Journal of Controlled Release 100 (2004) 97 – 109
www.elsevier.com/locate/jconrel
Encapsulation, stabilization, and release of BSA-FITC
from polyanhydride microspheres
Amy S. Determana, Brian G. Trewynb, Victor S.-Y. Linb,
Marit Nilsen-Hamiltonc, Balaji Narasimhana,*
a
Department of Chemical Engineering, Iowa State University, 2035 Sweeney Hall, Ames, IA 50011, USA
b
Department of Chemistry, Iowa State University, Ames, IA 50011, USA
c
Department of Biochemistry, Biophysics, and Molecular Biology, Iowa State University, Ames, IA 50011, USA
Received 28 May 2004; accepted 11 August 2004
Available online 21 September 2004
Abstract
In order to determine the efficacy of using polyanhydrides as a carrier for therapeutic proteins, the model protein bovine
serum albumin labeled with fluorescein isothiocyanate (BSA-FITC) was encapsulated in microspheres of poly sebacic
anhydride (poly(SA)), and random copolymers of poly(SA) and poly(1,6-bis-p-carboxyphenoxy)hexane (poly(CPH)). The
microspheres were fabricated via the double emulsion (water/oil/water) technique and were characterized using scanning
electron microscopy, gel permeation chromatography, confocal microscopy, and a Coulter counter. The effect of protein
loading, protein distribution, and change in polymer composition was examined in an in vitro release study. The secondary
structure of the encapsulated BSA-FITC was determined with Fourier transform infrared spectroscopy. The primary structure of
the released protein was analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis. Poly(SA) and 20:80
(CPH:SA) microspheres were found to conserve the primary structure of the released protein and the secondary structure of the
encapsulated protein, and showed a sustained delivery for approximately 15 and 30 days, respectively. As the CPH content in
the copolymer increased, the secondary structure of FITC-BSA was not conserved, as indicated by the steep decrease in the ahelix content.
D 2004 Elsevier B.V. All rights reserved.
Keywords: Polyanhydrides; Bovine serum albumin; Primary structure; Secondary structure; Stabilized
1. Introduction
* Corresponding author. Tel.: +1 515 294 8091; fax: +1 515
294 2689.
E-mail address: nbalaji@iastate.edu (B. Narasimhan).
0168-3659/$ - see front matter D 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.jconrel.2004.08.006
Between 1980 and 2001, the Food and Drug
Administration (FDA) approved 554 new therapeutic
drugs [1]. Small molecular weight drugs made up the
majority of the new therapeutics approved (504),
followed by recombinant proteins (40), and mono-
98
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
clonal antibodies (10). As recombinant DNA practices
have become a routine, the number of protein
products entering the market has increased. With the
increasing number of therapeutic proteins being
marketed in United States each year more research
is being directed towards developing superior methods of delivering proteins. Unlike small molecular
weight drugs, proteins are complex three-dimensional
molecules, whose functionality depends on their
higher-order structure [2]. Proteins are prone to
chemical (e.g., deamidation, oxidation) and physical
(aggregation, precipitation, and adsorption) alterations
[2–5]. The mechanisms by which proteins undergo
structural alterations is protein-specific; however,
there are known factors that decrease the stability of
proteins such as elevated temperature and moisture
[3,4,6,7]. Parenteral administration remains the most
common method of delivering proteins; however, it
provides no stabilization of the shelf life of the protein
or increase of the half-life of the protein in vivo [8].
A vehicle capable of encapsulating proteins,
minimizing the mechanisms of degradation and maximizing the in vivo activity, and providing controlled
release that can be delivered via parental administration is needed. The polymer that has received the
most attention as a protein delivery vehicle is
poly(d,l-lactide-co-glycolide) (PLGA). PLGA has
been used to encapsulate and release numerous model
and recombinant proteins, such as bovine serum
albumin (BSA) [9–16], lysozyme [15,17,18],
recombinant human growth hormone (rhGH) [19–
22], and recombinant human insulin like growth
factor-1 (rhIGF-I) [23–25]. Because PLGA is a
bulk-eroding polymer, in an aqueous environment
the encapsulated protein is exposed to elevated
moisture content. This increase in moisture can cause
aggregation of the encapsulated protein [6,7]. The
addition of excipients (i.e., trehalose or dextran) helps
prevent covalent aggregation of the encapsulated
protein [12,13,26]. Another issue with PLGA is that,
as it degrades, the pH within the polymeric device
drops significantly [27], which can be detrimental to
the protein. This problem can be overcome by the coencapsulation of basic compounds, which have
proven to help stabilize the encapsulated protein [9].
Polyanhydrides are biodegradable polymers that
have also been shown to stabilize and provide
sustained release of proteins [28]. Unlike PLGA,
polyanhydrides are surface eroding polymers, which
minimize the moisture level to which an encapsulated protein is exposed, thus reducing protein
aggregation due to moisture [29–32]. Another
benefit of polyanhydrides is that the pH of the
degrading polymeric material does not drop as
severely as for PLGA, thus providing a more
suitable microclimate for encapsulated (and released)
protein molecules [31].
In parenteral formulations, microspheres are the
most commonly used vehicle to encapsulate proteins
or small molecular weight drugs. Microspheres can be
fabricated by three different procedures: hot melt,
solvent removal, or spray drying [33–40]. The hot melt
procedure is not advantageous when encapsulating
proteins, because the elevated temperatures needed to
melt the polymer can denature the protein. Solvent
removal, either oil-in-oil (O/O) or water-in-oil-inwater (W/O/W) (also known as the double emulsion
method), and spray drying can both be performed at
room temperature. While spray drying requires the use
of an atomizer, the solvent removal technique requires
no special equipment; however, care must be taken
when encapsulating proteins via the solvent removal
technique because the presence of a water/oil interface
can cause protein inactivation [41].
