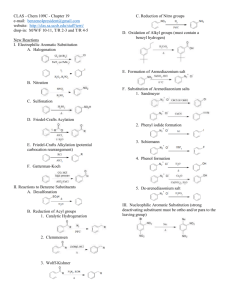
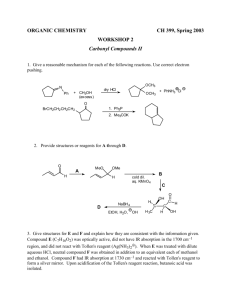
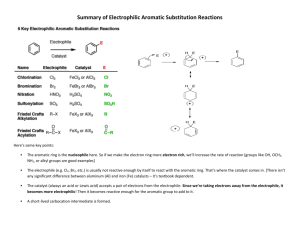
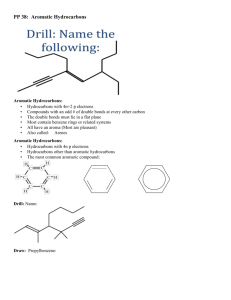
8 - Durham e-Theses
advertisement

Durham E-Theses Studies directed towards the step-growth photopolymerization of carbonyl compounds Spanomanolis, Constantinos How to cite: Spanomanolis, Constantinos (1978) Studies directed towards the step-growth photopolymerization of carbonyl compounds, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/8406/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: e-theses.admin@dur.ac.uk Tel: +44 0191 334 6107 http://etheses.dur.ac.uk 2 STUDIES DIRECTED TOWARDS THE STEP-GROWTH PHOTOPOLYMERIZATION OF CARBONYL COMPOUNDS by Constantinos Spanoraanolis, B.Sc. (Graduate Society) The copyright of this thesis rests with the author. No quotation from it should be published without his prior written consent and information derived from it should be acknowledged. A t h e s i s submitted t o the U n i v e r s i t y o f Durham f o r the Degree of Doctor o f Philosophy 197B 13 DEC 1978 si HOM Non s i e n l e g e n t i , ancor, troppo s i c u r e a g i u d i c a r , s i comme quel che stima l e blade i n campo p r i a che s i e n mature: ch' i ' ho veduto t u t t o i l verno prima l o prun m o s t r a r s i r i g i d o e f e r o c e , p o s c i a p o r t a r l a rosa i n su l a cima; e legno v i d i g i a d r i t t o e v e l o c e c o r r e r l o mar per t u t t o suo cammino, perire al fine a l l 1 i n t r a r de l a foce. Dante A l i g h i e r i , 'La D i v i n a Commedia Paradiso, Canto X I I I , 130 - 138. 'So a l s o l e t not the people be too sure i n judging, l i k e those t h a t reckon the corn i n the f i e l d before i t i s r i p e : for I have seen the b r i a r f i r s t show a l l harsh and r i g i d through the w i n t e r and l a t e r bear the rose upon i t s top; and once I saw a s h i p t h a t ran s t r a i g h t and s w i f t over the sea through a l l i t s course and p e r i s h a t the l a s t e n t e r i n g the harbour.' - ii - To the Memory of my Mother - iii - ABSTRACT Studies D i r e c t e d Towards the Step-Growth Photopolymerization of Carbonyl Compounds T h i s t h e s i s d e s c r i b e s some work c a r r i e d out i n the r e l a t i v e l y new area of step-growth photopolymerization of carbonyl compounds. The p o s s i b i l i t y of extending the photoreductive polymerization of aromatic diketones (leading to the s y n t h e s i s of polybenzopinacols) to the photochemical s y n t h e s i s of pentaphenylglycerols and higher analogues investigated; was however examination of the model photochemical r e a c t i o n between benzophenone and t r i p h e n y l g l y c o l e s t a b l i s h e d t h a t t h i s g l y c o l (and the r e l a t e d 1,2,2,2-tetraphenylethanol) i s not a hydrogen donor f o r t r i p l e t benzophenone. I n an attempt to examine the p o s s i b i l i t y of extending the photoreductive a d d i t i o n of benzophenone to diphenylmethane to polymer s y n t h e s i s , m-dibenzoylbenzene and m- and p-dibenzylbenzenes were prepared and c h a r a c t e r i z e d . I r r a d i a t i o n of the diketone i n the presence of an equimolar amount of e i t h e r of the dibenzylbenzenes afforded low molecular weight products, shown by ^"H and ^ C n.m.r. spectroscopy to c o n t a i n benzopinacol, tetraphenylethane and 1,1,2,2-tetraphenylethanol type of u n i t s , i n a d d i t i o n to benzophenone-type carbonyl and benzyl groups. R e s u l t s obtained from the p y r o l y t i c decomposition or treatment w i t h dehydrating agents of one of the products were c o n s i s t e n t with the proposed structure. I r r a d i a t i o n of t e r e - or iso-phthalaldehyde i n the s o l i d s t a t e y i e l d e d mostly s t a r t i n g m a t e r i a l s whereas branched low molecular weight products were obtained from the i r r a d i a t i o n of e i t h e r of these aromatic dialdehydes i n the presence of the hydrogen-donating isopropanol. An i n v e s t i g a t i o n on the s t r u c t u r e of the 'photopolymer i r r a d i a t i n g benzaldehyde 1 produced on i n the l i q u i d phase, by means of conventional - iv - spectroscopic 'Classical 1 analyses d i d not lead to a convincing s t r u c t u r a l hypothesis. chemical r e a c t i o n s proved to be more u s e f u l i n t h i s case; i t was thus shown t h a t the 'photopolymer', which has the same elemental composition as benzaldehyde but a molecular weight 9 - 1 1 times a s high, can be a c e t y l a t e d , oxidized and i s r e a c t i v e towards s u l p h u r i c a c i d ; i t i s furthermore s t a b l e to i r r a d i a t i o n i n benzene s o l u t i o n a t 300nm i n the absence o f a i r and non-photor e d u c i b l e i n isopropanol; i t i s however reduced by e i t h e r sodium borohydride or sodium bis(2-methoxyethoxy)-aluminium hydride g i v i n g r i s e to a d i f f e r e n t s t r u c t u r e i n each case. F i n a l l y an extension of the Paterno-Btichi r e a c t i o n of perfluoro compounds to polymer s y n t h e s i s was attempted by examining the c y c l o a d d i t i o n of p e r f l u o r o g l u t a r y l f l u o r i d e to two p e r f l u o r o o l e f i n s under a v a r i e t y of experimental c o n d i t i o n s . T h i s work was l a r g e l y u n s u c c e s s f u l and the r a t i o n a l i z a t i o n of the r e s u l t s obtained w i l l be found i n the l a s t Chapter o f this thesis. - V - ACKNOWLEDGEMENTS The author i s indebted to Dr. W.J. F e a s t f o r h i s continued help and encouragement u n f a i l i n g l y given during the s u p e r v i s i o n of t h i s work. His patience and understanding, both on the p r o f e s s i o n a l and personal l e v e l , have been i n v a l u a b l e to the author. Thanks are due to Dr. R.S. Matthews f o r running n.m.r. s p e c t r a , Dr. B.J. Cromarty f o r t r a n s l a t i n g papers from German, and Drs. G.M. Brooke and C D . B a r t l e t t f o r h e l p f u l suggestions. A s s i s t a n c e from members of the t e c h n i c a l and laboratory s t a f f i s g r e a t l y appreciated; i n p a r t i c u l a r a s s i s t a n c e from Mrs. S.E. Robson f o r running i . r . s p e c t r a and Mr. D. Hunter f o r p r a c t i c a l advice i s g r a t e f u l l y acknowledged. D i s c u s s i o n s with the other members o f the Research Laboratory '25' have been p a r t i c u l a r l y h e l p f u l and t h e i r c o l l a b o r a t i o n i s a l s o a p p r e c i a t e d . The author thanks the European Research O f f i c e o f the U.S. Array f o r the award of a maintenance grant and Mr. Apostolos Spanomanolis of 'Christos Spanomanolis Sons', Athens, Greece, f o r some f i n a n c i a l support. F i n a l l y thanks are due to Mrs. E . Duddy and Miss A. Laing f o r typing t h i s t h e s i s , and Mr. M. Strange f o r helping with some of the drawings. - vi - MEMORANDUM The work described i n t h i s t h e s i s was c a r r i e d out i n the Chemistry L a b o r a t o r i e s of the U n i v e r s i t y of Durham between September 1975 and August 1978. T h i s work has not been submitted f o r any other degree and i s the o r i g i n a l work of the author, except where acknowledged by r e f e r e n c e . - vii - STUDIES DIRECTED TOWARDS THE STEP-GROWTH PHOTOPOLYMERIZATION OF CARBONYL COMPOUNDS TABLE OF CONTENTS Page No. Abstract i i i Acknowledgements Memorandum v vi Table of Contents v i i CHAPTER 1: GENERAL INTRODUCTION AND BACKGROUND 1.1. Photopolymerization Redefined 1 1.2. Photopolymerization V i a the S i n g l e t Manifold 2 1.3. Photopolymerization V i a the T r i p l e t Manifold 4 1.4. Photopolymerization V i a a R e a c t i v e Ground S t a t e Species 8 Formed i n an I n i t i a l Photochemical Reaction 1.5. Step-Growth Photopolymerization i n the S o l i d S t a t e 9 1.6. O r i g i n s o f Current Work 10 1.7. B r i e f I n t r o d u c t i o n i n t o the Photochemistry of Carbonyl 13 Compounds CHAPTER 2; PHOTOREDUCTIVE POLYMERIZATION OF AROMATIC BISKETONES 2.1. Introduction 17 2.1.a. H i s t o r i c a l Background 17 2.1.b. Some Aspects of Relevant Photochemistry 32 2.1.b.i. Introductory Survey 2.1.b.ii Spin F l i p p i n g and Reactions w i t h i n the Solvent Cage 2.1.b.iii. The Yellow Intermediate Observed i n the Photoreduction 32 38 41 of Aromatic Ketones 2.1.b.iv. Complex Formation between K e t y l R a d i c a l and Ketone 43 2.1.b.v. E f f e c t o f S t r u c t u r e on Photoreduction o f Aromatic Ketones 47 - viii - Page No. 2.1.b.vi. S t e r i c E f f e c t s on Photoreduction 50 2.1.b.vii. The Temperature E f f e c t on Photoreduction 52 2.1.b.viii. Photoreductive Addition of Ketones to Methylene Groups 53 2.1. e. 2.2. O b j e c t i v e s of the Study 54 Attempts to Synthesize Poly(pentaphenylglycerols) and 57 Higher Analogues 2.2.a. I n t r o d u c t i o n to the Idea 57 2.2.b. How One Event Led to Another 59 2.2.c. The 'Prehistory' of Organic Photochemistry - the Nature of 61 a Dispute 2.2.d. The Resolution of the Dispute 64 2.2.e. Ground S t a t e S y n t h e s i s of T r i p h e n y l g l y c o l 67 2.2.f. The Benzophenone/Triphenylglycol 67 2.2.g. The P h o t o r e a c t i v i t y of 1,2,2,2-Tetraphenylethanol 72 2.2.h. Some Relevant Photoreactions 73 2.2.i. Attempts to Prepare Pentaphenylglycerol V i a Ground S t a t e 74 Photoreaction Chemistry 2.2.j . Experimental 2.2.j.i. Readily Available Materials 2.2.j . i i . 2.2.j.iii. 2.2. k. 2.3. Solvents M a t e r i a l s Synthesized from Simpler Chemicals D i s c u s s i o n and Conclusions Use of the Diphenylmethane/Benzophenone Reaction as the 79 79 80 80 85 89 Polymer B u i l d i n g Step 2.3.a. D e t a i l s of Model Reaction 90 2.3.a.i. D i s c u s s i o n of L i t e r a t u r e 90 2.3.a.ii. Experimental R e s u l t s 94 2.3.a.iii. Comments on L i t e r a t u r e Claims i n the L i g h t of Present R e s u l t s 97 - ix - Page No. 2.3.b. S y n t h e s i s of Monomers 104 2.3.b.i. D i s c u s s i o n of P o s s i b l e Routes 104 2.3.b.ii. S e l e c t i o n of Routes 105 2.3.b.iii. Reasons f o r F a i l u r e 114 2.3.b.iv. P u r i f i c a t i o n and Proof o f S t r u c t u r e 116 2.3.C. Polymerization Runs 117 2.3.c.i. A Preliminary Investigation 117 2.3.c.ii. Examination of the E f f e c t o f Concentration 118 2.3.c.iii. Large S c a l e Experiments 121 2.3.d. C h a r a c t e r i z a t i o n o f Products 123 2.3.e. Reactions o f Polymers 128 2.3.e.i. Depolymerization 128 2.3.e.ii. Treatment with Dehydrating Agents 129 2.3.e.iii. P a r a l l e l Model Compound S t u d i e s 131 2.3.f. Experimental 134 2.3.f.i. Readily Available Materials 134 2.3.f.ii. Solvents 134 2.3.f.iii. M a t e r i a l s Synthesized from Simpler Chemicals 135 2.3.g. D i s c u s s i o n and Conclusions CHAPTER 3: 135 THE PRODUCTS OF IRRADIATION OF BENZALDEHYDE AND ITS DIFUNCTIONAL ANALOGUES 3.1. Introduction 141 3.1.a. E a r l y Photochemical I n v e s t i g a t i o n s 141 3.1.b. Ground S t a t e Polymerization of Benzaldehyde and i t s 145 Difunctional Analogues 3.I.e. The Photochemistry of Phthalaldehyde 147 3.1.d. B r i e f I n t r o d u c t i o n i n t o the Photochemistry of Benzaldehyde 148 3.1. e. O r i g i n a l Ideas 151 3.2. The Photochemistry of Tere- and Iso-phthalaldehyde 3.2.a. Results 152 152 - x Page 3.2. b. 3.3. D i s c u s s i o n i n the L i g h t of Recent Knowledge The Benzaldehyde Photoproduct 155 156 3.3.a. Recent Preparation and S p e c t r a l C h a r a c t e r i s t i c s 156 3.3.b. Experimental 157 3.3.b.i. P u r i f i c a t i o n of Benzaldehyde 157 3.3.b.ii. Irradiations 158 3.3.b.iii. P u r i f i c a t i o n of Product 159 3.3.b.iv. Attempted C h a r a c t e r i z a t i o n of the 'Photopolymer' 160 of Benzaldehyde 3.3.c. Conclusion CHAPTER 4: and Suggestions f o r Further Work STUDIES ON THE 164 CYCLOADDITION OF PERFLUOROGLUTARYL- FLUORIDE TO SOME PERFLUOROOLEFINS 4.1. The Paterno-Buchi Reaction 168 4.1.a. Introduction 168 4.1.b. Mechanism 169 4.1. e. Limitations 171 4.2. A p p l i c a t i o n of the Paterno-Buchi Reaction to Polymer 173 Synthesis 4.3. A p p l i c a t i o n s of the Paterno-Buchi Reaction to Poly- 176 f l u o r i n a t e d and P e r f l u o r o Systems 4.4. The P h o t o l y s i s of A c y l F l u o r i d e s 183 4.5. O r i g i n a l Idea 186 4.6. Experimental 188 4.6.a. Starting Materials 188 4.6.b. Irradiations 190 4.6. c. Results 191 4.7. Discussion 191 4.8. Conclusions 196 No. - xi - Appendices and References Appendix A - Apparatus, Instruments and General Techniques Appendix B - I n f r a r e d Spectra Appendix C - L e c t u r e s , C o l l o q u i a , Seminars and Conferences Attended CHAPTER 1 General I n t r o d u c t i o n and Background The term photopolymerization i s commonly understood to mean the i n i t i a t i o n by l i g h t of a chain polymerization process, although more g e n e r a l l y i t i m p l i e s the i n c r e a s e i n molecular weight caused by l i g h t and therefore i n c l u d e s the 1 2 p h o t o c r o s s l i n k i n g o f macromolecules. ' The l i n k between these two aspects of photopolymerization i s the use o f l i g h t as energy source and polymeric materials as the o b j e c t of i n t e r e s t . P h o t o i n i t i a t e d v i n y l polymerization has been the o b j e c t o f r e s e a r c h 3 i n t e r e s t s i n c e the t u r n o f the century and a l a r g e volume of s c i e n t i f i c and 4-8 t e c h n o l o g i c a l information has been published. The area remains one o f i n t e n s e a c t i v i t y both i n academic and i n d u s t r i a l l a b o r a t o r i e s and i s a l s o of considerable commercial importance i n the s u r f a c e coatings, e l e c t r o n i c s , 9-12 p r i n t i n g and r e l a t e d i n d u s t r i e s . The use o f p h o t o c r o s s l i n k i n g o f polymeric m a t e r i a l s has been t r a c e d back 13 to the Babylonians, although the development o f t h e area r e a l l y begins with the work o f Minsk on the s y n t h e s i s o f p o l y ( v i n y l c i n n a m a t e ) . He a l s o showed how the i n c o r p o r a t i o n o f dyes i n t o coatings containing the polymer provided a 14 15 means o f p h o t o s e n s i t i z i n g the c r o s s l i n k i n g r e a c t i o n . ' This deliberate p h o t o c r o s s l i n k i n g i s , l i k e p h o t o i n i t i a t e d v i n y l polymerization, an area o f intense c u r r e n t a c t i v i t y and a t o p i c with considerable commercial , . 11,12,16 importance. C r o s s l i n k i n g i s commonly observed as a process competing with degradation i n polymers exposed to l i g h t . I n general terms such c r o s s l i n k i n g i s l a r g e l y due to r e a c t i o n s o f secondary polymer r a d i c a l s formed by cleavage o f groups or atoms from the main polymer c h a i n . A c t u a l c r o s s l i n k i n g may occur by r a d i c a l combinations or by additions to unsaturated s i t e s i n other c h a i n s . A number of examples o f photochemical c r o s s l i n k i n g of polymers and the r e l a t e d photochemical 13 17 18 g r a f t i n g o f one monomer to a polymer may be found i n the l i t e r a t u r e . ' ' - 2 - The preceding paragraphs very b r i e f l y d e s c r i b e r e a c t i o n s long c l a s s i f i e d under the general heading o f photopolymerization. Over the l a s t few y e a r s , however, there have been s e v e r a l r e p o r t s o f polymer syntheses of the s t e p growth type i n which photochemical r e a c t i o n s a r e used i n forming the bonds o f a l i n e a r polymer c h a i n i n each propagation s t e p . pointed out the need to re-examine De Schryver and Smets have the concept o f photopolymerization. They have suggested t h a t the term be reserved s p e c i f i c a l l y f o r polymerization processes i n which every chain propagation s t e p i n v o l v e s a photochemical 19 reaction, and i n t h i s sense photopolymerization i s d i s t i n c t from photo- i n i t i a t e d polymerization. De Schryver and Smets have pointed out t h a t photo- polymerizations can be d i v i d e d i n t o three c l a s s e s according t o the nature of the s p e c i e s involved i n the propagation step, which may be a s i n g l e t e x c i t e d s t a t e , a t r i p l e t e x c i t e d s t a t e , o r a r e a c t i v e ground s t a t e r e s u l t i n g from a 20 photochemical r e a c t i o n . 1.2. Photopolymerization V i a the S i n g l e t Manifold. The d i m e r i z a t i o n of anthracene a t the 9,10 p o s i t i o n s i s a w e l l e s t a b l i s h e d 21 photochemical r e a c t i o n proceeding v i a the s i n g l e t s t a t e . When b i s - anthracenes a r e i r r a d i a t e d polymers can be obtained and i t has been demonstrated t h a t t h i s r e a c t i o n proceeds v i a the s i n g l e t manifold, and g i v e s the head t o t a i l 22 23 product, a s shown i n F i g u r e 1.1. ' Two p o i n t s a r e p a r t i c u l a r l y worthy o f note, i n the f i r s t p l a c e the r e a c t i o n proceeds i n the d i r e c t i o n o f polymer formation when the i r r a d i a t i o n i s c a r r i e d out a t 350 nm and depolymerization at below 300 nm wavelength; t h i s s e r v e s to i l l u s t r a t e the importance o f s e l e c t i n g the appropriate energy range f o r any p a r t i c u l a r photopolymerization process. Secondly, the degree o f polymerization (d.p.) a t t a i n a b l e i n t h i s r e a c t i o n appears to depend on the nature o f the group l i n k i n g the two anthracene u n i t s i n the monomer, thus f o r monomers o f s t r u c t u r e I s i g n i f i c a n t l y lower d.p.'s were obtained on i r r a d i a t i o n than f o r monomers o f s t r u c t u r e I I 22 21 (Figure 1.2). ' * - 3 - / OIOIO 009) hy Dioro F i g u r e 1.1 I I I F i g u r e 1.2 The e x p l a n a t i o n o f the discrepancy l i e s i n t h e p o s s i b i l i t y o f a c h a i n growth terminating s i d e r e a c t i o n f o r s t r u c t u r e I which does not e x i s t f o r s t r u c t u r e I I , namely a 1,5-sigmatropic hydrogen s h i f t from t h e methylene attached t o C-9 to C-10. T h i s o b s e r v a t i o n s e r v e s to i l l u s t r a t e the Importance o f understanding a l l the d e t a i l s o f the p h o t o r e a c t i o n s o f monomers s e l e c t e d f o r use i n step-growth photopolymerization. - 4 - 1.3. Photopolymerlzatlon V i a the T r i p l e t Manifold. Populating the e x c i t e d s t a t e by way o f energy t r a n s f e r from an appropriate s e n s i t i z e r ensures r e a c t i o n on the t r i p l e t manifold and excludes I n t e r v e n t i o n of s i n g l e t s t a t e s o f the monomer. One o f the e a r l i e s t examples o f s o l u t i o n phase step-growth photopolymerization was the benzophenone s e n s i t i z e d polymerization o f tetrachloroblsmaleimldes (Figure 1.3, I I I , X = C l , 19 24 R = -fCHg)^) • ' A d e t a i l e d study o f the photochemistry o f the blchromophorlc system I I I by De Schryver and co-workers u n d e r l i n e s the p o i n t , made I n r e l a t i o n to the photopolymerlzatlon o f blsanthracenes, t h a t a l l p o s s i b l e a s p e c t s o f the r e a c t i o n and the p r o p e r t i e s o f the r e a c t a n t s and products must be considered i n * X X P a 6 m IV tr V F i g u r e 1.3 designing step-growth photopolymerization p r o c e s s e s . Thus, although f o r tetrachlorobismaleimides ( I I I , X = C l , R = -fCH.)- ) where n = 7, 9 o r 11 z n i r r a d i a t i o n o f a s o l u t i o n o f monomer and benzophenone i n methylene c h l o r i d e leads to genuine l i n e a r high polymers o f s t r u c t u r e I V , monomers where an even number o f methylenes separate the malelmide u n i t s f a i l t o reach high molecular - 5 - weight s i n c e the products p r e c i p i t a t e from s o l u t i o n a t a r e l a t i v e l y low degree o f polymerization. T h i s i s not the only r e s t r i c t i o n on the g e n e r a l i t y of the s y n t h e s i s o f polymers I V from monomers I I I . When the monomers having uns u b s t i t u t e d maleimi.de double bonds ( I I I , X = H) a r e i r r a d i a t e d i n s o l u b l e products a r e formed, presumably accounted f o r by c r o s s l i n k i n g v i a v i n y l polymerization a t the double bonds. F o r some lengths of methylene sequence R monomeric cyclomers V a r e formed (Figure 1.3); the r e l a t i v e proportions o f l i n e a r polymer ( I V ) , cyclomer (V), and i n s o l u b l e c r o s s l i n k e d product depend on the nature o f the v i n y l s u b s t i t u e n t s X, the l i n k i n g group R, and t h e r e a c t i o n conditions such a s the monomer concentration. A f u r t h e r f a c t o r which 24 was emphasized by these authors was how c r i t i c a l l y important very high reagent p u r i t y can be i n r e a c t i o n s of t h i s kind; thus, although no detectable d i f f e r e n c e was e s t a b l i s h e d between samples o f tetrachlorobismaleimides p u r i f i e d by s l i g h t l y d i f f e r e n t techniques one sample polymerized to genuine high polymer on i r r a d i a t i o n i n the presence o f benzophenone whereas another sample was recovered as unchanged monomer a f t e r i d e n t i c a l treatment; presumably t r a c e s o f an e f f e c t i v e t r i p l e t quencher c o n s t i t u t e d the d i f f e r e n c e between samples. Other examples o f photopolymerization v i a the t r i p l e t manifold i n c l u d e 25 the i r r a d i a t i o n o f biscoumarins i n the presence o f benzophenone s e n s i t i z e r and the s y n t h e s i s o f polyoxetanes by i r r a d i a t i o n o f bisbenzophenones i n the 26 27 presence o f equimolar amounts o f the d i o l e f i n s , t e t r a m e t h y l a l l e n e o r furan. ' The u n s e n s i t i z e d p h o t o l y s i s of biscoumarins with s h o r t c h a i n length (n < 8) 28 y i e l d s e x c l u s i v e l y endo cyclomers a r i s i n g from the e x c i t e d s i n g l e t manifold (Figure 1.4, VI) while benzophenone s e n s i t i z e d r e a c t i o n leads to high molecular weight polymers. The predominant exo head t o head s t r u c t u r e was e s t a b l i s h e d by nmr spectroscopy (Figure 1.4, V I I ) . The case of photochemical r e a c t i o n between benzophenone and tetramethyla l l e n e has been s t u d i e d i n d e t a i l and i n a d d i t i o n to the oxetane s t r u c t u r e s - 6 - n hv 0* o VI 9CO 9 12 «3 Q OC(CH„LC ii 212 c©6 hy H3 0 H VII Figure 1.4 a r i s i n g from c y c l o a d d i t i o n there are three t e r t i a r y a l c o h o l s formed v i a i n i t i a l a b s t r a c t i o n of hydrogens from the methyl groups of tetramethylallene by t r i p l e t benzophenone followed by coupling of the k e t y l and a l l y l r a d i c a l s 29 (Figure 1.5). mass balance. Products i n Figure 1.5 account f o r over 98% of the o v e r a l l Approximately 90% of benzophenone molecules r e a c t by c y c l o - a d d i t i o n , the r e s i d u a l 10% r e a c t i n g by i n i t i a l a b s t r a c t i o n of hydrogen from 29 tetramethylallene. of bisketones I n the l i g h t of t h i s evidence one would expect i r r a d i a t i o n i n the presence of equimolar amounts o f t e t r a m e t h y l a l l e n e y i e l d copolymers of s t r u c t u r e V I I I , structures). Figure 1.6 (together with isomeric to H © <0>O 0 tHA 3'2 n 3'2 H ( C H © H O 3^ tHJ3'2 Figure 1.5 © (CH,) 3'2 Ar' Ar' QK3 H H CH tHJ. © VIII Figure 1.6 m - 8 - Such copolymers were r e c e n t l y reported 26 I n which c a . 90% of the polymer chain l i n k s are oxetanes. The use of furan as the r e a c t i n g diene system ( i n s t e a d of t e t r a m e t h y l - a l l e n e ) presented s p e c i a l problems due to i t s v o l a t i l i t y which were s u c c e s s f u l l y 27 overcome i n the end r e s u l t i n g i n the s y n t h e s i s of l i n e a r polymers. A more d e t a i l e d examination o f these cases confirms the r e s t r i c t i o n s on g e n e r a l i t y and the p r a c t i c a l d i f f i c u l t i e s which apply to t h i s area of polymer synthesis. 1.4. Photopolymerizatlon V i a a R e a c t i v e Ground S t a t e Species Formed i n an I n i t i a l Photochemical Reaction. The photochemical p o l y a d d i t i o n of dibenzaldiimines i s an example o f t h i s class (Figure 1.7, I X ) . The r e a c t i o n i s benzophenone s e n s i t i z e d and proceeds v i a coupling of Ar-CH-NH-R type r a d i c a l s . " ' 0 Another example reported i s the photopolymerization o f b i s a z i d e s , the propagation s t e p of which i s b e l i e v e d to proceed through a r e a c t i v e n i t r e n e 31-34 intermediate" (Figure 1.7, X). f R Ar—CH—N+CHsWI—CH—Ar - ™ s P • - -CH—NH4<H,-H*KH— * (OijljCHOH * 2 2 340 nm IX Figure 1.7 n - 9 - Stevens and co-workers maleimide to benzene (Figure 1.8) . 37 ' made use of the photocycloadclition of and a l k y l benzenes 38 39 ' to s y n t h e s i z e l i n e a r polyimides Reaction i s thought to i n v o l v e 2+2 c y c l o a d d i t i o n o f photo- e x c i t e d maleimide to the aromatic r i n g followed by ground s t a t e D i e l s - A l d e r a d d i t i o n w i t h a second maleimide unit. "0 6-^0^0=6^ o N—R N n Figure 1.8 Another i n t e r e s t i n g example o f t h i s type of photopolymerization i s the 40 r e d u c t i v e coupling of aromatic diketones which a f f o r d s polybenzopinacols. Photoreductive polymerization w i l l be d e a l t w i t h i n d e t a i l i n Chapter 2 of t h i s thesis. 1.5. Step-Growth Photopolymerization i n the S o l i d S t a t e . 41-44 T h i s f i e l d was i n i t i a t e d by Hasegawa and co-workers. The photo- c y c l o d i m e r i z a t i o n of o l e f i n s to cyclobutane d e r i v a t i v e s was a p p l i e d to the s o l i d s t a t e photocycloaddition p o l y m e r i z a t i o n of 2 , 5 - d i s t y r y l p y r a z i n e and - 10 - r e l a t e d s t r u c t u r e s (Figure 1.9, X I ) . The s o l i d s t a t e photopolymerlzation was found to be topochemically c o n t r o l l e d . The double bonds should be a p p r o p r i a t e l y l o c a t e d one beside the other f o r the d l m e r l z a t l o n to take p l a c e . For I n s t a n c e , only one c r y s t a l l i n e form of o,a'-bis(4-acetoxy-3-methoxybenzylidene)-pb e n z e n e d l a c e t o n l t r l l e was found to polymerize i n the s o l i d s t a t e (Figure XII) 1.9, 45 ©CH^HQCH-<H0 hv X n 9 N N hv N N H XI _n Figure 1.6. 1.9 O r i g i n s of Current Work. The s u c c e s s f u l photoreductive coupling o f aromatic bisketones and the s y n t h e s i s o f polyoxetane copolymers from equimolar mixtures of diketones and - 11 - d i o l e f i n s are c h a r a c t e r i s t i c examples of the step-growth photopolymerizatlon of carbonyl compounds. The work to be d e s c r i b e d i n t h i s t h e s i s was intended as an attempt to extend the number of known examples i n t h i s a r e a . To t h i s end, a number of mono- and di-carbonyl compounds were s y n t h e s i z e d and c h a r a c t e r i z e d ; t h e i r p h o t o r e a c t i v i t i e s under a v a r i e t y of d i f f e r e n t c o n d i t i o n s were examined, and r e a c t i o n products were i d e n t i f i e d wherever possible. The main reasons f o r undertaking t h i s study were: (1) the d e s i r e to explore the r e l a t i v e l y new area of photopolymerization f o r i t s own (ii) step-growth sake; the p o s s i b i l i t y of preparing novel polymeric s t r u c t u r e s which might have i n t e r e s t i n g p h y s i c a l o r chemical p r o p e r t i e s . The o r i g i n a l o b j e c t i v e was to attempt e s t a b l i s h i n g the range of 46 a p p l i c a b i l i t y of the polymer syntheses formulated i n F i g u r e 1.10. rr hy n 1 • C | C ! r? n hy n Figure 1.10 I t was intended t h a t X and Y would be e i t h e r f l u o r i n e o r h i g h l y f l u o r i n a t e d groups but i n p r i n c i p l e a wide v a r i e t y of s t r u c t u r e s could be used. The experimental work c a r r i e d out on t h i s i n i t i a l p r o j e c t i n v o l v e d the study of a model r e a c t i o n between a p e r f l u o r o d i c a r b o n y l compound (namely hexafluorog l u t a r y l f l u o r i d e ) and two d i f f e r e n t perfluoromonoolefins (namely perfluorohept- - 12 - 1-ene and p e r f l u o r o c y c l o h e x e n e ) . The aim was to determine the conditions under which the model r e a c t i o n could g i v e a high y i e l d o f 1:1 o r 2:1 adducts, and then to attempt the a c t u a l polymer forming r e a c t i o n ( d i a c i d f l u o r i d e i n the presence of a d i o l e f i n ) under the appropriate conditions (Figure 1.11). F cr-v F hv hv ^C(CF) 0 2:1 2--1 OF |C(CF 2)4 h 2 :1 1:1 F i g u r e 1.11 T h i s work was l a r g e l y u n s u c c e s s f u l and the r e s u l t s obtained w i l l be d i s c u s s e d i n Chapter 4 o f t h i s t h e s i s . The main body o f work to be described i s concerned with an examination o f some a s p e c t s o f the photopolymerization o f aromatic carbonyl compounds. As a f i r s t aim the photochemistry o f t e r e - and iso-phthalaldehyde was examined both i n the s o l i d s t a t e and i n the presence o f hydrogen donors. No r e a c t i o n was detected i n the f i r s t case, whereas the l a t t e r experiments afforded i n s o l u b l e branched m a t e r i a l s o f a s y e t u n i d e n t i f i e d s t r u c t u r e s . I r r a d i a t i o n o f the mono- - 13 - f u n c t i o n a l compound, benzaldehyde, e i t h e r neat o r i n the presence o f hydrobenzoin y i e l d e d a telomer with i n t e r e s t i n g p h y s i c a l p r o p e r t i e s . Attempts to c h a r a c t e r i z e t h i s m a t e r i a l w i l l be d e s c r i b e d i n Chapter 3. F u r t h e r attempts to extend the w e l l known photochemical s y n t h e s i s o f 40 polybenzopinacols t o polymers with more than two a d j a c e n t C-H_-C-0H u n i t s i n D 5 the r e p e a t i n g block (polypentaphenylglycerols and higher analogues) (Figure 1.12) d i d not give p o s i t i v e r e s u l t s . Model monomer r e a c t i o n s were attempted f o r t h i s purpose and, as a r e s u l t , i n t e r e s t i n g p r o p e r t i e s o f c e r t a i n aromatic a l c o h o l s were e s t a b l i s h e d . 9 o where m > 3 m J nn F i g u r e 1.12 F i n a l l y , extension to polymer s y n t h e s i s o f a w e l l e s t a b l i s h e d r e a c t i o n , namely the photoreductive coupling o f t h e benzophenone-diphenylmethane system, afforded polymeric m a t e r i a l s o f r e l a t i v e l y low d.p. S p e c t r o s c o p i c and chemical 13 evidence and e s p e c i a l l y the use o f comparative C nmr s t u d i e s have been s u c c e s s f u l l y employed f o r the assignment o f product s t r u c t u r e s . A detailed account of t h i s work appears i n Chapter 2. Most o f the chemistry involved i n these s t u d i e s i s based on a s p e c t s o f the photochemistry o f the carbonyl group. 1.7. B r i e f I n t r o d u c t i o n i n t o the Photochemistry o f Carbonyl Compounds. The absorption o f l i g h t i n non-conjugated o r g a n i c molecules u s u a l l y i n v o l v e s the promotion o f a s i n g l e e l e c t r o n from a a, v o r n o r b i t a l i n the ground s t a t e to a p r e v i o u s l y vacant n* o r o* antibonding o r b i t a l . Molecular - 14 - o r b i t a l s , appropriate energy l e v e l s and e l e c t r o n i c t r a n s i t i o n s f o r the s i m p l e s t example of a molecule containing the carbonyl chromophore, formaldehyde, a r e 47 available. With simple ketones, the t r a n s i t i o n of lowest energy i s the weak n •*• I T * t r a n s i t i o n g e n e r a l l y r e s p o n s i b l e f o r the longest wavelength ( c a . 290 ran) absorption and fundamental i n most photochemical processes i n v o l v i n g s a t u r a t e d ketone p h o t o l y s i s . A very i n t e n s e absorption around 150 nm corresponds to a n -+• ir* e x c i t a t i o n . Conjugation s h i f t s both the n •*• wavelengths. TT* and IT -> I T * absorption bands to longer The lone p a i r o r b i t a l s a r e r e l a t i v e l y u n a f f e c t e d w h i l s t the h i g h e s t ir o r b i t a l i n the ground s t a t e i s r a i s e d i n energy, r e l a t i v e to a non-conjugated carbonyl ir o r b i t a l , and the lowest ir* o r b i t a l lowered i n energy. I n appropriately s u b s t i t u t e d unsaturated ketones the ir ir* c o n f i g u r a t i o n may become the lowest 47 r t r i p l e t s t a t e , the n -»• ir* s h i f t being l a r g e compared to the n •+ ir* s h i f t . Most of the carbonyl photochemistry occurs v i a the t r i p l e t e x c i t e d manifold, indeed v i a the n,ir* t r i p l e t , which r e s u l t s from the t r a n s i t i o n of one e l e c t r o n from the oxygen n o r b i t a l to the ir* o r b i t a l , followed by a s p i n f l i p system c r o s s i n g ) . Figure (inter- A common p i c t u r e of the carbonyl n,ir* t r i p l e t i s shown i n 1.13. t mm Figure 1.13 For most carbonyl compounds intersystem c r o s s i n g from the e x c i t e d s i n g l e t 29 t o the t r i p l e t s t a t e i s a v e r y e f f i c i e n t p r o c e s s . The quantum y i e l d f o r - 15 - intersystem c r o s s i n g of benzophenone i s 1.00. With some carbonyl compounds however, the r e l a t i v e energies of the e x c i t e d s t a t e s may intersystem c r o s s i n g . be unfavourable f o r For example 2-naphthaldehyde, 9-anthraldehyde and 9- 48 tetracenealdehyde a l l f l u o r e s c e on i r r a d i a t i o n i n ethanol intersystem c r o s s i n g . indicating inefficient I n non-polar s o l v e n t s the n -»• ir* t r a n s i t i o n i s s h i f t e d to longer wavelengths and the v •*• it* t r a n s i t i o n to s h o r t e r wavelengths, and i n heptane s o l v e n t 2-napthaldehyde and s i m i l a r aromatic aldehydes become f l u o r e s c e n t with e f f i c i e n t intersystem non- crossing. The commonly accepted p i c t u r e of the n, I T * e x c i t e d s t a t e of carbonyl compounds, i n v o l v i n g promotion of an 'n' e l e c t r o n to an antibonding ir* o r b i t a l , 29 r e s u l t s i n a lone e l e c t r o n l o c a l i z e d on the oxygen. analogous to an ordinary alkoxy r a d i c a l . reactions; The s t a t e i s thus formally T h i s analogy extends to observed thus, alkoxy r a d i c a l s are known to a b s t r a c t hydrogen and add to o l e f i n s , r e a c t i o n s which f i n d t h e i r counterparts i n photoreduction and photo29 49 50 c y c l o a d d i t i o n r e a c t i o n s of carbonyl compounds. ' ' The m a j o r i t y of known photochemical r e a c t i o n s of alkanones have been 1 c l a s s i f i e d under one of three r e a c t i o n t y p e s r ^ R R R \ hu ( i ) cleavage o f the bond to the carbonyl - Norrish type I cleavage: + group R" f 0 (ii) intramolecular (Norrish type I I ) or i n t e r m o l e c u l a r a b s t r a c t i o n of a hydrogen atom by the carbonyl oxygen atom: R R'-H + L H H •R- \ / c ! J /\ R i R / K - 16 - (ill) cycle-addition to an unsaturated carbon-carbon l i n k a g e : n hu An oxetane c—o R 3 \/ 4 An oxetene (unstable) V An o, B-unsaturated compound Aldehydes and aromatic ketones undergo r e l a t e d r e a c t i o n s and unsaturated carbonyl compounds (conjugated and non-conjugated) undergo a wide v a r i e t y o f cleavage, a b s t r a c t i o n , rearrangement and c y c l o a d d i t i o n r e a c t i o n s . The v a r y i n g 52 53 54 r e a c t i o n s a r e d i s c u s s e d i n standard t e x t s , r e l a t i n g t o carbonyl compound photochemistry those by Swenton and Turro e t a l . ^ ' and t o p i c s s p e c i f i c a l l y may be found i n reviews such a s 5 The types o f r e a c t i o n d i r e c t l y r e l e v a n t to these s t u d i e s a r e type 2 i n t e r m o l e c u l a r (photoreduction o f carbonyl) and type 3 (oxetane and p o l y oxetane formation). chapters. They w i l l be d i s c u s s e d i n more d e t a i l i n the appropriate CHAPTER 2 Photoreductlve polymerization of aromatic bisketones - 17 - 2.1. Introduction 2.1.a. H i s t o r i c a l background The photoreductive coupling of benzophenone i n ethanol i s a w e l l e s t a b l i s h e d r e a c t i o n (Figure 2.1) . 56 o r isopropanol 57 Reaction occurs by n,ir* 99 hv isopropanol i i OH OH F i g u r e 2.1 e x c i t a t i o n of benzophenone carbonyl followed by reduction and coupling of the resultant ketyl radicals. With benzophenone and isopropanol the y i e l d has 58 been reported to be q u a n t i t a t i v e and the r e a c t i o n s shown i n F i g u r e 2.2 have hv ii OH CH, I 3 + H—C + OH 3 I OH I CH, r >®-f-® OH OH OH H„C 3 \'X C C OH C H 3 ^ II F i g u r e 2.2 ^cT ^ ^ ^ - - 18 - been used to account f o r the observed p r o c e s s . A more d e t a i l e d d i s c u s s i o n of t h i s photoreduction w i l l be given i n 2.1.b. At the beginning of t h i s decade the r e a c t i o n was a p p l i e d to the s y n t h e s i s of polybenzopinacols from benzophenone-type aromatic diketones, according to 59—62 the scheme shown i n F i g u r e 2.3. Higgins and co-workers i n 1970; 0 0 II II n Ar.C.Ar.C.Ar The f i r s t r e s u l t s were p u b l i s h e d by they attempted the photoreductive p o l y m e r i z a t i o n OH OH I — •c * isopropanol/ cosolvent Figure Ar I Ar - cAr 2.3 of m- and p-dibenzoylbenzene and 4,4<-dibenzoyldiphenylether ( p r e p a r a t i v e method A, Table 2.1). I n the same i s s u e of the same j o u r n a l Pearson and 59 Thiemann d e s c r i b e d a s i m i l a r i n v e s t i g a t i o n of (preparative methods D-F, Table 2.1). weight oligomers. p-dibenzoylbenzene Both groups obtained only low molecular Modifications introduced by Higgins' group (preparative methods B, C) using an open system under a stream of nitrogen r a t h e r than a c l o s e d v e s s e l under a nitrogen atmosphere and s h o r t e r i r r a d i a t i o n times gave a s i g n i f i c a n t l y higher molecular weight polymer from 4,4'-dibenzoyldiphenylether. Polymers from 4,4'-dlbenzoyldiphenylsulphide / 4,4*-dibenzoyldiphenyl-methane and -ethane were a l s o obtained by the modified procedures B,C, but m- and p-dibenzoylbenzene and 4,4'-dibenzoyldiphenyl f a i l e d to give polymers. Higgins claimed t h a t use of a 450 watt lamp with Pyrex f i l t e r i n s t e a d of a 100 watt long wavelength lamp allowed much s h o r t e r i r r a d i a t i o n times but 61 produced polymers of lower inherent v i s c o s i t y . He suggested t h a t the lower molecular weight polymers may have been a r e s u l t of cleavage of C-C bonds by - 19 - O H O l vo vo m in to vo vo vo vo vo vo 0) Q) Q) 0) tt) 3 a ai 01 N 0) (0 o •H 01 a> u a OS 00 Q Q a) w o—o ^0> w 8 CN CN O ci id O vo H n « H O Pi a O 00 in in o> u CN s i n vo 00 IS CN rH •9 CN 0 CD <*> 8 00 H N •H U rH id rfHIQHh «c pa n < m u o=o o=o 6Q •d a D 6 6 - 20 - 0) S VO Vfi H H N vo vo vo 01 o> VO vo 0) CN CM CN 0) aa 10 8n Wo 2 TI ai 04 01 vo C CN CN 5* ro CN CN en 0) ft Pi CN ro ro r-t CN ro IX VO 0) dP 10 82 vo ro CN 00 8 VO 8 VO iH 0) 4J T J 01 H CN •9 Qi 0) 0) ffl U O m u CN 3 8 n o 2 8 CN 8 CN 8 - 21 o °o rH c O 4J a • s a; x +J 0 rH iH n >w 01 •H a> I c x M £-5 id id n > -a _ •P ts a •p 0 0 fH o m O _ 0 (N c -a 4J H o 0 a> •P id 0 M 4J m m CM •0 in 0 rH H 0 id Pi S N 0 a rH 1 82 8 rH «H TJ 1 V CM •5 9 8 H5 d 1 >-i c >i § iH > 0 01 01 3 3 § CM 0 rH •a 01 id >H •Hon 01 O in rH "2 > 0 c id 3 3 v id •O iH > 4J tn id m +J 0 •H Id B •a a 0 M 8 "H O D 01 01 W O 3 u 01 3 •p c 0 u M 0 -rt .P rt rl Q id •P 4-I Si id • 0 0 •P a' 5 0 rH rl 0 > S 41 Q 0 4H A c 01 C O 0 0 M c o £ 01 -P a o •rl 01 01 p Id i H O 0 •d u id -o «• CM •P a 0 >d rH 0 >i •d 0 T3 •-4 0 U 0 X! a ro ^ • O CM (O 01 •p •d id 0 c S ^ 0 U 01 0 0 4J rH iH > 0 N A c o -d M W p0 I 0 ,Q id • 4J c •a 01 rH \ rl 8 *s 4 a IT •rl . C -P O o id oi U 0 m •p id - 0 a rH O rH P C <d _ 8 C 0 § -2 U * tH rl 01 J3 0 -P U 0 0 id H T-I Oi c i rH •P _ id -O •d S 1% x5 Id 3 0 +J *d c X T» id *: > id o rH 5 55 •d c 0 » TH i2 c 11 3 - 3 C oi O 0 0 (3 •H _ H TJ O (U <H rH •p 0 o m M M e 0 X) H 01 r l «d 0 N 0 0 o o •H •rl 0 T) rH O •H 01 0 •H rH I t r l rH * >• •P 3 D» rH C O -rj V) -P 0 *H o C •rl 8 -° 01 U O > CD ^ s o 0 »H m a 0 •d n -a* id " o u id o O -H ig T3 3 S •H 01 oi *d in > rl •rt O id M H to u <o « •d n § (0 01 01 ft U o 0 > rH 3 01 0 0 55 •d § •P TJ a> «n C •rl O 01 01 o § 0 0 4J 10 +J 01 i H 0 id •a 3:2 c •d Q § &•po •d 8 SI § cn oi i *d Q< 6 tt) O O A H H n •d n «J 0 id a • o id _ -r( 9 3 0 _ O r l <0 •H 9 2 +J 01 rH 01 H H 0 J o o id O 0) 01 -H Id *J CO 01 4J 8 id3 +JS a u o •rl u ri 0 0 S O 1 I rH 3 rl r^ rH 0 0 O "W rH £ 2 a rH „ o oi • m a in CM O O o r*i -H 01 3 o 55 - 22 - 6 1 the higher i n t e n s i t y i r r a d i a t i o n ; * explanation may t h i s seems u n l i k e l y and a more p l a u s i b l e l i e i n the g r e a t e r heat output of the 450 watt lamp. The 1 reported f a i l u r e of m- and p-dibenzoylbenzenes and 4,4 -dibenzoyldiphenyl to form high polymers was attributed '" to a bathochromic s h i f t of the ir -»• I T * 6- absorption i n going from benzophenone to these diketones, r e s u l t i n g i n overlap I T * absorptions and l a c k of e f f i c i e n t hydrogen a b s t r a c t i o n from the with the n low-lying ir,ir* t r i p l e t . Although l a t e r work v a l i d a t e s t h i s explanation i n the 26 case of 4,4'-dibenzoyldiphenyl i t seems l e s s p l a u s i b l e f o r the other monomers. The higher molecular weight polymers produced by H i g g i n s 1 group, showed no carbonyl absorptions ( c a . 1650 cm "S i n t h e i r i n f r a r e d s p e c t r a but low molecular weight p o l y p i n a c o l s showed considerable r e s i d u a l carbonyl s t r e t c h , which was 60 attributed to unreacted monomer or to such s t r u c t u r e s as the one shown i n F i g u r e 2.4. , Strong bands around 3500 cm ^ were c h a r a c t e r i s t i c of the t e r t i a r y OH OH OH I 0 - c — A r — C C Ar I C- 6. 1-2 Figure -OH 2.4 groups i n the p o l y p i n a c o l s . The i n f r a r e d s p e c t r a of the polymers were compared with those of model p i n a c o l s obtained by photocoupling pbenzoyldiphenylether and p-benzoyldiphenylethane, and by coupling p-benzoyldiphenylmethane with magnesium and i o d i n e . I n f r a r e d s p e c t r a of the model p i n a c o l s and the polymers were q u a l i t a t i v e l y s i m i l a r . Likewise, infrared s p e c t r a of oligomers from m- and p-dibenzoylbenzene were q u a l i t a t i v e l y s i m i l a r to the i n f r a r e d spectrum of benzopinacol. N.m.r. s p e c t r a of the oligomers from m- and p-dibenzoylbenzenes 4,4'-dibenzoyldiphenylether 60 and showed a s m a l l peak a t S 5.3 assigned to - 23 - benzhydryl protons; the higher molecular weight samples showed no benzhydryl hydrogen i n t h e i r n.m.r. s p e c t r a . Higgins^ 1 has suggested t h a t i n p r e p a r a t i v e methods B,C t h i s may be a r e s u l t o f i n c r e a s e d monomer c o n c e n t r a t i o n compared to method A, and t h a t weaker reducing s o l v e n t s may a l s o y i e l d higher molecular weight polymer.^ 0 Pearson and Thiemann s t u d i e d the i r r a d i a t i o n o f p-dibenzoylbenzene i n 59 isopropanol i n some d e t a i l . Under 'normal' i r r a d i a t i o n c o n d i t i o n s (preparative method D, Table 2.1) a polymer comprising c a . 5 coupled monomer u n i t s was obtained. The n.m.r. spectrum o f t h i s polymer was assigned as f o l l o w s : fi 1.3 ( s i n g l e t CH >, 6 2.4 (broad s i n g l e t OH), 6 3.0 (broad s i n g l e t 3 OH), 6 5.8 (very weak benzhydryl hydrogen), 6 7.8 ( m u l t i p l e t , aromatic hydrogens The i n t e n s i t y o f the s i g n a l a t 6 5.8 i n d i c a t e d t h a t a t l e a s t h a l f the polymer ends were capped with benzhydryl hydrogen (Figure 2.5, X = H) OH OH F i g u r e 2.5 The r e l a t i v e i n t e n s i t i e s o f the s i g n a l s a t 6 5.8 and 6 1.3 suggested t h a t a few polymer c h a i n s terminated w i t h i s o p r o p y l a l c o h o l groups (Figure 2.5, X = (CH^)2 C0H c y c l i c form. ) w h i l s t the remainder o f the polymer was designated t o be i n When the r e a c t i o n was c a r r i e d out a t a higher temperature (100° p r e p a r a t i v e method E , Table 2.1) the product obtained was s a i d t o be capped almost e x c l u s i v e l y with hydrogen, the n.m.r. s i g n a l a t 6 5.8 being much stronger whereas t h e product obtained a t 25° ( p r e p a r a t i v e method F , Table 2.1) was assigned a c y c l i c s t r u c t u r e s i n c e only aromatic (6 7-7.5) and OH (6 3.0) proton - 24 - n.m.r. s i g n a l s were observed. I n these cases i n f r a r e d s p e c t r a o f the products 59 from p-dibenzoylbenzene showed the absence o f carbonyl groups. 59-61 Elemental analyses o f p o l y p i n a c o l s , where reported, were sometimes i n only f a i r agreement with the c a l c u l a t e d v a l u e s . 59 During i r r a d i a t i o n s o f m- and p-dibenzoylbenzenes a y e l l o w colour has 59 been observed. Pearson has suggested t h a t t h i s i s due t o the intermediate shown i n Figure 2.6 ( i n the case o f p-dibenzoylbenzene), the evidence on which t h i s assignment was based being a long wavelength OH absorption a t 416.4 nm, the OH F i g u r e 2.6 n o n - r a d i c a l nature o f the intermediate (which showed no e . s . r . s i g n a l s ) and its stability. However m-dibenzoylbenzene could not form an analogous quinone-dimethide s t r u c t u r e , which c a s t s doubt on t h i s r a t i o n a l i z a t i o n . 62 De Schryver and co-workers reported t h a t s i g n i f i c a n t l y higher molecular weight polybenzopinacols could be obtained by i r r a d i a t i o n o f degassed i s o p r o p a n o l - d i c h l o r o m e t h a n e s o l u t i o n s o f bisbenzophenone a t 350 nm (preparative method G, Table 2.1) . No benzhydryl hydrogen s i g n a l appeared 62 i n the n.m.r. s p e c t r a o f the products, d.p.'s ranging from 23 t o 70. The r e s u l t s o f the work reviewed above a r e summarised i n Table 2.1. The e f f e c t of s o l v e n t composition on the r e a c t i o n was examined by v a r i o u s 60,61,62 workers. De Schryver reported t h a t the r a t e o f disappearance o f the 62 carbonyl f u n c t i o n a l i t y depends on the nature of the cosolvent; f o r 0.05M s o l u t i o n s o f 4,4'-dibenzoyldiphenylmethane a t 32°C, r e l a t i v e r a t e s f o r a 1:5 molar r a t i o o f isopropanol:- c o s o l v e n t were, CHC1_ 1, C H_CH, 2.5 and benzene 4. C - 25 - Higgins claimed t h a t a 50:50 mixture was most s a t i s f a c t o r y f o r the benzeneisopropanol s o l v e n t system, arguing t h a t i n c r e a s i n g the proportion of isopropanol reduced monomer s o l u b i l i t y w h i l s t decreasing the proportion o f isopropanol n e c e s s i t a t e d longer i r r a d i a t i o n t i m e s . ^ He reported t h a t f o r p o l y p i n a c o l s prepared by method A (Table 2.1) i t made no d e t e c t a b l e d i f f e r e n c e whether the s o l v e n t was neat isopropanol or 50:50 mixtures of THF isopropanol, or benzene and ethanol, or benzene and and isopropanolthese r e s u l t s are s u r p r i s i n g i n the l i g h t of the work reported by the B e l g i a n group. As can be seen from the above review of the e a r l y p u b l i c a t i o n s i n t h i s f i e l d there was some confusion and disagreement between the v a r i o u s groups. 64 Work c a r r i e d out i n t h i s department ambiguities. was r e l e v a n t i n r e s o l v i n g some of the The e s s e n t i a l f e a t u r e s of t h i s work are summarized i n Table 2.2. Aromatic diketones were prepared by F r i e d e l - C r a f t s s y n t h e s i s and r i g o r o u s l y purified. Monomers were d i s s o l v e d i n mixtures of isopropanol and benzene and oxygen was excluded from the s o l u t i o n s by bubbling nitrogen through f o r about 30 mins. I r r a d i a t i o n s were c a r r i e d out using a Rayonet photochemical r e a c t o r i n c o r p o r a t i n g RUL-3500 lamps which emit between 320o8 and 4000A* with the g r e a t e s t i n t e n s i t y a t 3660A*. T h i s work was c a r r i e d out s o l e l y to check the p h o t o r e a c t i v i t y of the monomers and consequently a d e t a i l e d optimiz a t i o n of conditions was not undertaken, however a number of u s e f u l p o i n t s emerge from a c o n s i d e r a t i o n of the data presented i n Table 2.2. the r e a c t i o n s c a r r i e d out using 1:1 benzene-isopropanol Considering mixtures, i t i s c l e a r t h a t i n s e v e r a l cases s i g n i f i c a n t l y higher d.p.'s were obtained by Andrews than those p r e v i o u s l y reported; an improvement from d.p. for example, i n the case of m-dibenzoylbenzene 3 to d.p. 17 was observed. Since these r e a c t i o n s were c a r r i e d out under apparently i d e n t i c a l c o n d i t i o n s the most l i k e l y cause o f the observed d i f f e r e n c e must be the p u r i t i e s of monomer and/or s o l v e n t s and the explanation advanced p r e v i o u s l y f o r the l a c k of r e a c t i v i t y of - 26 - s id vo O •• id 0 id v D5 (U 8 H H IS G Oi H G Oi Qi O—CJ 8 (0 o—u S I 0 3 l/l H (0 id CM CN G 0 id a Oi 0 O N 04 0 o 0 I-I id « U N H 0 8 IS CO 8 8 8 0 3 0 •H TJ 0 O o=o 0 0 6 O ft a. 6 O 6 O CN SB 8 Ar Polypinacol Code Irradiation in 1:1 benzene:isopropanol 8 • t •0 CO CM 8 CN CM in rn rH CM CO T in 'CM CM a u <> 8 •ro <> CM 8 8 "~CN 8 CM in s o CM a u (25) ro 12600 • (18) (11) •r—N (23) • Oi 10300 Irradiation in 3 to 4:1 benzene:isopropanol 1 - 27 - •0 Ok r» 8 * rH O <» 'CM rH a u - 28 - m-dlbenzoylbenzene (namely, a bathochromlc s h i f t of the IT •* it* absorption and i n e f f i c i e n t hydrogen a b s t r a c t i o n by the l o w - l y i n g TT,TT* t r i p l e t ) must be i n v a l i d . T h i s i n t e r p r e t a t i o n i s confirmed by c o n s i d e r a t i o n of the u l t r a v i o l e t s p e c t r a of aromatic diketones shown i n F i g u r e 2 . 7 . ^ v L \t 9 C O T •5 X o i 230 , 250 1 270 J 0 1 290 Figure r 310 1 330 1 350 1 370 M n m ) 2.7 For the m a j o r i t y of diketones examined by Andrews the p o l y p i n a c o l s p r e c i p i t a t e d from s o l u t i o n when a 1:1 benzene-isopropanol mixture was whereas when a higher proportion of benzene was used the polymers used remained i n s o l u t i o n and consequently a higher d.p. was g e n e r a l l y a t t a i n e d . The b e s t r e s u l t s i n t h i s f i e l d have been achieved however by De Schryver 40 and co-workers who adopted a new approach to the s u b j e c t . They considered how the photoreductive coupling of d i a r y l ketones was i n f l u e n c e d by the d e t a i l e d mechanism f o r the photoreduction of benzophenone put forward by - 29 - Weiner i n 1971. The cage r e a c t i o n reported by Welner, t h a t i s t h e combination of a (C H ) COH and a (CH^COH r a d i c a l t o give the mixed p i n a c o l o f s t r u c t u r e g 5 2 (CgHg) C(OH)C(OH)(CH ) , c o n s t i t u t e s a c h a i n terminating step i n t h i s context. 