to be due to the sudden tensing of the chordae and leaflets on opening followed by low frequency decrescendo murmur 5. Echo shows stenotic leaflets iv. Treatment III. 1. Prophylaxis against ARF 2. Diuretics to prevent pulmonary congestion 3. Digoxin if LV function is impaired 4. B blockers if HR is up due to conducting AF b. Mitral Regurgitation – may result from structure, leaflets, chordae, or papillary muscles i. Etiology 1. Myxomatous degeneration (Mitral prolapse) can lead to MR 2. Myocardial infarct of papillary muscles 3. Left ventricular enlargement, specifically dilation ii. Pathophysiology – portion of LV systolic volume goes to low-pressure LA 1. Elevation of LA pressure and volume 2. Reduction in forward cardiac output 3. Volume related stress when regurgitant fluid comes back during diastole 4. Acute MR – caused by ruptured papillary muscles can cause immediate pulmonary congestion a. Prominent v wave in the LA tracing b. Increased LV output via the FS curve (increased EDP and increased stroke volume) 5. Chronic MR – caused by rheumatic heart disease – LA/LV allowed to undergo compensatory changes a. LA becomes low-pressure sink b. LV undergoes eccentric hypertrophy to volume overload c. Forward output is maintained for a while d. Eventually compensated state becomes decompensated and heart failure occurs iii. Clinical Manifestation 1. Acute shows pulmonary edema 2. Chronic shows low forward cardiac output 3. Holosystolic murmur radiating to the axilla 4. CXR shows increased LA and LV size 5. Echo shows increased LA and LV size as well as regurgitant flow in Doppler Echo iv. Treatment – augment forward cardiac output and reduce regurgitation 1. Acute MR – diuretics to relieve pulmonary congestion, nitro to reduce forward resistance 2. Chronic MR – oral arterial vasodilators (ACE or hydralazine) 3. Surgery to repair the leaky valve before heart failure sets in c. Mitral Valve Prolapse i. Characterized by asymptomatic billowing of the mitral leaflets into the LA during ventricular systole – shows myxomatous degeneration which means dense collagen matrix is replaced by loose connective tissue ii. Sometimes patients may reveal chest pain or palpitations due to associated arrhythmias iii. Clinical finding is a systolic click which is the sound of the mitral leaflet of the chordae as they tense – sound increases with squatting iv. Major complication is chronic progressive MR Aortic Valve Disease a. Aortic Stenosis – age related calcific changes of the valve – “senile” AS i. Pathology 1. Age related – calcification of normal tricuspid valve 2. Congenital – bicuspid valve provides irregular flow and damage ii. Pathophysiology – blood flow across the valve is obstructed 1. AS develops over a chronic course and so LV is able to compensate via concentric hypertrophy (similar to hypertension) 2. IV. Significant pressure gradient develops between the LV and aorta, resulting in gradients of 100mmHg 3. LA also hypertrophies due to increased diastolic pressure and provides an LA “kick” at the end of LV systole (beneficial ~25% of SV) 4. Normal valvular orifice is 3-4 cm. Clinical significance occurs when valve is 2cm. Moderate stenosis is 1-1.5 cm. Severe stenosis occurs at 0.8cm or less iii. Clinical Manifestation 1. Angina – a. Demand increased because of hypertrophy and increased wall stress due to higher ventricular pressures b. Decreased supply because increased diastolic pressure does not allow for adequate filling of coronary vessels 2. Syncope on exertion due to inability to maintain cardiac output with a fixed stenotic aortic valve 3. Congestive heart failure symptoms – increased LA pressures at the end of diastole causing pulmonary congestion 4. Coarse, late-peaking systolic ejection murmur heard best at the right upper sternal border 5. Weak and late carotid upstroke 6. S4 gallop 7. Echo and EKG show ventricular hypertrophy and increases pressure gradients between the LV and the aorta iv. Treatment 1. Severe – Valve replacement 2. Mild – endocarditis prophylaxis, avoidance of meds that may cause hypotension and worsened forward cardiac ouput b. Aortic Regurgitation i. Etiology – results from either disease of the leaflets (bicuspid, RF) or dilation of the root (Marfan’s, aneurysm) ii. Pathophysiology – depends on size of regurgitant orifice, pressure gradient across valve during diastole, duration of diastole 1. Acute – may cause immediate backup and increased LA pressure. Leads to pulmonary congestion and acute SOB 2. Chronic – LV undergoes compensatory dilation and eccentric hypertrophy a. Widened pulse pressure due to increased systolic pressure (via preload) and decreased diastolic pressure (via regurgitation) b. Decreased perfusion pressure (aortic diastolic P is low) may cause supply-side