QC - THE IDEA
James O. Westgard, Ph.D.
Outline of lesson
Need for QC?
A simple graphical tool - the QC chart
In the beginning, there was Shewhart
Learning the QC lingo?
Doing the deed
References
Please note: an updated version of this lesson is available in the Basic QC Practices, 2nd Edition.
Need for QC
The product of a testing process is a numerical result.
Unlike a physical product that can be inspected to assess
whether it looks good or bad, you can't look at a test
result and tell whether it's valid. 247 - what do you
think? If this is a patient sample, do you think the test
result is of good quality (meaning the correct value)?
If the value of 247 is measured on a sample that has
been analyzed before and has the values shown in the
accompanying histogram, do you think the test result is
of good quality? Because values between 240 and 260
have often been observed in past measurements, it is
expected that this new value should also fall in that
range if everything is working okay, therefore, the
patient test results included in this run of measurements
are also most likely correct.
A simple graphical tool - the QC chart
Click here to see an animation about the QC chart.
In the laboratory, control charts
are used to make it simple to
compare today's observed value
with what is expected based on
past history. As shown in the
second figure, by turning the
histogram sideways and
spreading the results out
according to the time they were
collected, it is easy to see how
each observation compares to the
expected distribution of past
observations, which are shown by
the central line and certain limits
calculated from the mean and
standard deviation (SD) of the of
the past control data. In this
figure, the limit lines correspond
to the mean plus/minus 1 SD, 2
SD, and 3 SD. Assuming a
gaussian or normal distribution, it
would be expected that about
68% of the points fall within 1
SD of the mean, 95% within 2 SD of the mean and 99.7% within 3 SD of the mean. Therefore, it
would be very unexpected (0.3% chance) to observe a control value greater than 3 SD from the
mean and such an observation usually indicates there is a problem with the method. It is somewhat
unexpected to observe a control value greater than 2 SD from the mean, but this will happen at least
5% of the time when analyzing 1 control per run, so it may indicate a real problem or it may be a
false alarm. It is very common (32% chance) to see individual values beyond 1 SD from the mean,
therefore this control limit is of no value for making a judgment about method performance based
on a single control value.
That's the idea behind statistical quality control. See if you can get the right answer for a known
sample. The right answer is actually a range of values that are calculated from the mean and
standard deviation of past results. That mean and control limits can be shown on a control chart to
make it simple to plot new control measurements and see how they compare with the expected
range of values.
In the beginning, there was Shewhart
Walter A. Shewhart was a statistician at Bell Telephone Laboratories who developed the scientific
basis for statistical process control. Shewhart stated that "the object of industry is to set up
economic ways of satisfying human wants and in so doing to reduce everything possible to routines
requiring a minimum amount of human effort. Through the use of the scientific method, extended to
take account of modern statistical concepts, it has been found possible to set up limits within which
the results of routine efforts must lie if they are to be economical. Deviations in the results of a
routine process outside such limits indicate that the routine has broken down and will no longer be
economical until the cause of trouble is removed." Shewhart made this statement in the preface to
his book on the "Economic Control of Quality of Manafactured Product" that was published in
1931.
Statistical process control, from the beginning, has been concerned with achieving the desired
quality (satisfying human wants) at minimum cost (economic control). Shewhart identified critical
elements such as the expected variation of a routine process, a way to set limits that will identify
when the routine has broken down, and the need to eliminate causes of trouble when the process
was observed to exceed those limits.
Almost twenty years passed before Levey and Jennings introduced statistical control methods in
clinical laboratories in 1950 [2]. Shewhart's original recommendations called for making a group of
measurements, calculating the average and range (maximum difference), then plotting the average
and the range on two different control charts. Levey and Jennings proposed making duplicate
measurements on a patient specimen. Because the actual level of the measured constitutent varied
from specimen to specimen, this was a more difficult application. Henry and Segalove [3]
developed an alternative procedure in which a stable reference sample was analyzed repeatedly and
individual measurements were plotted directly on a control chart. This reference sample type of QC
in which individual values or single values are plotted directly is commonly known today as a
Levey-Jennings chart.
Since that time, industry has developed stable control products that mimic patient samples, thus
today there are safe QC materials readily available for most established tests. A better
understanding of the performance characteristics of QC procedures has been developed [4], which
has led to refinements such as the multirule procedure for evaluating and interpreting control data
[5]. Strategies for cost-effective operation have been further refined [6]. Computer programs have
been developed to implement statistical control procedures by performing the necessary
calculations, preparing graphical displays, applying the desired control rules, and alerting analysts
to problem situations. Today, support for handling control results is provided by most automated
analyzers, information systems, and even point-of-care devices.
Learning the QC linguage
Statistical process control is the general term used to describe those aspects of a control system in
which statistics are applied to determine whether observed performance is within the expected
variation of the process, in contrast to other components of a total control system such as preventive
maintainence, instrument function checks, operator training, etc., that are included in CLIA's broad
definition of quality control.
Statistical control procedure is used here to refer to a specific protocol for analyzing a specific
number of control materials and interpreting a specific number of test results. In healthcare
laboratories, a control procedure is usually implemented by collecting test results on stable control
materials, then plotting those control observations on a control chart that has specified control
limits, or by evaluating those control results by data calculations employing specified decision
criteria or control rules.
Control chart is a graphical method for displaying control results and evaluating whether a
measurement procedure is in-control or out-of-control. Control results are plotted versus time or
sequential run number; lines are generally drawn from point to point to accent any trends,
systematic shifts, and random excursions.
Control limits are lines drawn on a control chart to provide graphical criteria for assessing whether a
measurement procedure is in-control or out-of-control. These control limits are usually calculated
from the mean and standard deviation (SD, or s) determined for a given control material. Typically
the interpretation is based on a specified number of results or points exceeding a certain control
limit when in-control patient test results are reported. When out-of-control, the run is rejected and
no test results can be reported.
Control rule means a decision criterion for judging whether an analytical run is in-control or out-ofcontrol. It is commonly defined by a symbol of the form AL, where A is an abbreviation for a
statistic or represents a number of control measurements, and L identifies the control limits, often
specified as the mean plus or minus a multiple of the standard deviation (s) or sometimes by a
specified probability for false rejection (Pfr). Some examples follow:
13s refers to a control rule that is commonly used with a LeveyJennings chart when the control limits are set as the mean plus
3s and the mean minus 3s. A run is rejected when a single
control measurement exceeds the mean plus 3s or the mean
minus 3s control limit.