2 3 2 Such a complication could be e l i m i n a t e d i f a bisbenzhydrol (Figure 2.8) was used as both a hydrogen donor and a monomer i n the polymerization, one would then 9^ 9 H— C - - @ — A r — C OH —H OH F i g u r e 2.8 expect higher molecular wieght polymers t o be obtained. Further t h i s approach allows the preparation o f a l t e r n a t i n g copolymers or homopolymers by choice o f appropriate monomer p a i r s whereas one would expect t h a t the i r r a d i a t i o n o f a binary mixture of bisketones i n the presence o f isopropanol 40 would l e a d t o s t a t i s t i c a l c o p o l y p i n a c o l s . De Schryver and co-workers prepared a number o f aromatic bisketonos and b i s k e t o t r i a z o l e s by w e l l established routes. bisbenzhydrols. Reduction o f some of the diketones afforded the corresponding I r r a d i a t i o n o f equimolar s o l u t i o n s (benzene solvent) o f b i s k e t o n e s / bisbenzhydrols a t 350 nm y i e l d e d as expected high molecular weight p o l y p i n a c o l s . Before i r r a d i a t i o n , oxygen was excluded from the s o l u t i o n s by degassing on a vacuum l i n e . R e s u l t s a r e summarized i n Table 2.3. I t can be seen t h a t the bisketone/bisbenzhydrol system provides the most v a l u a b l e method f o r obtaining good y i e l d s o f high molecular weight m a t e r i a l s . I n some cases d.p.'s as high as 430 were obtained. On the other hand, the p h o t o l y s i s o f some binary mixtures o f aromatic bisketones i n the presence o f isopropanol y i e l d e d s t a t i s t i c a l c o p o l y p i n a c o l s . I n some favourable c a s e s , the n.m.r. spectrum o f the copolymers obtained - 30 - Table 2.3 40 Photopolymerizatlon of B i 9 a r y l k e t o n e - Bisbenzhydrol System 0 0 OH Concentration OH b d.p. Cnl 430 0.36 Ketone Hydrol Ar Ar' (mole/litre ) time A A 0.300 4 days 1 1 0.170 5 days A 10 0.013 4 days A E 0.170 5 days 0.21 A P 0.100 4 days 0.08 K5 A 0.170 4 hrs 141,000 362 0.43 K6 A 0.170 5 hrs 140,000 354 0.43 Footnotes: a Irradiation M —n 174,000 0.18 93,000 102 0.33 a: combined ketone/hydrol c o n c e n t r a t i o n b: measured on the high molecular weight f r a c t i o n obtained by p r e c i p i t a t i o n A: Ar 1: Ar = 10: Ar E: Ar = P: Ar = CH, 31 Table 2.3 contd. N K5 N Ar V N (CH„) N / CH CH N **** N K6 Ar N~(CH_) CH N / N N N CH - 32 - showed three d i s t i n c t l i n k a g e s and from the i n f r a r e d spectrum intensities the copolymer's composition and the number average length of the sequences could be determined. De Schryver's work provided two very important c o n c l u s i o n s : i) the photopolymerization o f b i s a r y I k e t o n e s i s h i g h l y a f f e c t e d by every aspect o f the photoreduction o f the model ketones, ii) the bisarylketone/bisbenzhydrol system provides a means o f overcoming the 'cage' r e a c t i o n which i s a c h a i n terminating r e a c t i o n ; i t i s c l e a r l y the b e s t method f o r the s y n t h e s i s o f high molecular weight p o l y p i n a c o l s . 2.1.b. Some a s p e c t s o f r e l e v a n t photochemistry 2.1.b.i. I n t r o d u c t o r y survey The photoreduction o f carbonyl compounds i n s u i t a b l e hydrogen donating 67 s o l v e n t s has been widely i n v e s t i g a t e d f o r w e l l over h a l f a century 56 Ciamician and S i l b e r began r e s e a r c h i n the f i e l d . since Photoreductive coupling has already been d i s c u s s e d b r i e f l y i n 2.1.a w i t h r e f e r e n c e t o the p r e p a r a t i o n o f polybenzopinacols and a simple mechanism f o r the photoreduction o f benzophenone i n isopropanol was given (see F i g u r e 2.2). I n the presence o f a l k a l i , however, i t was d i s c o v e r e d t h a t benzhydrol 68 was formed, and i n a s e r i e s o f photoreductions o f benzophenone and s u b s t i t u t e d 68 benzophenones containing a s m a l l amount o f sodium-2-propanolate, Bachmann showed t h a t benzhydols could be obtained i n high y i e l d . He proposed t h a t as A f a s t as the p i n a c o l was formed i t decomposed to benzhydrol ( i n the case o f benzopinacol). However, the proposed mechanisms f o r the formation o f benzhydrol 69 from benzophenone have subsequently been questioned. The most i n t e n s i v e l y s t u d i e d r e a c t i o n i s t h e a c t u a l photoreductive coupling r e a c t i o n y i e l d i n g 1,2-ethane d i o l s . Aldehydes and ketones undergo the r e a c t i o n , ketones having r e c e i v e d by f a r the most a t t e n t i o n . that yield Some examples o f ketones p i n a c o l s on a p r e p a r a t i v e s c a l e i n c l u d e , a p a r t from benzophenone, 6 - 33 - 4,4'-dimethoxybenzophenone, acetophenone, 72 70 4,4'-dichlorobenzophenone, 2-, 3-, and 4 - a c e t y l p y r i d i n e s , 73 70 3-benzoylpyridine, 74 and a - t e t r a l o n e . Benzophenone i s the most thoroughly s t u d i e d ketone, y i e l d i n g in quantitative y i e l d . benzopinacol The quantum y i e l d f o r the formation o f acetone i n the photoreduction o f benzophenone i n isopropanol i s n e a r l y constant using 75 i r r a d i a t i o n a t s e v e r a l d i f f e r e n t wavelengths between 366 and 254 nm, suggesting t h a t both n -*• I T * e x c i t a t i o n and TT -*• v* e x c i t a t i o n (of the benzene 29 chromophore) may u l t i m a t e l y b r i n g about r e a c t i o n . Various s o l v e n t s have been used as hydrogen donors, i n p a r t i c u l a r a l c o h o l s and alkylbenzenes. 76 77 ' Many comparisons o f s o l v e n t s have been made with benzophenone as r e a c t i n g ketone. Some quantum y i e l d s o f benzophenone 78 disappearance i n v a r i o u s s o l v e n t s a r e shown i n Table 2.4. Table 2.4 Quantum Y i e l d s o f Benzophenone Disappearance Solvent Molar Concentration o f Benzophenone i n Hydrogen Donor Solvents Quantum Y i e l d of Disappearance Water lO" 4 0.02 Benzene icf 2 0.05 Toluene lo" 2 0.45 Hexane ID" 2 - ID" 4 1.00 Ethanol lo" 4 - icf 1 1.00 Isopropanol io" 5 - ID" 1 0.80 Where the quantum y i e l d s o f benzophenone disappearance t o 2.00 a r e almost zero (water and benzene), bond energies with r e s p e c t t o hydrogen a b s t r a c t i o n a r e high enough (> 100 k c a l mole "S to prevent hydrogen atom a b s t r a c t i o n by the t r i p l e t with anything other than very low c o l l i s i o n a l e f f i c i e n c y . 77 In 71 - 34 - hexane, toluene and ethanol, quantum y i e l d s are l e s s than or equal to u n i t y 77 and concentration Independent. I t I s suggested t h a t a l l benzophenone t r i p l e t s a b s t r a c t a hydrogen atom, the appropriate C-H bond energies i n hexane and toluene being approximately 85 and 80 k c a l mole' 1 respectively. The o v e r a l l quantum y i e l d s then depend on the thermal r e a c t i o n s o f the r a d i c a l 1 R formed on a b s t r a c t i o n from the s o l v e n t RH. Reversion o f the primary hydrogen a b s t r a c t i o n r e a c t i o n ( i . e . Ph &0H + R* 2 an o v e r a l l quantum y i e l d o f l e s s than u n i t y . y i e l d s tend to a l i m i t i n g value of 2, R* [ i . e . » PhjCO + RH) r e s u l t s i n I n isopropanol where quantum (CH^) C0H] t r a n s f e r s hydrogen 2 to benzophenone with the production of acetone and more Ph C0H. 2 Such a t r a n s f e r i s e n e r g e t i c a l l y improbable i n the benzyl or hexyl r a d i c a l s from toluene o r hexane. Where ketones are not photoreduced i n alcoholic solvents, a more powerful hydrogen donor such as t r i - n - b u t y l s t a n n a n e may 79 photoreduction, as i n the case of 2-acetonaphthone. 66 A r e c u r r i n g problem allow i n mechanistic s t u d i e s o f benzophenone photoreduction i n isopropanol was the f a i l u r e to d e t e c t the mixed p i n a c o l 2-methyl-l,1diphenyl-1,2-propanediol (Figure 2.9), whereas analogous cross-coupled 76 products were obtained f o r benzophenone photoreduction i n toluene and cumene, 0 T f OH 3 OH Figure 2.9 80 and primary a l c o h o l s o l u t i o n s . Weiner has shown t h a t t h i s mixed p i n a c o l i s i n f a c t produced i n the photoreduction of benzophenone i n isopropanol, and under conditions where a l l f r e e k e t y l r a d i c a l s are s c a v e n g e d . ^ Thus, the mixed p i n a c o l r e s u l t s from a cage r e a c t i o n and the o v e r a l l r e a c t i o n i s explained - 35 - by the modified sequence shown i n F i g u r e 2.10. The s i g n i f i c a n t f r a c t i o n o f cage reaction (0.11) r e q u i r e s t h a t , i n the p a i r o f r a d i c a l s comprising A OH + (CH ) CHOH 3 0 O".^© ^ 2 + < C a ) 3 2 (A) CH. ^ C—CH OH 3 OH H HC CH 3 3 0 (CH ) COH + 3 2 OH H,C ,CH. 3 (CH > COH + 3 3 c 2 * OH OH OH 66 OH OH HC 3 OH (B) F i g u r e 2.10 & H (where - 36 - the e l e c t r o n s p i n s are i n i t i a l l y p a r a l l e l ) , s p i n f l i p p i n g must occur i n some r a d i c a l p a i r s p r i o r to d i f f u s i o n out of the s o l v e n t cage. t h a t there has been much controversy It. must be noted over the nature o f intermediates 81 photoreduction o f benzophenones i n isopropanol. t h a t the observed yellow intermediate i n the 81 82 Filipescu ' has proposed has the s t r u c t u r e B (shown i n F i g u r e 2.10) i n the case o f benzophenone and 4 - a l k y l s u b s t i t u t e d benzophenone photoreductions 81 i n isopropanol. F o r benzophenone, F i l i p e s c u suggested a r e a c t i o n scheme shown i n Figure 2.11. * OH + ^ <Q>~C-<g) +(CH ) 60H (CH ) CHOH 3 3 2 2 > B (R=H) l B + QH OH Figure OE 2.11 I t was proposed t h a t B y i e l d e d products v i a t h e dark r e a c t i o n shown i n Figure 2.12. OH 66 However, Weiner s t a t e d t h a t the q u a n t i t a t i v e formation C "3 V/-3_. „ j . ^ ^ X ^ S / ^ N Ph-r-Cf*=V X. 0—H J 0 r 0 c § d a r / \ Figure * > reaction 2.12 Products - 37 - o f B from the p a i r Ph C(OH) + (CH ) COH i s i n c o n s i s t e n t w i t h the d e t e c t i o n 2 3 2 of the mixed p i n a c o l i n t h e product mixture. W h i l s t observed yellow c o l o u r a t i o n i n t h e r e a c t i o n may be due t o the presence o f B, W e i n e r ^ concluded t h a t the yellow intermediate cannot be transformed i n t o products a t a r a t e c o n s i s t e n t with h i s e a r l i e r s t u d i e s . The low e f f i c i e n c y o f photoreduction i n benzene has r e s u l t e d i n the s o l v e n t being used i n many s e n s i t i z a t i o n experiments. However, there i s a low quantum y i e l d of reduction even with benzene s o l v e n t and observations concerning the a c t u a l behaviour of benzophenone t r i p l e t s i n benzene s o l v e n t s 83 are confusing. The value given f o r the quantum y i e l d o f disappearance o f benzophenone i n Table 2.4 i s 0.05 and i s the upper l i m i t as determined by Beckett and P o r t e r . 7 7 B e l l and L i n s c h i t z 8 4 estimated a quantum y i e l d o f 0.1 f o r formation o f the primary k e t y l r a d i c a l PhjCOH compared to 0.022 +_ 0.0O6 85 found by Buettner and Dedinas. D e a c t i v a t i o n o f t r i p l e t s i n benzene has 86 87 ' to occur v i a formation o f b i r a d i c a l intermediates o f the 87 type shown i n F i g u r e 2.13 ( f o r benzophenone). Schuster and B r i z z o l a r a have been suggested H F i g u r e 2.13 pointed out t h a t t h i s provides a path f o r formation o f biphenyl according t o the scheme i n F i g u r e 2.14. 3 The major products from i r r a d i a t i o n o f benzophenone i n benzene (6 x 10*" M 14 s o l u t i o n ) a t 290 nm a r e biphenyl and benzopinacol and C s t u d i e s have 83 r e c e n t l y shown t h a t the biphenyl i s e s s e n t i a l l y s o l v e n t d e r i v e d . Schuster 38 C x2 C—O 6 H H 2 H H OH0> C OH F i g u r e 2.14 88 suggested t h a t the quenching o f benzophenone t r i p l e t s i n aromatic s o l v e n t s i n v o l v e s an i n t e r a c t i o n i n c o r p o r a t i n g a t l e a s t p a r t i a l charge t r a n s f e r from ketone t r i p l e t as donor towards aromatic a s acceptor. 2.1.b.ii. Spin f l i p p i n g and r e a c t i o n s w i t h i n the s o l v e n t cage The photoreduction o f benzophenone by toluene and cumene i s known 76,89 to y i e l d t e r t i a r y a l c o h o l s , c r o s s coupling products r e s u l t i n g from the combination o f the benzophenone k e t y l r a d i c a l and s u b s t r a t e r a d i c a l s (Figure 2.15). As a l r e a d y mentioned, most workers denied the e x i s t e n c e o f such a product f o r the benzophenone/isopropanol system u n t i l Weiner^ i d e n t i f i e d the mixed p i n a c o l by a n a l y t i c a l g . l . c . 6 The 2-methyl-l,1-diphenyl- 1,2-propanediol has been obtained i n 18% y i e l d i n the e l e c t r o c h e m i c a l reduction 90 of benzophenone i n acetone-water mixture. I r r a d i a t i o n o f benzophenone i n methanol r e s u l t s i n the formation o f the mixed p i n a c o l and Mauser obtained BO 91. the 1,1-diphenylglycol (Ph C(0H)-C(0H)H ) i n 5% y i e l d . ' I n isopropanol, 2 the c r o s s p i n a c o l was estimated t o be p r e s e n t i n the product to the extent o f 92 11% by Weiner and l e s s than 4% by Gotzmer. When camphorquinone was used to - 39 - OP f ©O S o OH 0 0 "«> OH OH „ H CH. CH, (trace) (24%) ©4—r@ n JO 0 9 CH W CH 3 Figure (24%) OH ^ CH OH 3 CH ( 5 2 % ) 3 2.15 scavenge a l l the f r e e k e t y l r a d i c a l s which d i f f u s e out of the s o l v e n t cage, i r r a d i a t i o n of benzophenone i n isopropanol with benzhydrol. be 0.8. y i e l d e d the c r o s s p i n a c o l along The r a t i o of benzhydrol to the c r o s s p i n a c o l was found to T h i s i s good evidence t h a t the mixed p i n a c o l r e s u l t s , a t l e a s t i n * p a r t*, from a cage r e a c t i o n .66,92 fcJ The i r r a d i a t i o n of benzhydrol i n acetone generated the same r a d i c a l s as the benzophenone-isopropanol system. However, a g r e a t e r amount o f the mixed p i n a c o l i s found among the products of the former r e a c t i o n , p o s s i b l y because of d i f f e r e n c e s i n the s o l v e n t s forming the cage. Both systems show the same 81 l i g h t - a b s o r b i n g intermediate. Complete reduction of the carbonyl group occurs when a benzophenone k e t y l r a d i c a l a b s t r a c t s a hydrogen atom from an acetone k e t y l r a d i c a l . A b s t r a c t i o n of a hydrogen atom from a benzophenone k e t y l r a d i c a l by acetone k e t y l r a d i c a l i s estimated an to be n e g l i g i b l e s i n c e the quantum y i e l d f o r benzophenone disappearance i s high (up to 2 ) ; a l s o the e n e r g e t i c s of the process agree with t h i s conclusion s i n c e the 0-H bond s t r e n g t h i n benzophenone - 40 - k e t y l r a d i c a l i s 35 k c a l mole whereas the bond strength i n an acetone k e t y l 1 r a d i c a l i s only 20 k c a l mole . The f a c t t h a t i n the i r r a d i a t i o n o f benzophenone 93 in (-)2-butanol the recovered s o l v e n t i s not racemized could be considered as evidence i n favour of t h e above hypothesis. Unlike benzophenone which undergoes a one e l e c t r o n reduction process t o y i e l d p i n a c o l , photolyses o f d i ( 2 - p y r i d y l ) k e t o n e , d i ( 4 - p y r i d y l ) k e t o n e and 4-pyridyl-phenylketone i n isopropanol l e a d to the formation o f the corresponding dipyridylmethanols and 4 - p y r i d y l p h e n y l c a r b i n o l a s t h e only products i n each 94 95 case. ' I t has been shown t h a t the lowest n,ir* t r i p l e t s t a t e o f these ketones i s involved i n the hydrogen a b s t r a c t i o n r e a c t i o n . The complete reduction o f the carbonyl chromophore i n the h e t e r o c y c l i c ketones may be r a t i o n a l i z e d by the d i f f e r e n c e i n e l e c t r o n e g a t i v i t y between nitrogen and carbon. Since there i s a higher p r o b a b i l i t y o f f i n d i n g the e l e c t r o n o f the k e t y l r a d i c a l on nitrogen, the t r a n s f e r of a hydrogen atom from the acetone k e t y l r a d i c a l y i e l d s the conjugated enol which r e v e r t s t o the c a r b i n o l by prototropism (Figure 2.16) . OH Ph— C — < — > Thus i t can be seen t h a t the r e d u c i b i l i t y o f OH Ph— C = GH ^ ) N + Me C0H — ^ P h — C 2 a = OH ^ ^ N H + Me C0-> P h — C — 2 F i g u r e 2.16 carbonyl compounds depends on the nature o f the carbonyl double bond and i t s s u b s t i t u e n t s , and i s s e n s i t i v e t o s t r u c t u r e m o d i f i c a t i o n s which a f f e c t the e l e c t r o n d i s t r i b u t i o n , p a r t i c u l a r l y the u - e l e c t r o n d e r e a l i z a t i o n . Similarly, aromatic ketones whose t r i p l e t s t a t e s possess pronounced ir,ir* c h a r c t e r a r e - 41 - more s u s c e p t i b l e to complete reduction. I n the case of the photoreductive coupling of blsbenzophenones, t h i s imposes some l i m i t s on polymer formation. 2.1.b.iii. The yellow Intermediate observed I n the photoreduction of aromatic ketones As has been pointed out already the formation of a yellow intermediate a y QC i s a c h a r a c t e r i s t i c f e a t u r e of the photoreduction of benzophenone i n a l c o h o l . S e v e r a l explanations have been put forward to account f o r t h i s intermediate, and these were reviewed by F i l i p e s c u who concluded, mainly on the b a s i s of s p e c t r o s c o p i c evidence t h a t the most l i k e l y s t r u c t u r e was the conjugated 81 enol d i s c u s s e d e a r l i e r (Figure 2.10). Nevertheless a d e f i n i t i v e proof of the s t r u c t u r e of t h i s frequently observed intermediate has eluded the many i n v e s t i g a t o r s who have worked on the problem. I n the photoreduction o f heteroaromatic ketones intermediates have a l s o been detected s p e c t r o s c o p i c a l l y , f o r example d i ( 4 - p y r i d y l ) k e t o n e g i v e s an 94 i n t e n s e l y absorbing s p e c i e s on photoreduction i n isopropanol. However, the d e t e c t i o n of intermediates during photoreduction i s by no means u n i v e r s a l , f o r example no intermediate could be detected during the photoreduction o f 94 di(2-pyridyl)ketone and the yellow colour a s s o c i a t e d w i t h the photoreduction 97 of benzophenone i n isopropanol i s not observed when benzhydrol i s the donor. Despite the d i f f i c u l t y i n unequivocally i d e n t i f y i n g the i n t e r m e d i a t e ( s ) involved i n photoreduction the s t r u c t u r e proposed by F i l i p e s c u seems a reasonable candidate s i n c e there i s a body of information i n the l i t e r a t u r e which can be r a t i o n a l i z e d i n terms of i n i t i a l coupling between the r a d i c a l derived from the hydrogen donor and the benzophenone k e t y l r a d i c a l , such coupling o c c u r r i n g a t the ortho or para p o s i t i o n s of a phenyl r i n g . Decafluoro98 photoreduced i n isopropanol e x c l u s i v e l y to decaf luorobenzhydrol (<j> = 0 . 6 ) . benzophenone provides a good example; t h i s ketone, u n l i k e benzophenone i s Although the primary photochemical step should be a hydrogen a b s t r a c t i o n by - 42 - the n,ir* t r i p l e t s t a t e s i n c e t h i s ketone shows the same p r o p e r t i e s as benzophenone ( s i m i l a r absorprion spectrum, i d e n t i c a l n,ir* lowest t r i p l e t energy, and e f f i c i e n t i n t e r s y s t e m c r o s s i n g ) . The f a i l u r e to i s o l a t e the p i n a c o l may a r i s e from t h e i n s t a b i l i t y o f t h e p e r f l u o r o p i n a c o l i t s e l f . 99 Dedinas reported t h a t t h e photoreduction o f decafluorobenzophenone i n cyclohexane y i e l d s , i n a d d i t i o n to the c r o s s coupling product, ortho and para s u b s t i t u t e d nonafluorobenzophenones. The formation o f these products was r a t i o n a l i z e d as shown i n F i g u r e 2.17. F (C F ) CO 6 5 2 + C H 6 1 2 -^^ ) pC 5 2 6 H l i n OH — F 0 F 4-^-F 5 C H,, n '6 ll + C f i F / C 5 V C F F F H hv <c F ) co q 6 5 2 + ' ( C F C OH OH OH H 6 5»^" 6 11 OH C.H 6 11 C.H 6 11 ° I H 6 l l - ^ . r F C — ( 0 ) - 6H11 H 6^ C C H F F + HF Figure 2.17 I r r a d i a t i o n o f acetophenone i n a mixture o f e t h e r and cyclohexane g i v e s acetophenone p i n a c o l s i n good y i e l d , together with 2-butanol d e r i v a t i v e s and 100 a p - s u b s t i t u t e d acetophenone (Figure 2.18). The acetophenone k e t y l r a d i c a l and ether r a d i c a l combine t o y i e l d the 2-butanol d e r i v a t i v e s i n a manner analogous t o the formation o f 1,1,2-triphenylethanol i n the photoreduction o f benzophenone i n toluene. The formation o f the p - s u b s t i t u t e d acetophenone can be r a t i o n a l i z e d by p o s t u l a t i n g t h a t the d e l o c a l i z e d k e t y l r a d i c a l i s attacked a t the para p o s i t i o n by t h e ethoxy e t h y l r a d i c a l t o give t h e e n o l i c intermediate which would be expected t o be o x i d i z e d by a i r t o y i e l d the observed product. - 43 - 1 \ CH 7 ._ . . />~A" 3 n v„. hv CH, CH, ^ CH I I 3 CH, CHCH CH, , 3 , 3 OH nil OH JQ j— O OH u H ° r> O (12%) H,C \ V OH HO 3 ' CH H 3 / HC 3 (3%) OEt / + H C~CH-OEf E t OEt H C H 3""~<0)~| CH. 3 Figure 2.18 The reduction o f benzophenone i n water I s a l s o c o n s i s t e n t with t h i s p a t t e r n 101,102 of r e a c t i o n . I n the absence of oxygen benzopinacol i s obtained whereas i n aerated s o l u t i o n hydroxybenzophenones are formed (<}> = 0.02) , and a t r a n s i e n t assigned as a hydroxy1 r a d i c a l adduct o f benzophenone was observed. 2.1.b.iv. Complex formation between k e t y l r a d i c a l and ketone The f a t e o f the benzophenone k e t y l r a d i c a l and the acetone k e t y l radical which d i f f u s e out of the s o l v e n t cage has been explained i n terms of two reactions. Hydrogen atom t r a n s f e r from the acetone k e t y l r a d i c a l to a ground s t a t e benzophenone molecule r e s u l t s i n the formation o f a new benzophenone ketyl radical. Benzopinacol i s then formed from the d i m e r i z a t i o n o f two benzophenone k e t y l r a d i c a l s . T h i s mechanism appears p l a u s i b l e k e t y l r a d i c a l has a high p r o b a b i l i t y which i s i n e x c e s s . s i n c e the acetone of encountering a benzophenone molecule The disappearance o f the benzophenone k e t y l i s found from e l e c t r o n second order k i n e t i c s . radical spin resonance and f l a s h p h o t o l y s i s s t u d i e s to follow Accumulating evidence shows t h a t the photoreduction o f - 44 - benzophenone i n benzhydrol does not always proceed through a d i r e c t combination of the two primary r a d i c a l s ; i n the photochemical r e a c t i o n between an excess of deuterated benzophenone and non-deuterated benzhydrol, perdeuterated, h a l f - d e u t e r a t e d and non-deuterated benzopinacols were not obtained i n the 1 0 expected s t a t i s t i c a l d i s t r i b u t i o n ; " ^ pinacol obtained. i n p r a c t i c e l a r g e amounts o f benzo- small q u a n t i t i e s of mixed p i n a c o l d ^ Q and p i n a c o l d Q were The r e s u l t can be r a t i o n a l i z e d by a r e v e r s i b l e hydrogen atom exchange o c c u r r i n g i n a benzophenone-ketyl r a d i c a l complex (Figure 2.19). J3> H—0—C 9 JO »;c—OH o >0 F i g u r e 2.19 S i n c e deuterated benzophenone was used i n e x c e s s , the e q u i l i b r i u m was d r i v e n towards the d i r e c t i o n o f the deuterated k e t y l r a d i c a l , r e s u l t i n g i n a g r e a t e r amount of benzopinacol being formed. 103 Schenck reported t h a t the i r r a d i a t i o n o f a s o l u t i o n o f benzophenone-d^ ( i n excess) and benzopinacol-d Q i n benzene r e s u l t e d i n the formation o f benzopinacol-djQ e x c l u s i v e l y (Figure 2.20). T h i s r e a c t i o n appears somewhat curious s i n c e benzopinacol i s known to be i n e r t to benzophenone n,ir* t r i p l e t 104 s t a t e a t the u s u a l wavelength (350 nm) employed i n the photoreduction. 10 The wavelength s e l e c t e d by Schenck was not s p e c i f i e d ; " * i t i s , however, probable t h a t s h o r t wavelength u l t r a v i o l e t l i g h t was used, s i n c e the C-C bond i n the p i n a c o l can be c l e a v e d by i r r a d i a t i o n a t 240 nm, or t h e r m a l l y . One way o f i n t e r p r e t i n g the r e s u l t s i s i n terms of the e q u i l i b r i a 0 - 45 - 6 K 99 o-h-o ^ or OH OH Figure shown i n F i g u r e OH OH 2.20 2.21. 240 nm OH OH OH C—0-H— 0=C H-0 Se5 D fe 5 D C C 6 D 5 - ^ OH Figure F u r t h e r reports by F r a n z e n 1 0 5 6°5 — C I OH C D ,6 5 : D 6 5 OH 2.21 tend to i n d i c a t e t h a t the p i n a c o l does not form from a d i r e c t combination o f the primary k e t y l r a d i c a l s . The photoreduction o f benzophenone i n the presence o f benzhydrol y i e l d s two i d e n t i c a l radicals 14 which however can be d i s t i n g u i s h e d by C labelling. Franzen i r r a d i a t e d a - 46 - benzene s o l u t i o n containing benzophenone C and benzhydrol C; a mixture o f benzophenone and benzopinacol as shown i n F i g u r e 2.22 was obtained. + H — r 14 - C—O c==o * 14 12 c a of OH OH OH Ti2 I 12 i; c- OH OH -c Figure The ^0> 2.22 experimental r e s u l t s are c o n s i s t e n t with a redox r e a c t i o n between benzophenone and diphenylhydroxymethyl radical. The r a d i o a c t i v i t i e s o f the p i n a c o l s formed were found to be l e s s than h a l f the a c t i v i t i e s of the i n i t i a l benzhydrols. Also, the r a d i o a c t i v i t y of the p i n a c o l s was found to decrease 12 with i n c r e a s i n g benzophenone or O + 14 H-c OH OH 9 Oh - r OH or OH T 12 l ^0> C- OH OH 12 -= C—0—H Figure 12 OH I 12 12 r=0---- H—0 — O 9 9. 99. 14 C=0 C concentration (Figure 2.23). 2.23 Po 14 C 14 OH - 47 - Furthermore, the e x i s t e n c e of the hydrogen atom exchange between benzophenone and a k e t y l r a d i c a l has been e s t a b l i s h e d by e l e c t r o n s p i n resonance s t u d i e s . form a 1 0 3 The acetone k e t y l r a d i c a l would a l s o be expected to hydrogen bridge w i t h a b a s i c oxygen atom. I n t h i s sense, a k e t y l r a d i c a l / k e t o n e complex should be envisaged to r a t i o n a l i z e the hydrogen atom t r a n s f e r i n the photoreduction of benzophenone i n i s o p r o p a n o l a. C=0- / CH / C— 0- H—- 0 = C H-0-C* \ or Of CH Figure 2.1.b.v. 1 0 3 (Figure 2.24). CH 3 CH 2.24 E f f e c t o f s t r u c t u r e on photoreduction of aromatic ketones Not a l l ketones are photoreduced to g i v e p i n a c o l s . The r e a c t i v i t y of aromatic ketones has been r a t i o n a l i z e d i n r e l a t i o n to the lowest l y i n g t r i p l e t l e v e l s of the k e t o n e . 1 0 6 When the lowest l y i n g t r i p l e t l e v e l has n,w* c h a r a c t e r , ketones are g e n e r a l l y r e a d i l y r e d u c i b l e with quantum y i e l d s f r e q u e n t l y approaching u n i t y . Ketones which possess a T T , T T * c o n f i g u r a t i o n f o r the lowest t r i p l e t s t a t e , however, are much l e s s disposed towards an example being p-phenyl benzophenone. photoreduction, I t i s suggested t h a t the higher e f f i c i e n c y i n photoreduction processes of the n,ir* s t a t e may a r i s e from the h i g h l y l o c a l i s e d n e l e c t r o n of the oxygen, whereas i n the ir,ir* t r i p l e t state the unpaired e l e c t r o n s are more d e l o c a l i z e d and hydrogen a b s t r a c t i o n i s thus 5 5 more e n d o t h e r m i c . ' 1 0 7 (Figure 2.25.) A t h i r d type of p o s s i b l e lowest l y i n g t r i p l e t s t a t e i s a charge t r a n s f e r 106 state. I n p-aminobenzophenone represented by the formula DRA, where D i s 48 Odd e l e c t r o n s delocalized over ir system Highly r e a c t i v e centre i n hydrogen a b s t r a c t i o n F i g u r e 2.25 the electron-donating amino group, R i s the aromatic r i n g , and A i s the e l e c t r o n - a c c e p t i n g carbonyl group, three important c h a r g e - t r a n s f e r s t a t e s + may be considered. These a r e DR A~ corresponding + o f benzophenone, D R A corresponding t o the c h a r g e - t r a n s f e r s t a t e t o the c h a r g e - t r a n s f e r s t a t e o f a n i l i n e + and D RA . The three s t a t e s i n t e r a c t with one another t o give a new low energy c h a r g e - t r a n s f e r (C-T) s t a t e not present i n benzophenone o r a n i l i n e . The negative charge on the oxygen i n t h i s C-T s t a t e r e s u l t s i n v i r t u a l l y zero r e a c t i v i t y towards hydrogen atom a b s t r a c t i o n . P o l a r s o l v e n t s such as isopropanol should enhance i n t r a m o l e c u l a r c h a r g e - t r a n s f e r from the e l e c t r o n donating group t o the carbonyl group, thus helping t o decrease the energy o f such c h a r g e - t r a n s f e r s t a t e s below t h a t o f the n,ir* s t a t e . Thus i n isopropanol where the C-T s t a t e o f p-aminobenzophenone i s the lowest l y i n g t r i p l e t the quantum y i e l d o f p i n a c o l i z a t i o n i s zero. cyclohexane, state, I n a l e s s p o l a r s o l v e n t such as where the c h a r g e - t r a n s f e r t r i p l e t i s no longer the lowest l y i n g t r i p l e t , p h o t o p i n a c o l i z a t i o n o f p-aminobenzophenone has been shown t o occur 108 q u i t e r e a d i l y with a quantum y i e l d o f 0.2. p-Hydroxybenzophenone has been - 49 - reported t o behave i n a s i m i l a r manner, the molecule r e a c t i n g i n the b a s i c form i n the e x c i t e d s t a t e where the pK o f i t s protonation e q u i l i b r i u m i s much lower than i n the ground s t a t e . I n accord with t h i s theory aminobenzophenones can be reduced i n isopropanol when converted t o t h e i r onium s a l t s . However, 1 0 many o f these r e s u l t s have r e c e n t l y been questioned and a r e now s u s p e c t . ^ ' 1 1 0 Thus i n r e l a t i o n t o s o l v e n t e f f e c t s on the s u b s t i t u t e d benzophenones, although the ketones are u n r e a c t i v e i n isopropanol, but r e a c t i n cyclohexane, the p r o c e s s , i n f a c t does not appear t o i n v o l v e hydrogen atom a b s t r a c t i o n leading 109 to p i n a c o l s . Pitts s t a t e s t h a t no p i n a c o l i s formed on i r r a d i a t i o n o f p-aminobenzophenone i n cyclohexane. He emphasizes t h a t spectrophotometric methods o f a n a l y s i s have f r e q u e n t l y been employed i n studying benzophenones and t h a t these methods i n v o l v e disappearance o f r e a c t a n t r a t h e r than o f product. appearance Thus a l t e r a t i o n s i n r e a c t i o n paths a r i s i n g from u n c o n t r o l l e d environmental changes and l e a d i n g t o products other than p i n a c o l s may not be observed. P i t t ' s contention i s underlined i n s t u d i e s reported by C o h e n 1 1 0 of s o l v e n t e f f e c t s on t h e i s o m e r i z a t i o n of t r a n s - s t i l b e n e i n the presence of benzophenone and aminobenzophenones, he suggested t h a t t h e f a i l u r e o f paminobenzophenone t o photoreduce i n p o l a r s o l v e n t s r e s u l t s from e s s e n t i a l l y the complete absence o f t r i p l e t s r a t h e r than the c h a r g e - t r a n s f e r s t a t e i t s e l f being u n r e a c t i v e . P i n a c o l s may only be i s o l a t e d f o l l o w i n g photochemical reduction o f a r y l ketones i n isopropanol, i f the p i n a c o l s a r e themselves s t a b l e i n i r r a d i a t e d 67 acetone s o l u t i o n s . Thus 9,9'-dihydroxy-9,9'-bixanthene i s converted i n t o 70 xanthen-9-one i n s u n l i g h t , and correspondingly xanthen-9-one i s not converted t o the p i n a c o l on i r r a d i a t i o n i n isopropanol (Figure 2.26). Ortho s u b s t i t u t e d aromatic ketones, o r those with other s u b s t i t u e n t s which can form a six-membered r i n g , hydrogen bonded to the carbonyl oxygen, may tautomerize i n an i n t r a m o l e c u l a r hydrogen a b s t r a c t i o n r e a c t i o n . Thus - 50 - CH OT l O H HO HO H.C \/ CH C II H.C 00® 3 CH \CH/ OH Figure 2.26 2-methylbenzophenone forms an enol under the i n f l u e n c e o f u l t r a v i o l e t l i g h t , 111 to the complete e x c l u s i o n o f photoreduction. " 2.1.b.vi. S t e r i c e f f e c t s on photoreduction The e x c i t e d ketone molecule must come i n t o c l o s e contact with the a l c o h o l molecule i n order f o r the a b s t r a c t i o n o f the a-hydrogen t o take p l a c e , and any bulky s u b s t i t u e n t s i n the aromatic ketone o r a l c o h o l which are c l o s e t o the r e a c t i o n centre could hinder the i n t e r a c t i o n o f the r e a c t a n t s and lower the r a t e o f hydrogen a b s t r a c t i o n . The p h o t o l y s i s o f a l k y l p h e n y l ketone i n isopropanol-benzene, y i e l d s a mixture o f acetone, d l and meso p i n a c o l s and the corresponding hydrols with 112 •acetone = •pinacol + •hydrol* ^ * u a n t u o v i e l d f o r h v d r o 1 formation i s found t o i n c r e a s e while t h a t o f p i n a c o l formation and the r a t e constant f o r hydrogen a b s t r a c t i o n decrease w i t h a - s u b s t i t u t i o n becoming b u l k i e r (Table 2.5). - 51 - 112 ^ Table 2.5 Comparative photolyses o f some a l k y l p h e n y l ketones 0 OH OH OH Rate constant 6, R pinacol Rate constant I n I n isopropanol * *hydrol k M r - 1 sec" 2,4-dimethyl- 1 3-heptanol k M _ 1 r -CH 3 -CH_CH, 2 3 -CH(CH ) 3 -C(CH ) 3 2 3 -0 sec" 0.34 0.007 7.5 x 1 0 5 2.8 x 1 0 5 0.19 0.033 4.4 x 1 0 5 2.5 x 1 0 5 0.07 0.049 1.3 x 1 0 5 0.5 x 1 0 5 0.00 0 0.20 unknown 1 c 1.0 x 10 The I d e n t i c a l r a t e constant f o r t r i p l e t decay k ( = 3 x 10 d sec ) found f o r the ketones i n Table 2.5 suggests t h a t I n c r e a s e d s t e r l c hindrance makes hydrogen a b s t r a c t i o n more d i f f i c u l t . i t y i e l d s benzaldehyde abstraction. t-Butylphenylketone does not undergo photoreduction, i n s t e a d by an a-cleavage followed by hydrogen Ortho s u b s t i t u t e d benzophenones a l s o f a i l to p i n a c o l i z e ; although orthomethylbenzophenone i s photostable prolonged i r r a d i a t i o n o f 2,4,6-trimethylbenzophenone i n isopropanol l e a d s to 2,4,6-trimethylbenzhydrol „ „j * - 113,114 as the reduction product. The s o l v e n t can a l s o c o n t r i b u t e to s t e r i c hindrance. The lower r a t e constant f o r hydrogen a b s t r a c t i o n i n 2,4-dimethyl-3-heptanol (Table 2.5) 52 - r e l a t i v e to isopropanol, r e f l e c t s a s u b s t a n t i a l decrease i n r e a c t i v i t y . The decreasing r a t e o f benzophenone photoreduction i n going from isopropanol ( r e l a t i v e r e a c t i v i t y 1.0), through m e t h y l - t - b u t y l c a r b i n o l (0.90), methyli s o b u t y l c a r b i n o l (0.39) to methylneopentylcarbinol (0.18), has been a t t r i b u t e d to s t e r i c hindrance as s o l v e n t chain branching 2.1.b.vii. increases. 1 1 5 ' 1 2 6 The temperature e f f e c t on photoreduction For aromatic ketones p o s s e s s i n g n e a r l y degenerate n,ir* and I T , i r * triplet s t a t e s , the r e l a t i v e populations of these s t a t e s a t thermal e q u i l i b r i u m are given by the Boltzmann d i s t r i b u t i o n N(n,ir*) N(ir,ir*) AE T _ e - ^T RT being the energy gap s e p a r a t i n g the two s t a t e s . i n t e n s i t y o f a non-conjugated The phosphorescence bichromophoric system such as the e s t e r shown i n F i g u r e 2.27 i s approximately the sum of the phosphorescence w w II of benzophenone II E Figure T = 2 kcal mol" 1 2.27 and t r i p h e n y l e n e i n a r a t i o l J l g = 1-5 ut 77K. At room temperature the r a t i o T 86 I :I T B = 0.20. The r e l a t i v e population of benzophenone t r i p l e t i n c r e a s e s v i a thermal a c t i v a t i o n and the emission occurs more r a p i d l y from the s h o r t lived n,IT* t r i p l e t . I t i s t h e r e f o r e expected t h a t f o r aromatic ketones e x h i b i t i n g a lowest n,ir* t r i p l e t with a nearby n i r * t r i p l e t , the r e a c t i v i t y f towards hydrogen a b s t r a c t i o n w i l l i n c r e a s e w i t h i n c r e a s i n g temperature. The - 53 - i n c r e a s e i n the r a t e o f hydrogen a b s t r a c t i o n with temperature when 2-acetonaphthone i s i r r a d i a t e d i n ethanol could be due to a s i g n i f i c a n t i n c r e a s e of the n,ir* t r i p l e t p o p u l a t i o n . 1 1 7 On the other hand, the use of high temperatures may have a dramatic e f f e c t on the f i n a l products. I r r a d i a t i o n o f benzophenone i n isopropanol a t s u f f i c i e n t l y high temperatures produces i n c r e a s i n g amounts o f benzhydrol; a t 150°C, benzhydrol becomes n e a r l y the e x c l u s i v e product, r e s u l t i n g from the 118 d i s p r o p o r t i o n a t i o n of two benzophenone k e t y l r a d i c a l s . 2.1.b.viii. Photoreductlve a d d i t i o n o f ketones to methylene groups Carbonyl compounds may undergo photoaddition to methylene groups adjacent to double bonds or aromatic systems. Benzophenone has been reported to r e a c t 119 with diphenylmethane i n s u n l i g h t to form 1,1,2,2-tetraphenylethanol whereas i r r a d i a t i o n of benzophenone with bis-(4-methoxyphenyl)methane g i v e s the c a r b i n o l and symmetrical dimers of the r a d i c a l s i n v o l v e d i n the r e a c t i o n Figure 2.28). 9 9H0> C3 CO 9H0> < \ P . . !H-C !—«"•-_ C O H OH + B CO-^-CB -^-OCH3—» 3 2 9 @ - C OH H.CO OCH 99 + H H—C OH H,CO OCH Figure OH 2.28 H f-Q-oca 3 D OCH 1 2 0 - 54 - Transannular i n t e r a c t i o n between the e x c i t e d carbonyl group of c y c l o dodecanone and one of the n e a r e s t methylene groups, with i n t r a m o l e c u l a r 121 c y c l i z a t i o n , yields a t e r t i a r y alcohol (Figure 2.29). Many other examples OH h\> Figure CO 2.29 122 of such photoadditions may be found i n the l i t e r a t u r e . Photoreductive a d d i t i o n of benzophenone to diphenyl methane w i l l be d e a l t with i n more d e t a i l i n S e c t i o n 2.2 of t h i s t h e s i s . 2.I.e. O b j e c t i v e s o f the study The photoreductive polymerization of aromatic bisketones has been thoroughly i n v e s t i g a t e d and high molecular weight polybenzopinacols have been s u c c e s s f u l l y prepared. The following s e c t i o n s d e s c r i b e attempts to extend t h i s area of s y n t h e t i c organic photochemistry. Section 2.2 i s concerned with an i n v e s t i g a t i o n of the p o s s i b i l i t y of preparing poly(pentaphenylglycerols) and, e v e n t u a l l y , higher analogues (see s e c t i o n 1.6) by photoreductive p o l y a d d i t i o n o f bisbenzophenones to s u i t a b l e hydrogen donors as i l l u s t r a t e d i n F i g u r e 2.30. For such a process to occur the requirements would be: i) i n i t i a l e x c i t a t i o n o f the carbonyl chromophore by 350 nm u l t r a v i o l e t light, ii) a b s t r a c t i o n of a t e r t i a r y hydrogen by the photoexcited carbonyl r e s u l t i n g i n the formation of the two primary r a d i c a l s shown i n F i g u r e 2.31, and - 55 - c— I I OH OH - c--Ar" I C—Ar—( OH OH m •cI hv solvent -Ar'-f— C—f- H -C— -Ar m Ar'- -C- I I OH L OHJm OH • C- I OH m OH m where m > 2 F i g u r e 2.30 111) combination o f the two primary r a d i c a l s to y i e l d the c r o s s coupled product. The f i r s t o b j e c t i v e of t h i s e x e r c i s e was therefore the c a r e f u l examination — A r —COH where m > 1 F i g u r e 2.31 o f the model r e a c t i o n (Figure 2.32) i n order to e s t a b l i s h whether processes o c c u r r i n g on photochemical e x c i t a t i o n o f benzophenone i n the presence of t r i p h e n y l g l y c o l (Figure 2.32, m = 1) would j u s t i f y f u r t h e r attempts to extend - 56 - t h i s r e a c t i o n to polymer s y n t h e s i s . OXQ hv H—C solvent I OH OH m where m Figure 2.32 Section 2.3 i s concerned with some i n v e s t i g a t i o n s on the p o s s i b i l i t y of extending a r e a c t i o n already mentioned i n 2 . 1 . b . v i i i , namely the photoreductive 119 a d d i t i o n of benzophenone to diphenylmethane to polymer s y n t h e s i s . This would i n i t i a l l y r e q u i r e a re-examination o f the model r e a c t i o n , s i n c e l i t e r a t u r e r e p o r t s on the formation and y i e l d of photoproducts are somewhat confusing l a t e r , s e c t i o n 2.3). (see Extention of the r e a c t i o n t o polymer formation would i n v o l v e the i r r a d i a t i o n , under conditions optimized during the model compound s t u d i e s of a bisbenzophenone i n the presence of a d i f u n c t i o n a l analogue o f diphenylmethane, f o r i n s t a n c e a bisbenzylbenzene. One obtaining f u r t h e r reason f o r attempting t h i s r e a c t i o n was the p o s s i b i l i t y of t e c h n o l o g i c a l l y i n t e r e s t i n g m a t e r i a l s by s t r u c t u r a l modification the r e s u l t i n g polymer. I t was of thought t h a t dehydration of the f i r s t formed . poly(1,1,2,2-tetraphenylethanol) under mild conditions conjugated system (Figure 2.33). interesting e l e c t r i c a l properties. could a f f o r d a t o t a l l y Such polyconjugated systems might possess Attempts to s y n t h e s i z e w e l l c h a r a c t e r i z e d m a t e r i a l s of t h i s general type have appeared frequently i n the l i t e r a t u r e i n 123 recent years. 57 A ? 9 OH HO c=c H 9 Figure 2.2. \ 2.33 Attempts to s y n t h e s i z e poly(pentaphenylglycerols) and higher 2.2.a. analogues I n t r o d u c t i o n to the i d e a As b r i e f l y mentioned i n 2.I.e., the o r i g i n a l i d e a was the s y n t h e s i s of perphenylated polymers with more than two adjacent C H COH groups i n the 6 i| c repeat u n i t . One conceivable way of e f f e c t i n g such a s y n t h e s i s could i n p r i n c i p l e be the photoreductive p o l y a d d i t i o n of an aromatic bisketone to a s u i t a b l e hydrogen donor, as i l l u s t r a t e d i n F i g u r e 2.30. On the b a s i s of l i t e r a t u r e information regarding the behaviour of photoexcited carbonyl groups of the benzophenone type i n the presence of hydrogen donors i t might be expected t h a t i r r a d i a t i o n of such a mixture a t the appropriate wavelength would r e s u l t i n the formation of the two primary r a d i c a l s shown i n Figure 2.31. question to be answered was whether the cage r e a c t i o n between the two would take p l a c e , and i f so to what extent; The radicals f u r t h e r i t was e s s e n t i a l to know what other p r o c e s s e s , i f any, would occur. P o s s i b l e r e a c t i o n paths f o r the two primary r a d i c a l s considered beforehand are i l l u s t r a t e d i n F i g u r e 2.34. I t can be seen t h a t the cage r e a c t i o n i s the only process which would b r i n g about formation of a homopolymer. D i f f u s i o n out of the cage and d i m e r i z a t i o n o f r a d i c a l K would r e s u l t i n the formation of a s t r u c t u r e with a p i n a c o l u n i t i n i t ; a s i m i l a r path f o r r a d i c a l L would give a s t r u c t u r e with 58 Q Ar 9 Ar I OH K) OH OH (L m x21 x2 IT QUO Ar Ar OH Ar OH Ar OH Cage ID OH OH OH m reaction Y Ar - C — HH Ar* Ar OH OH a . r 9 C — Ar II OH OH OH m (M) N Figure 2.34 2m+2 CgHgCX)H u n i t s . A hydrogen atom t r a n s f e r from r a d i c a l L to r a d i c a l K would r e s u l t i n complete reduction of the bisbenzophenone to the benzhydrolended product (M) and o x i d a t i o n of the a l c o h o l i c reagent t o s t r u c t u r e N. Formation of end groups o f the type M need not n e c e s s a r i l y be chain terminating for polymer growth s i n c e they can i n t e r a c t with aromatic ketones o f the benzophenone type to give p i n a c o l l i n k s . End groups o f type N however might c r e a t e more of a problem s i n c e they might be expected to undergo photochemical cleavage to give r a d i c a l s ; polymerization. benzoins are widely used as p h o t o i n i t i a t o r s f o r r a d i c a l A l l the other steps i n d i c a t e d i n Figure 2.34 are chain - 59 - propagating, although i f a l l three r a d i c a l combination processes shown i n the Figure occurred the r e s u l t i n g p o l y o l would be a copolymer containing repeating u n i t s with varying numbers o f adjacent CgHgCOH groups, 2, m + 2, 2m + 2. I t was decided t o proceed with t h i s i d e a i n the hope t h a t a t l e a s t some information on the photochemical behaviour o f these systems would be obtained; the r e a c t i o n shown i n F i g u r e 2.35 was chosen as an i n i t i a l o b j e c t i v e for t h i s i n v e s t i g a t i o n . OJlQ yO 350 nm polyol ? H + H solvent I OH OH OH OH Figure 2.35 The study o f the model r e a c t i o n i s an e s s e n t i a l p r e l i m i n a r y step i n the i n v e s t i g a t i o n o f any p o t e n t i a l step-growth polymerization. I t was i n t h i s context t h a t the photoreaction between benzophenone and t r i p h e n y l g l y c o l was undertaken, the product(s) of t h i s r e a c t i o n being appropriate model(s) f o r t h e repeat u n i t ( s ) o f the a n t i c i p a t e d p o l y o l (Figure 2.35). 2.2.b. How one event l e d t o another Appropriate s t a r t i n g m a t e r i a l s a r e e s s e n t i a l f o r chemical r e a c t i o n s . I n the case o f the i n v e s t i g a t i o n proposed i n the preceding paragraph, one o f the s t a r t i n g m a t e r i a l s , benzophenone, was r e a d i l y a v a i l a b l e . T r i p h e n y l g l y c o l , however, had to be s y n t h e s i z e d from simpler chemicals. 67 One o f the standard t e x t s o f organic photochemistry r e a c t i o n shown i n Figure 2.36. No mention o f by-products conditions appeared i n the t e x t . reported the or reaction A reference was made t o an o l d paper by - 60 - 0OJCQ 9 hv ^*C^ + H H C— H OH I I OH OH Figure 2.36 124 Ciamician and S i l b e r . When the r e a c t i o n reported was attempted on a small s c a l e (nitrogen streamed benzene s o l u t i o n , 350 nm u l t r a v i o l e t l i g h t ) a white s o l i d was obtained a t n e a r l y q u a n t i t a t i v e y i e l d , which, a f t e r r e c r y s t a l l i z a t i o n and drying was found t o be benzopinacol. R e p e t i t i o n o f the r e a c t i o n a t v a r i o u s concentrations and f o r d i f f e r e n t i r r a d i a t i o n times y i e l d e d c o n s t a n t l y the same product, benzopinacol, unequivocally i d e n t i f i e d by melting p o i n t elemental analyses and s p e c t r o s c o p i c examinations. determination, Even v a r i a t i o n o f the benzo- phenone-benzylalcohol molar r a t i o seemed t o produce no a l t e r a t i o n s i n the nature or y i e l d o f t h e f i n a l product. Examination o f the s o l v e n t a f t e r i r r a d i a t i o n by t . l . c . on s i l i c a revealed the presence o f t r a c e s o f benzopinacol and s e v e r a l 124 6V other products. Consultation o f the reference i n d i c a t e d i n the t e x t r e v e a l e d t h a t Ciamician and S i l b e r themselves appeared not t o be d e f i n i t e about the formation o f t r i p h e n y l g l y c o l from the benzophenone-benzylalcohol mixture on exposure t o s u n l i g h t . I n the same paper, however, r e f e r e n c e s were made t o e a r l i e r work by Ciamician and S i l b e r , and a l s o by the r i v a l group, headed by Paternb; view. these r e f e r e n c e s appeared t o be i n t e r e s t i n g from s e v e r a l p o i n t s o f These very e a r l y , almost p r e h i s t o r i c a l s t u d i e s (by modern standards) o f the 'actions o f s u n l i g h t 1 on organic substances, however p r i m i t i v e and crude, have s i g n i f i c a n t l y contributed t o the development o f modern organic photochemistry - 61 - so as to deserve a b r i e f mention i n t h i s t h e s i s . 2.2.C. The 'pre-history' o f s y n t h e t i c o r g a n i c photochemistry - the nature of a dispute S y n t h e t i c s t u d i e s on the photochemistry of carbonyl compounds go back to 1886 when Ciamician and S i l b e r t r i e d to ' e s t a b l i s h the p r i n c i p a l p r o c e s s e s 125 determined or, b e t t e r say, a c c e l e r a t e d by the s o l a r r a y s ' . A few y e a r s l a t e r , Loew t r i e d the f i r s t s y s t e m a t i c c l a s s i f i c a t i o n o f the 'actions of l i g h t ' on organic substances. 126 I n subsequent papers 56 127 ' Ciamician and S i l b e r examined the photochemistry of v a r i o u s carbonyl compounds and e s t a b l i s h e d the f a c t that i n s o l a t i o n of the ketone (or aldehyde)/alcohol system r e s u l t s i n the 'oxidation of the a l c o h o l to aldehyde whereas the ketones (or aldehydes) transform 127 themselves i n t o the corresponding p i n a c o l s ' . Thus, benzophenone and acetophenone y i e l d e d on i n s o l a t i o n i n ethanol benzopinacol and 'acetophenopinacol 127 respectively; acetaldehyde was produced as w e l l . The p h o t o r e a c t i v i t y of benzophenone i n aromatic a l c o h o l s was a l s o examined 128 by Ciamician and S i l b e r but the r e a c t i o n was found to be 'a l o t more complicated than i n the previous case, because more products are formed i n t h i s 1 reaction . Copious amounts of benzopinacol were i s o l a t e d from the r e a c t i o n of benzophenone with b e n z y l a l c o h o l on exposure to s u n l i g h t , together with some hydrobenzoin, (CgHgCHOH)^ The i n t e r e s t i n g f e a t u r e of t h i s r e a c t i o n was the i s o l a t i o n of a substance melting a t 168° and g i v i n g the f o l l o w i n g elemental analyses: I C 82.95% H 6.44% C B The v a l u e s c a l c u l a t e d f o r 2g ie°2 ^ (CgH ) CO + C H CH OH) were: 5 2 g 5 2 II 82.77% 6.44% t h e ' a d d i t i o n C 82.76%, H 6.21%. P r o d u c t ' o f t n e t w o reagents, On the b a s i s of t h i s evidence Ciamician and S i l b e r assigned to the product the s t r u c t u r e of t r i p h e n y l g l y c o l 1 - 62 - (Figure 2.36) , a compound a l r e a d y known a t t h a t time to have a melting p o i n t of 1 6 4 ° . 1 2 9 Subsequently another group of I t a l i a n workers i n v e s t i g a t i n g the a c t i o n of s u n l i g h t on organic substances, Paternb and h i s c o l l a b o r a t o r s of the U n i v e r s i t y of Rome, began p u b l i s h i n g t h e i r r e s u l t s . I n a paper published i n 1909 they d e s c r i b e d the p h o t o r e a c t i v i t y of benzophenone with v a r i o u s a l i p h a t i c 119 hydrocarbons. They confirmed what Ciamician and S i l b e r had claimed p r e v i o u s l y , namely t h a t i n a l l cases examined about two t h i r d s of the we! ght of benzophenone used was transformed i n t o benzopinacol. They a l s o reported the i s o l a t i o n of some 'addition products' formed i n the r e a c t i o n s examined. In the same paper Paterno and C h i e f f i a l s o described the behaviour of benzophenone on i n s o l a t i o n i n the presence of aromatic hydrocarbons, namely benzene, toluene, ethylbenzene, p-xylene e t c . They acknowledged C i a m i c i a n and 128 S i l b e r ' s work i n t h i s f i e l d but they pointed out t h a t the formation of benzopinacol i n the r e a c t i o n between benzophenone and cumene reported by their 128 rivals was 'an i s o l a t e d observation i n which the authors d i d not study what change d i d cumene undergo during the r e a c t i o n ' . Furthermore, Paternb and C h i e f f i reported the i s o l a t i o n of the a d d i t i o n product from the benzophenone/toluene i n s o l a t i o n ( ( C ^ H J _C(0H)CH„C^H,_) i n modest y i e l d , b b i 2 b 5 I t should a l s o be mentioned here t h a t i t was i n t h i s important paper t h a t the process now famous as the Paterno-Buchi r e a c t i o n and the photoreductive addition of benzophenone to diphenylmethane were f i r s t reported. F u r t h e r work by Paterno appeared i n the l i t e r a t u r e on the photoreactions of acids, e t h e r s ^ 0 and b e n z y l a c e t a t e ^ " ^ with benzophenone. Paterno and h i s team a l s o i n v e s t i g a t e d the behaviour of benzophenone on i n s o l a t i o n i n the presence 132 of various a l k a l o i d s . I n most of these s t u d i e s , benzopinacol was isolated i n s i g n i f i c a n t q u a n t i t i e s , o c c a s i o n a l l y s m a l l amounts of the 'addition products' were obtained. Paterno and C h i e f f i s t a t e d t h a t a d d i t i o n of benzophenone to - 63 molecules was only p o s s i b l e when the methylene group, -CH ~, was p r e s e n t . 