The polymers used in this study are poly(sebacic
anhydride) (poly(SA)) and random copolymers of
poly(SA) and poly(1,6-bis-p-carboxyphenoxy)hexane
(poly(CPH)) (see Fig. 1). These polymers are of
interest because they have vastly different degradation
rates. Poly(CPH) degrades on a time scale of years,
while poly(SA) degrades on a time scale of weeks
[42]. Random copolymers of SA and CPH have
degradation times that fall in between the degradation
time of the homopolymers [43]. By varying the
polymer chemistry, a suitable degradation time can
be achieved to meet delivery needs.
The release of p-nitroaniline (PNA), a small
molecular weight model drug, from microspheres of
homopolymers and copolymers of poly(CPH) and
poly(SA) has previously been studied [44]. The
release profile of PNA was shown to be a function
of polymer erosion rate and the interaction of the drug
with the polymer. It is the hypothesis of this study that
the release of proteins from polyanhydrides is also a
function of polymer erosion rate and the interaction of
the protein with the polymer.
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
99
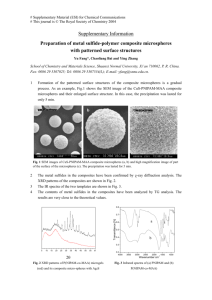
Fig. 1. Structure of repeating units of polymers: (a) poly(CPH); (b) poly(SA).
The focus of this work is to determine the
effectiveness of polyanhydride carriers such as poly(SA) and copolymers of poly(CPH) and poly(SA) for
stabilization and release of bovine serum albumin
labeled with fluorescein isothiocyanate (BSA-FITC).
The microspheres are fabricated via the double
emulsion technique and are characterized by various
techniques. Sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) is used to determine
the primary structure of BSA-FITC after it is released
from the eroding polyanhydride microspheres. The
secondary structure of BSA-FITC encapsulated in the
microspheres is assessed using Fourier transform
infrared spectroscopy (FTIR).
2. Materials and methods
2.1. Materials
Sebacic acid (99%), p-carboxy benzoic acid
(99+%), and 1-methyl-2-pyrrolidinone anhydrous
(99+%), and KBr (FTIR grade) were purchased from
Aldrich (Milwaukee, WI). 1,6-dibromohexane
(98+%) and poly(vinyl alcohol) (99+%) (PVA) were
purchased from Acros (Fairlawn, NJ). Fluorescein
isothiocyanate bovine serum albumin (BSA-FITC)
was purchased from Sigma (St. Louis, MO). The BCA
assay kit was purchased from Pierce Chemicals
(Rockford, IL). Forty percent polyacrylamide/bis
19:1, the SDS-PAGE silver staining kit, and molecular
weight standards for SDS-PAGE were purchased from
BioRad (Hercules, CA). Petroleum ether (hexane,
55% n-hexane) was purchased from Fisher Scientific
(Pittsburgh, PA) and dried and distilled over sodium
and benzophenone (Fisher) before use. All the other
chemicals were of analytical grade and were purchased from Fisher Scientific.
2.2. Polymer synthesis
The prepolymers of SA and CPH were synthesized by the methods described by Shen et al. [45],
and for the CPH prepolymer the method was similar
to the one described by Conix [46] for 1,3-bis( pcarboxyphenoxy)propane. The polymers were synthesized by melt polycondensation as described by
Kipper et al. [44].
2.3. Polymer characterization
Neat polymers were characterized by the procedures described by Kipper et al. [44]. The chemical
structure of the polymers was characterized with 1H
NMR. Samples were dissolved in deuterated chloroform (99.8% atom-d) and the chloroform peak was
used to calibrate the chemical shift of the polymer.
Polymer molecular weight was determined using gel
permeation chromatography (GPC). A PL Gel column
from Polymer Laboratories (Amherst, MA) and a
Waters GPC system (Milford, MA) were used to
separate the samples dissolved in chloroform. The
elution times of the samples were compared to
poly(methyl methacrylate) standards from Fluka
(Milwaukee, WI). Using differential scanning calorimetry (DSC) (DSC7, Perkin Elmer, Shelton, CT),
the melting point of the semicrystalline polymers was
compared to that reported by Shen et al. [45].
2.4. Microsphere fabrication
A double emulsion technique was employed to
encapsulate BSA-FITC in microspheres of varying
polymer composition. The following polymers were
used: poly(SA), 20:80 (CPH:SA), 50:50 (CPH:SA),
and 80:20 (CPH:SA). The fabrication technique used
was similar to the method reported by Tabata and
100
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
Langer [28]. Briefly, BSA-FITC (5 mg) was dissolved
in de-ionized water (200 Al) and was added to polymer
(100 mg) dissolved in methylene chloride (2 ml). The
two phases were emulsified with a homogenizer at
10,000 rpm for 30 s (Tissue-Tearek, Biospec Products, Bartlesville, OK) forming the inner emulsion. One
percent PVA (4 ml) saturated with methylene chloride
(80 Al) was immediately added to the inner emulsion
and homogenized for 30 s at 10,000 rpm to form the
double emulsion. The microspheres were then dispersed in 1% PVA (100 ml) and stirred for 2 h at 300
rpm using an overhead stirrer with a 3-in. impeller
(Wiarton, Ontario). The microspheres were collected
by centrifugation for 10 min at 1500g using an
Eppendorf Centrifuge 5403 (Westbury, NY). After the
first centrifugation, the supernatant was collected and
then replaced with de-ionized water to wash the
microspheres. The microspheres were washed two
additional times to ensure all of the free protein and
PVA had been removed. The microspheres were then
suspended in 4 ml of de-ionized water, flash frozen, and
dried under vacuum overnight.
2.5. Microsphere characterization
The mass of microspheres recovered divided by the
initial mass of polymer and protein was used to
calculate the yield. To determine the size distribution,
the microspheres were dispersed by sonication in
CoulterR Balanced Electrolyte Solution prior to using
a Beckman Model Multisizerk Three Coulter Counter (Fullerton, CA). Scanning electron microscopy
(SEM) (Hitachi S-2460 N) was used to study the
surface morphology of the microspheres. Confocal
microscopy (Nikon Eclipse TE200 microscope and
Chiu Technical 100 W mercury source) was used to
analyze the BSA-FITC distribution within the microspheres. The molecular weight loss during the
fabrication process was determined using GPC by
comparing the molecular weight of blank microspheres to that of neat polymer.
