LCAO-MO Correlation Diagrams
advertisement

LCAO-MO Correlation Diagrams
•
(Linear Combination of Atomic Orbitals to yield Molecular Orbitals)
For (Second Row) Homonuclear Diatomic Molecules (X2 ) - the following
LCAO-MO’s are generated:
LCAO
MO symbol
1sA + 1sB
σ1 s
1sA - 1sB
2sA + 2sB
2sA - 2sB
2px , A + 2px , B
σ1 s*
σ2 s
2px , A - 2px , B
2py , A + 2py , B
2py , A - 2py , B
2pz , A + 2pz , B
π 2 p*
π2 p
2pz , A - 2pz , B
σ2 p*
σ2 s*
π2 p
π 2 p*
σ2 p
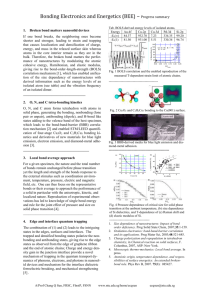
For the above LCAO-MO combinations, the coordinate system is chosen such that the
“z” axis is along the horizontal direction and is considered the internuclear
(“bond”) axis. Here, the y-axis is considered to be along the vertical direction and the
x-axis is considered to be perpendicular to the plane of the page. This choice is, of course,
arbitrary. The correlation diagrams showing the energy ordering and relationship
between the Atomic Orbitals (AO’s) and resultant Molecular Orbitals (MO’s) for
several situations are listed below. Each MO can maximally contain two (2) electrons
- with opposite spins (↑↓) - as required by the Pauli Exclusion Principle (PEP). Also,
degenerate MO’s - when occupied - will follow Hund’s Rule in order to achieve a ground
state (energy-preferred) electron configuration. There are two schemes - I and II below.
Scheme I applies for Li through N (and their ions), inclusive and Scheme II applies for O
through Ne (and their ions), inclusive. As can be seen, the difference lies in the relative
energy ordering of the π2p MO’s versus the σ2p MO’s. For Z ≤ 7 atoms, the (degenerate)
π 2p MO’s are lower in energy than σ2p MO. For Z ≥ 8 atoms, the (degenerate) π2p
MO’s are greater in energy than σ2p MO. The reason has to do with energy stabilization
by attenuation of electron-electron repulsion. In Scheme I, the π2p MO’s are filled before
the σ2p MO because the electron density in the π2p MO’s are concentrated (between the
atoms) away from (i.e., above and below) the internuclear axis. This leads to a reduction in
the electron-electron repulsions. This is particularly important since the electrons in the
already - occupied σ2s - σ2s* MO’s will interact less strongly with electrons in the π2p
MO’s than those in the σ2p MO’s (electron density also directed along the internuclear axis).
In Scheme II - followed by atoms toward the end of the second row - the already occupied
σ2s - σ2s* MO’s are drawn closer (“tighter”) due to the greater nuclear charge (Z). For
Z ≥ 8, this is enough so that the σ2p MO’s will interact less strongly with the σ2s - σ2s*
MO’s. Hence, the σ2p MO will be lower in energy than the π2p MO’s. [After discussing
Scheme II, your text sometimes follows Scheme I - for simplicity - for all second row
diatomics (and their ions) in some of the homework problems.] By paying attention to the
PEP and Hund’s Rule - as mentioned above - we fill the MO’s from “bottom - up” in an
“Aufbau” manner for our chosen MO scheme. This will give us ground state (MO)
electron configurations.
1
•
Homonuclear (Second Row) Diatomic Molecules (X2 ) - or their ions
Scheme I - X = Li, Be, B, C, N; i.e., Atomic # Z ≤ 7
(Energy increases vertically up the page)
σ*2p
π*
π*2p
2p
2pz,A
2py,A 2px,A
2pz,B
2py,B 2px,B
σ2p
π2p
π2p
σ*2s
2sA
2sB
σ2s
σ*1s
1sA
1sB
σ1s
•
Homonuclear (Second Row) Diatomic Molecules (X2 ) - or their ions
Scheme II - X = O, F, Ne; i.e., Atomic # Z ≥ 8
(Energy increases vertically up the page)
σ*2p
π*
π*2p
π2p
π2p
2p
2pz,A
2py,A 2px,A
2pz,B
2py,B 2px,B
σ2p
σ*2s
2sA
2sB
σ2s
σ*1s
1sA
1sB
σ1s
2
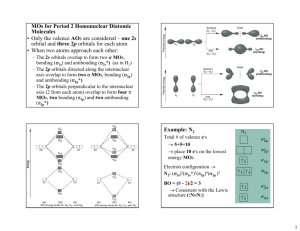
Boundary Surface Diagrams (BSD) for the LCAO-MO’s formed from
2s, 2px , 2py , & 2pz AO’s of second row homonuclear diatomic molecules.