2 S i m i l a r behaviour was observed when benzophenone was i n s o l a t e d i n the presence 1 1 of b e n z y l p h e n o l . ^ " ^ I n 1914, Paterno published h i s 11th paper on the 'Synthesis i n Organic 134 Chemistry by Means of L i g h t ' . I n i t he summarized work a l r e a d y done by him and coworkers and b i t t e r l y c r i t i c i z e d Ciamician and S i l b e r f o r 'not having c l e a r l y understood the concepts o f the r e a c t i o n s they were attempting' and f o r d e l i b e r a t e l y 'presenting Paternb's work under f a l s e l i g h t * . Furthermore Paterno 128 pointed out t h a t the evidence presented by Ciamician and S i l b e r f o r the formation o f t r i p h e n y l g l y c o l from benzophenone and b e n z y l a l c o h o l was insufficient, t h e r e f o r e , Paterno i n s i s t e d , no c o n c l u s i o n could be drawn as to whether the compound was a c t u a l l y formed i n the r e a c t i o n o r not. 135 Ciamician and S i l b e r r e p l i e d by saying t h a t a t the time o f 128 publication they, too, had t h e i r r e s e r v a t i o n s regarding the formation of triphenylglycol; they could not however, accept the charges o f d e l i b e r a t e l y stating false results. % 134 Paterno a l s o attempted an e a r l y c l a s s i f i c a t i o n of the photoreactions of carbonyl compounds as f o l l o w s : i) ' t r i m e t h y l e n i c condensation' C II c + c — ^ I o ii) — —c I — I c — o—c / \ I I 'doubling o f the carbon chain' C || O H C =»» H C C H - 64 iii) ' e n o l i c condensation' \ / c h _I > +H— S 1 0 iv) _l_l_ T~"i OH OH H 'dehydrogenation of alkanes' C C H II + ^ n 2n+l 0 - C C—+ | | OH OH C H. n 2n A comprehensive review of examples f o r each o f the above! c l a s s e s appears i n the paper. I t i s indeed hard to overestimate the importance o f these e a r l y workers' e f f o r t s on the development o f modern o r g a n i c photochemistry. Despite the l a c k of h e l p f u l t h e o r e t i c a l concepts and s p e c t r o s c o p i c and chromatographic t o o l s to analyse observed r e a c t i o n s , they p a t i e n t l y c o l l e c t e d data which l a t e r on served t h e i r purpose. Speaking a t the V I I Congress o f Applied Chemistry i n London i n 1909, Paternb t r i e d to a t t r a c t the a t t e n t i o n o f the young chemists of the time to the impressive r e s u l t s obtained i n the f i e l d o f photochemical synthesis. 'For us', he concluded, 'a much more modest f i e l d i s r e s e r v e d , the c o l l e c t i o n and p r e p a r a t i o n o f the m a t e r i a l which should serve i n the c o n s t r u c t i o n 136 of the m a j e s t i c e d i f i c e ' . A p o i n t , however, which was made c l e a r from the above information i s t h a t the two f a t h e r f i g u r e s o f organic photochemistry never r e a l l y agreed on whether t r i p h e n y l g l y c o l i s produced on i r r a d i a t i n g benzophenone i n b e n z y l a l c o h o l o r not. 2.2.d. The r e s o l u t i o n o f the dispute 121 I n the o r i g i n a l paper proposing the formation o f t r i p h e n y l g l y c o l , C i a m i c i a n and S i l b e r d e s c r i b e d t h r e e separate i n s o l a t i o n s of benzophenone - 65 - w d i s s o l v e d i n neat benzylalcohol (1:2 / ) f o r periods from 4 to 5 months. w main product i s o l a t e d by them was benzopinacol. hydrobenzoin, The Other products reported were t r i p h e n y l g l y c o l and a r e s i n , which the authors a t t r i b u t e d to the a c t i o n of l i g h t on benzaldehyde, i t s e l f a p o s s i b l e r e a c t i o n product a r i s i n g from the o x i d a t i o n of b e n z y l a l c o h o l . them. Benzaldehyde however was not i s o l a t e d by The scheme shown i n F i g u r e 2.37 was proposed by the authors f o r the C + H 9 9 Oh C — -c OH c- a OH OH H 2 C + H C- H OH OH OH eDJ> D 9 C + H C- H H II 0 OH Figure OH OB 2.37 benzophenone/benzylalcoho1 i n s o l a t i o n . The r e a c t i o n was r e - i n v e s t i g a t e d by us. w A 1:2 / w s o l u t i o n of benzophenone i n b e n z y l a l c o h o l (25g:50g) was t r a n s f e r r e d i n t o a Pyrex tube, n i t r o g e n streamed for 20 mins, the tube was q u i c k l y stoppered and the s o l u t i o n was for 18 h r s a t 350 run. irradiated At the end o f the i r r a d i a t i o n p e r i o d the s o l u t i o n was yellow and the i n s i d e s u r f a c e of the tube was covered with a l a y e r of white crystals. Examination of the tube contents by t . l . c . on s i l i c a (benzene, chloroform) and i . r . spectroscopy r e v e a l e d the presence o f unreacted s t a r t i n g m a t e r i a l s , benzopinacol and s m a l l amounts of benzaldehyde. No triphenylglycol was i s o l a t e d or detected by t . l . c . on s i l i c a ; concentrations g r e a t e r than 1% would have r e a d i l y been detected. I r r a d i a t i o n time to 89 h r s . The r e a c t i o n was repeated i n c r e a s i n g the Again as i n the previous case the s o l u t i o n was yellow and the i n s i d e s u r f a c e of the tube was covered with white c r y s t a l s . - 66 - Benzopinacol and benzaldehyde were e a s i l y i d e n t i f i e d by t . l . c . on s i l i c a (benzene, chloroform) and i . r . spectroscopy, along with unreacted s t a r t i n g m a t e r i a l s and hydrobenzoin. The i n t e r e s t i n g f e a t u r e o f t h i s experiment the d e t e c t i o n by t . l . c . on s i l i c a of t r i p h e n y l g l y c o l . was Attempts to i s o l a t e the product by f r a c t i o n a l p r e c i p i t a t i o n (ethanol s o l v e n t ) were, however, u n s u c c e s s f u l , probably due to the s m a l l amounts of m a t e r i a l p r e s e n t . A t h i r d i n v e s t i g a t i o n was undertaken under the same c o n d i t i o n s . This time, however, c o l o u r l e s s c r y s t a l s which formed on the i n s i d e w a l l of the r e a c t i o n tube were p e r i o d i c a l l y broken by means of a long s p a t u l a i n order to allow the u l t r a v i o l e t - l i g h t to penetrate the s o l u t i o n . A f t e r 180 h r s of i r r a d i a t i o n a crop o f c r y s t a l s and a v i s c o u s b r i g h t yellow s o l u t i o n had formed i n s i d e the tube. The c r y s t a l s were separated by f i l t r a t i o n and the y e l l o w o i l recovered was nitrogen streamed and r e - i r r a d i a t e d for a f u r t h e r 120 h r s . The crystals separated were found to c o n s i s t of a mixture of unreacted benzophenone, benzopinacol, hydrobenzoin chloroform). and t r i p h e n y l g l y c o l by t . l . c . on s i l i c a (benzene, The yellow o i l a l s o deposited a f r e s h crop of c r y s t a l s during the 120 h r s i r r a d i a t i o n p e r i o d . Examination of these c r y s t a l s ( c a . 1.5g) by i . r . spectroscopy showed the m a t e r i a l to be t r i p h e n y l g l y c o l by comparison w i t h the i . r . spectrum o f t r i p h e n y l g l y c o l obtained by ground s t a t e s y n t h e s i s (see 2.2.e). The yellow o i l was found to c o n s i s t of benzaldehyde, hydrobenzoin and t r a c e s o f benzopinacol, b e n z y l a l c o h o l and unreacted benzophenone. Attempts to o b t a i n a pure sample of t r i p h e n y l g l y c o l by repeated r e c r y s t a l l i z a t i o n s from v a r i o u s s o l v e n t s were l a r g e l y u n s u c c e s s f u l , s i n c e a very f a i n t spot corresponding to benzopinacol was always p r e s e n t on the t . l . c . p l a t e s (benzene). These experiments c e r t a i n l y confirmed the c o r r e c t n e s s of Ciamician's observations. I t would a l s o appear t h a t the formation of t r i p h e n y l g l y c o l from benzophenone/benzylalcohol was a l s o confirmed. i s time dependent. The formation of benzaldehyde F i n a l l y , i r r a d i a t i o n of an 1:1 molar mixture of benzophenone - 67 - and b e n z y l a l c o h o l i n benzene s o l u t i o n (0.0035 t o t a l concentration) f o r 160 h r s (350 run) afforded t r i p h e n y l g l y c o l i n c a . 8% y i e l d . 2.2.e. Ground s t a t e syntheses o f t r i p h e n y l g l y c o l The f i r s t attempt t o s y n t h e s i z e the above mentioned compound (method A) 137 was based on an e s t a b l i s h e d router namely the a c t i o n o f benzoin on an excess o f phenylmagnesiumbromide. The e x e r c i s e afforded 60% o f n e a r l y pure triphenylglycol. Repeated r e c r y s t a l l i z a t i o n s gave 45% o f a white s o l i d which on examination crystalline by t . l . c . on s i l i c a was found to be a s i n g l e compound (Figure 2.38). 0 OH OH (method A) Figure 2.38 T r i p h e n y l g l y c o l was a l s o prepared corresponding (method B) by reduction o f the carbonyl compound, a-phenylbenzoin (2,2-diphenyl-2- hydroxyacetophenone), i t s e l f prepared by the a c t i o n o f one mole o f b e n z i l on one mole o f phenylmagnesiumbromide (Figure 2.39). Pure t r i p h e n y l g l y c o l (85%) was obtained from t h i s preparation, M.pts and s p e c t r o s c o p i c data f o r the products obtained by the two methods were identical. Elemental analyses were s a t i s f a c t o r y . Preparation and p u r i f i c a t i o n procedures w i l l be d e s c r i b e d i n more d e t a i l i n 2 . 2 . j . i i i . 2.2.f. The benzophenone/triphenylglycol photoreactlon Following p r e p a r a t i o n , c h a r a c t e r i z a t i o n and rigorous p u r i f i c a t i o n o f t r i p h e n y l g l y c o l , i t was decided to proceed with the i r r a d i a t i o n o f a 1:1 molar 9 68 Et_0 OH EtOH NaBH (method B ) H OH OH Figure 2.39 r a t i o o f t h i s compound and benzophenone f o r reasons s t a t e d i n 2.2.a. Benzene was chosen a s an appropriate s o l v e n t . I n the f i r s t experiment t r i p h e n y l g l y c o l (2.90g, 0.01 mole) prepared by method A and benzophenone (1.82g, 0.01 mole) were d i s s o l v e d i n benzene (350 mis) i n a long Pyrex tube. The c l e a r , c o l o u r l e s s s o l u t i o n was nitrogen streamed, the tube was q u i c k l y stoppered and i r r a d i a t e d f o r 18.4 h r s a t 350 nm; This experiment y i e l d e d unchanged s t a r t i n g m a t e r i a l s , as i n d i c a t e d by t . l . c . on silica (benzene, chloroform, ethanol) and i . r . s p e c t r a l examination. R e p e t i t i o n o f e x a c t l y the same r e a c t i o n using t r i p h e n y l g l y c o l prepared by method B y i e l d e d i d e n t i c a l r e s u l t s . I n a subsequent experiment, benzophenone (0.273g, 0.0015 mole) and t r i p h e n y l g l y c o l (0.435g, 0.0015 mole) prepared by method B were d i s s o l v e d i n benzene (50 mis) i n a Pyrex tube. The s o l u t i o n was degassed by s e v e r a l f r e e z e -3 pump-thaw c y c l e s and tube was s e a l e d under reduced pressure (10 mmBg). I r r a d i a t i o n f o r 94 h r s a t 350 nm afforded q u a n t i t a t i v e recovery o f s t a r t i n g materials. During t h i s experiment a p a r a l l e l photoreaction between benzophenone - 69 - and benzhydrol (1:1 molar r a t i o ) was c a r r i e d out i n order to t e s t the e f f i c i e n c y of the u l t r a v i o l e t lamps: benzopinacol was q u a n t i t a t i v e l y obtained a f t e r 3 h r s of i r r a d i a t i o n . R e p e t i t i o n of t h i s r e a c t i o n using t r i p h e n y l g l y c o l prepared by method A gave the same r e s u l t . On the b a s i s of the l i t e r a t u r e information a v a i l a b l e on the o f benzophenone with a l c o h o l i c hydrogen donors, one would photoreactions expect some i f not a l l the processes shown i n F i g u r e 2.40 to occur upon i r r a d i a t i o n of the benzophenone/triphenylglycol system. materials i s a surprising r e s u l t . The q u a n t i t a t i v e recovery of s t a r t i n g I t seemed remarkable and l e d to a considerable e f f o r t i n p u r i f y i n g the t r i p h e n y l g l y c o l and seeking evidence f o r the presence o f an impurity which could a c t as an e f f i c i e n t quencher o f t r i p l e t benzophenone. Benzene s o l v e n t and benzophenone were a l s o e x h a u s t i v e l y searched f o r impurities. R e p e t i t i o n s of the i r r a d i a t i o n s using more r i g o r o u s l y p u r i f i e d m a t e r i a l s made no d i f f e r e n c e to the r e s u l t . One hypothesis considered was t h a t the hydrogen atom t r a n s f e r i l l u s t r a t e d i n Figure 2;40 d i d i n f a c t occur i n low y i e l d and t h a t the a-phenylbenzoin formed acted as a quencher i n very low c o n c e n t r a t i o n s . The hypothesis seemed worth i n v e s t i g a t i n g s i n c e one could p o s t u l a t e a degenerate i s o m e r i z a t i o n pathway i n v o l v i n g a photoenolization and phenyl migration which might have accounted f o r the p o s t u l a t e d quenching (Figure 2.41). The photochemistry o f a-phenylbenzoin i s not d i s c u s s e d i n the l i t e r a t u r e , depitc: the very 138 e x t e n s i v e i n v e s t i g a t i o n s reported on benzoins i n g e n e r a l . When i r r a d i a t e d under the conditions d e s c r i b e d p r e v i o u s l y , a-phenylbenzoin proved to be p a r t i c u l a r l y p h o t o l a b i l e , both on i t s own amounts o f and i n the presence of equimolar benzhydrol. F i n a l l y , the 'quencher theory' was excluded by the i r r a d i a t i o n of a nitrogen streamed benzene s o l u t i o n containing a 1:1:1 molar mixture of benzophenone, 70 Q Q hv » II 99 OH OH a ca x2 OH OH OH OH OH REACT! H ATOM TRANSFER OH OH OH hv 9\9 V \ OH OH « hv F i g u r e 2.40 OH - 71 - Figure 2.41 benzhydrol and t r i p h e n y l g l y c o l 12 h r s (Figure 2.42). (0.0035H t o t a l concentration) a t 350 nm f o r An 1:1 molar mixture of t r i p h e n y l g l y c o l and benzopinacol was obtained, benzophenone and benzhydrol having completely disappeared. 0 <§H- i i 9 ©4OH 1 mole OH OH 1 mole CH 0.0035M 12 h r s 1 mole 99 OH OH 350 runs OH Figure 2.42 T h i s r e a c t i o n confirmed the observations t h a t the t e r t i a r y hydrogen i n - 72 - t r i p h e n y l g l y c o l i s i n e r t towards a b s t r a c t i o n by t r i p l e t benzophenone. 2.2.g. The p h o t o r e a c t i v i t y of 1,2,2,2-tetraphenylethanol The observations described above concerning the photochemical behaviour of t r i p h e n y l g l y c o l c e r t a i n l y put an end to the hopes of o b t a i n i n g poly(pentaphenylglycerol)s by the photoreductive p o l y a d d i t i o n of bisketones to polyphenylated a l c o h o l i c hydrogen donors o f the type d i s c u s s e d . The new question which arose was whether the p h o t o s t a b i l i t y of t r i p h e n y l g l y c o l was due to the presence o f two adjacent CgH^COH u n i t s next to the t e r t i a r y hydrogen, which was expected to be a b s t r a c t a b l e . I n order to t e s t t h i s hypothesis i t was decided to s u b s t i t u t e the 2-hydroxy group i n t r i p h e n y l g l y c o l with a group of a t o t a l l y d i f f e r e n t nature, f o r i n s t a n c e an aromatic r i n g , and i n v e s t i g a t e the photochemical behaviour of the r e s u l t i n g system. To t h i s end 1,2,2,2- tetraphenylethanol (Figure 2.43) was s y n t h e s i z e d by reducing <0H D 6 benzopinacolone H EtOH Figure 6 OH 2.43 and subsequently i r r a d i a t e d with benzophenone (1:1 molar r a t i o ) i n benzene s o l u t i o n (0.0056M t o t a l concentration) f o r 12 hrs a t 350 nm. s p e c t r o s c o p i c examination and t . l . c . on s i l i c a Infrared (benzene, chloroform, ethanol) e s t a b l i s h e d t h a t no d e t e c t a b l e r e a c t i o n had taken p l a c e ; s t a r t i n g m a t e r i a l s were q u a n t i t a t i v e l y recovered. - 73 - As i n the case o f t r i p h e n y l g l y c o l , i r r a d i a t i o n o f a benzene s o l u t i o n containing an 1:1:1 molar mixture o f benzophenone, benzhydrol and 1,2,2,2tetraphenylethanol (0.0035H t o t a l concentration) a t 350 nm f o r 24 h r s r e s u l t e d i n the formation of an 1:1 molar mixture o f benzopinacol and 1,2,2,2tetraphenylethanol. No other products were detected by t . l . c . on s i l i c a , benzophenone and benzhydrol having disappeared completely. 2.2.h. Some r e l e v a n t photoreactions I t was so f a r e s t a b l i s h e d t h a t the t e r t i a r y hydrogen i n the s t r u c t u r e shown i n F i g u r e 2.44 i s a b s t r a c t a b l e when X = H o r CJA- but non-abstractable b 5 when X = ( C H J _C0H and (C-H..) ,C. 6 5 2 6 5 3 C 9 o H c X OH Figure 2.44 In an attempt to examine the p o s s i b i l i t y o f other v e r s i o n s o f the system shown being i n e r t to hydrogen a b s t r a c t i o n by benzophenone t r i p l e t , hydrobenzoin (X = (CgHjXH(OH)) and 1-phenylethanol (X = CH^)) were i r r a d i a t e d i n the presence of e i t h e r benzophenone o r i t s d i f u n c t i o n a l analogue, m-dibenzoylbenzene. I r r a d i a t i o n o f a 1:1 molar s o l u t i o n o f benzophenone and 1-phenylethanol i n benzene (350 nm) r e s u l t e d i n 100% disappearance o f both s t a r t i n g m a t e r i a l s a f t e r 140 h r s o f i r r a d i a t i o n . Benzopinacol and acetaldehyde were among the r e a c t i o n products immediately recognized. i d e n t i f i e d by t . l . c . on s i l i c a (benzene). At l e a s t three more products were - 74 - I r r a d i a t i o n of a 1:1 molar mixture of m-dibenzoylbenzene and hydrobenzoin i n benzene presented s p e c i a l problems due to the low s o l u b i l i t y of the d i o l . However the p o r t i o n o f i t t h a t d i d d i s s o l v e i n benzene a t 0.0035M t o t a l concentration r e a c t e d r e a d i l y with the photoexcited benzophenone-type carbonyl of m-dibenzoylbenzene y i e l d i n g hydroxy1 containing m a t e r i a l s (350 run, 160 h r s ) . Again a b r i g h t yellow colour developed s h o r t l y a f t e r the lamps were turned on. The i r r a d i a t i o n was repeated using methylene c h l o r i d e s o l v e n t i n order to ensure complete s o l u b i l i t y of both s t a r t i n g m a t e r i a l s . A f t e r only 38 h r s of i r r a d i a t i o n s o l u t i o n was again b r i g h t yellow and s l i g h t l y t u r b i d . Examination o f the tube contents r e v e a l e d t h a t some s t a r t i n g m a t e r i a l s had been l e f t unchanged but most of them had r e a c t e d to g i v e s e v e r a l products as shown by t . l . c . on s i l i c a (benzene). The products from the above r e a c t i o n s were not r i g o r o u s l y examined, the main o b j e c t i v e of the e x e r c i s e being to e s t a b l i s h whether photoreaction with benzophenone occurred or not. 2.2.1. Attempts to prepare pentaphenylglycerol v i a ground s t a t e chemistry I n v e s t i g a t i o n s so f a r r e v e a l e d t h a t the model r e a c t i o n f o r the polymer s y n t h e s i s formulated i n F i g u r e 2.30 does not occur due to a r a t h e r s u r p r i s i n g i n e r t n e s s of t r i p h e n y l g l y c o l towards benzophenone t r i p l e t s . f u r t h e r i n v e s t i g a t e t h i s behaviour e x h i b i t i n g the same remarkable Attempts to l e d to the discovery of another molecule inertness. At the same time, the p r e p a r a t i o n of the expected product, pentaphenyl- g l y c e r o l was undertaken v i a ground s t a t e s y n t h e s i s i n order to examine i t s s t a b i l i t y , determine i t s p r o p e r t i e s and provide a r e f e r e n c e f o r comparison. 139 The compound i s reported i n the l i t e r a t u r e and the r e c i p e f o r i t s p r e p a r a t i o n given i n the o r i g i n a l paper was followed i n every d e t a i l (see F i g u r e 2.45). The o r i g i n a l worker reported t h a t pentaphenylglycerol was obtained as a white c r y s t a l l i n e s o l i d a f t e r a number o f r e c r y s t a l l i z a t i o n s (m.pt. 163°), and t h a t - 75 - C H —O 2 5 OH | v ,0 C,H c—c—c II I II 0 OH * ©-?—f—=—©> Et 0 v=/ 2 | OH 0 | OH | OH F i g u r e 2.45 139 i t s elemental a n a l y s i s was s a t i s f a c t o r y . I n our hands, t h i s r e a c t i o n gave a white c r y s t a l l i n e s o l i d (5g), m.pt. 162°, which, a f t e r repeated r e c r y s t a l l i z a t i o n s from methanol and drying under vacuum over P ° 5 analysed as f o l l o w s : C 87.54, H 5.64. 2 ^2^2^^ C a l c u l a t e d f i g u r e s f o r pentaphenylglycerol a r e : C 83,33 H ' 5 T h i s m a t e r i a l was found t o be a s i n g l e component by t . l . c . on s i l i c a ethanol, methanol, a c e t o n i t r i l e ) . and drying under vacuum over P ° 5 I t melted a t 164°. f o r 4 8 n r s g a 2 which analysed a s f o l l o w s : C 87.44, H 5.87. v e a 97 * ' * (benzene, Further r e c r y s t a l l i z a t i o n s m a t e r i a l melting a t 164° Sublimation (120°, 0.005 mmHg), gave a m a t e r i a l i d e n t i c a l t o the one obtained a f t e r r e c r y s t a l l i z a t i o n s ( i . r . s p e c t r a were superimposable), melting a t 164.5°, which analysed: H 6.04. The h i g h e s t m/e peak observed i n the mass spectrum o f our was 260 (molecular weight o f pentaphenylglycerol = 472); to fragmentation i n the mass spectrometer source. C 87.15, product t h i s was a t t r i b u t e d The *H n.m.r. spectrum showed two peaks, one i n the aromatic hydrogen region (6 7.30) and another a t 6 2.73 which disappeared on a d d i t i o n o f D^O. The i n t e g r a t e d i n t e n s i t i e s were i n a r a t i o o f 15:1, f o r pentaphenylglycerol the c a l c u l a t e d r a t i o i s 8.33:1. The s o l i d s t a t e i n f r a r e d spectrum o f the m a t e r i a l (KBr d i s c ) showed a r a t h e r - 76 - broad OH band a t 3465 cm 3080 cm , a m u l t i p l e t i n the C-H aromatic region (3020 cm - and peaks i n the f i n g e r p r i n t region are g e n e r a l l y c o n s i s t e n t with 139 the product s t r u c t u r e proposed i n the paper. However, the s o l u t i o n phase (CCl^ solvent) of the m a t e r i a l showed only one peak a t j u s t over 3600 cm~^. Comparative i . r . s t u d i e s on some phenylated d i o l s and monoalcohols (see Table 2.6) one showed t h a t 1,2-diols g e n e r a l l y d i s p l a y two peaks i n d i l u t e s o l u t i o n s , above and one below 3600 cm the former being assigned s t r e t c h i n g and the l a t t e r to the i n t r a m o l e c u l a r l y bonded OH to the f r e e OH (Figure 2.46). I1 c H c I H H Figure 2.46 The s i n g l e OH peak i n the s o l u t i o n phase i . r . spectrum i n d i c a t i n g the absence of intramolecular hydrogen bonding i n the product from the a c t i o n of p h e n y l l i t h i u m on d i e t h y l mesoxalate seemed r a t h e r s u r p r i s i n g and with the formula of pentaphenylglycerol. inconsistent Furthermore, elemental a n a l y s i s data and n.m.r. i n t e g r a t e d i n t e n s i t y r a t i o s suggested t h a t the product obtained 139 i n our hands was not the one claimed i n the paper. T h i s thought l e a d to a systematic search f o r a compound which could be formed from the above r e a c t i o n i n good y i e l d and be c o n s i s t e n t with the data obtained; (CgHg)^COH looked a promising candidate. 164°-165°;^° I t s melting point i s reported i t s mass spectrum extends to m/e a n a l y s i s f i g u r e s (C, 87.65; H, 6.19) triphenylcarbinol, to be 260 and c a l c u l a t e d elemental agree w e l l w i t h those obtained i n t h i s - 77 - Table 2.6 Comparative s o l u t i o n phaae l . r . s p e c t r a of some phenylated 1,2-dlols and monoalcohols (OH s t r e t c h i n g region, CCl^ s o l u t i o n , 3mm Concentration range: 10 -3 M-8 KBr c e l l ) x 10 -4 M Absorptions S t r u c t u r a l formula Name 3900 ^Q Hydrobenzoin H- •C C H I I OH OH Triphenylglycol •C—H OH OH Benzopinacol < f - 0 OH Benzylalcohol H OH C—H I OH ? Benzhydrol CH Product obtained from the r e a c t i o n : C H Li + C H — O - C — C — C — OC,H_ 2 5 |, , , 2 5 6 O 5 OH O Proposed 139 formula: T IT Q-c—c-c-Q OH 3300 J OH OH L - 78 - work. F u r t h e r the p.m.r. and i . r . s p e c t r o s c o p i c r e s u l t s a r e i n complete accord with the product being t r i p h e n y l c a r b i n o l . T h i s hypothesis was confirmed 139 by comparison with an a u t h e n t i c specimen. I n the o r i g i n a l paper, following colour t e s t f o r 'pentaphenylglycerol' was suggested: 'The the product from pentaphenylglycerol + a c e t y l c h l o r i d e + d i m e t h y l a n i l i n e i n c o l d f o r 5 mins was t r e a t e d with a s o l u t i o n of hydroxylamine and the mixture made a l k a l i n e with a l c o h o l i c KOH, heated and cooled. A reddish-wine produced on a c i d i f i c a t i o n and a d d i t i o n of FeCl-j s o l u t i o n ' . colour The t e s t was was c a r r i e d out twice on a sample of the r e a c t i o n product and on a sample of pure triphenylcarbinol. I n both cases the same reddish-wine colour was produced. The author has been unable to a s c e r t a i n the s i g n i f i c a n c e attached by the o r i g i n a l worker to the colour t e s t , although c l e a r l y i t i s not c h a r a c t e r i s t i c of pentaphenylglycerol. results. C a r e f u l r e p e t i t i o n of the o r i g i n a l r e c i p e gave i d e n t i c a l - 79 - 2.2.j. Experimental 2.2.j.i. Readily a v a i l a b l e m a t e r i a l s Benzylalcohol ( t e c h n i c a l grade) was purchased from Hopkin & Williams L t d . , and s t o r e d over 4A molecular s i e v e s ; p r i o r t o use i t was twice f r a c t i o n a l l y distilled (15 cm x 4 cm d i a . , g l a s s h e l i c e s ) ; the f r a c t i o n d i s t i l l i n g 205° and 206° was c o l l e c t e d ( l i t . s i n g l e spot on t . l . c . on s i l i c a 1 4 0 between 205.03°/760 mmHg). The product gave a (chloroform, ethanol, benzene). Benzhydrol was obtained from departmental stock, r e c r y s t a l l i z e d twice from ethanol/water (70:30) and sublimed twice (95°/0.005 mmHg); had a melting p o i n t o f 69° ( l i t . silica 1 4 0 the product 69°) and gave a s i n g l e spot on t . l . c . on (ethanol, benzene, chloroform, toluene, a c e t o n i t r i l e ) . Benzophenone ( t e c h n i c a l grade) was purchased from Hopkin & W i l l i a m s L t d . and r e c r y s t a l l i z e d t w i c e from ethanol/water (70:30) and once from cyclohexane, melting a t 48.5° ( l i t . 1 4 0 49°). I t gave a s i n g l e spot on t . l . c . on s i l i c a (benzene, ethanol, chloroform, a c e t o n i t r i l e , toluene, e t h y l a c e t a t e , t e t r a c h l o r i d e , acetone/water carbon 50:50). 1-Phenyl ethanol ( t e c h n i c a l grade) was purchased from BDH Laboratory Reagents and s t o r e d over 4A molecular sieves,- p r i o r to use i t was d i s t i l l e d twice under reduced p r e s s u r e (15 cm x 4 cm d i a . , g l a s s h e l i c e s , 98°-100°/20 mmHg lit. (col. 1 4 0 , 98°-99°/20 mmHg). The product gave a s i n g l e peak on a n a l y t i c a l g . l . c . , A /140°). Diethyl ketomalonate (C H OCOCOOC H,.) was purchased from A l d r i c h 2 5 2 and used as obtained without p u r i f i c a t i o n . Chemicals, I t s m.s. and i . r . spectrum were c o n s i s t e n t with the assigned s t r u c t u r e . Benzoin, b e n z i l and benzopinacolone were a l l obtained from departmental stock, r e c r y s t a l l i z e d twice from appropriate s o l v e n t s , d r i e d under vacuum and i d e n t i f i e d by i n f r a r e d spectroscopy (KBr d i s c s ) and melting point, determinations P u r i t i e s were checked by t . l . c . on s i l i c a (benzene). - 80 - Sodium borohydride was purchased from BDH Laboratory Reagents and used as obtained. 2.2.j.ii. Solvents Benzene ( a n a l y t i c a l grade) was purchased from Hopkln & Williams L t d . , twice f r a c t i o n a l l y d i s t i l l e d sodium, wire f o r two weeks. i n t o a dry f l a s k . (15 cm x 4 cm d i a . , Dixon gauzes) and s t o r e d over P r i o r to use i t was r e d i s t i l l e d under nitrogen A n a l y t i c a l g . l . c . i n d i c a t e d a s i n g l e component ( c o l . 'A'/60°). Methylene c h l o r i d e ( t e c h n i c a l grade) was obtained from departmental stock, f r a c t i o n a t e d twice (40°-42°) and s t o r e d over anhydrous magnesium sulphate f o r two days. I t was r e d i s t i l l e d under nitrogen (41°) before use. Ethanol and 1,4-dioxane were obtained from departmental stock and used without p u r i f i c a t i o n . 2.2.j.iii. M a t e r i a l s s y n t h e s i z e d from simpler chemicals S y n t h e s i s o f hydrobenzoin from benzoin Sodium borohydride s o l u t i o n of benzoin (2.7g, 0.072 mole) i n water (50 mis) was added to a (15.Og, 0.072 mole) i n 1,4-dioxane (200 mis) contained i n a c o n i c a l f l a s k and the mixture was b o i l e d (20 mins). Following d e s t r u c t i o n of unreacted NaBH^ with d i l u t e CH^COOH and evaporation of the s o l u t i o n , a white s o l i d was obtained which was washed with water and r e c r y s t a l l i z e d twice ( e t h a n o l / water 60:40) to give hydrobenzoin (12.lg, 8 0 % ) , m.pt. 147°-148° ( l i t . 150°), i d e n t i f i e d by mass spectrometry ( c o r r e c t molecular ion a t m/e expected fragment ions) and i n f r a r e d spectroscopy. 1 4 0 214 149°and The substance was r e c r y s t a l l i z e d t h r e e more times from the same s o l v e n t and f i n a l l y from a l a r g e volume of ethanol/water (80:20); m.pt. on 147.5° (big p l a t e s , s i n g l e spot on t.l.c. silica). S y n t h e s i s of m-dibenzoylbenzene m-Dibenzoylbenzene was i n i t i a l l y prepared according to the published route. - 81 - To a s t i r r e d s o l u t i o n of i s o p h t h a l o y l c h l o r i d e benzene (400 m i s ) , i n small portions (50g, 0.246 mole) i n dry anhydrous aluminium c h l o r i d e (80g, 0.60 (ca. 60 mine). The s o l u t i o n was mole) was added r e f l u x e d f o r 2 h r s , cooled and hydrolysed by pouring i n t o 10% HCl/ice water s o l u t i o n (3 l i t r e s ) . evaporation of benzene, the r e s u l t i n g crude m a t e r i a l was After recovered by filtration and washed s u c c e s s i v e l y with a 5% aqueous s o l u t i o n of sodium carbonate, a 5% aqueous s o l u t i o n of HC1 and water p r i o r to r e c r y s t a l l i z a t i o n from cyclohexane. Repeated r e c r y s t a l l i z a t i o n (20 to 25 times) afforded spot on t . l . c . on s i l i c a (acetone, methanol, t o l u e n e ) . 103°-104° ( l i t . 6 0 chloroform, benzene, ethanol, The y i e l d of pure m a t e r i a l was 109°-110°). a material giving a single acetonitrile, j u s t under 21%; m.pt. The product had the c o r r e c t m.s. and i . r . spectrum. The u n s a t i s f a c t o r y y i e l d obtained by the above procedure to a more d e t a i l e d i n v e s t i g a t i o n of t h i s r e a c t i o n . by t . l . c . A n a l y s i s of the r e a c t i o n by-products (benzene, chloroform) revealed the presence of low mobility substances ( p o s s i b l e polymeric m a t e r i a l s ) , some u n i d e n t i f i e d products, and i s o p h t h a l i c acid. The l a t t e r must have r e s u l t e d from the h y d r o l y s i s o f unreacted isophthaloyl chloride. I n one preparation using the procedure given above i t was p o s s i b l e to i s o l a t e c a . lOg of i s o p h t h a l i c a c i d from the r e a c t i o n l i q u o r s by treatment with aqueous potassium carbonate and subsequent a c i d i f i c a t i o n o f the aqueous l a y e r . I n an attempt to improve the y i e l d of the r e a c t i o n , the following modified procedure was used: doubly d i s t i l l e d i s o p h t h a l o y l c h l o r i d e (200g) were d i s s o l v e d i n sodium d r i e d benzene (2O0O mis) with vigorous s t i r r i n g . anhydrous aluminium c h l o r i d e (400g) was added i n smaller portions than i n the previous method over a longer period of time ( c a . 6 h r s ) ; was complete, the mixture was r e f l u x e d for another 6 h r s . F i n e l y ground when the addition s t i r r e d a t room temperature f o r 6 hrs and Following h y d r o l y s i s of the mixture, then organic - 82 - m a t e r i a l s were e x t r a c t e d with benzene (2 x 1000 mis) and the e x t r a c t s b o i l e d three times with a c t i v a t e d animal c h a r c o a l . The c o l o u r l e s s s o l u t i o n obtained was evaporated and the recovered white s o l i d was r e c r y s t a l l i z e d three times from cyclohexane to give pure m-dibenzoylbenzene (196g, 70%) as shown by t . l . c . on s i l i c a using a v a r i e t y of e l u e n t s . T h i s product had the c o r r e c t m.s. and i . r . spectrum, m.pt. 103°-104°, and elemental a n a l y s i s (found C, 83.68; 5.29; calculated for C H 2 0 0 1 4 c 2 ' 8 3 9 - °/' H H, » 4.93). A small sample of the p u r i f i e d m-dibenzoylbenzene was d i s s o l v e d i n a l a r g e volume of b o i l i n g cyclohexane, and the s o l u t i o n was l e f t to cool slowly over a p e r i o d of s e v e r a l days. Large prisms of m a t e r i a l , some o f them n e a r l y 2.5 cm long were obtained, having a very sharp melting p o i n t o f 103.8°. Preparation o f 1,2,2,2-tetraphenylethanol To a s o l u t i o n of benzopinacolone (3.48g, 0.01 mole) i n ethanol (100 mis) a s o l u t i o n o f sodium borohydride (0.38g, 0.01 mole) i n water was s l o w l y added. A f t e r f r o t h i n g had subsided, the mixture was r e f l u x e d f o r 2 h r s . Addition of 2 l i t r e s of water caused the p r e c i p i t a t i o n of a white powdery m a t e r i a l which recovered by f i l t r a t i o n , was washed with copious amounts of water, r e c r y s t a l l i z e d from 50:50 methanol/water and d r i e d under vacuum to give 1,2,2,2-tetraphenylethanol (2.8g, 8 0 % ) , m.pt. for C H 0: 2 g 2 2 150° C, 89.14; (lit. 1 4 0 H, 6.29. 151°); found C, 88.89; H, 6.20; calculated The m a t e r i a l gave a s i n g l e spot on on s i l i c a (benzene, chloroform, ethanol, methanol). no molecular ion, the h i g h e s t peak being a t m/e t.l.c. The mass spectrum showed 332 (molecular i o n - 1 8 ) ; other main peaks were a t 224, 243, 182, 167, 165, 152, 105, 77. the The infrared spectrum of the r e c r y s t a l l i z e d product (nujol mull) showed a t r i p l e t of peaks i n the OH s t r e t c h i n g region, 3600 (strong-sharp), 3540 (weak-sharp), c a . 1 3450 (broad-very weak) cm . The product was r e - p u r i f i e d by sublimation a t 140°/0.005 mmHg; the i n f r a r e d spectrum of t h i s product (m.pt. 151.5°) showed a q u a r t e t o f peaks i n the OH stretching region a t 3590 (strong-sharp), - 83 - 3560 (weak-sharp), 3540 (medium-sharp), 3450 (medium-broad) cm to peaks a t v 3080, 3050, 3020, 2960, 2920, 1590, 1490, 1445, 1045, 7o5 cm max : (spectrum recorded as KBr disc) . , i n addition 1030, I n an attempt t o ensure the complete dryness o f the product, a benzene s o l u t i o n o f a sample o f the sublimed 1,2,2,2-tetraphenylethanol was r e f l u x e d i n a Dean-Stark apparatus; there was no change i n the i n f r a r e d spectrum or the m.pt. of the recovered sample. A f u r t h e r s u r p r i s i n g r e s u l t was obtained when the s o l u t i o n phase i n f r a r e d spectrum of the substance was examined under the conditions s p e c i f i e d i n Table 2.6. A doublet o f peaks was obtained, s i m i l a r t o the ones obtained f o r 1,2-diols i n the OH s t r e t c h i n g region, possibly i n d i c a t i n g the existance of some degree o f intermolecular hydrogen bonding i n an associated dimer. Preparation o f g-phenylbenzoin A saturated s o l u t i o n o f b e n z i l (16g) i n ether was slowly added (30 mins) t o a s t i r r e d ether s o l u t i o n (200 mis) o f phenylmagnesiumbromide (from 15g bromobenzene) a t 0°. The r e s u l t i n g s o l u t i o n was r e f l u x e d f o r 30 mins, then hydrolysed by pouring i n t o 1 l i t r e 10% HCl/ice water mixture. A brown viscous mass was obtained which was t r e a t e d several times w i t h ethanol t o give a w h i t i s h powder (65%); t h i s was r e c r y s t a l l i z e d several times from ethanol/water (70:30) g i v i n g a-phenylbenzoin (50%), a white c r y s t a l l i n e s o l i d m.pt. (lit. 1 4 0 C, 83.31; 88°); found: H, 5.59. C, 83.89; H, 5.95; c a l c u l a t e d f o r C_H,^0„: 20 16 2 This m a t e r i a l gave a s i n g l e spot on t . l . c . on s i l i c a (benzene, chloroform, ethanol, acetone/water, a c e t o n i t r i l e ) . f u r t h e r p u r i f i e d by sublimation (90°/0.005 mmHg); melted a t 88.8°. being m/e 87° The product was the sublimed m a t e r i a l The mass spectrum showed no molecular i o n the highest peak 184 w i t h prominent peaks at m/e i s extensive fragmentation i n the source. 183, 151, 106, 105, 77; c l e a r l y there The u.v. spectrum i n cyclohexane revealed three peaks a t 350 ( 3 8 ) , 347 (84), and 317 (258) nm (c) i n a d d i t i o n to strong aromatic bands. The i . r . spectrum (KBr disc) showed v : 3500, - 84 - 3050, 3040, 1675, 1600, 1580, 1500, 1450, 1350, 1250, 1185, 1030, 850, 700 1 (doublet) cm . The n.m.r. spectrum showed a m u l t i p l e t I n the aromatic proton region a t 6 7.7(15) and a s i n g l e t a t 6 4.91(1) which disappeared on a d d i t i o n of D2O assigned t o the hydroxy1 group. Preparation o f T r i p h e n y l g l y c o l 137 Method A; Finely ground benzoin (21.2g, 0.1 mole) was slowly added to a vigorously s t i r r e d ether s o l u t i o n o f phenylmagnesiumbromide (from 55g bromobenzene). Addition, over a period o f 1 h r , was c a r r i e d out a t 20°, the r e s u l t i n g mixture was s t i r r e d f o r 5 hrs a t room temperature and then gently refluxed f o r 6 h r s ; mixture. i t was then hydrolysed by pouring i n t o 5% HCl/ice water A powdery product was obtained i n 60% y i e l d which, a f t e r repeated r e c r y s t a l l i z a t i o n s from ethanol yielded pure t r i p h e n y l g l y c o l (45%) , m.pt. icc° tn- 128,129 ...-o . , . 0 , 165 (lit. 168 , 164 ) . Method B: A s o l u t i o n o f sodium borohydride (0.01 mole) i n water was slowly added t o a s t i r r e d s o l u t i o n o f a-phenylbenzoin (0.01 mole) i n ethanol (150 mis). A f t e r i n i t i a l vigorous f r o t h i n g had subsided, the mixture was refluxed f o r 3 hrs. water; A white m a t e r i a l p r e c i p i t a t e d on a d d i t i o n o f 1 l i t r e o f i t was recovered by f i l t r a t i o n , washed w i t h water, r e c r y s t a l l i z e d from ethanol/water (70:30) and d r i e d under vacuum t o give t r i p h e n y l g l y c o l (85%) , m.pt. 166°. Materials obtained by both methods gave, a f t e r repeated r e c r y s t a l l i zations from ethanol/water (70:30), single spots on t . l . c . on s i l i c a (benzene, toluene, chloroform, a c e t o n i t r i l e , methanol, acetone/water, ethanol, carbon t e t r a c h l o r i d e , ethylacetate, chloroform/carbon t e t r a chloride). Elemental analyses were as f o l l o w s : H, 5.94; method B: C, 82.73; method A: C, 82.91; H, 6.25; calculated f o r C H 0 : C, 82.38; 20 18 Z 0 H, 6.44. Both samples displayed the same s p e c t r a l c h a r a c t e r i s t i c s . i n f r a r e d spectrum (KBr disc) showed v The : 3570, 3470, 3090, 3070, 3040, 2940, 1500, 1450, 1190, 1065, 1045, 1030, 900, 760, 750, 700, 620 (doublet). - 85 - The H n.m.r. spectrum (CDCl /CD.jC0CD , TMS e x t . r e f . ) showed a 3 6 7.7(15), doublets at 6 a s i n g l e t at 6 3.88(1); 5.59 raultiplet 3 (J = 4 Hz, 1) and 6 4.04 the signals at 6 4.04 at (J = 4 Hz, 1 ) , and and 3.88 disappeared on a d d i t i o n of D^O allowing t h e i r assignment as hydroxyl protons attached t o the secondary and t e r t i a r y carbons r e s p e c t i v e l y . Sublimation o f t r i p h e n y l g l y c o l (130°/0.005 mmHg) afforded a m a t e r i a l (m.pt. 166.8°) whose i . r . spectrum i n the s o l i d s t a t e (KBr disc) displayed 1 3 peaks i n the OH s t r e t c h i n g region (v : max i n the C-H a l i p h a t i c s t r e t c h i n g region (v 3560, 3520, 3460 cm" ) : 1 2930, 2890 cm" ). and 2 peaks There were DlclX also some minor differences i n the f i n g e r p r i n t region o f the spectrum. Elemental 1 analysis f i g u r e s , and H n.m.r. spectrum o f the sublimed were not d i f f e r e n t from those o f the r e c r y s t a l l i z e d m a t e r i a l . The substance gave a s i n g l e spot on t . l . c . on s i l i c a (benzene, chloroform, ethanol, a c e t o n i t r i l e ) . The solution phase i n f r a r e d spectrum o f the sublimed m a t e r i a l i n the OH s t r e t c h i n g region (CCl solvent - see Table 2.6) was the same as the one f o r the r e c r y s t a l l i z e d 4 material. 2.2.k. Discussion and Conclusions The basic question asked i n t h i s Section, as already set out i n 2.2.a was: 'Can poly(pentaphenylglycerols) and higher analogues o f s t r u c t u r e shown i n Figure 2.30 be prepared by the photoreductive a d d i t i o n o f aromatic bisketones to s u i t a b l e a l c o h o l i c hydrogen donors?'. This question was answered i n 2.2.f. where i t was established t h a t the model r e a c t i o n i l l u s t r a t e d i n Figure 2.47 i s l i m i t e d t o the case where m = 1, benzopinacol being q u a n t i t a t i v e l y obtained; when m = 2 r e a c t i o n does not a f f o r d any detectable products. Cases where m > 2 have not been examined, p a r t l y i n view o f the absence o f reports on such compounds i n the l i t e r a t u r e and p a r t l y due t o the doubts on the p o s s i b i l i t y o f t h e i r existence a r i s i n g from the f a i l u r e t o i s o l a t e pentaphenylglycerol from the r e a c t i o n described i n 2.2.1. - 86 - 9 + H 350 nms solvent C_ OH _m=l or 2 Figure 2.47 The f a i l u r e o f the model r e a c t i o n (Figure 2.47, m = 2) t o proceed leads to the conclusion that the photoreductive polymerization o f bisketones cannot be extended beyond the polybenzopinacol stage, i n other words t h i s type o f polymerization cannot be applied t o the synthesis o f s t r u c t u r e s o f the k i n d shown i n Figure 1.12. Although the question regarding the synthesis o f poly(pentaphenylglycerols) and higher analogues was answered, some new questions have been uncovered: a) The o l d dispute between Ciamician and Paterno regarding the formation o f t r i p h e n y l g l y c o l was resolved i n favour o f Ciamician (2.2.d). However, when the reaction was repeated by us a b r i g h t yellow colour was observed a f t e r a few hours o f u l t r a v i o l e t i r r a d i a t i o n , i n d i c a t i n g the formation o f an intermediate. By analogy t o the benzophenone/isopropanol and benzophenone/ ethanol photoreductions (see 2.1.b) i t seems probable t h a t t h i s i s a species o f the type shown i n Figure 2.48. Such a species might act as an i n t e r n a l l i g h t f i l t e r and t h i s would account f o r the slow r e a c t i o n r a t e observed and the f a i l u r e o f the r e a c t i o n t o go i n t o completion even a f t e r 300 hrs o f i r r a d i a t i o n (see 2.2.d). A l t e r n a t i v e l y , one could a t t r i b u t e the b r i g h t yellow colour t o the photolysis o f benzaldehyde, i t s e l f a product o f the benzylalcohol/ benzophenone photoreaction; p h o t o l y s i s o f neat colourless benzaldehyde was 87 99 -C OH Ok *C CH C H = _9 \ _ / — HO H C ~ OH Figure 2.48 found t o y i e l d yellow coloured materials (see Chapter 3 ) . A b r i g h t yellow colour was also observed i n the photoreactions o f m-dibenzoylbenzene/hydrobenzoin mixtures and benzophenone/l-phenylethanol mixtures (2.2.g). Again, one could postulate the formation o f s i m i l a r coloured intermediates as shown i n Figure 2.49. However, the o r i g i n o f the yellow CH HO OH c II 0 c= ( )—c / c- I HO OH H I OH Figure 2.49 colour has not been r i g o r o u s l y examined and the nature o f the intermediates involved i s open t o question. - 88 - 3) The inertness o f t r i p h e n y l g l y c o l and 1,2,2,2-tetraphenylethanol towards benzophenone t r i p l e t i s another question not easy t o deal w i t h . In 2.1.b.vi the s t e r i c e f f e c t on photoreduction was examined and some examples were given showing t h a t s t e r i c hindrance can be important. Comparative molecular model studies o f the compounds examined here have shown t h a t f o r both structures the t e r t i a r y hydrogen, which was expected t o be a b s t r a c t a b l e , appears t o be only marginally more hindered than the t e r t i a r y hydrogen i n benzhydrol or hydrobenzoin. I n our view, i t seems u n l i k e l y t h a t the complete lack o f r e a c t i v i t y e x h i b i t e d by these compounds can be accounted f o r by s t e r i c hindrance; on the other hand, the C-C a bond separating the (OH)HC(CgH ) 5 group from the (CgHg)COH group ( i n t r i p h e n y l g l y c o l ) and the (CgH,.) 3C group 2 ( i n 1,2,2,2-tetraphenylethanol) should r u l e out any e l e c t r o n i c i n t e r a c t i o n s between the groups, at l e a s t o f the type known and understood by us. the Finally, 'quencher theory* having been r u l e d out (see 2 . 2 . f ) , we are l e f t w i t h no explanation t o suggest f o r these observations. Other authors have commented on the unexpected s e l e c t i v i t y of the benzophenone t r i p l e t i n hydrogen a b s t r a c t i o n r e a c t i o n s . Thus, although i t i s w e l l established t h a t hydrogen a b s t r a c t i o n occurs r e a d i l y from toluene and diphenylmethane i t has been reported t h a t the t e r t i a r y hydrogen o f t r i p h e n y l 97 methane i s not abstractable. I n the case o f these three hydrocarbons i t i s not d i f f i c u l t t o construct a r a t i o n a l i z a t i o n o f these observations i n terms of s t e r i c hindrance. On the other hand i f s t e r i c e f f e c t s are c o n t r o l l i n g the a b s t r a c t a b i l i t y o f the t e r t i a r y hydrogens i n benzhydrol, hydrobenzoin, t r i p h e n y l g l y c o l and tetraphenylethanol the e f f e c t s depend on rather s u b t l e d i f f e r e n c e s which are not r e a d i l y detected by examination o f space f i l l i n g molecular models. y) The f a i l u r e t o i s o l a t e pentaphenylglycerol or detect high molecular weight products from the r e a c t i o n between d i e t h y l mesoxalate and excess o f p h e n y l l i t h i u m i s another problem which arose during the course o f these - 89 - investigations. Evidence presented i n s e c t i o n 2.2.1 tends to suggest t h a t 139 the o r i g i n a l claim to have s y n t h e s i z e d pentaphenylglycerol was erroneous. The o r i g i n a l author's only evidence, apart from the 'colour t e s t ' which.has been shown to be meaningless, i s the good agreement found and c a l c u l a t e d analytical figures. However, the o c c l u s i o n of methanol s o l v e n t i n the molecule due to inadequate drying might have lowered elemental a n a l y s i s f i g u r e s , leading to the wrong c o n c l u s i o n s . The formation of t r i p h e n y l c a r b i n o l from the r e a c t i o n of a l a r g e excess of p h e n y l l i t h i u m with d i e t h y l ketomalonate i s not i n h e r e n t l y unreasonable and i t i s i n t e r e s t i n g to note t h a t the product y i e l d obtained i n t h i s work and by the o r i g i n a l author corresponds to the formation of a t h i r d of a mole of c a r b i n o l f o r each d i e t h y l ketomalonate r e a c t i o n was not undertaken; consumed. A d e t a i l e d study of t h i s however i f i t i s assumed t h a t the i n i t i a l a t t a c k of p h e n y l l i t h i u m takes p l a c e a t the c e n t r a l ketogroup and i s followed by e l i m i n a t i o n of carbon monoxide and ethoxide anion, the beginnings of a p o s s i b l e r a t i o n a l i z a t i o n can be c o n s t r u c t e d . Although i t may be t h a t pentaphenyl- g l y c e r o l i s i n h e r e n t l y unstable and fragmented as f a s t as i t i s formed; this would be s u r p r i s i n g s i n c e examination of space f i l l i n g models would not l e a d one to suspect t h a t the compound i s s i g n i f i c a n t l y more s t e r i c a l l y hindered than, say, benzopinacol. However, the observation t h a t benzopinacol g i v e s 118 r a d i c a l s by C-C bond homolysis under mild c o n d i t i o n s may be quoted i n support of the p o s t u l a t e d i n s t a b i l i t y of p e n t a p h e n y l g l y c e r o l . 6) F i n a l l y the d i f f e r e n c e i n the s o l i d s t a t e i n f r a r e d spectrum between r e c r y s t a l l i z e d and sublimed t r i p h e n y l g l y c o l , and the unexpected of exhibition a doublet of peaks i n the OH s t r e t c h i n g region by 1,2,2,2-tetraphenylethanol i n CC1 s o l u t i o n (see 2 . 2 . j . i i i ) are f u r t h e r questions which remain unanswered. 2.3. Use of the diphenylmethane-benzophenone r e a c t i o n as the polymer b u i l d i n g step 4 - 90 - 2.3.a. D e t a i l s of model reactions 2.3.a.l. Discussion o f l i t e r a t u r e The diphenylmethane-benzophenone photoreaction was f i r s t i n v e s t i g a t e d by 11 Paternb and C h i e f f i almost 60 years a g o . ^ Their experiment consisted o f i n s o l a t i n g 20g (0.109 mole) o f benzophenone and 25g methane i n a sealed tube. temperature, (0.148 mole) of diphenyl- Such a mixture c o n s t i t u t e s a l i q u i d mass a t ambient so the authors found i t more convenient not to use a solvent. Hard c r y s t a l s were observed forming on the i n s i d e w a l l o f the tube a f t e r only two days o f i n s o l a t i o n ; they were separated a f t e r ten days and the remaining s o l u t i o n was i n s o l a t e d again u n t i l r e a c t i o n was complete. The s o l i d m a t e r i a l obtained was dissolved i n b o i l i n g benzene and an 'almost pure' novel compound c r y s t a l l i z e d on cooling. Examination o f the r e c r y s t a l l i z a t i o n solvent revealed the presence o f traces o f the novel compound and the excess o f dlphenylmethane. No other materials were i d e n t i f i e d . I n order t o make sure t h a t the s o l i d m a t e r i a l obtained from the i n s o l a t i o n was i n f a c t pure, the authors t r e a t e d i t successively w i t h the f o l l o w i n g hot solvents: 1 ethanol, 'acetic e t h e r , 'acetic a c i d ' , 'acetic ether' again and f i n a l l y benzene, s t i l l leaving an i n s o l u b l e p o r t i o n ['acetic ether' i s the l i t e r a l t r a n s l a t i o n , we t h i n k t h i s was probably e t h y l a c e t a t e ] . A l l the samples thus obtained melted a t 212°-214°. The novel compound was found t o be only s l i g h t l y soluble i n the above solvents ( i n cold) and also i n d i e t h y l e t h e r , chloroform, carbon d i s u l p h i d e , p-xylene, fused phenol, fused thymol etc. I t was found t o be soluble i n hot solvents g i v i n g l a r g e , w e l l defined needles on cooling. The r e s u l t s o f elemental analysis and e b u l l i o s c o p i c molecular weight determination i n ethanol and benzene were i n good agreement w i t h the values calculated f o r C ^ I ^ O . t h e 1:1 a d d i t i o n product. Q Treatment o f the product w i t h ?2 5 ^ n 1 XY - 6 1 1 6 afforded tetraphenylethylene, i d e n t i f i e d by means of elemental analysis and m.pt. determination. Treatment w i t h phosphorous and iodine afforded tetraphenylethane s i m i l a r l y i d e n t i f i e d . - 91 - On the b a s i s o f the above evidence the authors assigned to the product 119 the s t r u c t u r e of 1,1,2,2-tetraphenylethanol. 