4. measurements of surface conductivity
advertisement

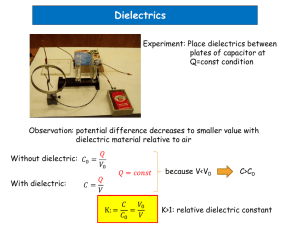
Conductivity at the Interface between Two Dielectrics Due to Image Forces Korobeynikov S. M.1, Melekhov A. V.2, Soloveitcik Yu.G.1, Royak M.E.1, Charalambakos V.P.3 and Agoris D.P.3 1 Novosibirsk State Technical University, 20, K. Marx av., Novosibirsk, 630092, Russia e-mail: kor_ser_mir@ngs.ru 2 Institute of Laser Physics SB RAS, 13, Institutskaya str., Novosibirsk, 630090, Russia 3 University of Patras, Patras-Rio, Gr 26500, Greece PACS 77.30; 73.25; 47.65 Abstract. In the present paper, the processes of recombination and dissociation of ion pairs in liquid dielectrics near the boundary of the other dielectrics (solid, gas, or liquid) are considered. Different ratio of the dielectric permittivities is proposed. It has been shown that if the permittivity of the liquid is less than the permittivity of the other dielectrics dissociation increases and recombination decreases. In the case of inverse ratio the recombination increases and the dissociation decreases. The experimental results of the electrical conductivity measurements in films, foams, and also the charge of colloidal particles, surfaces and bubbles are discussed. 1. INTRODUCTION Study of the charge carriers generation, movement and disappearance in two phase medium is important for electrical insulation [1], colloidal and electrical chemistry [2], electrohydrodymamics (EHD) [3] and other related fields. In the field of electrical insulation there is a problem of contact of two dielectrics where one of them is liquid. While devices like transformers and partition insulator are working and the oil is moving along the paper the oil electrization takes place. The stability of different colloidal systems like suspensions, foams, films is closely connected with the creation and behavior of the charges at the interfaces. EHD device converses the electrical energy into kinetic or hydrostatic energy of liquid. Usually EHD devices (pumps, heat exchanger, etc) have at least three systems: the first one produces space charge in dielectric liquids, the second one moves the liquid due to electrical force, and the third one removes space charges. The functioning of the second device is not a problem because it is a special HV electrode system but other ones concern to the problem of charge generation and recombination. The aim of the present paper is the study of charge creation due to image forces and surface conductivity at the interface between the liquid dielectrics and the dielectrics with another permittivity. 2. RECOMBINATION AND DISSOCIATION NEAR DIELECTRICS WITH DIFFERENT PERMITTIVITIES As it was shown earlier [4] the energy of ion pair dissociation W in the layer close to the interface of the dielectric with higher permittivity could be several times less than the energy of dissociation in the bulk of liquid. In the case of arbitrary ratio of permittivities of coexisting phases the obtained expression is following: ( e' / e) ( R1 R2 ) Ws Wb 1 (( R1 R2 ) 2 4 R1 R2 ) (1) Here index b is related to the bulk dissociation, index s – to the surface one, R1, R2 – radii of ions of the ion pair (fig.1), e – ion charge, e' – image ion charge e (1 2 ) e (1 2 ) (2) 1 – dielectric permittivity of liquid, 2 - dielectric permittivity of another coexistent dielectrics. It is clear that in the case of lower permittivity of the second dielectrics in comparison with the permittivity of the first one, the energy of the ion pair dissociation is more than the energy of the ion pair dissociation in the bulk of liquid. The ratio of the liquid dielectric permittivity to the permittivity of the solid dielectrics is very important for the dissociation-recombination equilibrium processes. The formulas, obtained in [4] for 2/1>>1, can be modified to find equilibrium constants in the close to boundary region for arbitrary ratio 2/1. k RS R12 k DS 4 ( D1 D2 ) e 2 1/ 2 -( LB / r ( e ' / e ) LB /( r 2 R12 ) ) (3a) /r 2 dr ) ( D1 D2 ) RB R12 e 2 1/ 2 LB / r ( e ' / e ) LB /( r 2 R12 ) 2 r dr ( RB r e 2 1/ 2 -( LB / r ( e ' / e ) LB /( r 2 R12 ) ) 2 /r dr ) (3b) D1, D2 – diffusion coefficients of ions, R12- sum of ion radii, kDS – constant of dissociation, kRS – constant of recombination, LB is doubled so called Bjerrum radius RB e2 801 kT The symbols used correspond to the previous paper [4]. Fig.2 and 3 demonstrate calculated o o dependences of kRS/kR and kDS/kD on the ratio 2/1 for transformer oil (R1=1 A , R2=5 A , o o o o Lb=250 A , 1= 2.3, D1 D2 1010 m2 s 1 ) and for ester (R1=1 A , R2=5 A , Lb=140 A , 1=4). One can see that the recombination constant doesn’t change very much and the change is similar for the both liquids. The dissociation constant changes more radical, formal treatment could lead to the difference between cases 2/1>>1 and 2/1<<1 more the 21 orders of magnitudes. Here one should note that dielectric permittivity can’t be less than 1, therefore constant values at 2/1< 1/2.3 for transformer oil and 2/1< ¼ for ester are meaningless and they should not be considered. If this circumstance is taken into account the constant change at transition from 2=1 to 2= is to be less. So the change of the dissociation constant is to be 13 orders of magnitude approximately, five orders in the case of 2<1, the other 8 orders - at 2>1 . 