• 2s Atomic Orbital (Bonding & Antibonding) Combinations:
2s Bonding:
+
2sA
+
+
+
Bonding Combo
σ2s
2sB
2s Antibonding:
+
2sA
−
−
Antibonding Combo
+
−
σ∗2s
2sB
3
• 2p Atomic Orbital Combinations along BOND AXIS:
2p Bonding (Along Bond Axis):
+
−
+
2pzA
−
+
2pzB
Bonding Combo
+
−
−
σ2p
2p Antibonding (Along Bond Axis):
+
−
−
2pzA
+
−
2pzB
Antibonding Combo
−
+
+
−
σ∗2p
4
• 2p Atomic Orbital Combinations PERPENDICULAR TO Bond Axis:
2p Bonding (Perpendicular to Bond Axis):
+
+
+
Bonding Combo
−
2pyA
−
−
+
π2p
2pyB
2p Antibonding (Perpendicular to Bond Axis):
+
−
+
Antibonding Combo
−
2pyA
+
−
−
−
+
π∗2p
2pyB
& SIMILARLY for 2px A & 2px B - Bonding & Antibonding M.O.’s
(These M.O.’s will be perpendicular to the plane of the paper.)
5
Where are the bonds??
In order to determine how many bonds will form between the atoms of the
diatomic molecule (in the spirit of the Valence Bond - “localized electron pair bond” model),
we define the parameter BOND ORDER:
BOND ORDER = B.O. ≡
({# of e− ‘s in Bonding MO’s} − {# of e− ‘s in Antibonding (* ) MO’s})
2
Prove for yourself that the Bond Order for N2 , with a ground state valence shell electron
configuration of: σ2 s2 (σ 2 s* ) 2 π2 p4 σ2 p2 is 3.0, i.e., a triple bond. Note, this is
exactly what the Valence Bond model would also predict!
If a molecule has a bond order of ZERO (0), then M.O. theory is telling us that the
MOLECULE WILL NOT FORM (i.e., no bonds will be “created”, since the number of
electrons in antibonding M.O.’s exactly “cancels” the number of electrons in bonding
M.O.’s). For example: He2 and Be2 have B.O. = 0, so these molecules “will not form”, i.e.,
they are not energetically stable. Finally, if a molecule has an odd number of electrons, such
as CN (a radical), then we can have a fractional bond order. Prove for yourself that CN, with
M.O. ground state valence shell configuration: σ2 s2 (σ 2 s* ) 2 π2 p4 σ2 p1 , has a bond order
of 2.5.
Bond orders can be used to predict the relative STRENGTH and relative LENGTHS
of BONDS. The relationship is: THE GREATER THE BOND ORDER, THE
SHORTER AND STRONGER THE BOND. Hence, B2 , C2 , and N2 ,which have
bond orders of 1.0, 2.0, and 3.0, respectively, have BOND LENGTHS of 159 pm,
124 pm, and 110 pm, respectively. The BOND ENERGIES of B2 , C2 , and N2 are:
289 kJ/mol, 599 kJ/mol, and 941 kJ/mol, respectively.
Paramagnetism & Diamagnetism:
It is important to realize that M.O. theory allows a molecule with an EVEN
(as well as an odd) number of electrons to have unpaired electrons.
According to the Valence Bond model all molecules with an EVEN number of
electrons will have the electrons either PAIRED in bonds or in lone PAIRS and are predicted to be DIAMAGNETIC. Thus, Valence Bond Theory predicts that
O2 (with 16 total electrons or 12 valence electrons) will have a double bond plus two (2) lone
pairs around each Oxygen atom. According to M.O. theory, the ground state valence shell
electron configuration is: σ2 s2 (σ 2 s* ) 2 σ2 p2 π2 p4 (π 2 p* ) 2 - where each of the two (2)
electrons in the π2 p* M.O. are UNPAIRED - according to Hund’s Rule. Hence, Valence
Bond theory would predict that the oxygen molecule is DIAMAGNETIC (no unpaired
electrons); whereas Molecular Orbital theory would predict that the oxygen molecule should
be PARAMAGNETIC (two (2) unpaired electrons). Experiment tells us that O2 is
“attracted” by a magnetic field, i.e., it is PARAMAGNETIC. Thus, in this case, M.O.
theory better rationalizes the “bonding picture” in O2. Note that both V.B. theory and M.O.
theory predict that the oxygen atoms are attached by a double bond - check this for yourself by
drawing the Lewis structure and also by calculating the bond order from the ground state M.O.
electron configuration (listed above).
6
Molecular Orbitals for CO
Atomic orbitals
for C
Less
electronegative
σ∗2p z
π∗2p x π∗2p y
E
N
E
R
G
Y
Atomic orbitals
for O
More
electronegative
2pC
2pO
σ2p z
π2p x
π2p y
σ∗2s
2sC
Bond Energy =
1074 kJ/mol
2sO
σ2s7
Hydrogen Fluoride (HF) LCAO-MO Diagram σ∗ (antibonding)
H atom
E
N
E
R
G
Y
1sH
F atom
2pxF
2pyF
(nonbonding)
2pF
σ (bonding)
2s (nonbonding)
8
2sF
ORBITALS FOR HYDROGEN FLUORIDE (HF):
+
+
−
σ∗ = 1sH - 2p zF
- 2pzF
1sH
+
+
Antibonding
−
+ 2pzF
σ = 1s H + 2pzF
1sH
Bonding
+
+
Nonbonding
(phases cancel)
−
1sH
2pyF or 2pxF
+
&
Nonbonding
(negligible overlap far apart in energy)
+
2sF
1sH
9