141 The compound was f u r t h e r studied by Bergmann and Engel who isolated two forms of i t , one melting a t 235° (a) and another melting a t 216° ($) , the l a t t e r prepared by the i n s o l a t i o n o f the benzophenone-diphenylmethane mixture. They a t t r i b u t e d t h i s phenomenon to isomerism of 1,1,2,2-tetraphenyl- ethanol, r e s u l t i n g from i n a b i l i t y of the (CgH,.) C0H and (CgHg) CH groups 141 2 2 present i n the molecule to r o t a t e f r e e l y about the ethane C-C bond. 142 Wegler r e - i n v e s t i g a t e d the i n s o l a t i o n experiment and obtained a product which, a f t e r three r e c r y s t a l l i z a t i o n s from acetone melted a t 215°-217°. Attempts to r e p l a c e the OH group by c h l o r i n e never gave halogenated d e r i v a t i v e s , and only a f t e r repeated r e c r y s t a l l i z a t i o n s was a homogeneous product obtained, i d e n t i f i e d as tetraphenylethylene. By repeating the r e c r y s t a l l i z a t i o n o f the photochemically prepared 1,1,2,2-tetraphenylethanol f i f t e e n times, i t s m.pt. was r a i s e d to 229°. The same r e s u l t was obtained when treatment with the s o l v e n t was c a r r i e d out i n c o l d , so the author concluded t h a t h i s r e s u l t s could not be i n t e r p r e t e d as the B-form being rearranged to the a-form. When the $-form was heated f o r s e v e r a l hours i n p y r i d i n e a t 135°, i t s melting p o i n t was lowered somewhat and r e c r y s t a l l i z a t i o n of the product y i e l d e d the a-form. Wegler concluded t h a t the 3-form was impure a and t h a t heating w i t h p y r i d i n e changed the i m p u r i t i e s to m a t e r i a l s r e a d i l y removed by recrystallization. subsequent The author considerad the reported cases of benzophenone photoreduction i n the presence of a l i p h a t i c and aromatic hydrocarbons, where benzopinacol and the dimerized hydrocarbons are formed and concluded t h a t i m p u r i t i e s i n the B-form were probably benzopinacol and tetraphenylethane. He then prepared an a r t i f i c i a l mixture of a and 20% benzopinacol and t e t r a phenyl ethane and claimed t h a t i t behaved e x a c t l y l i k e the s o - c a l l e d B-form. He a t t r i b u t e d the easy p u r i f i c a t i o n o f the B-form by b o i l i n g i t i n p y r i d i n e to the decomposition o f benzopinacol present i n t o benzophenone and benzhydrol - 92 - which could then be e a s i l y removed by r e c r y s t a l l i z a t i o n . When the cx-form was t r e a t e d w i t h sodium i n benzene, CH (CgH,-) , (CgHg^CO, (CgH,.) CHOH, 2 2 2 CgHgCOOH and predominantly (CgH > C = C(CgH ) were obtained, whereas 5 2 5 2 treatment o f the 0-forra w i t h b o i l i n g benzene i n the presence o f sodium gave 142 an intense blue-green colour a t t r i b u t e d by the author t o the presence o f benzopinacol. I n the same year t h a t Wegler published h i s r e s u l t s , 1,1,2,2-tetraphenylethanol was independently prepared by Richard v i a ground s t a t e synthesis and i t s melting p o i n t was determined t o be 2 3 6 . 5 ° . T h r e e years e a r l i e r the same compound was prepared by the action o f phenylmagnesiumbromide ono, ao o 144 diphenylacetophenone and the reported melting p o i n t was 232 -233 . In 1937, a s i m i l a r r e a c t i o n , namely the action o f phenylmagnesiumbromide on o o 145 CgHgCHClCOCl afforded 1,1,2,2-tetraphenylethanol melting at 232.5 -233 . 1,1,2,2-Tetraphenylethanol was also obtained i n low y i e l d by b o i l i n g benzopinacol i n cyclohexanone; 1 t h i s sample had a m.pt. o f 232°-233°. ^^ The f o l l o w i n g year the same author reported the r e p e t i t i o n o f the benzophenonediphenylmethane i n s o l a t i o n experiment i n i t i a l l y performed by Paterno and 119 Chieffi; he obtained a product which, a f t e r r e c r y s t a l l i z a t i o n s , melted a t 217°-218° on a copper block whereas m.pt. determination using a Kofler apparatus caused sublimation t o minute needles which became opaque a t 230°-232° and melted sharply a t 243°-244° Further studies o f the photochemical 1 reaction by B a n c h e t t i ^ y i e l d e d a product which melted at 206°-210°; treatment of the product w i t h a large volume o f acetone yielded a compound m e l t i n g a t 217°-218°. Considerable reduction i n the volume o f acetone yielded small amounts o f benzopinacol, i d e n t i f i e d oil the basis of i t s 'behaviour under the 147 microscope'. Further r e c r y s t a l l i z a t i o n of 1,1,2,2-tetraphenylethanol from acetic acid caused a r i s e i n the melting p o i n t t o 223°-224° whereas r e c r y s t a l l i z a t i o n from acetic anhydride yielded a m a t e r i a l melting at 227°228°. F i n a l l y , b o i l i n g the m a t e r i a l i n cyclohexanol f o r one hour and - 93 - r e c r y s t a l l i z a t i o n o f the nearly pure material thus obtained from chloroform147 acetone and then chloroform-ethanol yielded a product melting a t 244 . Having s a t i s f i e d himself on the question o f the p u r i t y o f 1,1,2,2-tetraphenylethanol thus obtained, and having established the formation o f benzopinacol from the photochemical experiment, Banchetti d i d not i n v e s t i g a t e the possible 147 formation o f any other by-products. He concluded t h a t the best way t o obtain 1,1,2,2-tetraphenylethanol i n high y i e l d was the i n s o l a t i o n o f the benzophenone-diphenylmethane mixture; p u r i f i c a t i o n o f the m a t e r i a l from the l a s t traces o f benzopinacol ( i t s e l f a low y i e l d by-product o f the photoreaction) necessitated however heating the ethanol w i t h a high b o i l i n g solvent preferably cyclohexanol - followed by r e c r y s t a l l i z a t i o n . This method afforded o 147 a non-specified high y i e l d o f pure m a t e r i a l melting at 244 . 148 Another preparation o f the ethanol appeared i n the l i t e r a t u r e i n 1956: treatment o f 0.01 mole o f the ketoester C H C(0)-CH(CgH )-0-C(0)CH i n d i e t h y l g 5 5 3 ether w i t h 0.01 mole phenylmagnesiumbromide, r e f l u x i n g f o r 30 mins, c o o l i n g , adding another 0.05 mole phenylmagnesiumbromide during 40 mins and refluxing f o r 15 mins gave a f t e r evaporation of d i e t h y l e t h e r solvent and p u r i f i c a t i o n from chloroform 38% pure 1,1,2,2-tetraphenylethanol. The melting p o i n t o f the compound was however reported t o be 231°-232.5°.^^ A few years l a t e r a group o f American workers obtained 1,1,2,2-tetraphenylethanol by the a d d i t i o n reaction o f sodium diphenylmethide w i t h benzophenone i n l i q u i d ammonia solvent followed by r e c r y s t a l l i z a t i o n from methylene c h l o r i d e ; o o 149 were obtained, melting a t 243 -244 . 86% o f pure compound The l i t e r a t u r e i n t h i s area already presents a r a t h e r confused p i c t u r e , i t was therefore necessary t o re-examine the photoreaction of benzophenone w i t h diphenylmethane before proceeding t o the proposed polymerization o f bifunctional monomers. Results o f t h i s re-examination are presented i n the next section and the l i t e r a t u r e claims reviewed t h e r e a f t e r . - 94 - 2.3.a.ii. a) Experimental Results I r r a d i a t i o n s without solvent A 1:1 molar mixture o f benzophenone and diphenylmethane ( s t a r t i n g w i t h 15g benzophenone) forming a homogeneous l i q u i d mass was t r a n s f e r r e d i n t o a c y l i n d r i c a l Pyrex tube and nitrogen streamed f o r % n r . The tube was q u i c k l y stoppered and i r r a d i a t e d a t 350 nm (ca. 40°). A f t e r 30 hrs o f i r r a d i a t i o n the inside w a l l s o f the tube were covered w i t h a t h i c k layer o f colourless c r y s t a l s and remaining s o l u t i o n was b r i g h t yellow. The tube was opened, c r y s t a l s were broken by means o f a long spatula and l e f t as a s o l i d p r e c i p i t a t e a t the bottom o f the tube. The s o l u t i o n was nitrogen streamed again f o r h hr and r e - i r r a d i a t e d f o r another 60 h r s . c r y s t a l s had formed i n the i n s i d e w a l l o f the tube. Again colourless Following opening o f the tube, d e s t r u c t i o n o f the c r y s t a l l i n e l a y e r , n i t r o g e n streaming and stoppering, s o l u t i o n was r e - i r r a d i a t e d f o r a f u r t h e r 14 hr period; a new layer o f c r y s t a l s had formed and yellow colour appeared somewhat more intense. At t h a t stage the tube was opened, the c r y s t a l s were f i l t e r e d o f f and the b r i g h t yellow s o l u t i o n obtained was nitrogen streamed and r e - i r r a d i a t e d f o r 29 hrs y i e l d i n g another layer o f hard colourless c r y s t a l s . At t h a t p o i n t the reaction was stopped, the c r y s t a l s were f i l t e r e d o f f , combined w i t h the crop obtained from the previous f i l t r a t i o n and dissolved i n b o i l i n g benzene. Slow cooling o f the benzene s o l u t i o n y i e l d e d 17.3g (ca. 60%) o f a white c r y s t a l l i n e material i n w e l l defined needles. Examination o f the needles by t . l . c . on s i l i c a (benzene) revealed the presence o f one component only, whereas examination o f the yellow s o l u t i o n revealed the presence o f the same m a t e r i a l and also benzophenone and diphenylmethane. Examination o f the r e c r y s t a l l i z a t i o n solvent by the same method revealed the presence o f the c r y s t a l l i n e m a t e r i a l , benzophenone and diphenylmethane. was detected. No benzopinacol Melting p o i n t o f the c r y s t a l s obtained was found t o be - 95 - o o 228 -229 . Subsequent r e c r y s t a l l i z a t i o n s from chloroform caused only a very small r i s e i n the melting p o i n t , not exceeding 2°. The same r e a c t i o n was attempted using a 20% excess o f diphenylmethane. This time however colourless c r y s t a l s formed i n the inside w a l l o f the tube were not broken but were l e f t t o accumulate. A f t e r 130 hrs o r i r r a d i a t i o n a b r i g h t yellow colour was observed i n s i d e the tube. Working-up the r e a c t i o n products as before afforded ca. 40% o f the white c r y s t a l l i n e s o l i d which, a f t e r r e c r y s t a l l i z a t i o n s from benzene and chloroform was found t o melt a t 229°-230°. Examination o f the white s o l i d , yellow s o l u t i o n and r e c r y s t a l l i z a t i o n solvents by t . l . c . on s i l i c a (benzene, chloroform) f a i l e d again t o reveal the presence o f benzopinacol or other by-products. B o i l i n g the white c r y s t a l l i n e m a t e r i a l i n p y r i d i n e f o r 1.5 h r s , p r e c i p i t a t i o n w i t h water and r e c r y s t a l l i z a t i o n o f the product thus obtained from benzene large c r y s t a l s (70%), m.pt. 231°. 0) Photoreactions i n benzene solvent An 1:1 molar mixture o f benzophenone and diphenylmethane (starting w i t h lOg benzophenone) was dissolved i n benzene (0.01M w i t h respect t o both reagents) i n a c y l i n d r i c a l Pyrex tube. The c l e a r , colourless s o l u t i o n was nitrogen streamed f o r 1 h r , tube was q u i c k l y stoppered and i r r a d i a t e d a t 350 : f o r 110 h r s . A white p r e c i p i t a t e had formed a t the end o f the i r r a d i a t i o n p e r i o d , the s o l u t i o n being again b r i g h t yellow. was recovered by f i l t r a t i o n , m.pt. 230°-231°. showed v The c r y s t a l l i n e m a t e r i a l I . r . spectroscopy (KBr disc) : 3540, 3080, 3060, 3020, 2920, 1600, 1580, 1480, 1445, 1340, UlcLX 1160, 1075 (doublet), 1050, 1030, 740, 700, 650 (doublet), 620 ( t r i p l e t ) , 580, 560 cm and was c l e a r l y d i s t i n c t from the i . r . spectrum o f benzopinacol 1 (KBr disc) v max : 3568-3540 ( c h a r c t e r i s t i c doublet i n the 0-H s t r e t c h i n g r e g i o n ) , 3080, 3060, 3020, 1600, 1580, 1495, 1445, 1340, 1320, 1160, 1030, 1 760, 740, 700, 650 (doublet), 610 cm" . Analysis o f the c r y s t a l l i n e m a t e r i a l - 96 - by t . l . c . on s i l i c a (benzene, chloroform) r e v e a l e d the presence o f one r e a c t i o n product only, together with two very f a i n t spots corresponding with s t a r t i n g materials. R e c r y s t a l l i z a t i o n o f the product from chloroform d i d not cause a s i g n i f i c a n t a l t e r a t i o n i n the melting point (231°-232°), or the s o l i d s t a t e i . r . spectrum (KBr d i s c ) . The h i g h e s t peak i n the mass spectrum occurred a t m/e = 332 (parent i o n (350)-18, most probably l o s s o f w a t e r ) , with major peaks a t 343, 342, 339, 183, 182, 177, 152, 105, 77. The m a t e r i a l was r e c r y s t a l l i z e d once more from benzene and d r i e d under vacuum f o r 24 h r s (m.pt. 232°). 14 Treatment o f t h e m a t e r i a l according t o B a n c h e t t i ' s p r o c e d u r e ^ resulted i n p a r t i a l (20%) l o s s o f m a t e r i a l but no s i g n i f i c a n t i n c r e a s e i n the melting p o i n t was observed C, 88.86; (m.pt. 232°-233°). Elemental a n a l y s i s gave: H, 6.60; c a l c u l a t e d f o r C H 0 , C, 88.70; o c 0 0 H, 6.75. The F.T. n.m.r. spectrum o f the m a t e r i a l i n CDCl^/CD-jCOCD-j ( e x t . TMS r e f . ) showed a s i n g l e t a t 6 7.13 (aromatic H) and four s i n g l e t s a t 6 5.23(A), 6 4.73(B), 6 3.02(C) and 6 2.81(D); of D 0; 2 1:1. peaks (C) and (D) disappeared on a d d i t i o n the i n t e g r a t e d i n t e n s i t i e s were i n the r a t i o (A) + (B):(C) + (D) = On the b a s i s o f the above evidence peaks (A) and (B) were assigned to the t e r t i a r y hydrogen i n 1,1,2,2-tetraphenylethanol and peaks (C) and (D) to the hydroxyl hydrogen. I n a l a t e r experiment, diphenylmethane (2.l0g, 0.0125 mole) and benzo- phenone (2.275g, 0.0125 mole) were d i s s o l v e d i n benzene (50 mis, 0.5M w i t h r e s p e c t to both r e a g e n t s ) . The c l e a r s o l u t i o n was nitrogen streamed f o r 1 h r , the Pyrex tube was q u i c k l y stoppered and the s o l u t i o n i r r a d i a t e d a t 350 run for 309.2 h r s . The s o l u t i o n became b r i g h t yellow and a white p r e c i p i t a t e was formed a t the bottom o f the tube. The p r e c i p i t a t e was separated by f i l t r a t i o n , washed with a l i t t l e c o l d benzene and d r i e d under vacuum. Both the white p r e c i p i t a t e and the yellow s o l u t i o n were examined by t . l . c . on silica (benzene, chloroform, e t h a n o l ) . I n both s o l v e n t and p r e c i p i t a t e - 97 - 1,1,2,2-tetraphenylethanol was i d e n t i f i e d . S t a r t i n g m a t e r i a l were not present, i n d i c a t i n g t h a t r e a c t i o n had gone t o completion. Analysis o f the solvent, however, revealed the presence o f spots corresponding t o 1,1,2,2tetraphenylethane and biphenyl (CgHg-CgH,-). A very f a i n t spot corresponding to benzopinacol was also observed. Analysis of the q u a n t i t a t i v e l y recovered p r e c i p i t a t e , m.pt. 216°-218° showed the presence o f some benzopinacol spot). (faint R e c r y s t a l l i z a t i o n o f the product from chloroform y i e l d e d pure 1,1,2,2-tetraphenylethanol, m.pt. 230°-231°. I s o l a t i o n o f pure benzopinacol from the r e c r y s t a l l i z a t i o n l i q u o r s was not possible, probably due t o small amounts o f m a t e r i a l present. Over 95% o f pure tetraphenylethanol was obtained from the r e c r y s t a l l i z a t i o n . 2.3.a.iii. Comments on l i t e r a t u r e claims i n the l i g h t o f present r e s u l t s Results from present experiments lead t o the f o l l o w i n g conclusions: a) the diphenylmethane-benzophenone photoreaction a t 350 nm i s a slow one, both i n benzene solvent and without the use o f solvent. 3) I r r a d i a t i o n o f a neat benzophenone-diphenylmethane mixture i s d i f f i c u l t t o study because o f the hard c r y s t a l s which keep forming on the i n s i d e w a l l s o f the c y l i n d r i c a l Pyrex i r r a d i a t i o n tube. These c r y s t a l s considerably diminish the amount o f l i g h t reaching the i n t e r i o r o f the tube, i t follows t h a t i r r a d i a t i o n times reported should be t r e a t e d w i t h caution. Also e s p e c i a l l y i n the study o f the 1:1 molar mixture, frequent opening of the tube, could have r e s u l t e d i n the upsetting of the precise 1:1 molar r a t i o . Y) I r r a d i a t i o n of neat reagents f o r up t o ca. 150 hrs ( j u s t over 6 days) under the above mentioned conditions r e s u l t e d i n only p a r t i a l . r e a c t i o n , w i t h 1,1,2,2-tetraphenylethanol as the only detectable product. pinacol nor 1,1,2,2-tetraphenylethane 6) Neither benzo- were detected by t . l . c . on s i l i c a . During a l l i r r a d i a t i o n s a b r i g h t yellow colour has been observed. On the basis o f e a r l i e r r a t i o n a l i z a t i o n s (see 2.1.b, also 2.2.k) one could - 98 - a t t r i b u t e the yellow colour t o the formation o f structures such as those shown i n Figure 2.50. Formation o f a s i m i l a r s t r u c t u r e from the combination 99. JK OH . ^ Q H HO c H and/or H Figure H 2.50 of two benzophenone k e t y l r a d i c a l s can be excluded since i n the photoreduction of benzophenone i n benzhydrol where two such primary r a d i c a l s are i n i t i a l l y formed, no yellow colour i s observed (see 2 . 1 . b . i i i ) . Again, as i n the case o f the benzophenone-benzylalcohol photoreaction examined i n 2.2.d such coloured species could act as i n t e r n a l l i g h t f i l t e r s and could account f o r the low reaction r a t e . c) I r r a d i a t i o n o f a d i l u t e benzene s o l u t i o n (0.01M t o t a l concentration) of benzophenone-diphenylmethane f o r 110 hrs resulted i n incomplete r e a c t i o n , y i e l d i n g 1,1,2,2-tetraphenylethanol as the only detectable product. On the other hand, the reaction went t o completion a f t e r 309.2 hrs o f i r r a d i a t i o n o f a more concentrated s o l u t i o n (0.5M); the l a t t e r experiment however afforded benzopinacol, 1,1,2,2-tetraphenylethane and biphenyl, a l l i n minute amounts. These r e s u l t s suggest t h a t the formation of benzopinacol and t e t r a p h e n y l ethane occurs only under f a i r l y r e s t r i c t e d conditions; thus n e i t h e r are found i n any o f the i r r a d i a t i o n s o f mixtures o f benzophenone and diphenylmethane e i t h e r neat or i n d i l u t e s o l u t i o n (0.01M) whereas at higher concentration (0.5M) these out-of-cage products were formed i n detectable q u a n t i t i e s . - 99 - C l e a r l y the f a c t o r s which c o n t r o l whether the in-cage product (1,1,2,2- tetraphenylethanol) or the out-of-cage products are formed must be subtle. A d e t a i l e d i n v e s t i g a t i o n of the e f f e c t s of concentration fairly and duration of i r r a d i a t i o n have not been undertaken s i n c e i t i s e s t a b l i s h e d t h a t under a wide range of conditions the r e q u i r e d product, t e t r a p h e n y l ethanol i s formed i n > 95% y i e l d ; t h i s would allow polymers of d.p. > 20 to be obtained from i r r a d i a t i o n of s u i t a b l e b i f u n c t i o n a l analogues. Never- t h e l e s s i t i s p o s s i b l e to speculate about the explanation of these observations. I n the f i r s t i n s t a n c e i t may be t h a t the r e l a t i v e proportions of in-cage and out-of-cage r e a c t i o n are c o n t r o l l e d by the d e t a i l s of the medium's composition. A l t e r n a t i v e l y i t may be t h a t the out-of-cage products from a r e a c t i o n of an intermediate (such as A, F i g u r e 2.50) arise with ground s t a t e benzophenone, t h a t i s by a process somewhat analogous to t h a t suggested 81 p r e v i o u s l y by F i l i p e s c u , and shown i n F i g u r e 2.51. Such a r e a c t i o n could only occur when the concentration of (A) becomes high enough and t h e r e f o r e the p r o b a b i l i t y of i t coming i n contact with a 9 j® 6 H-C 0 r J L . I - . ==( n 0 X x2 x2 H—C Figure 2.51 C H HO 6o C—C OH - loo - benzophenone molecule becomes s i g n i f i c a n t . I n c r e a s i n g the c o n c e n t r a t i o n o f the s o l u t i o n would a l s o make the r e a c t i o n more probable by i n c r e a s i n g the chance of intermediate (A) coming i n contact with a ground s t a t e benzophenone molecule. I r r a d i a t i o n o f the neat benzophenone/diphenylmethane system.may not l e a d to the formation o f intermediate A i n s u f f i c i e n t l y high c o n c e n t r a t i o n because of the f i l t e r e f f e c t o f the p r e c i p i t a t i n g t e t r a p h e n y l e t h a n o l . Thus the coloured s t r u c t u r e (A) p o s t u l a t e d i n F i g u r e 2.50 could be important i n the r a t i o n a l i z a t i o n o f the observed slow r e a c t i o n r a t e , the appearance o f the yellow colour and the minute amounts o f benzopinacol and 1,1,2,2-tetraphenylethane obtained under some c o n d i t i o n s . I t could not however e x p l a i n the formation o f b i p h e n y l , which i s most probably due t o the prolonged of benzophenone i n benzene s o l u t i o n . 77 84—88 ' irradiation Such a r e a c t i o n i s known to 83 88 y i e l d benzopinacol and biphenyl as the main products, ' and by i t s e l f would account f o r the formation o f both products mentioned above, would not however e x p l a i n the formation o f 1,1,2,2-tetraphenylethane. The almost e x c l u s i v e formation o f 1,1,2,2-tetraphenylethanol i n these r e a c t i o n s i s a marked exception t o the normal behaviour o f t r i p l e t benzophenone i n the presence o f hydrogen donors, where benzopinacol formation i s the main process. Even with aromatic hydrocarbon hydrogen donors which a r e s t r u c t u r a l l y f a i r l y c l o s e l y r e l a t e d t o diphenylmethane the in-cage d i m e r i z a t i o n o f the primary r a d i c a l s formed from i n i t i a l hydrogen a b s t r a c t i o n appears t o be a r e l a t i v e l y low y i e l d process. As a l r e a d y mentioned toluene and cumene y i e l d only ca. 50% o f the corresponding cage products when i r r a d i a t e d i n the presence 7 6»89 of benzophenone. On the other hand, the cage r e a c t i o n product o f the benzophenonediphenylmethane r e a c t i o n i s a p e c u l i a r compound i n i t s e l f , being i n s o l u b l e or only s l i g h t l y s o l u b l e i n a very wide range o f laboratory s o l v e n t s i n the c o l d , t h i s being a property not shared by c l o s e l y r e l a t e d s t r u c t u r e s such as benzopinacol, 1,2,2,2-tetraphenylethanol, t r i p h e n y l g l y c o l e t c . 1 The H F.T. - 101 - n.m.r. spectrum o f the m a t e r i a l d i s p l a y s two d i s t i n c t t e r t i a r y hydrogens and two hydroxy1 hydrogen resonances. T h i s could be a t t r i b u t e d t o the e x l s t a n c e of two s t a b l e conformational isomers o f the m a t e r i a l , as shown i n F i g u r e 2.52, although i t i s worth noting t h a t the r e l a t e d s t r u c t u r e s l i s t e d above do not show t h i s e f f e c t . Due t o the low s o l u b i l i t y o f the m a t e r i a l i n most c o l d 0 II H OH OH Figure 2.52 s o l v e n t s i t was not p o s s i b l e to i n v e s t i g a t e i t s s o l u t i o n phase i . r . spectrum or examine i t s p h o t o r e a c t i v i t y with benzophenone i n benzene s o l u t i o n a t concentrations comparable t o those o f previous experiments. Deciding whether the experimental r e s u l t s obtained i n our hands a r e i n agreement with e a r l i e r r e p o r t s i s a r a t h e r d i f f i c u l t t a s k . p a p e r s * " ^ r e c o r d e d Earlier l i m i t e d experimental d e t a i l s ; the i r r a d i a t i o n source was the sun r a t h e r than a lamp of defined output, the temperature was not recorded and may w e l l have been s i g n i f i c a n t (ground temperatures i n the North A f r i c a n d e s e r t may reach 70°C) and almost a l l these e a r l y papers are devoid o f q u a n t i t a t i v e information. S p e c t r a l and chromatographic evidence not being a v a i l a b l e t o these e a r l y workers, the melting p o i n t o f the product acquired a c e n t r a l r o l e i n the determination of p u r i t y . Ways of drying o f 1,1,2,2-tetraphenylethanol however were r a r e l y s p e c i f i e d and o c c l u s i o n o f t r a c e s o f s o l v e n t can be c r i t i c a l i n melting p o i n t determinations. Once these p o i n t s a r e taken i n t o account, an i d e a o f the d i f f i c u l t i e s arising - 102 - i n a s s e s s i n g previous r e s u l t s becomes c l e a r . 142 Wegler's claim t h a t as much as 20% benzopinacol and hydrocarbon can be present as the by-products o f the i n s o l a t i o n experiment seems t o be g r o s s l y exagerated. Wegler must have expected t o observe t h e formation of benzopinacol i n view o f the reported behaviour o f benzophenone on i n s o l a t i o n i n the presence o f v a r i o u s hydrogen donors; i t i s however s u r p r i s i n g t h a t Paterno c a t e g o r i c a l l y excluded the formation o f benzopinacol or any other 119 by-products i n t h i s case benzopinacol formation. s i n c e he had reported so many examples o f A p o s s i b l e explanation could be t h a t any benzopinacol or tetraphenylethane formed i n Paterno's experiments was destroyed by b o i l i n g the r e a c t i o n product i n benzene before examination. I n our hands, b o i l i n g a toluene s o l u t i o n containing an 1:1 molar mixture o f benzopinacol and 1,1,2,2-tetraphenylethane f o r 3 h r s r e s u l t e d i n the t o t a l disappearance o f s t a r t i n g m a t e r i a l s and formation of 1,1,2,2-tetraphenylethanol with t r a c e s of benzophenone and benzhydrol as by-products. 147 Banchetti claimed to have i s o l a t e d benzopinacol out o f h i s own i n s o l a t i o n experiment, by c o n s i d e r a b l y reducing the volume o f the mother l i q u o r s o f r e c r y s t a l l i z a t i o n o f the crude 1,1,2,2-tetraphenylethanol. 147 d i d not however s p e c i f y t h e percentage o f benzopinacol i s o l a t e d . He On the other hand, the evidence f o r the p i n a c o l formation (behaviour under the microscope) i s open to question. 147 Banchetti'a p u r i f i c a t i o n technique should a l s o be considered with great c a u t i o n s i n c e 1,1,2,2-tetraphenylethanol i t s e l f i s not very s t a b l e a t 150 high temperatures, decomposing by cleavage o f the ethane C-C bond. The reported by-products, benzopinacol and 1,1,2,2,-tetraphenylethane are very s o l u b l e i n s o l v e n t s l i k e acetone and chloroform even a t room temperature; i t should t h e r e f o r e be adequate t o wash and then r e c r y s t a l l i z e the product from one o f these s o l v e n t s . I n our hands, p u r i f i c a t i o n o f an 1,1,2,2- tetraphenylethanol sample (already r e c r y s t a l l i z e d from chloroform and benzene) using B a n c h e t t i ' s method ( b o i l i n g with cyclohexanol) d i d not s i g n i f i c a n t l y - 103 - a f f e c t the m.pt. or the l . r . spectrum of the m a t e r i a l recovered. however I n t e r e s t i n g to n o t i c e t r a c e s of (CgHg) 2 C 0 C H I t was ^ 6 5^ 2 ^ 2 P r e s e n t * n cyclohexanol ( t . l . c . on s i l i c a , benzene) probably r e s u l t e d from p a r t i a l thermal decomposition temperatures. of the tetraphenylethanol a t r e l a t i v e l y high I n one of our i n i t i a l i r r a d i a t i o n s o f neat benzophenone and diphenylmethane, crude product obtained was r e c r y s t a l l i z e d from benzene and chloroform only; t . l . c . on s i l i c a , 13 C F.T. n.m.r. and evidence however showed the product to be pure. 1 H F.T. n.m.r. No benzopinacol or 1,1,2,2- tetraphenylethane were detected which d i r e c t l y c o n t r a d i c t s B a n c h e t t i ' s assertions. The long arguments concerning melting p o i n t s demonstrate the shortcomings of t h i s p a r t i c u l a r c r i t e r i o n of p u r i t y ; i t i s worth noting t h a t 1,1,2,2-tetraphenylethanol prepared v i a ground s t a t e chemistry has a l s o given • ,. . . . n-,,-0 varying melting p o i n t s , 235 , 232°-233°, 144 231°-232.5°, 148 141 „,„o _-,-,o 146 -...o . . o 149 _o 143 232 -233 , 243 -244 , 236.5 , n 232.5°-233°. 145 I t seems q u i t e l i k e l y t h a t the compound e x i s t s i n two isomeric forms, p o s s i b l y c i s o i d and t r a n s o i d conformational isomers, having d i f f e r e n t m.pts.; supports t h i s p r o p o s i t i o n . F.T. n.m.r. evidence The v a r y i n g m.pts. recorded could thus r e s u l t from v a r y i n g proportions of the two i s o m e r i c forms. On the b a s i s of the r e s u l t s obtained from the model r e a c t i o n , one would expect the i r r a d i a t i o n of an aromatic bisketone i n the presence of an equimolar amount of a bisbenzylbenzene to y i e l d a l i n e a r polymer containing at l e a s t 95% tetraphenylethanol u n i t s i n the backbone. By analogy with the model r e a c t i o n the polymer formation would be expected to be slow and the product would be expected to contain a s m a l l percentage of benzopinacol tetraphenylethane u n i t s . and The formation of these two types of u n i t would be chain propagating r a t h e r than chain terminating and indeed no r e a c t i o n s which would be chain terminating were observed i n t h i s study of the model r e a c t i o n , apart from the formation of t r a c e s of biphenyl which presumably arose from - 104 - r e a c t i o n o f benzophenone with the s o l v e n t . F i n a l l y , c r o s s l i n k i n g and/or branching might occur v i a the t e r t i a r y hydrogen i n the tetraphenylethanol and tetraphenylethane types of u n i t of the proposed polymer. o For the a c t u a l polymer forming r e a c t i o n , m-dibenzylbenzene and mA and p-dibenzylbenzenes were s e l e c t e d as appropriate monomers. Benzene was chosen as the s o l v e n t . 2.3.b. 2.3.b.i. S y n t h e s i s of monomers D i s c u s s i o n of p o s s i b l e routes The preparation, p u r i f i c a t i o n and c h a r a c t e r i z a t i o n of m-dibenzoylbenzene has already been described i n s e c t i o n 2 . 2 . j . i l l . The preparation of a r y l a l k y l a t e d aromatic hydrocarbons has been the s u b j e c t o f many r e p o r t s . Zincke obtained 1,4-, 1,2-dibenzylbenzene and diphenylmethane by the a c t i o n o f m e t a l l i c z i n c on a mixture of b e n z y l c h l o r i d e and b e n z e n e . S i m i l a r r e s u l t s were obtained by T h i e l e and Balhom, who 152 reacted benzene with formaldehyde i n the presence of s u l p h u r i c a c i d . Huston and Friedmann showed t h a t the aluminium c h l o r i d e c a t a l y s e d r e a c t i o n of b e n z y l c h l o r i d e with benzene gave a complex mixture from which they i s o l a t e d diphenylmethane, anthracene, anthraquinone, a l i t t l e 1,2-dibenzylbenzene and 1,4-dibenzylbenzene, m.pt. 85°-86° ( a l c o h o l ) . Japanese workers reported the s y n t h e s i s of p-phenylenedibenzyl i n a very impure s t a t e (melting point range: 20°) by s t i r r i n g a mixture of benzene and b e n z y l c h l o r i d e i n t e t r a c h l o r o e t h a n e with z i n c powder under n i t r o g e n ; 154 the impure m a t e r i a l was found to be photoconductive. Profft, Drechsler and Oberender,^^ obtained low y i e l d s of 1,4-dibenzylbenzene (m.pt. 86.5°) from the r e a c t i o n of tetrachlorodurene with benzene under F r i e d e l - C r a f t s conditions. - 105 - Pure 1,4-dibenzylbenzene, m.pt. 84°-85° (acetone) was prepared by the one-step h y d r o l y s i s and decarboxylation of C g H g - O ^ - p - ( C H - C H ( C C l ) C g H ) ; g 4 3 156 5 the same compound was a l s o obtained from the a c t i o n of N^/KOH a t over 190° 156 on 1,4-(p-bromobenzoyl)benzene. P o s s i b l e routes f o r alkane preparation from the corresponding carbonyl 157 compound i n c l u d e the Clemensen reduction and the Wolff-Kishner r e d u c t i o n . The Clemensen reduction i n v o l v e s t r e a t i n g the carbonyl compound with amalgamated z i n c and concentrated HC1, y i e l d i n g hydrocarbon as the main product, but a l s o v a r i a b l e q u a n t i t i e s of the secondary case of ketones) and unsaturated substances. do not give s a t i s f a c t o r y r e s u l t s ; a l c o h o l s ( i n the P u r e l y aromatic ketones however p i n a c o l s and resinous products o f t e n 157 predominate. The Wolff-Kishner reduction a f f o r d s hydrocarbons upon heating the corresponding hydrazone or semicarbazone of the carbonyl compound with 157 158 potassium hydroxide or with sodium ethoxide i n a s e a l e d tube. ' The Huang-Minion modification of the r e a c t i o n (formation and decomposition hydrazone i n one r e a c t i o n v e s s e l ) i s experimentally more convenient of the and reported to proceed i n b e t t e r y i e l d . F i n a l l y a r y l a l k a n e s may a l s o be s y n t h e s i z e d by reduction of the 158 corresponding a r y l a l k y l h a l i d e s or a r y l a l k y l t o s y l e s t e r s . This synthetic route would i n v o l v e reduction of the corresponding carbonyl compound to the a l c o h o l , c o n v e r s i o n of the a l c o h o l to the a r y l a l k y l h a l i d e or to the t o s y l e s t e r , and f i n a l l y reduction with an e f f i c i e n t reducing agent, such as LiAlH . 1 5 8 4 2.3.b.ii. S e l e c t i o n of routes I n our hands, attempts to prepare m-dibenzylbenzene by reducing the corresponding dicarbonyl compound, m-dibenzoylbenzene v i a e i t h e r the Wolff-Kishner reduction (Huang-Minion m o d i f i c a t i o n ) , or the Clemensen reduction gave u n s a t i s f a c t o r y r e s u l t s . A mixture of products was obtained i n the l a t t e r - 106 - case which was found d i f f i c u l t t o separate, whereas the former route r e s u l t e d i n the formation o f dark i n t r a c t a b l e t a r s . 154 R e p e t i t i o n o f the Japanese workers' experiment yielded a small quantity of a mixture comprising a t l e a s t 10 components ( t . l . c . on s i l i c a , benzene). The f a i l u r e t o obtain the arylalky.Lated hydrocarbons by the above d e s c r i b e d routes l e d to the i n v e s t i g a t i o n o f a l t e r n a t i v e syntheses o f 1,4and 1,3-dibenzylbenzene, shown s c h e m a t i c a l l y i n Figure 2.53. I n a d d i t i o n to these routes once could a l s o attempt the p r e p a r a t i o n o f the dibromide d e r i v a t i v e of the bisbenzhydrol by t r e a t i n g i t w i t h constant b o i l i n g HBr followed by reduction using an e f f i c i e n t H source i n an appropriate s o l v e n t . Another a t t r a c t i v e i d e a would be t o u t i l i s e a r e a c t i o n reported by G r i n t e r and Mason; they obtained triphenylmethane by r e f l u x i n g t r i p h e n y l 159 c a r b i n o l i n formic a c i d . of benzhydrol I f t h i s process could be extended t o the conversion t o diphenylmethane, an easy s y n t h e s i s o f the r e q u i r e d monomers might be a v a i l a b l e from bisbenzhydrols (Figure 2.54). The experimental d e t a i l s o f these v a r i o u s attempts to s y n t h e s i z e the r e q u i r e d monomers a r e given below. i n Figure 2.53. The steps r e f e r r e d t o a r e those i n d i c a t e d - 107 - H ^ H H II B OH CI OH F/vi CI ( C iii V 0 O 0 = S = 0O 00== SS = =0 —<0H H H H MgCl 6 6o H MgCl v IE CH CH iv H H -?-©-r H vii) > H H H H B II OH OH / / v iv i) iii E v). H H H MgBr S0C1 Key i solvent (v)- H 2 °° r ""2™" ^ ^ (ii) ^ iii) ^ (g) chloride O R A Q LiAlH Mg solvent (iv) H a B H f v i n . OH" Figure 2.53 4 solvent 5 > (v) 108 Q Q H II HCOOH ^ OH H H Figure 2.54 Q - 109 - Experimental Step A; Magnesium turnings (48.60g) i n a 3 necked round bottom f l a s k f i t t e d with a mechanical s t i r r e r , p r e s s u r e e q u a l i z i n g dropping f u n n e l , double s u r f a c e r e f l u x condenser, nitrogen gas i n l e t rjid thermometer w e l l were covered with 1.8 l i t r e s f r e s h l y d i s t i l l e d THF. The: dropping funnel was f i l l e d with dry bromobenzene (314.00g) and a few drops were added with e f f i c i e n t s t i r r i n g . The r e a c t i o n s t a r t e d smoothly a f t e r about 5 mins, without the use of c a t a l y s t . Addition of bromobenzene was r e g u l a t e d by monitoring the pot which was maintained between 70°-8o°. temperature The s o l u t i o n got p r o g r e s s i v e l y darker and was dark brown a f t e r about 2h h r s when most of the magnesium had been consumed. A f t e r a d d i t i o n of bromobenzene had f i n i s h e d , pot contents were s t i r r e d f o r a f u r t h e r 1 hr period, a t the end of which temperature to 30°. Slow a d d i t i o n of terephthalaldehyde (67.Og) on 0.7 l i t r e THF was s t a r t e d with e f f i c i e n t s t i r r i n g , taking care to maintain the pot below 40°. dropped then temperature A f t e r a l l the terephthalaldehyde s o l u t i o n had been added, the pot contents were dark red. The mixture was b o i l e d under r e f l u x f o r 2 h r s , cooled to room temperature, and c a r e f u l l y poured i n t o ice/water (2.5 l i t r e s ) a c i d i f i e d with HC1 and s t i r r e d f o r 1 hr. The organic l a y e r was separated and the water l a y e r was e x t r a c t e d twice with benzene (2 x 300 m i s ) . The combined THF/benzene s o l u t i o n s were d r i e d over anhydrous magnesium sulphate and f i n a l l y v o l a t i l e s o l v e n t s were removed under vacuum to give l l 6 g of crude product (80%). R e c r y s t a l l i z a t i o n s from ethanol/water (75:25) afforded 70% o f pure product (lit. V max : ) 1 6 0 171°) 3 2 7 0 1280, 1240 ' ( t . l . c . on s i l i c a , benzene, chloroform), m.pt. 169°-170° d i s p l a y i n g the f o l l o w i n g i n f r a r e d absorptions (KBr d i s c , 3 0 8 0 ' 3 0 4 0 ' 3 o 2 0 ' 2 9 0 0 ' 1 6 0 0 ' 1 5 L 0 ' (doublet), 1200, 1185, 1155, 1120, 870, 830, 790, 740, 700, 635, 570, 495 1 cm" . 1 4 9 0 ' L 4 5 0 ' 1 4 2 °' 1 3 4 0 ' 1080, 1030, 1020, 1010, 920, - 110 - The r e a c t i o n d i d not proceed i n ether s o l v e n t due to the low solubility of terephthalaldehyde i n t h a t medium. Step B; p-Bisbenzhydrol (40g) obtained from step A and f r e s h l y d i s t i l l e d t h i o n y l c h l o r i d e (180 mis) were heated under r e f l u x f o r 2 h r s . The excess o f t h i o n y l c h l o r i d e was d i s t i l l e d o f f under reduced p r e s s u r e and 100 mis of a c e t i c a c i d p l u s 2 drops of concentrated h y d r o c h l o r i c a c i d were added. The white s o l i d which p r e c i p i t a t e d was f i l t e r e d o f f and t r e a t e d again with 60 mis of g l a c i a l a c e t i c a c i d p l u s two drops of concentrated h y d r o c h l o r i c a c i d . The s o l u t i o n was b o i l e d with d e c o l o u r i z i n g c h a r c o a l , f i l t e r e d hot and the c o l o u r l e s s f i l t r a t e allowed to c o o l . Cooling caused the p r e c i p i t a t i o n of the c o l o u r l e s s d i c h l o r i d e which was f i l t e r e d and d r i e d by pumping under vacuum for 48 hrs a t 60°. m.pt. 190° 1600, (lit. 1 6 1 White needles were obtained from chloroform/hexane, 197°-198°), \ > max (KBr d i s c ) : 3080, 3060, 3035, 2960, 1585, 1510, 1500, 1450, 1425, 1345, 1260, 1225, 1120, 1080, 1030, 1 1020, 1000, 835, 785, 735, 700, 635, 615, 495 cm" . Steps C,E: p-Bisbenzhydrylchloride (lO.Og) obtained from step B were slowly added under nitrogen to 200 mis f r e s h l y d i s t i l l e d THF and magnesium turnings (5.0g) with e f f i c i e n t s t i r r i n g . Reaction was allowed to proceed f o r 1 hr and then h a l f of the r e s u l t i n g s o l u t i o n was s t i r r e d with 100 mis isopropanol f o r 8 h r s , whereas the other h a l f was poured i n t o 500 mis of i c e / w a t e r mixture and s t i r r e d f o r 5 h r s . Both r e a c t i o n s gave l a r g e l y p-bisbenzhydrol, unreacted p-bisbenzhydrylchloride and a few other minor products but no p-bisbenzylbenzene was isolated. Step F: p-Bisbenzhydrol (lO.Og) obtained from step A was d i s s o l v e d i n f r e s h l y d i s t i l l e d p y r i d i n e (300 mis) and toluene p-sulphonylchloride (10.Og) was slowly added. A f t e r 3 hrs of s t i r r i n g under nitrogen, s o l u t i o n heated to 60° f o r 1 hr and then cooled to room temperature. was Addition of 0.5 l i t r e s of water caused the p r e c i p i t a t i o n of a yellow o i l which was - Ill - separated and pumped under vacuum f o r 48 h r s . was Resulted resinous m a t e r i a l found to be a mixture o f a t l e a s t 8 components by t . l . c . on s i l i c a (benzene). Most of i t d i s s o l v e d i n hot water and stayed i n s o l u t i o n even on cooling t o 5°. P r e p a r a t i o n o f p-bisbenzhydrylbromide: p-Bisbenzhydrol (20g) obtained from step A was slowly added t o constant b o i l i n g HBr (400 mis) and the r e s u l t a n t brown s o l u t i o n was b o i l e d under r e f l u x f o r 3 h r s . Most o f the water was taken o f f by d i s t i l l a t i o n under reduced p r e s s u r e , the residue was d i s s o l v e d i n 500 mis o f benzene and b o i l e d using a Dean-Stark apparatus. When n e a r l y a l l the water had come o f f , benzene s o l u t i o n was d r i e d over anhydrous magnesium sulphate and then volume o f benzene was reduced to 100 mis. Addition o f 600 mis o f hexane caused the p r e c i p i t a t i o n o f a m a t e r i a l which was f i l t e r e d and d r i e d by pumping under reduced pressure f o r 24 h r s . (85%) thus obtained, m.pt. 105°-108° ( l i t . bromine by Lassaigne's t e s t ; 1490, 775 ^ m a x 1 6 0 The s o l i d 112.5°), was found t o contain (nujol mull): 3080, 3040, 3020, 1510, 1420, 1300, 1250, 1200, 1165, 1120, 1075, 1030, 1020, 1000, 830, 1 (doublet), 730, 700, 675, 605, 495 (doublet) cm" . m a t e r i a l by t . l . c . on s i l i c a Examination o f the (benzene) revealed the presence o f one component only. Attempted reduction of the p-bisbenzhydrylbromide: from the previous preparation NaAlH (OCH CH OCH ) 2 2 2 3 2 The bromide obtained (15.0g) was d i s s o l v e d i n dry benzene (200 m i s ) ; ( V i t r i d e ) i n benzene (10% excess) was added and the mixture was r e f l u x e d f o r 2 h r s . A f t e r cooling and d e s t r u c t i o n o f t h e excess V i t r i d e with water, the benzene l a y e r was separated, d r i e d over anhydrous magnesium sulphate and the s o l v e n t was removed under reduced p r e s s u r e t o y i e l d a w h i t i s h powder (13.Og), m.pt. 185°-194° which was found to be i n s o l u b l e i n hot ethanol o r c o l d hexane. I t was d i s s o l v e d i n toluene and p r e c i p i t a t e d by slow a d d i t i o n o f 60°-80° petroleum e t h e r . The product - 112 - thus obtained was d r i e d by pumping under vacuum f o r 24 hrs a t 40 examined by i . r . spectroscopy (Nujol m u l l , v : m a x and 3080, 3060, 3025, 1600, 1580, 1510, 1490, 1450, 1420, 1375, 1160, 1110, 1075, 1030, 1020, 790, 700, 625 cm ^) . form; 730, I t was not p o s s i b l e to obtain the product i n a c r y s t a l l i n e i t appeared to be a r e l a t i v e l y high molecular weight m a t e r i a l . Attempted reduction of the p-bisbenzhydrol i n formic a c i d : p-Bisbenzhydrol (35,Og) obtained from step A was added to a l a r g e volume (2 l i t r e s ) of 98% formic a c i d a t 60°. A f t e r each a d d i t i o n vigorous e f f e r v e s c e n c e was and the evolved gases gave a white p r e c i p i t a t e with lime water. observed A f t e r the o a d d i t i o n was complete, the r e s u l t a n t brown s o l u t i o n was heated a t 60 for 6 h r s with s t i r r i n g and product was p r e c i p i t a t e d a f t e r c o o l i n g by pouring the s o l u t i o n i n t o 5 l i t r e s of water. A yellow o i l (22.Og) was obtained which was separated, d r i e d by pumping under reduced p r e s s u r e a t 60° f o r 24 h r s and examined by i . r . spectroscopy (contact f i l m , v 2920, 1740, 1510, 1490, 1450, 1370, 1160, 1080, 1030 -1 850, 815, 760, 730, 700, 630 c m . m a x : 3060, 3040, ( d o u b l e t ) , 945, 920, T h i s yellow o i l was d i s t i l l e d under vacuum (15-20 mmHg, 90°-260°) to y i e l d a y e l l o w i s h o i l which c r y s t a l l i z e d slowly on standing i n a sample b o t t l e . The i . r . spectrum was the same as the one of the yellow o i l before d i s t i l l a t i o n . essentially The high b o i l i n g f r a c t i o n which remained i n the d i s t i l l a t i o n pot was e x t r a c t e d with chloroform and p r e c i p i t a t e d with 60°-80° petroleum ether to y i e l d a w h i t i s h powder. Neither the yellow nor the white m a t e r i a l appeared to be the d e s i r e d product. Step G: To m-dibenzoylbenzene (lOOg) (obtained as d e s c r i b e d i n 2 . 2 . j . i i i ) i n 2000 mis ethanol i n a 5 l i t r e round bottom f l a s k f i t t e d w i t h r e f l u x condenser was slowly added a s o l u t i o n of NaBH^ (45g) i n water (300 m i s ) . The r e s u l t a n t s o l u t i o n was r e f l u x e d f o r 5 h r s and cooled to room temperature. Following the d e s t r u c t i o n of unreacted borohydride with 5% a c e t i c a c i d s o l u t i o n , the volume of s o l v e n t was reduced to 200 mis. Addition of a l a r g e - 113 - volume of water caused the p r e c i p i t a t i o n of a white m a t e r i a l which was c o l l e c t e d on a f i l t e r , washed s u c c e s s i v e l y with water, 5% HC1 solution and 5% aqueous sodium carbonate s o l u t i o n , and then b o i l e d with a c t i v a t e d animal c h a r c o a l i n ethanol s o l u t i o n . The white s o l i d obtained a f t e r r e c r y s t a l l i z a t i o n s from ethanol/water (70:30) spectroscopy (KBr d i s c , v : (80%) was examined by i . r . 3300, 3060, 3020, 2880, 1600, 1490, 1475, max 1450, 1335, 1270, 1235, 1195, 1070, 1030, 1020, 950, 920 1 800, 780, 760, 740, 700 ( d o u b l e t ) , 600, 545 cm" , Step B': ( d o u b l e t ) , 830, m.pt. 151°-153°. was c a r r i e d out i n e x a c t l y the same way as s t e p B. A dark o i l was obtained i n 90% y i e l d which was examined by L a s s a i g n e ' s method and found to contain c h l o r i n e . obtained f o r step D*. film, v m : T h i s product was not p u r i f i e d but was used as The i n f r a r e d spectrum of the o i l was recorded (contact 3080, 3060, 3020, 2940, 1600, 1590, 1490, 1450, 1210, 1075, 1030, 1000, 830, 790, 760, 730, 700, 625, 580 Step D: LiAlH 4 was 1150, 1 cm" . (lOg, 0.26 mole) i n a 2 l i t r e 3 necked round bottom f l a s k f i t t e d with a mechanical s t i r r e r , p r e s s u r e e q u a l i z i n g dropping n i t r o g e n gas i n l e t and double s u r f a c e r e f l u x condenser s u l p h u r i c a c i d bubbler was funnel, l e a d i n g to a concentrated covered w i t h 1 l i t r e sodium d r i e d e t h e r ; the mixture was s t i r r e d v i g o r o u s l y f o r 30 mins and then cooled to 0° by means of an i c e / w a t e r / s a l t bath. p-Bisbenzhydrylchloride (32.7g, 0.1 mole) obtained from step B was shaken with e t h e r (0.5 l i t r e s ) and the r e s u l t e d s l u r r y s l o w l y added to the mixture i n the f l a s k with vigorous s t i r r i n g . of the mixture changed e v e n t u a l l y from grey to l i g h t green. was complete, the mixture was gently r e f l u x e d f o r 12 h r s . was The colour After addition The excess l i t h i u m aluminium hydride was destroyed by s u c c e s s i v e slow a d d i t i o n s of 162 water (lOg), 10% NaOH (15g) and water (30g), r e s u l t i n g i n the formation o f a granular p r e c i p i t a t e which was f i l t e r e d o f f , washed with two 200 mis p o r t i o n s o f benzene and destroyed by slowly adding i t i n t o a l a r g e volume of - 114 - 6N HC1 u n t i l a c l e a r s o l u t i o n was formed. The combined e t h e r e a l and benzene s o l u t i o n s were d r i e d over anhydrous magnesium sulphate and finally the s o l v e n t s were removed under reduced p r e s s u r e . was A whitish solid obtained which was r e c r y s t a l l i z e d s e v e r a l times from hot ethanol, b o i l e d with a c t i v a t e d animal c h a r c o a l and then r e c r y s t a l l i z e d s e v e r a l times from methanol and ethanol to give a y e l l o w i s h c r y s t a l l i n e m a t e r i a l impure b i s b e n z y l b e n z e n e (30%) , melting a t 83°-85° ( l i t . 1 4 0 1,4- 86°). Step D'; was c a r r i e d out i n the same way as step D apart from the a d d i t i o n of the m-bisbenzhydrylchloride which was e a s i e r than i n D, s i n c e the compound i s s o l u b l e i n e t h e r . Reaction afforded a brown o i l which was d i s t i l l e d from a short column, b o i l e d with a c t i v a t e d animal c h a r c o a l i n benzene and d i s t i l l e d again to y i e l d a yellow o i l ; the o i l was e s t a b l i s h e d by Lassaigne's t e s t . absence of c h l o r i n e from Finally a yellowish o i l was obtained by d i s t i l l a t i o n a t 220°-239°/14 mmHg ( l i t . 1 4 0 229°-231°/14 mmHg 226°-227°/19 mmHg). 2.3.b.ill. Reasons f o r f a i l u r e The r e s u l t s obtained i n 2 . 3 . b . i i i n d i c a t e t h a t the only s u c c e s s f u l step out of those shown i n F i g u r e 2.53 are A, G, B, and B', D and D'. Treatment of 1,4-bisbenzhydrol with constant b o i l i n g HBr d i d i n f a c t give a bromide, but reduction of the bromide with V i t r i d e (NaAlH (OCH CH OCH 2 2 2 i n benzene s o l u t i o n a t 80° r e s u l t e d i n the formation of polymeric m a t e r i a l s . On the other hand reduction of the corresponding d i c h l o r i d e with l i t h i u m aluminium hydride i n ether a t 0°-37° r e s u l t e d i n the formation of d i b e n z y l benzene. Both V i t r i d e and l i t h i u m aluminium hydride are e x c e l l e n t sources of hydride anion. The two s t a r t i n g m a t e r i a l s though bear d i f f e r e n t h a l i d e s u b s t i t u e n t s , bromine being b u l k i e r and not as s t r o n g l y attached to the carbon atom as c h l o r i n e . One could t h e r e f o r e r a t i o n a l i z e the d i f f e r e n c e i n behaviour between the two b e n z y l h a l i d e s by p o s t u l a t i n g p a r t i a l i o n i z a t i o n o f - 115 - the p-bisbenzhydrylbromide y i e l d i n g a s t a b l e carbenium ion which could then i n i t i a t e polymerization, by e l e c t r o p h i l i c a t t a c k on phenyl r i n g s . Alternatively the polymerization may have occurred during the formation o f the bromide; again a carbenium intermediate i s i n v o l v e d and t h i s might have attacked an aromatic r i n g o r been quenched by a hydroxy1 group. The treatment o f p-bisbenzhydrol with a l a r g e excess o f formic a c i d would appear t o have y i e l d e d the formate e s t e r and polymeric m a t e r i a l s , p o s s i b l y v i a the route shown i n Figure 2.55. The reasons why t r i p h e n y l c a r b i n o l i s converted t o triphenylmethane, whereas benzhydryl hydroxyl i s not r e p l a c e d 9 o HCOOH H H OH ^ H OH H + H„0 + HCOO OH HCOO H H OH ® OH formate e s t e r 9 H H OH polyether H 6D OH H Figure 2.55 - 116 - by hydrogen under the same conditions probably l i e I n a combination of s t e r l c and e l e c t r o n i c e f f e c t s . 2.3.b.vi. P u r i f i c a t i o n and proof o f s t r u c t u r e 1,4-dlbenzylbenzene was p u r i f i e d by two more r e c r y s t a l l l z a t l o n s from absolute ethanol, b o i l i n g with a c t i v a t e d animal c h a r c o a l I n benzene, sublimation a t 106°/0.01 mmHg, another r e c r y s t a l l i z a t i o n from ethanol and washing with the same s o l v e n t (10°). I t was then pumped under reduced p r e s s u r e (0.01 mmHg) a t 40° f o r 48 h r s . product, m.pt. T h i s procedure gave 20% of pure 85.8° (lit.^° 86°) i n b i g p l a t e s a n a l y z i n g as f o l l o w s : C 92.56, H 7.12; calculated for C 2 o H 1 8 C 92.98, H 7.02. was found t o give a s i n g l e spot on t . l . c . on s i l i c a v max (KBr d i s c ) : The substance (benzene, chloroform); 3070,. 3060, 3020, 2900 (doublet), 2430, 1950, 1915, 1600, 1595, 1510, 1490, 1450, 1430, 1415, 1335, 1320, 1210, 1175, 1160, 1105, 1 1030, 930, 855, 760, 725, 700, 600, 485, 460 cm" . to m/e = 258 corresponding to the parent i o n ; 121, 120, 90, 77. 1070, The mass spectrum extended main peaks a t 192, 167, 150, Lassaigne's t e s t on the sample showed the absence of chlorine, as d i d the mass spectrum. 1 The H n.m.r. spectrum i n CDCl^ ( i n t . TMS r e f . ) showed two peaks i n the aromatic hydrogen region (6 7.10, 6 6.96) and a s i n g l e t a t 6 3.80. 