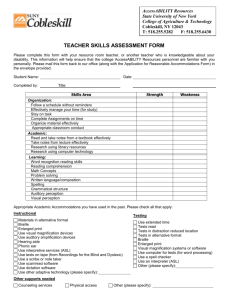
Structural Features of tRNALys Favored by Anticodon Nuclease as
advertisement

THE JOURNAL OF BIOLOGICAL CHEMISTRY © 2002 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 277, No. 6, Issue of February 8, pp. 3836 –3841, 2002 Printed in U.S.A. Structural Features of tRNALys Favored by Anticodon Nuclease as Inferred from Reactivities of Anticodon Stem and Loop Substrate Analogs* Received for publication, October 18, 2001, and in revised form, November 20, 2001 Published, JBC Papers in Press, November 26, 2001, DOI 10.1074/jbc.M110072200 Yue Jiang‡§, Shani Blanga‡, Michal Amitsur‡, Roberto Meidler‡, Eli Krivosheyev‡, Mallikarjun Sundaram¶, Ashok C. Bajji¶, Darrell R. Davis¶, and Gabriel Kaufmann‡储 From the ‡Department of Biochemistry, Tel Aviv University, Ramat, Aviv 69978, Israel and the ¶Department of Medicinal Chemistry, University of Utah, Salt Lake City, Utah 84112 The bacterial tRNALys-specific PrrC-anticodon nuclease efficiently cleaved an anticodon stem-loop (ASL) oligoribonucleotide containing the natural modified bases, suggesting this region harbors the specificity determinants. Assays of ASL analogs indicated that the 6-threonylcarbamoyl adenosine modification (t6A37) enhances the reactivity. The side chain of the modified wobble base 5-methylaminomethyl-2-thiouridine (mnm5s2U34) has a weaker positive effect depending on the context of other modifications. The s2U34 modification apparently has none and the pseudouridine (⌿39) was inhibitory in most modification contexts. GC-rich but not IC-rich stems abolished the activity. Correlating the reported structural effects of the base modifications with their effects on anticodon nuclease activity suggests preference for substrates where the anticodon nucleotides assume a stacked A-RNA conformation and base pairing interactions in the stem are destabilized. Moreover, the proposal that PrrC residue Asp287 contacts mnm5s2U34 was reinforced by the observations that the mammalian tRNALys-3 wobble base 5-methoxycarbonyl methyl-2thiouridine (mcm5s2U) is inhibitory and that the D287H mutant favors tRNALys-3 over Escherichia coli tRNALys. The detection of this mutation and ability of PrrC to cleave the isolated ASL suggest that anticodon nuclease may be used to cleave tRNALys-3 primer molecules annealed to the genomic RNA template of the human immunodeficiency virus. A tRNALys-specific anticodon nuclease (ACNase)1 in latent form is encoded by the optional Escherichia coli prr locus (1). It comprises the core ACNase polypeptide PrrC and type IC DNA restriction-modification enzyme EcoprrI that masks PrrCs activity (2–5). The phage T4-coded peptide Stp inhibits EcoprrI DNA restriction and activates ACNase (6). Cleavage of * This work was supported in part by grants from the Israeli National Science Foundation, the Israeli Ministry of Science Canadian-Israeli Cooperation Fund, and work at the University of Utah was supported by National Institutes of Health Grant GM55508. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. § Supported in part by postdoctoral fellowship of the Israeli Council for Higher Education. 储 Incumbent of the Louise and Nahum Barag Chair in Cancer Molecular Genetics. To whom all correspondence should be addressed. Tel.: 972-3-640-9067; Fax: 972-3-640-6834; E-mail: gabika@tauex.tau.ac.il. 1 The abbreviations used are: ACNase, anticodon nuclease; ASL, anticodon stem and loop; t6A, threonylcarbamoyl adenosine; mnm5s2U, 5-methylaminomethyl-2-thiouridine; mcm5s2U, 5-methoxycarbonyl methyl-2-thiouridine. tRNALys 5⬘ to the wobble base ensues, yielding 2⬘:3⬘-cyclic phosphate (2⬘:3⬘-p⬎) and 5⬘-OH termini. However, the lesion is normally offset by the phage T4-encoded polynucleotide kinase and RNA ligase (7). The active form of the core polypeptide PrrC has not been purified to homogeneity. Yet PrrC certainly harbors the ACNase function, judged from the manifestation of ACNase activity by E. coli (8) and mammalian cells overexpressing PrrC (9). Moreover, a tRNALys anticodon recognition site has been pinpointed in a cluster of selected PrrC mutations, two adjacent members of which (D287Y, S288P) alter the cleavage site specificity (10). Other mutations directed into this site (D287H, D287Q, or D287N) alter the specificity so that tRNALys forms with a hypomodified wobble base, otherwise poor substrates, are rendered more reactive than the natural. These compensatory effects suggest the existence of a specific interaction between the Asp287 residue of PrrC and the modified wobble base (11). Comparing bacterial and mammalian tRNALys substrates of ACNase has suggested that the specificity determinants reside in the anticodon loop and lower part