chem ethanol 1998
advertisement

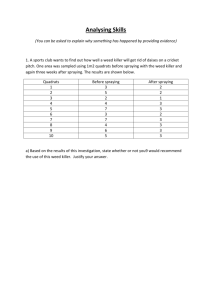
"Anaerobic production of ethanol by Saccharomyces cerevisiae in continuous culture" Group 3: Jenn-hui Khong, Shane Peterson, Nerida Rixon, Wun Long Wang. Abstract: D-glucose was used as the sole carbon source for ethanol production anaerobically by Saccharomyces cerevisiae in a chemostat system. Problems were experienced with media contamination by S.cerevisiae in the first chemostat set-up. In the second chemostat, a new media type (YPD broth) was used and ethanol production was achieved for approximately 30 hours, with a maximum ethanol concentration of 200mM being obtained. Ethanol production then ceased possibly because of microbial contamination of the reactor vessel. Steady-state was not obtained in either of the chemostat runs because of problems with contamination. Introduction: Production of alcohol by the action of yeast on malt or fruit extracts has been carried out on a large scale for many years and is recognized as the first 'industrial' process for the production of a microbial metabolite (Stanbury and Whitaker, 1984). Aside from the use of ethanol in alcoholic beverages, much research is being performed on the possibility of using ethanol as an automobile fuel. In some countries ethanol is being used to produce ethylene and other petrochemicals. These studies are being performed in agricultural areas of Brazil, South Africa and the United States, using a carbohydrate substrate such as sucrose or starch for ethanol production. There is massive growth in this area; In Brazil in the 1980's 20% of the petroleum imports were replaced with ethanol production from sugar cane. (Crueger, 1984) Saccharomyces cerevisiae is the most commonly used yeast for the production of ethanol. Under aerobic conditions and high glucose concentrations S.cerevisiae grows well but produces little ethanol, whilst under anaerobic conditions growth slows and ethanol production is increased. Pyruvate produced from glucose via glycolysis is split into acetaldehyde and CO2 by pyruvate decarboxylase. Reduction of the acetaldehyde by alcohol dehydrogenase produces ethanol. (Crueger, 1984) This pathway, like all fermentation processes is an energy-generating process in which organic compounds act as both electron donors and terminal electron acceptors (Stanbury and Whitaker, 1984); the NADH produced through glycolysis donates electrons when it is reoxidized to NAD+, and pyruvate acts as the electron acceptor (Prescott et al, 1993). The overall equation for the fermentation of D-glucose in anerobic conditions is: Glucose + 2ADP + 2Pi 2Ethanol + 2CO2 +2ATP (Becker & Deamer, 1991). Continuous culture systems (chemostats) offer many advantages over batch and fed-batch processes. This is because the steady-state nature of a chemostat (operating at or near Dmax) gives a relatively constant, and maximum productivity, and because most of the parameters remain constant, the chemostat system should be much easier to maintain over a batch system. The major disadvantage of chemostats is their susceptibility to contamination by 'foreign microorganisms'. Therefore, although ethanol production would be more efficient in a chemostat system, the problem of contamination has led to limited success and batch processes are most commonly used for ethanol production. (Walker and Gingold, 1993) Batch systems for ethanol production are set-up aerobically to obtain maximum biomass so that when the system is switched over to an anaerobic, continuous culture there will be enough biomass present to obtain a high glucose to ethanol conversion rate (Crueger, 1984). The aim of this experiment was to set-up and run a chemostat for the anaerobic production of ethanol and obtain steady-state conditions where all (or most) of the reactor parameters (such as biomass concentration, substrate concentration etc.) remain constant. Experience in the difficulties of setting up a chemostat and the problems which occur during the running of the chemostat was hoped to be gained. Materials & Methods: Materials: Inoculum: Yeast (Saccharomyces cerevisiae) 2.6g D-glucose 18.6g warm water 250mL (Glosz and Stephanopolous, 1990) Dissolve the glucose in warm water and sprinkle yeast on top of warm water to activate. The yeast strain used in the first chemostat run was Tandaco's Dry yeast and in the second chemostat Homebrew Australia's beer yeast was used because of the nonavailability of Tandaco's dry yeast. Media for Chemostat #1 (start 1/5 pm): D-glucose 90g (later lowered to 40g on 5/5 pm - see discussion) yeast extract 8.5g NH4Cl 1.3g MgSO4.7H2O 0.11g CaCl2 0.06g H20 1000mL (Glosz and Stephanopolous, 1990) Media for chemostat #2 (start 9/5 am): YPD medium: D-glucose yeast extract peptone 20g 10g 20g Dissolve ingredients in 1000mL of water and adjust the pH to 5.6 (O'Brien, 1997). The pH was adjusted to pH 5.0 in chemostat #2. See discussion for reasons why this new medium was used and why it was made more acidic. Equipment: For chemostat set-up: 1. 