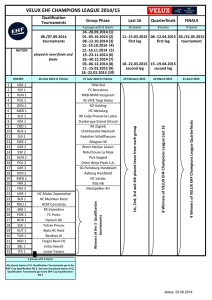
Hartree-Fock and Thomas-Fermi approximations
advertisement

Hartree-Fock and Thomas-Fermi
approximations
notes by L. G. Molinari
revised 10 October 2015
Introduction
The ground state properties of interacting systems may be studied by variational methods. In the Hartree - Fock approximation the energy is minimised
in the subset of Slater determinants. In the Thomas - Fermi approximation
the total energy is written as a functional of the unknown particle density,
and minimized. The two approximations are still used and useful. They
found their completion in the Density Functional Theory, by which most of
the progress in the study of electronic properties of solids molecules or atoms
was made [10].
1
The Hartree-Fock approximation
The Hartree-Fock equations provide an approximate evaluation of the ground
state of the Hamiltonian for a system of N interacting particles
X
X
v(i, j)
(1)
H=
h(i) + 12
i
i6=j
h(i) is a single particle operator, depending on the set i of fundamental
operators of particle i, and v(i, j) = v(j, i) is the two-particle interaction.
The Hartree1 equation was studied first [1]; it is a single-particle equation
1
Douglas Hartree, Cambridge 1897 - Cambridge 1958. In 1926 he obtained his PhD
with numerical studies of Bohr’s model of the atom. His advisor was Ernest Rutherford.
In the same year Schrödinger’s equation appeared, and in 1927 Hartree derived the Hartree
equations for multi-electron atoms. He was a pioneer in mechanical solvers for differential
equations (he built one with his student A. Porter, with Meccano parts) and then in
computer numerical applications to physics (from Wikipedia).
1
where the interaction with other particles enters as mean field (Hartree)
potential:
(hui )(~x, ω) + UH (~x)ui (~x, ω) = i ui (~x, ω),
i = 1 . . . N.
(2)
The Hartree potential is evaluated with the ground state density, to be calculated self-consistently:
Z
UH (~x) =
3 0
0
0
d x v(~x, ~x ) n(~x ),
n(~x) =
N X
X
j=1
|uj (~x, ω)|2 .
(3)
ω
A problem arises because it includes the self-interaction of the particle, and
the multi-particle ground state is not antisymmetric in the exchange of particles. A radical improvement was done by Fock2 , by adding the non-local
exchange term that guarantees the statistics [2].
There are several ways to deduce the HF equations. An elegant one is
to obtain them from a variational principle: instead of minimizing hΨ|H|Ψi
on all normalized antisymmetric vectors for N particles, the viable search is
restricted to Slater3 determinants of orthonormal one-particle trial states. As
such, the HF ground state energy EHF is the best (lowest) upper bound to the
exact ground state energy EGS , in a description with independent particles.
The difference is the correlation energy, and is negative by definition:
Ec = EGS − EHF ≤ 0
1.1
(4)
The HF equations
In the variational approach one starts from a set of N orthonormal trial
one-particle states |ui i. Their antisymmetrized product
1 X
(−1)σ Pσ |u1 , . . . , uN i
|HF i = √
N! σ
(5)
0
is
P a normalized Slater determinant. Note that the transformation |ui i =
j Uij |uj i, where U a matrix in SU(N ), preserves orthogonality and the
Slater state |HF 0 i built with the new set coincides with |HF i (appendix 1).
2
Vladimir A. Fock, St. Petersburg 1898, Leningrad 1974. He contributed to theoretical
physics and geophysics: he introduced occupation number states and the Fock space, and
developed the HF method in 1930. He independently obtained the Klein-Gordon equation,
described the degeneracy of H-atom as a SO(4) symmetry (1935), studied Dirac’s equation
in an external gravitational field (1929). He was arrested in 1937 during Stalinian terror
and freed a week later, thanks to Kapitza’s letter to Stalin.
3
John Slater, Oak Park (Illinois) 1900 - Sanibel Isl. (Florida) 1976.
