Synthesis of 3-FmocNH-2-(o-NO2Ph)-propionic acid
advertisement
-propionic acid")
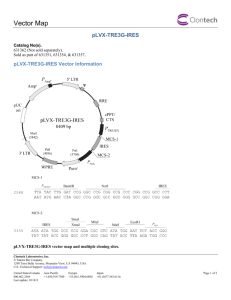
Supporting information for the article entitled “Generation of peptide-MHC class I complexes through UV-mediated ligand exchange.” Boris Rodenko,1 Mireille Toebes,2 Sine Reker Hadrup,2 Wim J. E. van Esch,3 Annemieke M. Molenaar,3 Ton N. M. Schumacher2 & Huib Ovaa1 1 Division of Cellular Biochemistry, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands. 2 Division of Immunology, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands. 3 Sanquin, Plesmanlaan 125, 1066 CX, Amsterdam, The Netherlands. Correspondence should be addressed to T.N.M.S. (t.schumacher@nki.nl) or H.O. (h.ovaa@nki.nl) Synthesis of N-fluorenylmethyloxycarbonyl-(2-nitro)phenylglycine The title compound is obtained in five steps from commercially available 2nitrophenylacetic acid 3 (Figure S-1). 2-Nitrophenylacetic acid methyl ester (4). This compound was synthesised according to a modified literature procedure.1 To a solution of 2-nitrophenylacetic acid 3 (45 g, 0.25 mol) in 500 mL of methanol was added thionyl chloride (1.9 mL; 25 mmol) over 30 min while maintaining the temperature at 0–4 oC. The reaction mixture was stirred at 0 oC for 30 min and then at room temperature for 24 h. After removal of the solvent in vacuo, the residue was partitioned between ethyl acetate (500 mL) and water (250 mL). The organic phase was washed with another 250 mL of water and then dried over anhydrous Na2SO4 and filtered. Removal of ethyl acetate in vacuo gave 50 g of 2-nitrophenylacetic acid methyl ester 4 as a yellow oil in quantitative yield. 1H NMR (400MHz, CDCl3) 8.10 (dd, 1H, J = 1.1 and 7.6 Hz) 7.59 (dt, 1H, J = 1.1 and 7.6 Hz) 7.47 (dt, 1H, J = 1.1 and 7.6 Hz) 7.35 (d, 1H, J = 7.6 Hz), 4.02 (s, 2H), 3.70 (s, 3H). MS(ESI): [M+H]+ calculated: 196.06, found: 196.03. 1 1’-Bromo-2-nitrophenylacetic acid methyl ester (5). This compound was synthesised according to a modified literature procedure.2 NBS (19.7 g, 0.11 mol) was added to a solution of 2-nitrophenylacetic acid methyl ester (18.0 g, 0.092 mol) in 200 mL of CCl4 in a reaction flask equipped with a reflux condenser. The vigorously stirred solution was irradiated with a 100 W tungsten lamp. The flask and lamp set-up was wrapped in aluminum foil to ensure efficient heating of the reaction mixture. After 6 h the white succinimide precipitate was filtered off and the filtrate was concentrated in vacuo yielding an orange solid. Recrystallisation with ethanol and washing of the off-white crystals with cold ethanol furnished 14.0 g (56 % yield) of brominated product 5. 1H NMR (400MHz, CDCl3) 8.01 (dt, 2H, J = 1.2 and 7.8 Hz) 7.70 (dt, 1H, J = 1.2 and 7.8 Hz) 7.54 (dt, 1H, J = 1.2 and 7.8 Hz), 6.10 (s, 1H), 3.82 (s, 3H). MS(ESI): [M+H]+ calculated: 274.0 and 276.0, found: 274.0 and 276.0. 1’-Phtalimido-2-nitrophenylacetic acid methyl ester (6). This compound was synthesised according to a literature procedure.3 A mixture of 1’-bromo-2nitrophenylacetic acid methyl ester (8.6g, 0.031 mol) and potassium phthalimide (5.8 g, 0.031 mol) in 100 ml of DMF was stirred at rt for 6 hr. The KBr was removed by filtration, and the filtrate was partitioned between CH2Cl2 (80 mL) and water (200 mL). The CH2Cl2 layer was separated and the aqueous layer was extracted twice with 80 mL portions of CH2C12. The CH2Cl2 extracts were combined, dried over anhydrous Na2SO4, and taken to dryness in vacuo to give a dark-red oil. Treatment of the oil with hot EtOH effected the separation of a white solid. The precipitate was filtered, washed with cold H2O, and dried in vacuo to yield 7.8 g (74 %) of product 6. 