6 – 'Hydrophobic' interactions
advertisement

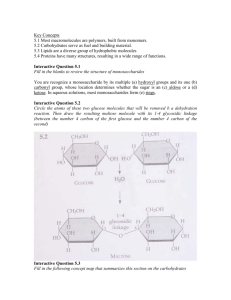
”The Physics and Chemistry of Water” 6 – ’Hydrophobic’ interactions A non-polar molecule in water disrupts the Hbond structure by forcing some water molecules to give up their hydrogen bonds. As a result, water near the solute will rearrange in order to minimise the number of lost Hbonds. This imposes a new structure on neighbouring water molecules, which is entropically unfavourable. The differences in dispersion interaction between water and e.g. hydrocarbons is rather small, and so the size and shape of the solutes are critical in determining the change in free energy upon dissolution. A comment on terminology Hydrophobic substances – Non-polar substances with low miscibility in water, such as hydrocarbons or fluorocarbons. Hydrophobic surfaces – Surfaces which are not wetted by water, and against which water subtends a large contact angle. The hydrophobic effect – The immiscibility of non-polar substances in water, and the mainly entropic nature of this incompatibility. Hydrophobic hydration (solvation) – The distortion of water structure in the vicinity of non-polar solute molecules or surfaces. Hydrophobic interaction – An unusually strong interaction between hydrophobic molecules and surfaces in water. Effect of temperature on the hydrophobic effect Free energy of transfer of pentane from the bulk liquid to aqueous solution (from Creighton). The strong temperature dependence of of both ∆G and ∆S is a result of the different heat capacities of the two phases. Over the whole T range there is a large positive heat capacity change upon transfer to water; d∆G d∆H d∆S ∆Cp = −T = =T dT 2 dT dT Some comments on the temperature dependence • In cold water (room temperature) the effect is mainly due to entropy; −T ∆S À 0, ∆H ≈ 0. • In hot water (near boiling) it is mainly enthalpic; ∆H À 0, −T ∆S ≈ 0. Although the entropic term approaches zero, which may be taken to suggest that water-ordering has disappeared, ∆Cp is still considerable, so much of the ordering is still remaining. Anyhow, water behaves much more like a normal solvent at high temperatures. • There are two different measures of ”maximum hydrophobicity”: i) The free energy of transfer ∆G has a maximum where ∆S = 0. ii) The equilibrium constant for transfer (which is proportional to −∆G/T ) is at a maximum where ∆H = 0. Driving forces require interpretation in terms of free energy, while the solubility is lowest at room temperature. Thus between 300 and 380 K the free energy becomes more positive, yet the solubility of oil in water increases. Effect of hydrocarbon chain length on the hydrophobic effect Free energy of transfer (µ◦HC − µ◦W ) of hydrocarbons from water to liquid hydrocarbon at 25◦C (from Tanford). Effect of hydrocarbon surface area Free energy of transfer ∆G = ∆H − T ∆S from bulk liquid to water. Thermodynamics of dissolution in water at 25◦C as a function of accessible hydrocarbon surface area, dashed line: aliphatics, solid line: aromatics (from Creighton). ∆G (kJ/mol) Methane 14.5 n-butane 24.5 Area* (nm2) ∼0.5 ∼1.0 ∆G/area (mJ/m2) 48 41 *With a 0.2 nm van der Waals radius, the surface area/molecule for methane is 4πa2 ≈ 0.5 nm2 , the area of n-butane is about 4πa2 + 2πa(3 × 0.1275) ≈ 1.0 nm2 . Compare with free energies of water-hydrocarbon interfaces, which typically are 40-50 mJ/m2. Solubility minima for small non-polar solutes Solubility of liquid benzene in water, as mole fraction X2 of dissolved solute (data from Ha et al., J. Mol. Biol. 209, 801 (1989)). The hydration has two contributions: a) The endothermic creation of a cavity. b) The exothermic addition of a molecule. At low temperatures, cavities are formed at a relatively low cost, so the solubilization is exothermic, and solubility decreases with increasing temperature (Le Chatelier’s principle). At higher temperatures, the structure in the liquid is less open, so creating voids in the water becomes more difficult, the process is endothermic, and (Le Chatelier’s principle again) solubility increases with increasing temperature after having passed a minimum. Isotope effects Hummer et al., Chem. Phys. 258, 349 (2000) D2O is generally a better solute for small nonpolar solutes than H2O, despite forming stronger H-bonds and being a more strongly associated liquid. Experimental relative free energy of transfer from H2O to D2O. The dashed line is the relative change in