2
The expectation value of the Hamiltonian is a functional of the N trial
states (appendix 2)
hHF |H|HF i =
X
hui |h|ui i +
i
1X
hui uj |v|ui uj − uj ui i
2 i,j
(6)
and does not change under the SU(N ) transformation on single particle
states. Note that the expression involves only one or two particles at a time,
the others being spectators. The interaction term contains both a direct
term and an exchange term. The negative sign results from Fermi statistics
(particles are exhanged), and cancels the self-term i = j.
Minimization of the energy must be done enforcing orthogonality and
normalization, that are required for the equation (6) to hold. Thus the
functional to minimize is
X
EHF (N ) = hHF |H|HF i −
ij (hui |uj i − δij )
(7)
ij
with Lagrange parameters ij that are the entries of a Hermitian matrix
(reality of EHF ).
The first variations arising from infinitesimal variations of states ui into ui +ηi
are4 ,
δi EHF = EHF [u1 , ..., ui + ηi , .., uN ] − EHF [u1 , .., ui , .., uN ]
X
= hηi |h|ui i +
hηi uj |v|ui uj − uj ui i − ij hηi |uj i + c.c.
j
Lemma 1.1. In a Hilbert space, if hη|ui + hu|ηi = 0 for all η, then u = 0.
Proof. In a real space the inner product is symmetric, then the result is
obvious. In a complex space, if η is replaced by iη it is: −ihη|ui + ihu|ηi = 0
i.e. hη|ui = 0. Then u = 0.
The first variations must vanish independently for all variations |ηi i. Since
the N −particle Slater state is left unchanged by a unitary transformation,
there is a degeneracy in the solutions uP
i . We may always pick the set that
diagonalizes the Hermitian matrix ij :
j ij |uj i = i |ui i. Then we obtain
the N Hartree-Fock equations:
N
X
h|ui i +
h · uj |v|ui uj − uj ui i = i |ui i,
i = 1...N
(8)
j=1
4
the term hui ηj |v|ui uj −uj ui i is identical to hηj ui |v|uj ui −ui uj i because of the exchange
symmetry of the potential h12|v|34i = h21|v|43i. The dummy indices ij are then redifined.
3
The presence of the exchange term replaces the single (self-consistent)
Hartree equation with a system of N coupled equations, which is computationally much more difficult.
Projection of the equation on the solution |ui i gives:
X
i = hui |h|ui i +
h ui uj |v|ui uj − uj ui i
(9)
j
The identity simplifies the evaluation of the ground state energy (6):
X
X
X
EHF (N ) =
hui uj |v|ui uj − uj ui i = 21
[hui |h|ui i + i ] (10)
i − 12
i<j
i
i
Proposition 1.2 (Koopman’s lemma (1934)). The highest eigenvalue is the
chemical potential of the system:
µ ≡ EHF (N + 1) − EHF (N ) = N +1
Proof. Let |u1 i . . . |uN +1 i be the solutions of HF equations for N +1 particles.
The Hartree Fock energy is
EHF (N + 1) =
N +1
1X
hui uj |v|ui uj − uj ui i
huj |h|uj i +
2 i6=j
N
+1
X
j
=huN +1 |h|uN +1 i +
N
X
huN +1 uj |v|uN +1 uj − uj uN +1 i+
j=1
+
N
N
X
1X
huj |h|uj i +
hui uj |v|ui uj − uj ui i
2
j
i6=j
= N +1 + EHF [u1 . . . uN ]
For N particles, the solution of HF equations are a different set |u01 i . . . |u0N i
that give the total energy EHF (N ). Therefore
µ = EHF (N + 1) − EHF (N ) = N +1 + EHF [u1 . . . uN ] − EHF (N ) ≥ N +1
because EHF (N ) is a stationary point. Let |u0N +1 i be a normalized state
orthogonal to the vectors |u0i i, then:
EHF [N + 1] ≤ EHF [u01 , . . . , u0N +1 ] = 0N +1 + EHF [N ]
where 0N +1 is the expression (9) evaluated with primed states. Therefore
N +1 ≤ µ ≤ 0N +1 .
The total energy is of order N , and single particle energies are of order 1.
If we assume that, for large N , the difference of two energies evaluated with
HF states for N + 1 and N particles (plus one) is O(1/N ) then µ = N +1 .