2.6. Protein loading
The amount of BSA-FITC encapsulated in the
microspheres was indirectly determined using a
procedure similar to the one described by Bouillot et
al. [11]. During the fabrication process, the micro-
spheres were collected by centrifugation, and the
supernatant was collected. The BCA protein assay
was utilized to determine the concentration of BSAFITC present in the supernatant, thus providing the
mass of protein not encapsulated. A mass balance was
performed to determine the amount of BSA-FITC that
was loaded into the microspheres. The protein that
was lost while washing the microspheres during the
fabrication process was not accounted for when
calculating the loading. Hence the loading and loading
efficiency are slightly overestimated. The encapsulation efficiency of the protein was determined by
dividing the mass of the loaded protein by the initial
mass of protein.
2.7. In vitro protein release
Microspheres (15 mg) were suspended in 1 ml of
water containing 3% (w/w) sodium dodecyl sulfate
(SDS). The samples were placed in an incubator at
37 8C and continuously agitated at 100 rpm. To
determine the mass fraction of BSA-FITC released,
the supernatant from the samples was collected at
predetermined times and fresh release media was
added to maintain perfect sink conditions. The
concentration of protein in each sample was
determined using the BCA protein assay upon
removal. The amount of protein released was
normalized by the amount of protein initially loaded
into the microspheres. The experiments were done
in triplicate.
2.8. SDS-PAGE
SDS-PAGE was used to determine the primary
structure of the BSA-FITC released from microspheres
in vitro. Microspheres were suspended in water
containing 3% (w/w) SDS under the same conditions
as the microspheres used for the in vitro release study.
The microspheres were allowed to degrade until the
concentration of BSA reached at least 30 Ag/ml. The
samples were mixed with an equal volume of a SDS
(1% w/v), Tris–HCl (pH 6.8) (0.06 mM), glycerol (3
mM), bromophenol blue (0.01% w/v) solution with and
without h-mercaptoethanol (0.05% v/v) for staining
with silver. h-Mercaptoethanol is a reducing agent that
breaks covalent disulfide bonds [47], thus by comparing the samples with and without h-mercaptoethanol,
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
Table 1
Characteristics of microsphere yields and loading efficiency of
BSA-FITC
Polymer
Yield (%)
Loading
efficiency (%)a
SA
20:80
50:50
80:20
68F11
56F9
67F8
57F10
54F14
68F13
48F16
54F8
a
F values are standard deviation values calculated from a
minimum of five batches.
the formation of inter-protein disulfide bonds between
BSA-FITC molecules could be determined. The
samples were resolved through a 10% polyacrylamide
gel with a 5% polyacrylamide stacker. Electrophoresis
was performed using a BioRad Mini-Protean II electrophoresis setup at a constant voltage (120 V) as
described by the manufacturer. The gels were stained
with silver using a Silver Stain kit (BioRad). The
molecular weight of the detected bands was compared
to standards. BSA-FITC samples were also run on the
gel to verify the accuracy of the standards. The silverstained gels were immediately photographed and dried
overnight.
2.9. FTIR
FTIR spectroscopy was performed using a Nicolet
Nexus 470 (Madison, WI), equipped with a cooled
MCT/A detector and an Ever-Glo source. Omnic 5.2
101
software was used to collect the data and to perform
initial data analysis. Dry air was purged through the
optical bench throughout data acquisition in order to
reduce IR absorbance due to water. Samples were
prepared by mixing microspheres (either BSA-loaded
or blank) with KBr (4% w/w). In order to compare the
native structure of the protein with that of the
encapsulated protein, BSA-FITC samples were also
mixed with KBr (4% w/w). Prior to collecting FTIR
spectra, all samples were stored at 50 8C under
vacuum overnight to eliminate any residual moisture.
The dry powder was then placed in a 7-mm dye and a
pellet was formed with a hand press (Spectra-Tech.,
Sheldon, CT).
Each spectrum was collected by running a total of
256 scans at a resolution of 4 cm 1. The spectra of
the encapsulated BSA-FITC were obtained by subtracting the spectra of the blank microspheres from
the spectra of the BSA-FITC-loaded microspheres. A
subtraction was considered successful if the resulting
spectra had a straight baseline in the region of 1800–
2500 cm 1 [48]. Fourier self-deconvolution was then
performed on the amide I region of the subtracted
spectra using the Omnic 5.2 software with an
enhancement factor of 1.2 and a bandwidth of 20
kHz [49]. Gaussian curves were fit to the Fourier
self-deconvoluted spectra [15,49]. The same procedure was also carried out on the spectra of the native
protein. The assignment of secondary structure either
a-helix, h-sheet, or unordered was done using the
assignment of peaks for BSA listed by Fu et al. [15].
Fig. 2. Size distribution based on volume percent of microspheres as a function of particle diameter for poly(SA) microspheres.
102
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
Table 2
Molecular weight characteristics of polymer microspheres as determined by GPC
Polymer
M w/M n of neat polymer
M w/M n of microspheres
M n loss (%)
Poly(SA)
CPH:SA (20:80)
CPH:SA (50:50)
CPH:SA (80:20)
74,000/15,000
21,000/10,000
15,000/7000
20,000/10,000
37,000/11,000
18,000/9000
14,000/7000
20,000/10,000
25
11
0
0
The area of each peak in the amide I region was
calculated and used to determine the secondary
structure of the protein using procedures described
elsewhere [12–15,22,48–51].