3. NUMERICAL SIMULATION In the previous section the consideration from the physical point of view was carried out, it was assumed that the forces acting on charges are considered to depend on the distance between the charges. The eccentricity of the forces was negligible. Numerical simulation permits to take into account the forces between ions and images of other ions more accurately. Let's construct the numerical simulating procedure for the two charges dissociation process when there is a plane separating two half-spaces with different dielectric permittivities: (of the upper half-space - 1=2.3, of the lower half-space 2 = ). Let's consider, that the first charge o o has radius 1A , the second - 5 A . When the first charge position is fixed, the second charge center distribution function f = f (r , z ) can be found from the equation of continuity of r ( current density i = - D1 + D2 r grad f + LB f F : )( ) r div grad f + LB f F = 0 , ( r ) (4) r where F = F (r , z ) - total force operating on the second charge, r , z - cylindrical coordinates of the second charge center when the first charge is located on R axis. The equation (4) is valid in area W with the following boundary: G1 - geometrical locus of the points describing the location of the second charge center when the second charge contacts the first charge, G2 geometrical locus of the points describing the location of the second charge center when the second charge touches two half-spaces with different dielectric permittivity separating plane (i.e. o G2 - part of plane z = 5 A ), G3 - axis of symmetry r = 0 , G4 - remote boundary o o r = 1000 A , G5 - remote boundary z = 1000 A . Let's consider that on the boundaries G1 , G2 and G3 the condition of the normal currents non-flowing is ( ) in = - (D1 + D2 ) ¶ f ¶ n + LB f Fn = 0 , (5) and on the remote boundaries G4 and G5 the condition f = 0 (6) is . Furthermore, in one of boundary G1 points (with maximum r , in our case it is the point of G1 and G2 intersection) let’s set a value f = 1. (7) Let's solve a boundary problem for the current balance equation (4) using finite element method (FEM). To derive equivalent variational formulation let’s multiply the first and the second members of the equation (4) on a trial function r, z and integrate results over the calculated area W. Applying the Green formula (integration by parts) and taking into consideration the boundary conditions (6)-(7) we’ll take: r grad f ×grad Y + LB f F ×grad Y d W= 0 ò( W ) (8) To build finite-element approximation let’s decompose W on the triangular finite elements and also let’s define the piecewise linear finite basic functions Y i on them. Let’s represent the function f as a linear combination of these basic functions f = å q j Y j . By substituting a trial function Y in turn for all basic functions Y i , a set of the finite-element equations will be obtained: å j r q j ò grad Y j ×grad Yi + LB Y j F ×grad Yi d W= 0 ( ) (9) W Thus, we have got a set of the linear algebraic equations (SLAE) å aijq j = bi , (10) j where matrix cells are defined ò( aij = or from the ratio r grad Y j ×grad Yi + LB Y j F ×grad Yi d W, or from the boundary conditions ) W (6,7), and the right-part member components bi all are zero, except the one component corresponding to node, where condition (7) is . To solve this SLAE LU-factorization method taking into account the special profile SLAE matrix data storage format was used. The dissociation problem was solved using mesh containing 130 015 nodes and 257 842 triangles. In addition, the mesh was essentially condensed near the boundaries G1 and G2 . The numerical solution accuracy was checked by some mesh subdivisions. Fig.4. displays the allocation of function f (r , z ) near the boundary G1 . The axes are graduated in Angstrom unit. To calculate the dissociation constant, the function f (r , z ) obtained as a solution of the boundary problem was scaled with constant g : f g (r , z ) = f (r , z ) g , (11) where g= ò f (r , z )d W (12) WB and WB - intersection of the calculated area W with a sphere of radius R B òf g (i.e. (r , z )d W= 1 ) WB Let’s consider the function LB f R (r ) = e r - 1 LB e R12 - 1 1 × R, g RB g R = 4p ×ò R 12 LB e r - 1 LB ×r 2dr » 7.2 ´ 10- 29 m 3 , (13) e R12 - 1 that defines a probability density of the second charge distribution to the first charge in homogeneous space with a relative permittivity e . Fig.5 shows the graphs of behavior of the function f g function f R along the boundaries G2 (curve 3) and G3 (curve 2) in comparison with the (r ) g (curve 1). Calculated using function f (r , z ) the dissociation constant kDS = ò i dS n » 2.4 ´ 105 s - 1 , (14) Sg where S g - surface of the cylinder, that completely contains the boundary G1 , differs from the corresponding dissociation constant kD » 3.8 ´ 10 - 6 s - 1 for homogeneous space almost 11 orders of magnitude. 