of the anticodon stem (9). These determinants must include the anticodon sequence, judged from the following observations. First, the anticodon of tRNALys resembles the anticodons of most secondary substrates cleaved when PrrC is overexpressed (10). Second, most single anticodon base substitutions in unmodified tRNALys abolish ACNase reactivity. Third, a substrate analog with a tRNALys anticodon transplanted in an otherwise tRNAArg sequence is as reactive as the wild type tRNALys sequence (11). The anticodon stem and loop (ASL) domain of E. coli tRNALys contains three modified bases that profoundly affect its conformation (12). They include the doubly modified wobble base 5-methylaminomethyl-2-thiouridine (mnm5S2U34), 6-threonyl carbamoyladenosine 3⬘ to the anticodon (t6A37) and the pseudouridine of the lower base pair of the stem (⌿39). Comparing the NMR solution structures of a synthetic, fully modified tRNALys ASL and hypomodified counterparts has suggested that mnm5s2U34 and t6A37 rigidify the anticodon in a predominantly A-RNA stacked conformation. In addition, t6A37 modestly destabilizes the base pairing interactions of the ASL while mnm5s2U34 and ⌿39 counteract this effect (12). It is conceivable that these conformational effects influence ACNase reactivity. However, some of the modifying groups could also interact with PrrC directly (11). Although the ASL seems the prime target of ACNase, weaker interactions with other substrate regions, e.g. the acceptor domain, are not excluded. This is suggested by the observations that substituting the discriminator base A73 partially inhibits ACNase and truncating the ACCA 3⬘-overhang relaxes the cleavage site specificity (11). 3836 This paper is available on line at http://www.jbc.org Substrate Conformation Favored by Anticodon Nuclease These results indicate that the tRNA acceptor domain helps position ACNase for the native substrate and that alternative RNA-protein interactions may form in the ACCA truncated mutants as well as for A73 mutants. Here we report that ACNase efficiently cleaves a synthetic tRNALys ASL containing the base modifications of E. coli tRNALys. Comparing it with various hypomodified and unmodified analogs revealed opposing effects of the base modifications on ACNase reactivity. Correlating these data with the observed contributions of the base modifications to the ASL solution structure (12, 14, 15) leads us to suggest that ACNase prefers substrates where the anticodon nucleotides assume a stacked ARNA conformation and where the base pairing interactions in the ASL stem are relatively destabilized. We also show that the PrrC D287H mutation renders human tRNALys3 relatively more reactive than the natural substrate. This finding and the inhibitory effect of the side chain of the tRNALys-3 modified wobble base (5-methoxycarbonylmethyl-2-thiouridine, mcm5s2U34) reinforce the previously proposed interaction between PrrC residue Asp287 and the side chain of the E. coli tRNALys wobble base (11). This result and the ability to cleave isolated ASLs raise the interesting prospect of directing ACNase against tRNALys-3 annealed to the genomic RNA template in the priming complex of the human immunodeficiency virus. EXPERIMENTAL PROCEDURES Materials—All ASL oligonucleotides used in this study are listed in Table I. The ASL oligonucleotides 1–15 in Table I were chemically synthesized. The methods have been described for oligonucleotides 1, 3, 4, 6, and 7 (13) oligonucleotides 14 and 15 (16) and the syntheses of oligonucleotides 8 –11 will be described elsewhere.2 ASL oligonucleotides 16 –23 were transcribed in vitro using T7 RNA polymerase according to an established procedure (17). Those containing inosine (ASLs 20 –22) were transcribed using GMP as primer and ITP instead of GTP. The in vitro transcribed ASLs were dephosphorylated by calf intestinal alkaline phosphatase. All ASLs were 5⬘-end labeled using T4 polynucleotide kinase. Bovine tRNALys-3 was a gift from Dr. Roland Marquet, CNRS Strasbourg. Purified E. coli tRNALys labeled with 32P at phosphate 33p34 was prepared by ligating fragments 1–33 and 34 –76 of tRNALys purified from T4-infected E. coli prr⫹ cells (7). T7 RNA polymerase and T4 polynucleotide kinase were purchased from U. S. Biochemical Corp., T4 RNA ligase from New England Biolabs, [␥-32P]ATP from Amersham Biosciences, Inc., poly(U) and trimethylamine-N-oxide from Sigma. Calf intestinal alkaline phosphatase and Protease Inhibitor Mixture Tablets (Complete Mini) were purchased from Roche Molecular Biochemicals and DNA oligonucleotides from Invitrogen. Bacteria and Plasmids—E. coli K38:pGP1–2 encoding thermo-inducible T7 RNA polymerase (18) served as host cell for