4
In the representation of position and spin, the Hartree-Fock equations
gain the familiar form
(h + UH )ui (~x, ω) −
XZ
d3 x0 Jω,ω0 (~x, ~x0 )ui (~x0 , ω 0 ) = i ui (~x, ω)
(11)
ω0
where UH is the self-consistent Hartree potential (3) and the exchange integral
contains the kernel
Jωω0 (~x, ~x0 ) =
N
X
uj (~x, ω)v(~x, ~x0 )u∗j (~x0 , ω 0 )
j=1
If the Hamiltonian does not depend on spin, the solutions factorize as
ui (~x)vmi (ω),Pwhere vm is an eigenstate of Sz . The completeness relation
for spinors, ω vm (ω)vm0 (ω) = δmm0 , simplifies the HF system of equations:
Z
X
(h + UH )ui (~x) − d3 x0 v(~x, ~x0 )
δmi ,mj uj (~x)uj (~x0 )∗ ui (~x0 ) = i ui (~x) (12)
j
Slater [4] (1951) suggested to approximate the exchange term with a local
average. This approximation restores locality, and anticipates the progress
of Density Functional Theory.
1.2
Helium atom
Neglecting spin, the Hamiltonian of the Helium atom with fixed nucleus is:
X p2
2e2
e2
i
−
+
(13)
H=
2m |~xi |
|~x1 − ~x2 |
i=1,2
The ground state is spherically symmetric: ψ(r1 , r2 , ξ)v0,0 (ω1 , ω2 ) where
ψ(r1 , r2 , ξ) = ψ(r2 , r1 , ξ), ξ = cos θ (θ is the angle formed by the vectors
~r1 and ~r2 ) and v0,0 (ω1 , ω2 ) = √12 [v↑ (ω1 )v↓ (ω2 ) − v↑ (ω2 )v↓ (ω1 )] is the singlet
state of total spin, antisymmetric in ω1 , ω2 . The eigenvalue equation for the
ground state
~2 1 ∂ 2 ∂
1 ∂ 2 ∂
r12 + r22 ∂
2 ∂
−
r
+
r
+ 2 2
(1 − ξ )
ψ
2m r12 ∂r1 1 ∂r1 r22 ∂r2 2 ∂r2
r1 r2 ∂ξ
∂ξ
"
#
2e2 2e2
e2
−
+
−p 2
ψ = EGS ψ
(14)
r1
r2
r1 + r22 − 2r1 r2 ξ
5
cannot be solved analytically. In the HF approximation one employs trial
functions u(r)v↑ (ω) and u(r)v↓ (ω), which give the state u(r1 )u(r2 )v00 (ω1 , ω2 ).
The single particle function u(r) solves the HF equation
−
Z(r)
~2 d 2 du
r
− e2
= u(r)
2
2mr dr dr
r
(15)
The equation describes an electron in the potential of an effective charge
Z ∞
Z r
Z
0 2
02
0
0 2
3 0 u(r )
r0 dr0 u(r0 )2
r dr u(r ) − 4πr
= 2 − 4π
Z(r) = 2 − r d r
|~r − ~r0 |
r
0
Two terms measure the total charge (in units of e) in the sphere of radius r.
For large r the screened nucleus charge is Z(∞) = 1, while for r → 0 it is
Z(0) = 2. The total Hartree-Fock energy is:
2
Z ∞
Z ∞
Z r
~ 0 2 2e2 2
2
2 2
2
r dr
EHF [u] = 8π
u −
u − 32π e
r dr u
r02 dr0 u2
2m
r
0
0
0
The HF equation is non-linear and is not solved analytically. A viable approximation (the standard calculation found in textbooks) is to impose a
Hydrogen-like solution with a variational parameter Z:
s
Z 3 −Zr/a0
e
,
u(r) =
πa30
and minimize EHF (Z) = (e2 /a0 )(Z 2 − 4Z + 85 Z). The minimum is at Z = 27
,
16
which gives the ground state estimate (an upper bound) EHF = −77.5 eV.
Exercise. Show that:
1.3
R
0
(r )
d3 r d3 r0 f (r)f
= 32π 2
|~
r−~
r0 |
R∞
0
r dr f (r)
Rr
0
r02 dr0 f (r0 )
Homogeneous systems
The HF equations are solved analytically if the system is translation invariant: the two particle interaction is a function v(~x −~y ) (for simplicity we avoid
spin dependence) and there is no external potential. We consider h = p2 /2m.