3. Results and discussion
3.1. Microsphere characterization
The yield for the microspheres was 56–67%, with
the centrifugation step being the most pivotal in the
overall recovery of microspheres (see Table 1). The
yield was calculated from no less than five batches of
microspheres of all compositions. The size distribution of all compositions of microspheres was found to
be uni-modal. As an example, the size distribution of
poly(SA) is shown in Fig. 2. The maximum volume
percent of all the microspheres used in this study was
found to occur at a particle diameter of ca. 20 Am. The
anhydride bond is labile in an aqueous environment
and the fabrication of the polyanhydride microspheres
was done in the presence of water, therefore the
amount of degradation that the polymers underwent
during the fabrication process was quantified by GPC
(Table 2). Poly(SA) showed the greatest M n loss,
while 80:20 CPH:SA showed the least. With increasing amounts of CPH in the copolymers the number of
CPH:CPH bonds increases. It is known that
CPH:CPH bonds are not as labile as CPH:SA or
SA:SA bonds [52,53], therefore it makes sense that
M n loss decreases in copolymers with increasing
amounts of CPH; these results are similar to those
obtained by Kipper et al. [44]. Fig. 3 shows the
Fig. 3. Scanning electron micrographs of polyanhydride microsphere surface morphologies: (a) poly(SA); (b) 20:80 (CPH:SA); (c) 50:50
(CPH:SA); (d) 80:20 (CPH:SA).
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
surface morphology of the microspheres obtained by
using SEM. The surface morphology of the microspheres varies with the polymer composition; the
more hydrophobic the polymer (i.e., the higher the
CPH content), the smoother the surface of the
microspheres. As the content of SA increases, the
surface appears to get rougher and dark circles below
the surface become apparent. The rough surface
morphology of the SA-rich compositions is a consequence of the fast degradation of SA:SA bonds [54–
57]. The dark circles that are present in microspheres
containing higher contents of SA are attributed to the
inner emulsion breaking near the surface. Fig. 4
shows the distribution of BSA-FITC within the
microspheres obtained by confocal microscopy. The
protein is uniformly distributed in the poly(SA)
microspheres. As the CPH content increases, the
BSA-FTIC is distributed less evenly throughout the
microspheres, as is apparent by both the protein-rich
(identified by the intense color) and protein-void
regions (identified by the lack of color). The uneven
distribution of BSA-FITC in the more hydrophobic
polymers is attributed to unfavorable protein–polymer
interactions.
103
3.2. Protein loading
The encapsulation efficiency of BSA-FITC was ca.
50%, which is similar to previous reports that use the
W/O/W method, see Table 1 [58,59]. Due to the
variation in loading from batch to batch, the protein
content of each batch of microspheres was determined
a priori in order to perform the normalization
correctly.
3.3. In vitro protein release
The in vitro release was conducted in an aqueous
solution containing 3% SDS. The SDS acted as a
surfactant to ensure that the (sticky) protein did not
bind to the reaction vessel and the sampling equipment, or adsorb to the degrading microspheres [10].
Additionally, the presence of SDS in the media rules
out complications of differential adsorption to the
different polymers that would then cause different
release rates. This enables us to directly relate the
protein release rate to the rate of degradation of the
polymers. Since SDS is a detergent that denatures the
protein, the secondary structure of the released protein
Fig. 4. Fluorescence micrographs of the distribution of BSA-FITC in polyanhydride microspheres: (a) poly(SA); (b) 20:80 (CPH:SA); (c) 50:50
(CPH:SA); (d) 80:20 (CPH:SA).
104
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
could not be assessed. The addition of SDS to the
release media did slightly increase the amount of
BSA-FITC released (data not shown) and aided in
more accurate BCA readings and resulted in protein
concentrations detectable by SDS-PAGE.
The release profile of BSA-FITC-loaded microspheres is a strong function of the polymer chemistry
as shown in Fig. 5. The Bonferroni multiple comparison adjustment was used to determine that the pvalues for all the multiple comparisons, all of the pvalues were less than 0.001, indicating that release
profiles of each polymer composition are statistically
different [60]. The release of BSA-FITC from
poly(SA) and 20:80 (CPH:SA) shows a small initial
burst followed by a period of zero order release. The
initial burst seen from the SA-rich microspheres is due
to the accelerated surface degradation that occurred
during the fabrication. The release of BSA-FITC from
50:50 (CPH:SA) and 80:20 (CPH:SA) did not show
an initial burst. The lack of an initial burst is attributed
to the fact that each microsphere had a slightly
different distribution of BSA-FITC as seen with the
confocal microscopy studies. It is hypothesized that
the varying distribution of BSA-FITC within the
microspheres counteracted any fluctuations that may
have resulted from the protein-rich regions or proteinvoid spaces near the surface, as seen in Fig. 4. As the
hydrophobicity of the polymer increases, the duration
of release increases. After 10 and 20 days, respectively, the majority of the protein encapsulated in
poly(SA) and 20:80 (CPH:SA) is released. After 50
days, only 50% of the encapsulated protein is released
from 50:50 (CPH:SA) and only 10% is released from
80:20 (CPH:SA). This data suggests that by altering
the copolymer composition, both the release rate of
BSA-FITC as well as the shape of the release profile
can be controlled. However, it is possible that some of
the BSA-FITC has been released as insoluble aggregates undetected by the BCA assay.
3.4. SDS-PAGE
Fig. 6 shows the SDS-PAGE results of the released
BSA-FITC and native BSA-FITC under both reduc-
Fig. 5. In vitro release of BSA-FITC from polyanhydride microspheres.
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
Fig. 6. (a) SDS-PAGE of BSA-FITC released from polyanhydride
microspheres under non-reducing conditions. Lane 1: BSA-FITC;
lane 2: molecular weight ladder; lane 3: poly(SA); lane 4: 20:80
(CPH:SA); lane 5: 50:50 (CPH:SA); lane 6: 80:20 (CPH:SA). (b)
SDS-PAGE of BSA-FITC released from polyanhydride microspheres under reducing conditions. Lane 1: BSA-FITC; lane 2:
molecular weight ladder; lane 3: poly(SA); lane 4: 20:80 (CPH:SA);
lane 5: 50:50 (CPH:SA); lane 6: 80:20 (CPH:SA).
ing and non-reducing conditions. Any protein aggregates present in the BSA-FITC would be conserved
when analyzed without h-mercaptoethanol. Because
BSA is known to form inter-protein multimers via
105
disulfide bonds [61], h-mercaptoethanol was used in
order to break any disulfide bonds, thus providing
insight on the possible formation of protein multimers.