expressing plasmid pRRC6 (8) and derivatives of plasmid pRRC11 encoding the PrrC mutants D222E (10) or D287H (11). ACNase Assays—E. coli tRNALys and mammalian tRNALys-3 were assayed as substrates of wild type and mutant alleles of ACNase using crude (S-150) fractions essentially as described (11). Assays involving ASLs required in general the removal of nonspecific nucleases. Therefore, they were performed with a partially purified enzyme fraction isolated from E. coli K38:pGP1–2:pRRC11-D222E cells by Superdex 200 gel filtration in a procedure to be detailed elsewhere.3 Additional protection against nonspecific degradation was afforded by including in the reaction mixture carrier poly(U), increasing the trimethylamine-N-oxide concentration to 1.5 M and lowering the reaction temperature from 10 to 0 °C. The standard reaction mixtures (10 l) contained 5 l of the partially purified core ACNase (D222E allele, Superdex 200 peak activity fraction of ⬃270 kDa) containing 6 ng of PrrC, 36 mM NH4Cl, 6 mM Tris-HCl buffer, pH 7.5, 9 mM MgCl2, 3 mM 2-mercaptoethanol, 50 –100 ng of poly(U), 1.5 M trimethylamine-N-oxide, 6% glycerol, protease inhibitor mixture (Complete, Mini, Roche Molecular Biochemicals, diluted according to manufacturers instructions), 2–3 fmol of 5⬘32 P-labeled ASL or E. coli tRNALys labeled with 32P at the ACNase cleavage junction. The products were deproteinized and separated by 2 A. C. Bajji and D. R. Davis, manuscript in preparation. S. Blanga, R. Meidler, M. Amitsur, Y. Jiang, G. Kaufmann, and A. Azem, manuscript in preparation. 3 3837 denaturing polyacrylamide-urea gel electrophoresis. Each assay was replicated 3– 4 times. Kinetic analyses were performed with E. coli tRNALys and several of the ASL substrates found most reactive (listed in Table II). The kinetic analyses entailed measurements of initial reaction rates during incubation times reaching 120 min at 1–50 fmol substrate concentrations. Determinations of cleavage extents were carried out with all the ASL substrates following 3 h incubation. The amounts of product were determined by counting the radioactivity or by densitometric tracing of the gel autoradiogram using Hewlett Packard ScanJet 3p and TINA software (Raytest Isotopenmessgeräte GmbH), compatible with the TINA-PCBAS and TIFF files of the scanner. RESULTS Specific and Efficient Cleavage of the Fully Modified tRNALys ASL by ACNase—An ASL 17-mer containing the three modified bases of E. coli tRNALys (henceforth the reference ASL, Fig. 1), various hypomodified and unmodified derivatives and ASLs containing modified bases of mammalian tRNALys3 (Table I) were examined as ACNase substrates. The reference ASL and other chemically synthesized ASLs (ASLs 1–15) matched the mammalian tRNALys-3 sequence, which differs from that of E. coli tRNALys in the upper three base pairs of the anticodon stem (compare the left and right panels in Fig. 1). However, the upper portion of the stem was considered nonessential for recognition by ACNase (9). The synthetic ASLs 5, 12, and 13 contained an extra 3⬘-dT residue since a labile linker was utilized for the chemical synthesis of the ASLs containing mcm5s2U and ms2t6A.2 The unmodified ASL 15 featuring the mammalian tRNALys3 sequence was also chemically synthesized. Unmodified ASLs 16 –23 were transcribed in vitro and all of them contained a 5⬘ terminal G residue dictated by the constraints of transcription with T7 RNA polymerase (17). The in vitro transcribed ASL 16 corresponded in sequence to E. coli tRNALys (Fig. 1b). ASL 19 resembled the E. coli tRNAVal-3 ASL sequence (19) except for a reversed 5⬘ to 3⬘ terminal GC base pair. The DNA templates of ASL 19 and of two other ASLs with arbitrary GC-rich stem sequences (ASLs 17 and 18) were also transcribed into inosine-rich versions (ASLs 20 –22). ASL 23 was a mutant of E. coli tRNALys sequence in which A37 was replaced by G. Incubation of the 5⬘-32P-labeled reference ASL with a partially purified ACNase preparation under conditions that minimize nonspecific degradation (“Experimental Procedures”) yielded a heptanucleotide-like product missing from the ACNase-negative control (Fig. 2, compare lanes 2 and 3 with lane 4). This product had a slightly reduced electrophoretic mobility compared with a heptanucleotide containing a (2⬘) 3⬘-terminal phosphate within a marker ladder generated by partial alkaline hydrolysis of the reference ASL (Fig. 2, compare lanes 1 and 4). This retardation of the ACNase product could be accounted for by the lower negative charge of the 2⬘:3⬘-cyclic phosphate end-group expected to be found in the product. Incubating the ACNase product with T4 polynucleotide kinase, endowed with 2⬘:3⬘-cyclic-phosphodiesterase and 