issue to the myocardium iii. Clinical manifestations – 1. Sensation of forceful heartbeat 2. Stigmata of widened pulse pressure (“water-hammer” pulse, head bobbing, Duroziez’s sign of capillary pulsations) 3. AR murmur best heard at right sternal border during early diastole (blowing quality, which ends prior to S1) 4. Austin Flint murmur which is a Mitral murmur (apex) caused by high regurgitant flow forcing the anterior mitral valve down iv. Treatment – usually asymptomatic and have normal contractile function 1. Endocarditis prophylaxis 2. ACE inhibitors to reduce afterload 3. Ca++ channel blockers, especially nifedipine Infective Endocarditis – classified by clinical course, substrate (native, prosthetic, IVD), or organism a. Pathogenesis i. Endocardial surface injury cause platelets to adhere and a sterile thrombus forms b. ii. This serves as a good place for the bacteria to land because fibrin is irregular, and it also protects the bacteria from host immune responses iii. Bacteria get there anytime a mucosal surface is ruptured, however, only those bacteria (~90% gram positive) that can survive can cause endocarditis iv. Complications once bacteria have colonized include mechanical cardiac injury, thrombotic or septic emboli, immune mediated injury Clinical – acute shows up as a very high fever and shaking chills, subacute is a low-grade fever with nonspecific constitutional symptoms of weight loss, fatigue, anorexia, etc i. May find heart murmurs consistent with the appropriate valve ii. Septic emboli may present as neurological findings if left heart is involved iii. Specific skin findings include splinter hemorrhage and Janeway lesions on palms and soles of the feet Cardiomyopathies (Chapter 10, Lilly) Classified either as dilated (ventricular enlargement with systolic dysfunction), hypertrophic (ventricular thickening with diastolic dysfunction), or restrictive (stiff myocardium because of fibrosis) I. Dilated Cardiomyopathies – dilation with only mild hypertrophy a. Etiology – genetic, toxic, metabolic, and infectious causes i. Acute viral myocarditis may lead to DCM 1. Though to be immune related, but immunosuppression doesn’t help 2. Often due to Coxsackie group of viruses ii. Alcoholic cardiomyopathy – EtOH impairs cellular function by inhibition mitochondrial oxidative phosphorylation iii. Familial forms – autosomal dominant and recessive, X-linked, mitochondrial. Genes involved code for structural proteins like actin, myosin, troponin, etc b. Pathology – all four chamber enlargement. Some concentric hypertrophy, but dilation is out of proportion to thickening c. Pathophysiology – ventricular dilation with decreased contractile function i. As V function falls, F-S mechanism is recruited such that increased diastolic volume improves SV ii. Sympathetics increase HR and contractility, helping to buffer falls in CO iii. As renal blood flow drops, RAS gets activated and volume retention occurs iv. Eventually, these compensatory mechanisms backfire and you get symptoms of CHF (pulmonary congestion, LA enlargement with AF, volume overload) d. Clinical findings – exactly the same as Heart Failure e. Physical Exam – exactly the same as Heart Failure f. Diagnosis i. ECG shows atrial and ventricular hypertrophy with a wide variety of arrhythmias – L or R BBB’s, AF, etc ii. Echo shows all four chambers are enlarged iii. Cardiac Cath is performed to rule out CAD as the source of the ventricular dysfunction g. Treatment i. ACE inhibitors are 1st line Tx ii. B-blockers have been shown to increase LF ejection fraction and mortality iii. Digitalis – may improve symptoms iv. K+ sparing diuretics (aldosterone blockers) v. Anti-arrhythmics – Amiodarone may help with AF and other SVT’s vi. Anti-coagulation b/c of AF, stasis in ventricles, venous stasis, etc. vii. Heart transplant II. Hypertrophic Cardiomyopathy a.k.a. IHSS results in thickened, stiffened ventricle a. Etiology i. Familial disease that arises as a new mutation that gets passed down via an autosomal dominant inheritance ii. Genes include myosin heavy chain, cardiac troponin, etc. Pathology – asymmetric hypertrophy of the ventricles (esp. the septum ~90%) i. Short widened myocytes that are oriented chaotically ii. Surrounded by fibroblasts and extracellular matrix c. Pathophysiology – significant VH which reduces diastolic function i. May also have systolic dysfunction if upper interventricular septum is involved. Basically, fast flow through a thickened aortic aperture may cause a low pressure suction of the anterior mitral valve and subsequent obstruction ii. W/O outflow tract obstruction (i.), the thickened myocardium creates an increased diastolic pressure, which causes backup in the LA and characteristic dyspnea on exertion iii. w/ outflow tract