4. MEASUREMENTS OF SURFACE CONDUCTIVITY DC measurements of the surface conductivity were carried out. Ceramics based on BaTiO3 and CaTiO3 having 2=40, >1013 Ohmcm according to the producer’s data were used. Rectangular-shaped samples were of 2.5 x 1 cm2 x 0.7mm. The surface was polished. Transformer oil with 1=2.3, =1012 Ohmcm was used as a liquid dielectrics. Measurements of the surface conductivity were performed by means of the direct current voltage power supply B5-50 with the voltage from 1 V till 300 V and nanoamperemetre EMG-1352. The samples with the attached coaxial cables were screened by the copper box. The background noise of the measuring device was several picoamperes. The resistance of the liquid was measured in a special microcell with the rectangular electrodes 2 x 1 cm made of stainless steel. Measurements of the surface conductivity were carried out in the ad lib cell made of two polish surfaces of ceramics with two plate-shaped foil metal electrodes between them (fig.6). Tightly pressed each other the plates had the clearance which size determined by the thickness of the foil that the electrodes were made of. Bulk resistance Rv and surface resistance Rs were determined from the ratios: d l d Rs s 2l Rv v (15) where v is the specific space resistance of the liquid, s is the specific surface resistance, - foil electrode thickness, d - distance between electrodes, l - their length. The bulk ceramic resistance that is two orders more than the oil’s one was neglected. The ratio of the bulk resistance to the surface one depends on the electrode thickness. Changing the part responsible for the surface conductivity could be separated from the whole resistance that is parallel connection of the bulk resistance and the surface one. Testing of the method was realized by the measuring of the standard transformer oil using electrodes of 12 m, 60 m and 200 m thickness. The width of every electrode was 1 mm, the length was 2.5 cm, the inter electrode gap -1mm, the material- stainless steel, gap voltage 100 V. The testing results are shown in the fig.7. The surface resistivity could be found from the graph having been directed the electrode thickness to zero. It can be realized that the resistance value is 2.11012 Ohm. Recounting obtained results for the case of the surface resistivity one can obtain 1014 Ohm. To estimate the surface conductivity from the side of ceramic sample and its possible contribution to the conductivity of the whole system the following measurements were carried out. All above-cited measurements were made in the air. Obtained surface conductivity was about 1015 Ohm. As far as the solid body conductivity and the surface conductivity from the side of the solid body contribution to the whole conductivity according to given experiment is only a methodological error their combined contribution was estimated and this one was unimportant. Our efforts to input organic compounds (C4H9)3NC2H5Br, (C4H9)4NCl, ((C2H5)2N)4PBr, (C6H5)3P C2H5Br were unsuccessful. Their solubility was so negligible that there were not any visible changes or changes of conductivity. The surface conductivity was also unvarying. This fact could be an additional confirmation of the measured value s 1014 Ohm. A new attempt to measure the surface conductivity in the more polarized conductive liquid isoamyl alcohol C5H11OH was made. Its dielectric permittivity is 16, the specific resistance is 106 Ohmcm. The surface conductivity measured with the method described above is s 4108 Ohm. 5. DISCUSSION Experimental data. Let’s estimate the electrical parameters of the surface layer by mean of the measured values of the surface resistance. One takes the mobility of the charge carriers near the interface equal to the bulk one ~10-4 cm2/(Vs). So for the case of ceramics in transformer oil one could get the surface charge carriers density 109 1/cm2. Taking into consideration the fact that the charge carriers are in the layer of 10 A depth the concentration has been approximately 10 16 1/cm3. For comparison: according to the measurements of the bulk conductivity the ionic concentration was 1011 1/cm3. In the case of isoamyl alcohol one can also see the great difference between ionic concentrations in the bulk of liquid and close to the ceramics surface. Thus the theoretical model, computations and experimental results show