2L Schott bottle (feed) 2. 1L Schott bottle (reactor) 3. 1L Schott bottle (outflow) 4. 6mm thick plastic tubing (with 3mm internal diameter) 5. 4mm thick plastic tubing (with 3mm internal diameter) 6. 2 drippers 7. 3 rubber bungs 8. 2 cotton wool bungs 9. 1 IEC magnetic stirrer and stirring rod 10. 1 EYELA Micro Tube Pump MP-3 with 4 channels 11. 1 Thermal Compact Submersible aquarium heater 12. 1 thermometer 13. 3 bosshead clamps 14. 2 retort stands 15. bucket containing water (for reactor) 16. aluminium foil For Sampling: 1. Eppendorf tubes (1.5mL) 2. P200 and P1000 Gillson pipettes 3. yellow and blue pipette tips 4. Measuring cylinder (100mL) 5. Haemocytometer and cover slip 6. portable pH probe and calibration solutions 7. marking pen 8. microcentrifuge 9. YSI Model 2700 Select Biochemistry analyzer (for glucose analysis) 10. Varian Star 4000 Gas Chromatography equipment 11. Spectrophotometer (used with visible light lamp) 12. Sharp Scientific Calculator EL-556G Methods: The chemostat was set-up as shown in Appendix 1. This set-up was based on the 1996 Ethanol Report "Chemostat Set-up 2" except for a few minor changes. A double-dripper system was used as the reactor vessel inflow device instead of the single dripper system. This was hoped to eliminate the possibility of yeast contamination of the media: the yeast would have to spread up two fine (0.75mm diameter) needle tips. The pump was calibrated by running the chemostat with water. On speed level 1, 45mL of water flowed through into the outflow vessel in 1 hour. It was decided to run the pump at level 1 for 1 minute on and 2 minutes off to give a flowrate of 15mL/h (D=0.03 h-1). All of the equipment in the set-up was sterilised by autoclaving, making sure that any outlets were wrapped in aluminium foil. 2L of media was autoclaved initially. The inoculum was covered and left overnight as a batch culture. The following day the inoculum was aseptically poured into the sterile reactor vessel, and media added to make the reactor vessel volume up to 500mL. The pump, timer and heater were then turned on and continuous culture began. Sampling: Sampling was performed twice a day in most cases. Each time sampling was done we collected tubes for G.C., glucose analysis, and for spectrophotometry. Two 1.5mL Eppendorf tubes were filled with outflow, one labelled "glucose" and the other "ethanol". A "1/10 glucose" tube was also collected by taking 0.1mL from the outflow and adding 0.9mL deionized water. All tubes were labelled with the group number, date and time. For measuring optical density, one 1.5mL eppendorf tube was filled with media from the feed vessel, and another filled with outflow. Ethanol determination: Preparation of samples: 1. Spin "ethanol" labelled 1.5mL eppendorf tube samples in microcentrifuge at 13000rpm for 10 minutes 2. Dilute 1/50 in water by adding 20uL of supernatant and make up to 1mL total volume with deionized water and put into G.C. vials 3. Add 20uL of 50% phosphoric acid into 1mL sample (do this in fume cupboard) The G.C. vials were given to Carol, who subjected the samples to gas chromatography on Tuesday 20/5: the results were assessed on Wednesday 21/5 to determine both how much ethanol was produced and whether any acetic acid or propionic acid was produced (sign of possible contamination). A Varian Star 4000 machine was used for gas chromatography. Fig 1. Plot of OD540nm vs yeast concentration (g/L) 1.05 Glucose determination: 1 The "glucose" and "1/10 glucose" samples were spun in a microcentrifuge at 13000rpm for 10 minutes. For measurement of glucose concentration, a YSI Model 2700 Select Biochemistry Analyzer was used. This analyzer takes 25uL samples, and we sampled from the "glucose" tubes. Biomass: Cell counts were performed of the outflow using a haemocytometer and microscope. The optical density (OD) of the medium in the feed vessel was measured at the time of sampling. The optical density was read at a wavelength of 540nm. To determine biomass (g/L) from the OD measured for the different outflow samples, a standard curve was plotted of OD540nm with respect to biomass (g/L). 