obstruction, the mitral valve remains open during systole (recall, it’s blocking the Ao aperture), causing MR. Furthermore, the outflow obstruction provides even more impetus for LVH, which increases myocardial demand. Note: situations which decrease the LV volume actually exacerbate the MV outflow obstruction. I.e. Valsalva exacerbates murmur d. Clinical Findings – Angina, syncope (arrhythmias), ventricular fibrillations and sudden death e. Physical Examination i. Commonly hear an S4 sound (LA pumping into stiff LV) ii. LV outflow obstruction results in AS-like murmur, MR murmur heard at apex iii. Valsalva maneuver typically reduces venous return to the heart, exacerbating the murmur – helps to differentiate between AS and HCM f. Diagnosis – Echocardiography can show the position of the valves and thickening of the ventricular walls g. Treatment i. B-Blockers reduce myocardial O2 demand by 1) decreasing HR/contractility, 2) decreased outflow gradient by dropping contraction, 3) increase diastolic filling time, 4) decrease frequency of ectopic beats ii. Ca++ blockers reduce ventricular stiffness and improve symptoms. Be careful b/c vasodilation may actually cause exacerbations of symptoms iii. Anti-arrhythmics iv. Implantable defibrillator – cardioverter v. Antibiotic prophylaxis to prevent endocarditis of aortic valve Restrictive Cardiomyopathy – less common and characterized by rigid, but not thickened ventricle with abnormal filling but no outflow obstruction a. Etiology – caused by fibrosis or scarring, infiltration of abnormal substance (amyloid). Many causes including scleroderma, amyloid, sarcoid, hemochromatosis, etc. b. Pathophysiology – like HCM w/o outflow obstruction c. Clinical Findings – signs of left and right sided heart failure are expected d. PE – similar to CHF, however, you may see Kussmaul’s sign, which is paradoxical worsening of JVD during inspiration, because RV can’t accommodate increased return e. Diagnosis – Echo, EDG, CT, MRI f. Treatment – Usually have poor prognosis – need to treat underlying cause. Must reduce volume to prevent symptoms of CHF b. III. Arrhythmias – (Chapter 11, Lilly) Arrhythmias can be generally classified as disorders of impulse formation, impulse conduction or both. I. Normal impulse formation is generated by heart’s pacemaker cells in the SA node. a. These cells are under the influence of the autonomic nervous system, which, via Badrenergic receptors increases the probability of the slow leak (phase 4) Na+ and (phase 0) Ca++ channels of being open. b. Cholinergic stimulation does the opposite by decreasing probability of Ca++ and increasing the probability of K+ (hyperpolarizing) channels to be open c. Pharmacologic agents (B-blockers, atropine) take advantage of these principles II. III. IV. Altered Impulse formation a. Escape Rhythms – when the SA node is too slow, a latent pacemaker will take over generating an escape beat if spontaneous and an escape rhythm if chronic b. Ectopic Rhythm – when latent pacemaker exceeds rate of SA node. Happens in situations of high circulating catecholamines c. Tissue Injury leads to pathologic changes in impulse formation outside of normal conduction system – caused by leaky membranes in injury Altered Conduction a. Conduction Block – impulse gets blocked when it hits a region that is not excitable. Caused by ischemia, fibrosis, and trauma and can be unidirectional or bidirectional b. Unidirectional block and reentry – repeated depolarization of a part of tissue due to unidirectional block i. Unidirectional block stops impulse conduction along one limb ii. Impulse travels down healthy end and reenters from opposite end of the unhealthy limb iii. If retrograde velocity is sufficiently slow to allow for the initial depolarization (on the healthy side) to repolarize, a reentrant circuit can be established causing an excessively high HR c. Bypass Tracts – in some individuals there is an alternative pathway than the normal conduction system called the bundle of Kent i. EKG findings include a ventricular depolarization that occurs earlier than normal (Kent conducts faster), which shows up as a shortened PR interval ii. EKG also shows widened QRS complex and a delta wave which is a nick in the Q upstroke Treatment of arrhythmias a. Bradyarryhthmia – i. anticholinergic drugs such as atropine ii. B1 agonists like isoproterenol iii. Pacing – 1. Transthoracic stimulation, which stimulates the heart via a large external patch applied to the chest 2. Transvenous – the more common type in which an electrode is deposited directly to the SA node, AV node or the ventricles b. Tachyarrythmias – both stop rapid beat and treat the sequelae i. Pharmacologic – based on the cause of the increased rate 1. Increased automaticity - reduce phase 