1,3-Dibenzylbenzene was p u r i f i e d by f u r t h e r d i s t i l l a t i o n s between 220°-235°/15 mmHg and f i n a l l y by a d i s t i l l a t i o n a t 140°/3 mmHg under nitrogen. A c o l o u r l e s s o i l was obtained, a n a l y z i n g as f o l l o w s : (calculated for C 2 Q H 1 Q C 92.88, H 7.02); chloroform) gave a s i n g l e spot; v max C 92.73, H 7.23 t . l . c . on s i l i c a (contact f i l m ) : (benzene, 3100, 3080, 3020, 2900, 2840, 1940, 1870, 1800, 1600, 1490 (doublet), 1445 3040, (doublet), 1 1090, 1075, 1030, 1000, 770, 750, 730, 700, 600, 550, 460, 430 cm" . L a s s a i g n e ' s t e s t on the product showed the absence of c h l o r i n e . spectrum extended to m/e = 258, corresponding to the parent i o n ; The mass main - 117 - peaks a t 181, 180, 168, 166, 153, 91, 2.3.c. 77. Polymerization runs 2.3.C.I. A preliminary Investigation 1,4-Dlbenzylbenzene (1.2918g, 0.005 mole) and 1,3-dlbenzoylbenzene (1.4316g, 0.005 mole) were d i s s o l v e d i n benzene (50 mis, 0.2M to both reagents) i n a c y l i n d r i c a l Pyrex v e s s e l . with r e s p e c t The c l e a r , c o l o u r l e s s s o l u t i o n was nitrogen streamed f o r 30 mins, the tube was q u i c k l y stoppered and i r r a d i a t e d a t 350 nm f o r 24 h r s . A b r i g h t yellow colour developed a t an e a r l y stage during the i r r a d i a t i o n and a t r a c e of a white p r e c i p i t a t e appeared a t the bottom of the tube. Benzene s o l v e n t was removed by f r e e z e - d r y i n g and a yellow m a t e r i a l was q u a n t i t a t i v e l y recovered showing both hydroxyl and benzophenone-type carbonyl bands i n the i . r . region. The presence of the strong carbonyl band was a t t r i b u t e d to e i t h e r unreacted monomer or to low molecular weight m a t e r i a l formed (telomer of poly(1,1,2,2-tetraphenylethanol)); thus the e a r l i e r expectation t h a t the polymerization r e a c t i o n might be a slow one was confirmed. On the other hand, the presence of the hydroxy1 band i n d i c a t e d t h a t , as expected, hydrogen a b s t r a c t i o n was t a k i n g p l a c e and t h a t the benzophenone-type k e t y l r a d i c a l of m-dibenzoylbenzene was i n f a c t y i e l d i n g products e i t h e r by d i f f u s i o n out of the cage and d i m e r i z a t i o n or by in-cage combination with the r a d i c a l r e s u l t i n g from p-bisbenzylbenzene. The i . r . spectrum of the y e l l o w i s h powder d i s p l a y e d the f o l l o w i n g absorptions (KBr d i s c , v c a . 3400 (broad), 3080, 3050, 3020 ( s t r o n g ) , 2900, 1655, 1480, 1445, 1420, 1320, 1275, 1160, 1 (doublet), 700, 640, 620, 555 cm . polybenzopinacol 1090, 1030, 1000, :) 3540, 3520 - 1600, 1580, 845, 830, 780, 1510, 740 Comparison with the i . r . spectrum of M (prepared by i r r a d i a t i o n of m-dibenzoylbenzene i n isopropanol/benzene, see Table 2.2), s y n t h e s i z e d by Andrews and 64 Feast (KBr d i s c , v : max 3560, 3500 - c a . 3400 (broad), 3060, 3020 (weak), - 118 - 2960 ( p o s s i b l y occluded s o l v e n t o r Incorporated iaopropanol m o i e t i e s ) , 1600, 1560, 1495, 1450, 1420, 1330, 1265, 1155, 1100, 1030, 920, 790, 765, 750, 1 700, 645, 615 cm ) r e v e a l e d t h a t the two s p e c t r a d i f f e r i n many regions; d i f f e r e n c e s i n the r e l a t i v e i n t e n s i t i e s o f t h e 0-H and aromatic C-H absorptions were evident, t h e yellow m a t e r i a l having a much stronger C-H aromatic group o f peaks. were a l s o observed. D i f f e r e n c e s i n the f i n g e r p r i n t region o f t h e spectrum • Washing the m a t e r i a l obtained w i t h c o l d hexane i n order t o e l i m i n a t e any unreacted bisbenzylbenzene, followed by drying under vacuum, d i d not a l t e r the colour, weight or the s o l i d s t a t e i . r . spectrum."* 2.3.c.ii. Examination of the e f f e c t of concentration The i r r a d i a t i o n s shown i n Table 2.7 were c a r r i e d out i n the same way as i n the previous experiment ( c y l i n d r i c a l Pyrex tubes, nitrogen streaming followed by s t o p p e r i n g , i r r a d i a t i o n a t 350 nm). I n a l l c a s e s , s o l u t i o n went b r i g h t yellow and a t r a c e o f a white p r e c i p i t a t e was observed a t the bottom of the tube; i t was separated from the yellow s o l u t i o n by f i l t r a t i o n , washed with a few drops o f c o l d benzene, d r i e d under vacuum and weighed; i n a l l cases i t amounted t o c a . 5% o f the t o t a l weight o f the two r e a c t a n t s used, and was i n s o l u b l e i n a wide range o f l a b o r a t o r y s o l v e n t s . Benzene s o l v e n t was removed from yellow s o l u t i o n s by f r e e z e - d r y i n g and yellow s o l i d s were obtained which were d r i e d by pumping under reduced p r e s s u r e and examined by i . r . spectroscopy experiments (KBr d i s c s ) . The s p e c t r a obtained from A, B and C were very s i m i l a r and comparable t o the i . r . spectrum of t h e yellow product obtained .from the 24 h r s i r r a d i a t i o n ( 2 . 3 . c . i . ) , but the benzophenone-type carbonyl band was much weaker, i n d i c a t i n g t h a t longer i r r a d i a t i o n times r e s u l t e d i n i n c r e a s e d comsumption o f a t l e a s t one o f the monomers (m-dibenzoylbenzene). The r e l a t i v e i n t e n s i t i e s o f the carbonyl band i n the s p e c t r a o f the t h r e e products was roughly the same, i n d i c a t i n g t h a t i n the concentration range 0.1M-1M r e a c t i o n r a t e (as monitored by - 119 - Table 2.7 I r r a d i a t i o n s o f egulmolar s o l u t i o n s of m-dlbenzoylbenzene/m- and pdibenzylbenzene of v a r y i n g concentrations Expt. Bisketone Alkane Solvent Total concn. Irradiation time A m-dibenzoylbenzene 0.7158g 0.0025 mole p-bisbenzyl benzene 0.6459g 0.0025 mole benzene 50 mis 0.1M 240 h r s B m-dibenzoylbenzene 0.7158g 0.0025 mole p-bisbenzyl benzene 0.6459g 0.0025 mole benzene 25 mis 0.2M 240 h r s C m-dibenzoylbenzene 0.7158g 0.0025 mole p-bisbenzyl benzene 0.6459g 0.0025 mole benzene 12.5 mis 0.4M 240 h r s D m-dibenzoylbenzene 2.860g 0.01 mole m-bisbenzyl benzene 2.580g 0.01 mole benzene 20 mis 1M 240 h r s carbonyl consumption) was not d r a m a t i c a l l y changing. A t y p i c a l i . r . spectrum of the white i n s o l u b l e p r e c i p i t a t e obtained from experiments A, B and C d i s p l a y e d the f o l l o w i n g absorptions (KBr d i s c , V max :) 3550 (very s t r o n g ) , 3090, 3060, 3020, 2960, 1660, 1600, 1580, 1495, 1475, 1445, 1420, 1340-1270 (broad), 1260, 1175, 1160, 1100, 1035, 1020, 960, 925, 910, 845, 805, 775, 745, 710, 700, 680, 645, 635, 610, 570, 540, 470 1 cm" . The i . r . spectrum of the white i n s o l u b l e p r e c i p i t a t e obtained from experiment D was not d i f f e r e n t from the one d e s c r i b e d i n the preceding paragraph. The benzene-soluble m a t e r i a l was d r i e d by pumping under reduced pressure following removal of s o l v e n t ; a p o r t i o n of i t was p r e c i p i t a t e d from benzene/60°-80° petroleum ether and a white powder was thus obtained a n a l y z i n g - 120 - as f o l l o w s : C 85.98, H 5.92. recorded (KBr d i s c , v ): The l . r . spectrum of the white powder was 3550, 3520 - ca. 3400 (broad), 3080, 3040, max 3020, 2920-2845 (possibly occluded petroleum e t h e r ) , 1655 (strong), 1600, 1580, 1495, 1445, 1420, 1320, 1275, 1175, 1160, 1140, 1045, 1030, 1000, 1 845, 815, 750, 700, 640, 620 cm . 920, The carbonyl peak appeared c o n s i d e r a b l y stronger than i n the i . r . s p e c t r a of the yellow products obtained from experiments A, B and C, i n d i c a t i n g t h a t consumption of carbonyl i n the r e a c t i o n between the two meta isomers was q u a l i t a t i v e l y slower than i n the r e a c t i o n between m-dibenzoylbenzene and p-dibenzylbenzene. Another p o r t i o n (2.0g) of the crude yellow product i s o l a t e d from experiment D was d i s s o l v e d i n benzene (10 m i s ) , nitrogen streamed, i r r a d i a t e d a t 350 nm f o r 435 h r s . and Again, a white s o l i d p r e c i p i t a t e d during i r r a d i a t i o n (ca. 6 % ) , which d i s p l a y e d an i . r . spectrum s i m i l a r to those already d e s c r i b e d f o r the white p r e c i p i t a t e s p r e v i o u s l y obtained. The yellow benzene s o l u t i o n was separated, and the s o l v e n t was removed by f r e e z e drying y i e l d i n g a yellow s o l i d which was p r e c i p i t a t e d from benzene/60°-80° petroleum e t h e r . The white powder obtained from p r e c i p i t a t i o n was examined by i . r . spectroscopy; a very s m a l l absorption a t c a . 1660 cm * was i n d i c a t i n g t h a t most of the m-dibenzoylbenzene had been consumed. phase i . r . spectrum of t h i s product c e l l ) showed a s i n g l e t a t 3560 cm 1 observed The solution (CCl^ s o l v e n t , s a t u r a t e d s o l u t i o n , 0.1 mm (0-H) , a doublet a t 3060-3020 cm (aromatic C-H), and a broad s i n g l e t a t 2920 cm 1 ( a l i p h a t i c C-H); the r e l a t i v e i n t e n s i t i e s of the 0-H and aromatic C-H peaks were roughly the same (aromatic C-H s l i g h t l y stronger). The s o l u t i o n phase i . r . spectrum of 64 polybenzopinacol M (see Table 2.2).examined under the same c o n d i t i o n s showed - a s i n g l e t a t 3560 cm * and a shoulder a t 3600 cm * (0-H), a very broad absorption between 3500-3400 cm * (hydrogen bonded 0-H), two s i n g l e t s a t 3060-3030 cm 1 (aromatic C-H), and a strong s i n g l e t a t 2960 cm 1 (aliphatic - 121 - C-H, p o s s i b l y due to occluded s o l v e n t ) . stronger than the two aromatic C-H The 0-H peak was c o n s i d e r a b l y peaks. The above p r e l i m i n a r y i n v e s t i g a t i o n s l e d to the c o n c l u s i o n t h a t the product obtained from the m-dibenzoylbenzene/m-dibenzylbenzene r e a c t i o n was not a polybenzopinacol (by comparative s o l i d s t a t e and s o l u t i o n phase i . r . evidence). T h i s was an encouraging r e s u l t and gave the i n c e n t i v e f o r a more thorough examination o f the p o l y a d d i t i o n r e a c t i o n . 2.3.c.iii. a) Large s c a l e experiments m-Dibenzoylbenzene/p-dlbenzylbenzene m-Dibenzoylbenzene (28.631g, 0.1 mole) and p-dibenzylbenzene 0.1 mole) were d i s s o l v e d i n sodium-dried benzene (500 mis, 0.4M to both r e a g e n t s ) . (25.834g, with r e s p e c t The s o l u t i o n was nitrogen streamed f o r 30 mins, the c y l i n d r i c a l Pyrex tube was q u i c k l y stoppered and i r r a d i a t e d a t 350 nm f o r 263 h r s . During i r r a d i a t i o n a white m a t e r i a l p r e c i p i t a t e d , remaining s o l u t i o n became b r i g h t yellow. whereas The white m a t e r i a l (A) was recovered by f i l t r a t i o n , washed with a l i t t l e c o l d benzene, d r i e d by pumping under reduced p r e s s u r e , weighed (KBr disc); (2.482g) and examined by i . r . spectroscopy spectrum obtained was e s s e n t i a l l y the same as the ones recorded for p r e v i o u s l y obtained white p r e c i p i t a t e s , already d e s c r i b e d i n 2 . 3 . c . i i i . The yellow s o l u t i o n separated y i e l d e d , following removal of s o l v e n t by f r e e z e - d r y i n g and pumping under reduced p r e s s u r e , a yellow m a t e r i a l (B) (51.980g) which was s p e c t r o s c o p i c a l l y examined ( i . r . , KBr d i s c ) ; i t was then r e d i s s o l v e d i n benzene (500 mis) and r e - i r r a d i a t e d f o r a f u r t h e r 144 h r s p e r i o d , y i e l d i n g another white p r e c i p i t a t e (C) (0.832g) and a yellow s o l i d (D) (51.140g). S o l i d (D) was then d i v i d e d i n t o 2 x 25g p o r t i o n s . were made: Two benzene s o l u t i o n s - 122- 25g o f s o l i d (D) I n 100 mis benzene 25g of s o l i d (D) I n 250 mis benzene The two s o l u t i o n s were r e - i r r a d l a t e d f o r 118 h r s , y i e l d i n g only s l i g h t white p r e c i p i t a t e s ( c a . 1% o f the weight o f s o l i d (D) used) and yellow s o l i d s (E) (from 25g (D)/100 mis CgH ) and (F) (from 25g (D)/250 mis C Hg). g $) g m-Dibenzoylbenzene/m-dlbenzylbenzene m-Dibenzoylbenzene (28.631g, 0.1 mole) and m-dibenzylbenzene (25.834g, 0.1 mole) were d i s s o l v e d i n sodium d r i e d benzene (500 mis, 0.4M with r e s p e c t to both r e a g e n t s ) . The s o l u t i o n was nitrogen streamed f o r 30 mins, the c y l i n d i c a l Pyrex tube was q u i c k l y stoppered and i r r a d i a t e d a t 350 nm f o r 263 h r s . During i r r a d i a t i o n a white m a t e r i a l p r e c i p i t a t e d , whereas remaining s o l u t i o n became b r i g h t yellow. The white m a t e r i a l (G) was recovered by f i l t r a t i o n , washed with a l i t t l e c o l d benzene, d r i e d by pumping under reduced p r e s s u r e , weighed (2.482g) and examined by i . r . spectroscopy (KBr d i s c ) ; spectrum obtained was the same as the ones recorded f o r p r e v i o u s l y obtained white p r e c i p i t a t e s , described i n 2 . 3 . c . i i i . The yellow s o l u t i o n separated y i e l d e d , following removal of s o l v e n t by freeze-drying and pumping under reduced p r e s s u r e , a yellow m a t e r i a l (H) (51.980g) which was s p e c t r o s c o p i c a l l y examined ( i . r . , KBr d i s c ) ; i t was then red!ssolved i n benzene (500 mis) and r e - i r r a d i a t e d f o r a f u r t h e r 144 h r s period, y i e l d i n g another white p r e c i p i t a t e ( I ) (1.044g) and a yellow s o l i d (J) (50.920g). S o l i d (J) was then d i v i d e d i n t o 2 x 25g p o r t i o n s . Two benzene s o l u t i o n s were made: 25g o f s o l i d (J) i n 100 mis benzene 25g of s o l i d (J) i n 250 mis benzene The two s o l u t i o n s were r e - i r r a d i a t e d f o r 118 h r s , y i e l d i n g only s l i g h t white p r e c i p i t a t e s , and yellow s o l i d s (K) (from 25g (J)/ICO mis CgHg) and (L) (from 25g (J)/250 mis CJH ). fi Following s p e c t r a l examinations, s o l i d s - 123 - (K) and (L) were combined, d i s s o l v e d i n 250 mis of benzene and r e - i r r a d i a t e d (350 ran) f o r 171 h r s , y i e l d i n g again very s l i g h t white p r e c i p i t a t e s (< 1% of the combined (K) + (L) weight) and a yellow benzene-soluble s o l i d (M) (48.932g). 2.3.d. C h a r a c t e r i z a t i o n of products A l l products obtained from experiments d e s c r i b e d i n 2 . 3 . c . i l l were d r i e d by pumping under reduced p r e s s u r e before examination. I . r . s p e c t r a (KBr d i s c s ) were the! same as the ones obtained for corresponding products d e s c r i b e d i n 2 . 3 . c . i and 2 . 3 . c . i i ; (D), ( E ) , spectra of (B), ( F ) , (H), ( J ) , ( K ) , ( K ) , ( L ) , (M), a l l d i s p l a y e d absorptions a t 1660 cm *, i n d i c a t i n g the presence of benzophenone-type carbonyl groups a t t r i b u t a b l e to e i t h e r unreacted monomer or low molecular weight polymer chains capped with aromatic carbonyl ends. Products (E) and (F) were combined, d i s s o l v e d i n benzene and the b r i g h t yellow s o l u t i o n s thus obtained were added dropwise excess of 60°-80° petroleum e t h e r with e f f i c i e n t s t i r r i n g . to a 10-fold A white s o l i d (N) was obtained i n 89% y i e l d , which was c o l l e c t e d and d r i e d under vacuum. (N) was r e d i s s o l v e d i n benzene forming a b r i g h t yellow s o l u t i o n . Second p r e c i p i t a t i o n from 60°-80° petroleum e t h e r afforded a white s o l i d (0) (80%) which was d r i e d by pumping under vacuum f o r 6 days at 40°; follows: C 86.54, H 7.18 i n the i . r . spectrum. (CB^Clg s o l v e n t ) . i t analyzed as (0) s t i l l d i s p l a y e d a weak absorption a t 1660 cm 1 I t d i d not form f i l m s on c a s t i n g on a c l e a n Hg s u r f a c e Small, b r i t t l e f i b r e s were drawn out of the melt; it became tacky between 188°-207° and melted between 207°-220°. S i m i l a r l y , product (M) was p r e c i p i t a t e d from benzene/60°-80° petroleum e t h e r y i e l d i n g a powder (P) (80%) which was d r i e d i n the same way as ( 0 ) . No f i l m s were obtained, but s m a l l , b r i t t l e f i b r e s were drawn out of the melt. (P) became tacky between 165°-177° and melted between 177°-192°. - 124 - The number average molecular weights o f the samples were determined by the i s o p i e s t i c method i n chloroform and are recorded below: n Total I r r time (hrs) Sample n M n Sample Total I r r time (hrs) M n (B) 263 2780 (H) 263 2030 (D) 407 3560 (J) 407 2130 (E) 525 3590 (K) 525 3320 (F) 525 3530 (D 525 3410 (0) 525 3690 (M) 525 3510 (P) 696 3670 Polymer c h a r a c t e r i z a t i o n was attempted by F.T. H n.m.r. spectroscopy (see Table 2.8). S p e c t r a o f samples (0) and (P) were run i n CDCl-j s o l v e n t ( e x t . TMS 64 reference) and compared to the s p e c t r a o f p-bisbenzylbenzene, p o l y p i n a c o l M, 1,1,2,2-tetraphenylethanol, benzopinacol and 1,1,2,2-tetraphenylethane. The f i r s t observation was the group o f peaks between 6 1.48 - 6 0.60 i n d i c a t i n g the presence of occluded s o l v e n t containing a l i p h a t i c C-H bonds, apparently petroleum ether. Polybenzopinacol M showed s i m i l a r peaks a t 6 1.61, 6 1.28. T h i s observation was somewhat s u r p r i s i n g s i n c e both (0) and (P) were pumped under reduced p r e s s u r e f o r a long time before the s p e c t r a were run, and t h a t should ether. for have e l i m i n a t e d any occluded v o l a t i l e m a t e r i a l s such as petroleum Evaporation o f a l a r g e volume o f the same batch o f petroleum e t h e r used p r e c i p i t a t i o n o f (0) and ( P ) , d i d not y i e l d any high b o i l i n g r e s i d u e s . Both (0) and (P) d i s p l a y e d peaks a t S 3.81 - 6 3.82 a s s i g n e d t o the methyl protons i n the -C H -CH -C H group, and peaks a t 6 4.80 - 6 4.81, assigned 6 4 2 g 5 to the t e r t i a r y hydrogen i n a 1,1,2,2-tetraphenylethane type o f u n i t . peaks a t 6 4.38 - 6 4.39 however could not be unequivocally Broad assigned t o any - 125 - Table 2.8 Comparative F.T. H n.m.r. s p e c t r a o f some phenylated substances (CDC1 s o l u t i o n s , e x t . TMS r e f . ) . 3 705 «.J9 3*2 Product (P) 'I 2 o** A^A_/VJA_ T-o2 Product (O) TOM 3 55 i-foi i-ag Polybenzopinacol M 64 6 96 p-Bisbenzylbenzene A 3-80 1 706 U-9o A 1,1,2,2-Tetraphenylethane 115 1A U_ 1,1,2,2-Tetraphenylethanol f I f f t i M PPm s c a l e i - 126 - s t r u c t u r e s i n c e the t e r t i a r y proton resonance i n 1,1,2,2-tetraphenylethanol occurs a t & 5.23 and <5 4.73. 13 Comparative F.T. C n.m.r. spectroscopy proved a more u s e f u l technique for the c h a r a c t e r i z a t i o n o f the m a t e r i a l s (see Table 2.9). The t r i p l e t s observed i n the 6 77 t o 78 region i n some o f the s p e c t r a a r e due t o the s o l v e n t CDClj. They a r e not seen where i t was p o s s i b l e t o prepare very concentrated s o l u t i o n s . Spectrum o f (0) showed a group o f peaks between 6 144.25 - 8 128.65 assigned t o the carbon atoms o f the aromatic r i n g s , and peaks a t 6 83.95, 6 81.35, 6 61.08, 6 57.44 and <S 42.44. Peaks a t 6 57.44 and 6 42.44 were 3 assigned by the sp h y b r i d i z e d carbons i n u n i t s analogous to 1,1,2,2tetraphenylethane and bisbenzylbenzene r e s p e c t i v e l y . The peak a t 6 83.95 3 was assigned t o the sp h y b r i d i z e d carbon of polybenzopinacol and peak a t 6 61.08 t o the carbon bonded to hydrogen i n the 1,1,2,2-tetraphenylethanol type o f u n i t present i n the polymeric m a t e r i a l . There remained the peak a t 6 81.35 which should correspond t o the carbon bonded t o the OH group i n the 1,1,2,2-tetraphenylethanol type o f u n i t , although i t has to be acknowledged t h a t such a peak i s not v i s i b l e i n the spectrum o f the model compound 1,1,2,2-tetraphenylethanol. T h i s may be a consequence o f the very low s o l u b i l i t y of the model compound and i n p r a c t i c e i t was only with considerable p e r s i s t a n c e i n the face o f d i f f i c u l t y t h a t a *^C spectrum was recorded. 1 13 Thus, combined H and C n.m.r. evidence would suggest t h a t , as p r e d i c t e d , the polymeric m a t e r i a l contains three major types o f groups as shown i n F i g u r e 2.56 and a l s o t h a t some polymer ends are capped with benzophenone-type aromatic carbonyl (CgHg-C(O)-) and benzyl (CgHg-C^-) groups. I n view o f t h i s conclusion i t i s r a t h e r s u r p r i s i n g t h a t prolonged did not r e s u l t i n the formation o f higher molecular weight irradiation products. - 127 - Table 2.9 13 Comparative F.T. C n.m.r. s p e c t r a of some phenylated substances (CDClj s o l u t i o n s , e x t . TMS r e f . ) . flU6 urns 8395 Product (P) CDC1- Polybenzoplnacol M aA. 64 CDC1U0O8 [iM-56 ittJM ^275 M'« « p-Bisbenzylbenzene J. |iX8'» IOT-35 12410 1,1,2,2-Tetraphenylethanol CDC1. S6-6? (16 06 JL 1,1,2,2-Tetraphenylethane n.<9 Benzopinacol CDCl. 150 140 I I 130 120 I I 110 100 I I 90 80 70 60 50 I I I I I ppra s c a l e 40 I - 128 - OH H H OH ra- or p OH OH H Figure 2.3.e. DJ CQ H 2.56 Reactions of the polymers 2.3.e.i. Depolymerization A sample o f the polymeric m a t e r i a l (O) was introduced i n t o a 3 necked round bottom f l a s k f i t t e d with water condenser, thermometer pocket and n i t r o g e n gas inlet. The f l a s k was s l o w l y heated by means o f a f r e e flame u n t i l the polymer had melted and then temperature was r a i s e d up to 350° and maintained t h e r e f o r 5 mins. A b l a c k t a r was formed which was l e f t to c o o l under n i t r o g e n . Examination o f the black t a r by i . r . spectroscopy gave the f o l l o w i n g r e s u l t s (contact f i l m , v 1510, 1495, 1450 max :) 3080, 3040, 3020, 2900, 2840, 1660 ( s t r o n g ) , 1600, (doublet), 1430, 1420, 1320, 1300, 1280, 1255, 1205, 1580, 1180, 1170, 1120, 1075, 1030, 1000, 985, 910, 850, 830, 800, 780, 720, 710, 700, 620, 600, 485 cm \ Examination of the black t a r by t . l . c . on s i l i c a r e v e a l e d the presence of m-dibenzoylbenzene, 640, (benzene) p-bisbenzylbenzene, diphenylmethane and benzophenone amongst the p y r o l y s i s products. A s i m i l a r treatment of polybenzopinacol M (see Tables M (see T a b l e s 2.4 64 2.6) prepared by Andrews and F e a s t spectrum (contact f i l m , v 2920, 1660 gave a black t a r with a d i f f e r e n t i.r. :) c a . 3480 (broadband), 3299, 3060, 3020, max ( s t r o n g ) , 1660, 1580, 1495, 1445, 1340, 1290, 1270, 1170, 1120, and - 129 - 1075, 1020, 1000, 985, 970, 950, 930, 910, 820, 780, 760, 700, 650, 640 can . m-Dibenzoylbenzene, m-bisbenzhydrol, benzophenone and benzhydrol were i d e n t i f i e d amongst the p y r o l y s i s products by t . l . c . on s i l i c a (benzene). These r e s u l t s are c o n s i s t e n t with the s t r u c t u r e s a s s i g n e d to these materials. 2.3.e.ii. Treatment w i t h dehydrating agents. The following experiments were performed w i t h product (0) a) The polymeric m a t e r i a l (20.Og) was d i s s o l v e d i n dry benzene (0.5 l i t r e s ) i n a 2 l i t r e 3 necked round bottom f l a s k f i t t e d w i t h gas i n l e t , r e f l u x condenser and thermometer pocket. Dry HC1 was bubbled through the straw-yellow s o l u t i o n a t room temperature (26°). A f t e r c a . 10 mins the temperature rose t o 28° and s o l u t i o n went r e d d i s h , p r o g r e s s i v e l y darkening t o brown-red; temperature o stayed constant a t 28 a f t e r 3 h r s of bubbling. S o l u t i o n was subsequently r e f l u x e d f o r a f u r t h e r 3 h r s period under an atmosphere of HC1. A f t e r cooling to room temperature, a s m a l l p o r t i o n of the s o l u t i o n was taken out, benzene s o l v e n t was removed under vacuum and the s o l i d obtained was examined by i . r . spectroscopy (KBr d i s c ) . No change was observed, the spectrum being super- imposable w i t h t h a t of product ( 0 ) . HC1 was bubbled through the benzene s o l u t i o n f o r a f u r t h e r 30 hr period a t room temperature. the of the E x p u l s i o n o f HC1 by p a s s i n g a stream of nitrogen through s o l u t i o n , removal o f the s o l v e n t under reduced p r e s s u r e and p r e c i p i t a t i o n the s o l i d obtained from benzene/60°-80° petroleum t h e r y i e l d e d a m a t e r i a l w i t h same i . r . spectrum (KBr d i s c ) and melting p o i n t as ( 0 ) . 3) The polymeric m a t e r i a l (2.00g) was d i s s o l v e d i n dry benzene (70 mis) i n a 250 ml 2 necked round bottom f l a s k f i t t e d with a Dean-Starke apparatus and a r e f l u x condenser. 80 mis of g l a c i a l a c e t i c a c i d and a few drops of concentrated s u l p h u r i c a c i d were added and the s o l u t i o n was r e f l u x e d f o r 4 h r s . A f t e r cooling the pot contents down t o room temperature, a drop of the s o l u t i o n - 130 - was p l a c e d on a NaCl p l a t e ; pumping under reduced pressure f o r s e v e r a l hours r e s u l t e d I n the removal o f v o l a t i l e m a t e r i a l s and the d e p o s i t i o n of a t r a n s p a r e n t f i l m on the p l a t e , v ^ ^ : 3080, 3060, 3030, 2970, 2920-2850 (strong, assigned to t r a c e s of apiezon grease e x t r a c t e d by r e f l u x i n g benzene from the ground g l a s s j o i n t s ) , 1680 (strong), 1600, 1580, 1505, 1495, 1480, 1455, 1445, 1410, 1220, 1 1180, 1160, 1075, 1035, 1020, 1000, 915, 850, 790, 760, 740, 700, 680, 620 cm" . The bulk of the s o l u t i o n was washed with 5% aqueous potassium carbonate, then plenty o f water; benzene l a y e r was separated, d r i e d over anhydrous magnesium sulphate and v o l a t i l e m a t e r i a l s were removed under reduced p r e s s u r e . A grey powder was obtained (1.85g) which was pumped under reduced p r e s s u r e f o r 6 h r s a t 40°. The s o l i d s t a t e i . r . spectrum of the m a t e r i a l (KBr d i s c ) was the same as the i . r . of the c o l o u r l e s s f i l m described above. o y) The polymeric m a t e r i a l (4.00g) was heated a t 250 excess of ortho-phosphoric a c i d . f o r 30 mins with an A f t e r cooling to room temperature, polymeric m a t e r i a l was e x t r a c t e d with benzene. Following washing of the benzene l a y e r with aqueous potassium carbonate, then plenty o f water and drying over anhydrous magnesium sulphate, a drop o f the benzene s o l u t i o n was p l a c e d on a NaCl p l a t e ; the t h i n f i l m deposited a f t e r evaporation of s o l v e n t under reduced p r e s s u r e was examined by i . r . spectroscopy; v : 3080, 3060, 3030, 2910, 1660 (strong) IDclX 1600, 1575, 1510, 1490, 1470, 1445, 1430, 1340, 1280, 1250, 1170, 1155, 1110, 1075, 1030, 1020,'1000, 980, 915, 850, 790, 765, 730, 710, 700, 680, 640, 620, 555, 490 6) 1 cm" . Product (0) (3.00g) was d i s s o l v e d i n 80 mis o f dry benzene; 90 mis o f g l a c i a l a c e t i c a c i d and a small c r y s t a l of iodine was added and the s o l u t i o n was t r e a t e d s u c c e s s i v e l y with an excess o f water, d i l u t e aqueous potassium carbonate, water again, 5% t h i o s u l p h a t e s o l u t i o n , and f u r t h e r water. The yellow benzene l a y e r was d r i e d over anhydrous magnesium sulphate, then v o l a t i l e m a t e r i a l s were removed by pumping under reduced p r e s s u r e and a y e l l o w i s h powder - 131 - was obtained (3.85g) which was p r e c i p i t a t e d from benzene/60°-80° petroleum ether (white powder, 8 0 % ) . I . r . examination (KBr d i s c ) showed the presence o f a weak 0-H band and a weak band a t 1680 -1 cm . Refluxing the same s o l u t i o n f o r 4.5 h r s followed by e x a c t l y the same treatment y i e l d e d 80% of a w h i t i s h powder with the following i . r . absorptions (KBr d i s c , v nicix ): 3080, 3060, 3030, 2920, 1680 ( s t r o n g ) , 1600, 1580, 1505, 1495, 1480, 1445, 1410, 1220, 1180, 1160, 1075, 1035, 1020, 1005, 915, 860, 840, 790, 765, 740, 700, 680, 620 cm ^. T h i s i . r . spectrum was found to be very n e a r l y superimposable to the one obtained from experiment B. c) The polymeric m a t e r i a l (1.20g) was d i s s o l v e d i n dry benzene (100 m i s ) ; toluene p-sulphonic a c i d (0.80g) was added and the s o l u t i o n was r e f l u x e d f o r 30 mins. A f t e r cooling, benzene was washed with aqueous potassium carbonate ( 5 % ) , then water and d r i e d over anhydrous magnesium s u l p h a t e . Removal of the s o l v e n t under reduced pressure y i e l d e d 1.09g o f a m a t e r i a l whose i . r . spectrum was found to be superimposable with the one obtained f o r products from experiments 0 and 6. The same r e s u l t was obtained when the experiment was c a r r i e d out s t i r r i n g the polymer/toluene p-sulphonic a c i d s o l u t i o n a t room temperature f o r 4 hrs. F i n a l l y , heating the polymer with an excess o f cone, s u l p h u r i c a c i d f o r P 30 mins.gave water s o l u b l e t a r s , whereas r e f l u x i n g with 2 ° 5 * n xylene f o r 2 h r s gave a m a t e r i a l d i s p l a y i n g v a r i o u s types of carbonyl bands i n the i n f r a r e d , i n c l u d i n g one a t 1660 cm ^. 2.3.e.iii. a) P a r a l l e l model compound s t u d i e s 1,1,2,2-tetraphenylethanol (2.00g, 0.0057 mole) i n hot g l a c i a l a c e t i c a c i d (150 mis) was b o i l e d under r e f l u x f o r 1 h r i n the presence o f a c r y s t a l of iodine. 0.1M A f t e r cooling, s o l u t i o n was t r e a t e d s u c c e s s i v e l y with water (50 m i s ) , aqueous potassium carbonate (50 m i s ) , d i l u t e aqueous t h i o s u l p h a t e (30 mis) and water (50 m i s ) . A y e l l o w i s h p r e c i p i t a t e was obtained which was d i s s o l v e d - 132 - i n benzene (100 mis) and kept over anhydrous magnesium sulphate overnight. Following removal of benzene s o l v e n t and drying under reduced p r e s s u r e , the y e l l o w i s h m a t e r i a l (1.82g) was examined by i . r . spectroscopy (KBr d i s c , v» ): nicLx 3075, 3060, 3020, 1680, 1600, 1580, 1490, 1445, 1340, 1320, 1280, 1185, 1175, 1160, 1075, 1030, 1015, 1000, 980, 945, 925, 840, 780, 760, 745, 700, 655, 635, 1 629, 620, 610, 575, 470 cm . Examination of the product by t . l . c . on s i l i c a (benzene) showed the presence of tetraphenylethylene, benzopinacolone 1,1,2,2-tetraphenylethane. and R e c r y s t a l l i z a t i o n s of the product from chloroform/ methanol afforded a y e l l o w i s h m a t e r i a l i n the form o f elongated p l a t e s , s i n g l e spot on t . l . c . on s i l i c a (benzene), m.pt. 220°-221°, having the f o l l o w i n g i . r . spectrum (KBr d i s c , v ) : 3079, 3065, 3020 (doublet), 1600, 1575, 1490, 1190, 1160, 1080, 1035, 1005, 985, 920 ( t r i p l e t ) , 1 575, 470, 450 cm . 1445, 780, 765, 750, 700, 630, 620, R e c r y s t a l l i z a t i o n o f the product from benzene/ethanol gave 1.5g o f a white c r y s t a l l i n e s o l i d , m.pt. 222°-223°. The i . r . spectrum of the c r y s t a l l i n e s o l i d was found to be superimposable with the one recorded f o r the product obtained from s i m i l a r treatment of 1,1,2,2-tetraphenylethanol, and i s t h e r e f o r e tetraphenylethylene, m.pt. 3) 223°-224° (benzene/ethanol) 1,1,2,2-tetraphenylethanol (lO.OOg, 0.028 mole) i n hot benzene (200 mis) was b o i l e d under r e f l u x f o r 3 h r s i n the presence of P p 2 cooling, benzene s o l u t i o n was f i l t e r e d and excess 2 ° 5 a d d i t i o n of water. °5 (20.00g). w destroyed by c a r e f u l a s After Evaporation of the benzene s o l v e n t y i e l d e d a white m a t e r i a l (9.0g) d i s p l a y i n g no 0-H band but C-H a l i p h a t i c and benzopinacolone-type carbonyl bands im the i . r . region (KBr d i s c ) . R e c r y s t a l l i z a t i o n s of the white m a t e r i a l from chloroform/methanol and benzene/ethanol y i e l d e d c a . 7g of pure tetraphenylethylene, m.pt. 223°-224°, i d e n t i f i e d by s o l i d s t a t e i . r . spectroscopy (KBr d i s c ) and mass spectrometry (m/e extended to 332 corresponding to the parent i o n . The mother l i q u o r s from r e c r y s t a l l i z a t i o n s were found by t . l . c . on s i l i c a (benzene) to contain benzopinacolone and 1,1,2,2tetraphenylethane . - 133 - 1,1,2,2-tetraphenylethanol was recovered unchanged a f t e r being b o i l e d under r e f l u x (1 hr) i n e i t h e r benzene or toluene (0.028M). T r a c e s of benzo- phenone and diphenylmethane were observed on t . l . c . on s i l i c a r e s u l t i n g probably from p a r t i a l thermal decomposition (benzene), of the ethanol. Refluxing an equimolar toluene s o l u t i o n of benzopinacol and 1,1,2,2tetraphenylethane (0.1M with r e s p e c t to both reagents) f o r 3 h r s r e s u l t e d i n t o t a l consumption of both s t a r t i n g m a t e r i a l s and formation of 1,1,2,2" tetraphenylethanol, i d e n t i f i e d by t . l . c . on s i l i c a spectroscopy (KBr d i s c ) . (benzene) and i . r . Traces of benzophenone, diphenylmethane and benzhydrol were a l s o observed on the t . l . c . p l a t e s . F i n a l l y , heating 1,1,2,2-tetraphenylethanol under nitrogen a t 350° f o r 5 mins (round bottom f l a s k , f r e e flame) r e s u l t e d i n t o t a l decomposition of the m a t e r i a l and q u a n t i t a t i v e formation of benzophenone and diphenylmethane, i d e n t i f i e d by t . l . c . on s i l i c a (benzene) and i . r . spectroscopy (contact f i l m ) . - 134 - 2.3.f. Experimental 2.3.£.l. Readily available materials The o r i g i n and p u r i f i c a t i o n procedure (where appropriate) f o r benzophenone, m-dibenzoylbenzene and NaBH^ used i n thisi have a l r e a d y been d e s c r i b e d i n 2 . 2 . j . Vitride (NaAlH (OCH CH OCH ) 2 2 2 3 2 70% benzene s o l u t i o n ) was purcahsed from A l d r i c h Chemical8 L t d . and used as obtained. Diphenylmethane was purchased from BDH Laboratory Reagents. I t was d i s t i l l e d t w i c e from a s h o r t column and then f r a c t i o n a t e d under n i t r o g e n o o (760 mmHg, 264 -266 ) . I n a second f r a c t i o n a t i o n under an atmosphere o f nitrogen a t 760 mmHg, product d i s t i l l i n g a t 266° was c o l l e c t e d i n a dry f l a s k (lit.^^ silica 265.6°/760 mmHg). The m a t e r i a l was a s i n g l e component by t . l . c on (benzene, ethanol, a c e t o n i t r i l e , carbon t e t r a c h l o r i d e ) ; the i . r . spectrum was recorded (contact f i l m , v ) : 3080, 3060, 3020, 2900, 2840, 1600, 1580, max 1490, 1450, 1430, 1380, 1320, 1200, 1180, 1155, 1110, 1075, 1030, 940, 920, 890, 845, 810, 785, 700, 610, 550, 460, 445 cm The pure compound c r y s t a l l i z e d slowly on standing forming b i g c o l o u r l e s s prisms, m.pt. 26°-27° ( l i t . * " ^ 26°-27°). Benzhydrylchloride v:as obtained from departmental s t o c k . twice under n i t r o g e n before use; I t was distilled the f r a c t i o n d i s t i l l i n g between 170°-176°/ 20 mmHg was c o l l e c t e d ( b . p t . ^ ^ 173°/19 mmHg) . Terephthalaldehyde was purchased from Koch-Light L a b o r a t o r i e s L t d . and r e c r y s t a l l i s e d twice from methanol/water 70:30. a s i n g l e spot on t . l . c . on s i l i c a (lit.^^ 116°). parent i o n ; 2.3.f.ii. White needles obtained gave o (benzene, chloroform) and melted a t 117 The mass spectrum extended to m/e = 134 corresponding t o the main peaks a t 133, 105, 84, 77, 57, 51. Solvents Benzene was p u r i f i e d as d e s c r i b e d i n 2 . 2 . j . i i ; THF ( f r e s h l y d i s t i l l e d ) was obtained from departmental stock and used immediately; p y r i d i n e and - 135 - toluene ( A n a l y t i c a l grades) were obtained from BDH Laboratory Reagents and used without f u r t h e r p u r i f i c a t i o n ; isopropanol ( t e c h n i c a l grade) was obtained from F i s o n s Laboratory Reagents L t d . ; i t was p u r i f i e d by f r a c t i o n a l d i s t i l l a t i o n p r i o r to use. 2.3.f.iii. M a t e r i a l s s y n t h e s i z e d from s i m p l e r chemicals 1,1,2,2-tetraphenylethane was prepared by the Wurtz coupling o f a l k y l . ... 157,158 halides: R e - d i s t i l l e d benzhydrylchloride (10.125g, 0.05 mole) was d i s s o l v e d i n sodium-dried d i e t h y l e t h e r (500 mis) i n an 1 l i t r e round bottom f l a s k with nitrogen i n l e t , double s u r f a c e r e f l u x condenser equipped (leading to a concentrated s u l p h u r i c a c i d b u b b l e r ) , and mechanical s t i r r e r . T h i n l y s l i c e d sodium (4.6g, 0.2 mole) was added and f l a s k contents were s t i r r e d a t room temperature f o r 24 h r s under an atmosphere of n i t r o g e n , and then r e f l u x e d f o r a f u r t h e r 12 hr p e r i o d . A f t e r c o o l i n g , the e t h e r e a l s o l u t i o n was f i l t e r e d and excess sodium was c a r e f u l l y destroyed using isopropanol. E t h e r was removed under reduced p r e s s u r e and a white s o l i d was obtained c o n s i s t i n g of 1,1,2,2-tetraphenylethane, diphenylmethane benzene). and benzhydrol ( t . l . c . on A f t e r s e v e r a l r e c r y s t a l l i z a t i o n s from ethanol and a c e t i c a c i d , 1,1,2,2-tetraphenylethane was i s o l a t e d i n a pure s t a t e as shown by t . l . c . silica silica, (benzene, chloroform, ethanol) i n 85% y i e l d , m.pt. 213° on 1 ( l i t . * ' * 212.5°). Lassaigne's t e s t on the product showed t h a t c h l o r i n e was absent. 2.3.g. D i s c u s s i o n and Conclusions The bisbensophenone/bisbenzylbenzene photopolymerization was found to be s i m i l a r to the model r e a c t i o n i n some r e s p e c t s , e.g. the slow r e a c t i o n r a t e , the e a r l y appearance o f the yellow colour during i r r a d i a t i o n s and the formation of products c o n t a i n i n g three main types of u n i t as shown i n F i g u r e 2.56. - 136 - However some new questions have a r i s e n : a) the o r i g i n and s t r u c t u r e of the white I n s o l u b l e p r e c i p i t a t e s obtained i n a l l photoreaction, $) the f a i l u r e of the polymer forming r e a c t i o n to proceed to completion even a t prolonged i r r a d i a t i o n s , c o n t r a r y to the behaviour of the benzophenone/ diphenylmethane system and d e s p i t e the presence of unreacted -C_H -CH„-C,H_ o 4 2 b 5 and -CgH ~C(0)-CgHg r e s i d u e s i n the polymeric m a t e r i a l s , detected by n.m.r. 4 and i . r . spectroscopy. y) the slower r a t e o f molecular weight i n c r e a s e observed f o r the m-dibenzoylbenzene/m-dibenzylbenzene system, r e l a t i v e to the corresponding m-/p- one, although both systems gave m a t e r i a l s of rougly the same molecular weight a f t e r 525 h r s o f i r r a d i a t i o n (see p. 124). A t y p i c a l i . r . spectrum of the white p r e c i p i t a t e from the above photor e a c t i o n s has already been described i n 2 . 3 . c . i i . Attempts to measure M r of such m a t e r i a l s i n s o l u t i o n f a i l e d due to extremely low s o l u b i l i t y i n a wide v a r i e t y of solvents. S i m i l a r l y , low s o l u b i l i t y i n deuterated s o l v e n t s prevented examination by n.m.r. spectroscopy. I t i s p o s s i b l e t h a t white p r e c i p i t a t e s might be e i t h e r branched or c r o s s l i n k e d m a t e r i a l s , formed by the i n t e r a c t i o n of a photoexcited bisbenzophenone carbonyl and t e r t i a r y hydrogen i n e i t h e r a 1,1,2,2-tetraphenylethanol type of u n i t or a u n i t , as shown i n Figure 2.57 1,1,2,2-tetraphenylethane type ( i n the case of b r a n c h i n g ) . The f a i l u r e of the polymerization r e a c t i o n to proceed to completion i s a d i f f i c u l t question to deal with. I n p a r t i c u l a r the e s t a b l i s h e d presence of both benzoyl and benzyl groups i n the r e a c t i o n products r u l e s out the p o s s i b i l i t y of one type o f f u n c t i o n a l group being t o t a l l y consumed by some k i n d of s i d e r e a c t i o n , thus l e a v i n g the other i n excess. One could a t t r i b u t e t h i s phenomenon to the formation of e i t h e r an e f f i c i e n t t r i p l e t quencher or a strong l i g h t absorbing intermediate which does e f f e c t i v e l y stop the r e a c t i o n a t a p a r t i c u l a r stage. - 137 - Q X H 5 H or OH n / n OHO Q n OH Figure I 2.57 The o r i g i n of the yellow colour observed during i r r a d i a t i o n s i s d i f f i c u l t to account f o r . P r e c i p i t a t i o n of the yellow crude polymeric m a t e r i a l s from benzene/60°-80° petroleum ether afforded white powders whose i . r . s p e c t r a and M n were not d i f f e r e n t from those of the crude m a t e r i a l s . Treatment of the white powders w i t h benzene a t room temperature b r i g h t yellow s o l u t i o n s . r e s u l t e d i n the formation of Evaporation of benzene s o l v e n t gave yellow m a t e r i a l s , whereas a d d i t i o n of excess 60°-80° petroleum ether to the yellow benzene s o l u t i o n s caused the p r e c i p i t a t i o n of white m a t e r i a l s ; the i . r . s p e c t r a of these products were superimposable. I t was f i r s t thought t h a t some k i n d of a s s o c i a t i o n between benzene s o l v e n t and the product occurs which g i v e s r i s e to - 138 - the coloured s p e c i e s . However the same c y c l e of c o l o u r l e s s to yellow changes can be e f f e c t e d using chloroform, which excludes such an e x p l a n a t i o n . Treatment o f the polymeric m a t e r i a l s with dehydrating agents e l i m i n a t e d the hydroxy1 peak i n the i . r . region and gave r i s e to a new peak a t 1680 cm a t t r i b u t e d to a benzopinacolone-type carbonyl group. 1 T h i s was observed both i n r e a c t i o n s c a r r i e d out a t high temperature and room temperature, and i s c o n s i s t e n t w i t h the expected r e a c t i o n scheme shown i n Figure 2.58. The OJQ I OH H H OH OH I OH H H 11 0 Figure o 2.58 1,1,2,2-tetraphenylethanol type of u n i t would be expected to loose HOH giving a conjugated s t r u c t u r e ; the benzopinacol type of u n i t would be expected to rearrange to a benzopinacolone one^"* and the 1,1,2,2-tetraphenylethane s t r u c t u r e would be expected to be u n r e a c t i v e . The model compound 1,1,2,2-tetraphenylethanol appears to be p a r t i a l l y d i s s o c i a t i n g a t temperatures of c a . 80° or over y i e l d i n g r a d i c a l s by rupture o f the ethane C-C a bond. I n the absence o f dehydrating agents these r a d i c a l s appear to recombine forming the i n i t i a l product, or d i f f u s e out o f the cage - 139 - and diraerize y i e l d i n g benzopinacol and 1,1,2,2-tetraphenylethane, a l l the steps are r e v e r s i b l e and s i n c e the ethanol d e r i v a t i v e i s very i n s o l u b l e , i t i s normally the predominant product. Addition of dehydrating agent a t high temperatures causes the rearrangement of the benzopinacol component of t h i s system to benzopinacolone (Figure 2.59). 99 OH T h i s hypothesis would e x p l a i n A •^2 inert solvent H 0 <0H= r<0> ! w OH // 9 J® — c OH Figure OH 2.59 the observed behaviour of the ethanol and i t s formation from benzopinacol and tetraphenylethane. At room temperature 1,1,2,2-tetraphenylethanol i s known to dehydrate q u a n t i t a t i v e l y to tetraphenylethylene ( p e r c h l o r i c a c i d / 166 methylenechloride s o l v e n t ) , no by-products were reported. I t can be concluded t h a t the photoreductive a d d i t i o n of m-dibenzoylbenzene to p- or m-dibenzylbenzene a t 350 run leads to the formation of low molecular weight m a t e r i a l s (M ca. 3500) c o n t a i n i n g u n i t s analogous to 1,1,2,2n tetraphenylethanol, benzopinacol and 1,1,2,2-tetraphenylethane. In contrast to the model r e a c t i o n , polymerization does not proceed to completion even a t prolonged (over 700 h r s ) i r r a d i a t i o n s . The products obtained decompose - 140 - thermally a t high temperatures to y i e l d s t a r t i n g m a t e r i a l s together with t r a c e s of diphenylmethane and benzophenone. Treatment with dehydrating agents r e s u l t s i n the e l i m i n a t i o n of water and the occurrence of rearrangements,both r e s u l t s being e n t i r e l y c o n s i s t e n t with the s t r u c t u r e s assigned to the polymers. I t would seem on the b a s i s of the r e s u l t s obtained i n t h i s work t h a t t r u l y s u c c e s s f u l photoreductive polymerization i s l i k e l y to be l i m i t e d to the case of benzopinacols, although the h i s t o r y of the a r e a suggests t h a t i t would be f o o l i s h to make dogmatic statements to t h i s e f f e c t . CHAPTER 3 The products of i r r a d i a t i o n of benzaldehyde and i t s d i f u n c t l o n a l analogues - 141 - 3.1. Introduction 3.1.a. E a r l y photochemical investigations I n v e s t i g a t i o n s i n t o the nature of the products formed on benzaldehyde, exposing e i t h e r neat or i n the presence of other substances to a c t i n i c r a d i a t i o n go back to the beginning of the century, i n 1901, C i a m i c i a n and 127 S i l b e r i n s o l a t e d a mixture of benzaldehyde and ethanol i n a s e a l e d g l a s s tube; they obtained acetaldehyde, a r e s i n , and an u n s p e c i f i e d q u a n t i t y of hydrobenzoin. Steam d i s t i l l a t i o n of the r e s i n gave q u a n t i t i e s of m a t e r i a l s described as 'benzoic ether', 'isohydrobenzoin' (the two forms of reported may hydrobenzoin have been the d l - and meso-diastereomers), and a 'yellow, hard and f r a g i l e m a t e r i a l ' which was separated, d r i e d and p r e c i p i t a t e d from benzene/ petroleum e t h e r . A white powder was thus obtained melting a t 100° - 101°; was found to have the same composition as hydrobenzoin four times as high. but a molecular it weight On the b a s i s of these f i n d i n g s the authors assigned to the white powder the formula 4(C,„H,.0„). 14 14 2 S i m i l a r r e s u l t s were obtained on 127 i n s o l a t i o n of an e t h e r e a l s o l u t i o n of benzaldehyde. F i v e y e a r s l a t e r the same workers reported r e s u l t s from the i n s o l a t i o n of 128 neat benzaldehyde i n a sealed g l a s s tube f o r a period of over 8 months. The r e a c t i o n product was described as a t r a n s l u c e n t mass s i m i l a r to p i t c h ' which i s a deep red v a r n i s h ; observed on the w a l l s of the tube. 'greek a few c r y s t a l s of benzoic a c i d were The t r a n s l u c e n t mass was e x t r a c t e d with ether and steam d i s t i l l e d to y i e l d a l i t t l e unreacted benzaldehyde and a yellow r e s i n which, a f t e r repeated treatments with b o i l i n g water and drying, gave a material'. 'fragile Following treatment of an e t h e r e a l s o l u t i o n of the r e s i n with aqueous carbonate and r e p r e c i p i t a t i o n from benzene/petroleum ether a white powder was obtained which was found to become tacky a t 120° and melt between 125° - 130°. Elemental a n a l y s e s and molecular weight determinations i n benzene gave r e s u l t s pointing to the formula 9C_H 0. C i a m i c i a n and S i l b e r suggested t h a t the r e s i n 7 6 obtained from the i n s o l a t i o n of neat benzaldehyde might i n f a c t be i d e n t i c a l to - 142 - t h e one o b t a i n e d from t h e I n s o l a t i o n o f t h e b e n z a l d e h y d e / e t h a n o l d i f f e r e n c e i n the e m p i r i c a l formulae, they proposed, mixture; m i g h t j u s t be due the to 128 difficulties i n h e r e n t i n t h e a n a l y s i s o f amorphous s u b s t a n c e s . I n s o l a t i o n of a benzaldehyde/benzylalcohol mixture 5 months a f f o r d e d s m a l l q u a n t i t i e s o f h y d r o b e n z o i n 4.4g W ( l O g , 1:1 /w) f o r and i s o h y d r o b e n z o i n o f a r e s i n a p p a r e n t l y t h e same a s t h a t a l r e a d y d e s c r i b e d . O n w hand, i n s o l a t i o n o f a benzaldehyde/benzophenone m i x t u r e r e s u l t e d i n t h e f o r m a t i o n o f a r e s i n and m a t e r i a l which, (15g, 2:1 and the other /w, small q u a n t i t i e s of a white 3 months) crystalline f o l l o w i n g e x t r a c t i o n and p u r i f i c a t i o n from a c e t i c a c i d melted a t 236° - 237° and a n a l y s e d a s f o l l o w s : C, 81.02; H, 5.65 ( c a l c u l a t e d f o r 41 34°5 ' ' 5 . 6 1 ) . On t h e b a s i s o f t h e s e r e s u l t s t h e a u t h o r s 128 C H : C 8 1 , 1 8 ; H C H assigned to the c r y s t a l l i n e product the formula 4 ^ 3 4 ° 5 ' an a d d u c t between f o u r m o l e c u l e s o f b e n z a l d e h y d e corresponding to and one o f benzophenone. C i a m i c i a n and S i l b e r a l s o i n v e s t i g a t e d t h e p r o d u c t s formed on i n s o l a t i n g a 167 benzaldehyde/nitrobenzene mixture. Two compounds were i s o l a t e d a s s i g n e d t h e 0 Figure 3.1 o s t r u c t u r e s shown i n F i g u r e 3.1 a l o n g w i t h some r e s i n m e l t i n g a t 125 o - 130 . I n an a t t e m p t t o p r e p a r e n o v e l compounds c o n t a i n i n g n i t r o g e n - i o d i n e bonds, M a s c a r e l l i i n s o l a t e d a b e n z a l d e h y d e / i o d o x y b e n z e n e (CgHglOg) m i x t u r e (250g, 5:1 w /w, 10 months, s e a l e d t u b e ) . 168 The y e l l o w - b r o w n w i t h e t h e r , l e a v i n g a w h i t i s h r e s i d u e ( c a . 2g, m.pt. p r o d u c t was 240° - 245°) w h i c h treated was r e c r y s t a l l i z e d from a c e t i c a c i d t o g i v e w e l l d e f i n e d w h i t e c r y s t a l s m e l t i n g a t 250°. T h i s p r o d u c t was s l i g h t l y s o l u b l e i n most o r d i n a r y s o l v e n t s and was found - 143 - to have the same composition as benzaldehyde and a molecular weight three times as high. The author described t h i s new compound as a 'trimer of benzaldehyde' without f u r t h e r examining i t s p r o p e r t i e s . Other products i s o l a t e d and c h a r a c t e r i z e d were benzoic a c i d , s t i l b e n e , iodobenzene, t r a c e s of i o d i n e and the usual r e s i n melting between 125° - 130°; obtained which were not i d e n t i f i e d . s e v e r a l high b o i l i n g l i q u i d s were a l s o The i n t e r e s t i n g f e a t u r e of t h i s experiment 167 i n the author's view 1 was the formation of the r e s i n and the c r y s t a l l i n e t r i m e r ' on i n s o l a t i o n of benzaldehyde i n the presence of an o x i d i z i n g agent (C H I0 ). 6 5 2 Three years l a t e r , Ciamician and S i l b e r r e i n v e s t i g a t e d the i n s o l a t i o n of 169 neat benzaldehyde i n order to e s t a b l i s h whether the •trimer' d e s c r i b e d by 168 Mascarelli did i n f a c t form i n the absence of iodinated compounds or not. Neat benzaldehyde (lOOg) was i n s o l a t e d i n a sealed tube f o r over 29 months so that the r e a c t i o n could proceed to completion. At the end of the i r r a d i a t i o n period an almost s o l i d red-brown m a t e r i a l had formed i n s i d e the tube, together with a few drops of water. T h i s experiment afforded 0.8g of the 1 trimer 1 described by M a s c a r e l l i (m.pt. 245° - 246° ( a c e t i c a c i d ) ) separated by v i r t u e of i t s i n s o l u b i l i t y i n d i e t h y l e t h e r , c a . 12g of an o i l ( b o i l i n g between 177° 200°) composed mainly of benzaldehyde, hydrobenzoin, ' s m a l l q u a n t i t i e s of benzoic a c i d and and 82.5g of the r e s i n a l r e a d y described i n previous e x p e r i - 127 128 131 ments. - ' 167 ' The r e s i n was r e d i s s o l v e d i n ether, t r e a t e d with aqueous sodium carbonate, then the e t h e r e a l l a y e r was separated and d r i e d over calcium chloride. P r e c i p i t a t i o n from petroleum ether y i e l d e d a y e l l o w i s h amorphous material; a portion of i t was p r e c i p i t a t e d s e v e r a l times from benzene/40° - 70° petroleum ether u n t i l a white powder was obtained which, a f t e r d r y i n g a t melted between 160° - 170°. 100° T h i s m a t e r i a l was found to have the same elemental composition as benzaldehyde and molecular weight determination i n a c e t i c a c i d i n d i c a t e d a tetramer. T h i s 'tetramer' of benzaldehyde was r e a c t i v e towards phenylhydrazine and s e m i c a r b a z i d e hydrochloride; treatment with semicarbazide - 144 - hydrochloride i n a c e t i c a c i d afforded a compound with an elemental composition C H N corresponding to the formula 9 2 7 ° 4 3 m e l t i n a 9 2 t 2 3 2 ° which the authors assigned to the semicarbazone of the tetramer 4C_H 0. 