Proposition 1.3. The Hartree-Fock equations for N particles in a box with
periodic b.c. are solved by an arbitrary set of N eigenvectors |~ki , mi i of p~
and Sz .
Proof. Let’s show that
X
~ki2 ~
|ki , mi i +
h · , ~kj mj |v|~ki mi , ~kj mj − ~kj mj , ~ki mi i = i |~ki mi i
2m
j
6
A resolution of identity is inserted in the matrix element: h · , ~kj mj |v| . . .i =
P
~
~ ~
~k,m |kmihkm, kj mj |v| . . .i. Translation invariance enforces conservation of
total momentum, ~k + ~kj = ~ki + ~kj , spin conservation requires m = mi .
Therefore the sum reduces to the single term (~k, m) = (~ki , mi ), and the
vectors |~ki mi i solve the HF equations.
The matrix element is evaluated:
i
1 h
h~ki mi , ~kj mj |v|~ki mi , ~kj mj − ~kj mj , ~ki mi i =
ṽ(0) − δmi ,mj ṽ(~ki − ~kj )
V
R
~
where ṽ(~k) = d3 x v(~x)e−ik·~x . The eigenvalues and the total energy per
particle are obtained:
i =
1 X
~2 ki2
δmi mj ṽ(~ki − ~kj )
+ n ṽ(0) −
2m
V j
N
EHF
1 X ~2 ki2 1
n X
=
+ n ṽ(0) −
δmi mj ṽ(~ki − ~kj )
N
N i=1 2m
2
2N 2 ij
(16)
(17)
The optimal set {(~ki , mi )} that minimizes EHF is not obvious.
1) The kinetic energy is minimized by taking the vectors ~ki as small as
possible, i.e. filling the Fermi sphere |~k| ≤ kF , with both values of ms . This
choice gives:
~2 k 2
1 X
(~k) =
+ nṽ(0) −
θ(kF − k 0 )ṽ(~k − ~k 0 )
2m
V 0
(18)
3 ~2 kF2
1
n X
EHF
=
+ nṽ(0) −
θ(kF − k)θ(kF − k 0 )v(~k − ~k 0 ).
N
5 2m
2
2N 2 0
(19)
~k
~k~k
This total HF energy coincides with the energy evaluated by first order perturbation theory: hF |H|F i (|F i is the Slater state with filled Fermi sphere).
2) If ṽ(~k) > 0, the (negative) exchange term in (17) can be lowered by
requiring mi = mj for all particles. This increases the kinetic energy, but
may be advantageous at low density. The HF ground state would then be
spin polarized.
Exercise. Consider a homogeneous system of N particles in a volume V ,
with total energy E, density n = N/V , mean energy per particle = E/N .
7
If only depends on n, show that the chemical potential and the pressure
are given by:
∂E
d
∂E
µ=
=
[n(n)], p = −
= n[µ − (n)].
∂N V
dn
∂V N
Since at equilibrium it is p = 0, then µ = (n).
2
The homogeneous electron gas (HEG)
Consider N electrons in a box of volume V , in a uniform background of positive charge density eN/V . The system is neutral, and the electrons interact
via Coulomb forces. Periodic b.c. are considered, in view of the thermodynamic limit that makes the system translation-invariant.
The HEG models the conduction electrons of metals, where the crystal structure is smoothed to a uniform background. The overall effect of the background is to remove the ~k = 0 Fourier component of the Coulomb potential
(i.e. the Hartree, or direct, term of the two body interaction exactly cancels
the background electrostatic self-energy and the interaction energy of electrons with background. The background then disappears from the Hamiltonian, together with the Hartree potential UH ).
In the HF approximation with ground state given by the filled Fermi sphere,
the single particle energy is
Z 3 0
dk
4πe2
~2 k 2 2e2 kF
k
~2 k 2
0
−
θ(kF − k )
−
F
, (20)
=
(k) =
3
0
2
~
~
2m
(2π)
2m
π
kF
|k − k|
F (x) =
1 1 − x2
1+x
+
log
2
4x
|1 − x|
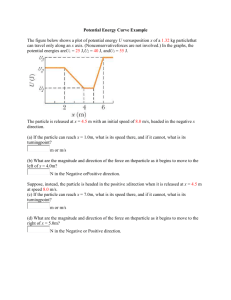
The function F is continuous but with singular derivative in x = 1: this
result is bad because d/dk diverges at k = kF . It implies that, in this
approximation, the level density (per unit volume and spin component)
ρ() =
4π
k 2 1 X
δ( − (k)) =
V
(2π)3 |0 (k)| (k)=
(21)
~k
vanishes at the Fermi energy, with sharp disagreement with experimental
data of several physical quantities at low temperature, such as the specific
heat5 , the conductivity of metals, or the critical temperature for the su5
2
For the electron gas cV = 32 π 2 kB
ρ(F )T .