3⬘-phosphatase activities (7, 20), shifted the product to the position of the 3⬘-dephosphorylated heptanucleotide marker (compare lanes 5, 6), consistent with removal of the cyclic phosphate end group. Hence, ACNase appeared to cleave the ASL similar to the natural substrate, targeting the same site and producing the same cleavage termini. Kinetic comparison of the reference ASL and full sized tRNALys (Table II) yielded respective Kcat (for a PrrC-D222E hexamer) of ⬃0.04 min⫺1 and ⬃0.02 min⫺1 and Km of 2.6 ⫻ 10⫺7 M and 0.8 ⫻ 10⫺7 M. A 2-fold faster cleavage of the ASL was also demonstrated by digesting the two substrates in the same reaction mixture (Fig. 3). Contributions of ASL Base Modifications to ACNase Reactivity—In vitro transcribed, unmodified tRNALys is less reactive 3838 Substrate Conformation Favored by Anticodon Nuclease FIG. 1. Composition of the tRNA ASLs. The sequence of the reference ASL is derived from mammalian tRNALys-3, which differs from E. coli tRNALys in the upper three bases of the anticodon stem. The mammalian tRNALys-3 isoacceptor contains the modified wobble base 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U or U9), the modified base 3⬘ to the anticodon is 2-methylthio-6-threonylcarbamoyl-adenosine (ms2t6A or A9), and the C-nucleoside pseudouridine (⌿) stabilizes the A31-⌿39 base pair. The single E. coli tRNALys isoacceptor has a related modification pattern with 5-methylaminomethyl-2-thiouridine (mmm5s2U or U8), 6-threonylcarbamoyl-adenosine (t6A or A7), and pseudouridine (⌿). The modified nucleoside abbreviations are those originally described by Sprinzl et al. (29). The updated nucleoside modification data base is maintained by McCloskey and co-workers (30). TABLE I Extents of cleavage of ASL substrates by ACNase ASL Stema Modified bases and extra dT % Cleavageb 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 Lys3 EC GC-rich 1 GC-rich 2 Val3-like (G) IC-rich 1 IC-rich 2 Val3-like (I) EC Ala37 3 Gly mnm2s2U34/t6A37/⌿39 ms2t6A37/⌿39 t6A37/⌿39 t6A37 mcm5s2U34/ms2t6A37/⌿39/3⬘-dT s2U34/⌿39 mnm5s2U34/⌿39 mcm5s2U34 mcm5s2U34/⌿39 mcm5U34 mcm5U34/⌿39 ms2t6A37/3⬘dT ms2t6A37/⌿39/3⬘-T ⌿39 60 ⫾ 3 73 ⫾ 4 70 ⫾ 2 73 ⫾ 5 8⫾1 2 ⫾ 0.3 7.2 ⫾ 0.8 1.1 ⫾ 0.2 0.2 ⫾ 0.1 1.1 ⫾ 0.3 0.2 ⫾ 0.1 43 ⫾ 2 30 ⫾ 3 1.8 ⫾ 0.3 3.8 ⫾ 0.4 4 ⫾ 0.5 NDc ND ND 3 ⫾ 0.5 2 ⫾ 0.3 0.4 ⫾ 0.1 4 ⫾ 0.6 a The unmodified Lys3 ASL sequence is 5⬘-UCAGACUUUUAAUCUGA-3⬘. The unmodified E. coli ASL sequence is pGUUGACUUUUAAUCAAC. The arbitrary GC-rich ASLs 1 and 2 are pGUUGACUUUUAAUCAAC and pGGCGGCUUUUAACCGCC, respectively. The Val3-like ASL resembles that of E. coli tRNAVal3 except for reversal of the 5⬘ to 3⬘ base pair. The IC-rich counterparts of the GC-rich and Val3-like ASL contain I instead of G except for the 5⬘ most G. EC A37 3 G is a mutant of the unmodified ASL corresponding to E. coli tRNALys. b Extents of cleavage were measured in standard reaction mixtures incubated for 3 h. c ND, not detectable. than the natural, fully modified ACNase substrate, suggesting a role for at least some of the base modification in substrate recognition and/or reactivity. The tRNALys base modifications relevant to ACNase reactivity could be confined to the ASL domain, judged by the high reactivity of the reference ASL (Fig. 2, Table II). The importance of at least one of the wobble base modifications to ACNase reactivity has been previously suggested by inefficient cleavage of hypomodified tRNALys lacking the mnm5U34 modification (both in vivo and in vitro) or s2U34 (in vivo) and suppression of the wobble base lesions by certain PrrC Asp287 replacement mutations (11). To investigate the contribution of all the ASL base modifications to ACNase reactivity under more defined conditions, we compared the reference ASL with the analogs listed in Table I in an in vitro ACNase assay. Apparent Km and Kcat values were obtained for the most reactive ASLs (Table II). Weaker substrates were characterized by extents of cleavage under standard assay conditions (Table I). Below we use these data to assess the contributions of individual base modifications to ACNase reactivity, comparing matched ASL pairs differing by the presence of a given base modification. The most pronounced effect was exerted by the bacterial t6A37 and mammalian ms2t6A37 modifications. Hypomodified ASLs containing either as the only modified base were cleaved to a 17–18-fold greater extent than the corresponding unmodified ASL. These two partially modified ASLs resembled the reference ASL in Km but had at least 2-fold larger Kcat values (Table II). Hence, the mnm5s2U34 and/or ⌿39 base modifications could attenuate ACNase reactivity of the reference ASL. The specific contributions of the wobble base modifications to