12s refers to the control rule that is commonly used with a LeveyJennings chart when the control limits are set as the mean
plus/minus 2s. In the original Westgard multirule QC procedure,
this rule is used as a warning rule to trigger careful inspection of
the control data by the following rejection rules.
22s - reject when 2 consecutive control measurements exceed the
same mean plus 2s or the same mean minus 2s control limit.
R4s - reject when 1 control measurement in a group exceeds the
mean plus 2s and another exceeds the mean minus 2s.
Run, analytical run, or run length refer to the interval, which could be a period of time or group of
samples, for which a decision on control status is to be made. CLIA defines a maximum run length
of 24 hours for chemistry analytes and 8 hours for hematology tests. Many laboratories define a
shorter period based on changes that may affect the performance of the testing process, such as
changing operators, changing reagents, recalibration, or other factors that may make the process
susceptible to problems. Run length varies from system to system and laboratory to laboratory. For
random access automated systems, a run is usually defined as the time interval at which controls are
reanalyzed. For manual systems and batch instruments, a run is often defined as a group (or batch)
of samples that are all analyzed at the same time.
Doing the deed
The idea is simple, but the application can be complicated.
First, you need to obtain control materials that are appropriate for the tests of interest and the
methods in use. See QC - the Materials for a discussion of important factors, such as matrix effects,
stability, vial to vial variation, assayed versus unassayed materials, analyte levels, and pre-treatment
problems.
Then you must assay the selected control materials under routine operating conditions to
characterize the expected measurement variation and establish the expected distribution of values.
This usually involves obtaining at least 20 values and calculating the mean and standard deviation.
There are a number of pitfalls from using bottle values or other estimates of the means, standard
deviations, and control limits, so you need to be careful with this step. See QC - the Calculations for
more information about data calculations.
Next you need to define appropriate control rules, numbers of control measurements (N), and the
analytical run length. See QC - the Regulations for the legal requirements for laboratory QC. See
QC - the Planning Process for a brief description of QC planning and links to other materials on this
website.
You must also define how you will implement these rules and Ns - manual plotting, or computer
assessment by the analyzer, a PC workstation, or a laboratory information system. For manual
implementation, see QC - the Levey-Jennings Control Chart for directions on how to prepare the
control chart, plot control results, and interpret control data.
Finally, you should prepare written guidelines to define the QC procedure in detail. This written
document is important for teaching laboratory analysts the QC procedure and establishing a uniform
practice. It is also necessary for meeting US regulatory requirements.
Now you're ready to implement QC. See QC - the Practice for a summary and review of the whole
approach. See FAQs about QC for additional information about some of the nitty- gritty problems
and issues with routine QC.
References
1. Shewhart WA. Economic Control of Quality of Manufactured Product. New York; D. Van
Hostrand Company, Inc., 1931.
2. Levey S, Jennings ER. The use of control charts in the clinical laboratory. Am J Clin Pathol
1950;20:1059-66.
3. Henry RJ, Segalove M. The running of standards in clinical chemistry and the use of the
control chart. J Clin Pathol 1952;27:493-501.
4. Westgard JO, Groth T, Aronsson T, Falk H, deVerdier C-H. Performance characteristics of
rules for internal quality control: probabilities for false rejection and error detection. Clin
Chem 1977;23:1857-67.
5. Westgard JO, Barry PL, Hunt MR, Groth T. A multi-rule Shewhart chart for quality control
in clinical chemistry. Clin Chem 1981;27:493-501.
6. Westgard JO, Barry PL. Cost-Effective Quality Control: Managing the Quality and
Productivity of Analytical Processes. Washington, DC:AACC Press, 1986.
Copyright © 2000. All rights reserved.
Westgard QC, Inc., 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's new? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
QC - THE MATERIALS
Elsa F. Quam, BS, MT(ASCP)
Control solutions, control materials
Matrix effects
Stability
Vial to vial variability
Assayed versus unassayed
Analyte levels
Pretreatment steps
Conclusions
References
PLEASE NOTE: An updated version of this lesson is available in Basic QC Practices, Second Edition
The purpose of a statistical quality control procedure is to monitor the analytical quality of the
measurement during stable operation, detect changes from the stable operation, and eliminate
reporting of results with medically important errors [1]. From the technologist’s standpoint, the
objectives of the control procedure are simply to "alert me when the method has a problem" and
"don't alert me when the method is working okay." These correspond to "true alarm" and "false
alarm" situations, which are characteristics of the QC procedure. In short, technologists want to
know about real problems, but can't afford to waste time when the method's working okay. Any
additional information that can aid in the troubleshooting the method is an "extra bonus."
Much has been written about how to perform the statistical calculations needed for quality control
procedures, how to choose control rules, how to apply the rules, how to construct control charts, and
how to interpret results from the control procedures, all of which assume we have found appropriate
stable control materials. We need to pay careful attention to the selection of the control materials.
Important attributes are the stability, vial to vial variability, assayed versus unassayed, appropriate
analyte levels, and pretreatment procedures. The very success of the control procedure depends
upon these attributes!
Control solutions, control materials
The International Federation of Clinical Chemistry defines a control solution or control material as
a "specimen or solution which is analyzed solely for quality control purposes, not for calibration"
[2]. We use the term control material or control product to refer to a control solution that is
available, usually commercially, in liquid, frozen, or lyophylized form, packaged in small bottles
suitable for use on a daily basis. Such control materials are widely available today for most
laboratory tests. They can be obtained from manufacturers who specialize in the production of
control materials, and also are often provided by the same companies who are selling you the
reagents, methods, and instrument systems. It is common today to purchase complete testing
packages that include the necessary control materials.
Matrix
Matrix refers to the substance or base from which the control material is prepared in addition to all
the additives such as spiking materials, preservatives, etc. added to make the product desirable to
the user. The American Society for Testing and Materials has defined matrix as " the principal
element or elements in a sample" and a matrix interference as "an effect due to the presence of a
constituent or characteristic."[3]
Ideally, control materials should have the same matrix as the specimens being tested so that they
will behave the same as the real specimen. For example, choose a whole blood control for point-ofcare blood glucose analyzers and for blood gas and whole blood electrolyte analyzers to maintain a
similar matrix; use a serum/protein based control for analyzers that perform tests on serum or
plasma. Control materials are also available having matrices of urine and spinal fluid. In general,
materials prepared from human sources have been preferred in the past, however because of the
potential biohazard risk today, bovine based control products have become more popular.