Samples with and without h-mercaptoethanol were
compared. A small percentage of the commercially
available BSA-FITC used for the study contained
multimers, as indicated by the higher molecular
weight bands seen in lane 2 of Fig. 6a. These same
bands are seen in lanes 3–6 of Fig. 6a, the protein
released from the polyanhydride microspheres. In the
absence of h-mercaptoethanol, the released BSA
showed no additional indications of inter-protein
multimers as compared to the unencapsulated protein,
which would have been represented by a larger
percentage of higher molecular weight bands. With
the addition of h-mercaptoethanol, the disulfide bonds
that were present in the unencapsulated BSA were
eliminated as were the higher molecular weight bands
seen in the BSA released from the polyanhydride
microsphere. This data is shown in Fig. 6b.
The molecular weight of albumin is 66,400 Da [61]
and is represented in lane 1 of Fig. 6. If the released
protein is still intact, it should have a band corresponding to 66,400 Da. As shown in Fig. 6, bands
corresponding to 66,400 Da are present in all the lanes.
However, faint low molecular weight bands are also
observed. These low molecular weight bands indicate
that a small portion of the protein undergoes hydrolysis
due to the experimental conditions. The pH of the
release medium drops slightly as the polyanhydride
microspheres degrade, especially in the absence of a
buffering solution. Because protein concentrations
high enough to be detected by silver staining can only
be obtained by allowing the microspheres to degrade
for several days, the released protein is exposed for a
long time to a high temperature and mildly acidic
conditions which leads to hydrolysis [62]. This
observation is consistent with the data reported by
Igartua et al. [16] that while BSA incubated at 37 8C for
1 day is intact, BSA incubated for several days is
hydrolyzed. To verify this, the native protein was
incubated with 15 mg of polymer in a 3% SDS solution.
After several days, the media was sampled and run on a
gel. The protein did undergo cleavage as a result of the
acidic conditions and high incubating temperature
(data not shown). Because the darkest bands (indicating the highest protein content) correspond to 66,400
Da for all polymer compositions, this indicates that the
106
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
primary structure of BSA-FITC encapsulated within
polyanhydride microspheres was generally conserved.
3.5. FTIR
The secondary structure of BSA-FITC encapsulated within polyanhydride microspheres was determined in order to assess what effects the microsphere
fabrication process and the presence of the polymer
had on the storage stability of the protein. The two
typical signatures of protein secondary structure are
a-helices and h-sheets [63]. A protein that is
lyophilized will have a higher content of h-sheets
than a protein that is in aqueous solution due to
intermolecular interactions [49]. Because the h-sheet
content of lyophilized powder is not always indicative of the integrity of protein structure, only the ahelix content was used to determine the secondary
structure of BSA-FITC [49]. Three sets of spectra
were collected: blank microspheres, BSA-FITCloaded microspheres, and the native protein. The
spectra of the encapsulated protein were obtained by
subtracting the spectra of the blank microspheres
from that of the BSA-loaded microspheres. This
spectrum was then compared to the spectra of the
native protein.
The a-helix content of the lyophilized BSA-FITC
is 45% (Fig. 7), which is comparable to the values
reported previously for unlabeled BSA [12,13,15,
49,50]. To determine if the fabrication process or the
presence of the polymer altered secondary structure
of BSA-FITC, the spectrum of the encapsulated
protein was compared to the native protein. The
BSA-FITC encapsulated in poly(SA) and 20:80
CPH:SA microspheres showed little or no deviation
in the a-helix content when compared to the native
protein (see Fig. 7). This indicates that neither the
polymer nor the microsphere fabrication process had
an effect on the protein secondary structure. Thus,
BSA-FITC can be stabilized in poly(SA) and 20:80
CPH:SA and released continuously for 3 weeks.
However, as the CPH content in the microspheres
was increased to 50% and beyond, the % a-helix
content decreased steadily, to a point at which no ahelices were detected for the protein encapsulated in
80:20 (CPH:SA). This decrease in the a-helix content
is attributed to the polymer hydrophobicity, and not
to the microsphere fabrication process, since no
Fig. 7. Histogram showing the secondary structure of native BSAFITC vs. BSA-FITC encapsulated in polyanhydride microspheres.
The F values are calculated from a minimum of three spectra.
change in a-helix content was observed in the
poly(SA) microspheres, which were fabricated by
the same process. To further verify this contention,
lyophilized protein was physically mixed with each
polymer (poly(SA), 20:80 (CPH:SA), 50:50
(CPH:SA), and 80:20 (CPH:SA)) and analyzed as
before. The spectrum of the protein was obtained by
subtracting the spectrum of the polymer from the
spectrum of the polymer/protein blend. Fourier selfdeconvolution was then performed on the amide I
region and Gaussian curves were fit to the spectra.
The secondary structure of protein mixed with
polymer was found to match the secondary structure
of the encapsulated protein (data not shown). Further,
to verify that this phenomenon was not an artifact of
the polymer peaks interfering with the subtraction
process, protein was added to a polymer pellet
without physically mixing the two and analyzed as
before. The subtracted spectra of the polymer from
the protein/polymer spectra (when not physically
mixed) showed a-helices, indicating that the polymer
was not interfering with the data analysis. Hence the
change in protein secondary structure due to
increased polymer hydrophobicity is a result of the
polymer–protein interactions.