ACNase reactivity were evaluated similarly. The extent of cleavage of the mnm5s2U34/⌿39-ASL was 3– 4-fold larger than that of the s2U34/⌿39-ASL (Table I). This suggested that the mnm5U34 side chain exerts a positive effect on ACNase reactivity. In contrast, the mcm5U34-ASL and mcm5s2U34-ASL, both containing the side chain of the tRNALys-3 wobble base, were less reactive than the corresponding unmodified ASL (Table I). The opposing effects of mnm5U34 and mcm5U34 were underscored by the large difference in the extents of cleavage between the mnm5s2U34/⌿39 and mcm5s2U34/⌿39 containing ASLs (7.2 versus less than 0.2%, respectively). The negative effect of the mcm5U34 was further demonstrated by the different kinetic values of the ms2t6A37/⌿39/3⬘-dT ASL (Km ⫽ 3.0 ⫾ 0.6 ⫻ 10⫺8 M, Kcat ⫽ 0.014 min⫺1) and mcm5s2U34/ms2t6A37/ Substrate Conformation Favored by Anticodon Nuclease FIG. 2. ACNase specifically cleaves the reference ASL. The 5⬘P-labeled reference ASL was incubated with partially purified ACNase (Superdex 200 fraction) and the products were separated by denaturing 20% polyacrylamide-urea gel electrophoresis as such (lane 4) or after treatment with T4 polynucleotide kinase (lane 5). A marker “ladder” (lane 1) was obtained by partial alkaline hydrolysis of the ASL. Part of the digest was further treated with polynucleotide kinase that effectively dephosphorylates tri- and higher oligonucleotides (lane 6). The gap between the heptamer and octamer markers in lanes 1 and 6 is due to the presence of the basic mnm5s2U34 residue in the octamer. Numbers on the right are chain length in nucleotides; numbers on the left marked by an asterisk represent the slower migrating 3⬘-OH containing oligonucleotides. Lanes 2 and 3 are respective controls of untreated ASL or ASL incubated with inactivated ACNase (Superdex fraction preincubated 30 min at 37 °C). 3839 32 TABLE II Kinetic parameters of different ASL substrates of ACNase Substrate Kcata min E. coli tRNALys mnm5s2U34/t6A37/⌿39-ASL ms2t6A37/⌿39-ASL t6A37/⌿39-ASL t6A37-ASL ms2t6A37/3⬘-dT-ASL ms2t6A37/⌿39/3⬘-dT-ASL mcm5s2U34/ms2t6A37/⌿39/3⬘-dT-ASL a ⫺1 0.018 ⫾ 0.005 0.044 ⫾ 0.009 0.199 ⫾ 0.091 0.145 ⫾ 0.055 0.109 ⫾ 0.073 0.016 ⫾ 0.004 0.014 ⫾ 0.0005 0.004 ⫾ 0.0005 KM M 10⫺8 8⫾2 26 ⫾ 6 30 ⫾ 10 17 ⫾ 7 60 ⫾ 30 3.8 ⫾ 0.8 3.0 ⫾ 0.6 6.0 ⫾ 2.0 Reaction mixtures (20 l) contained 12 ng of PrrC (2.2 nM hexamer). ⌿39/3⬘-dT ASL (Km ⫽ 6.0 ⫾ 2.0 ⫻ 10⫺8 M, Kcat ⫽ 0.004 min⫺1). Thus, mcm5s2U34 reduced the Kcat by more than 3-fold and the binding affinity by ⬃2-fold and the overall catalytic efficiency ⬃7-fold. On the other hand, adding mnm5s2U34 to the t6A/⌿39 background increased Kcat by more than 3-fold and reduced the affinity ⬃2-fold (Km ⫽ 2.6 ⫾ 0.6 ⫻ 10⫺7 M, Kcat ⫽ 0.044 min⫺1 versus Km ⫽ 1.7 ⫾ 0.7 ⫻ 10⫺7 M, Kcat ⫽ 0.144 min⫺1, respectively). Hence, the negative changes in catalytic efficiency con- FIG. 3. E. coli tRNALys and the reference ASL as ACNase substrates. The two substrates were incubated in the same reaction mixture. RNA extracted from aliquots withdrawn at the indicated times was separated by denaturing polyacrylamide-urea gel electrophoresis. ASL frag, heptanucleotide cleavage product of the ASL. ferred by mcm5s2U34 may be ascribed to the side chain rather than 2-thiol group. This inference is supported by the similar inhibitions caused by mcm5U34 and mcm5s2U34 when introduced into the unmodified background (Table I, compare ASLs 8 and 10 with 15). It is noteworthy that the lack of the s2U34 modification from E. coli tRNALys had no detectable effect on the cleavage efficiency in vitro although in vivo this lesion was synthetically lethal with PrrC and severely inhibited tRNALys cleavage (11). The fully modified E. coli tRNALys mnm5s2U34/t6A37/⌿39ASL was cleaved with a 10-fold higher Kcat and exhibited a 3– 4-fold weaker binding compared with mcm5s2U34/ms2t6A37/ ⌿39/3⬘-dT-ASL carrying the base modifications of tRNALys-3 (Table II). However, confounding this comparison is the large negative effect of the extra 3⬘-dT residue in the latter substrate. Namely, against the ms2t6A37/⌿39 backdrop, the extra 3⬘-dT reduced the Km and Kcat each by about an order of magnitude (Table II). The presence of ⌿39 instead of U39 rendered the ASL in certain cases less reactive. The unmodified ASL was cleaved to a 2-fold higher extent than the ⌿39-containing counterpart. Similarly, introducing ⌿39 into ASLs containing mcm5U34 or 3840 Substrate Conformation Favored by Anticodon Nuclease FIG. 4. Mutation D287H renders tRNALys-3 more reactive than E. coli tRNALys. The two tRNA substrates were incubated in the standard reaction mixture containing the indicated PrrC alleles. The products were separated by denaturing polyacrylamide