0.5g of dry yeast was dissolved in 2mL deionized water and then serial dilutions performed by taking 0.5mL of this stock solution and adding 1mL H20 (Hence a 1:3 dilution). This procedure was continued until 6 different dilutions were obtained. Another series of dilutions was also prepared and the OD540 measured for all samples. Both sets of data (the 2 dilution series) were entered into the statistical mode of a scientific calculator to determine whether there is a relationship between OD540 and yeast concentration. Results and Discussion: The recorded data and subsequent calculations for chemostat runs 1 and 2 are shown in spreadsheet form (Appendix 2). Both of the chemostats had problems with contamination and hence no steady state was obtained for either set-up. For the standard curve of OD540 with respect to yeast concentration (g/L), two sets of serial dilutions were performed and OD values obtained (Appendix 3). The correlation coefficient r2 was calculated for both data sets to determine whether there is a linear relationship between OD540 and yeast concentration (g/L). For the first series of dilutions, r2=0.0152 and for the second dilution series r2=0.866. The first set of data cannot be relied upon, yet the high correlation obtained in the second dilution series means that linearity can be assumed. The results of the second dilution series were plotted, and a trend line drawn in (Fig 1). OD540 0.95 0.9 0 100 200 300 400 yeast concentration (g/L) (g/L) The OD540 readings in this standard curve are all within a very small range. It was very difficult to dissolve enough dry yeast in water to give high absorbance readings in the vicinity of 1.10 or greater. This practical aspect of the experiment was overlooked; it was initially hoped to get a wide range of absorbance readings up to around OD=1.7. This would mean that OD readings for the various time samples could be read straight off the standard curve (Fig 1) without any need for extrapolation. To read OD values in a range outside that of the standard curve, linearity outside the data range was assumed and extrapolation used to get a yeast concentration (g/L) from the measured OD reading. Instead of extrapolating the curve by hand, for a higher degree of accuracy the statistical mode of the calculator was used to give corresponding x-values (yeast concentration in g/L) from the measured yvalue (OD540). Hence, these biomass concentrations (X in g/L) read off the standard curve for the different samples were recorded in the spreadsheet (Appendix 2). In some cases, the OD due to biomass was too low and gave negative biomass values when read off the standard curve (on data recording a -ve value for X was assumed to be approximately 0g/L). The standard curve method therefore provides an estimate of biomass concentration, but using this method did not give many "useable" results, many X is approximately 0g/L results being obtained (Appendix 2). Another possible method for biomass concentration detemination was established by Guerts et al (1980). This is a simple method which involves taking 3060mL samples of the outflow, and centrifuging two aliquots of each sample at 3000 g for 10 minutes. The supernatants are discarded and the pellets washed twice with demineralized water, and dried for 48h at 100 C. (Guerts et al, 1980) The dried biomass can then be weighed, and biomass concentration (g/L) determined. The chemostat beginning on 1/5 ran until the 7/5 when a group decision to terminate chemostat #1 was made due to severe contamination of the reactor vessel with chains of green cocci microorganisms (and some rod-shaped microorganisms). These cooci-shaped microorganisms were clustered close together in chains which made it impossible to count them using a haemocytometer and microscope. The decision to cease this chemostat was hence not based on direct cell counts of the contaminant microorganisms but on an overall impression of the contaminant population. For this first chemostat run, there was a serious lack of adequate data entry into the spreadsheet (Appendix 2). Many parameters such as pH measurement, and OD540 readings were being ommitted. Hence, all members of the group made a concerted effort to spend a bit more time in recording data for Chemostat #2. All of the equipment was autoclaved, and it was decided to change the media for chemostat #2. It had already been noted during chemostat #1 that a 90g/L glucose concentration was too high (on reading past ethanol reports revealing glucose wasteage). Glucose concetration was reduced to 40g/L on the second day of chemostat #1. The glucose concentration was reduced further for chemostat #2 to 20g/L as this concentration is used with success in Molecular Biology I and IIa and Microbiology I for yeast growth. The media chosen (YPD media) has not been used in past ethanol chemostat experiments, but it is a proven growth media for S.cerevisiae (O'Brien, 1997). It was decided to make the media a less hospitable environment for possible contaminant microorganisms. Although stated in O'Brien (1997) that the YPD media should have pH5.6, it was decided to make the media more acidic at pH 5.0. S.cerevisiae is known to survive at even lower pH values than this - in Guerts et al (1980) the pH was maintained at pH 4.0 for a chemostat growing S.cerevisiae CBS 426. As the strain and pH requirements of the Beer Yeast used in Chemostat #2 are unknown, it was decided not be make the media highly acidic at pH 4.0 in case the particular yeast strain was not capable of surviving. The glucose and G.C. analysis was performed