4, make diastolic potential more negative, make the threshold less negative (last two make it harder to reach threshold) 2. Reentry – decrease conduction velocity, increase refractory period 3. Triggered activity – shorter AP duration, correct Ca++ overload ii. Vagotonic – carotid sinus increases vagal tone iii. Cardioversion and Defibrillation – apply a large external current to depolarize all of the myocytes at one time. Cardioversion is timed to the QRS to prevent fibrillation, whereas defib, because the heart is already in fibrillation, is simply applied iv. Ablation therapy – if electrophysiology can localize the area of the myocardium responsible for the tachycardia, that region can be destroyed with a catheter Specific Types of Arrhythmias and their Treatment (Chapter 12, Lilly) I. Bradyarryhthmias a. Sinus Bradycardia – slowing of the heart rhythm, not always pathologic b. Sick Sinus Syndrome – SA node dysfunction that causes intermittent changes in CO leading to hypotension and dizziness/syncope/neuro changes. Can be coupled with tachycardia to due to a atrial fibrillation/flutter. I.e. atrial changes cause both a slowed SA node and a abnormal pacemakers II. III. IV. Escape Rhythms – cardioprotective for SA node malfunction a. Junctional rhythm is when the AV node becomes the pacemaker b. Slowed HR with no P wave in front of the QRS c. If the pacemaker is lower than the AV node, you get EKG findings similar to BBB’s. I.e. pacer in the LV spreads slowly to the RV AV Conduction abnormalitites a. 1st Degree Block – Prolongation of PR interval due to slowed conduction; usually benign i. Reversible causes – vagal tone, transient ischemia, drugs (digitalis, B-blockers, non-dihydropyridine Ca++ channel blockers ii. Structural causes – myocardial infarct, degeneration of the conduction system b. 2nd Degree Block – two types characterized by intermittent failure of conduction i. Mobitz Type I – each beat shows a progressively increasing PR interval until finally a beat is skipped – usually asymptomatic and is not great cause for concern, but pacing is sometimes necessary ii. Mobitz Type II – Sudden, unpredictable loss of AV conduction without preceding change in PR interval – often show widened QRS and may progress to type III c. 3rd Degree Block – complete failure of conduction system (AV dissociation) i. Caused by infarction, drug toxicity, and chronic degeneration due to age ii. No relation between P waves and ventricular contraction iii. Often show an escape or junctional rhythm that may resemble a BBB Tachyarrythmias a. Supraventricular Arrhythmias i. Sinus Tachycardia – characterized by SA node firing at a rate above 100. P waves appear normal and vagal stimulation reduces rate ii. Atrial Premature Beats - atrial reentry due to sympathetic stimulation and are often non-pathologic. These beats are often blocked because they arrive while the ventricle is refractory. If they do get through, the QRS is wide iii. Atrial Flutter – atria are at 180 bpm or above 1. Many P waves find a refractory AV node and thus the signal is not transmitted (e.g. 2:1 block) 2. P waves show a sawtooth pattern 3. Carotid massage and drugs that slow conduction through the AV node alleviate probs with A. Flutter 4. Treatment includes cardioversion, burst pacing using a permanent pacemaker, pharmacologic treatment (B-blockers, Ca++ channel blockers, digoxin, conduction prolongers), radiofrequency catheter ablation iv. Atrial Fibrillation – chaotic rhythm with extremely fast atrial rate 1. Caused by chronic diseases such as hypertension, CAD, alcohol intoxication, etct 2. Especially dangerous because stiff ventricle needs atrial contraction to provide enough diastolic filling and because atrial contraction prevents stasis and thrombus formation 3. Treatment is the same as that for flutter with the addition of anticoagulants v. Multifocal Atrial Tachycardia 1. EKG shows an irregular rhythm with multiple P waves (>3) and an average atrial rate greater than 100 2. A flat baseline between P waves distinguishes this from AF b. Ventricular Arrhythmias i. Ventricular Premature Beats (PVC’s) 1. Show up as non-P wave related, widened QRS complexes that arise from ectopic foci in the ventricles 2. When the occur in doublets or triplets, there is an increased risk 3. If symptomatic, treat with B-blockers V. ii. Ventricular Tachycardia 1. Very wide QRS complexes (greater than 3 small boxes) 2. Can exists as monomorphic (homogenous) or polymorphic 3. Polymorphic variety includes “Torsades de Pointes,” which can occur due to a number of causes iii. Ventricular Fibrillation – cause of “dropping dead” in MI 1. The only treatment is prompt ventricular defibrillation 2. Usually preceded by a period of VT Easy Classification scheme: a. Insert here