8
Figure 1: The function F(x).
perconducting transition6 . Nevertheless, the total energy is of interest and
reliable at high density:
e2 3
3
EHF
2
=
(kF a0 ) −
(kF a0 )
(22)
N
2a0 5
2π
The energy scale is fixed by the ionization energy of the H-atom, e2 /2a0 =
13.61 eV (the Hartree energy unit is often used: 1 Ha = e2 /a0 ). If Seitz’s
parameter rs is used7 :
2.210 0.916
EHF
≈ 13.61 eV
−
.
(23)
N
rs2
rs
The negative exchange energy explains the cohesion of conduction electrons in
metals: it balances the kinetic energy and stabilizes the electron gas at finite
densities. The minimum is achieved at rmin = (4π/5)(9π/4)1/3 ≈ 4.83, with
an energy per particle equal to -1.29 eV. The value rmin is within the range of
rs -values of metals: rs (Li)= 3.25, rs (Na)= 3.93, rs (K)= 4.86, rs (Rb)= 5.20,
rs (Cs)= 5.62, rs (Al)= 2.07, rs (Au)= 3.01.
The HF energy per particle at the value rs (Na) is −1.23 eV, which compares
well with the experimental value −1.13 eV of the binding energy measured
as heat of vaporization of the metal.
The HF expression (23) is an upper bound for the exact ground-state
energy per particle. A lower bound was found by Lieb and Narnhofer, by
minimisinig T and V separately [3]:
" 2
#
91
e2 3 9π 3 1
EGS
>
−
N
2a0 5 4
rs2 5 rs
6
In the BCS model it is TC ≈ TD exp(1/ρ(F )g), where TD is Debye’s temperature and
g is the coupling of electrons to phonons
7
rs measures the radius available per particle in units of Bohr’s radius: (4/3)π(rs a0 )3 =
V /N . For spin 1/2: kF = (3π 2 N/V )1/3 , then: (a0 kF ) = (9π/4)1/3 (1/rs )
9
The minimum kinetic value is that of the ideal gas, the Coulomb minimum
corresponds to delta-localized electrons.
Bloch (1929) conjectured that, at low density, the gas prefers to polarize, because parallel spins lower the Coulomb energy. However, Overhauser showed
that neither the para or ferro-magnetic HF ground states are stable against
the formation of spin density waves (particle density is uniform, but with
spatial modulation of spin density) [5]. The subject was reconsidered by
Ceperley et al. [6], with numerical simulations.
At very low densities, the total energy is minimized by a state with the
electrons localized in lattice sites (Wigner crystal), where they perform zero
point motion.
The relation dE = −pdV + µdN gives the pressure8 and the chemical
potential in HF approximation at T = 0, as functions of the density:
dE drs e2 0.352 0.073
∂E − 4
(24)
p=−
=−
= 4
∂V N
drs dV N
2a0
rs5
rs
e2 3.683 1.222
∂E −
µ=
(25)
= (kF ) =
∂N V
2a0
rs2
rs
At rmin the pressure vanishes, µ = 1.29 eV (it coincides with the average
energy per particle).
The bulk modulus is an important parameter of solids; it measures the volume response to pressure and is always positive
∂V (26)
B = −V
∂p N
Exercise. Evaluate the bulk modulus for the HEG. Show that at rmin = 4.83
it is B = 2.1×1010 erg/cm3 ; for Potassium (rs = 4.86) the experimental value
is B = 2.81 × 1010 erg/cm3 . Other values are: Al) B = 76.0 × 1010 erg/cm3 ,
Na) B = 6.42 × 1010 erg/cm3 (from Ashcroft-Mermin, Solid State Physics,
Holt Saunders 1976). Show that B < 0 for rs > 6.03, where HF badly fails.