Control materials, even when selected to have the appropriate matrix, undergo a substantial number
of manipulations during their production which can alter the properties of the matrix. These
alterations include human and non-human additives for achieving specific concentrations and/or
stability as well as physical changes to the material such as freeze drying or lyophilization. These
alterations may in turn cause interferences in the testing process which may not be present in fresh
human samples.
Certain test methodologies may also influence the selection of control materials. For example, a
bovine based control material will usually assay low by a bromcresol purple albumin method,
which has been optimized for human albumin. In contrast, bovine controls are acceptable for use
with the less specific bromcresol green albumin method. For some assays, such as those used in
lipoprotein testing, fresh or frozen human pools may be the most appropriate control materials [4].
Careful consideration to the control material matrix is an important consideration in the QC
planning process.
Stability
When possible, it is desirable to purchase at least a one year supply of the same lot or batch number.
Many products are now available with expiration dates of more than two years. The desired
expiration date of the control product should be included in the specifications listed at the time of
purchase. This planning step will pay off in the ability to provide a continuous monitor of the
analytical process through many method and instrument changes, while reducing costs by
minimizing the crossover testing necessary during the checking out period of new control lot
numbers. It usually isn't necessary to purchase and store the entire lot for the duration of the
expected time of use because most vendors are willing to sequester specified lot numbers for the
desired period and set up automatic shipment and billing schedules at monthly, every other month,
or quarterly intervals. This strategy also has the advantage of not requiring payment or inventory
of the control materials until they are needed for use.
Vial to vial variability
The variation observed when monitoring a method is almost entirely due to measurement
imprecision and vial to vial variability of the control materials themselves, which is usually a small
part of the total variation observed. Commercial control materials that have been lyophilized or
freeze dried must be reconstituted with water or specialized diluent, therefore, it is very important to
standardize the reconstitution step. Use Class A volumetric pipettes, deonized Type 1 water, and
instructions that specify the mixing time and the reconstitution time to minimize the vial to vial
variability due to the preparation process.
Many liquid control products which eliminate the reconstitution process are now available. These
products are generally more expensive and sometimes contain additives or preservatives which
could introduce sources of error due to matrix problems with certain methods. Depending upon the
analytical methods to be monitored, the benefits of the reduced vial to vial variability may outweigh
any increased costs. In addition, liquid control products are generally stable for 14 to 30 days after
the vial has been opened whereas lyophilized products are usually only stable for less than 48 hours
after reconstitution. Liquid controls may therefore, in some cases, be a better buy because of
reduced waste due to stability, elimination of vial to vial to variability, and reduction in operator
errors due to the reconstitution process.
Assayed versus unassayed controls
Control products are available as assayed or unassayed materials. Assayed control materials
generally come with an assay sheet of expected values for analytes assayed by various methods and
instrument systems. These assay sheets usually list, for each constituent present, expected mean
values as well as expected ranges. Values may even be available for reference methods used to
measure certain analytes. These ranges are provided only as guidelines until the laboratory has
established its own statistical limits. Assayed controls are generally more expensive than unassayed
controls due to the cost incurred with the value-assignment process. They may, however, be
valuable for the smaller laboratory, for meeting CLIA regulations, and for troubleshooting method
problems.
Analyte levels
Constituent levels of quality control materials should be chosen at medical decision concentrations
and/or at critical method performance limits such as upper and lower linearity limits. Two or three
different concentrations are often needed for each analyte. Choosing control materials at critical
concentrations (medical and/or performance) will allow the analyst to estimate the random error at
critical levels of the method during stable operation and to provide a monitor at the most important
performance levels for that analyte. Statland has provided recommendations for decision levels for
many tests [5], which are summarized in tabular form on this website.
Vendors manufacture sets of materials to have critical concentration levels and also to monitor the
working range of a method. Purchase of these materials will often include the services of a quality
control program. Services provided by these quality control programs include statistical
calculations of your laboratory data, preparation of Levey Jennings plots using your mean and
standard deviation, and a comparison report which compares your monthly and cumulative statistics
to those of a peer group of users using same lot number of materials. Costs for these programs are
usually included in the cost of the control materials. An alternate option is to select materials from
different vendors in order to minimize possible matrix problems that may be inherent in all
materials from a single vendor.
Pretreatment steps
Many laboratory tests such as digoxin, hemoglobin A1C, and total iron binding capacity require
pretreatment of the specimen prior to determination by the analytical process. These procedures
often require manual pipetting and mixing steps which are more problem prone that the analytical
determination. It is best, for these procedures, to have control materials that are submitted to the
pretreatment steps and are therefore treated exactly as the specimen being tested. If the analytical
method is also problem prone, it may be advantageous to select one or two control materials which
do not undergo the pretreatment process in addition to the one or two materials which are included
in the pretreatment. This strategy will aid the analyst, when errors occur, by isolating the analytical
process from the pretreatment process.
Conclusions
The selection of appropriate control materials requires consideration of many factors and should be
part of the planning process for the selection and implementation of SQC. The process becomes
even more complicated when selecting control materials suitable for a multiconstituent analyzer.
Compromises may have to be made in order to limit the number of control materials used. There is
no right or wrong way to choose control materials for a given method, just as there is no perfect
control which behaves exactly the same as a fresh human sample. Selection of appropriate control
materials is a balancing act in which cost, stability, ease of use, performance due to matrix effects,
and constituent levels must be considered and weighted for each laboratory’s own application and
use.
Control materials being used should be reassessed yearly. Product vendors are continually making
changes and enhancements in their products so that these products are more marketable. Presently,
it is hard to keep abreast of all the changes in immunoassay control products as vendors are
scurrying to provide materials with constituents to fit the test menus of these new analyzers. Other
quality control services and enhancements are also being provided by instrument vendors. Some
instrument manufacturers will be offering quality control troubleshooting services by using a
modem hookup to review QC measurements from the instrument at the laboratory site. Of course,
control materials purchased from the instrument vendor must be used to obtain the service. The
selection of control materials is an important consideration in the statistical quality control planning
process and must be reviewed as our needs change.