for the samples taken from chemostat #2 only. This was because of a lack of samples from Chemostat #1 and because of a lack of time to set-up the tubes for analysis and perform the glucose analysis. In hindsight, it would have been of great benefit to determine substrate and product concentrations during chemostat #1. Although we could smell ethanol in the outflow, the actual amount produced remained unknown for the entire experiment. It would have been worthwhile having these results because even though microbial contamination of the reactor occurred, a comparison between the ethanol concentration of chemostat #1 and #2 over time would have shown whether the YPD media used in chemostat #2 gave a higher product yield and was better utilised by the S.cerevisiae. From the G.C. and glucose analysis results, the ethanol concentration (mM) and glucose concentration (g/L) were plotted at various times after the start of chemostat #2 at t=0h (Fig 2). Fig 2. Ethanol concentration (mmol/L) and glucose concentration (g/L) in outflow with respect to time 200 150 100 50 0 0 50 100 150 60 50 40 30 20 10 0 200 time (h) The ethanol levels were noted to increase until a maximum of 200mM was attained at around t=60h (Fig 2). Subsequent samples had no ethanol production. The glucose concentration (g/L) shows a decrease from 20g/L at t=0h (glucose concentration in the media is 20g/L) to trace amounts (close to 0g/L) from t=60h onwards (Fig 2). These results seem to indicate that a lack of glucose led to the rapid reduction in ethanol production from 200mM to 0mM within a time period of less than 2 days. The glucose concentration in the media was possibly too low as it was reduced to close to 0g/L in the reactor within 60h, around 98% of the glucose being utilised. This reduction in glucose concentration is expected though, Glick and Pasternak's experiment (1994) showing that 96% of sugars were converted to ethanol and other byproducts. Another possible reasons for the sudden loss of ethanol production in the chemostat could be that there are other microorganisms either utilising the glucose substrate to produce a product other than ethanol, or either contaminant microorganisms or S.cerevisiae using ethanol as a carbon and energy source for growth. An experiment performed by Barford and Hall (1979) revealed that S.cerevisiae can continue growth when ethanol is the limiting carbon and energy source. The major end-products are carbon dioxide and water, though a vast range of endproducts are possible such as acetic and lactic acids. (Barford and Hall, 1979) In both chemostat #1 and #2, there were microorganisms which were present as chains of green cocci in the reactor vessel and outflow (Appendix 2). These microorganisms were obviously able to tolerate a low pH reaching as low as pH4.0 in the reactor on the 14/5 and 15/5 (Appendix 2), and could utilise either glucose or ethanol as a substrate. As there were only trace amounts of acetic and propionic acid present in the outflow in the chemostat #2 samples, it is unlikely that acetic acid or propionic producing microorganisms (such as Clostridium propionicum) (Cord-Ruwisch, 1997) are the source of this contamination. The G.C. equipment may have been malfunctioning and providing innaccurate results giving 0mM ethanol production when in fact there was ethanol being produced. There was possibly some error incurred during sampling, such as mixing up Eppendorf tubes containing diluted outflow and undiluted outflow samples. The trend in biomass concentration over time for chemostat #2 was ascertained by looking at both the cell count using a haemocytometer and the biomass concentration, X (g/L). This was necessary because many of the X values were 0g/L as obtained from the standard curve (as previously discussed). The biomass was not plotted as a function of time because the curve had too many innaccurate 0g/L points, the results can be seen in Appendix 2. The biomass tended to increase as time progressed. We would expect this to occur, biomass concentration increasing as utilisation of the glucose substrate by the yeast for growth occurs. The biomass concentration would then be expected to level off as steady-state is reached and the rate of biomass increase equalling the biomass washout rate. Chemostat #2 was terminated because of yeast contamination of the media. The likely source of this contamination was a non-sterile tip used on the Gillson pipette when sampling. Because of the shortage of pipette tips in the laboratory, tips which had been used for sampling the outflow were being rinsed and re-used. The cocci-chains and occasional rod-like microorganism contamination in both chemostat #1 and #2 had a number of possible sources. There may have been a loose stopper on one of the Schott bottles used in the chemostat setup which allowed for contaminant entry. For measurement of outflow volume, some members of the group were pouring the outflow into a measuring cylinder, and then returning the outflow to the outflow bottle. It is highly probable that this process allowed some microorganism entry into the outflow vessel, from which it could travel through the tubing and contaminate the reactor vessel. Suggestions for future years: 1. 