surface tension. The stronger H-bonds in D2O leads to a more icelike structure with a lower (number) density, with an increased number of (small) molecule sized cavities and thus an increased solubility. Experimental evidence for hydrophobic hydration I Soper & Finney, PRL 71, 4346 (1993) Neutron diffraction from 1:9 molar ratio methanolwater mixtures (6 different isotopic combinations, resulting in 10 distinct partial structure factors). Left: H–H pair correlation functions for the 1:9 MeOH/H2O mixture (line) and for pure water (circles). Right: Pair correlation function for the MeOH and H2O molecular centers. The peak at ∼3.7 Å indicates a shell of water around the methanol molecules in solution. • Water forms a loose H-bonded cage around the methanol molecule. • The water shell around the methanol molecules is achieved without significant modification of the order between water molecules. • Results contradict speculations about enhancement of water structure by alcohols. Experimental evidence for hydrophobic hydration II Bowron et al., PRL 81, 4164 (1998) EXAFS characterization of hydrophobic hydration of Krypton in water and ice. Kr-O partial pair-correlation function in the liquid state at ∼ 5◦, and in ice. Numbers refer to densities at the location of the first peak. The hydration ”cage” is more loosely defined in the liquid, but the number of molecules in the first coordination shells are similar; ∼13 in the liquid and ∼12.5 in the solid. Diffusion of atomic hydrogen (H*) I Kirchner et al., PRL 89, 215901 (2002) Par-Carrinello simulation of 63 H2O molecules in a 12.6 Å cubic periodic box. Partial pair-correlation functions (left ordinate) and running coordination numbers (right ordinate) for an H atom (H*) in water at 315 K. A hydration shell is formed with H2O molecules oriented with an H atom closer to the H* than the oxygen. The average distance of H* from the H2O molecules is similar to O–O-distances in the bulk. Diffusion of atomic hydrogen (H*) II Kirchner et al., PRL 89, 215901 (2002) Dynamical properties of the solvation shell. Mean-square displacements of the H* atom, the cavity center, ”bulk” water and H* relative to the cavity center, indicating anomalously fast diffusion of H* in water. • The cavity trajectory suggests that H2O in the solvation shell are exchanged, as opposed to diffusion of small cations which travel with their hydration shell attached. • The fast migration of the cavity appears to be caused by rapid fluctuations in the H-bond network, permitting rapid exchange. • Results do not support the idea of a ”frozen” solvation shell. Hydrophobic interactions • Attraction between hydrophobic molecules in water is often stronger than their attraction in free space; the van der Waals energy for two contacting methane molecules is −2.5 × 10−21 J in free space, but −14 × 10−21 J in water. • Continuum theories predict a reduced interaction in water. • This phenomenon is the result of H-bond configurational rearrangement in two overlapping hydration shells as they approach; it is thus longer ranged than chemical bonds. • Driving force for self-assembly of biological membranes, protein folding, polymer association, micellization, etc. Amphiphilic association Evans & Miller, Water Science Reviews 4, 1 (1989) Thermodynamics of micellization of C14TAB. Over the range 25 to 160◦C, the free energy remains almost constant, while the CMC increases by a factor of 10, and the aggregation number decreases from 72 to 8. Interaction between a methane molecule and a paraffin wall in liquid water Wallqvist et al., Chem. Phys. Lett. 145, 26 (1988) Monte Carlo simulation of 216 molecules, RWK2M (RWK2 + internal vibrations) modeling of water-water, different LJ interactions for watermethane and wall-methane, water-wall potential V (z) = a/z 9 − b/z 3. Distribution functions of methane molecules relative to the wall for the gas and solvent phase. The wall is at 11.8 Å. The hydrophobic solute is not forced into contact with the wall by the water, but is stabilized in a solvent-separated configuration by the hydrogenbonded network. General references • A. Ben-Naim, Water and aqueous solutions, New York: Plenum 1974. • T. Creighton, Proteins: Structure and molecular properties, New York: Freeman (1993). • J. N. Israelachvili, Intermolecular and surface forces, London: Academic Press 1992. • C. Tanford, The hydrophobic effect: Formation of micelles and biological membranes, New York: Wiley 1973.