References
1. Westgard JO, Barry PL. Cost-Effective Quality Control: Managing the Quality
andProductivity of Analytical Processes. Washington DC;AACC Press, 1986.
2. Buttner J, Borth R, Boutwell JH, Broughton PMG. International Federation of Clinical
Chemistry provisional recommendation on quality control in clinical chemistry. I. General
principles and terminology. Clin Chem 1976;22:532-40.
3. ASTM Committee on Terminology. Compilation of ASTM standard
definitions. Philadelphia, PA: American Society for Testing and Materials,
1990.
4. Handbook of Lipoprotein Testing, edited by Nader Rifai, G. Russell Warnick, Marek H.
Dominiczak, Chapter 11: Matrix Effects in the Measurement and Standardization of Lipids
and Lipoproteins written by W. Greg Miller.
5. Statland BE. Clinical Decision Levels for Lab Tests. Oradell, NJ;Medical Economic Books,
1983.
Copyright © 2000. All rights reserved.
Westgard QC, 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's new? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
QC - THE CALCULATIONS
James O. Westgard, Ph.D.
Outline of lesson
What calculations?!
Mean, SD, CV
Control Limits
Cumulative or lot-to-date calculations
Z-scores, SDI's
PLEASE NOTE: an updated version of this lesson is available in Basic QC Practices, 2nd Edition
What Calculations?
Are any calculations necessary if the control material has an assay sheet that
lists the range of acceptable values for my method?
Yes, you still need to collect your own control measurements and calculate the control limits that
apply in your own laboratory. Values and limits found on assay sheets often describe the
performance observed by a specific method in several different laboratories, which means that the
figures are likely to include variations that occur between laboratories. Therefore, those limits are
likely to be too wide for an individual method in your laboratory. If the control limits are too wide,
you won't be able to detect problems in your own laboratory.
Note that US CLIA regulations require that the laboratory determine it's own mean and standard
deviation. [493.1218(5d) "When calibration or control materials are used, statistical parameters
(e.g., mean and standard deviation) for each lot number of calibration material and each lot of
control material must be determined through repetitive testing."]
What statistics need to be calculated to establish my own control limits?
You need to calculate the mean and standard deviation from the control results that have been
collected for each control material. It's also common to express the standard deviation in percent by
calculating a coefficent of variation, or CV.
Mean, SD, CV
How many control measurements should be collected before making these
calculations?
The rule of thumb is to collect at least 20 measurements over at least 2 weeks or 10 working days,
and preferably over at least 4 weeks or 20 working days. You do this by including control materials
as part of your daily work for a long enough period to observe the variation expected in your
laboratory. Too short a period leads to too small an estimate of the standard deviation. Longer is
usually better because the estimates will include more operators and more method changes, such as
pre and post maintenance performance, changes in reagent lot numbers, sample probes or pipettes,
etc., thus even one month might be too short a period. In practice, calculations of the mean and
standard deviation are often made monthly and then the monthly data are added to data from
previous months to calculate the cumulative or lot-to-date mean and standard deviation that are then
used for setting control limits. These cumulative or lot-to-date control limits are a better
representation of long term test performance.
How many significant figures are needed in the control results that are used to
estimate the mean and standard deviation?
Control results should have at least one more significant figure than the values reported for patient
test results in order to get good estimates of the mean and standard deviation and to be able to set
appropriate control limits. With some instrument systems where test results are rounded for clinical
significance, only whole numbers end up being reported for control results, thereby giving a
discrete distribution of control values with only a few possible results, rather than the continuous
gaussian distribution that is expected. This may lead to some practical problems in setting control
limits because the calculated control limits may not correspond to the discrete integer values being
reported.
What is the equation for the mean?
The mean is determined by adding a group of measured values, then dividing the total by the
number of measurements in the group. This is often written as:
where the mean may be symbolized by, an x with a bar over it (hence the term x-bar), xi
represents an individual measurement, represents the operation of summation or addition of all
these xi values, and n is the number of xi values in the group. Using just 3 numbers for an example
(which is not sufficent data according to the current laboratory practice of obtaining a minimum of
20 results), for the values of 100, 105, and 98, xi is the total of these three or 303, and the mean or
average is 303/3 or 101.
What's a practical way to calculate the mean?
Hand held calculators can be used to easily calculate the total of a group of measurements, then
divide that total by the number of measurements included. Scientific calculators usually have a
built-in program for both the mean and standard deviation. Electronic spreadsheets, such as Lotus 12-3 and Excel, usually have built-in functions for calculating the mean and standard deviation from
a column of data. Statistical programs, such as Minitab, SPSS, SAS, and Systat have functions for
calculating the mean and standard deviation, as well as describing the population in terms of the
observed median, mode, range, lowest value, highest value, etc.
In most laboratories, the QC program in the laboratory computer system will calculate the control
data captured on-line or through manual entry. The QC programs incorporated in instrument
systems and some Point-of-Care devices have similar capabilities. Stand alone QC programs on
personal computers are also available and offer complete support for calculations, graphic displays
of control charts, and storage of results. Participants in external survey programs offered by
instrument or control manufacturers can also submit their control data for analysis by the vendors,
though the data analysis may require up to a month for return of the results.
What does the mean tell me about method performance?
The mean value for a control material provides an estimate of the central tendency of the
distribution that is expected if method performance remains stable. Any change in accuracy, such as
a systematic shift or drift, would be reflected in a change in the mean value of the control, which
would be shown by a shift or drift of the distribution of control results. Always keep in mind that
the mean is related to accuracy or systematic error and the standard deviation is related to precision
or random error. See QC - The Idea for a review of how the mean of the distribution of control
results is related to the mean and control limits on a control chart.
What is the equation for the standard deviation?
The standard deviation is determined by first calculating the mean, then taking the difference of
each control result from the mean, squaring that difference, dividing by n-1, then taking the square
root. All these operations are implied in the following equation:
where s represents the standard deviation, means summation of all the (xi - )2 values, xi is an
individual control result, is the mean of the control results, and n is the total number of control
results included in the group.
For computerized calculations and for estimating the cumulative standard deviation, the form of the
equation that is commonly used is:
where xi2 is the summation of
all the squared individual values,
and ( xi)2 is the square of the
sum of all the individual values.