7 6 On the b a s i s of t h i s evidence they proposed the s t r u c t u r a l formula shown i n Figure 3.2 f o r the photoproduct of benzaldehyde. H 0 H 0 Ok i i C C— H 0 1 C — C—H o Figure 3.2 The i n s o l a t i o n of benzaldehyde i n the presence of a l i p h a t i c hydrocarbons 119 v was examined by Paterno and C h i e f f i . They reported obtaining the r e s i n o u s product a l r e a d y described by Ciamician and S i l b e r i n a d d i t i o n to the 'trimer' described by M a s c a r e l l i . Heating the ' t r i m e r 1 i n sealed tubes a t 300° r e s u l t e d i n the formation of benzaldehyde whereas the amorphous r e s i n gave under i d e n t i c a l treatment a tarry l i q u i d ; no inflammable gases or v o l a t i l e l i q u i d s were observed. D i s t i l l a t i o n of the tarry l i q u i d afforded benzaldehyde and a high b o i l i n g f r a c t i o n which c r y s t a l l i z e d on standing and was i d e n t i f i e d as hydrobenzoin by melting point determination. I n 1914, Ciamician and S i l b e r repeated the i n s o l a t i o n of a benzaldehyde/ benzophenone mixture and i s o l a t e d a compound melting a t 245° whose elemental a n a l y s i s and molecular weight determinaticn suggested t h a t i t could be the 'addition product' of the two reagents ( C H 2 0 7 2 2 3 ^ ' t h a t i s an adduct of two molecules of benzaldehyde with one of benzophenone. The r e p o r t s on these e a r l y i n v e s t i g a t i o n s a r e confusing. Paterno d i d not miss the chance to indulge i n s c o r n f u l c r i t i c i s m of Ciamician and h i s team f o r what he considered to be t h e i r i n a c c u r a t e and i n c o n s i s t e n t r e p o r t s on the resinous product formed on i n s o l a t i o n of benzaldehyde, 'which melted i n i t i a l l y - 145 - a t 100° - 101° then a t 125° - 130° and now melts a t 160° - 170° and which was i n i t i a l l y an octamer or nonamer of benzaldehyde and i t i s now recognized as a tetramer .. 1 3 4 However confusing they might be, these e a r l y experiments r e p r e s e n t the only reported attempts to i n v e s t i g a t e the s t r u c t u r e of t h i s ' r e s i n ' formed on i r r a d i a t i o n of benzaldehyde. I n subsequent y e a r s the p r e p a r a t i v e photochemistry of benzaldehyde i n the presence of a wide v a r i e t y o f substances was examined by v a r i o u s workers; i t was found to photooxidize y i e l d i n g benzoic a c i d , add to unsaturated l i n k a g e s , quinones or methylene groups, a b s t r a c t hydrogen from s u i t a b l e donors e t c . A comprehensive review of these processes may be found i n 167 standard t e x t s such as the one by Schonberg. The r e a c t i o n s a r e g e n e r a l l y c o n s i s t e n t with the expected behaviour of a t r i p l e t e x c i t e d carbonyl group l i n k e d to an aromatic r i n g . F i n a l l y , the ' r e s i n ' reported by the e a r l y workers was r e c e n t l y prepared by the gas phase i r r a d i a t i o n of benzaldehyde a t X > 300nm and was found to possess i n t e r e s t i n g photophysical p r o p e r t i e s ; 171 e l u c i d a t e i t s s t r u c t u r e were however reported. 3.1.b. no attempts to Ground s t a t e polymerization of benzaldehyde and i t s d i f u n c t i o n a l analogues Benzaldehyde was found not to polymerize under the a c t i o n of y - i r r a d i a t i o n 172 or by treatment i n s o l u t i o n or i n bulk with a wide range of c a t i o n i c initiators. However, when benzaldehyde was heated with b o r o n t r i f l u o r i d e etherate a t 150° i n the absence of s o l v e n t a polymeric product was formed i n low y i e l d (1.4%) . I t was r e l a t i v e l y non-polar and f l u o r e s c e n t softening a t 79° and melting a t 88°. Compression moulding a t 90° gave a g l o s s y , b r i t t l e d i s c . n e i t h e r boron nor f l u o r i n e ; The product contained e b u l l i o m e t r i c determinations both i n carbon t e t r a - c h l o r i d e and i n benzene i n d i c a t e d a polymer of d.p. 133. The polymer was i d e n t i f i e d as polybenzyl on the b a s i s of elemental a n a l y s i s and i n f r a r e d 172 o spectroscopy. Mixtures of benzaldehyde and styrene t r e a t e d a t 0 with boront r i f l u o r i d e etherate copolymerized through the carbonyl group of benzaldehyde to y i e l d a product i s o s t e r i c with polystyrene but which may not have a d e f i n i t e - 146 - repeat u n i t . Elemental a n a l y s i s of the copolymer produced from a 50:50 mixture of monomers i n d i c a t e d a product i n c o r p o r a t i n g approximately 80% styrene and 20% benzaldehyde, and having a molecular weight of 14,600. Both i s o - and tere-phthalaldehyde have been polymerized under a v a r i e t y o f conditions to give benzoin copolymers, 173 174 ' which are claimed to be very u s e f u l as coatings which can be c r o s s - l i n k e d t o form binders f o r g l a s s - f a b r i c laminates. The copolymers were s a i d to possess e x c e l l e n t thermal s t a b i l i t i e s and t e a r 174 strength. Terephthalaldehyde has a l s o been polymerized i n the presence o f 175 176 ketene and b o r o n t r i f l u o r i d e e t h e r a t e , a pale yellow powder o f d.p. and DMSO. The l a t t e r method y i e l d e d < 9, s o l u b l e i n DMSO, DMF and ethylene carbonate, i d e n t i f i e d by i . r . spectroscopy as polybenzoin (Figure 3.3,1); autooxidation i n 176 DMSO s o l u t i o n gave the high-melting and i n s o l u b l e p o l y b e n z i l (Figure 3.3,11). Reduction of terephthalaldehyde i n dioxane with Zn and Ct^(SO^)^ afforded polymer with HNO^ r e s u l t e d i n the formation o f 3.3,111). poly(p-phenylenedicarbonyl), poly(p-phenylenebis(hydroxymethylene)) (Figure Treatment o f t h i s 177 Condensing the polycarbonyl with o-phenylenediamine and/or i.e. polybenzil H ~€H-irOH " © - s - s - In II I OH I OH III Figure 3.3 - 147 - 3,3",4,4'-tetraminobiphenyl y i e l d s heat r e s i s t a n t r e s i n o u s p o l y q u i n a x o l i n e s u s e f u l i n the preparation of p r o t e c t i v e c o a t i n g s and laminated r e s i n s having 177 heat s t a b i l i t y , s o l v e n t r e s i s t a n c e , f l e x i b i l i t y and low s o f t e n i n g p o i n t s . 178 Copolymers of terephthalaldehyde with v a r i o u s substances l i k e ferrocene, 179 180 poly(vinylalcohol) and polyamide a c i d s have been reported. F a i r l y r e c e n t l y , s e l e c t i v e chemical r e a c t i o n s on one aldehydic group o f t e r e - and i s o 181 phthalaldehyde were made p o s s i b l e by use o f polymer supports. 3.I.e. The photochemistry of phthalaldehyde Schttnberg and Mustafa reported t h a t a benzene s o l u t i o n o f phthalaldehyde exposed to s u n l i g h t f o r 1 day deposited c o l o u r l e s s c r y s t a l s of a dimer, m.pt. o ca. 184 (decomposition) ,? 182 (Figure 3.4). H i H OH i H C-H hu 7" » ©C° 11 »a/or » o Figure 3.4 Kagan i r r a d i a t e d phthalaldehyde (200 mg) i n carbon t e t r a c h l o r i d e (50 mis) for 1 h r . under nitrogen and reported obtaining c r y s t a l l i n e p h t h a l i d e , i . e . the lactone of 2-hydroxymethylbenzoic a c i d , m.pt. 70° i n c a . 80% y i e l d . H e obtained s i m i l a r r e s u l t s a t d i f f e r e n t concentrations, i n the presence of a i r , and with i r r a d i a t i o n a t e i t h e r 253.7nm or 350nm. t h a t i r r a d i a t i o n of phthalaldehyde On the other hand, he showed (0.727g) i n pentane (250 mis) a t 350nm f o r 1 hr. (Nj atmosphere) y i e l d e d 67% o f the pure photodimer shown i n F i g u r e 3.5; and F i g u r e 3.5 - 148 - i r r a d i a t i o n of neat phthalaldehyde (N^ atmosphere) a t 350nm f o r 3 h r s . y i e l d e d phthalide (15%), the photodimer (55%), and 30% of unchanged F i n a l l y , i r r a d i a t i o n of phthalaldehyde (0.172g) i n benzene under starting material. (50 mis) a t 350nm atmosphere gave phthalide (45%) the dimer (51%) and unchanged starting 183 material (4%). The l a c k of dimer formation i n c h l o r i n a t e d s o l v e n t s was a t t r i b u t e d by Kagan to e f f i c i e n t quenching of the d i m e r i z a t i o n s t e p i n the proposed mechanism by C I * r a d i c a l s from the s o l v e n t . T h i s was the f i r s t example of a photochemical Canizzaro r e a c t i o n i n which both photo-oxidation and -reduction was shown to occur simultaneously on i r r a d i a t i o n of an aldehyde. Cohen and co-workers showed t h a t the d i m e r i z a t i o n of phthalaldehyde i s s t e r e o s p e c i f i c i n c o n t r a s t to the d i m e r i z a t i o n of o-phthalaldehydic a c i d (2-formylbenzoic acid) which i s n o n - s t e r e o s p e c i f i c ; 184 two quite d i f f e r e n t paths. 185 Further work by Kagan the r e a c t i o n s proceed v i a on the p h o t o l y s i s of phthalaldehyde a t 77K pointed to the conclusion t h a t a benzoyl r a d i c a l intermediate was involved i n the r e a c t i o n which was assumed to proceed from a short l i v e d phthalaldehyde e x c i t e d s t a t e to form phthalide and/or the photodimer. 3.1.d. B r i e f i n t r o d u c t i o n i n t o the photochemistry of benzaldehyde Benzaldehyde, one of the few molecules which e x h i b i t phosphorescence i n the 186 187 gas phase ' has been e x t e n s i v e l y used as a t r i p l e t energy donor both i n the gas phase^*^ and i n s o l u t i o n . T h e molecule has been the o b j e c t of numerous t h e o r e t i c a l and spectroscopic s t u d i e s and a s u b s t a n t i a l amount of data appear i n the l i t e r a t u r e . D i r e c t photolyses of gaseous benzaldehyde reported upto the e a r l y s e v e n t i e s 192-195 have only been p r e l i m i n a r y i n nature. I t was e s t a b l i s h e d t h a t n •* I T * e x c i t a t i o n i n the gas phase r e s u l t e d i n polymer formation on the c e l l w a l l and t h a t a t elevated temperatures the quantum y i e l d of benzaldehyde consumption 195 as high as 40. I t was a l s o reported t h a t a t X < 270nm benzaldehyde was - 149 - decomposed to benzene and carbon monoxide. B l a c e t and Vanselow claimed that hydrogen gas, b e n z i l , benzophenone and diphenyl were formed i n a d d i t i o n to benzene and carbon monoxide upon i r r a d i a t i o n of benzaldehyde a t 313nm. I n an attempt to obtain absolute quantum y i e l d s f o r the primary photop h y s i c a l and photochemical processes and to study the e f f e c t o f wavelength on the system, S t e e l and coworkers c a r r i e d out a d e t a i l e d examination of the photo196 chemistry of benzaldehyde. The quantum y i e l d s of benzene and carbon monoxide formation, benzaldehyde consumption and phosphorescence emission were studied. These data, together with information from following the phorphorescence l i f e time over a range o f pressure and e x c i t a t i o n wavelengths, and t r i p l e t transfer studies allowed a p i c t u r e of the primary photophysical processes to be b u i l t up (Figure 3.6) and r a t e constants f o r these processes to be determined. authors i n t e r p r e t e d t h e i r data as i n d i c a t i n g t h a t with 276nm l i g h t , S The Q > e x c i t a t i o n , r e s u l t e d i n the population of the higher v i b r a t i o n a l l e v e l s of two different t r i p l e t states (T^ and D . At low p r e s s u r e s , d i s s o c i a t i o n occurs from both these v i b r a t i o n a l l y e x c i t e d t r i p l e t s y i e l d i n g benzene and carbon monoxide. c H,. + CO C-H_ 40 v decomposition o 30 cm x 10 20 365nm hu 10 276nm 0 O' F i g u r e 3.6 - 150 - C o l l i s i o n a l d e a c t i v a t i o n of these s t a t e s feeds the lower v i b r a t i o n a l l e v e l s of the lowest t r i p l e t ; from here there are no chemical decay channels and phos- phorescence can be observed. S Q > S 1 I n c o n t r a s t to S excitation, Q e x c i t a t i o n (X i> 365nm) r e s u l t s i n e f f e c t i v e l y zero benzene or carbon monoxide formation, s i n c e only the lower v i b r a t i o n a l l e v e l s of one t r i p l e t were populated, and the phosphorescence y i e l d s were i n s e n s i t i v e to pressure. There was however a s i g n i f i c a n t quantum y i e l d of benzaldehyde consumption and polymer formation could be observed. The products of i r r a d i a t i o n of benzaldehyde i n s o l u t i o n have r e c e n t l y been 197 examined by Bradshaw and coworkers. When i r r a d i a t e d i n benzene s o l u t i o n f o r r e l a t i v e l y s h o r t periods, benzaldehyde y i e l d e d almost equal amounts of benzoin, deoxybenzoin and hydrobenzoin; i n ethanol s o l u t i o n the main product was benzoin with only a t r a c e of benzoin p r e s e n t . A more d e t a i l e d hydro- investigation showed that benzoin was the major i n i t i a l product of the r e a c t i o n i n d i l u t e benzene s o l u t i o n and t h a t i t was slowly consumed on prolonged i r r a d i a t i o n . The p i c t u r e i s complicated by the observation t h a t i r r a d i a t i o n of benzoin i n benzene apparently g i v e s f i r s t benzaldehyde and then deoxybenzoin and hydrobenzoin a t 197 longer i r r a d i a t i o n times. Other workers showed by means of c h e m i c a l l y induced dynamic n u c l e a r s p i n p o l a r i z a t i o n (CIDNP) t h a t on i r r a d i a t i o n of benzaldehyde i n s o l u t i o n a t r i p l e t benzaldehyde a b s t r a c t e d a hydrogen atom from a ground s t a t e benzaldehyde molecul 198 to give a r a d i c a l p a i r which c o l l a p s e d to form benzoin. The r e a c t i o n was r e v e r s i b l e as shown by the f a c t t h a t benzoin on i r r a d i a t i o n formed the same radical pair. No products were however i s o l a t e d from these s t u d i e s . The photochemistry of benzoin has r e c e n t l y been i n v e s t i g a t e d i n more d e t a i l I t was found t h a t i r r a d i a t i o n of the compound (3.18g) i n benzene s o l u t i o n (300 mis) f o r 6 h r s . r e s u l t e d i n only 28% conversion. V.p.c. showed benzaldehyd and benzoin benzoate were the only v o l a t i l e products formed. Column chromato- graphy on s i l i c a g e l afforded unreacted benzoin (2.1g, 66%) and benzoin benzoate - 151 - (0.21g, 1 3 % ) . I r r a d i a t i o n of benzoin i n benzene i n the presence of dodecanethiol (an e f f i c i e n t scavenger of benzoyl r a d i c a l s ) r e s u l t e d i n an i n c r e a s e d y i e l d of 199 benzaldehyde and t o t a l supression of benzoin benzoate 200 Kornis and De Mayo formation. a t t r i b u t e d the e f f i c i e n t formation o f benzaldehyde from benzoin to hydrogen t r a n s f e r from the hydroxybenzyl r a d i c a l to the benzoyl r a d i c a l (Figure 3.7), although benzaldehyde could a l s o be formed v i a an i n t r a - molecular H a b s t r a c t i o n such a s t h a t postulated by P i t t s and coworkers f o r the F i g u r e 3.7 201 formation o f acetaldehyde i n the vapour phase p h o t o l y s i s of a c e t o i n . p i c t u r e i s by no means c l e a r ; The thus although both the benzoyl and hydroxybenzyl r a d i c a l have been trapped by i r r a d i a t i n g benzoin i n the presence of N-oxides and 138 n i t r o s o compounds, the i n c r e a s e i n benzaldehyde quantum y i e l d with added t h i o l i s c o n s i s t e n t with H-transfer p r i o r to or a t the same time as a-cleavage. I t has been shown t h a t b e n z i l i s formed by combination of two benzoyl r a d i c a l s ; a n d i t i s known t h a t b e n z i l y i e l d s benzoin benzoate as the major product of i r r a d i a t i o n i n cumene s o l u t i o n , t h a t i s i n the presence o f a hydrogen donor. 3.I.e. O r i g i n a l ideas The photoreductive polymerization of aromatic diketones i s w e l l documented (see Chapter 2 ) . However, there have been no r e p o r t s on the i r r a d i a t i o n s o f - 152 - aromatic dialdehydes i n the presence of hydrogen donors. I t was t h e r e f o r e decided to i n v e s t i g a t e the behaviour of two of these dialdehydes, namely t e r e and iso-phthalaldehyde, on exposure to u l t r a v i o l e t l i g h t both i n the s o l i d s t a t e and i n s o l u t i o n i n the presence of hydrogen donors. I t was a l s o decided to attempt a c l o s e r examination of the 'photopolymer* formed on i r r a d i a t i o n of neat benzaldehyde i n the l i q u i d phase i n the hope of obtaining some information on the s t r u c t u r e of t h i s m a t e r i a l which has been the s u b j e c t of s e v e r a l previous investigations. 3.2. The photochemistry of t e r e - and iso-phthalaldehyde 3.2.a. Results A summary of experimental procedures and r e s u l t s obtained may Table be found i n 3.1. I r r a d i a t i o n s were c a r r i e d out i n s e a l e d tubes under reduced p r e s s u r e i n order to exclude oxygen from the system, s i n c e terephthalaldehyde i s known to 204 y i e l d t e r e p h t h a l i c a c i d on i r r a d i a t i o n i n the presence of oxygen. D i s t i l l e d water and perfluoro(methylcyclohexane) were used as the suspending agents f o r the s o l i d s t a t e i r r a d i a t i o n s s i n c e the dialdehydes were not d e t e c t a b l y soluble i n e i t h e r of these media a t 25°. I n both c a s e s e f f i c i e n t s t i r r i n g mixing was provided by means of an e x t e r n a l mechanical and stirrer. Whereas i r r a d i a t i o n s i n the s o l i d s t a t e afforded q u a n t i t a t i v e recovery of s t a r t i n g m a t e r i a l s , i r r a d i a t i o n s i n the presence of a hydrogen donor afforded i n i t i a l l y i n s o l u b l e products which showed both carbonyl and hydroxyl absorptions i n the i . r . region. modifications: I r r a d i a t i o n s were repeated under the following (a) the s o l u t i o n s were made as concentrated as p o s s i b l e , (b) H- donor and s o l v e n t were very c a r e f u l l y p u r i f i e d before use, (c) s o l u t i o n s were c a r e f u l l y degassed on a g r e a s e - f r e e , Hg-free vacuum l i n e and tubes were s e a l e d under lower p r e s s u r e ( c a . 10 ^ mm Hg), (d) 350nm r a d i a n t source was used i n s t e a d of the HANOVIA 450-W Hg lamp i n an attempt to e f f e c t g r e a t e r o v e r l a p between ft 3 H Donor Medium i < ro < < < O & a CN ® o n u rH o B CM m *r O & i CJ ro a ro u ro a © ® u u a o a a § o 1 f g h Not characterised Not characterised Not characterised 1:4 v/v 1:4 v/v j 1:4 v/v in O rH ro ro 61.5 in Not characterised < 1 01 ro ro a 8 u a a o s 4J in 0.351 1 1:3 v/v < 1 1 1 f Comments 98% starting materials j back Products •o 0.448 i Solvent Ratio H-donor o 0.0369 m 1 26.5 rH 0.0486 u — ro Suspension c u CHO r^^j quantitatively recovered CHO 1 Temp. Method u Suspension j (Hrs.) j .9 Carbonyl : Compound • Starting Materials - 153 - < m PQ in ^> m m M< m n ©> Q a ? a ? ro a a o ro a u i ro u a a S a @ o a ro - 154 - Table 3.1 (cont.) Footnotes: a. LAMP A: LAMP B: b. HANOVIA 450-W Hg Lamp. RUL-350 mm Photochemical Reactor. METHOD A: I r r a d i a t i o n s c a r r i e d out i n pyrex tubes; solutions degassed by s e v e r a l freeze-pump-thaw c y c l e s on conventional vacuum l i n e s and sealed under reduced pressure ( r e s i d u a l p r e s s u r e i n tube ^ 0.005 mm Hg). METHOD B: I r r a d i a t i o n s c a r r i e d out i n pyrex tubes; solutions degassed by s e v e r a l freeze-pump-thaw c y c l e s on high vacuum grease-free/mercury f r e e vacuum l i n e and s e a l e d under reduced p r e s s u r e ( r e s i d u a l p r e s s u r e i n tubes 10~5 mm Hg). METHOD C: I r r a d i a t i o n c a r r i e d out i n 1L f l o r e n t i n e f l a s k ; s o l u t i o n nitrogen streamed f o r 1 h r . and q u i c k l y stoppered. c. I d e n t i f i e d by i . r . spectroscopy/recovered m a t e r i a l gave one major spot corresponding t o therephthalaldehyde on s i l i c a t . l . c . examination (CHCl^ e l u e n t ) . d. 2% o f a yellow m a t e r i a l was a l s o obtained (soluble i n a c e t o n e ) . (u : 3450, 2930, 2860, 2740, 1700, 1605 c m . ) . max - 1 e. Product e x t r a c t e d by f r e e z e - d r y i n g (112.5% of the t e r e p h t h a l aldehyde used, r e a c t i o n with donor and/or s o l v e n t ( ? ) ) . B r i t t l e f i b r e s drawn out o f the melt. Displayed c o n s i d e r a b l e thermal s t a b i l i t y (no change i n i . r . a f t e r strong h e a t i n g on bunsen flame). Soluble i n DMF, NMP, P y r i d i n e , e t h a n e d i o l , ethylmethylketone. R e p r e c i p i t a t i o n from DMF ( s o l v e n t ) / C C 1 55% o f a yellow m a t e r i a l . f. 4 (nonsolvent) afforded Product e x t r a c t e d by f r e e z e - d r y i n g (116.4% o f the i s o p h t h a l aldehyde u s e d ) . B r i t t l e f i b r e s drawn out of the melt. Soluble i n DMF, NMP, P y r i d i n e , ethylmethylketone. R e p r e c i p i t a t i o n from DMF ( s o l v e n t ) / C C 1 61% o f a yellow m a t e r i a l . 4 (nonsolvent) afforded g. i . r . spectrum o f product obtained superimposable with i . r . of product obtained from expt. 3 - s i m i l a r p h y s i c a l p r o p e r t i e s . h. i . r . spectrum o f product obtained superimposable with i . r . of product obtained from expt. 4 - s i m i l a r p h y s i c a l p r o p e r t i e s . - 155 - the lamp emission and the carbonyl n length p h o t o l y s i s e f f e c t s , modified, the use of DMF v* absorption and to exclude s h o r t wave- (e) work-up of the s o l u t i o n s a f t e r i r r a d i a t i o n was or any other high b o i l i n g s o l v e n t being avoided. The products however were the same as those obtained i n the previous experiments. Treatment of the products w i t h DMF a t room temperature f o r 7 days r e s u l t e d i n s u b s t a n t i a l degradation and i n c o r p o r a t i o n of DMF by both i . r . spectroscopy and mass spectrometry. i n t o the products, as shown Heating of the DMF-treated products under high vacuum (100°) for 1 day r e s u l t e d i n p a r t i a l e l i m i n a t i o n of the DMF peaks from the i . r . spectrum. stability The products showed c o n s i d e r a b l e thermal (no change i n the i . r . spectrum a f t e r heating a t 250° f o r 10 mins.). B r i t t l e f i b r e s could be drawn from the melt. Comparison of the i . r . spectra of the products with that of polybenzoin prepared according to e s t a b l i s h e d 173 route revealed considerable d i f f e r e n c e i n the p o s i t i o n s and i n t e n s i t i e s of the main bands. Both photoproducts were found to be s o l u b l e i n (CH^^SO g i v i n g b r i g h t yellow s o l u t i o n s . However, i n c o n t r a s t with the behaviour of poly- 173 benzoin no autooxidation was detected, the s o l u t i o n s being s t i l l c l e a r a f t e r 2 months of standing whereas polybenzoin s o l u t i o n s p r e c i p i t a t e p o l y b e n z i l ; i n t r o d u c t i o n of oxygen gas i n the s o l u t i o n s a f f e c t e d no change; of the photoproducts a f t e r 3 months of standing using water as the non-solvent y i e l d e d unchanged photoproducts, 3.2.b. precipitation as shown by i . r . spectroscopy. D i s c u s s i o n i n the l i g h t of r e c e n t knowledge The n o n - r e a c t i v i t y of the dialdehydes i n the s o l i d s t a t e should not be considered as a s u r p r i s i n g r e s u l t , s i n c e the most l i k e l y i n i t i a l product (a 138 benzoin-type s t r u c t u r e ) would be expected to c l e a v e r a p i d l y , s t a r t i n g m a t e r i a l as the main product. The s m a l l y i e l d y i e l d i n g the ( c a . 2%) of a coloured m a t e r i a l obtained from the r e l a t i v e l y longer i r r a d i a t i o n of isophthalaldehyde i n perfluoro(methylcyclohexane) non-solvent could be the r e s u l t of an out of cage d i f f u s i o n and d i m e r i z a t i o n of the r a d i c a l s r e s u l t e d from cleavage of the - 156 - initially formed benzoin s t r u c t u r e ; such a process would give p i n a c o l and b e n z i l - t y p e s t r u c t u r e s which could f u r t h e r r e a c t to form the coloured m a t e r i a l ( s ) . The behaviour of the dialdehydes on i r r a d i a t i o n i n the presence of a H-donor i s vastly different. S t a r t i n g m a t e r i a l s disappear completely and new products are formed, the p h y s i c a l c h a r a c t e r i s t i c s and s p e c t r a l parameters of which were c o n s i s t e n t with t h e i r being branched low molecular weight p i n a c o l s . The branching occurs p o s s i b l y v i a the t e r t i a r y benzhydryl hydrogen i n the polymer backbone which would be expected to be more e a s i l y a b s t r a c t a b l e than the t e r t i a r y hydrogen i n isopropanol. 3.3. The benzaldehyde 3.3.a. photoproduct Recent preparation and s p e c t r a l c h a r a c t e r i s t i c s Benzaldehyde was r e c e n t l y i r r a d i a t e d i n the gas phase r e s u l t i n g i n the 171 formation of a polymeric f i l m with i n t e r e s t i n g p r o p e r t i e s . The r e a c t i o n v e s s e l c o n s i s t e d of a c y l i n d r i c a l Pyrex (or quartz) sleeve placed c o n c e n t r i c a l l y around a medium pressure mercury vapour lamp ( P h i l i p s p h i l o r a HPK 125). experiments were performed a t 50.0 +_ 0.2°. The The lamp (protected by a quartz tube) and the r e a c t i o n c e l l were immersed i n 1M CuSO. which served as t h e r m o s t a t i c 4 f l u i d and a l s o , i n a l a y e r 0.5 cm t h i c k between the lamp and the c e l l , as a f i l t e r to cut o f f a l l l i g h t with wavelengths < 300nm. T h i s s e l e c t i o n was necessary to minimize benzaldehyde decomposition to benzene and carbon monoxide. I n every p h o t o l y s i s , the p r e s s u r e drop observed corresponded to a t l e a s t 70% of the benzaldehyde polymerizing. The gas chromatographic a n a l y s e s of the r e s i d u a l gases showed peaks with the same r e t e n t i o n time as benzene and benzaldehyde, the l a t t e r being present i n t r a c e q u a n t i t i e s . F r e e z i n g down of the r e s i d u a l gases showed the presence of an important f r a c t i o n t h a t was non- condensable a t -196° assigned by the authors^"^ as carbon monoxide formed by secondary p h o t o l y s i s of the photopolymer. The product was removed from the c e l l s mechanically or by d i s s o l u t i o n i n v a r i o u s s o l v e n t s . I t was found to d i s s o l v e - 157 - very slowly i n tetrahydrofuran, dioxane and methanol a t room temperature, q u i c k l y i n benzene a t 20° and methanol a t 60°. and The u l t r a v i o l e t absorption spectrum of the polymer i n methanol s o l u t i o n showed a maximum a t 234nm, a 0 shoulder a t 248nm (corresponding to the absorption maximum of benzoin^ ^) and a s t r u c t u r e l e s s t a i l out to a t l e a s t 450nm. a l s o recorded The i . r . spectrum of the polymer was (KBr d i s c ) and was shown to be d i f f e r e n t from t h a t of benzoin. Two absorption bands a t 1685 and 1723 cm * were i n d i c a t i v e of two d i f f e r e n t carbonyl environments. Luminescence e x c i t a t i o n s p e c t r a of the product s o l u t i o n , room temperature) (benzene taken with 366, 406, and 436nm e x c i t a t i o n were c o n s i s t e n t with the presence of two a c t i v e chromophores i n the s t r u c t u r e . polymer f i l m on the c e l l w a l l was found to s e n s i t i z e c i s _ The , trans isomerization of 1,3-pentadienes and to be a c t i v e even a f t e r long periods of s e r v i c e . Its a c t i v i t y decreased by a f a c t o r of 15 a f t e r 3 h r s . of i r r a d i a t i o n i n the presence of 50.7 t o r r of oxygen; t h i s treatment a l s o caused the disappearance of the 171 y e l l o w i s h c o l o u r a t i o n of the polymer. established; 3.3.b. 3.3.b.i. The s t r u c t u r e of the polymer was the aim of t h i s work was to f i l l t h a t not gap. Experimental P u r i f i c a t i o n of benzaldehyde 300 mis. portions of t e c h n i c a l grade benzaldehyde (BDH Chemicals Ltd.) K C were washed w i t h f i v e 200 mis. p o r t i o n s of 1M 2 ° 3 (aqueous s o l u t i o n ) . The m a t e r i a l thus obtained was l e f t to stand over lOOg anhydrous MgSO^ f o r 5 h r s . I t was then t r a n s f e r r e d through a s i n t e r i n t o a 500 mis 3-necked f l a s k (operation c a r r i e d out under a nitrogen b l a n k e t ) . T h i s product was f r a c t i o n a l l y d i s t i l l e d (vacuum j a c k e t e d column, 3' x 1", packed with g l a s s h e l i c e s ) . The apparatus used i n the f r a c t i o n a l d i s t i l l a t i o n was d r i e d i n the oven before assembly purged with dry nitrogen f o r s e v e r a l hours before the f r a c t i o n a t i o n was and started; a nitrogen atmosphere was maintained throughout by means of a nitrogen l i n e . E q u i l i b r a t i o n of the column took about 24 h r s . (pot temp. 178°, column temp. - 158 - 210 , head temp. 170 178°. - 178 ) ; head temperature was f i n a l l y e q u i l i b r a t e d a t Take o f f r a t e was kept a t approximately 1 : 120; the f i r s t 30 and last 50 mis were d i s c a r d e d . Benzaldehyde thus p u r i f i e d was stored under dry nitrogen No s i g n s of benzoic a c i d were noticed a t the neck of the f l a s k . a n a l y s i s f i g u r e s , mass spectrum and i . r . spectrum were i n d i c a t i v e of pure benzaldehyde. by t . I . e . on s i l i c a (recorded as a l i q u i d f i l m ) A s i n g l e spot was obtained on examination (benzene, chloroform, e t h y l a c e t a t e , acetone/water, carbon t e t r a c h l o r i d e , propanol); one peak only. Elemental analytical g.l.c. ( C o l . A, 150°, C o l . 0, 110°) showed The u l t r a v i o l e t spectrum showed the following a b s o r p t i o n s : 370 ( 2 ) , 353 (8.4), 338.7 (10.4), 327.5 (10.4), 317 (9.4), 306 (7.3), 288 285 (318.4), 278.5 (412), 248 (2906), 241 3.3.b.ii. (360), (3618) nm (e) (cyclohexane). Irradiations Benzaldehyde was t r a n s f e r r e d i n t o dry c y l i n d r i c a l Pyrex v e s s e l s under nitrogen, degassed by s e v e r a l freeze-pump-thaw c y c l e s on a conventional vacuum -3 l i n e , and s e a l e d under reduced p r e s s u r e (10 mm Hg). I n some i r r a d i a t i o n s a Hg-free, g r e a s e - f r e e vacuum l i n e was used and the tubes were s e a l e d under lower pressure ( c a . 10 ^ mm Hg). 350nm lamps. A l l i r r a d i a t i o n s were c a r r i e d out a t c a . 40° using The r e a c t i o n was found to be very slow. A yellow colour u s u a l l y developed a f t e r 50 h r s . of i r r a d i a t i o n and the i n i t i a l l y water white mobile l i q u i d turned i n t o a brown v i s c o u s mass a f t e r 200 h r s . of i r r a d i a t i o n . Benzaldehyde was i n i t i a l l y i r r a d i a t e d f o r periods of 250 -*360 h r s . In a l a t e r experiment four p o r t i o n s of the pure m a t e r i a l were i r r a d i a t e d f o r 100, 128 660 and 1110 h r s . Contrary to previous r e p o r t s observed on the w a l l s of the tube. 350 no c r y s t a l l i n e m a t e r i a l s were The v i s c o u s mass formed appeared homo- geneous and no signs of water or other immiscible l i q u i d s were detected which c a s t s doubts on the v a l i d i t y of e a r l i e r arguments. The tubes were frozen i n l i q u i d a i r before the s e a l was broken and the open end was immediately connected to a concentrated s u l p h u r i c a c i d bubbler. pressure was noticed on opening the tubes, even a f t e r prolonged No irradiations, - 159 - I n d i c a t i n g the absence of non-condensable gases. 3.3.b.ii. P u r i f i c a t i o n of product The crude products appeared as very v i s c o u s masses containing s u b s t a n t i a l amounts of unreacted benzaldehyde. They were g e n e r a l l y p u r i f i e d by repeated p r e c i p i t a t i o n s from benzene/60° - 80° petroleum e t h e r . The r e s u l t a n t yellow powders were d i s s o l v e d i n d i e t h y l e t h e r , washed with d i l u t e aqueous carbonate, then with water and p r e c i p i t a t e d again using petroleum ether as the non-solvent. Some samples were a l s o p r e c i p i t a t e d from ethanol/water. 127 128 previous r e p o r t s ' 169 ' i t was not p o s s i b l e to obtain white powders even a f t e r a g r e a t number of p r e c i p i t a t i o n s . a f f o r d any c r y s t a l l i n e compounds. Concentration of mother l i q u o r s d i d not I n some e a r l y experiments, steam d i s t i l l a t i o n was used as a p u r i f i c a t i o n technique; benzaldehyde was the only d e t e c t a b l e s t e a m - v o l a t i l e m a t e r i a l ( t . l . c . on s i l i c a 100°, I n c o n t r a s t with (benzene, chloroform), C o l . A, 150°, 60°). The products were found to be s o l u b l e i n carbon t e t r a c h l o r i d e , methylenechloride, toluene, acetone chloroform, ( i n cold) and i n hot a l c o h o l i c s o l v e n t s ; they were i n s o l u b l e i n a l i p h a t i c hydrocarbons, methanol/water/potassium carbonate mixtures. aqueous potassium carbonate or On slow c o o l i n g of hot concentrated or d i l u t e a l c o h o l i c s o l u t i o n s , yellow amorphous powders were obtained. long (sometimes over 100 cm.) f i b r e s were drawn out of the melt; Fairly they were however very b r i t t l e . T . l . c . a n a l y s i s of the p r e c i p i t a t e d products (benzene/petroleum ether, ethanol/water) showed the presence of one component only, sometimes with a small f a i n t t a i l which i s however not uncommon f o r polymeric m a t e r i a l s (benzene, chloroform, carbon t e t r a c h l o r i d e / c h l o r o f o r m 50:50, ethanol/chloroform 50:50, chloroform/methanol/carbon t e t r a c h l o r i d e , acetone/methanol/ethanol, e t h a n o l / acetone 50:50, benzene/chloroform, ethanol/acetone/water). D e c o l o u r i z a t i o n of the amorphous m a t e r i a l was attempted using a c t i v a t e d animal c h a r c o a l i n hot methanol s o l u t i o n . Repeated treatments gave an o f f white - IbO - material (60%) which could not be f u r t h e r decolourized by t h i s method; the same s p e c t r a l c h a r a c t e r i s t i c s as the y e l l o w i s h i t had powder obtained from the precipitations. A l l products melted between 130° - 140° and elemental a n a l y s i s correponded to those c a l c u l a t e d f o r benzaldehyde. figures Photopolymer y i e l d s ( a f t e r p r e c i p i t a t i o n and drying) were found to vary with i r r a d i a t i o n time as follows: 32% (100 h r s . ) , 48% (350 h r s . ) , 59% (660 h r s . ) , 68% (1110 h r s . ) . 3.3.b.iv. Attempted c h a r a c t e r i z a t i o n of the 'photopolymer' of benzaldehyde The s o l i d s t a t e i . r . spectra (KBr d i s c s ) and the u l t r a v i o l e t spectra (methanol) of the photoproducts obtained from i r r a d i a t i o n s v a r y i n g i n d u r a t i o n from lOO to 1100 h r s . were i d e n t i c a l and the same as those reported p r e v i o u s l y The ^"H n.m.r. spectrum (CDCl^, i n t . TMS (6 7.3). r e f . ) displayed o n l y one broad peak The "^C n.m.r. spectrum showed a peak a t 6 198.9 1 ( p o s s i b l y carbonyl carbon), a s i n g l e t a t 6 128.3 (aromatic r i n g carbons) and a s i n g l e t a t 6 42.38 which i s d i f f i c u l t to assign; i t s value i s however c l o s e to the one recorded for the sp"' hybridised carbon i n 1,4-dibenzylbenzene (see Chapter 2 ) . spectrum of the m a t e r i a l The mass m extended to / e 930 (d.p. of c a . 9) with prominent peaks a t i r r e g u l a r i n t e r v a l s , namely a t 8lO, 722, 706, 690, 616, 600, 583, 569, 553, 511, 493, 477, 465, 405, 389, 375, 361, 351, 339, 315, 301, 283, 271, 252, 239, 221, 210, 208, 180, 168, 152, 122, 105, 90, 77 (spectrum recorded a t 70 eV). Determinations of molecular weights i n chloroform s o l u t i o n gave d.p.'s i n the range 9 - 1 1 ( i s o p i e s t i c method) f o r p u r i f i e d samples, independent of i r r a d i a t i o n time. The s o l u t i o n phase i . r . spectrum of the photoproduct saturated s o l u t i o n ) was recorded i n the region 4000 to 2500 cm. Appendix B ) . indicated (carbon t e t r a c h l o r i d e , 1 (Spectrum 39, The spectrum showed some a l t e r a t i o n on s u c c e s s i v e d i l u t i o n and the presence of f r e e hydroxyl (3620 cm. ^, sharp), hydrogen bonded hydroxyl (3200 - 3500 cm. \ internally broad and s t r u c t u r e l e s s ) and - 161 - aromatic carbon - hydrogen s t r e t c h i n g (3030 - 3090 cm. ) . The shoulder v i s i b l e a t higher concentrations (D, Spectrum 39, Appendix B) might a r i s e from intermolecular hydrogen bonding and the band i n the region 2800 - 3000 cm. ^ d i s p l a y s s t r u c t u r e which a l s o a l t e r s somewhat on d i l u t i o n ; t h i s band would be expected to a r i s e from a l i p h a t i c carbon - hydrogen s t r e t c h i n g modes, however the band shape a l t e r a t i o n on d i l u t i o n i s p u z z l i n g . of the benzaldehyde The s o l i d s t a t e i . r . spectrum 'photopolymer' (No. 31, Appendix B) d i s p l a y s two absorptions i n the carbonyl r e g i o n , 1685 and 1723 cm. *; the carbonyl frequencies observed f o r benzoin benzoate, one of the reported benzaldehyde photoproducts, were 1712 cm. ^ ( e s t e r carbonyl) and 1698 cm. * (keto group); i t seems u n l i k e l y therefore t h a t the benzoin benzoate u n i t i s a p a r t of the photopolymer s t r u c t u r e , although i t must be admitted t h a t t h i s i s not a p a r t i c u l a r l y rigorous argument. I r r a d i a t i o n of the m a t e r i a l (3.0g) i n benzene (20 mis) (nitrogen streamed s o l u t i o n , 24 h r s . , 300nm) e f f e c t e d no change i n e i t h e r the molecular weight or the s p e c t r a l c h a r a c t e r i s t i c s . These usual p h y s i c a l approaches to s t r u c t u r e e l u c i d a t i o n d i d not y i e l d a convincing s t r u c t u r a l hypothesis although i t i s c l e a r t h a t the product contains hydroxyl, carbonyl and phenyl groups. I n view of t h i s f a c t i t was decided to attempt some i n v e s t i g a t i o n s o f chemical r e a c t i o n s on the benzaldehyde 'photo- polymer' i n the hope t h a t t h i s ' c l a s s i c a l ' approach might y i e l d some s t r u c t u r a l evidence. An account o f these preliminary s t u d i e s i s given below. Attempted photoreduction of the 'photopolymer' (3.0g, benzene/isopropanol 2:3 (50 m i s ) , nitrogen streamed s o l u t i o n , 24 h r s . , 300nm) y i e l d e d unchanged s t a r t i n g m a t e r i a l as evidenced by i . r . and n.m.r. spectroscopy. Attempts to obtain c r y s t a l l i n e d e r i v a t i v e s of the *photopolymer' v i a the carbonyl groups by r e a c t i n g small portions of i t with 2,4-dinitrophenylhydrazine i n ethanol or hydroxylamine hydrochloride i n the presence o f e i t h e r p y r i d i n e o r sodium a c e t a t e gave i n c o n c l u s i v e r e s u l t s . The l a t t e r experiment - 162 - a f f o r d e d r e c o v e r y o f s t a r t i n g m a t e r i a l w h e r e a s t h e former r e s u l t e d formation o f a m a t e r i a l w h i c h was u n s t a b l e and c o u l d n o t be T r e a t m e n t o f t h e p h o t o p r o d u c t w i t h bromine i n c a r b o n s o l u t i o n a t room t e m p e r a t u r e as evidenced purified. tetrachloride e f f e c t e d no change i n t h e s t r u c t u r e o f t h e m a t e r i a l by i . r . s p e c t r o s c o p y . s l o w a b s o r p t i o n o f bromine was However on h e a t i n g t h e s o l u t i o n a t 50° observed with simultaneous e v o l u t i o n o f an g a s , p o s s i b l y r e s u l t i n g from s u b s t i t u t i o n o r o x i d a t i o n r e a c t i o n s . the photoproduct did i n the (2.0g) w i t h bromine ( l . O g ) i n a c e t i c a c i d a acid Refluxing (20 m i s ) f o r 2 h r s . n o t e f f e c t i n c o r p o r a t i o n o f bromine i n t o t h e o r i g i n a l s t r u c t u r e a s shown by Lassaigne's test. The operation resulted t h e i . r . s p e c t r u m a t 1770 h y d r o x y l band was for cm. observed; i n t h e a p p e a r a n c e o f a new peak i n ^ b u t no change i n t h e shape o r i n t e n s i t y o f the t h e s e l a t e r r e s u l t s f a v o u r an o x i d a t i o n p r o c e s s the r e a c t i o n s w i t h bromine. R e f l u x i n g the 'photopolymer 1 (5.0g) w i t h a c e t y l c h l o r i d e (100 m i s ) f o r 3 h r s . under a n i t r o g e n b l a n k e t a f f o r d e d a m a t e r i a l whose i . r . s p e c t r u m showed a c o n s i d e r a b l y d i m i n i s h e d h y d r o x y l band i n t e n s i t y and cm. p r e s u m a b l y due to acetate group(s). i n t e n s i t y o f the a l i p h a t i c carbon R e f l u x i n g the in n e u t r a l acetone 1 photopolymer 1 No a new absorption a t 1740 s u b s t a n t i a l i n c r e a s e i n the - hydrogen a b s o r p t i o n was (5.0g) w i t h p o t a s s i u m however apparent. permanganate (750 m i s ) f o r 3 h r s . y i e l d e d a p r o d u c t (lO.Og) whose i . r . spectrum showed no m a j o r change from t h a t o f t h e o r i g i n a l m a t e r i a l . P y r o l y t i c decomposition aldehyde tar. The i n an i n e r t atmosphere y i e l d e d p r i m a r i l y b e n z - ( o v e r 90% y i e l d ) a l o n g w i t h s e v e r a l minor p r o d u c t s an intractable v e r y h i g h y i e l d o f b e n z a l d e h y d e from t h i s p y r o l y s i s s u g g e s t s u n i t i s n o t s u b s t a n t i a l l y changed on i n c o r p o r a t i o n i n t o the polymer. examination o f t h e n o n - v o l a t i l e r e s i d u e s by t . l . c . chloroform, acetone, carbon on s i l i c a t h a t the Attempted (benzene, t e t r a c h l o r i d e ) d i d not y i e l d d e f i n i t i v e s i n c e t h e r e were many o v e r l a p p i n g components. i t was and ethanol, results Contrary to previous r e p o r t s n o t p o s s i b l e t o i s o l a t e h y d r o b e n z o i n from t h i s experiment. 119 - 163 Reduction - w i t h a n e x c e s s o f sodium b o r o h y d r i d e i n 1,4-dioxane s o l u t i o n (40 h r s . a t r e f l u x ) r e s u l t e d i n a l m o s t c o m p l e t e e l i m i n a t i o n o f t h e c a r b o n y l f u n c t i o n ( s ) and spectrum (No. i n c r e a s e i n t h e i n t e n s i t y o f t h e h y d r o x y l bands i n t h e i . r . 32, Appendix B ) . m o l e c u l a r w e i g h t may Mass s p e c t r a l a n a l y s i s i n d i c a t e d t h a t h a v e been s i g n i f i c a n t l y r e d u c e d . r e d u c t i o n w i t h e x c e s s V i t r i d e i n benzene s o l u t i o n On the t h e o t h e r hand, ( n i t r o g e n b l a n k e t , 18 h r s . a t r e f l u x ) r*f.ar&i(i an a l m o s t q u a n t i t a t i v e r e c o v e r y o f a m a t e r i a l whose i . r . spectrum (No. 33, Appendix B) was s i g n i f i c a n t l y d i f f e r e n t from t h e one from t h e r e d u c t i o n w i t h sodium b o r o h y d r i d e a b s o r p t i o n s were t o t a l l y e l i m i n a t e d . rationalized i f the 1 does r e f l u x i n g of the and a s i g n i f i c a n t change spectrum this reaction Such a d i f f e r e n c e i n b e h a v i o u r (No. (Found: i n t h e i n t e n s i t y and i n t h e number and 38, Appendix B ) . C, 5.70%) was be reduces 'photopolymer' w i t h aqueous s u l p h u r i c a c i d 84.94; H, shape o f the h y d r o x y l (6N) bands i n t e n s i t i e s o f t h e c a r b o n y l bands i n Elemental a n a l y s i s of the product 5.07%; C a l c u l a t e d f o r (C_H.O) : / b H, might not. r e s u l t e d i n a marginal c h a n g e the i . r . i n t h i s case the carbonyl photopolymer• c o n t a i n e d e s t e r groups s i n c e V i t r i d e them w h e r e a s b o r o h y d r i d e Prolonged and obtained C, from 79.22? n c o n s i s t e n t w i t h t h e o v e r a l l change b e i n g r e p r e s e n t e d a s : - + (C.H.0) 7 6 n T h a t i s an a p p a r e n t l o s s o f one r e s i d u e s i n the polymer. " » molecule The p r o d u c t (C-H_0, ) 7 5 h n of water of t h i s r e a c t i o n although extremely nauseous s m e l l which p r e c l u d e d f u r t h e r Treatment of the dichromate f o r e v e r y two a s o l i d had (80 m i s ) ) w i t h (10 m i s ) ) and c o n c e n t r a t e d s u l p h u r i c a c i d potassium (15 m i s ) f o r 40 h r s . a t r e f l u x , r e s u l t e d i n a c o n s i d e r a b l e d e c r e a s e i n t h e i n t e n s i t y the bands i n the i . r . spectrum of a s s o c i a t e d w i t h h y d r o x y l and an i n c r e a s e i n t h e i n t e n s i t y and number o f bands i n t h e c a r b o n y l r e g i o n . s i g n i f i c a n t a l t e r a t i o n i n t h e p o s i t i o n and a l i p h a t i c carbon an examination. 'photopolymer' (3.0g i n benzene (20.Og i n w a t e r benzaldehyde T h e r e was also a i n t e n s i t y o f t h e bands i n t h e - hydrogen s t r e t c h i n g r e g i o n . Elemental a n a l y s i s of the - 164 product (Found: being represented C, 64.98; H, - 4.56%) was c o n s i s t e n t w i t h the o v e r a l l as:2- , + /H Cr 0 (C H O) » (C,H,0_ 7 6 n T h a t i s , an a p p a r e n t 7 6 2.5 n 'photopolymer'. p a r t i c u l a r l y r e p r o d u c i b l e and a t t e m p t s solvents (6.27%). _) i n c o r p o r a t i o n o f t h r e e oxygen atoms f o r e a c h p a i r benzaldehyde r e s i d u e s i n the product change T h i s experiment, to prepare of however, was larger q u a n t i t i e s of not the r e s u l t e d i n t h e i s o l a t i o n o f a g r e e n i s h powder, s o l u b l e i n o r g a n i c ( e t h a n o l , benzene, chloroform) w h i c h was found t o c o n t a i n chromium T r e a t m e n t o f t h i s m a t e r i a l w i t h d i e t h y l e t h e r a f f o r d e d an s o l u b l e m a t e r i a l which contained 1.28% chromium. ether- Treatment of both m a t e r i a l s w i t h d i l u t e s u l p h u r i c a c i d i n e t h e r s o l u t i o n d i d n o t e f f e c t r e m o v a l o f chromium; t h e s e m a t e r i a l s were p r o b a b l y The chromium c o m p l e x e s o f t h e r e s u l t s of the r e a c t i o n s o f the 'photopolymer'. *photopolymer' w i t h a c i d and o x i d i z i n g a g e n t s u n d e r a c i d c o n d i t i o n s a r e n o t amenable t o a s i m p l e e x p l a n a t i o n . hydroxyl d i m i n i s h i n g / c a r b o n y l i n c r e a s i n g e f f e c t of the a c i d reminiscent of a pinacol - pinacolone molecule obvious of water t y p e o f r e a c t i o n and The treatment i s the l o s s of a i s , of course, a l s o c o n s i s t e n t w i t h such a sequence. An c a n d i d a t e f o r t h e o x i d a t i o n r e a c t i o n w h i c h r e s u l t s i n an i n c r e a s e i n t h e oxygen c o n t e n t b u t no change i n t h e c a r b o n / h y d r o g e n r a t i o would be o x i d a t i o n o f aldehyde to c a r b o x y l i c a c i d u n i t s ; Appendix B) d o e s n o t s u p p o r t u n f o r t u n a t e l y the i n f r a r e d spectrum s u c h a r a t i o n a l i z a t i o n and p r o c e s s i s a c c o m p a n i e d by a marked d e c r e a s e p r o d u c t ' and 3.3.c. The 35, i n f a c t the o x i d a t i o n i n the hydroxyl intensity. I t has not proved p o s s i b l e , a t the time o f w r i t i n g to r a t i o n a l i z e r e s u l t s d e s c r i b e d above; (No. the p o s s i b l e nature of the benzaldehyde the 'photo- s u g g e s t i o n s f o r e l u c i d a t i o n o f i t s s t r u c t u r e a r e d i s c u s s e d below. C o n c l u s i o n s and s u g g e s t i o n s f o r f u r t h e r work p r e s e n t e x p e r i m e n t a l work adds t o t h e f a c t s known a b o u t t h e b e n z a l d e h y d e 'photopolymer• w i t h o u t s o l v i n g the problem o f i t s s t r u c t u r e . I t i s perhaps - 165 - s u r p r i s i n g t h a t i n t h e l i g h t o f a v a i l a b l e knowledge and remains unsolved techniques t h e problem d e s p i t e the i n t e r m i t t e n t a t t e n t i o n s of d i f f e r e n t workers a period of seventy y e a r s . I n attempting t o s e t up h y p o t h e s e s for test i t becomes c l e a r t h a t an enormous v a r i e t y o f s t r u c t u r e s c o u l d r e a s o n a b l y be d e r i v e d by of 'paper c h e m i s t r y ' f o r t h e p r o d u c t benzaldehyde; In Chapter this point i s i l l u s t r a t e d the photochemistry photochemical below. the combination process. I f we o f r a d i c a l p a i r s p r o d u c e d from c o n s i d e r the primary known t o be formed on i r r a d i a t i o n o f b e n z a l d e h y d e we o f p o s s i b i l i t i e s c a n be g e n e r a t e d in F i g u r e 3.8. p r o c e s s e s , and o f one The i t may products. products. be t h a t the I f , on these are illustrated 'photopolymer 1 a r i s e s from f u r t h e r r e a c t i o n s t h e o t h e r hand, t h e r a d i c a l - r a d i c a l r e or other o f the phenyl I I drawn i n t h e l o w e r h a l f o f t h e F i g u r e would be rings the Both t h e s e s p e c i e s c o u l d l e a d t o a v a r i e t y o f f u r t h e r However t h e s e a r e by no means t h e o n l y p l a u s i b l e i n i t i a l r a d i c a l - r a d i c a l combination, p o s i t i o n s of phenyl radicals. radicals s e e t h a t a v e r y l a r g e number from t h e i r c o m b i n a t i o n ; o c c u r r e d a t t h e p a r a - p o s i t i o n s o f one t h e s t r u c t u r e s I and initial pair of top p a r t o f t h e F i g u r e s u m m a r i z e s some o f t h e e s t a b l i s h e d of these m a t e r i a l s . combination irradiation o f a r o m a t i c c a r b o n y l compounds, d i s c u s s e d i n 2, a p r e v a l e n t theme was an i n i t i a l of prolonged over products of which could e q u a l l y reasonably occur a t ortho r i n g s (generating conjugated d i e n e s ) and between C l e a r l y t h e s t r u c t u r e s w h i c h c o u l d be drawn a r e a l m o s t identical endless, t h i s a p p r o a c h t o a s t r u c t u r a l h y p o t h e s i s i s u n l i k e l y t o be v e r y h e l p f u l a t and this stage. It The i s p e r h a p s w o r t h d i s c u s s i n g p o s s i b l e ways around t h e p r e s e n t t e c h n i q u e s a v a i l a b l e i n t h e p r e s e n t work s u g g e s t p o l y m e r o f b e n z a l d e h y d e , and a s i n g l e component ( t . l . c . on t h a t i t h a s a d.p. silica) be r e g e n e r a t e d on p y r o l y s i s a l t h o u g h in the polymer. Probably and t h a t the product i n the range 9 - 1 1 , difficulties. i s a homothat i t i s t h a t the b a s i c benzaldehyde u n i t s there are several d i f f e r e n t s t r u c t u r a l the f i r s t p o i n t to e s t a b l i s h i n any t h i s i n v e s t i g a t i o n would be t h e homogeneity o f t h e m a t e r i a l ; can units continuation of t h i s might be - 166 - H H // IOH O II hu C OH H i c O hu c C C- i // o o / OH C / H Vc H HO 4 KETONIZATION 6o F i g u r e 3.8. H I I l l u s t r a t i o n o f some p o s s i b l e modes o f c o m b i n a t i o n o f t h e p r i m a r y r a d i c a l p a i r formed on of irradiation benzaldehyde. t e s t e d u s i n g high p r e s s u r e l i q u i d chromatography. Assuming t h e p r o d u c t i s shown t o be homogeneous, t h e l i n e s o f s t r u c t u r a l i n v e s t i g a t i o n r e p o r t e d h e r e a r e probably capable of f u r t h e r e x p l o i t a t i o n . the p o s s i b i l i t i e s . Two examples w i l l serve to i l l u s t r a t e The p r o d u c t o f r e d u c t i o n w i t h V i t r i d e w i l l be a t l e a s t a two component m i x t u r e i f t h e 'photopolymer' backbone c o n t a i n s e s t e r links; - 167 - the components m i g h t be s e p a r a b l e by h i g h p r e s s u r e l i q u i d c h r o m a t o g r a p h y and amenable t o c h a r a c t e r i z a t i o n . The p y r o l y t i c f r a g m e n a t i o n o f t h e p r o d u c t s o b t a i n e d by t r e a t i n g t h e 'photopolymer' w i t h s u l p h u r i c a c i d and a g e n t s may the oxidizing y i e l d products other than benzaldehyde which might provide c l u e s to overall structure. The above s u g g e s t i o n s a r e i l l u s t r a t i v e o f t h e c h e m i c a l a p p r o a c h , and i t i s still p o s s i b l e t h a t s p e c t r o s c o p i c t e c h n i q u e s m i g h t y i e l d more d e t a i l e d information. the even n.m.r. s p e c t r a m i g h t p r o v e much more i n f o r m a t i v e i f a s a m p l e o f benzaldehyde if T h u s , improved "^C n.m.r. f a c i l i t i e s may w e l l be u s e f u l ; 'photopolymer' d e r i v e d from CgD^CHO were t o be p r e p a r e d . Finally, a s y s t e m a t i c s t u d y o f p o s s i b l e model compounds were c a r r i e d o u t t h e r e c e n t r e s o l u t i o n improvements i n t h e ESCA t e c h n i q u e m i g h t a l l o w u s e f u l i n f o r m a t i o n t o be deduced. structural CHAPTER 4 S t u d i e s on t h e c y c l o a d d i t i o n o f p e r f l u o r o g l u t a r y l f l u o r i d e t o some perfluoroolefins - 168 - 4.1. The P a t e r n o - B u c h i 4.1.a. reaction Introduction The p h o t o c y c l o a d d i t i o n o f c a r b o n y l compounds t o o l e f i n s l e a d i n g formation o f oxetanes ( F i g u r e 4.1) was f i r s t to the r e p o r t e d by P a t e r n o and C h i e f f i 119 at the beginning o f the century. u \ R / c C the reaction i n R \ ^ Bttchi r e i n v e s t i g a t e d R C C < hu R' Figure 4.1 1954^°^ and a s a r e s u l t o f h i s work t h e c y c l i c f o u r member e t h e r t h e r e a c t i o n p r o d u c t was v e r i f i e d . However, i t was n o t u n t i l structure for fourteen l a t e r t h a t t h e f i r s t a t t e m p t t o d e m o n s t r a t e t h e s c o p e and u s e f u l n e s s of the 29 r e a c t i o n was made. years ^ The r e a c t i o n i s frequently referred to as the Paterno- B u c h i r e a c t i o n and i t i s r e c o g n i s e d a s t h e most u s e f u l method f o r t h e s y n t h e s i s o f c y c l i c f o u r membered e t h e r (carbonyl Both s t a r t i n g materials compounds and o l e f i n s ) a r e r e a d i l y a v a i l a b l e e i t h e r c o m m e r c i a l l y o r through w e l l e s t a b l i s h e d nearly compounds ( o x e t a n e s ) . quantitative, laboratory syntheses. Y i e l d s v a r y from v e r y low t o depending on a v a r i e t y o f f a c t o r s , t h e number and n a t u r e o f s i d e r e a c t i o n s b e i n g one o f them. Comprehensive r e v i e w s o f t h e P a t e r n o - 29 207 Btlchi r e a c t i o n a r e a v a i l a b l e . In ' t h e o r i g i n a l e x p e r i m e n t , P a t e r n o and C h i e f f i r e p o r t e d t h a t insolation o f benzophenone i n a p e t r o l e u m m i x t u r e r i c h i n 2 - m e t h y l - 2 - b u t e n e r e s u l t e d i n the f o r m a t i o n o f a n o x e t a n e i n good y i e l d (Figure 4.2). Btichi's assignment o f o x e t a n e s t r u c t u r e s t o t h e p r o d u c t s o b t a i n e d by i r r a d i a t i o n o f a c e t o p h e n o n e , b e n z a l d e h y d e and b u t a n a l i n t h e p r e s e n c e o f p u r e 2 - m e t h y l - 2 - b u t e n e was b a s e d on t h e i d e n t i f i c a t i o n o f c a r b o n y l compounds formed from a c i d c a t a l y s e d cleavage - 169 - H_C 9 CH f c 9 H CH cu 3<0)- sunliqht C H H CH CH H CH CH F i g u r e 4.2 of these products. A c o m p r e h e n s i v e t a b l e o f o x e t a n e s p r e p a r e d by t h e v 29 P a t e r n o - B u c h i r e a c t i o n h a s been p r o d u c e d by A r n o l d . s u c c e s s f u l l y undergoing C a r b o n y l compounds p h o t o c y c l o a d d i t i o n i n c l u d e a l i p h a t i c and a r o m a t i c a l d e h y d e s and k e t o n e s , fluorocompounds, p - q u i n o n e s a s w e l l a s compounds c o n t a i n i n g f u n c t i o n a l groups i n a d d i t i o n t o t h e c a r b o n y l g r o u p . embrace l i n e a r and c y c l i c s y s t e m s , i n c l u d i n g f l u o r o o l e f i n s . used I n a d d i t i o n to o l e f i n s o t h e r u n s a t u r a t e d s y s t e m s have been employed i n c l u d i n g a c e t y l e n e s and k e t e n i m i n e s . Olefins allenes, The c y c l o a d d i t i o n may be i n t r a m o l e c u l a r o r i n t e r - molecular. 