Fig. 7 also shows the h-sheet content of the
encapsulated BSA-FITC in each of the polymer
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
microspheres. Native BSA-FITC has a h-sheet content of 23%. Once encapsulated, the h-sheet content
increases, indicating that the encapsulated protein
aggregates and forms pockets of protein-rich regions
in the microspheres, trying to minimize exposure to
the polymer. These observations are consistent with
the confocal microscopy images in Fig. 4.
4. Conclusions
It has been shown that polyanhydrides are capable
of releasing BSA-FITC for an extended time period,
and by varying the copolymer composition the
release profile of the protein can be altered. Polyanhydride microspheres rich in SA fabricated by the
double emulsion technique were successful in
preserving the primary structure of the encapsulated
BSA-FITC without the addition of excipients or
lyoprotectants. The hydrophobic polyanhydride
microspheres also prevented the BSA-FITC from
undergoing covalent inter-protein multimers via the
formation of disulfide bonds. The primary structure
of the released protein was intact and underwent
hydrolysis only after it was allowed to incubate at a
high temperature (37 8C) in mildly acidic media for
several days. Though the protein has been shown to
be stable while encapsulated and capable of being
released at its native molecular weight, the secondary
structure of the protein could be altered upon release
from the microspheres. Because BSA has no
measurable biological activity and the secondary
structure of the released protein could not be
determined, further work needs to be done to further
support the conclusion that poly(SA) and 20:80
(CPH:SA) microspheres can deliver biologically
active therapeutic proteins.
Though polyanhydride microspheres offer a sanctuary against the formation of covalent disulfide bonds
and provide a method of prolonged delivery, not all
polyanhydrides are suitable vehicles for protein
stabilization. From this study, it is evident that the
increased hydrophobicity of CPH provides a harsh
climate for the BSA-FITC, and the secondary structure
of the protein, quantified by the a-helix content,
undergoes significant perturbation. The increased
degradation time (few months) of the CPH-rich
copolymers also limits the usefulness of such copoly-
107
mers for medical applications. The data indicates that
polyanhydrides such as poly(SA) and 20:80 (CPH:SA)
are well suited for protein stabilization and delivery.
Acknowledgments
The authors would like to acknowledge financial
support from the Whitaker Foundation and from the
USDA. ASD was supported by a USDA Multidisciplinary Graduate Education Training Grant
(2001-52100-11506). The authors would like to thank
Lee Bendickson for his assistance with the SDSPAGE experiments and Justin Recknor for his help
with the statistical analysis of the release kinetics data.
Finally, the authors would also like to thank the
undergraduate students at Iowa State University who
participated in this project: Quentin Leigh, Elizabeth
Schmerr, Angie Clark, Jennifer Graham, and Katie
Pfeifer.
References
[1] J.M. Reichert, Trends in development and approval times for
new therapeutics in the United States, Nat. Rev., Drug Discov.
2 (2003) 695 – 702.
[2] S.P. Schwendeman, M. Cardamone, A. Klibanov, R. Langer,
M.R. Brandon, in: S. Cohen, H. Bernstein (Eds.), Microparticulate Systems for the Delivery of Proteins and Vaccines,
Marcel Dekker, New York, 1996, pp. 1 – 49.
[3] S.L. Nail, in: K. Park (Ed.), Controlled Drug Delivery
Challenges and Strategies, American Chemical Society,
Washington, DC, 1997, pp. 185 – 203.
[4] C. Schoneich, M.J. Hageman, R.T. Borchardt, in: K. Park (Ed.),
Controlled Drug Delivery Challenges and Strategies, American
Chemical Society, Washington, DC, 1997, pp. 205 – 227.
[5] J.L. Cleland, M.F. Powell, S.J. Shire, The development of
stable protein formulations: a close look at protein aggregation, deamidation, and oxidation, Crit. Rev. Ther. Drug Carr.
Syst. 10 (4) (1993) 307 – 377.
[6] W.R. Liu, R. Langer, A.M. Klibanov, Moisture-induced
aggregation of lyophilized proteins in the solid state,
Biotechnol. Bioeng. 37 (1991) 177 – 184.
[7] H.R. Costantino, R. Langer, A. Klibanov, Solid-phase
aggregation of proteins under pharmaceutically relevant
conditions, J. Pharm. Sci. 83 (12) (1994) 1662 – 1669.
[8] J. Hanes, J.L. Cleland, R. Langer, New advances in microsphere-based single-dose vaccines, Adv. Drug Deliv. Rev. 28
(1997) 97 – 119.
[9] G. Zhu, S.R. Mallery, S.P. Schwendeman, Stabilization of
proteins encapsulated in injectable poly(lactide-co-glycolide),
Nat. Biotechnol. 18 (2000) 52 – 57.
108
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
[10] G. Crotts, T.G. Park, Stability and release of bovine serum
albumin encapsulated within poly(d,l-lactide-co-glycolide)
microparticles, J. Control. Release 44 (1997) 123 – 134.
[11] P. Bouillot, N. Ubrich, F. Sommer, T.M. Duc, J.-P. Loeffler, E.
Dellacherie, Protein encapsulation in biodegradable amphiphilic microspheres, Int. J. Pharm. 181 (1999) 159 – 172.
[12] K.G. Carrasquillo, J.C.A. Carro, A. Alejandro, D. Diaz, K.
Griebenow, Reduction of structural perturbations in bovine
serum albumin by non-aqueous microencapsulation, J. Pharm.
Pharmacol. 53 (2001) 115 – 120.
[13] K.G. Carrasquillo, A.M. Stanley, J.C. Aponte-Carro, P. De
Jesus, H.R. Costantino, C.J. Bosques, K. Griebenow, Nonaqueous encapsulation of excipient-stabilized spray-freeze
dried BSA into poly(lactide-co-glycolide) microspheres results
in release of native protein, J. Control. Release 76 (2001)
199 – 208.
[14] I.J. Castellanos, K.G. Carrasquillo, J.D.J. Lopez, M.