What's a practical way to
calculate the standard deviation?
It is easy to use a scientific calculator, an electronic spreadsheet, or a statistics program, all of which
have built-in functions for calculating the standard deviation of a group of measurements. This
function for calculating the standard deviation is often labeled "SD". Specialized QC software in
laboratory information systems, instruments, and personal computer workstations will automatically
calculate the standard deviation for the data being accumulated. External quality assessment
programs offered by manufacturers of instruments and control materials will also process the data
of participants and provide reports that include the calculated results.
What does the standard deviation tell about method performance?
The standard deviation is related to the spread or distribution of control results about the expected
mean. Whereas the mean is an indicator of central tendency and therefore related to accuracy or
systematic error, the standard deviation is a measure of the width of the distribution and is related to
imprecision or random error. The bigger the standard deviation, the wider the distribution, the
greater the random error, and the poorer the precision of the method; the smaller the standard
deviation, the narrower and sharper the distribution, the smaller the random error, and the better the
precision of the method.
For a measurement procedure, it is generally expected that the distribution of control results will be
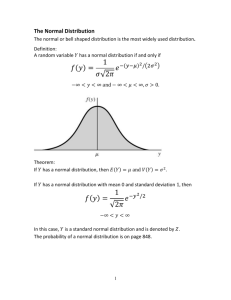
normal or gaussian, as shown above. For a gaussian distribution, the percentage of results that are
expected with certain limits can be predicted. For example, for control results that fit a gaussian
distribution, it would be expected that 68.2% of the observed results will be within plus/minus 1s of
the mean; 95.5% within plus/minus 2s of the mean, and 99.7% within plus/minus 3s of the mean.
What's a CV?
CV refers to the "coefficient of variation," which describes the standard deviation as a percentage of
the mean, as shown in the following equation:
CV = (s/ )100
where s is the standard deviation,
ratio to a percentage.
is the mean, and the multiplier of 100 is used to convert the s/
Why is a CV useful?
The standard deviation of a method often changes with concentration, i.e., the larger the
concentration, the larger the standard deviation, therefore it is usually necessary to estimate the
standard deviation at the concentration level of interest. Because the CV reflects a ratio of the
standard deviation to the concentration, it is often provides a better estimate of method performance
over a range of concentrations.
For example, you may be interested in planning a QC procedure on the basis of the performance
needed at a critical decision concentration of 200 mg/dL, but the nearest control available has a
mean of 190 mg/dL. Therefore, it is best to calculate the CV from the observed results at 190
mg/dL, then apply that CV to the 200 mg/dL decision level. This is the reason that QC planning
applications with the QC Validator program use a percentage figure for the imprecision of the
method.
Control limits
How do you calculate control limits?
Given the mean and standard deviation for a control material, control limits are calculated as the
mean plus and minus a certain multiple of the standard deviation, such as 2s or 3s. For cholesterol
where a control material has a mean of 200 mg/dL and a standard deviation of 4 mg/dL, the 2s
control limits would be 192 and 208 mg/dL, and the 3s control limits would be 188 and 212 mg/dL.
See a web-based Control Limit calculator in the lesson, QC - The Levey-Jennings Chart
How many significant figures should be used in control limit calculations?
As a rule of thumb, the control results and the calculated standard deviation should have at least one
more significant figure than needed for clinical significance of the patient test result; the mean of a
control material should include at least two more significant figures than needed for clinical
signficance of the patient test result. When in doubt, carry more significant figures than necessary
and round at the end when the control limits have been calculated. Most calculators and computers
carry plenty of extra figures so you can round at the end.
Cumulative or lot-to-date calculations
What's a cumulative or lot-to-date control limit?
Typically, control results are summarized by calculating the mean, standard deviation, CV, and N
on a monthly basis. In order to establish longer term estimates of the mean and standard deviation,
the control data or calculated results need to be accumulated to describe performance observed over
a longer periods of time. Longer term limits are often described as "cumulative limits," which
indicates they have been calculated from cumulative means and standard deviations. These may
also be referred to as "lot to date" limits when these calculated values are provided by a
manufacturer or supplier who processes the control data for a group of laboratories in order to
provide information about the comparative performance between laboratories and between
methods.
What's a cumulative standard or lot-to-date deviation?
This is a long term estimate of a method's precision performance based on a large number of control
measurements collected over a long period of time. A long period here is at least two months and
could be several months, even a year.
How is a cumulative or lot-to-date standard deviation calculated?
These calculations are often automatically performed by the QC programs in laboratory computer
systems, personal computer work stations, and in many automated instruments and even some
point-of-care devices.
If you need to perform these calculations yourself, one practical approach is to calculate monthly
statistics, then tabulate the month n's, xi and xi2, which can then be totaled and used in the
equation below to provide the cumulative estimate:
where nt( xi)t2 is the total of the sums of all the squared individual values, and ( xi)t2 is the square
of the total of the sums of all the individual values, and nt is the total number of measurements in
the time period of interest.
What's a cumulative or lot-to-date mean?
This is a long term estimate of the central tendency observed for a control material based on a large
a number of control measurements collected over a long period of time. A long period here is at
least two months and could be several months, even a year. Changes in the accuracy of a method
could lead to shifts or drifts in the mean observed for a control material.
How is a cumulative or lot-to-date mean calculated?
From the monthly statistics that are calculated, tabulate the monthly n's and xi's, which can then be
totaled for the period of interest (two months, several months), and used in the equation below to
provide the cumulative mean:
where ( xi)t is the total of the monthly sums of individual values and nt is the total of the monthly
ns for the period of interest.
How are cumulative or lot-to-date control limits calculated?
The estimates for the cumulative or lot-to-date mean and standard deviation, as calculated above,
are used to calculate cumulative or lot-to-date control limits. Here's a table that illustrates the whole
procedure.
(Cumulative results are show in parentheses.)