1 H NMR (400MHz, CDCl3) 8.16 (dd, 1H, J = 1.4 and 7.8 Hz), 7.93 (dd, 2H, J = 3.3 and 5.3 Hz), 7.80 (dd, 2H, J = 3.3 and 5.3 Hz), 7.60 (dt, 1H, J = 1.5 and 7.8 Hz), 7.53 (dt, 1H, J = 1.5 and 7.8 Hz), 7.43 (dd, 1H, J = 1.5 and 7.8 Hz), 6.93 (s, 1H), 3.78 (s, 3H). MS(ESI): [M+H]+ calculated: : 341.1, found: 341.0. N-Fluorenylmethyloxycarbonyl-(2-nitro)phenylglycine (8). A solution of 7.8 g (0.023 mol) of 1’-phtalimido-2-nitrophenylacetic acid methyl ester was hydrolysed in 50 mL of refluxing concentrated HCl-AcOH (3:2) for 5 hr. The phthalic acid was removed by filtration and the filtrate was taken to dryness in vacuo, yielding crude (2- 2 nitro)phenylglycine-HCl salt 7 as a yellow foam (5.4 g; quant.), which was used without further purification in the next reaction. Fluorenylmethylchloroformate (5.7 g, 22 mmol) dissolved in dioxane (75 mL) was slowly added over a 10 min period to a cooled (4 oC) and rapidly stirring suspension of crude (2-nitro)phenylglycine-HCl salt (4.6 g, 20 mmol) in 10% (w/w) aqueous sodium carbonate (75 mL) in dioxane (75 mL). The solution was allowed to warm to room temperature and stirred for an additional 2 h. The reaction mixture was poured into 600 mL of water and extracted with Et2O (200 mL, 2x). The aqueous layer was cooled (4 oC) and the pH was adjusted to 2 by the addition of 2N aqueous HCl. The acidified aqueous layer was extracted with Et2O (3x150 mL). The combined organic layers were dried with Na2SO4 and concentrated in vacuo, yielding the crude product as a yellow foam. Flash chromatography over silica gel using a gradient of ethylacetate-hexane (4:1) to ethylacetate to ethylacetate-methanol (4:1) yielded racemic title compound 8 as an off-white powder (4.6 g, 56 % yield over 2 steps). This compound is stored in the dark at –20 oC. MS(ESI): [M+H]+ calculated: 419.1 found 419.0 1H-NMR (400 MHz, d6-DMSO) 12.12 (bs, 1H), 8.22 and 7.91 (2 x d, 1H, J = 7.8 Hz, rotamers), 7.88 (d, 2H, J = 7.4 Hz), 7.84 (d, 2H, J = 7.4 Hz), 7.76-7.58 (m, 2H), 7.43-7.31 (m, 6H), 6.28 (s, 1H), 4.27-4.16 (m, 3H, rotamers). CRITICAL. Fmoc-(2-nitro)phenylglycine was found to be unstable in NMP and DMF, solvents routinely used in automated peptide synthesis. We therefore recommend the use of dichloromethane during peptide coupling procedures. 3 Expression and purification of biotin ligase Materials Equipment Incubator/orbital shaker Centrifuge 250 mL – 1 L buckets Spectrophotometer (for measuring OD at 600 nm and 280 nm) Plastic cuvettes Quartz cuvette Sonicator Reagents Wash buffer: 0.5 % Triton X-100, 1 mM EDTA LB medium LB agar plates + antibiotics Expression construct: BirA biotin ligase was cloned in a pET21B vector (amino acids 1- 321, in frame with a His-tag). Mw 36,4 kDa Bacterial strain BL21 (DE3) pLysS (cat. # 69451, Novagen) Carbenicillin (cat. #: C1389, Sigma-Aldrich) http://www.sigmaaldrich.com/catalog/search/ProductDetail/SIAL/C1389 Chloramphenicol (cat. #: C0378, Sigma-Aldrich) http://www.sigmaaldrich.com/catalog/search/ProductDetail/SIGMA/C0378 1-S-Isopropyl--(D)-thiogalactopyranoside (IPTG) (cat #: 11 411 446001, Roche) http://www.roche-applied-science.com Talon Co2+-resin (cat. #8901-2, Clontech) http://www.clontech.com/AIT/Ecommerce/Clontech/ProductCatalog.aspx?enc =true&idcategory=&idsearch=|242|188|26|67|28|101|212 Procedure 1 Transform biotin ligase expression plasmid into BL21(DE3)pLysS. Plate on LB plates containing 50 µg/mL carbenicillin and 34 µg/mL chloramphenicol. 