Alvarez, K. Griebenow, Encapsulation of bovine serum
albumin in poly(lactide-co-glycolide) microspheres by the
solid-in-oil-in-water technique, J. Pharm. Pharmacol. 53
(2001) 167 – 178.
[15] K. Fu, K. Griebenow, L. Hsieh, A. Klibanov, R. Langer, FTIR
characterization of the secondary structure of proteins encapsulated within PLGA microspheres, J. Control. Release 58
(1999) 357 – 366.
[16] M. Igartua, R.M. Hernandez, A. Esquisabel, A.R. Gascon,
M.B. Calvo, J.L. Pedraz, Stability of BSA encapsulated into
PLGA microspheres using PAGE and capillary electrophoresis, Int. J. Pharm. 169 (1998) 45 – 54.
[17] T.G. Park, H.Y. Lee, Y.S. Nam, A new preparation method for
protein loaded poly(d,l-lactic-co-glycolic acid) microspheres
and protein release mechanism study, J. Control. Release 55
(1998) 181 – 191.
[18] M. van de Weert, R. van’t Hof, J. van der Weerd, R.M.A.
Heeren, G. Posthuma, W.E. Hennink, D.J.A. Crommelin,
Lysozyme distribution and conformation in a biodegradable
polymer matrix as determined by FTIR techniques, J. Control.
Release 68 (2000) 31 – 40.
[19] O.L. Johnson, J.L. Cleland, H.J. Lee, M. Charnis, E. Duenas,
W. Jaworowicz, D. Shepard, A month-long effect from a
single injection of microencapsulated human growth hormone,
2 (7) (1996) 795–799.
[20] H.K. Kim, T.G. Park, Microencapsulation of human growth
hormone within biodegradable polyester microspheres: protein
aggregation stability and incomplete release mechanism,
Biotechnol. Bioeng. 65 (6) (1999) 659 – 667.
[21] J.L. Cleland, A.J.S. Jones, Stable formulations of recombinant
human growth hormone and interferon-gamma for microencapsulation in biodegradable microspheres, Pharm. Res. 13
(10) (1996) 1464 – 1475.
[22] T.-H. Yang, A. Dong, J. Meyer, O.L. Johnson, J.L. Cleland,
J.F. Carpenter, Use of infrared spectroscopy to assess
secondary structure of human growth hormone within
biodegradable microspheres, J. Pharm. Sci. 88 (2) (1999)
161 – 165.
[23] X.M. Lam, E.T. Duenas, A.L. Daugherty, N. Levin, J.L.
Cleland, Sustained release of recombinant human insulin-like
[24]
[25]
[26]
[27]
[28]
[29]
[30]
[31]
[32]
[33]
[34]
[35]
[36]
[37]
[38]
[39]
growth factor-I for treatment of diabetes, J. Control. Release
67 (2000) 281 – 292.
L. Meinel, O.E. Illi, J. Zapf, M. Malfanti, H.P. Merkle, B.
Gander, Stabilizing insulin-like growth factor-I in poly(d,llactide-co-glycolide) microspheres, J. Control. Release 70
(2001) 193 – 202.
J. Elisseeff, W. McIntosh, K. Fu, T. Blunk, R. Langer,
Controlled-release of IGF-I and TGF-b1 in a photopolymerizing hydrogel for cartilage tissue engineering, J. Orthoptera
Res. 19 (2001) 1098 – 1104.
H.R. Costantino, K. Griebenow, P. Mishra, R. Langer, A.
Klibanov, Fourier-transform infrared spectroscopic investigation of protein stability in the lyophilized form, Biochim.
Biophys. Acta 1253 (1995) 69 – 74.
A. Brunner, K. Mader, A. Goepferich, pH and osmotic
pressure inside biodegradable microspheres during erosion,
Pharm. Res. 16 (6) (1999) 583 – 847.
Y. Tabata, S. Gutta, R. Langer, Controlled delivery systems for
proteins using polyanhydride microspheres, Pharm. Res. 10
(4) (1993) 487 – 496.
A. Goepferich, Mechanisms of polymer degradation and
erosion, Biomaterials 17 (1990) 103 – 114.
J.A. Tamada, R. Langer, Erosion kinetics of hydrolytically
degradable polymers, Proc. Natl. Acad. Sci. U. S. A. 90 (1993)
552 – 556.
A. Goepferich, R. Langer, The influence of microstructure and
monomer properties on the erosion mechanism of a class of
polyanhydrides, J. Polym. Sci., A, Polym. Chem. 31 (1993)
2445 – 2458.
A. Brunner, A. Goepferich, in: S. Cohen, H. Bernstein (Eds.),
Microparticulate Systems for the Delivery of Proteins and
Vaccines, Marcel Dekker, New York, 1996, pp. 169 – 201.
E. Mathiowitz, H. Bernstein, S. Giannos, P. Dor, T. Turek, R.
Langer, Polyanhydride microspheres: IV. Morphology and
characterization of systems made by spray drying, J. Appl.
Polym. Sci. 45 (1992) 125 – 134.
E. Mathiowitz, R. Langer, Polyanhydride microspheres as
drug carriers: I. Hot-melt microencapsulation, J. Control.
Release 5 (1987) 13 – 22.
E. Mathiowitz, E. Ron, G. Mathiowitz, C. Amato, R. Langer,
Morphological characterization of bioerodible polymers: 1.
Crystallinity of polyanhydride copolymers, Macromolecules
23 (13) (1990) 3212 – 3218.
E. Mathiowitz, W.M. Saltzman, A. Domb, P. Dor, R. Langer,
Polyanhydride microspheres as drug carriers: II. Microencapsulation by solvent removal, J. Appl. Polym. Sci. 35 (1988)
755 – 774.
C. Bindschaedler, K. Leong, E. Mathiowitz, R. Langer,
Polyanhydride microsphere formulation by solvent removal,
J. Pharm. Sci. 77 (8) (1988) 696 – 698.
N.B. Viswanathan, P.A. Thomas, J.K. Pandit, M.G. Kulkarni,
R.A. Mashelkar, Preparation of non-porous microspheres with
high entrapment efficiency of proteins by a (water-in-oil)-inoil emulsion technique, J. Control. Release 58 (1999) 9 – 20.