3
The Thomas-Fermi approximation
Thomas [11] and Fermi [12] introduced an approximate method, variational
in character. The method aims at expressing the total energy as a functional
8 2 −4
e a0
= 2.94 × 1013 Pa.
10
of the density. The ground state energy and density are obtained by minimization.
A main difficulty is to construct an effective functional for the kinetic energy.
The Thomas-Fermi functional is based on the total energy of the ideal Fermi
gas, adjusted to allow for a position-dependent density:
Z
5
3 ~2
TF
2 32
T [n] =
(3π )
(27)
d3 x n(~x) 3
5 2m
For a uniform density N/V , the total energy of free electrons is recovered.
The Thomas-Fermi energy functional for an atom with Z electrons is:
Z
Z
x) e2
n(x)n(y)
TF
TF
2
3 n(~
E [n] = T [n] − Ze
dx
+
(28)
d3 xd3 y
|~x|
2
|~x − ~y |
The electrostatic terms are classical.
The
is minimized with the
R 3 functional
additional Lagrange multiplier µ Z − d x n(~x) , to fix the number of electrons. The density at minimum solves:
Z
2
δE T F
Ze2
~2
n(~y )
2 23
2
0=
=
(3π ) n(~x) 3 −
+e
d3 y
−µ
δn(~x)
2m
|~x|
|~x − ~y |
and µ = ∂ET F /∂Z. The equation can be rewritten with the aid of Poisson’s
formula:
2
2
~2
(3π 2 ) 3 n(~x) 3 − eϕ(~x)
µ=
2m
∇2 ϕ(~x) = −4πe2 [Z δ(~x) − n(~x)]
A solution is found with the assumption of spherical symmetry, but the result
is not good: the atom is too large. Dirac (1930) [13] introduced an extra term,
the exchange energy functional, given by the expression for HEG, i.e. the
second term in eq.(22), but with local density:
4 Z
3
4
2 3
(29)
Ex [n] = −e
d3 x n(~x) 3 .
1
4π 3
A term dependent on the gradient of the density was introduced by von
Weizsacker (1935) [14], to improve the kinetic functional:
Z
~2
|∇n(~x)|2
vW
T [n] =
d3 x
.
8m
n(~x)
This form can be justified
on P
the Rbasis of HF approximation: the kiPN
~2
netic energy is hT i = 2m
d3 x|grad ui (~x, m)|2 . Put ui (~x, m) =
i=1
m
p
P P
n(~x)fi (~x, m) where n(~x) is the total density; then N
x, m)|2 = 1.
i=1
m |fi (~
R
P
P
~2
One evaluates: hT i = T vW [n] + 8m
d3 x n(~x) m N
x, m)|2 . The
i=1 |∇fi (~
second term is the “exchange kinetic energy”.
11
Making approximations exact
DFT originated in 1964 from a theorem by Hohenberg and Kohn [7] who
established that for of a quantum system of particles with given interaction,
there is a one-to-one correspondence among the external potential (up to a
constant) and the ground state density. This implies that the total energy
is a sum of the potential energy and a universal (unknown) functional of the
density. This would be the exactification of the Thomas-Fermi approach.
Soon after, Kohn and Sham [8] wrote a Schrödinger equation which made
DFT usable. The many particle problem is associated to a single particle
problem with an appropriate exchange-correlation local potential
(h + UH )ui (~x) + vxc (~x)ui (~x) = i ui (~x),
i = 1...N
(30)
In Kohn’s words [9], the equation is the exactification of the Hartree equation. The potential vxc is unknown, but useful approximations to it were
obtained from the theory of the homogeneous electron gas (HEG), that has
been studied in depth by perturbative methods and Montecarlo simulations.
Appendix 1
Let |u1 i . . . |uN i be orthonormal single particle states. Define the new orthonormal states |u0i i = Uij |uj i (summation is implicit), where U is a SU(N )
matrix. The two sets produce the same N -particle fermionic Slater state.
√
0
N ! S(N )− |u01 , . . . , u0N i =
Proof.