2 Pick colony and grow a 10 mL culture (LB + antibiotics) for approx. 6 hrs. 4 3 PAUSE POINT. Store culture at 4 oC overnight. 4 Next day, inoculate 1 L with 10 mL culture and grow at 37 oC until OD600nm= 0.5. Allow the culture to cool down to 30 oC before proceeding with the next step. 5 Add IPTG (0.4 mM final concentration) and grow culture overnight at 30 oC! 6 Spin at 4 oC, 4,000 rpm for 15-20 min. 7 Resuspend pellet in 80 mL 20 mM Tris pH 8, 100 mM NaCl, divide over two 50 mL falcon tubes and sonicate (4 times 10 sec). Minimize sonication time, as prolonged sonication may destroy protein functionality. 8 Spin samples at 12,000g for 10 min at 4 °C to pellet all insoluble material. 9 From here Clontech protocol B for Co2+-batch/gravity column purification is followed (starting page 30): http://www.clontech.com/clontech/techinfo/manuals/PDF/PT1320-1.pdf 10 Prepare 2 mL of Co2+ beads, which is sufficient for approx. 5 mg of protein. 11 Use sonicated supernatant for Co2+-batch/gravity column purification step 8 page 30. 12 Wash the resin with 10 bed volumes of wash buffer (page 31, step 12-14 Clontech) and repeat this step. 13 Prepare a column (page 31, step 17). 14 Wash with 5 bed volumes of wash buffer. 15 Wash the protein by adding 3 bed volumes of 5 mM imidazole, 20 mM Tris pH 8, 100 mM NaCl. Collect 1 mL fractions and analyze by SDS-PAGE. 16 Repeat step 15 with 10 mM imidazole, 20 mM Tris pH 8, 100 mM NaCl. Collect 1 mL fractions and analyze by SDS-PAGE. 17 Elute the protein by adding 6 bed volumes of 50 mM imidazole/ 20 mM Tris pH 8, 100 mM NaCl. Collect 1 mL fractions and analyze by SDS-PAGE. Most protein will be in fraction 1 and 2. 18 Add β-mercaptoethanol to 5 mM and glycerol to 5 %. Measure OD280nm (ε = 47,440) and make 20 μL aliquots of 100 μM. Mw 36.4 kDa. One aliquot is sufficient for the biotinylation of 3 μM heavy chain in 20 mL. PAUSE POINT. Store the protein at –20 oC. 19 Optionally, the activity of the purified BirA enzyme can be tested using the following assay. To a 1.5 mL Sarstedt polypropylene microtube add: - x L BirA substrate (GGGLNDIFEAQKIEWH, Mw 1813) 50 M 5 final concentration. - 50 L 10X ligase buffer (200 mM Tris pH 7.5, 50 mM MgCl2) - x L purified BirA (approx. 2 M BirA is needed for 50 M peptide) - 10 L biotin (5 mM stock) - 10 L ATP (500 mM stock) - x L H2O 500 L end volume 20 Incubate overnight at RT. 21 Add TFA to samples to 0.1 % final concentration. Analyze by reverse phase HPLC (e.g. Waters Delta-Pak, C18, 100 Å, 15 m, 3.9 X 300 mm). As a reference, analyze peptide without BirA exposure. Run a 60 minute gradient: H2O: CH3CN (80:20), 0.1% TFA to H2O:CH3CN (65:35), 0.1 % TFA. Anticipated results The expected yield of BirA enzyme from 1 L of bacterial culture is 2 mL of a 100 μM solution. Timeline (4 to 5 days) Step 1 1 day Step 2 6h Step 3 16 h (overnight) Step 4 6h Step 5 16 h (overnight) Step 6-18 3h Step 19 30 min Step 20 16 h (overnight) Step 21 2.5 h 6 ELISA titration curve References 1. Liu, B. & Hu, L. 5’-(2-Nitrophenylalkanoyl)-2’-deoxy-5-fluorouridines as potential prodrugs of FUDR for reductive activation. Bioorg. Med. Chem. 11, 3889–3899 (2003). 2. Zhu, Q., Girish, A., Chattopadhaya, S.& Yao, S. Q. Developing novel activitybased fluorescent probes that target different classes of proteases. Chem. Commun. 1512-1513 (2004). 3. Davis, A. L., Smith, D. R. & McCord, T. J. Synthesis and microbiological properties of 3-amino-1-hydroxy-2-indolinone and related compounds. J. Med. Chem. 16, 1043-1045 (1973). 7