2. 3. The method for determination of biomass concentration needs to be improved as the standard curve method gives only a "rough" estimate and many X=0g/L results are obtained. Weighing the dried biomass as in Guerts et al (1980) may be a more effective method as previously discussed in "Results and Discussion" section of this report. Although the chemostat set-up can never be completely sterile when the outflow and feed vessels are opened regularly for sampling, the sampling procedure should be as sterile as possible. The yeast contamination in the media which occurred on two occasions during chemostat #2 was almost certainly attributed to a yeast contaminated pipette tip. It is of vital importance to read the past chemostat reports and literature before beginning the chemostat project. There are so many valuable suggestions in how to set-up the chemostat, and time can be saved if students have some knowledge of when and where problems with the chemostat are likely to occur. 4. It is up to the manager to make sure the group members realise that as much data as possible must be recorded each time they come in for data analysis. This aids in determining whether there are any problems with the chemostat, and improvements can be made if necessary, or the chemostat terminated if the problems are beyond repair. 5. Instead of performing both the glucose and G.C. analysis after the chemostat has been terminated, it is much wiser to obtain results during the running of the chemostat. Students then know the exact state of the chemostat, whether results are as expected or whether the chemostat conditions need to be altered. For example, if G.C. analysis was performed during chemostat #2, it would have been noted that there was no ethanol being produced and changes or termination could have occurred. 6. When making changes to the chemostat from past reports, extensive research must be done so that reasons for making such a change can be stated when writing the report. 7. If one group member decides to make a change to the chemostat (such as changing the media or running conditions), they should consult with the other group members first. 8. It would be of significant advantage to have some form of carbon dioxide measurement apparatus incorporated into the chemostat setup. 9. Make sure that glucose and G.C. analysis are performed on all samples, including those from failed chemostat set-ups. Knowing the trends of glucose concentration and ethanol concentration are important in determining when and/or why the chemostat failed. 10. The possibility of experimenting with different substrates exists and there are many utilisable substrates for S.cerevisiae to grow on. The effect of using a mixed substrate such as glucose and ethanol on ethanol production, or the use of a starch-based substrate could prove to be interesting experiments. References: Barford, J.P. and Hall, R.J. (1979) "Investigation of the Significance of a Carbon and Redox balance to the measurement of Gaseous Metabolism of Saccharomyces cerevisiae." Biotechnology and Bioengineering 21: pp 609-626. Becker, W.M. and Deamer, D.W (1991) The World of The Cell. The Benjamin/Cummings Publishing Company, California, USA. Cord-Ruwisch, R (1997) N301: Industrial Microbial Physiology. Murdoch University Press, Murdoch, Perth, Western Australia. Crueger, W (1984) Biotechnology: a textbook of industrial microbiology. Sinauer Associates, Inc., MA, USA. Glick and Pasternak (1994) Molecular Biotechnology: Principles and Applications of Recombinant DNA. ASM Press, Washington, USA. Glosz, R. And Stephanopolous, G (1990) "Microaerobic ethanol fermentation." Biotechnology and Bioengineering 36: p1006. Guerts, T., Hermine, E., and J. Roels (1980) "A Quantitative Description of the Growth of Saccharomyces cerevisiae CBS 426 on a mixed substrate of Glucose and Ethanol." Biotechnology and Bioengineering 22: pp 2031-2043. O'Brien, P (1997) Molecular Biology IIa/IIb Lecture Guide and Laboratory Manual. Murdoch University Pree, Perth, Western Australia. Prescott, L.M., Harley, J.P and D.A. Klein (1993) Microbiology. Wm. C. Brown Publishers, Dubuque, Iowa, USA. Stanier, R.Y., Doudoroff, M and E.A. Adelberg (1971) General Microbiology. MacMillan and Company Limited, London. Stanbury, P.F and Whitaker, A. (1984) Principles of Fermentation Technology. Pergamon Press, New York, USA. Walker, J.M and Gingold, E.B. (1993) Molecular Biology and Biotechnology. The Royal Society of Chemistry, Cambridge, UK. Appendix 1: Chemostat set-up (kept the same for chemostat #1 and #2). Appendix 3: Standard Curve Raw Data.