urea gel electrophoresis as described in the legend to Fig. 3. The ratios between the reactivities of the two substrates were calculated from the initial rates of cleavage. EC, E. coli tRNALys; Lys3, tRNALys-3. mcm5s2U34 reduced their activity more than 5-fold (Table I). However, ⌿39 had no apparent effect on either kinetic parameter when introduced in the ms2t6A37/3⬘-dT context and over the t6A37 background it even reduced the Km ⬍3-fold (Table II). ASL Reactivity and Stem Stability—Unmodified ASLs with stems (underlined) corresponding in sequence to mammalian tRNALys3 (5⬘-UCAGACUUUUAAUCUGA-3⬘) or E. coli tRNALys (5⬘-GUUGACUUUUAAUCAAC-3⬘) were similarly reactive as ACNase substrates, confirming the assumption that the top three base pairs are not critical for ACNase specificity (9). Arbitrary, all GC stems of the respective ASLs 17 and 18 (5⬘-GGCGGCUUUUAACCGCC-3⬘ and 5⬘-GCCGGCUUUUAAGCGGC-3⬘) abolished the reactivity. A similar effect was exerted by the GC-rich stem of ASL 19, resembling that of E. coli tRNAVal-3 except for a reversed 5⬘ to 3⬘ terminal pair (5⬘-GCACCCUUUUAAGGUGC-3⬘). Replacing the G residues of these ASLs with inosine residues (except for the 5⬘-terminal) restored the reactivity almost to the level of the unmodified reference ASLs in the case of the two arbitrary stem sequences. However, the tRNAVal-3-like I-rich ASL was an order of magnitude less reactive. A PrrC Mutation Renders tRNALys-3 More Reactive Than the Natural Substrate—The D287H, D287N, or D287Q mutations have rendered hypomodified E. coli tRNALys forms containing s2U34 or mnm5U34 more reactive than the natural substrate, opposite to the preference of wild type PrrC (11). These effects, attributed to some interaction of Asp287 with the wobble base, have also raised the prospect of designing new ACNase cleavage specificities (10), especially for a differently modified wobble base such as that of mammalian tRNALys-3. In fact, whereas wild type PrrC or the pseudo-wild type allele D222E (11) cleaved E. coli tRNALys 2–3 faster than mammalian tRNALys-3, D287H featured the opposite preference (Fig. 4). DISCUSSION The tRNALys ASL as a Minimal Essential Substrate of ACNase—The reference ASL with the three modified bases of E. coli tRNALys was cleaved by ACNase twice as fast as the natural substrate but featured a 3– 4-fold reduced affinity to the enzyme, suggesting that the ASL domain contains all or nearly all of the ACNase recognition determinants. The small differences between the ASL and full-sized tRNA in Kcat and Km are attributed to greater conformational freedom of the ASL. Such flexibility could facilitate attainment of a productive transition-state conformation by the ASL but weaken initial binding to the enzyme. However, the weaker binding of the ASL also leaves open possible interactions between PrrC and other portions of tRNALys such as the acceptor region. Namely, mutating the A73 discriminator base inhibits ACNase and truncating the 3⬘-terminal ACCA sequence relaxes the cleavage site specificity (11). These outcomes could be explained by a weak interaction between PrrC and the acceptor region of tRNALys. Accordingly, the A73 discriminator could contribute to substrate specificity, directly or by counter-selecting other tRNAs. We also speculate that truncation of the substrate to contain just the ASL eliminates these interactions that cause misalignment and therefore allows for specific and efficient recognition of the cleavage site. Structural ASL Features Favored by ACNase—Comparing the NMR solution structures of the reference ASL and hypomodified counterparts has suggested that individual base modifications play distinct and partially opposing roles in transforming the disordered, unmodified tRNALys ASL into the highly ordered native structure (12). Thus, t6A37 improves stacking with A38, and strengthens the interaction of U33 N3H with the 35p36 phosphate, a hallmark of the anticodon loop U-turn. On the other hand, t6A37 partially destabilizes the ASL stem base pairing, reducing the Tm by about 2 °C. The wobble base side chain mnm5U34 also stabilizes the anticodon base stacking and U-turn conformation but partially counteracts the base pair disrupting effect of t6A37. Destabilization of the stem and/or the C32-A38 base pair (21) by t6A37 also opposes the effect of the ⌿39 modification (12, 14, 15). Here we have shown that the ASL base modifications also exert distinct effects on ACNase reactivity (Tables I and II). The underlying causes of these complicated and opposing effects may be understood by considering the contributions of the individual base modifications to the ASL solution structure, as described above. Such a correlation leads us to propose