4.1.b. Mechanism P h o t o r e d u c t i o n and c y c l o a d d i t i o n r e a c t i o n s a r e c h a r a c t e r i s t i c o f t h e 29 carbonyl n ir* s t a t e and c a r b o n y l compounds u n d e r g o i n g 208 g e n e r a l l y undergo p h o t o r e d u c t i o n i n i s o p r o p a n o l . photocycloaddition The i m p o r t a n t s t e p s i n t h e r e a c t i o n f o r e l e c t r o n r i c h o l e f i n s may f r e q u e n t l y be r e p r e s e n t e d by t h e scheme shown i n F i g u r e 4.3. R e a c t i o n may be b r o u g h t about by i r r a d i a t i o n i n r e g i o n s where o n l y t h e c a r b o n y l compounds a b s o r b ; thus i n i t i a l c a r b o n y l chromophore c a n be d e m o n s t r a t e d . Deactivation of the excited without oxetane e x c i t a t i o n of the state f o r m a t i o n may o c c u r by a v a r i e t y o f c o m p e t i n g p r o c e s s e s , t h e -5 85 n,ir* t r i p l e t e x i s t i n g i n f l u i d s o l u t i o n f o r approximately 10 sec. o f c y c l o a d d i t i o n must be r a p i d enough t o o c c u r b e f o r e t h e e x c i t e d compound r e t u r n s t o t h e ground s t a t e by, f o r example, r a d i a t i v e The r a t e carbonyl deactivation. S i n g l e t and t r i p l e t e x c i t e d s t a t e s may be quenched b y i n t e r a c t i o n w i t h some - 170 - hi) i) Excitation ii) RCOR • Intersystem Crossing RCOR* (singlet) RCOR* ( s i n g l e t ) * RCOR* (triplet) radiative or non-radiative iii) D e a c t i v a t i o n RCOR* with or without with molecular iv) > RCOR (ground state) quenchers rearrangement Reaction 1 -R O •R' RCOR* R— • R' K" R F i g u r e 4.3: Reaction •R' — R' R' R scheme f o r o x e t a n e other molecule such a s an unsaturated system 29 R' formation The actual photocycloaddition g e n e r a l l y i n v o l v e s a t t a c k on t h e ground s t a t e o l e f i n by t h e n,ir* triplet 29 excited carbonyl compound. The r e a c t i o n s e q u e n c e i n F i g u r e 4.3 i n v o l v e s a d d i t i o n o f t h e l o n e e l e c t r o n o f t h e oxygen atom t o a n u n s a t u r a t e d giving a 1,4-biradical intermediate oxetane. which subsequently With unsymmetrical u n s a t u r a t e d system r i n g c l o s e s t o form t h e o l e f i n s two i s o m e r s may be formed; t h e s t e r e o c h e m i s t r y o f t h e m a j o r a d d u c t c a n be p r e d i c t e d from a c o n s i d e r a t i o n of t h e r e l a t i v e s t a b i l i t y of the possible b i r a d i c a l intermediates. n,rt* t r i p l e t s p i n i n v e r s i o n must o c c u r i s involved, T h e r e i s good e v i d e n c e f o r the formation before I f the 209 bonding. of the b i r a d i c a l intermediate 29 during the reaction, and a s i g n i f i c a n t number o f c y c l o a d d i t i o n p r o d u c t s c a n be r a t i o n a l i z e d by t h e b i r a d i c a l mechanism. T h e r e a r e however a l t e r n a t i v e mechanisms p r o p o s e d w h i c h i n c l u d e c h a r g e - t r a n s f e r complex f o r m a t i o n , a n d 211 212 e x c i p l e x formation. ' F o r i n s t a n c e i n t r a m o l e c u l a r c y c l o a d d i t i o n i n 5213 hept-2-one i s t h o u g h t t o p r o c e e d v i a s i n g l e t e x c i p l e x f o r m a t i o n . I t has b u t a d i e n e p r o c e e d s by a d d i t i o n o f d i e n e t r i p l e t t o ground s t a t e benzophenone. a l s o been s u g g e s t e d t h a t c y c l o a d d i t i o n o f benzophenone t o 2 , 3 - d i m e t h y l - l , 3 - 214 - 171 Other r e p o r t s - o f o x e t a n e f o r m a t i o n from e x c i t e d o l e f i n s and ground s t a t e • i , , , 215,216,217 c a r b o n y l compounnf. h.wo a p p e a r e d . 4.I.e. Limitations A m a j o r r e s t r i c t i o n on extent s u c c e s s f u l high y i e l d photocycloaddition is the n a t u r e and of competing p r o c e s s e s , by t h e e x c i t e d c a r b o n y l from t h e o l e f i n , m a i n l y hydrogen The c a s e may cyclohexene be exemplified (Figure 4.4), by where t h r e e p r o d u c t s i n a d d i t i o n ho H H 3 6 01' X2 99 *9 H H OH abstraction. to t h e o x e t a n e 9 6 H H & itself; the r e a c t i o n of p h o t o e x c i t e d benzaldehyde w i t h isolated. H abstraction s o l v e n t , o r even t h e o x e t a n e t h e hydrogen a t o e t h e r oxygen b e i n g p a r t i c u l a r l y s u s c e p t i b l e t o reactions OH Figure H H OH 4.4 H are - 172 Cyclobutane f o r m a t i o n may - be an a l t e r n a t i v e r e a c t i o n t o o x e t a n e formation, 218 a s w i t h 1,4-quinones s u c h a s c h l o r a n i l (Figure 4.5). and o R hu J* Figure t y p e I I p r o c e s s e s may N o r r i s h type I 4.5 compete w i t h o x e t a n e f o r m a t i o n . N o r r i s h type I rupture o f a c y l - h a l i d e bonds o f p e r f l u o r o a c y l c h l o r i d e s and b r o m i d e s competes w i t h 219 oxetane formation Products from t h e s e c a r b o n y l compounds. must be s t a b l e to the i r r a d i a t i o n c o n d i t i o n s used. a r e t r a n s p a r e n t i n t h e l o n g w a v e l e n g t h r e g i o n b u t may 220 at short Oxetanes r i n g open on irradiation wavelengths. P h o t o c h e m i c a l l y a c t i v e chromophores p r e s e n t i n a d d i t i o n t o t h e group o f t h e c a r b o n y l compound may l e a d t o c o m p l i c a t i o n s , f o r example i n f u r t h e r r e a c t i o n beyond o x e t a n e f o r m a t i o n 29 221 tetramethylallene (Figure 4.6). ' (CH ) C=C=C(CH ) 3 2 carbonyl 3 as with i r r a d i a t i o n of fluorenone 2 Figure 4.6 and - 173 - O x e t a n e p r o d u c t s though d e t e c t a b l e and n.m.r. s p e c t r o s c o p y may be c h a r a c t e r l z a b l e by Infrared and too u n s t a b l e f o r p u r i f i c a t i o n , an example b e i n g o x e t a n e from 4,4'-dimethyoxybenzophenone and i s o b u t y l e n e which fragments i n t o 29 formaldehyde and d i a r y l e t h y l e n e the (Figure 4.7). 208 ' OCH. I—T V2 hu CH =C(CH ) 2 3 2 l(4-CH.,0(C6 H 4'2 0 (4-CH OC H ) C=(CH ) 3 Figure 4.2. 6 4 2 3 + HCHO 2 4.7 A p p l i c a t i o n o f t h e P a t e r n o - B u c h i r e a c t i o n t o polymer s y n t h e s i s The p h o t o c h e m i c a l r e a c t i o n o f a c a r b o n y l group w i t h an o l e f i n y i e l d i n g an o x e t a n e h a s been u s e d f o r c r o s s l i n k i n g , c h a i n e x t e n s i o n and modification of p o l y m e r s . ^ I n p r i n c i p l e , t h e r e a r e two structural p o s s i b l e ways i n w h i c h the r e a c t i o n c a n be u s e d i n t h e s y n t h e s i s o f l i n e a r p o l y m e r s w i t h u n i t s i n the p o l y m e r c h a i n ; they a r e r e p r e s e n t e d by e q u a t i o n s ( i ) and ( i i ) 26 i n F i g u r e 4.8. The l o w e r r o u t e was examined by Andrews and oxetane Feast; h» 27 ' (i) \ 0 c c — c- \ + c Li Figure 4.8 i_L c c — y (ii) a v a r i e t y of aromatic bisketones m e t h y l a l l e n e and - 174 - and two d i f f e r e n t d i o l e f i n i c systems, f u r a n were used a s monomers. several c r i t e r i a : T h e i r c h o i c e was based the model r e a c t i o n s w i t h m o n o f u n c t i o n a l r e a g e n t s tetraon were 29 thoroughly s t u d i e d and u n d e r s t o o d ; photostable solvents; monomers and p o l y m e r s were s o l u b l e i n r e a c t i o n s p r o c e e d e d i n h i g h y i e l d and conversion without c h a i n t e r m i n a t i n g s i d e r e a c t i o n s and b o t h r e a c t a n t s and p r o d u c t s were s t a b l e t o r a d i a t i o n i n t h e e n e r g y iung<= r e q u i r e d f o r p h o t o p o l y m e r i z a t i o n . These r e s t r i c t i o n s eliminated a large proportion reactions as candidates o f t h e documented P a t e r n o - B t i c h i f o r photopolymerization studies. I n the case of the b i s k e t o n e - t e t r a m e t h y l a l l e n e photopolymerizations, i n v e s t i g a t i o n s c a r r i e d out e s t a b l i s h e d t h a t under a p p r o p r i a t e c o n d i t i o n s p o l y m e r s i n w h i c h c a . 90% o f the polymer c h a i n l i n k s a r e o x e t a n e s c a n prepared by i r r a d i a t i o n of equimolar s o l u t i o n s of aromatic 26 methylallene i n benzene. k e t o n e s seem t o be I n t h i s r e s p e c t , the behaviour i n l i n e with the behaviour of t h e i r raonofunctional co- be d i k e t o n e s and of aromatic the tetra- bisanalogue, benzophenone, w h i c h a f f o r d s c a . 90% o x e t a n e s upon i r r a d i a t i o n i n t e t r a m e t h y l a l l e n e , and 10% h y d r o x y l - c o n t a i n i n g formed i n an i n i t i a l products, a r i s i n g p r e s u m a b l y from hydrogen a b s t r a c t i o n r e a c t i o n by the radicals photoexcited 29 carbonyl ( s e e C h a p t e r 1, S e c t i o n 1 . 3 ) . The r e a c t i o n between benzophenone and f u r a n h a s been t h e s u b j e c t o f 222-229 numerous i n v e s t i g a t i o n s . I n 1963 i t was r e p o r t e d t h a t i r r a d i a t i o n benzophenone i n e x c e s s o f f u r a n g i v e s t h e 1:1 adduct, of 6,6-diphenyl-2,7-dioxa- 222,223,224 bicyclo[3.2.0Jhept-3-ene the f o r m a t i o n o f 2:1 i n near q u a n t i t a t i v e y i e l d . Subsequently a d d u c t s from t h e p h o t o c y c l o a d d i t i o n o f two molecules of 225 benzophenone t o one o f f u r a n was reported, and a d d u c t s i s o l a t e d were a s s i g n e d the s t r u c t u r e s shown i n t h e upper p a r t o f F i g u r e 4.9. chemical behaviour By a n a l o g y t o t h e p h o t o - o f the b i s k e t o n e - t e t r a m e t h y l a l l e n e system, i r r a d i a t i o n e q u i m o l a r amounts o f b i s k e t o n e s and f u r a n i n benzene were e x p e c t e d p o l y m e r s o f t h e s t r u c t u r e shown i n t h e l o w e r p a r t o f F i g u r e 4.9. to of yield Several - 175 O II - Q H it II Q o H H 5 H Q O. Ar i 3: 3 Ar Ar n Q Figure n 4.9 a t t e m p t s to e f f e c t t h e e x p e c t e d r e a c t i o n were l a r g e l y u n s u c c e s s f u l , y i e l d i n g o n l y low m o l e c u l a r w e i g h t m a t e r i a l s s u g g e s t i n g t h a t t h e p r o b l e m s i n h e r e n t i n t h e q u a n t i t a t i v e m a n i p u l a t i o n o f f u r a n (b.p. 32°C) were p r e v e n t i n g p r e p a r a t i o n o f p r e c i s e l y equimolar s o l u t i o n s . the Finally this difficulty was overcome by p r e p a r i n g 2:1 a d d u c t s between t h e d i k e t o n e s and f u r a n a c c o r d i n q t o the e q u a t i o n i n F i g u r e 4.10. These a d d u c t s b e i n g s o l i d s c o u l d be quantitatively Q \ ho Ar Figure manipulated Ar without d i f f i c u l t y , s o l u t i o n s o f 2:1 4.10 and s u b s e q u e n t i r r a d i a t i o n of equimolar f u r a n - d i k e t o n e a d d u c t s and d i k e t o n e s i n b e n z e n e gave r i s e 27 l i n e a r polymers o f d.p.'s upto 23. m a t e r i a l s r e v e a l e d both oxetane Spectroscopic examinations of these and h y d r o x y l bands i n t h e i n f r a r e d r e g i o n . to - 176 - P r e v i o u s s t u d i e s o f t h e r e a c t i o n between benzophenone and f u r a n had n o t revealed any t e n d e n c y f o r hydrogen a b s t r a c t i o n from f u r a n by benzophenone 222-229 triplets; i n d e e d i n t h e r e a c t i o n s o f benzophenone w i t h m e t h y l f u r a n s , where a b s t r a c t i o n from t h e a l l y l i c C - H bonds m i g h t be e x p e c t e d t o be favourable, 228 t h e a u t h o r s s p e c i f i c a l l y s t a t e t h a t o x e t a n e s were t h e o n l y p r o d u c t s . since the experimental d e t a i l s o f the polymerizations r e a c t i o n s were d i f f e r e n t i n s e v e r a l r e s p e c t s relatively could slow oxetane formation), be p o s t u l a t e d . For instance be and t h o s e o f t h e model (due t o low monomer s o l u b i l i t y and p o s s i b l e s o u r c e s o f t h e h y d r o x y l hydrogen s l o w p h o t o r e a c t i o n o f benzophenone w i t h 77 83—87 benzene i s known t o y i e l d b e n z o p i n a c o l and d i p h e n y l ; contain However, ' a l s o , a l l polymers a C - H bond a t o two e t h e r o x y g e n s and s u c h h y d r o g e n s a r e known t o p a r t i c u l a r l y s u s c e p t i b l e to a b s t r a c t i o n . 232 Such a b s t r a c t i o n s could lead to c r o s s l i n k i n g and p r e c i p i t a t i o n . It i s thus apparent t h a t a p p l i c a t i o n o f the Paterno-Btichi r e a c t i o n to polymer s y n t h e s i s i n t h e s y s t e m s i n v e s t i g a t e d was o n l y p a r t l y s u c c e s s f u l , m a i n l y due t o competing p r o c e s s e s ( e s p e c i a l l y hydrogen a b s t r a c t i o n and c r o s s l i n k i n g ) , r e l a t i v e l y low monomer s o l u b i l i t y and p r o l o n g e d i r r a d i a t i o n t i m e s . 4.3. Applications perfluoro o f t h e P a t e r n o - B u c h l r e a c t i o n t o p o l y f l u o r i n a t e d and systems C y c l i c ethers a s a c l a s s have b e e n known f o r a l o n g t i m e and h a v e been w e l l recognized as useful organic Generally ethers, preparation and p l a s t i c i z e r s . because o f I n 1961 however, a new c l a s s o f f o u r membered f l u o r o s u b s t i t u t e d o x e t a n e s , appeared i n t h e l i t e r a t u r e and t h e i r from f l u o r o o l e f i n s and f l u o r o a l d e h y d e s v i a t h e P a t e r n o - B t i c h i r e a c t i o n was d e s c r i b e d . ethers and a s s o l v e n t s s u c h e t h e r s have been l i m i t e d i n t h e i r u s e f u l n e s s r e l a t i v e l y low s t a b i l i t y . cyclic intermediates More s p e c i f i c a l l y t h i s new c l a s s o f p o l y f l u o r o c y c l i e c a r r i e d one h y d r o g e n atom on t h e r i n g c a r b o n a d j a c e n t to ether oxygen; t h e i r o u t s t a n d i n g c h e m i c a l and p h y s i c a l s t a b i l i t y was e m p h a s i z e d by t h e 233 author. - 177 - The new 2 - h y d r o p o l y f l u o r o o x e t a n e s r e p o r t e d r a n g e d l i q u i d s t o low m e l t i n g s o l i d s , depending carbons i n the molecule. from c l e a r , colourless g e n e r a l l y on t h e t o t a l number o f T h o s e c o n t a i n i n g l e s s t h a n about were c l e a r , c o l o u r l e s s l i q u i d s b o i l i n g from 100° - 300°C. eighteen carbons They e x h i b i t e d high h y d r o l y t i c s t a b i l i t y , b o t h under aqueous a c i d and aqueous b a s e c o n d i t i o n s . They were s o l u b l e i n a l k a n o l s , e t h e r s a n d v a r i o u s p e r f l u o r o c a r b o n s o l v e n t s b u t i n s o l u b l e i n w a t e r and s o l u t i o n s c o n t a i n i n g h i g h p e r c e n t a g e o f w a t e r . They were n o n - i n f l a m m a b l e and e x h i b i t e d o u t s t a n d i n g r e s i s t a n c e a g a i n s t t h e r m a l and oxidative degradation. T h e s e p r o p e r t i e s made t h e new compounds u s e f u l a s ' s t a b l e l i q u i d ' m a t e r i a l s , f o r example, a s t r a n s f o r m e r f l u i d s , f l u i d s for high t e m p e r a t u r e power t r a n s m i s s i o n s , o r h y d r a u l i c s y s t e m s , o r l i q u i d mechanical d r i v e s . coupled V i g o r o u s c h l o r i n a t i o n c o u l d s u b s t i t u t e t h e 2-hydrogen w i t h c h l o r i n e , and t h e 2 - c h l o r i n e c o u l d be r e a d i l y c o n v e r t e d t o o t h e r i n t e r e s t i n g c h e m i c a l i n t e r m e d i a t e s by c o n v e n t i o n a l methods o f r e p l a c e m e n t and modification of the r e l a t i v e l y reactive ring chlorine substituent using s t a n d a r d methods employed i n s y n t h e t i c o r g a n i c c h e m i s t r y . T h e s e p o l y f l u o r o - 2 - h y d r o o x e t a n e s were g e n e r a l l y p r e p a r e d by d i r e c t i r r a d i a t i o n o f t h e two r e a c t a n t s i n c y l i n d r i c a l q u a r t z r e a c t o r s approximately f o u r d i a m e t e r s l o n g , m a i n t a i n e d a t a t m o s p h e r i c p r e s s u r e and u n d e r r e f l u x a s o l i d carbon dioxide/acetone cooled condenser. r e a c t a n t s were u s e d . mercury resonance No s o l v e n t s f o r t h e two U l t r a v i o l e t l i g h t from a l o w - p r e s s u r e , 10 w a t t , q u a r t z , lamp f i t t e d the source o f energy. from i n a s p i r a l around I r r a d i a t i o n t i m e s ranged t h e r e a c t o r was employed a s from t h r e e d a y s (chlorotri- fluoroethylene/w-hydroperfluorovaleraldehyde) t o twelve days (hexafluoropropene-l/perfluoro-n-butyraldehyde). Y i e l d s ranged from 27% t o 6 6 % o f t h e o r y . 233 No mention o f b y - p r o d u c t s Oxetanes o r s i d e r e a c t i o n s was made by t h e a u t h o r . s u c c e s s f u l l y s y n t h e s i z e d by t h i s method a r e i l l u s t r a t e d i n F i g u r e 4.11 and i n c l u d e 3 , 4 , 4 - t r i f l u o r o - 3 - t r i f l u o r o m e t h y l - 2 - p e r f l u o r o - n propyloxetane (I), 3,4,4-trifluoro-2,3-bis(trifluoromethyl)oxetane (II), - 178 - 2-(4H-octafluoro-n-butyl)-3,4,4-trifluoro-3-trifluoromethyloxetane Vapour p h a s e chromatography and (III). n.m.r. s t u d i e s r e v e a l e d t h e p r e s e n c e CF CF 3 of two C,F 3 6 3 CF„ CF II II 2 Figure isomers f o r the o x e t a n e s o b t a i n e d , products. I I I 4.11 the s t r u c t u r e s g i v e n a r e f o r the m a j o r S i m i l a r p o l y f l u o r i n a t e d o x e t a n e s were r e p o r t e d i n the following 233,235,236 years. 236 I t was (450 w a t t r e p o r t e d by Cook and Hanovia h i g h - p r e s s u r e Landrum t h a t under a c t i n i c m e r c u r y lamp) h e x a f l u o r o a c e t o n e gave good y i e l d s of o x e t a n e s w i t h e t h y l e n e , v i n y l f l u o r i d e and With v i n y l and v i n y l i d e n e f l u o r i d e i t was p o s s i b l e a d d u c t s and irradiation vinylidene fluoride. p o s s i b l e t o i s o l a t e and a l s o determine the isomer identify distribution. Free r a d i c a l addition of a l i p h a t i c aldehydes to f l u o r i n a t e d o l e f i n s been shown t o y i e l d k e t o n e s d e r i v e d t e r m i n a l c a r b o n o f t h e d o u b l e bond. under c e r t a i n circumstances from a d d i t i o n o f an a c y l r a d i c a l t o 237,238 k e t o n e s and both B i s s e l l and o x e t a n e s can be has the F i e l d s have shown t h a t produced 239 simultaneously. phase of mixtures formation They r e p o r t e d of acetaldehyde o f complex m i x t u r e s that u l t r a v i o l e t irradiation and a fluorinated ethylene i n the gas r e s u l t e d i n the from w h i c h t h e k e t o n e d e r i v e d from a d d i t i o n an a c e t y l r a d i c a l to t h e CF^ group o f the o l e f i n and c y c l o a d d i t i o n of the aldehyde to the o l e f i n could the oxetane d e r i v e d be o b t a i n e d . Four tetrafluoroethylene, chlorotrifluoroethylene, bromotrifluoroethylene l , l - d i c h l o r o - 2 , 2 - d i f l u o r o e t h y l e n e were s t u d i e d . The of from olefins, and major product i n each case - 179 was the ketone. The r e a c t o r c o n s i s t e d of a 5 l i t r e c o o l e d q u a r t z w e l l was was - i n s e r t e d , a 100 f l a s k i n t o which a water- w a t t h i g h - p r e s s u r e m e r c u r y v a p o u r lamp suspended i n t h e w e l l a t t h e c e n t r e of the f l a s k . i n t r o d u c e d t h r o u g h a vacuum m a n i f o l d a t t a c h e d Gaseous r e a c t a n t s t o a s i d e arm The r e a c t a n t s were i n t r o d u c e d from s u p p l y c y l i n d e r s u n t i l was obtained. The m i x t u r e was h a l f i t s i n i t i a l value; component, b u t t'niis d i d n o t represent telomerlc materials extended f u r t h e r . reactor. the d e s i r e d i r r a d i a t e d u n t i l the pressure t h e y i e l d s o f b o t h k e t o n e and i r r a d i a t i o n t i m e was of the had were pressure fallen to complete consumption of either oxetane a c t u a l l y decreased i f the S i z e a b l e q u a n t i t i e s of high b o i l i n g a c c u m u l a t e d i n t h e bottom o f the r e a c t o r . Perfluorinated o x e t a n e s , i n o t h e r words o x e t a n e s w i t h e i t h e r f l u o r i n e o r p e r f l u o r i n a t e d attached to t h e r i n g c a r b o n s were p h o t o c h e m i c a l l y s y n t h e s i z e d f o r the groups first time 240 by H a r r i s and Coffman i n 1961. B e f o r e t h e p u b l i c a t i o n o f t h e i r work o n l y known example o f t h i s c l a s s was hexafluorooxetane i t s e l f , the p r e p a r e d by the 241 electrolytic c a r r i e d out f l u o r i n a t i o n o f t h e p a r e n t compound, o x e t a n e . e i t h e r a t atmospheric pressure the b.p.'s o f t h e r e a c t a n t s . o r i n g l a s s ampoules d e p e n d i n g I n the c a s e o f a t m o s p h e r i c p r e s s u r e the r e a c t o r c o n s i s t e d o f a v e r t i c a l q u a r t z tube magnetic s t i r r e r , a gas i n l e t a d a p t o r and ( 2 " x 10") the e x i t o f w h i c h was stream of n i t r o g e n h e l i x shaped passed. reactions, carbon acetone/solid carbon u l t r a v i o l e t r a d i a t i o n source consisted pressure m e r c u r y lamp w h i c h was filled of the r e a c t i o n mixture. w i t h the s h a k e n , t h e ampoule and p r o d u c t s were s e p a r a t e d attached to a s h a k e r . i t s c o n t e n t s were i r r a d i a t e d w i t h two H - 85 C - 3 lamps p l a c e d techniques. A l t e r n a t i v e l y , a g l a s s ampoule was f l u o r o c a r b o n y l compound and as c l o s e to the b o t t l e as p o s s i b l e . c h r o m a t o g r a p h i c a l l y and examined by of f i t t e d around q u a r t z r e a c t i o n t u b e so t h a t i t s r a d i a t i o n impinged p r i m a r i l y upon t h e portion dioxide f i t t e d w i t h a T - t u b e through w h i c h a s l o w The (4" x 2*1") low on f i t t e d with a a large acetone/solid c o o l e d c o n d e n s e r v e n t e d through a t r a p , a l s o c o o l e d by dioxide, S y n t h e s e s were a the liquid partially While being General I n both Electric cases, spectroscopic - 180 - Some p o r f l u o r o o x e t a n e s p r e p a r e d by t h e methods d e s c r i b e d listed i n T a b l e 4.1. of oxetanes cases prepared I t : c a n be s e e n from t h i s from a c i d o r d i a c i d a s h i g h a s 9 1 % . No b y - p r o d u c t s above a r e table that percentage y i e l d s f l u o r i d e s a r e v e r y h i g h , i n some from t h e s e r e a c t i o n s were r e p o r t e d by 240 the a u t h o r s . I n c o n t r a s t to t h i s behaviour, i r r a d i a t i o n o f sym-di- c h l o r o t e t r a f l u o r o a c e t o n e and h e x a f l u o r o p r o p e n e f o r two d a y s lead t o the formation o f t h e corresponding oxetane i n an ampoule (12%) , two u n i d e n t i f i e d i n 240 v o l a t i l e compounds, CF2CI2 ( 1 % ) , CO, CO^ and S i F In a following publication, Harris investigated 4 the photoreactions of 219 a c y l f l u o r i d e s i n the presence of f l u o r o o l e f i n s ; that he s t a t e d c a t e g o r i c a l l y ' i r r a d i a t i o n o f m i x t u r e s o f p o l y f l u o r o a c y l f l u o r i d e s and f l u o r o o l e f i n s leads p r i m a r i l y to the formation o f oxetanes; f l u o r i d e and h e x a f l u o r o p r o p y l e n e g i v e f o r example, p e r f l u o r o b u t a n o y l t h e c i s and t r a n s i s o m e r s o f 2 - p e r f l u o r o - n-propyl-3-trifluoromethyltetrafluorooxetane (Figure 4.12). V i r t u a l l y no C n-C F COF + 3 7 CF =CFCF 2 F " 3 7~ hu n 3 -CF. F F F i g u r e 4.12 o t h e r products a r e formed. 1 When t h e p h o t o r e a c t i o n o f h e x a f l u o r o p r o p y l e n e w i t h was perfluoroacylchlorides examined however, a v a r i e t y o f p r o d u c t s was formed. . T h u s p e r f l u o r o - b u t a n o y l c h l o r i d e and h e x a f l u o r o p r o p y l e n e y i e l d e d a t l e a s t t e n p r o d u c t s 240 (Figure 4.13). terminal In contrast to the reactions of fluoroacyl fluorides with fluoroolefins ( i n which 1:1 a d d u c t s were v i r t u a l l y p r o d u c t s ) o n l y s m a l l y i e l d s o f 1:1 a d d u c t s were f o u n d . the exclusive T h e r e were a l s o - 181 - 240 T a b l e 4.1. U l t r a v i o l e t c y c l e - a d d i t i o n s o f p e r f l u o r o c a r h o n y l compounds t o perfluoroolefins C a r b o n y l compound Olefin Period of irradiation (days) Oxetane (% y i e l d ) (CF ) 3 CF =CFCF CF^COCF^ 39g 2 (0.235m) 43g CF =CFCF C F C0C F CF =CFCF 3 7 45g 3 7 3 7 45g 3 7 40g C F (0.123m) 2 45g ( C 3 (0.233m) 2 (0.131m) C F COC F 2 35g (50) CF, (0.286m) C F COC F 2 5 2 6 43g (0.162m) 0—,(C F ) 3 F 5 11 (0.129m) 2 17g (62) 0 — 12 (C F ) 3 l c F,| ,1 7 2 ( 3 2 ) F ' 5 11 3 F CT (33, 2 CF. (0.133m) CF =CFCF 56g 95g 2 (0.483m) CF =CFCF 35g 68g 7 2 (0.162m) C O F 7 15 40g (O.H3m) F I 3 2 (38) |-CF F ° - f (0.453m) 2 hCF 3 3 CF =CFCF 50g O 3 (0.633m) C F C0F F 2 2 3 C 7 .CF. (0.233m) CF COF 3 (46) 3 CF =CFCF 2 } .CF„ 2 35g F 2 5 2 (0.267m) = C F C 2 3 L i ^ C F 3 F 3 7 (7 3) 3 15 13 (0.333m) 2 (91) |-CF ; r ~ 3 F F F0C(CF ) C0F CF =CFCF 26g 60g 2 3 (0.107m) 2 (CF ) COF 2 3 3 (34) (0.40m) CF. F F (CF ): 2 and 1(31) IF - 182 n-C,F_COCl 37 + C F =CFCF, 2 3 h " > CF CF 3 n-C F (CF -CF)Cl 3 7 - n-C,F_Cl 37 (trace) n-C,.F, + 6 14 21% A 3 O CI C F -n 3 C O ? + 2 + F h i 7 - n and/or CF 32% 3 g % 2 C 1 [ C F CF C F ( C F ) ]C1 + n-C.F C F ( C F _ ) C _ F -n 38% + CF,CFC1CF„C1 11% Figure trace 4.13 r e l a t i v e l y s m a l l amounts o f t h e p r o d u c t s o b t a i n e d from t h e p h o t o l y s i s o f acyl chloride i t s e l f . sequences The b u l k o f t h e p r o d u c t s was thought to a r i s e beginning w i t h a d d i t i o n s of the a c y l c h l o r i d e p h o t o l y s i s from fragments 240 (i.e. t h e c h l o r i n e atom and A s i m i l a r behaviour the p e r f l u o r o a l k y l r a d i c a l ) was observed b u t a n o y l b r o m i d e and p e r f l u o r o p r o p y l e n e n-C F CBr 3 n - C + ? F 4 9 C F ( C F 3 CF =CFCF 2 ) C F 3 7 C F . CF_ , 3 | 3 BrCF -C CFCF Br 2 2. _ n + + h " 3 n - c » F 3 7 c (Figure 4.14). BrCCF^F(CF > I C ^ 3 2 + 3 + + 2 C. „ F „ 12 26 Figure The olefin. i n t h e p h o t o r e a c t i o n between p e r f l u o r o - CCF CF(CF )] Br n-C F. 6 14 to the (?) + n-C.F_Br 3 7 4.14 a c y l h a l i d e / h e x a f l u o r o p r o p y l e n e r e a c t i o n s were a l l c a r r i e d o u t i n t h e same manner. The r e a c t o r c o n s i s t e d o f a v e r t i c a l q u a r t z tube f i t t e d with a magnetic s t i r r e r , a l a r g e acetone-dry (2 x l O i n . ) i c e cooled condenser through a d r y i c e c o o l e d t r a p , t h e e x i t o f w h i c h was f i t t e d w i t h a T-tube through w h i c h a slow s t r e a m o f n i t r o g e n p a s s e d . ultraviolet s o u r c e c o n s i s t e d o f a h e l i x shaped resonance (4 x 2.5 The vented radiation i n . ) low p r e s s u r e m e r c u r y lamp w h i c h f i t t e d around t h e q u a r t z r e a c t o r so t h a t i t s r a d i a t i o n - 183 - impinged p r i m a r i l y upon t h e l i q u i d p o r t i o n 4.4. The p h o t o l y s i s o f a c y l of the reaction mixture. fluorides A knowledge o f t h e band p o s i t i o n s and i n t e n s i t i e s forms t h e s t a r t i n g point f o r any photochemical i n v e s t i g a t i o n . I n the u l t r a v i o l e t spectra o f p e r f l u o r o a c y l h a l i d e s a marked b a t h o c h r o m i c s h i f t o f t h e maximum o f t h e n tr* t r a n s i t i o n i s o b s e r v e d on p a s s i n g from t h e f l u o r i d e s t h r o u g h chlorides 219 to t h e b r o m i d e s . The s p e c t r a l c h a r a c t e r i s t i c s o f t h e p e r f l u o r o b u t a n o y l h a l i d e s a r e r e c o r d e d i n T a b l e 4.2 a s an example. As e a r l y a s 1959, H a s z e l d i n e and Nyman d e m o n s t r a t e d t h a t i r r a d i a t i o n o f c h l o r o d i f l u o r o a c e t y l f l u o r i d e gave m a i n l y 1 , 2 - d i c h l o r o t e t r a f l u o r o e t h a n e and d i c h l o r o d i f l u o r o m e t h a n e w i t h s m a l l amounts o f s e v e r a l o t h e r materials T a b l e 4.2 A b s o r p t i o n bands o f p e r f l u o r o b u t y r y l h a l i d e s i n t h e u.v. r e g i o n n-C^COX Gaseous p h a s e Cyclohexane X A (nm) (e) max A F 215 (66) CI 258 fluoride (nm) ( e ) - ( 3 8 ) ; 266 (37) Br including max 259 (46.5); 217 (692); 266 (46) 273 (56) c a r b o n y l f l u o r i d e , c a r b o n y l c h l o r i d e f l u o r i d e , and s i l i c o n (Figure C1CF tetra- 4.15). COF h " » C1CF C F C I + C I CF + C0FC1 + COF + SiF F i g u r e 4.15 More d e t a i l e d p h o t o l y t i c s t u d i e s o f p o l y f l u o r o a c y l f l u o r i d e s were reported 219 by Harris; t h e e x p e r i m e n t a l p r o c e d u r e was t h e same a s i n t h e a c y l h a l i d e / p e r f l u o r o p r o p e n e i r r a d i a t i o n s (see S e c t i o n 4.3). P o l y f l u o r o a l k a n e s were the - 184 main p r o d u c t s obtained - from t h e s e p h o t o l y s e s ; f o r example, from t h e of p e r f l u o r o b u t a n o y l f l u o r i d e , the major product hexane. was i s o l a t e d was I n a d d i t i o n a h i g h e r b o i l i n g f r a c t i o n was i s o l a t e d having perfluoro-n- obtained from w h i c h an ether t h e s t r u c t u r e (n-C^F^)^CFOC^F^-n. A n a l y s i s o f the i n f r a r e d s p e c t r a o f t h e e f f l u e n t g a s e s c o n s i s t e d o f 5 - 15% C C ^ , t r a c e s o f S i F and CO^. irradiation and 4 C 3 F I J and 7 indicated that t h e r e m a i n d e r was they CO T h e s e r e s u l t s wore c o n s i s t e n t w i t h the scheme p r o p o s e d by H a r r i s (Figure 4.16). I n t h i s scheme t h e p h o t o e x c i t e d C F COF 3 C F d!bF ? 3 C F COF 3 2 ? • C F * ? 3 C P 3 a c y l f l u o r i d e molecule c l e a v e s + 7 *COF > 7 C C F * 3 + 7 F 3 7°v C F COF 3 XT' ? C F T 7 C F " 3 7 C F C^ + 3 7 CF" • C F OCF(C F ) 3 7 3 7 2 C F T 7 2 'COF F i g u r e 4.16: • Scheme a c c o u n t i n g COF + 2 CO for r e s u l t s of i r r a d i a t i n g p e r f l u o r o - butanoy1fluoride t o g i v e a p e r f l u o r o p r o p y l r a d i c a l and p e r f l u o r o p r o p a n e was observed a fluoroformyl r a d i c a l . Since no among t h e r e a c t i o n p r o d u c t s , i t f o l l o w s t h a t t h e p e r f l u o r o p r o p y l r a d i c a l does n o t a b s t r a c t a f l u o r i n e atom from the fluoride. acyl- I t i s a l s o p r o b a b l e t h a t t h e a l t e r n a t i v e mode o f c l e a v a g e o f the * e x c i t e d a c y l f l u o r i d e molecule ( i . e . C F C O F 3 occur. C F CO" 7 3 + 7 F") does not To a m a j o r e x t e n t t h e p e r f l u o r o p r o p y l r a d i c a l r e a c t s w i t h a n o t h e r r a d i c a l to give perfluorohexane; however i t c a n a l s o a t t a c k a m o l e c u l e o f a c y l f l u o r i d e a t t h e oxygen t o g i v e t h e r a d i c a l C F O C F C F 3 combines w i t h a n o t h e r such 7 3 7 the which e v e n t u a l l y p e r f l u o r o p r o p y l r a d i c a l t o g i v e an e t h e r . The f a t e of - 105 - the "COF r a d i c a l produced i n t h e p h o t o l y s i s was i n d i c a t e d by t h e d e t e c t i o n o f C02» CO and COF2 i n t h e e f f l u e n t g a s e s . T h e r a d i c a l may d i r e c t l y t o COF2 and CO a s i n d i c a t e d i n F i g u r e disproportionate 4.16 o r p o s s i b l y to g i v e o x a l y l f l u o r i d e which then undergoes d i s p r o p o r t i o n a t i o n . and i t dimerizes The CO2 S i F ^ p r e s u m a b l y a r i s e by r e a c t i o n o f COF2, o x a l y l f l u o r i d e o r t h e "COF r a d i c a l with the w a l l s o f the r e a c t i o n v e s s e l . From t h e i r r a d i a t i o n o f a m i x t u r e o f p e r f l u o r o b u t a n o y l f l u o r i d e and p e r f l u o r o o c t a n o y l f l u o r i d e , perfluoro- n-hexane, -decane, and - t e t r a d e c a n e were i s o l a t e d a s e x p e c t e d on t h e b a s i s o f t h e r e a c t i o n scheme g i v e n abovei I r r a d i a t i o n o f a d i a c y l f l u o r i d e , p e r f l u o r o g l u t a r y l f l u o r i d e was c a r r i e d out under t h e same e x p e r i m e n t a l c o n d i t i o n s f o r four days. t i m e , t h e r e a c t i o n m i x t u r e was c o m p l e t e l y s o l i d . evident: i ) hard b r i t t l e m a t e r i a l stuck A t t h e end o f t h i s Two k i n d s o f w h i t e s o l i d t o t h e s i d e s o f t h e r e a c t o r and ii) somewhat 'mushy' s o l i d from t h e c e n t r e p o r t i o n The t o t a l y i e l d o f r e c o v e r e d s o l i d was r o u g h l y 4 5 % on a w e i g h t b a s i s . solids reacted 190° not with moist a i r to give, of the reaction v e s s e l . Both a f t e r s e v e r a l days, products melting a t - 300° and 100° - 195° r e s p e c t i v e l y . extensively were The n a t u r e o f t h e s e m a t e r i a l s was i n v e s t i g a t e d by t h e a u t h o r , who assumed t h a t t h e y were l a r g e l y l o n g - c h a i n d i c a r b o x y l i c a c i d f l u o r i d e s o f s t r u c t u r e FOC ( C F 2 ) n C O F . 3 I n c o n t r a s t t o t h e b e h a v i o u r o f p e r f l u o r o a c y l f l u o r i d e s , the. i r r a d i a t i o n of a p e r f l u o r o a c y l c h l o r i d e leads to decarbonylation r e s u l t i n g p r i m a r i l y i n the 240 formation o f a p o l y f l u o r o a l k y l c h l o r i d e . C l e a v a g e o f t h e C - C I bond seems t o o c c u r w i t h t h e f o r m a t i o n o f R-CO* and C I " r a d i c a l s . hexafluoroglutarylchloride The p h o t o l y s i s o f a p p a r e n t l y p r o c e e d s i n a manner a n a l o g o u s t o t h a t of monofunctional a c i d c h l o r i d e s y i e l d i n g 1,3-dichloro-hexafluoropropane a s t h e m a j o r p r o d u c t a l o n g w i t h some 1 , 6 - d i c h l o r o p e r f l u o r o h e x a n e . The r e l a t i v e p r o p o r t i o n o f t h e p r o d u c t r e s u l t i n g from d i m e r i z a t i o n o f r a d i c a l s i s s i g n i f i c a n t l y l a r g e r than f o r monofunctional a c y l c h l o r i d e s . No p r o d u c t s w i t h 240 more t h a n s i x c a r b o n s were i s o l a t e d and no p e r f l u o r o c y c l o p r o p a n e was f o u n d . - 186 - From t h e p h o t o l y s e s o f p o l y f l u o r o a c y l b r o m i d e s o n l y t h e d e c a r b o n y l a t i o n p r o d u c t s , p e r f l u o r o a l k y l b r o m i d e s were i s o l a t e d . F o r example, from 5 H - o c t a - fluorovalerylbromide, 4H-octafluorobutylbromide (93%) and CO were o b t a i n e d . The d i f f e r e n c e s i n behaviour e x h i b i t e d by t h e t h r e e t y p e s o f 240 acyl h a l i d e s upon i r r a d i a t i o n can be r e l a t e d t o t h e r e l a t i v e s t r e n g t h s o f t h e C - X bonds i n t h e compounds. I n the p h o t o l y s i s o f the a c y l f l u o r i d e s , i n which t h i s bond i s t h e s t r o n g e s t , no p r o d u c t s a r e found whose f o r m a t i o n would demand i t s r u p t u r e t o y i e l d a f l u o r i n e atom. 4.5. Original idea A p p l i c a t i o n o f t h e P a t e r n o - B u c h i r e a c t i o n t o polymer s y n t h e s e s i n t h e a r o m a t i c d i k e t o n e / d i o l e f i n s e r i e s was d i s c u s s e d i n S e c t i o n 4.2. behaviour On only p a r t l y successful for reasons t h e o t h e r hand, l i t e r a t u r e i n f o r m a t i o n on the o f t h e p e r f l u o r o a c y l f l u o r i d e / p e r f l u o r o o l e f i n s y s t e m on e x p o s u r e ultraviolet irradiation a t t r a c t i v e one ( S e c t i o n s 4.3 and 4.4) made t h i s s y s t e m f o r u s e i n l i n e a r polymer s y n t h e s i s . appear Thus, the use o f an liquid p e r f l u o r o r e a c t a n t s would e l i m i n a t e t h e c o m p l i c a t i o n s i n t r o d u c e d by u s e solvents. I r r a d i a t i o n s of p r e c i s e l y equimolar f l u o r i d e s / p e r f l u o r o d i o l e f i n s would be e x p e c t e d weight mixtures of to of perfluorodiacid- to y i e l d l i n e a r high molecular p e r f l u o r o p o l y o x e t a n e s s i n c e no s i d e r e a c t i o n s seem t o compete w i t h 240 oxetane formation. Such p r e c i s e l y m e t e r e d m i x t u r e s would be e x p e c t e d be e a s i l y formed and l i n e techniques. t h e n be e x c l u d e d transferred to i n t o q u a r t z v e s s e l s u s i n g c o n v e n t i o n a l vacuum Oxygen, w h i c h d i s s o l v e s v e r y r e a d i l y i n f l u o r o c a r b o n s , could from t h e m i x t u r e by s u c c e s s i v e f r e e z e - pump - thaw c y c l e s . P r o d u c t s would be e x p e c t e d photopolymerization. t o be t r a n s p a r e n t i n t h e e n e r g y The main a d v a n t a g e o f t h e p e r f l u o r o o l e f i n s y s t e m however, l i e s perfluorodiacidfluoride/ i n the absence a b s t r a c t i o n r e a c t i o n s by t h e p h o t o e x c i t e d c a r b o n y l . c o u l d i n t e r f e r e w i t h polymer formation range r e q u i r e d f o r of competing hydrogen The o n l y r e a c t i o n i s the t e l o m e r i z a t i o n of the which acid - 187 - fluoride, ' w h i c h would d e s t r o y t h e r e q u i r e d o l e f i n and a c y l f l u o r i d e g r o u p s . d.p. would be l i m i t e d ; As reaction.^ however t h e r e was a c a t e g o r i c a l s t a t e m e n t i n t h e o x e t a n e f o r m a t i o n was t h e 0 b r i e f l y mentioned i n S e c t i o n compounds i s complex, i t f.n a l s c therefore of the I f t h i s reaction occurred the attainable l i t e r a t u r e t h a t under a p p r o p r i a t e c o n d i t i o n s exclusive 1:1 s t o i c h i o m e t r y 1.7 t h e p h o t o c h e m i s t r y o f c a r b o n y l an a r e a n e c e s s a r y f o r workers using of considerable a c t i v i t y and i t i s carbonyl photochemistry as a s y n t h e t i c method t o be r e a d y t o modify p r o c e d u r e s i n t h e l i g h t o f new i n f o r m a t i o n . is f o r example, e s t a b l i s h e d reaction that the d e t a i l e d course o f a photochemical i s dependent on t h e n a t u r e o f i n c i d e n t l i g h t c h r o m a t i c ! t y ) and t h e p h y s i c a l c o n d i t i o n s solution, nature of solvent, I t ( w a v e l e n g t h and mono- of the reagents (gas, solid, presence of s e n s i t i z e r s or quenchers). T h u s an approach to t h e i n v e s t i g a t i o n o f step-growth p h o t o p o l y m e r i z a t i o n s l e a d i n g t o perfluoro or highly f l u o r i n a t e d p o l y o x e i t a n e s would i n v o l v e a compromise between choosing systems which could y i e l d i n t e r e s t i n g polymers i f a p p r o p r i a t e reaction conditions c o u l d be found and c h o o s i n g s y s t e m s known t o p r o c e e d i n h i g h y i e l d and c o n v e r s i o n and would t h e r e f o r e within a reasonable The be more l i k e l y t o y i e l d p o l y m e r s time. f i r s t o b j e c t i v e o f t h e e x e r c i s e was t h e r e f o r e the photochemical e x a m i n a t i o n o f s i m p l e s y s t e m s , by i r r a d i a t i n g a w e l l - c h a r a c t e r i z e d and vigorously As p u r i f i e d d i a c i d f l u o r i d e i n t h e presence o f an o l e f i n . mentioned i n S e c t i o n 1.6, p e r f l u o r o g l u t a r y l f l u o r i d e was c h o s e n , p a r t l y b e c a u s e o f t h e f a c t t h a t i t s p h o t o c h e m i s t r y h a s been documented and t h e r e no already r e a s o n s w h i c h would e x c l u d e i t a s a c a n d i d a t e f o r p o s s i b l e seem t o be photo- 240 polymerization attempts, in i t s preparation and p a r t l y b e c a u s e o f t h e c o n s i d e r a b l e a v a i l a b l e i n t h i s Department. Two d i f f e r e n t o l e f i n s were a l s o u s e d , a c y c l i c one and a s t r a i g h t c h a i n Results o b t a i n e d w i l l be d i s c u s s e d i n Section 4.6. expertise perfluoro- terminal one. - 188 - 4.6. Experimental 4.6.a. Starting materials P e r f l u o r o g l u t a r y l f l u o r i d e was p r e p a r e d by t h e r o u t e o u t l i n e d i n F i g u r e 4.17. Typical r e s u l t s f o r the various stages i n t h i s synthesis are as Cl./AlCl. STAGE 1 STAGE 2 STAGE 3 F KMn0 /acetone 4 KF/CH..CN 3 4 F F STAGE 5 STAGE 4 F i g u r e 4.17 follows:Stage 1. trichloride mechanical initially P e r c h l o r o c y c l o p e n t a d i e n e (780g, 2.85 m o l e s ) and a l u m i n i u m (121g, 0.92 m o l e s ) were mixed i n a 1 1. R.B. f l a s k u s i n g a stirrer. C h l o r i n e was p a s s e d i n t o t h e m i x t u r e ; h e a t was a p p l i e d t o s t a r t t h e r e a c t i o n and t h e r e a f t e r c h l o r i n e was p a s s e d a t s u c h o a r a t e t h a t t h e temperature o f t h e m i x t u r e was m a i n t a i n e d a t ^ 45 C by t h e exothermicity of the reaction. r e a c t i o n temperature The f l o w o f c h l o r i n e was s t o p p e d when t h e f e l l below 30°C. The b l a c k s o l i d o b t a i n e d was t r e a t e d with l a r g e e x c e s s o f water u n t i l a y e l l o w o i l appeared. Recrystallization o f t h i s o i l from e t h a n o l g a v e 649g o f o c t a c h l o r o c y c l o p e n t e n e ( 6 6 % ) i d e n t i f i e d by i.r. spectroscopy (u 1620, 1200, 770 cm. . max Stage potassium 2. Octachlorocyclopentene f l u o r i d e were mixed w i t h 2.5 l i t r e s o f s u l p h o l a n e i n a 5 1. R.B. f l a s k using a mechanical 180° (350g, 1.02 m o l e s ) and 942g o f d r y stirrer. - 185° f o r a b o u t l * j d a y s . and c o l l e c t e d T e m p e r a t u r e was r a i s e d and m a i n t a i n e d a t P r o d u c t formed was c a r r i e d v i a n i t r o g e n s t r e a m i n liquid a i r traps. F r a c t i o n a l d i s t i l l a t i o n o f the crude - 189 p r o d u c t gave 129g spectroscopy (u S t a g e 3. - of octafluorocyclopentene 1780, max 1720, 1410, 1340, (60%) i d e n t i f i e d by 1310, 300g o f p o t a s s i u m permanganate 1235, 1180, (1.90 m o l e s ) lOOO, 9 0 0 ) . were added i n small p o r t i o n s to a s t i r r e d s o l u t i o n of octafluorocyclopentene 0.47 moles) i n 2 l i t r e s of dry acetone. Temperature was and then r e f l u x f o r 2 hours. m i x t u r e was w i t h SO2 i n a p o l y t h e n e bag. I t was then f i l t e r e d , a c i d i f i e d with d i l . An e t h e r e a l s o l u t i o n was c o n t i n u o u s e x t r a c t i o n o v e r n i g h t , d r i e d o v e r MgSO^ and r e m o v a l o f e t h e r , m a t e r i a l was S H2 °4 a n ( lOOg were k e p t f o r t h e n e x t s t a g e . s p e c t r o s c o p y (u the acetone was * decolourised then obtained filtered. o b t a i n e d by vacuum d i s t i l l a t i o n by After (0.01 mm. Perfluoroglutaric acid 3500, 1780, 1180 cm. 1 of temperature 2 l i t r e s o f w a t e r were t h e n added and removed, t h e aqueous s u s p e n s i o n was i d e n t i f i e d by i . r . On c o m p l e t i o n a l l o w e d t o warm up t o room allowed to stand o v e r n i g h t . Hg/140°C). (lOOg, l o w e r e d by means o f an a c e t o n e / d r i c o l d b a t h and m a i n t a i n e d a t below -20°C. the p'sriranganate a d d i t i o n , s o l u t i o n was i.r. was ). max S t a g e 4. 350g 140g (1.79 m o l e s ) flask (0.73 moles) o f p e r f l u o r o g l u t a r i c a c i d were mixed w i t h o f p r e v i o u s l y d i s t i l l e d b e n z o t r i c h l o r i d e i n a 1 1. f i t t e d w i t h a water c o n d e n s e r / a c e t o n e - d r i c o l d c o l d f i n g e r connected H to a 2 S 0 6 1 4 bubbl - Temperature was system r a i s e d up t o 212° u s i n g a l i s s a p o l nx b a t h w h i l e s t i r r i n g was p r o v i d e d by means o f a m a g n e t i c A f t e r about 5 h r s . of b o i l i n g , distillation 78° - 160° g.l.c. was f r a c t i o n was r e t a i n e d and i d e n t i f i e d by i . r . S t a g e 5. (3 f o o t c o l u m n / g l a s s h e l i c e s ) . found t o c o n t a i n m a i n l y one P e r f l u o r o g l u t a r y l c h l o r i d e thus obtained spectroscopy ( u m a x Perfluoroglutarylchloride 1810, 1200, (258g, 1.14 lllO, moles) to a s o l u t i o n o f 1 Kg. o f d r y p o t a s s i u m f l u o r i d e i n 2.5 nitrile stirrer. ( f r a c t i o n a t e d from P ° 5 ^ i n a 5 1. R.B. 2 On stirrer. t h e a p p a r a t u s was m o d i f i e d f o r f r a c t i o n a l and m i x t u r e was d i s t i l l e d ( C o l . A/100°C). R.B. 990, was The component by (144g, 890, 90%) 805 cm." s l o w l y added l i t r e s of dry aceto- f l a s k using a mechanical a d d i t i o n of the a c i d c h l o r i d e , the pot temperature r o s e ; the - 190 - r e a c t i o n was assumed t o have f i n i s h e d when, a f t e r a d d i n g c h l o r i d e t h e p o t temperature a l l the acid went down t o room t e m p e r a t u r e . then m o d i f i e d f o r f r a c t i o n a t i o n (3 f o o t c o l u m n / g l a s s T h e a p p a r a t u s was helices); t h e 45° - 48° f r a c t i o n was found t o c o n t a i n m a i n l y a c i d f l u o r i d e and some a c e t o n i t r i l e by g.l.c. ( C o l . A/room t e m p e r a t u r e ) . I n f r a r e d a n a l y s i s of t h e product obtained (156g, 6 9 % ) r e v e a l e d t h e p r e s e n c e o f some a c i d c h l o r i d e - peak a t 1810 cm. (u max : 1 1 11B0, 1200, 1135, 3.010, 900 c m . " ) , P e r f l u o r o h e p t - l - e n e was s y n t h e s i z e d by p y r o l y s i n g t h e c o r r e s p o n d i n g 244 perfluorocarboxylic acid salt: CF., ( C F J , C 0 0 H 3 2 6 N S - ° H » CF„ ( C F j COONa 3 2 6 CF (CF ) CF=CF 3 Perfluoro-octanoic acid o f NaOH i n 100 m i s o f w a t e r . 8 hrs. Product obtained about 1 h r . 4 2 (lOOg, 0.24 m o l e s ) was n e u t r a l i s e d u s i n g 9.66g S a l t was d r i e d b y pumping under vacuum f o r (103g, 9 9 % ) was p y r o l y s e d (40O°C/760 mm Hg) R e s u l t i n g c o l o u r l e s s l i q u i d was washed w i t h a q . l^CO^, P and vacuum t r a n s f e r r e d from 2 ° 5 ' t h e p r o d u c t t o be p u r e . i.r. 2 spectroscopy (u c 9-1- - a t room t e m p e r a t u r e for degassed ( C o l . A) showed T e r m i n a l o l e f i n t h u s o b t a i n e d was i d e n t i f i e d b y : 1 1780, 1370, 1320, 1200, 1070, 940 c m . " ) . Yield nicix after purification: 71g ( 8 6 % ) . P e r f l u o r o c y c l o h e x e n e was p r o v i d e d . f l u o r i n a t i o n of undecafluorocyclohexane. ( C o l . A/70°C) and c h a r a c t e r i z e d b y i . r . I t was o b t a i n e d from t h e d e h y d r o I t was found t o be p u r e b y g . l . c . spectroscopy (u : 1740, 1370, nicix 1300, 1260, 1190, 1150, 1110, 1010, 4.6.b. 1 980 c m . " ) . Irradiations General Procedure S t a r t i n g m a t e r i a l s were a c c u r a t e l y weighed s o a s t o g i v e a 2:1 o l e f i n / P a c i d f l u o r i d e m o l a r r a t i o and were i n t r o d u c e d i n t o a f l a s k c o n t a i n i n g 2 ° 5 " - 191 They were d e g a s s e d - by means o f 4 - 5 f r e e z e - pump - thaw c y c l e s on c o n v e n t i o n a l vacuum l i n e and t h e n vacuum t r a n s f e r r e d a into c y l i n d r i c a l v e s s e l s and s e a l e d under r e d u c e d p r e s s u r e (10 ^ t o r r ) . quartz After irradiations, t u b e s were f r o z e n i n l i q u i d a i r , h o t s p o t t e d , opened and c o n n e c t e d through t r a p s t o a c o n v e n t i o n a l vacuum l i n e . to the vessel in series, b a t h ) and 4.7. two t r a p s were c o n n e c t e d t h e one n e a r e s t t o i t b e i n g a t -78°C t h e o t h e r one a e i n g a t -178°C s e p a r a t e d by 4.6.c. The two (acetone/dricold (liquid a i r ) . P r o d u c t s were t h u s volatility. R e s u l t s (see Table 4.3) Discussion The r e s u l t s o b t a i n e d i n d i c a t e t h a t d e s p i t e l i t e r a t u r e r e p o r t s on 4= i *i , *i * 4-v. 219,233,234,235,236,240 s u c c e s s f u l p e r f l u o r o and p o l y f l u o r o o x e t a n e s y n t h e s e s o f t e n i n h i g h y i e l d s , none o f t h e r e a c t i o n s a t t e m p t e d amount o f o x e t a n e . v e r y low y i e l d , h e r e gave a d e t e c t a b l e I n most c a s e s telomeirs o f a c i d f l u o r i d e were o b t a i n e d i n i n a d d i t i o n t o low v o l a t i l i t y products. I r r a d i a t i o n o f p e r f l u o r o h e p t - l - e n e by i t s e l f u s i n g t h e 5-w Hanovia low p r e s s u r e lamp which d e f i n i t e l y h a s an o u t p u t a t 185nm where t h e m i g h t be e x p e c t e d to absorb to a product which, seemed l i k e l y ( E x p t . 