Let
us
evaluate
the
new
state,
|Ψ
i
=
√
N ! U1j1 . . . UN jN S(N )− |uj1 . . . ujN i. The operator S(N )− produces a non
zero state only if {j1 . . . jN } is a permutation σ of {1 . . . N }. Therefore, the
summation on {j1 . . . jN } is a sum over permutations Pσ on N -particle states
√ X
|Ψ0 i = N !
U1σ1 . . . UN σN S(N )− Pσ |u1 . . . uN i
σ
X
√
=
(−1)σ U1σ1 . . . UN σN N !S(N )− |u1 . . . uN i = det U |Ψi
σ
Appendix 2
Let |u1 i . . . |uN i be orthonormal√one-particle states, and construct the N particle fermionic state |Ψi
. , uN i. For one and twoPN= N !S(N )− |u1 , 1. .P
particle operators O1 =
i=1 o(i) and O2 = 2
i6=j o(i, j), with o(i, j) =
12
o(j, i), it is:
hΨ|O1 |Ψi =
N
X
hui |o|ui i,
hΨ|O2 |Ψi =
i=1
X
hui uj |o|ui uj − uj ui i
(31)
ij
Proof. Since O1 commutes with S(N )− and S(N )2− = S(N )− :
X
hΨ|O1 |Ψi =N !
hu1 . . . uN |O1 S(N )− |u1 , . . . , uN i
i
=
XX
(−1)σ hu1 |uσ1 i . . . hui |o|uσi i . . . huN |uσN i
σ
i
The inner products are zero unless σk = k for all k 6= i. Then necessarily
σi = i, and only the identity permutation contributes to the sum. Similarly:
X
hΨ|O2 |Ψi =N !
hu1 . . . uN |o(i, j)S(N )− |u1 . . . ui . . . uj . . . uN i
ij
=
XX
ij
(−1)σ δ1σ1 . . . hui uj |o|uσi uσj i . . . δN σN
σ
the inner products are non-zero if σk = k for k 6= i, j. Then, the only
permutations that contribute are the identity and the exchange σi = j, σj =
i. The last one has a factor (−1)σ = −1.
References
[1] D. R. Hartree, The wave mechanics of an atom with a non-Coulomb
central field, Proc. Cambridge Phyl. Soc. 24 (1928) 89.
[2] V. Fock, Näherungsmethode zur Lösung des quantenmechanischen
Mehrkörperproblems, Z. Phys. 61 (1930) 126.
[3] E. H. Lieb and H. Narnhofer, The thermodynamic limit for jellium, J.
Stat. Phys. 12 (1975) 291-310; see also the book, Lieb-Seiringer, The
stability of matter in quantum mechanics, (2010) Cambridge University
Press.
[4] J. C. Slater, A Simplification of the Hartree-Fock Method, Phys. Rev.
81 (1951) 385.
[5] A. W. Overhauser, Spin density waves in an electron gas, Phys. Rev.
128 (1962) 1437.
13
[6] S. Zhang and D. M. Ceperley, The Hartree-Fock ground state of the
three-dimensional electron gas, Phys. Rev. Lett. 100 (2008) 236404;
arXiv:0712.1194.
[7] P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev.
136 (1964) B864.
[8] W. Kohn and L. J. Sham, Self-consistent equations including exchange
and correlation effects, Phys. Rev. 140 (1965) A1133.
[9] W. Kohn, Nobel Lecture: Electronic structure of matter-wave functions
and density functionals, Rev. Mod. Phys. 71 (1999) 1253.
[10] E. Engel and R. M. Dreizler, Density Functional Theory, an advanced
course, Springer (2011)
[11] L.H. Thomas, The calculation of atomic fields, Proc. Cambridge Phil.
Soc. 23 (1926) 542.
[12] E. Fermi, Un metodo statistico per la determinazione di alcune proprietà
dell’atomo, Rendiconti dell’Accademia Nazionale dei Lincei 6 (1927)
602; Eine statistische Methode zur Bestimmung einiger eigenschaften des
Atoms und ihre Anwendung auf die Theorie des Periodischen Systems
der Elemente, Z.Phys. 48 (1928) 73.
[13] P.A.M. Dirac, Note on exchange phenomena in the Thomas atom, Proc.
Cambridge Phil. Soc. 26 (1930) 376-85.
[14] C. F. von Weizsäcker, Zur Theorie der Kernmassen, Zeitschrift für
Physik 96 (7-8) (1935) 431-458.
14