E. Vasheghani-Farahani, M. Khorram, Hydrophilic drug
release from bioerodible polyanhydride microspheres, J. Appl.
Polym. Sci. 83 (2002) 1457 – 1464.
A.S. Determan et al. / Journal of Controlled Release 100 (2004) 97–109
[40] C. Berkland, M.J. Kipper, B. Narasimhan, K.K. Kim, D. Pack,
Microsphere size, precipitation kinetics, and drug distribution
control drug release from biodegradable polyanhydride microspheres, J. Control. Release 94 (2003) 129 – 141.
[41] H. Sah, Protein behavior at the water/methylene chloride
interface, J. Pharm. Sci. 88 (12) (1999) 1320 – 1325.
[42] A.J. Domb, R. Langer, Solid-state and solution stability of
poly(anhydrides) and poly(esters), Macromolecules 22 (1989)
2117 – 2122.
[43] E. Shen, M.J. Kipper, B. Dziadul, M.-K. Lim, B. Narasimhan,
Mechanistic relationships between polymer microstructure and
drug release kinetics in bioerodible polyanhydrides, J. Control.
Release 82 (2002) 115 – 125.
[44] M.J. Kipper, E. Shen, A. Determan, B. Narasimhan, Design of
an injectable system based on bioerodible polyanhydride
microspheres for sustained drug delivery, Biomaterials 23
(2002) 4405 – 4412.
[45] E. Shen, R. Pizsczek, B. Dziadul, B. Narasimhan, Microphase
separation in bioerodible copolymers for drug delivery,
Biomaterials 22 (2001) 201 – 210.
[46] A. Conix, Poly[1,3-bis( p-carboxyphenoxy)propane anhydride], Macromol. Synth. 2 (1966) 95 – 98.
[47] R.H. Garrett, C.M. Grisham, Biochemistry, 2nd ed., Harcourt
College Publishers, Fort Worth, TX, 1999.
[48] K. Griebenow, A.M. Klibanov, Lyophilization-induced reversible changes in the secondary structure of proteins, Proc. Natl.
Acad. Sci. U. S. A. 92 (1995) 10969 – 10976.
[49] K. Griebenow, A.M. Klibanov, On protein denaturation in
aqueous–organic mixtures but not in pure solvents, J. Am.
Chem. Soc. 118 (1996) 11695 – 11700.
[50] I.J. Castellanos, W.L. Cuadrado, K. Griebenow, Prevention of
structural perturbations and aggregation upon encapsulation of
bovine serum albumin into poly(lactide-co-glycolide) microspheres using the solid-in-water technique, J. Pharm. Pharmacol. 53 (2001) 1099 – 1107.
[51] H.R. Costantino, B. Chen, K. Griebenow, C.C. Hsu, S.J. Shire,
Fourier-transform infrared spectroscopic investigation of the
secondary structure of aqueous and dried recombinant human
deoxyribonuclease I, Pharm. Pharmacol. Commun. 4 (1998)
391 – 395.
109
[52] K.W. Leong, B.C. Brott, R. Langer, Bioerodible polyanhydrides as drug-carrier matrices: I. Characterization, degradation, and release characteristics, J. Biomed. Mater. Res. 19
(1985) 941 – 955.
[53] D. Larobina, G. Mensitieri, M.J. Kipper, B. Narasimhan,
Mechanistic understanding of degradation in bioerodible
polymers for drug delivery, AIChE J. 48 (2002) 2960 – 2970.
[54] K.J. Leach (Pekarek), E. Mathiowitz, Degradation of doublewalled polymer microspheres of PLLA and P(CPP:SA)20:80:
I. In vitro degradation, Biomaterials 19 (1998) 1973 – 1980.
[55] C.A. Santos, B.D. Freedman, K.J. Leach, D.L. Press, M.
Scarpulla, E. Mathiowitz, Poly(fumaric-co-sebacic anhydride)
a degradation study as evaluated by FTIR, DSC, GPC, and Xray diffraction, J. Control. Release 60 (1999) 11 – 22.
[56] Y. Tamada, R. Langer, The development of polyanhydrides for
drug delivery applications, J. Biomater. Sci., Polym. Ed. 3 (4)
(1992) 315 – 353.
[57] K.J. Leach (Pekarek), E. Mathiowitz, Degradation of doublewalled polymer microspheres of PLLA and P(CPP:SA)20:80:
II. In vivo degradation, Biomaterials 19 (1998) 1958 – 1981.
[58] Y.-Y. Yang, T.-S. Chung, N.P. Ng, Morphology, drug
distribution, and in vitro release profiles of biodegradable
polymeric microspheres containing protein fabricated by
double-emulsion solvent extraction/evaporation method, Biomaterials 22 (2001) 231 – 241.
[59] S.W. Sun, Y.I. Jeong, S.W. Jung, S.H. Kim, Characterization
of FITC-albumin encapsulated poly(dl-lactide-co-glycolide)
microspheres and its release characteristics, J. Microencapsul.
20 (4) (2003) 479 – 488.
[60] J. Neter, M.H. Kutner, C.J. Nachtsheim, W. Wasserman,
Applied Linear Statistical Models, 4th ed., WCB McGrawHill, Boston, MA, 1996.
[61] T.J. Peters, All About Albumin, Academic Press, San Diego,
CA, 1996.
[62] W. Votsch, H.A. Wagner, F.A. Anderer, Non-enzymatic
cleavage of serum albumin from horse (Equus caballus),
Comput. Biochem. Physiol. 66B (1980) 87 – 91.
[63] B.R. Singh, in: B.R. Singh (Ed.), Infrared Analysis of Peptides
and Proteins: Principles and applications, American Chemical
Society, Washington, DC, 2000, pp. 2 – 37.