that ACNase favors substrates where the anticodon nucleotides are held in a helical A-RNA conformation and the U-turn conformation is retained but the ASL stem base pairing interactions are modestly destabilized. This assumption is backed by the following arguments. First, t6A37 or ms2t6A37 that dramatically enhanced the ACNase reaction rate also stabilize the anticodon A-RNA and the loops U-turn conformations and destabilize the anticodon stem and bifurcated C32-A38 base pair (12, 14), suggesting that ACNase favors these structural features. In agreement, ⌿39, which inhibited ACNase activity in certain cases, exerts the opposite effect on ASL base pairing interactions. As for the mild positive effect of mnm5U34 on ACNase reactivity, it could reflect a balance between a positive contribution to ACNase reactivity of the enhanced anticodon stacking by this modification and a negative contribution due to the stabilized base pairing. However, part of the ACNase-stimulating effect of mnm5U34 may be attributed to a direct interaction with Asp287, the anticodon-recognizing residue of PrrC (an issue to be elaborated below). Abolition of ACNase reactivity by the GC-rich stems, but not the IC-rich, reinforces the notion that base pairing stability of the ASL negatively affects the reactivity. As already mentioned, the higher Kcat and lower Km of the reference ASL compared with the full-sized tRNALys may be attributed to the fact that the upper base pairs of the stem are more constrained within the intact tRNA structure. Hence, the destabilization of the anticodon stem, needed for the productive interaction with ACNase, may be inhibited with intact tRNALys. However, as we have seen for the ASLs, the more rigid conformation of the intact tRNA could promote tighter initial binding to the enzyme. Substrate Conformation Favored by Anticodon Nuclease Transplanting the tRNALys UUU anticodon within an otherwise tRNAVal-3(UUU) sequence does not elicit detectable ACNase reactivity whereas tRNAArg(UUU) is highly reactive (11). The difference between the two chimerical tRNA sequences has been attributed to ill-defined ACNase determinants found outside the anticodon loop and shared by tRNALys and tRNAArg but missing from tRNAVal-3. However, in view of the current data, a new interpretation becomes apparent. Namely, the failure to cleave tRNAVal-3(UUU) may reflect the greater stability of its GC-rich anticodon stem (4 GC pairs) and the reversed purine-pyrimidine configuration of the two base pairs at the bottom of the stem. The importance of this configuration is suggested by the weaker reactivity of the IC-rich version of the tRNAVal-3-ASL, compared with the counterparts with arbitrary stem sequences that shared the tRNALys/ tRNAArg configuration. Presumably, the reversed base pair configuration further stabilizes the stem by enhancing G39-A38 base stacking. A Distinct Wobble Base Side Chain Causes the Reduced Reactivity of tRNALys-3—The relative ACNase reactivities of natural substrate, the completely unmodified version and mutants thereof as well as of the mammalian tRNALys isoacceptors have suggested the following order of wobble base preference of ACNase: mnm5s2U⬎U⬎C⬎mcm5s2U (11). However, such an order has been derived from eclectic data collected in vivo and in vitro and comparisons of substrates differing in more that just their wobble bases. The current comparison uses defined in vitro assay conditions of matched ASLs differing only in a wobble base modification (Tables I and II). These results confirm the opposing contributions of the bacterial mnm5U34 and mammalian mcm5U34 side chains to ACNase reactivity, whereas both modifications similarly influence the overall ASL conformation (12, 24). Therefore, the inhibitory effect of mcm5U34 on ACNase reactivity suggests a specific interaction between residue Asp287 of PrrC and the mnm5U34 side chain of the natural substrate (11). Since replacing mnm5U34 with mcm5U34 reduces both Km and Kcat (Table II), the Asp287mnm5U34 interaction could influence both substrate binding and the catalytic step. The detection of PrrC mutations that alter the substrate cleavage specificity and compensate for a missing wobble base modification (11) raised the possibility that some of these mutations would also cause ACNase to prefer mammalian tRNALys-3 over the natural substrate. One interest in such an outcome stems from the role human tRNALys-3 plays as the primer tRNA of reverse transcription in human immunodeficiency viruses (22, 23). There are several characteristics of the tRNALys-3 primer that raise the prospect of using ACNase as a model for developing anti-HIV therapeutics. The tRNALys-3 primer is indifferent to viral