9, T a b l e 4.3) r e s u l t e d i n a 26% on t h e b a s i s o f g . l . c . a n a l y s i s and t o be a m i x t u r e o f i s o m e r s and infrared olefin conversion spectroscopy telomers of the s t a r t i n g olefin. I s o m e r i z a t i o n o f f l u o r o o l e f i n s v i a f l u o r i n e atom s h i f t s h a s b e e n r e p o r t e d r e c e n t l y by H a s z e l d i n e f o r t h e i r r a d i a t i o n o f perfluoro-2,3-dimethylbut- 245 2-ene. The low t e m p e r a t u r e some e a r l i e r a t t e m p t s had reaction ( E x p e r i m e n t 5) was that almost a l l examples r e p o r t e d i n the l i t e r a t u r e o c c u r r e d a t t e m p e r a t u r e s well route to oxetane o x e t a n e was I t seemed p o s s i b l e t h a t i n o u r e x p e r i m e n t s f o r m a t i o n may formed i t may i t was (after noted below a m b i e n t . f a i l e d to g i v e oxetanes) because c a r r i e d out t h e d i r a d i c a l on have e i t h e r r e v e r t e d t o monomers o r i f t h e have been i n a h i g h v i b r a t i o n a l e n e r g y state and th - 192 - II Dl II u *> 1) o > o • I in t; -i • o I-I o -rl .11 > in rH HI .-I •ft •U •u o X <d 3 c C-i fl .c: w -I •rl x; c S •H in •* v o o •iH in n» o nl !H (V O U C (U •H \ o o r-l • id c u IT El .(.: ID D< U |fl ^ - X id i) o> 01 4J in at K 0 •-! A) 1 or - 01 IF K O • id rH IJ1 .n a o m •o r-l in 4J •O -rt •rl O •H ••-I rH <M Ul o O >1 •d ID 3 0) •rl r-l 7 J > •d G X 11 rQ in id (1 <u •-I •U P, 4-> c o in <H <U !>i 2-.MH m o •O ID O M' as at u nl c 0 •i HI •ii o y n. U o u (J •H #-* I i/i i) i: (V -H c li l» o id Pi O ki • O tr IJ rl 0) Q > O m c [I 0 O •H id £1 CT- a r: rH s o I-I .c u in r-i • in iv nl >" 10 H id o o y. • •a a 3 rH o <u u a> t: u •d -rl il u O id •.-I f-H •rl 4-> in nl r-l .-I fO O -H > n (V -u s o o d (N IN 0\ 10 m r-i in i-i o CN r-l O ll IN u id •1) 111 i: Vl -H U ll 111 *J 0J r* t 0 u CO O o m r- li) to K in in Jo c-« — O in in O m in in o I CO Ul in r- 00 in lO 1.0 o u, u o r>. u O in 0) V) en tri ~ O + o 8 O 8 CO cn t f4 •u -rH •a O ti. o o U u U ft, (J U o U b, O U ti, O U In O rl -.-I o -j O r-l i-C bi o u r>. o (ii U U O n CM U W »i O o o fu U n U OI fc. U U II II fc. O Ii. U uII [., U u u ro fu G 0 o fi. CJ II b. O *r CM CN U bi U U U - 193 - Table 4.3 ( c o n t . ) Footnotes: a. Olefin/acid fluoride r a t i o : b. Lamp Lamp Lamp Lamp c. Using r e f r i g e r a t e d methanol b a t h . d. As % o f t o t a l w e i g h t o f s t a r t i n g m a t e r i a l s e. Plus isomers by i . r . evidence (peak a t 1720 cm. ^ A B C D F HANOVIA RAYONET HANOVIA HANOVIA 450-W Hg Lamp RPR-2O0 253.7nm PCR 1L/5W Low P Hg Lamp 125W medium P Hg Lamp F \ / C=C / R f. 2:1. f ) . \ R f Gc/MS examination o f t h e 8 component m i x t u r e r e v e a l e d t h e presence o f a substance (m/e = 469) which c o u l d be e i t h e r OFC(CF-) CFO o r t h e 1:1 adduct. I . r . evidence ( s t r o n g - COF -1 a b s o r p t i o n 1880 cm. ) seems t o f a v o u r OFC(CF_)-CFO. 6 g. I . r . spectrum o f o i l showed s t r o n g -COF a b s o r p t i o n s t r o n g R--C00H a b s o r p t i o n Hi lOOO cm. h. (1880 cm. ^) (1780 cm. ^) and s t r o n g bands around , c h a r a c t e r i s t i c o f oxetanes. Waxy s o l i d o b t a i n e d by t r e a t i n g i n v o l a t i l e m a t e r i a l s w i t h CCl^. I . r . spectrum f a i l e d t o r e v e a l s t r o n g -COF band. M a t e r i a l s l i g h t l y s o l u b l e i n H-0 g i v i n g s t r o n g l y a c i d s o l u t i o n (pH = 1 -»• 2) . k. Gc t r a c e ( C o l . A/30°). starting materials. I . r . spectrum s i m i l a r t o t h a t o f 1. Orange wax s o l u b l e i n h o t H^O ( s t r o n g l y a c i d i c s o l u t i o n ) , i n acetone, i n s o l u b l e i n Et^O, CCl^. I . r . spectrum shows t h e presence o f s t r o n g -COF a b s o r p t i o n (1880 cm. . m. Low v o l a t i l i t y m a t e r i a l s heated a t 200°/0.01 mm Hg t o y i e l d a y e l l o w o i l and a r e m a i n i n g r e d brown v i s c o u s l i q u i d . I . r . s p e c t r a o f b o t h m a t e r i a l s appeared v e r y s i m i l a r . No -COF a b s o r p t i o n around 1880 cm. ^, s t r o n g band a t 1720 cm. ^. Probably t e l o m e r and/or isomers o f p e r f l u o r o o l e f i n . n. I . r . e x a m i n a t i o n o f low v o l a t i l i t y f r a c t i o n showed a much s t r o n g e r peak a t 1720 cm. * and a s m a l l e r peak a t 1880"* ( t e r m i n a l perfluoroolefin). F r a c t i o n heated a t 280°/0.01 mm Hg f o r 2 h r s . t o y i e l d a c o l o u r l e s s f a i r l y m o b i l e l i q u i d and a y e l l o w i s h o i l d i s p l a y i n g s i m i l a r i . r . and n.m.r. s p e c t r a . Gc t r a c e s ( C o l . 0^ a t - 165°) show 4 and 7 components r e s p e c t i v e l y - p r o b a b l y telomers o f t h e p e r f l u o r o o l e f i n . different - 194 - o i t s e l f have c l e a v e d . was formed. However, even a t -20 no d e t e c t a b l e amount o f oxetane The I n t e r e s t i n g f i n d i n g was t h a t a t low t e m p e r a t u r e s no o l e f i n i s o m e r i z a t i o n was observed e i t h e r (Experiment 10, Table 4 . 3 ) . Since l i t e r a t u r e r e p o r t s o r i g i n a t e f r o m r e p u t a b l e e s t a b l i s h m e n t s , a r e w e l l documented and have been r e p e a t e d and extended by s e v e r a l o t h e r w o r k e r s , the p o s s i b i l i t y o f u n r e l i a b l e r e p o r t s can be d i s m i s s e d , and t h e reasons f o r our f a i l u r e r e q u i r e c a r e f v l a n a l y s i s . An o b v i o u s q u e s t i o n i s whether lamps used had s u f f i c i e n t o u t p u t a t t h e r e q u i r e d wavelengths and t h i s i s n o t easy t o deal w i t h . The f a c t t h a t t e l o m e r i z a t i o n r e a c t i o n s o f b o t h a c i d f l u o r i d e and o l e f i n components have been observed can be t a k e n as good evidence t h a t was absorbed by monomers t o some e x t e n t . light N e v e r t h e l e s s , i t would be more s a t i s f a c t o r y i f a h i g h i n t e n s i t y l i g h t source i n c o r p o r a t i n g a broad band monochromator were a v a i l a b l e f o r p r e p a r a t i v e work which r e q u i r e s wavelengths s h o r t e r t h a n t h e 254nm band o f t h e mercury lamp; equipment. we were unable t o o b t a i n such The a b s o r p t i o n o f p e r f l u o r o a c y l f l u o r i d e s i s r e p o r t e d as X 215nm 219 (e - 6 6 ) , whereas t h e s h o r t w a v e l e n g t h end o f t h e spectrum o f a mercury lamp c o n s i s t s p r e d o m i n a n t l y o f l i n e s a t 185, 238, 248 and 254nm. between t h e e m i s s i o n o f t h e lamp and t h e COF Overlap a b s o r p t i o n w i l l t h e r e f o r e be poor, which p o s s i b l y accounts f o r t h e l o n g ( s e v e r a l days) i r r a d i a t i o n p e r i o d s r e p o r t e d for s u c c e s s f u l p e r f l u o r o - o x e t a n e syntheses d e s p i t e t h e use o f h i g h powered 219,233,234,235,240 „ . ' _ _ . lamps. On b a l a n c e , i t seems t h a t s i n c e o t h e r s have J4 t s u c c e s s f u l l y used mercury lamps, o v e r l a p between lamp e m i s s i o n and COF a b s o r p t i o n was n o t t h e main cause o f t h e f a i l u r e t o s y n t h e s i z e o x e t a n e s , however a more r e a d i l y r e g u l a t e d system would o f f e r advantages t o any f u t u r e worker i n this field. Having c o n s i d e r e d q u e s t i o n s r e l a t e d t o t h e ' F i r s t Law o f P h o t o c h e m i s t r y ' , doubts about t h e p u r i t y o f r e a c t a n t s need a t t e n t i o n . was i d e n t i f i e d by i . r . , m.s., r e l i a b l y pure. and n.m.r. s p e c t r o s c o p y . Perfluorohept-l-ene G.l.c. showed i t t o be P e r f l u o r o c y c l o h e x e n e was a l s o found t o be p u r e by g . l . c . - 195 - As f a r as t h e p u r i t y o f a c i d f l u o r i d e i s concerned, i t was known from t h e beginning t o contain traces o f a c e t o n i t r i l e ( g . l . c . e v i d e n c e ) , which appears 29 n o t t o quench t h e c a r b o n y l t r i p l e t . Careful fractionation under U (using a 3 f o o t column packed w i t h 'Dixon gauzes') b e f o r e use, a f f o r d e d m a t e r i a l , which, on g . l . c . e x a m i n a t i o n , appeared t o be f r e e from a c i d c h l o r i d e c o n t a m i n a t i o n . However, s i n c e p u r i f i c a t i o n o f a c i d f l u o r i d e prepared by exchange f l u o r i n a t i o n i s a ver> i i f t i c u l t t a s k :.i.i£inly flue t o t h e f o r m a t i o n o f a c i d h a l i d e azeotropes t h e p o s s i b i l i t y o f a c i d c h l o r i d e t r a c e s b e i n g p r e s e n t can n o t be e x c l u d e d . I f t h i s were t h e case, i t would be expected t h a t R.C0C1 (A 258nm, e ^ r max 219 40) would o v e r l a p s t r o n g l y w i t h t h e i n t e n s e 254nm band o f t h e mercury lamp; consequently a c y l c h l o r i d e s o r t h e i r p h o t o l y s i s p r o d u c t s c o u l d be a c t i n g as quenchers f o r t h e e x c i t e d s t a t e r e q u i r e d f o r oxetane f o r m a t i o n , o r i n t e r f e r i n g w i t h oxetane f o r m a t i o n a t some o t h e r stage. I n most cases, a c i d f l u o r i d e s used f o r s u c c e s s f u l oxetane syntheses were 219 prepared e i t h e r from a c i d s u s i n g SF^ o r by e l e c t r o c h e m i c a l f l u o r i n a t i o n . One case o f a s u c c e s s f u l use o f an a c i d f l u o r i d e o r i g i n a t i n g from an a c i d 219 c h l o r i d e has been r e p o r t e d . However m a t e r i a l was subsequently d i s t i l l a t i o n t h r o u g h a spinning-band still, p u r i f i e d by as opposed t o t h e f r a c t i o n a l d i s t i l l a t i o n procedure used by u s . Quartz g l a s s used f o r t h e p r e p a r a t i o n o f t h e i r r a d i a t i o n tubes was examined by u l t r a v i o l e t spectroscopy. I t was found t o be t r a n s p a r e n t i n t h e r e g i o n 220 - 300nm and v i r t u a l l y t r a n s p a r e n t i n t h e r e g i o n below 220nm; some s l i g h t a b s o r p t i o n was observed a t 215nm (A = 0.13) where t h e COF group i s r e p o r t e d t o 219 absorb. T h i s however should n o t be s i g n i f i c a n t f o r t h e r a t i o n a l i z a t i o n o f the f a i l u r e t o o b t a i n t h e d e s i r e d adducts. F i n a l l y , a sample o f p e r f l u o r o g l u t a r y l f l u o r i d e used f o r i r r a d i a t i o n s was examined f o r C I c o n t e n t by shaking i t i n a sealed tube w i t h aqueous s i l v e r nitrate. them. S i l v e r s a l t s o b t a i n e d a f t e r t r e a t m e n t d i d n o t c o n t a i n AgCl amongst I t should be k e p t i n mind however t h a t an e f f i c i e n t quencher can t o t a l l y 24 i n h i b i t r e a c t i o n a t e x t r e m e l y low c o n c e n t r a t i o n s indeed, as t h e bismaleiraide , - 196 - p h o t o p o l y m e r i z a t i o n experiments by De S c h r y v e r , Feast and Smets have shown (Section 1.3). 4.8. Conclusions The p r e l i m i n a r y i n v e s t i g a t i o n s c a r r i e d o u t on t h e c y c l o a d d i t i o n o f p e r f l u o r o g l u t a r y l f l u o r i d e t o t h e two p e r f l u o r o o l e f i n s under a v a r i e t y o f e x p e r i m e n t a l c o n d i t i o n s have served t o i l l u s t r a t e s y n t h e t i c o r g a n i c c h e m i s t r y can be. how d i f f i c u l t t h i s area o f The i s o l a t i o n o f h i g h p u r i t y FOC (CF2) . j C O F f o l l o w i n g i t s p r e p a r a t i o n from t h e c o r r e s p o n d i n g a c i d c h l o r i d e i n CH^CN s o l v e n t appears t o be a s e r i o u s problem n o t easy t o d e a l w i t h by u s i n g s t r a i g h t f o r w a r d fractional distillation techniques. High temperature i r r a d i a t i o n s seem t o promote o l e f i n t e l o m e r i z a t i o n , whereas low temperature i r r a d i a t i o n s seem t o l e a v e t h e o l e f i n unchanged. of I t i s d i f f i c u l t t o be d e f i n i t e about t h e causes f a i l u r e t o o b t a i n t h e oxetanes. High p u r i t y s t a r t i n g m a t e r i a l s a r e r e q u i r e d f o r t h e s e r i o u s i n v e s t i g a t i o n o f any chemical r e a c t i o n . This c o n d i t i o n i s i m p e r a t i v e i n t h e case o f photochemical i n v e s t i g a t i o n s . Bearing t h i s i n mind, and g i v e n t h e suspect p u r i t y o f p e r f l u o r o g l u t a r y l f l u o r i d e any f u t u r e examiner o f t h e proposed syntheses r e p o r t e d i n t h i s c h a p t e r m i g h t do w e l l t o s y n t h e s i z e p e r f l u o r o g l u t a r y l f l u o r i d e by an a l t e r n a t i v e r o u t e so as t o exclude t h e p o s s i b i l i t y o f c h l o r i d e contamination. APPENDICES AND REFERENCES APPENDIX A APPARATUS, INSTRUMENTS AND GENERAL TECHNIQUES Vacuum system Freeze d r y i n g , p y r o l y s e s , freeze-pump-thaw c y c l e s , s u b l i m a t i o n s and v a c u u m d i s t i l l a t i o n s r e q u i r i n g p r e s s u r e s o f t h e o r d e r o f 10 mm Hg were undertaken w i t h a c o n v e n t i o n a l vacuum system i n c o r p o r a t i n g a r o t a r y o i l pump and a mercury d i f f u s i o n pump. Pressures as low as 10 ^ mm Hg were a c h i e v e d (where r e q u i r e d ) by means o f a grease f r e e , m e r c u r y - f r e e vacuum l i n e i n c o r p o r a t i n g a r o t a r y o i l pump, an Edwards vapour pump, e l e c t r o n i c p r e s s u r e measuring d e v i c e s and ' t e f l o n ' t a p s . I n f r a r e d s p e c t r a - see Appendix B. Mass s p e c t r a were measured w i t h an A.E.I. MS9 s p e c t r o m e t e r . U l t r a v i o l e t s p e c t r a were r e c o r d e d on a Unicam SP800 s p e c t r o p h o t o m e t e r . N.m.r. s p e c t r a were recorded on a Bruker S p e c t r o s p i n HX 90E High R e s o l u t i o n NMR s p e c t r o m e t e r o p e r a t i n g a t 90MHz f o r *H and 22.635MHz f o r ^ C (ambient temperature). M o l e c u l a r w e i g h t s were r e c o r d e d on a Perkin-Elmer 115 M o l e c u l a r Weight Apparatus u t i l i z i n g t h e vapour p r e s s u r e ( i s o p i e s t i c ) method o f d e t e r m i n i n g t h e number average m o l e c u l a r w e i g h t o f a s o l u t e i n s o l u t i o n . i n chloroform solution; A l l measurements were made b e n z i l was used as s t a n d a r d f o r t h e d e t e r m i n a t i o n of the instrument constant. An o p e r a t i n g temperature o f 37° was employed. M o l e c u l a r w e i g h t s c o u l d be measured w i t h + 2% s t a n d a r d d e v i a t i o n . A n a l y t i c a l vapour phase chromatography A Perkin-Elmer 452 Gas Chromatograph was used w i t h a column o f d i - n d e c y l p h t h a l a t e / C e l i t e (2.2m x 7mm d i a . ) , hydrogen as c a r r i e r gas and a h o t wire detector. Carbon and hydrogen a n a l y s e s were c a r r i e d o u t w i t h a Perkin-Elmer 240 E l e m e n t a l Analyser. Chromium a n a l y s i s was c a r r i e d o u t on a Perkin-Elmer 403 Atomic Absorption Spectrophotometer. T h i n l a y e r chromatography was c a r r i e d o u t u s i n g Merck 60F-254 s i l i c a g e l p l a t e s and c a r e f u l l y p u r i f i e d s o l v e n t s a t room t e m p e r a t u r e . M e l t i n g p o i n t s were r e c o r d e d u s i n g a R e i c h e r t m e l t i n g p o i n t microscope and a r e uncorrected. Irradiations. Unless o t h e r w i s e i n d i c a t e d , i r r a d i a t i o n s were c a r r i e d o u t w i t h Rayonet Type RS P r e p a r a t i v e Photochemical Reactors (Models RPR-204 o r RPR-208). sources were e i t h e r RUL-3500& lamps o r RUL-3000A*. Temperatures were ca. 30°. R e a c t i o n v e s s e l s were g e n e r a l l y c y l i n d r i c a l i n shape and 7 diameters long. Light approximately APPENDIX B INFRARED SPECTRA I n f r a r e d s p e c t r a were r e c o r d e d on a P e r k i n - E l m e r 457 I n f r a r e d Spectrophotometer (PE457) o r a P e r k i n - E l m e r Spectrophotometer (PE577). conditions designated Spectrum No. S p e c t r a were r u n u s i n g KBr Grating Infrared c e l l s under t h e by: (A) KBr (B) Nujol (C) Thin contact f i l m disc mull Instrument PE457 577 Grating Discussed on page; 66 Material Conditions triphenylglycol isolated from i r r a d i a t i o n o f Ph CO + PhgCHOH 2 PE457 84 triphenylglycol obtained v i a ground s t a t e syntheses (recrystallized) PE577 85 triphenylglycol obtained v i a ground s t a t e syntheses (sublimed) PE577 74 crude product from n ±zr of m-dibenzoylbenzene + hydrobenzoin 5. PE577 76 product i n CU^Cl^ from d i e t h y l - mesoxalate + PhLi PE457 84 a-phenylbenzoin 7. PE457 69 p r o d u c t from i r rn o f a-phenylb e n z o i n i n benzene s o l u t i o n 8. PE457 69 p r o d u c t f r o m i r rn o f a-phenylb e n z o i n + b e n z h y d r o l i n benzene PE457 82 1,2,2,2-tetraphenylethanol B vi-ecrystallized) 10. PE457 83 1,2,2,2-tetraphenylethanol (sublimed) 11. PE457 82 12. PE577 113 m-bisbenzhydrylbenzene 13. PE457 109 p-bisbenzhydrylbenzene 14 PE457 112 m-dibenzoylbenzene o i l d i s t i l l e d from products o f p-bisbenzhydrylbenzene + HCOOH 15 PE457 111 (PhCBrH)„C-H. p a r a isomer 2 6 4 B 16. PE457 112 p o l y m e r i c m a t e r i a l formed on B C H t r e a t m e n t o f (PhCBrH) 6 4» P a r a 2 isomer, w i t h V i t r i d e i n benzene 17. PE577 110 (PhCClH) „C,.H . p a r a isomer Z b H 18. PE457 113 (PhCClH)„C.H. meta isomer 19. PE457 116 1,4-dibenzylbenzene 20. PE457 116 1,3-dibenzylbenzene 21. PE577 95 ^ 6 4 1,1,2,2-tetraphenylethanol I r r a d i a t i o n s o f equlmolar amounts o f m-dlbenzoylbenzene (DBB) and e i t h e r p-dibenzylbenzene (p-dbb) o r m-dibenzylbenzene (m-dbb). 22. PE457 117 DBB/p-dbb: yellow product from preliminary investigation (24 h r s . i r r a d i a t i o n ) 23. PE457 119 DBB/p-dbb: t y p i c a l spectrum o f white p r e c i p i t a t e 24. PE457 119 DBB/m-dbb: t y p i c a l spectrum o f white p r e c i p i t a t e 25. PE457 121 DBB/p-dbb: t y p i c a l spectrum o f yellow polymeric m a t e r i a l (578 h r s . i r r a d i a t i o n ) 26. PE457 122 DBB/m-dbb: t y p i c a l spectrum o f yellow polymeric m a t e r i a l (578 h r s . i r r a d i a t i o n ) 27 PE457 128 b l a c k t a r o b t a i n e d from p y r o l y s i s of yellow polymeric m a t e r i a l from DBB/p-dbb (sample 0) 28. PE457 131 p r o d u c t from t r e a t m e n t o f sample (0) w i t h a c e t i c a c i d / i o d i n e i n benzene B Photoreduction o f dlaldehydes 29. PE457 155 p r o d u c t from i r r 1 1 o f tere- phthalaldehyde i n isopropanol/ benzene 30. PE457 155 product from i r r 1 1 of iso- phthalaldehyde i n isopropanol/ benzene The benzaldehyde p h o t o p r o d u c t 31. PE457 160 T y p i c a l spectrum o f t h e benzaldehyde p h o t o p r o d u c t (BPP) NaBH, 32. PE457 163 p r o d u c t from BPP dioxane 33. PE457 163 ^ ^ „„„ p r o d u c t f r o m BPP 34. PE457 162 p r o d u c t f r o m BPP ffif^v-j—* chloride 35. PE577 163 j A. B „„„ p r o d u c t f r o m BPP dichromate • • •> H c Vitride _ > Benzene v (no Cr found) 36. PE457 164 p r o d u c t f r o m BPP d l c h r ° H m a t e l c h r ° H m a t e ^ (Cr c o n t e n t 6.27%) 37. PE457 164 p r o d u c t from BPP d ) B (Cr c o n t e n t 1.28%) 6N H SO. -—2-» 38. PE457 163 p r o d u c t f r o m BPP 39. PE457 160 BPP i n CC1. s o l u t i o n 4 Pure CCl. 4 75% Pure CCl /25% s a t u r a t e d s o l n . o f BPP i n C C l 4 4 50% Pure CCl^/50% s a t u r a t e d s o l n . o f BPP i n CCl^ s a t u r a t e d s o l u t i o n o f BPP i n CC1. I I I I I I I I I I I 1 I I I — \ N*2 I I I I I I I I I I I I I I N"5 «no^a» a DO 2yo 2000 an SB am mn m u nm u » «mn ion am I' 600 am too 250 N*7 in Ml y i NT2 I IF i i i ii i ^^^^^ 25)0 20X) 18D i It X) KOO 1210 KB 803 600 q p 250 r 1 i 25P nVM:...JMI ayo tfa «pD UP an 10 p aoo 600 «jO N-21 I vrn urn \ I N*a 1 I OT7 mo r*T T T T T TT T T T T Ti I I J I I n Jo 44 A A AlUUi , m A 40 r36 WW i jUOO^SOO '3000 2500 ••1.1,1.1.1.1.1.1,1.1.1.1,l,I,I W I 1 1600 1400 1200 I I I TO I 800 1 400 I i 400 250 I I APPENDIX C The Board o f Studies i n Chemistry r e q u i r e s t h a t each postgraduate r e s e a r c h t h e s i s contain an appendix (a) listing a l l research c o l l o q u i a , research seminars and l e c t u r e s (by e x t e r n a l speakers) arranged by the Department o f Chemistry s i n c e 1 October 1976; and (b) a l l research conferences attended by the w r i t e r of the t h e s i s , during the p e r i o d when the research f o r the t h e s i s was c a r r i e d out. 1. Research C o l l o q u i a , Seminars and L e c t u r e s 1.1. 1976-77 a) U n i v e r s i t y o f Durham Chemistry C o l l o q u i a Wednesday, 20th October P r o f e s s o r J.B. Hyne ( U n i v e r s i t y o f C a l g a r y ) , "New Research on an Old Element - Sulphur". Wednesday, 10th November Dr. J.S. Ogden (Southampton U n i v e r s i t y ) , "The C h a r a c t e r i z a t i o n o f High Temperature Species by Matrix I s o l a t i o n " . Wednesday, 17th November Dr. B.E.F. Fender ( U n i v e r s i t y o f Oxford), " F a m i l i a r but Remarkable Inorganic S o l i d s " . Wednesday, 24th November Dr. M.I. Page (Huddersfield P o l y t e c h n i c ) , "Large and Small Rate Enhancements o f I n t r a m o l e c u l a r C a t a l y s e d R e a c t i o n s " . Wednesday, 8th December P r o f e s s o r A.J. Leadbetter ( U n i v e r s i t y o f E x e t e r ) , " L i q u i d Crystals". Wednesday, 26th January Dr. A. Davis (ERDR), "The Weathering o f Polymeric M a t e r i a l s " . Wednesday/ 2nd February Dr. M. F a l k , (NRC Canada), " S t r u c t u r a l Deductions from the V i b r a t i o n a l Spectrum o f Water i n Condensed Phases". Wednesday, 9th February P r o f e s s o r R.O.C. Norman ( U n i v e r s i t y o f Y o r k ) , " R a d i c a l C a t i o n s ; Intermediates i n Organic R e a c t i o n s " . Wednesday, 23rd February Dr. G. H a r r i s ( U n i v e r s i t y o f S t . Andrews), "Halogen Adducts o f Phosphines and A r s i n e s " . F r i d a y , 25th February P r o f e s s o r H.T. Dieck (Frankfurt U n i v e r s i t y ) , "Diazadienes - New Powerful Low-Valent Metal Liganda". Wednesday, 2nd March Dr. F. Hibbert (Birkbeck College, London), " F a s t Reaction S t u d i e s of Slow Proton T r a n s f e r s I n v o l v i n g Nitrogen and Oxygen A c i d s " . F r i d a y , 4 t h March Dr. G. B r i n k (Rhodes U n i v e r s i t y , R.S.A.), " D i e l e c t r i c S t u d i e s of Hydrogen Bonding i n A l c o h o l s " . Wednesday, 9 t h March Dr. 1.0. Sutherland ( S h e f f i e l d U n i v e r s i t y ) , "The Stevans* Rearrangement O r b i t a l Symmetry and R a d i c a l P a i r s " . F r i d a y , 18th March P r o f e s s o r Hans Bock (Frankfurt U n i v e r s i t y ) , "Photoelectron S p e c t r a and Molecular P r o p e r t i e s : A Vademecum f o r the Chemist". Wednesday, 30th March Dr. J.R. MacCallum ( U n i v e r s i t y o f S t . Andrews), of Polymers". "Photooxidation Wednesday, 20th A p r i l Dr. D.M.J. L i l l e y (G.D. S e a r l e , Research D i v . ) , " T a i l s o f Chromatin S t r u c t u r e - Progress towards a Working Model". Wednesday, 27th A p r i l Dr. M.P. Stevens ( U n i v e r s i t y o f H a r t f o r d ) , "Photocycloaddition Polymerisation". Wednesday, 4th May Dr. G.C. T a b i s z ( U n i v e r s i t y o f Manitoba), " C o l l i s i o n Induced L i g h t S c a t t e r i n g by Compressed Molecular Gases". Wednesday, 11th May Dr. R.E. Banks (UMIST), "The Reaction o f Hexafluoropropene with H e t e r o c y c l i c N-Oxides". Wednesday, 18th May Dr. J . Atwood ( U n i v e r s i t y o f Alabama), "Novel S o l u t i o n Behaviour o f Anionic Organoaluminium Compounds: the Formation o f L i q u i d Clathrates". Wednesday, 25th May P r o f e s s o r M.M. Kreevoy ( U n i v e r s i t y o f Minnesota), "The Dynamics of Proton T r a n s f e r i n S o l u t i o n " . Wednesday, 1 s t June Dr. J . McCleverty ( U n i v e r s i t y o f S h e f f i e l d ) , "Consequences of Deprivation and Overcrowding and on the Chemistry o f Molybdenum Tungsten". Wednesday, 6th J u l y P r o f e s s o r J . Passmore ( U n i v e r s i t y o f Brunswick), "Adducts Between + Group V Pentahalides and a P o s t s c r i p t on S _ I " . b) Durham U n i v e r s i t y Chemical S o c i e t y Tuesday, 19th October Dr. J.A. Salthouse ( U n i v e r s i t y o f Manchester), "Chemistry and Energy". Tuesday, 26th October Dr. R.E. Richards ( U n i v e r s i t y o f Oxford), "NMR Measurements on Intact Biological Tissue". Tuesday, 2nd November Dr. B. S u t c l i f f e ( U n i v e r s i t y of Y o r k ) , "The Chemical Bond as a Figment of the Imagination". Tuesday, 16th November Mr. R. F i c k e n (Rohm & Haas), "The Graduate i n I n d u s t r y " . Tuesday, 30th November Dr. R.J. Donovan ( U n i v e r s i t y of Edinburgh), "The Chemistry of the Atmosphere". Tuesday, 18th January Professor I . F e l l s ( U n i v e r s i t y of Newcastle), "Energy Storage: the Chemists' Contribution to the Problem". Tuesday, 8th February Dr. M.J. C l e a r e (Johnson Matthey Research C e n t r e ) , "Platinum Group Metal Compounds as Anti-Cancer Agents". Tuesday, 1 s t March Professor J.A.S. Smith (Q.E. C o l l e g e , London), "Double Resonance". Tuesday, 8th March P r o f e s s o r C. Eabom ( U n i v e r s i t y of S u s s e x ) , " S t r u c t u r e and Reactivity". 1.2. 1977-78 a) U n i v e r s i t y of Durham Chemistry C o l l o q u i a Tuesday, 27th September Dr. T . J . Broxton (La Trobe U n i v e r s i t y , A u s t r a l i a ) , " I n t e r a c t i o n of Aryldiazonium S a l t s and A r y l a z o a l k y l E t h e r s i n B a s i c Alcoholic Solvents". Wednesday, 19th October Dr. B. Heyn ( U n i v e r s i t y o f Jena, D.D.R.), "o-Organo-Molybdenum Complexes as Alkene Polymerisation C a t a l y s t s " . Thursday, 27th October Professor R.A. F i l l e r ( I l l i n o i s I n s t , o f Technology, U.S.A.), "Reactions o f Organic Compounds with Xenon F l u o r i d e s " . Wednesday, 2nd November Dr. N. Boden ( U n i v e r s i t y o f Leeds), "NMR Spin-Echo Experiments f o r Studying S t r u c t u r e and Dynamical P r o p e r t i e s of M a t e r i a l s 1 Containing I n t e r a c t i n g Spin- ! P a i r s " . Wednesday, 9th November Dr. A.R. B u t l e r ( U n i v e r s i t y of S t . Andrews), "Why I l o s t F a i t h i n L i n e a r Free Energy Relationships". Wednesday, 7th December Dr. P.A. Madden ( U n i v e r s i t y of Cambridge), "Raman Studies o f Molecular Motions i n L i q u i d s " . Wednesday, 14th December Dr. R.O. Gould ( U n i v e r s i t y o f Edinburgh), "Crystallography t o the Rescue i n Ruthenium Chemistry". Wednesday, 25th January Dr. G. Richards ( U n i v e r s i t y o f Oxford), "Quantum Pharmacology". Wednesday, 1 s t February Professor K.J. I v i n Metathesis Reaction: of Cycloalkenes". (Queen U n i v e r s i t y , B e l f a s t ) , "The O l e f i n Mechanism o f Ring-Opening Polymerisation F r i d a y , 3rd February Dr. A. Hartog (Free U n i v e r s i t y , Amsterdam, Holland), " S u r p r i s i n g Recent Studies i n Organo-Magnesium Chemistry". Wednesday, 22nd February Professor J.D. B i r c h a l l (Mond D i v i s i o n , I . C . I . L t d . ) , " S i l i c o n i n the Biosphere". Wednesday, 1 s t March Dr. A. Williams ( U n i v e r s i t y of K e n t ) , "Acyl Group T r a n s f e r Reactions". F r i d a y , 3rd March Dr. G. van Koten ( U n i v e r s i t y o f Amsterdam, H o l l a n d ) , " S t r u c t u r e and R e a c t i v i t y of Arylcopper C l u s t e r Compounds". Wednesday, 22nd March P r o f e s s o r H. Vahrenkamp ( U n i v e r s i t y of F r e i b u r g , Germany), "Metal-Metal Bonds i n Organometallic Complexes". Wednesday/ 19th A p r i l Dr. M. Barber (UMIST), "Secondary Ion Mass S p e c t r a of Surfaces and Absorbed S p e c i e s " . Tuesday, 16th May Dr. P. Ferguson (C.N.R.S., Grenoble), "Surface Plasma Waves and Absorbed Species on Metals". Thursday, 18th May P r o f e s s o r M. Gordon ( U n i v e r s i t y of E s s e x ) , "Three C r i t i c a l P o i n t s i n Polymer S c i e n c e " . Monday, 22nd May P r o f e s s o r D. Tuck ( U n i v e r s i t y of Windsor, O n t a r i o ) , " E l e c t r o chemical S y n t h e s i s of I n o r g a n i c and Organometallic Compounds". Wednesday - Thursday 24th, 25th May P r o f e s s o r P. von R. S c h l e y e r ( U n i v e r s i t y of E r l a n g e n , Nllrnberg), I. "Planar Tetra-coordinate Methanes, P e r p e n d i c u l a r E t h y l e n e s , and P l a n a r A l l e n e s " . II. Ill. "Aromaticity i n Three Dimensions". " N o n - c l a s s i c a l Carbocations". Wednesday, 2 1 s t June Dr. S.K. T y r l i k (Academy o f Science, Warsaw), "Dimethylglyoximec o b a l t Complexes - C a t a l y t i c Black Boxes". F r i d a y , 23rd June P r o f e s s o r W.B. Pearson ( U n i v e r s i t y o f F l o r i d a ) , "Diode L a s e r Spectroscopy a t 16 pm". F r i d a y , 30th June P r o f e s s o r G. Mateescu (Case Western Reserve U n i v e r s i t y ) , "A Concerted Spectroscopy Approach t o the C h a r a c t e r i z a t i o n o f Ions and Ion P a i r s : b) F a c t s , P l a n s , and Dreams". Durham U n i v e r s i t y Chemical S o c i e t y Thursday, 13th October Dr. J.C. Young, Mr. A.J.S. Williams ( U n i v e r s i t y o f Aberystwyth) , "Experiments and Considerations Touching Colour". Thursday, 20th October Dr. R.L. Williams (Metropolitan P o l i c e F o r e n s i c Science Dept.), "Science and Crime". Thursday, 3rd November Dr. G.W. Gray ( U n i v e r s i t y o f H u l l ) , " L i q u i d C r y s t a l s - T h e i r O r i g i n s and A p p l i c a t i o n s " . Thursday, 24th November Mr. G. R u s s e l l (Alcan), "Designing f o r S o c i a l A c c e p t a b i l i t y " . Thursday, l e t December Dr. B.F.G. Johnson ( U n i v e r s i t y of Cambridge), "Chemistry of Binary Metal Carbonyls". Thursday, 2nd February P r o f e s s o r R.A. Raphael ( U n i v e r s i t y o f Cambridge), " B i z a r r e Reactions of A c e t y l e n i c Compounds". Thursday, 16th February P r o f e s s o r G.W.A. Fowles ( U n i v e r s i t y of Reading), "Home Winemaking". Thursday, 2nd March P r o f e s s o r M.W. Roberts ( U n i v e r s i t y of B r a d f o r d ) , "The Discovery of Molecular Events a t S o l i d S u r f a c e s " . Thursday, 9th March P r o f e s s o r H. Suschitzky ( U n i v e r s i t y of S a l f o r d ) , " F r u i t f u l F i s s i o n s o f Benzofuroxans". Thursday, 4th May P r o f e s s o r J . Chatt ( U n i v e r s i t y of S u s s e x ) , "Reactions of Coordinated Dinitrogen". Tuesday, 9th May P r o f e s s o r G.A. Olah (Case Western Reserve U n i v e r s i t y , C l e v e l a n d , Ohio), " E l e c t r o p h i l i c Reactions o f Hydrocarbons". 2. Conferences Attended Photopolymerization and Polymer Photodegradation, Southampton, September 1976. 1 s t Polymer Surfaces Symposium, Durham, March 1977. REFERENCES 1. G. Oster and N.-L. Yang, Chem. Rev., 1 9 6 8 , 6 8 , 1 2 5 . 2. N.G. Gaylord, Tech. Papers, Reg. Conf. Soc. P l a s t . Eng., Mid-Hudson S e c t . , Nov. 6 - 7 , 1 9 6 7 , 3 2 . 3. J . B l y t h and A.W. Hofmann, Annalen, 1 8 4 5 , 53_, 2 8 9 . 4. J . Hutchinson and A. Ledwith, Adv. Polymer S c i . , 1 9 7 4 , 1 4 , 4 9 . 5. H.-G. Heine, H.-J. Rosenkranz and H. Radolph, Angew. Chem. I n t . E d i t . , 1972, 11, 974. 6. A. Ledwith, I n t e r n . Conf. on E x c i p l e x e s , London, Ontario, May 1 9 7 4 . 7. M.R. Sandner and C.L. Osborn, Tetrahedron L e t t . , 1 9 7 4 , 4 1 5 . 8. C.H. Carder, P a i n t and Varnish Production, 1974 (August), 1 9 . 9. G. I r i c k , J . Appl. Polymer S c i . , 1 9 7 2 , 1 6 , 2387. 10. R. N i l s s o n and D.R. Reams, Photochem. Photobiol., 1 9 7 4 , 19_, 1 8 1 . 11. M. Tsuda, J . Polymer S c i . , P a r t A - l , 1 9 6 9 , 1_, 2 5 9 . 12. A.D. Hay, D.A. Bolon, K.R. Leimer and R.F. C l a r k , J . Polymer S c i . , P a r t B, 1 9 7 0 , £ , 9 7 . 13. J.L.R. W i l l i a m s , Tech. Papers, Reg. Conf. Soc. P l a s t . Eng., Mid-Hudson S e c t . , Nov. 6 - 7 , 1 2 3 . 14.. L.M. Minsk, J.G. Smith, W.P. van Deuson and J . F . Wright, J . Appl. Polymer S c i . , 1 9 5 9 , 2, 3 0 2 . 15. E.M. Robertson, W.P. van Deuson and L.M. Minsk, J . Appl. Polymer S c i . , 1959, 16. 2, 308. R.C. S c h u l t z , L. Rohe and H. Adler, Eur. Polymer J o u r n a l (Bratislava Supplement) , 1 9 6 9 , 5_, 3 0 9 . 17. H. Tanaka, M. Tsuda and H. Nakanishi, J . Polymer S c i . , P a r t A - l , Polymer Chem., 1 9 7 2 , 1 0 , 1 7 2 9 . 18. H. Jacobs and R. S t e e l e , J . Appl. Polymer S c i . , 1 9 6 0 , _3, 2 3 9 . 19. P.C. De Schryver, N. Boens and G. Smets, J . Polymer S c l . , P a r t A - l , Polymer Chem., 1972, 10, 1687 and r e f s . c i t e d t h e r e i n . 20. F.C. De Schryver e t a l . , J . Polymer S c l . , Polymer Chem., 1975, 13_, 215. 21. J.B. B i r k s , 'Photophysics o f Aromatic Molecules', I n t e r s c i e n c e , New York, 1970, pp. 320, 369. 22. F.C. De Schryver, L. Anand, G. Smets and J . Switten, J . Polymer S c i . , P a r t B, Polymer L e t t e r s , 1971, 9_, 777. 23. F.C. De Schryver and G.A. Delzenne, Ger. Offen. 2,212,427/1972. 24. F.C. De Schryver, W.J. F e a s t and G. Smets, J . Polymer S c i . , P a r t A - l , Polymer Chem., 1970, 8_, 1939. 25. F.C. De Schryver i n 'Microsymposium on Photochemical Processes i n Polymer Chemistry*, Leuven, Belgium, June 1972. 26. D.J. Andrews and W.J. F e a s t , J . Polymer S c i . , Polymer Chem., 1976, 14, 319. 27. D.J. Andrews and W.J. F e a s t , i b i d . , 331. 28. L.H. Leenders, E . Schouteden and F.C. De Schryver, J . Org. Chem., 1973, 38, 957. 29. D.R. Arnold, Adv. Photochem., 1968, 6_, 301. 30. H. Leblanc, Doctoral T h e s i s , Leuven, Belgium, 1972. 31. R.C. S c h u l t z and H.H. B o s s i e r , Umschau Wlss. Tech., 1971, 71, 673. 32. H.H. B o s s i e r and R.C. S c h u l t z , Macromol. Chem., 1972, 158, 113. 33. L. Homer and A. Christmann, Angew. Chem., 1963, 75, 707. 34. R.C. S c h u l t z , Pure & Appl. Chem., 1973, 34, 305. 35. N. Kardush and M.P. Stevens, J . Polymer S c i . , P a r t A - l , 1972, 1093. 36. Y. Musa and M.P. Stevens, i b i d . , 10, 319. 37. D. Bryce-Smith and M.A. Hems, Tetrahedron L e t t e r s , 1966, 1895. 38. J.S. Bradshaw, Tetrahedron L e t t e r s , 1966, 2039. 39. U.S.P. 40. F.C. De Schryver, Tran Van Thien, S. Toppet and G. Smets, J . Polymer 3,366,642/1968. S c l . , Polymer Chem. E d i t . , 1975, 13_, 227. 41. M. I g u c h i , H. Nakanishi and M. Hasegawa, J . Polymer S c i . , P a r t A - l , 1968, 6, 42. 1055. M. Hasegawa, Y. Suzuki, F. Suzuki and H. Nakanishl, J . Polymer S c i . , P a r t A - l , 1 9 6 9 , J_, 7 4 3 . 43. M. Hasegawa and Y. Suzuki, Polymer L e t t e r s , 1967, 5_, 8 1 3 . 44. M. Hasegawa Y. Suzuki and T. Tamalci, B u l l . Chem. Soc. Japan, 1 9 7 0 , 43, :020. 45. M.J. Helm and F.B. Z i e n t y , J . Polymer S c i . , P a r t A - l , 1 9 7 2 , 1 0 , 1 3 1 1 . 46. W.J. F e a s t , Research Proposal to European Research O f f i c e , U.S. Army, March 1974, p. 2 . 47. J.G. C a l v e r t and J.N. P i t t s , J r . , 'Photochemistry', J . Wiley, New York, 1966, p. 2 5 2 . 48. K. Bredereck, Th. F o e r s t e r and H.G. O e s t e r l i n , Chem. Abs. , 1 9 6 3 , 5 8 , 75146. 49. C. Walling and M.J. G i b i a n , J . Amer. Chem. S o c , 1 9 6 5 , 8 2 , 50. A. Padwa, Tetrahedron L e t t e r s , 1 9 6 4 , 3 4 6 5 . 51. N.J. Turro, J.C. Dalton, K. Dawes, G. F a r r i n g t o n , R. Hautala, D. Morton, 3361. M. Niemczyk and N. Shore, Accounts Chem. Res., 1 9 7 2 , 5_, 9 2 . 52. N.J. Turro, 'Molecular Photochemistry', W.A. Benjamin, I n c . , New York, 1965. 53. R.O. Kan, 'Organic Photochemistry', McGraw-Hill, New York, 1 9 6 6 . 54. R.B. Cundall and A. G i l b e r t , 'Photochemistry', T. Nelson & Sons L t d . , London, 1970. 55. J.S. Swenton, J . Chem. Educ., 1 9 6 9 , 4 6 , 2 1 7 . 56. G. Ciamician and P. S i l b e r , Ber., 1 9 0 0 , 57. W.D. Cohen, Rec. Trav. Chim., 1 9 2 0 , 3 9 , 2 4 3 . 58. L.F. F i e s e r , 'Organic Experiments', D.C. Heath & Co., Boston, 1 9 6 4 , Ch. 4 0 . 59. D.E. Pearson and P.D. Thiemann, J . Polymer S c i . , P a r t A - l , Polymer Chem., 1970, 8, 2103. 2911. 60. J . Higgins, A.H. Johannes, J . F . Jones, R. S c h u l t z , D.A. McCombs and C.S. Menon, J . Polymer S c l . , P a r t A - l , Polymer Chem., 1970, £ , 61. 1987. D.A. McCombs, C.S. Menon and J . Higgins, J . Polymer S c i . , P a r t A - l , Polymer Chem., 1971, 9_, 1261. 62. F.C. De Schryver, T. Tran Van and G. Smets, J . Polymer S c l . , P a r t B, Polymer Chem., 1971, 9_, 425. 63. M. Gomberg and W.E. Bachmann, J . Amer. Chem. Soc., 1927, 49, 236. 64. D . J . Andrews, Ph.D. T h e s i s , Durham U n i v e r s i t y , 1973. 65. Q u a l i t a t i v e measurements by the author confirmed Andrews' o b s e r v a t i o n . 66. S.A. Weiner, J . Amer. Chem. Soc., 1971, 93_, 424. 67. A. Schonberg, 'Preparative Organic Photochemistry', S p r i n g e r - V e r l a g , B e r l i n , 1968 (Ch. 2 2 ) . 68. W.E. Bachmann, J . Amer. Chem. Soc., 1933, 55, 391 and r e f e r e n c e s t h e r e i n . 69. Reference 67, Ch. 20. 70. A. Schonberg and A. Mustafa, J . Chem. Soc., 1944, 71. M.R. Kegelman and E.V. Brown, J . Amer. Chem. Soc., 1953, 75, 4649. 72. C. Weizmann, E . Bergmann and Y. Hirshberg, J . Amer. Chem. Soc., 1938, 60, 67. 1530. 73. W.L. Bencze, C.A. Burckhardt and W.L. Yost, J . Org. Chem., 1962, 2T_, 2865. 74. F. Bergmann and Y. Hirshberg, J . Amer. Chem. Soc., 1943, 65, 75. J.N. P i t t s , J r . , R.L. L e t s i n g e r , R.P. T a y l o r , J.M. P a t t e r s o n , 1429. G. Recktenwald and R.B. Martin, J . Amer. Chem. Soc., 1959, 81, 76. G.S. Hammond, W.P. Baker and W.M. 83, 1068. Moore, J . Amer. Chem. Soc., 1961, 2795. 77. A. Beckett and G. P o r t e r , Trans. Farad. S o c , 1963, 59_, 2038. 78. J.N. P i t t s , J r . , and J.K.S. Wan i n 'Chemistry o f the Carbonyl Group', Ed. S. P a t a i , I n t e r s c i e n c e , New York, 1966, Ch. 14. 79. G.S. Hammond and P.A. Leermakers, J . Amer. Chem. Soc., 1962, 84, 207. 80. H. Mauser and V. B i h l , Z. Naturforsch (B) , 1967, 2!2, 1077. 81. N. F i l i p e s c u and F.L. Minn, J . Amer. Chem. Soc., 1968, 90, 1544. 82. N. F i l i p e s c u , L.M. Kindley and F.L. Minn, J . Org. Chem., 1971, 36_, 861. 83. J . S a l t i e l , H.C. C u r t i s and B. Jones, Molec. Photochem., 1970, 2, 331. 84. J.A. B e l l and H. L i n s c h i t z , J . Amer. Chem. Soc., 1963, 85_, 528. 85. A.V. Buettner and J . Dedinas, J . Phys. Chem., 1971, 75_, 187. 86. J . S a l t i e l , H. C u r t i s , L . Metts, J.M. Miley, J . Winterle and M. Wrighton, J . Amer. Chem. Soc., 1970, 92_, 410. 87. D.I. Schuster and D.F. B r i z z o l a r a , J . Amer. Chem. Soc., 1970, 92, 4357. 88. D.I. Schuster, T.M. Weil and A.M. Halpern, J . Amer. Chem. Soc., 94, 8248. 89. G.L. C l o s s and L.E. C l o s s , J . Amer. Chem. Soc., 1964, 91, 4550. 90. M. N i c o l a s and R. P a i l l a n d , 91. J.S. Bradshaw, Chem. Comm., 1973, 504. 92. G. Gotzmer, J r . , K.F. Muller and M.J. C z i e s l a , J . Org. Chem., 1973, Ind. Chiro. Beige, 1971, T36, 117. 38, 2964. 93. M.J. G i b i a n , Tetrahedron L e t t e r s , 1967, 5331. 94. F.L. Minn, C.L. T r i c h i l o , C.R. Hurt and N. F i l i p e s c u , J . Amer. Chem. Soc., 1970, 92, 3600. 95. W.L. Bencke, C.A. Burckhart and W.L. Yost, J . Org. Chem., 1962, 27, 2865. 96. H.L.J. Backstrom and R.J.V. N i c k l a s s o n , Acta Chem. Scandln., 1966, 20, 2617. 97. W.M. Moore and M.D. Ketchum, J . Phys. Chem., 1964, 86, 214. 98. N. F i l i p e s c u , J.P. P i n i o n and F.L. Minn, Chem. Comm., 1970, 1413. 99. J . Dedinas and T.H. Reagan, J . Phys. Chem., 1972, 76, 3926. 100. B.D. Challand, Can. J . Chem., 1969, 4T_, 687. 101. M.B. Ledger and G. P o r t e r , Faraday Trans., I , 1972, 68, 539. 102. P.S. Eugel and B.M. Monroe, Adv. Photochem., 8_, 253. 103. G.O. Schenck, G. Kotzenburg and E . R o s e l i u s , Tetrahedron L e t t e r s , 1970, 5185. 104. R.S. Davison and F.A. Younis, Chem. Comm., 1969, 826. 105. V. Franzen, J u s t u s L l e b l g s , Ann. Chem., 1960, 633, 1. 106. G. P o r t e r and G. Suppan, Pure Appl. Chem., 1964, 9_, 499. 107o F. Wilkinson, Adv. Photochem., 1964, 108. G. P o r t e r and G. Suppan, Proc. Chem. Soc., 1964, 191. 109. E . J . Baum, J.K.S. Wan and J.N. P i t t s , J r . , J . Amer. Chem. Soc., 3_f 2 4 1 « 1966, 88, 2652 and r e f e r e n c e s c i t e d t h e r i n . 110. S.G. Cohen, M.D. Saltzman and J.B. Guttenplan, Tetrahedron L e t t e r s , 1969, 4321. 111. N.C. Yang and C. R i v a s , J . Amer. Chem. Soc., 1961, 83, 2213. 112. F.D. Lewis, Tetrahedron L e t t e r s , 1970, 1373. 113. M.Y. Moss, D i s s . Abstr., 1968, B29, 944. 114. N.D. Heindel, E.W. S a r r e r and H.A. Pfau, Tetrahedron L e t t e r s , 1968, 3579. 115. D.E. Pearson and M.Y. Moss, i b i d . , 1967, 3791. 116. F.A. Lamb and B.M. Vittimberga, J . Org. Chem., 1973, 38, 3520. 117. P. Suppan, Ber. Bunseng. Phys. Chem., 1968, 72, 321. 118. D.C. Neckers and D.P. Colebrander, Tetrahedron L e t t e r s , 1968, 119. E . Paterno and G. C h i e f f i , Gazz. Chim. I t a l . , 1909, I I , 39, 415. 120. E . Bergmann and S . - I . F u j i s e , Annalen, 1930, 483, 65. 121. M. Barnard and N.C. Yang, Proc. Chem. S o c , 1958, 302. 122. Reference 67, Ch. 21. 123. See f o r example: a) A.A. B e r l i n , J . Macromol. Sci.-Chem., 1971, A5, 1187. b) I . Schopov e t a l . , 1976, 177, 1653. Macromol. Chem., 1975, 5045. 176, 511 and i b i d . , D. Braun e t a l . , Macromol. Chem., 1976, 177, 1373, 1387, 1673, 2221. 123. c) 124. G. Clamiclan and P. S i l b e r , Ber., 1915, I_ 193. 125. G. Ciamician and P. S i l b e r , A t t i Accad. L l n c e l , 3 J a n 1886. 126. D. Loew, 'Die Chemischen Energie der lebenden Z e l l e ' , 127. G. Ciamician and P. S i l b e r , A t t l Accad. L l n c e l , 1901, 10, 92. 128. G. Ciamician and P. S i l b e r , Gazz. Chlm. I t a l . , 1904, 3 4 ( 1 1 ) , 129. 129. A. Gardeur, Chemisettes C e n t r a l b l a t t , 1897, 11^, 662. 130. E. Paterno and G. C h i e f f i , Gazz. Chim. I t a l . , 1910, 4 0 ( 1 1 ) , 321. 131. E . Paterno and G. F o r l i - F o r t i , Gazz. Chim. I t a l . , 1910, 40(11), 332. 132. E . Paterno, Gazz. Chim. I t a l . , 1914, 44(11), 99. 133. E . Paterno, i b i d . , 1914, 44, 151. 134. E . Paterno, i b i d . , 1914, 4 4 ( 1 1 ) , 463. 135. G. Ciamician and P. S i l b e r , Ber., 1915, I_, 193. 136. E . Paterno, Gazz. Chlm. I t a l . , 1909, 39(11), 213. 137. McKenzie and Wren, J . Chem. S o c , 97, 481. 138. A. Ledwith, P.J. R u s s e l l and L.H. S u t c l i f f e , J . Chem. Soc.,Perkin ( I I ) , f 1972, (1899). 1925. 139. J.M. Wilson, J . Chem. Soc., 1951, 2297. 140. D i c t i o n a r y o f Organic Compounds, EyneB & Spottinswoode ( P u b l i s h e r s ) L t d . , London, 1965. 141. Bergmann and Engel, Z e i t . Phys. Chem., 1930, B8, 133. 142. R. Wegler, Ber., 1934, 67B, 35. 143. G. Richard, Chem. Zentr., 1934, 1051, 3744. 144. F. Koelsch, J . Amer. Chem. Soc., 1931, 5_3, 1147. 145. J.S.W. Boyle, A. McKenzie and W. M i t c h e l l , 146. A. B a n c h e t t i , Ann. Chim. Appl., 1941, 31, 430. 147. A. B a n c h e t t i , Gazz. Chim. I t a l . , 1942, 72_, 74. 148. 0. Polansky, E . S c h i n z e l and F. Wessely, Monatsch., 1956, 87, 24. Ber., 1937, 70B, 2153. 149. P.J. Hamrick, J r . , and C.R. Houser, J . Amer. Chem. Soc., 1959, 81, 2096. 150. S.H. Metzger, J r . , D i s s . Abs., 1963, 23_, 2702. 151. H. Zlncke, Ber., (5, 119 and 9_, 31. 152. T h i e l e and Balhorn, Ber., 37, 1467. 153. R.C. Huston and T.E. Frledmann, J . Amer. Chem. S o c , 1916, 38, 2531. 154. Japanese Patent 6,902,708/1969. 155. E . P r o f f t , G. D r e c h s l e r and H. Oberender, Wlss. Z. Tech. Hochsch. Chem. Leuna-Merseburg 2,1959/60, 259. 156. A.B. Galun, A. Kaluszyner and E.D. Bergmann, J . Org. Chem., 1962, 27, 2372. 157. A. Vogel, ' P r a c t i c a l Organic Chemistry', 3rd E d i t i o n , Longman, London 1974 158. R.T. Morrison and R.N. Boyd, 'Organic Chemistry', 3rd E d i t i o n , A l l y and Bacon I n c . , Boston, 1974. 159. R. G r i n t e r and S.F. Mason, Trans. Faraday Soc., 1964, 60, 889. 160. B e i l s t e i n ' s Hundbuch der Organischen Chemie. 161. D.S. T a r b e l l and J.C. Petropoulos, J . Amer. Chem. Soc., 1952, 74, 244. 162. C.K. S t e i n h a r d t , p r i v a t e communication t o the author i n F i e s e r & F i e s e r , 'Reagents f o r Organic S y n t h e s i s ' , John Wiley & Sons, I n c . , 1967, p. 584. 163. R.S. Matthews, p r i v a t e communication. 164. 'Handbook o f Chemistry and P h y s i c s ' , 46th E d i t i o n , The Chemical Rubber Co. r Ohio, 1965. 165. D.A. McCombs, C.S. Menon and J . Higgins, J . Polymer. S c i . , P a r t A - l Polymer Chem., 1971, £, 1799. 166. A. Gandini and P.H. P l e s c h , Proc. Chem. Soc., 1964 ( A p r i l ) , 113. 167. G. Ciamician and P. S i l b e r , Rend. Acc. L i n c e i , 1905, I_, 265. 168. L . M a s c a r e l l i , Gazz. Chim. I t a l . , 1906, 169. G. C i a m i c i a n and P. S i l b e r , Rend. Acc. L i n c e i , 1909, I , 216. 670. 170. G. Ciamician and P. S i l b e r , i b i d . , 1914, X X I I I , 112. 171. G.R. De Mare, J.R. Fox, M. Termonia and B. T s h i b a n g i l a , European Polymer J . , 1975, 12_, 119. 172. R. Raff, J . L . Cook and B.V. E t t i n g , J . Polymer S c i . , 1965, Part A 3(10), 3511. 173. U.S.P. 3,419,462 (1968). 174. U.S.P. 3,414,466 (1968). 175. T.B. Fomina, N.V. Artem'eva, Yu. N. Sazarov and M.M. Koton, Dokl. Akad. Nauk. SSSR, 1970, 1 3 9 ( 4 ) , 838. 176. K e n j i Yokota and Yoahio I s h i i , Kogyo Kagaku Z a s s h l , 1963, 66_, 130. 177. U.S.P. 178. Bilow, Norman and Rosenberg, J . Polymer S c i . , 3,509,097/1970. P a r t A - l , 1969, 7(9), 2689. 179. J . F . Jackson and S . J . G i l l , J . Polymer S c i . , P a r t A - 2 , 1967, 5(4) , 663. 180. U.S.P. 3,442,861 (1969). 181. Leznoff, C l i f f o r d and Wong, Can. J . Chem., 1973, 5 1 ( 2 2 ) , 182. A. Schonberg and A. Mustafa, J . Amer. Chem. S o c , 1955, 77, 183. J . Kagan, Tetrahedron L e t t e r s , 1966, 49, 6097. 184. K.F. Cohen, J.T. Pinhey and R.J. Smith, Tetrahedron L e t t e r s , 1968, 4729 185. D.A. H a r r i s o n , R.N. Schwartz and J . Kagan, J . Amer. Chem. Soc., 1972, 3756. 5755. 92, 5793. 186. P. Longia, C.R. Acad. S c i . , 1962, 255, 865. 187. M. Stockburger, Z. Phys. Chem. (Frankfurt am Main) , 1962, 31_, 350. 188. G.R. De Mare, P. Goldfinger, G. Huybrects, E . Jonas and M. Toth, Ber. Bunsenges Phys. Chem., 1969, 73, 867. 189. N.C. Yang, Photochem. Photobiol., 1968, T_, 767. 190. N.C. Yang, J . I . Cohen and A. Shani, J . Amer. Chem. Soc., 1968, 90, 3264 191. See f o r example: a) J.M. H o l l a s , E . Gregorek and L . Goodman, J . Chem. Phys., 1968, 49, 1745. b) F. Dora, 2. Elektrochem., 1957, 61^, 950. c) J.R. Majer, S.-A. M.A. Naman and J.C. Robb, T r a n s . Faraday Soc., 1969, 65, 1846. d) K. Znuzuka and T. Yokota, B u l l . Chem. Soc. Japan, 1965, 38, 1055. e) K. Klmura and S. Nagakura, Theor. Chem. Acta, 1965, 3^, 164. f) R. Shlmada and L. Goodman, J . Chem. Phys., 1965, 43, 2027. g) O.A. Ponomarev, I z v . Vyssh. Ucheb. Zared. F l z . , 1966, 9 ( 4 ) , 21. 192. F. Almasy, J . Chem. Phys., 1933, 30, 528, 634, 713. 193. G.R. De Mare, M.C. Fontaine and P. Goldfinger, J . Org. Chem., 1968, 33, 2528. 194. 195. a) M. de Hemptinne, J . Phys. Radium, 1929, 9_, 357. b) C.R. Acad. S c i . , 1928, 186, 1295. F.E. B l a c e t and D. Vanselow, A b s t r a c t s , 131th National Meeting of the Amer. Chem. Soc., Miami Beach, F l o r i d a , 1957. 196. M. Berger, I . L . G o l d b l a t t and C. S t e e l , J . Amer. Chem. Soc., 1973 95, 197. 1717. J.S. Bradshaw, R.D. Knudsen and W.W. P a r i s h , J . Chem. Soc., Chem. Comm., 1972, 1321. 198. 199. a) M. Cocivera and A.M. Trozzolo, J . Amer. Chem. Soc., 1970, 92_, 1772. b) G.L. C l o s s and D.R. Paulson, i b i d . , 7229. F.D. Lewis, R.T. Lauterbach, H.-G. Heina, W. Hartmann and H. Rudolph, J . Amer. Chem. Soc., 1975, 9J7, 1519. 200. Kornis and De Mayo, Can. J . Chem., 1964, 42_, 2822. 201. J.N. P i t t s e t a l . , J . Amer. Chem. S o c , 1969, 91, 2461. 202. Heine, Hartmann, Kory, Maggar, Hoyle, McVery and L e w i s , J . Org. Chem., 1974, 39, 691. 203. Bunbury and Chuang, Can. Chem. J . , 1969, 47_, 2045. 204. a) H. Sulda, Monatsch., 1912, 33/ 1173. 205. b) C.J. K o t h a r i and H.E. Watson, J . I n d i a n I n s t . S c l . , 1931, A ( I I ) , 14. c) H.L.J. Blackstrom, Medd. Vetenstapsakad. NobelInst., 1927, 6, No. 15. d) M. T r a u t z , Eders Jahrbuch, 1909, 57, 421. a) P. Preiswerk and H. Erlenmeyer, Helv. Chlm. Acta, 1934, 17_, 329. b) 206. E . Schauenstein and M. Stampter, Ber., 1944, 77B, 19. G. Blichi, C.G. Biichi, C.G. Inman and E.S. L i p i n s k y , J . Amer. Chem. Soc., 1954, 76, 4327. 207. L.L. H u l l e r and J . Hamer, * 1 , 2 - C y c l o a d d l t i o n R e a c t i o n s ' , I n t e r s c i e n c e , New York, 1967. 208. D.R. Arnold, R.L. Hinman and A.H. G l i c k , Tetrahedron L e t t e r s , 1964, 209. N.J. Turro, P. Wriede, J.C. Dalton, D.R. Arnold and A.H. G l i c k , J . Amer. Chem. Soc., 1967, 89, 210. 1425. 3950. R.A. C a l d w e l l , G.W. Sovocool and R.P. Gajewski, J . Amer. Chem. Soc., 1973, 95, 2549. 211. Y. Shigemitsu, Y. Katsuhara and Y. Odaira, Tetrahedron L e t t e r s , 1971, 2887. 212. S. P a r i d and S.E. S c h e i l e r , J . Chem. Soc. ( D ) , Chem. Comm., 1973, 296. 213. S.R. Kurowsky and H. Morrison, J . Amer. Chem. Soc., 1972, 94, 507. 214. J . S a l t i e l , R.M. Coates and W.G. Dauben, J . Amer. Chem. Soc., 1966, 88, 2745. 215. N. Nozaki, I . O t a n i , R. Noyari and M. Kawanisi, Tetrahedron, 1968, 24, 2183. 216. C. DeBoer, Tetrahedron L e t t e r s , 1971, 4977. 217. S. F a r i d , J.C. Doty and J.L.R. W i l l i a m s , J . Chem. Soc. (D), 1972, 711. 218. D. Bryce-Smith and A. G i l b e r t , Proc. Chem. S o c , 1964, 87; Tetrahedron L e t t e r s , 1964, 3471. 219. J . F . H a r r i s , J r . , J . Org. Chem., 1965, 30, 2182. 220. J.D. Margerum, J.N. P i t t s , J r . , J.G. Rutgers and S. S e a r l e s , J . Amer. Chem. Soc., 1959, 81, 1549. 221. H. Gotthardt, R. Steinmetz and G.S. Hammond, Chem. Comm., 1967, 480. 222. G.O. Schenk, W. Hartmann and R. Steinmetz, Chem. Ber., 1963, 96_, 498. 223. G.S. Hammond and N.J. Turro, S c i e n c e , 1963, 142, 1451. 224. D. Gagnaire and E . Payo, Subiza, B u l l . Soc. Chim. France, 1963, 11, 2623. 225. M. Ogata, H. Watanabe and H. Kano, Tetrahedron L e t t e r s , 1967, 533. 226. J . L e i t i c h , Tetrahedron L e t t e r s , 1967, 1937. 227. S. T o k l and H. S a k u r a i , Tetrahedron L e t t e r s , 1967, 4119. 228. C. R i v a s and E. Payo, J . Org. Chem., 1967, 32_, 2918. 229. S.H. Schroeter and CM. Orlando, J r . , J . Org. Chem., 1969 , 34_, 1181. 230. J . Dedinas, J . Phys. Chem., 1971, 75_, 181. 231. D.I. Schuster, T.M. Weil and M.A. Halpem, J . Amer. Chem. Soc. , 1972, 94, 8248. 232. R.J. G r i t t e r , i n "The Chemistry o f the E t h e r Linkage', S. P a t a i Ed., I n t e r s c i e n c e , New York, 1967, Ch. 9. 233. U.S.P. 2,995,572/1961. 234. Dutch Patent 5,404,598/1964. 235. French Patent 1,391,493/1965. 236. E . J . Cook and B.F. Landrum, J . H e t e r o c y c l i c Chem., 1965, _5, 327. 237. J.D. L a Z e r t e and R.J. Koshar, J . Amer. Chem. Soc., 1955, 77, 910. 238. H. Muramatsu and K. I n u k a i , J . Org. Chem., 1962, 2J_, 1572. 239. E.R. B i s s e l l and D.B. F i e l d s , J . Org. Chem., 1964, 29, 249. 240. J . F . H a r r i s , J r . , and D.D. Coffman, J . Amer. Chem. Soc., 1962, 84, 1553. 241. U.S.P. 2,594,272/1952. 242. R.N. Haszeldine and F. Nyman, J . Chem. Soc., 1959, 1084. 243. J . L . Z o l l i n g e r , J.R. Throckmorton, S.T. Ting, R.A. Mitsch and D.E. E l r i c k , J . Macromol. S c i . (Chem.), 1969, 3_, 1443. 244. J.D. L a Z e r t e , L . J . H a l l , T.S. Reid and G.H. Smith, J . Amer. Chem. Soc., 1953, 75, 245. 4525. A.N. B e l l , R. F i e l d s , Chem. Comm., 1975, R.N. Haszeldine and I . Kumadaki, J . Chem. Soc., 866. 113 Of c 1978