that near the boundary between two dielectrics special the layers which conductivity differs from the bulk conductivity should be formed. There are some discrepancies in the rate of dissociation: the numerical simulation leads to less change of near interface dissociation rates than the physical consideration. Nevertheless both in the model and in computations the layer conductivity near the interface and in the bulk of liquid is to be rather different. What properties of these layers one should expect? Boundary with dielectrics of higher permittivity. In these cases 2/1>>1, so as the dissociation increases, the recombination decreases, image forces attract charges from the bulk of liquid. The ion concentration could increase, in comparison with the bulk one, near the metal surfaces (e.g. electrodes) and some dielectric samples with the higher permittivity. What kind of the layer structure could be then? It could be the double electrical layers, when the sizes of ions in dissolved dissociation pollution in the form of ion pair differ one another. In the case of equal sizes of the ions the layer has both positive and negative ions. In the electrical insulation systems the presence of dielectrics with the other permittivity could be dangerous. It leads to the additional conductivity, the energy losses, electric field redistribution and therefore probability of breakdown increasing in this region. Moreover, if in the contact of liquid dielectrics with the solid one, the double layer is formed, then at the forced movement of liquid along the solid boundary the charges are to be separated and the induced charge is to appear on some parts of the system. This fact could explain some problems in streaming electrification of the oil in transformers [1]. Boundary with dielectrics of lower permittivity. In these cases 2/1<<1, so dissociation decreases, recombination increases, image forces repulse charges from the boundary to the bulk of liquid. The ion concentration decreases in comparison with the bulk one, near some dielectric samples with the lower permittivity (e.g. bubbles, foams, thin films etc). Experiments in the conductivity measurements in the thin water films show that the thinner film is – the less o conductivity is [6]. This effect appears if the film is thinner than 400 A . It was explained by the charge carriers mobility decreasing in the films. In our opinion it is doubtful supposition. Both in [6] and in other papers, it was shown that the charge carriers mobility in the dilute part of the double electric layer is the same as the bulk mobility. As the size of the dilute part (tens of Angstrom) is less than the thickness of the film, it is obviously that this effect should not be taken into consideration for explanation. In our opinion one of the reasons of such kind of behavior could be electrolyte dissociation decrease in the film due to the image forces. In accordance with (1), the image charge is equal 0.98 of the ion charge. Besides, in this case one should take into account the influence of two bounds between the liquid and another dielectrics – the air. Moreover images of the images are to form the infinite row of influenced charges here, which value weakly decreases with the transition to the next charge in the row. The action of every charge could lead to the decrease of the dissociation degree in the film. The less film thickness is – the less dissociation degree is. This consequence has qualitative coincidence with the data of the work [6]. The charge of the surface. By the influence of image forces in some two-phase systems one could explain the appearance and the sign of the interface charge. For this case one should take in account the structure of the double electric layer. It is assumed that the surface doesn’t content active groups; liquid doesn’t have surfactants and specific adsorbed ions. If the ions of the ion pair have equal sizes the double layer could not be formed. Another situation is to happen if the ions have different sizes. Then if 2/1>>1 the smaller ions are attracted to the surface, while the bigger ions form the second plate of the double electric layer. So the surface will acquire the charge of the smaller ion. In the case when 2/1<<1, ions repulse from the interface. In the equilibrium the current that attributes with repulsion is to be compensated by the diffusion current and the induced current, that appears due to the field of the formed double electric field. It is obviously that the diffusion current in the case of smaller ions balances the repulsion current at the less gradient, because of the less diffusion coefficient. Therefore the ion concentration profiles could form higher concentration of the bigger ions near the surface than the concentration of the smaller ions. It is clear that the surface acquires the charge which sign corresponds