genetic drift (25) and the tRNA anticodon interaction with the HIV A-loop is critical for transcription initiation (26). Provided that the tRNA anticodon is accessible to ACNase in the primer-template complex, it may be possible to tailor ACNase to discriminate between the free form of tRNALys-3 and the annealed tRNA. Detection of ACNase derivatives more proficient in cleaving tRNALys-3 would constitute a step toward the goal of developing a model system for therapeutics targeted at the primer-template complex. Since the reduced reactivity of tRNALys-3 is likely due to the presence of mcm5U34 instead of mnm5U34 in the natural substrate, it was expected that tRNALys-3 will be rendered 3841 more reactive by some of the PrrC mutations that compensate for the absence of mnm5U34 (11). This expectation was confirmed by the in vitro behavior of the D287H allele, which cleaved tRNALys-3 relatively faster than wild type and pseudowild type alleles, whereas the latter two alleles were relatively more reactive with E. coli tRNALys (Fig. 4). Mechanistically, the reversal of substrate preference may be explained in that the mnm5U34-Asp287 and mcm5U34-D287H pairs form saltbridges or hydrogen bond interactions of opposite polarities that are vital for substrate binding and/or reactivity. Interestingly, among the PrrC D287 replacement mutants expressed in mammalian cells D287Q showed the highest activity with tRNALys-3, cleaving it to a severalfold higher extent than wild type PrrC.4 The efficient cleavage of the isolated ASL domain by ACNase also suggests that partially melted tRNALys-3 annealed to the HIV-1 genomic RNA will be recognized by ACNase since the native ASL conformation would be retained in the primer-template complex (27, 28). This expectation has been recently confirmed.5 REFERENCES 1. Kaufmann, G. (2000) Trends Biochem. Sci. 25, 70 –74 2. Levitz, R., Chapman, D., Amitsur, M., Green, R., Snyder, L., and Kaufmann, G. (1990) EMBO J. 9, 1383–1389 3. Linder, P., Doelz, R., Gubler, M., and Bickle, T. A. (1990) Nucleic Acids Res. 18, 7170 4. Amitsur, M., Morad, I., Chapman-Shimshoni, D., and Kaufmann, G. (1992) EMBO J. 11, 3129 –3134 5. Tyndall, C., Meister, J., and Bickle, T. A. (1994) J. Mol. Biol. 237, 266 –274 6. Penner, M., Morad, I., Snyder, L., and Kaufmann, G. (1995) J. Mol. Biol. 249, 857– 868 7. Amitsur, M., Levitz, R., and Kaufmann, G. (1987) EMBO J. 6, 2499 –2503 8. Morad, I., Chapman-Shimshoni, D., Amitsur, M., and Kaufmann, G. (1993) J. Biol. Chem. 268, 26842–26849 9. Shterman, N., Elroy-Stein, O., Morad, I., Amitsur, M., and Kaufmann, G. (1995) Nucleic Acids Res. 23, 1744 –1749 10. Meidler, R., Morad, I., Amitsur, M., Inokuchi, H., and Kaufmann, G. (1999) J. Mol. Biol. 287, 499 –510 11. Jiang, Y., Meidler, R., Amitsur, M., and Kaufmann, G. (2001) J. Mol. Biol. 305, 377–388 12. Sundaram, M., Durant, P. C., and Davis, D. R. (2000) Biochemistry 39, 12575–12584 13. Sundaram, M., Crain, P. F., and Davis, D. R. (2000) J. Org. Chem. 65, 5609 –5614 14. Stuart, J. W., Gdaniec, Z., Guenther, R., Marszalek, M., Sochacka, E., Malkiewicz, A., and Agris, P. F. (2000) Biochemistry 39, 13396 –13404 15. Durant, P. C., and Davis, D. R. (1999) J. Mol. Biol. 285, 115–131 16. Bajji, A. C., and Davis, D. R. (2000) Org. Lett. 2, 3865–3868 17. Milligan, J. F., and Uhlenbeck, O. C. (1989) Methods Enzymol. 180, 51– 62 18. Tabor, S., and Richardson, C. C. (1985) Proc. Natl. Acad. Sci. U. S. A. 82, 1074 –1078 19. Sprinzl, M., Horn, C., Brown, M., Ioudovitch, A., and Steinberg, S. (1998) Nucleic Acids Res. 26, 148 –153 20. Cameron, V., and Uhlenbeck, O. C. (1977) Biochemistry 16, 5120 –5126 21. Auffinger, P., and Westhof, E. (1999) J. Mol. Biol. 292, 467– 483 22. Mak, J., and Kleiman, L. (1997) J. Virol. 71, 8087– 8095 23. Arts, E. J., Miller, J. T., Ehresmann, B., and Le Grice, S. F. (1998) J. Biol. Chem. 273, 14523–14532 24. Benas, P., Bec, G., Keith, G., Marquet, R., Ehresmann, C., Ehresmann, B., and Dumas, P. (2000) RNA 6, 1347–1355 25. Wakefield, J. K., Wolf, A. G., and Morrow, C. D. (1995) J. Virol. 69, 6021– 6029 26. Huang, Y., Shalom, A., Li, A., Wang, J., Mak, J., Wainberg, M. A., and Kleiman, L. (1996) J. Virol. 70, 4700 – 4706 27. Puglisi, E. V., and Puglisi, J. D. (1998) Nat. Struct. Biol. 5, 1033–1036 28. Elgavish, T., VanLoock, M. S., and Harvey, S. C. (1999) J. Mol. Biol. 285, 449 – 453 29. Sprinzl, M., Hartmann, T., Weber, J., Blank, J., and Zeidler, R. (1989) Nucleic Acids Res. 17, r1–r172 30. Rozenski, J., Crain, P. F., and McCloskey, J. A. (1999) Nucleic Acids Res. 27, 196 –197 4 M. Perlman, M. Amitsur, O. Elroi-Stein, L. Kleiman, and G. Kaufmann, manuscript in preparation. 5 M. Perlman, C. Isel, R. Marquet, and G. Kaufmann, unpublished results.