reagents
advertisement

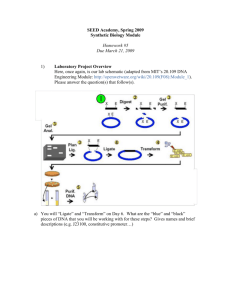
EXPERIMENT 12 ISOLATION AND QUANTIFICATION OF GENOMIC DNA FROM MOUSE LIVERS PRINCIPLE Genomic DNA molecules exist as nucleoprotein in cells. The isolation principle of DNA is not only to separate DNA from complex with proteins, lipids and sugars, but to maintain its integrity. In the extraction solution, SDS serves as detergent to dissolve plasma and nuclear membrane and separate DNA from histone. EDTA chelates Ca2+ and Mg2+ to inhibit degradation of DNA by DNase. Then phenol/chloroform precipitate proteins and dissolve lipids. Thereafter chloroform extracts trace phenol from DNA solution. Finally, DNA is precipitated by 100% ethanol. REAGENTS 1. 1×TE buffer (pH 8.0): 10 mmol/L Tris-HCl pH 8.0, 1 mmol/L EDTA-Na2 pH 8.0. 2. 10% SDS 3. Saturated distilled phenol 4. Chloroform/isoamyl alcohol (24:1, v/v) 5. 10 mmol/L NH4Ac 6. 100% ethanol PROCEDURE Part I Preparing of liver homogenate 1. Sacrifice and dissect rat to get livers. 2. Weigh 1 gram of liver after rinsing with cold physiological saline. 3. Put the tissue in a grass homogenizer and add 5 ml of cold TE buffer. Then grind the liver to a homogenate. PART II ISOLATION DNA 1. Pipette 0.5 ml of liver homogenate to a 1.5 ml eppendorf tube, and add 50μl of 10% SDS. Mix it by hand with a medium up and down motion. Then incubate at room temperature for 10 min to form a sticky solution. 2. Mix an equal volume of saturated distilled phenol and chloroform/isoamyl alcohol (24:1, v/v) and add 0.5 ml of this solution to the tube. Mix up by hand with a medium up and down motion for 10 min and then centrifuge at 1 3. 4. 5. 6. 12000 g at 4 ℃ for 10 min. Remove upper aqueous layer carefully with an adjustable pipette and transfer it to a new 1.5 ml eppendorf tube. Add 0.5 ml of chloroform/isoamyl alcohol (24:1, v/v). Mix up by hand with a medium up and down motion for 10 min and then centrifuge at 12000 g at 4 ℃ for 10 min. Remove upper aqueous layer carefully and transfer to a new 1.5 ml eppendorf tube. Adjust the volume to 0.4 ml of DNA solution by either adding TE buffer or removing DNA solution. Add 120μl of 10 mmol/L NH4Ac and mix gently by inversion. Then add 1 ml of 100% ethanol and mix gently by inversion, incubate the solution for 10 min at 4 ℃. Centrifuge at 12000 g at 4 ℃ for 5 min after incubation, discard the supernatant and wash the pellet by adding about 1 ml of 70 % ethanol. Centrifuge at 12000 g at 4 ℃ for 2 min, discard the supernatant and wash the pellet once again. Remove the last trace liquid on the inner wall of tube by pipette or filter paper. Evaporate the ethanol at room temperature for 15 min. Add 1.0 ml TE buffer to dissolve the pellet by plucking the tube gently. If this does not work, bath the tube in water at 56 ℃ and pluck the tube gently at intervals. PART Ⅲ QUANTIFICATION OF DNA Pipette 0.5 ml of DNA sample and add 3.5 ml distilled water. Mix them well. Set the ultra-violet spectrophotometer to zero with distilled water at 260 nm or 280 nm and measure the absorbance (A) of the diluted DNA sample . CALCULATION 1. DNA content (μg/ml) = A260×50μg/ml×4ml×÷0.5ml 2. Calculated the value of A260/A280 SPECIAL NOTE 1. Severely shaking will break DNA chain, so shaking should be gently either by hand or by vortexer. 2. If high purity of DNA should be obtained, adding RNase and proteinase K is recommended to digest RNA and protein respectively. 3. The peak absorbance of DNA is at 260nm, whereas that of protein is at 280nm. Based on this principle, the purity of DNA can be estimated. The A260/A280 value of pure DNA is about 1.8. If A260/A280<1.8, the DNA sample may be contaminated by protein or phenol; if A260/A280>1.8, the DNA sample may be contaminated by RNA. The A260/A280 value 1.8±0.5 could be accepted for most experiments including sequencing and transfection. 2