to the sign of the bigger ion charge. What kind of ions is to take part in these processes in the real two-phase system? It is well known that independently of the components nature, in the absence of electrolytes, the component with higher permittivity obtains positive charge in the two-phase system [6-9]. Where from could the ions in the absence of electrolytes appear? In our opinion the water molecules play the part of the ion provider. After dissociation of the water molecules the ions H+ and OH- present in the liquid. Their sizes differ significantly, hydroxyl ion is two times bigger than the proton one. It is doubtless that some two-phase systems contain water as contamination, which removing requires special hard efforts. Therefore water molecules are in the liquid. Consequently the proton is to lead to the positive charging of the particles with higher 2 while the hydroxyl ion is to lead to the negative charging of species with lower 2. Particularly, the bubbles are to have got negative charge. This fact was registered in experiments in water [6,7], and freon [9]. The explanation, proposed in [7] is based on the decreasing dielectric permittivity near the boundary with the air due to decrease of the liquid density close to the surface layer. But the fact of density decrease is insignificant, and related decrease of the permittivity is assumed insignificant. Nevertheless, the mechanism of the negative surface charge creation due to decrease of the permittivity is ideologically similar to our mechanism. In reality the presence of the coexistence phase with low 2 is equivalent to decrease of permittivity from the position of decreasing of the charges interaction energy. A series of experiments in the fields of dielectric composite materials, colloid chemistry, boiling and cavitation show this effect. Experimental data [8] point that in composition “ferroelectric ceramic dust – liquid”, the ceramics which permittivity is 10000, obtains the positive charge, but the liquid dibutylphthalat which permittivity is 7, obtains the negative charge. In another experiment [9] it is shown that the bubbles in boiling freon have the negative charge. Special experiments on measurements of -potential of the bubble in water (1~80) show that both the bubble and the surface of glass cell (2~5) acquire the negative charge. 6. CONCLUSION As a result of consideration one could propose the new mechanism of the surface charge and the surface conductivity creation due to the action of image forces near the interface between liquid dielectrics and the dielectrics with another permittivity. One can suppose that it is possible to control the dissociation-recombination equilibrium due to inserting another dielectrics with different permittivity into liquid, e.g. non-soluble liquid, gas etc. ACKNOWLEDGEMENTS This work was supported by Russian Foundation for Basic Researches (grant N 01-0216932) and NATO Fellowship Program. REFERENCES [1] M. A. Brubaker, J. K. Nelson, “A Parametric Study of Streaming Electrification in a FullScale Core-Form Transformer Winding Using a Network-Based Model”. IEEE Trans. on Power Delivery, Vol.15, pp. 1188-1200, 2000. [2] I.A. Razilov, F. Gonzalez-caballero, A.V. Delgado, S.S. Dukhin, “The Influence of pH on Low-temperature Dielectric Dispersion. The Participation of Admixture in the Polarization of the Stern Layer”. Colloid Journal of the Russian Academy of Sciences, Vol. 58, pp. 222-229, 1996. [3] J. Seyed-Yagoobi, J. Didion, J.M. Ochterbeck, J.Allen, “Thermal Control and Enhancement of Heat Transport Capacity of Two-Phase Loops with Electrohydrodynamic Conduction Pumping”, Fifth Microgravity Fluid Physics and Transport Phenomena Conference, NASA Glenn Research Center, Cleveland, OH, CP-2000-210470, pp. 542-565, August 9, 2000. [4] S. M. Korobeynikov, A. V. Melekhov, G. G. Furin, V. P. Charalambakos and D. P. Agoris “Mechanism of surface charge creation due to image forces”. Journal of Phys.D.: Applied Physics, Vol.35, p.1193-1196, 2002. [5] E. Swayne, John Newman, C. Radke, “Surface Conductivity and Disjoining Pressure of Common Black Films Stabilized with Sodium Dodecyl Sulfate”. Journal of Colloid and Interface Science, Vol. 203, pp. 69-82, 1998. [6] A. Graciaa, G. Morel, P. Saulner, et al, “The -Potential of Gas Bubbles” Journal of Colloid and Interface Science. Vol. 172, pp. 131- 136, 1995. [7] R. Schechter, A. Graciaa, J. Lachaise, “The Electrical State of a Gas\Water Interface Source”. Journal of Colloid and Interface Science, Vol. 204, pp398-399, 1998. [8] E. M. Belokurov, V.M. Kopylov, S.M. Korobeynikov et al, “Study of dielectric Media with Relatively High Permittivity”, Colloid Journal. V. 63, pp. 395-402, 2001. [9] Asch V. “ELECTROKINETIC PHENOMENA IN BOILING "FREON-113”, Journal of Applied Physics V.37, P. 2654-2656, 1966. FIGURES http://sermir.narod.ru/tryd/figures.htm