Monthly total (cumulative total)
Month
1
Calculated statistics
Control Limits
n
x
x2
Mean
s
Mean +/- 3s
20
3983
793465
199.15
3.63
188.3 - 210.0
20
3993
797537
199.65
4.20
187.1 - 212.2
2
(40)
20
(7976)
4002
(1591002)
801138
(199.40)
200.10
(3.86)
4.22
(187.8 - 211.0)
187.5 - 212.7
3
(60)
20
(11978)
4020
(2392140)
808182
(199.63)
201.00
(3.97)
2.92
(187.7 - 211.6)
192.2 - 209.8
4
(80)
20
(15998)
3995
(3200322)
798259
(199.96)
199.75
(3.77)
3.68
(188.7 - 211.3)
188.7 - 210.8
5
(100)
(19993)
(3998581)
(199.93)
(3.73)
(188.7 - 211.1)
See a web-based QC calculator that performs these calculations.
Z-scores and SDI's
What's a z-score?
A z-score is a calculated value that tells how many standard deviations a control result is from the
mean value expected for that material. It is calculated by taking the difference between the control
result and the expected mean, then dividing by the standard deviation observed for that control
material. For example, if a control result of 112 is observed on a control material having a mean of
100 and a standard deviation of 5, the z-score is 2.4 [(112- 100)/5]. A z-score of 2.4 means that the
observed control value is 2.4 standard deviations from its expected mean, therefore this result
exceeds a 2s control limit but not a 3s control limit.
Why is a z-score useful?
It is very helpful to have z-scores when you are looking at control results from two or more control
materials at the same time, or when looking at control results on different tests and different
materials on a multitest analyzer. You can quickly see if any result exceeds a single control limit,
for example, a z-score of 3.2 indicates that a 3s control limit has been exceeded. You can also look
for systematic changes or trends occurring across different control materials, for example,
consecutive z-scores of 2 or greater on two different control materials.
What's an SDI?
If you participate in an external quality assessment program or a proficiency testing program, you
are asked to analyze a series of unknown specimens and submit your test results for comparison
with those obtained by other laboratories. The data from all the laboratories are usually
analyzed to determine an overall average and standard deviation for the group. The program will
generally report your performance relative to the group. The difference between your test results
and the overall average is often expressed by a standard deviation index, or SDI, which expresses
the difference in terms of the number of standard deviations from the overall mean. For example, an
SDI of 1.0 would indicate your result fell one standard deviation from the mean. On a series of
specimens, if you observe SDIs such as +1.5, +0.8, +2.0, +1.4, and +1.0 (all positive), this suggests
that your method is generally running on the high side and is biased, on average, by +1.3 SDI. To
figure the size of this average bias in concentration units, you need to multiply by the actual value
of the group SD.
Note the similarity between the calculation of the SDI and the z-score. They're basically the same
thing, but the z-score tends to be used in internal QC programs to compare an individual QC result
with the expected values for that material, whereas the SDI tends to be used in external QC
programs to compare the performance of the lab with the overall mean for a defined comparative
group or with an established target value.
Why's an SDI useful?
One advantage is that it allows you to inspect results from many different tests at the same time,
without having to think about different units and the actual magnitude of the change in the units of
the test. In general, any SDI of 2.0 or greater deserves some special concern, regardless what the
test is. Any test whose average SDI is 1.0 or greater deserves some special attention because your
method shows a systematic difference from the group. In the future, this bias might lead to
unacceptable results.
Copyright © 2000. All rights reserved.
Westgard QC, 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's new? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
QC - THE REGULATIONS
Sharon S. Ehrmeyer, Ph.D.
HCFA and COLA
CLIA QC Requirements based on Test Complexity
Waived tests
Provider Performed Microscopy
Moderate Complexity Tests
High Complexity Tests
What if a control fails to meet criteria for acceptability?
JCAHO
Standards for Quality Control
CAP
Guidelines for Quality Control
References
If you perform laboratory tests in the US, the government's got you covered! One way or another,
you have to comply with the Clinical Laboratory Improvement Amendments of 1988 (CLIA’88),
which establish the minimum standards for all laboratory testing, including specific regulations for
quality control [1]. You can, however, select the organization that will administer these regulations
or administer professional standards that are equivalent to these regulations:
The Health Care Financing Administration (HCFA) will inspect any size laboratory,
including physician office laboratories, for adherence to the CLIA standards;
The Commission of Office Laboratory Accreditation (COLA) inspects only physicians
office laboratories for adherence to COLA standards which closely parallel the CLIA
regulations;
The Joint Commission on Accreditation of Healthcare Organizations (JCAHO) inspects
laboratories as part of an overall inspection of a hospital or healthcare organization;
The College of American Pathologists Laboratory Accreditation Program (CAP-LAP)
mainly inspects large laboratories directed by pathologists.
There are other government approved organizations that have standards for laboratories to follow
and some states impose specific requirements [2].
Health Care Financing Administration (HCFA) and Commission of Office
Laboratory Accreditation (COLA) Requirements for QC
The first part of this discussion focuses on CLIA’88 and COLA regulations [1,3] since COLA's 200
question checklist adheres to the CLIA’88 QC requirements almost exactly. Professional standards
administered via CAP and JCAHO are considered later.
CLIA QC Requirements based on Test Complexity
CLIA'88 regulations are based on four categories of test complexity: waived, provider performed
microscopy (PPM), moderate complexity, and high complexity. Current information on test
complexity can be obtained from the CDC’s web site (http://www.cdc.gov/phppo/dls/testcat.htm).
Each testing category has different regulatory requirements for personnel, quality control, quality
assurance, proficiency testing, etc.
Waived testing
This category requires the least regulation. Under CLIA and COLA, the minimum requirement for
anyone performing waived testing is to follow the manufacturers’ directions for QC, and if no
directions are included, to follow good laboratory practices. No specific QC requirements are
identified. Good laboratory practice would dictate that controls be run and results documented and
reviewed for correctness before reporting patient results. The tests that are waived include the
following (current September 29, 1997 list of Waived Tests):
Dipstick or tablet reagent urinalysis for bilirubin, glucose, hemoglobin, ketones, leukocytes,
nitrite, pH, protein, specific gravity, and urobilinogen
Fecal occult blood
Ovulation tests - visual color comparison tests
Urine pregnancy tests - visual color comparison tests
Erythrocyte sedimentation rates - non-automated
Blood glucose - all glucose monitoring devices cleared by FDA for home use; HemoCue BGlucose System, and Cholestech L.D.X.
Hemoglobin - (non-automated) by copper sulfate; (automated) by HemoCue Hemoglobin
System
Hematacrit - all spun microhematocrit procedures, Wampole STAT-CRIT, Separation
Technology STI HemataSTAT II and Model C70, StatSpin Technologies CritSpin, and
Vulcon Technologies Microspin 24, Protime - ITC (International Techidyne Corp.) and
BMC CoaguChek
H. pylori -- Serim Pyloritek Test Kit, GI Suppy HP-FAST, Quidel QuickVue One-Step for
whole blood, Delta West CLOtest, Abbott FlexPack HP, ChemTrak AccuMeter, and
SmithKline FlexSure
Strep A (throat only) Quidel QuickVue In-Line One-Step, Binax NOW, Abbott Signify and
SmithKline ICON Sx
Cholesterol -- Cholestech L.D.X., BMC Accu-Chek InstantPlus, ChemTrak AccuMeter, and
J & J ADVANCED CARE
HDL Cholesterol -- Cholestech L.D.X.
Triglyceride -- Cholestech L.D.X
Gastric occult blood -- SmithKline Gastroccult
Micro albumin -- BMC Chemstrip Micral
All qualitative color comparison pH testing for body fluid (other than blood)
To keep up to date with all the new test additions and new methodologies for waived tests, you can
check the CDC Web site [http://www.cdc.gov/phppo/dls/clia.htm].
Provider Performed Microscopy (PPM)
As of January 1993, the category "Physician-Performed Microscopy" was established under
CLIA’88. In the April 24, 1995 Federal Register, the category was renamed to "ProviderPerformed Microscopy." This category is a subset of moderate complexity and is exclusively for
physicians, dentists, nurse practitioners and midwives, and physician assistants performing the
testing as part of a patient examination. The primary instrument for performing the test(s) is the
microscope.
The 9 tests identified under a PPM CLIA certificate can be performed, as well as waived tests.
When these tests are provided, the practitioners are expected to follow the manufacturers’ directions
for QC or follow good laboratory practices. Good laboratory practice would dictate that controls be
run whenever possible and results documented and reviewed for acceptability before reporting
patient results. The PPM category includes:
all direct wet mount preparations (suspended in saline or water) for the presence or absence
of bacteria, fungi, parasites, and human cellular elements
KOH preps
pinworm exams
fern tests
post-coital direct qualitative exams of vaginal or cervical mucous
urine sediment exams
nasal smears for granulocytes
fecal leukocyte exams
qualitative semen analysis (limited to the presence or absence of sperm and detection of
motility).
Moderate Complexity Tests
Approximately 75% of all tests performed in laboratories today fall in the category of moderate
complexity. For this category, the QC requirements are identified in subpart K for CLIA’88 -Quality Control. Originally, in the February 28, 1992 Federal Register, HCFA stated that as of
September 1, 1994, laboratories would meet most of CLIA'88 QC requirements simply by
following manufacturers' labeling, provided the FDA approved manufacturers' QC instructions.
This never happened and the implementation date (9/1/94) has now been suspended, in the May 12,
1997 Federal Register, until at least 7/31/98.[5] Until HCFA implements new QC requirements,
laboratories performing moderate complexity tests will meet the CLIA QC requirement by
following sections §493.1201 and §493.1202(c) only.
Section §493.1201 (general QC: moderate or high complexity testing, or both) states: The
laboratory must establish and follow written QC procedures for monitoring and evaluating the
quality of the analytical testing process of each method to assure the accuracy and reliability of
patient test results and reports.
Section §493.1202(c) identifies the specific requirements for unmodified moderate complexity
tests: [1,4]
follow manufacturer directions;
have a procedure manual for the method that identifies how to perform the testing and
reporting of results;
perform and document calibration procedures or check calibration at least once every six
months;
assay at least two levels of control materials each day of testing (a run cannot exceed 24
hours) and keep records;
perform and document any applicable specialty and subspecialty control procedures;
perform and document remedial actions as specified in §493.1219;
maintain records of all QC activities for 2 years or 5 years for immunohematology and 10
years for pathology as specified in §493.1221.[4]
As you can see, CLIA QC includes a lot more than traditional statistical QC and defines standards
for calibration, procedure manuals, remedial actions and record keeping. Concerning statistical QC,
for most moderate complexity tests, the general requirement is to analyze two levels of QC
materials on each day of testing. However, for certain tests, i.e., blood gases, hematology and
coagulation tests, etc., CLIA requires additional QC:
Blood gases require, at a minimum, one control sample every 8 hours of testing and a
calibrator or control in each run unless the instrument "autocals" at least every 30 minutes.
Automated hematology and coagulation test systems require two levels of controls every 8
hours of testing and each time a change in reagent occurs.
Manual cell counts using a hemocytometer must be tested in duplicate and one control is
required every 8 hours of operation.
For manual coagulation testing, each analyst must perform two levels of controls before
testing patient samples and with each change in reagent. In addition, patient and control
samples must be tested in duplicate.
With electrophoresis, one control needs to be included in each electrophoretic cell and the
control must contain fractions representative of those routinely reported in patient samples.
For toxicology, each thin layer chromatogrophy (TLC) plate must be spotted with at least
one sample of calibration material containing all drug groups reported by the laboratory with
TLC, one control must be included in each chamber, and the sample must be processed
through each step of patient testing including extraction.
Qualitative tests with built-in controls are adequate provided the kit has been qualified with
at least one positive and negative control. CAP requires one positive (external) and one
negative (external) control per day.
For those methodologies where the manufacturer specifies, surrogate or electronic controls
may be used to fulfill the daily QC requirement. In all cases, the appropriate number of
controls (two levels per day for most analytes) must be included and the results must be
documented and reviewed to ensure the adequacy of the testing process.
In all cases, documentation of both the QC results and the specific remedial action to "out of control
results" must be available to the inspector.
High Complexity Tests
This category includes those tests that are modified by the laboratory, developed by a laboratory, or
a test classified as high complexity under CLIA. Laboratories under CLIA and COLA also must
comply with Section §493.1201 (general QC): The laboratory must establish and follow written QC
procedures for monitoring and evaluating the quality of the analytical testing process of each
method to assure the accuracy and reliability of patient test results and reports. Section §493.1202
states for each test of high complexity performed, the laboratory must meet all applicable standards
of this subpart (subpart K). For statistical QC, laboratories must be in compliance with the
following sections:
§493.1218: Control procedures are performed on a routine basis to monitor the stability of
the method or test system; control and calibration materials provide a means to indirectly
assess accuracy and precision of patient test results. Control procedures must be performed
as defined in this section unless otherwise specified in sections §493.1223 through
§493.1285 (these state specific QC requirements for blood gases, hematology, etc.).
§493.1218(b) for each method that is developed in-house, is a modification of the
manufacturer’s test procedure, or is a method that has not been cleared by the FDA as
meeting the CLIA requirements for general QC (all highly complex methods), the laboratory
must evaluate instrument and reagent stability and operator variance in determining the
number, type, frequency of testing calibration or control materials and establish criteria for
acceptability used to monitor test performance during a run of patient specimen(s).
§493.1218(d)(1) The stated values of an assayed control material may be used as the target
values provided the stated values correspond to the methodology and instrument employed
by the laboratory and are verified by the laboratory.
§493.1218(d)(2) Statistical parameters for unassayed materials must be established over
time by the laboratory through concurrent testing with calibration materials or control
materials having previously determined statistical parameters. (The statistical parameters,
e.g. mean and SD, for each lot number must be determined through repetitive testing).
§493.1218(e) Control results must meet the laboratory's criteria for acceptability prior to
reporting patient test results. Laboratory criteria for acceptability refers to the particular
control limits or control rules chosen by the laboratory.
What if a control fails to meet criteria for acceptability?
Regardless how a test has been classified, the regulations require "remedial action and
documentation of this activity. Section 493.1219(b) Remedial actions states that when: "Results of
controls and calibration materials fail to meet the laboratory's established criteria for
acceptability, all patient test results obtained in the unacceptable test run or since the last
acceptable test run must be evaluated to determine if patient test results have been adversely
affected and the laboratory must take remedial action necessary to ensure the reporting of accurate
and reliable patient test results.
Joint Commision on Accreditation of Healthcare Organizations (JCAHO)
Requirements for QC
The requirements identified for CLIA’88 and COLA also are applicable to JCAHO. Under JCAHO
[6], the goal of QC is to achieve quality in laboratory testing and produce the best possible test
results and outcomes. Additional JCAHO requirements are found for the different test classification
categories.
JCAHO and Waived Testing
JCAHO recognizes waived tests as defined by CLIA'88, but identifies additional QC requirements
that include:
Defined QC checks that at least meet the minimum manufacturer's recommendations (when
no QC requirement is specified, the testing institution must define a policy);
Maintenance of appropriate QC and test records;
Proof of training and continued competence of all testing personnel;
Proof all testing personnel have access to current written procedures for QC and remedial
actions.
Maintenance of QC records, including a mechanism to correlate or link analyst, QC records,
instrument and instrument problems, and individual patient test results.
For glucose, at a minimum two levels of controls are required each day a glucose is
performed. QC is focused on each meter used, not each individual performing the test.
Therefore, not all testing personnel need to routinely perform QC, but each meter must be
validated by QC before testing patient samples.
JCAHO and Moderate Complexity Tests
For moderate complexity tests, JCAHO, for the most part, follows CLIA'88 and mandates the same
seven QC requirements as CLIA’88 and COLA and also accepts electronic controls for now.
However, for use of electronic controls, JCAHO requires that the laboratory verify the
manufacturer’s QC claims and run external (usually liquid) controls periodically to validate that no
change occurred with the testing system.
JCAHO and High Complexity Testing
Testing sites must follow all the CLIA QC requirements for high complexity tests as well as for any
modified moderate complexity tests (this includes not following the manufacturer’s directions) and
tests developed in-house.
JCAHO Standards for Quality Control
In addition to the requirements identified above, JCAHO requires that all testing sites meet the
following standards associated with quality control.
General QC Requirements:
(QC.1) Each specialty and subspecialty (of testing) has a documented quality control
program.
(QC.1.3) The laboratory’s QC system includes daily surveillance of results by appropriate
personnel.
(QC.1.4) The laboratory takes remedial action for deficiencies identified through QC
measures or authorized inspections and documents such actions.
(QC.1.5) The laboratory ensures that QC results meet its criteria for acceptability before it
reports patient test results.
Clinical Chemistry QC Requirements:
(QC.6) Using appropriate controls, the laboratory verifies each procedure in clinical
chemistry at least once each day of use.
(QC.6.2) Using repetitive testing, the laboratory establishes control ranges with valid
statistical measurements for each procedure in chemistry.
(QC.6.3) The laboratory has established and makes available to its staff acceptable limits for
all standard and reference QC samples, as well as the action to take when results are outside
satisfactory control limits.
(QC.6.4) The laboratory established test control limits to provide results with meaningful
clinical applications.
Hematology and Coagulation QC Requirements:
(QC.7.1) The laboratory verifies each procedure and test parameter against known standards
or controls within the range of clinically significant values each day of use.
(QC.7.3) The laboratory has established and makes available to its staff acceptable limits for
all standard and reference QC samples, as well as the action to take when results are outside
satisfactory control limits.
(QC.7.4) The laboratory has established test control limits to provide results with
meaningful clinical applications.
College of American Pathologists Laboratory Accreditation Program
(CAP-LAP) Requirements for Quality Control
CAP-LAP's philosophy is that all clinical laboratory tests need to follow the requirements defined
for high complexity testing under CLIA'88 [7]. Requirements for routine analysis of QC follow the
CLIA requirements in terms of number and frequency, except controls must be included with all
tests (even those identified as waived tests under CLIA'88). For now, CAP only allows "acceptable"
alternative (electronic) QC for point of care testing. The latest POCT Checklist (Checklist 30,
1997.0) states that certain unit-use devices/kits may warrant some combination of instrument,
procedural, and/or electronic controls. Question 30.0550 (Checklist 30) states that POCT sites using
alternative QC must have scientifically acceptable evidence (documented information) that the
entire analytical process is being evaluated correctly. Except for electronic controls, CAP requires
that control specimens be tested in the same manner as patient samples.
Qualitative tests (even those with built-in controls) need to be evaluated with both a positive and
negative control each day of use. For all tests, CAP-LAP requires an audit trail that ties the patients’
results with the analyst, instrument and QC. In addition, the QC program should show evidence of
documented review on the next shift, if no supervisor is on site, and at least weekly review by the
technical supervisor and monthly secondary review by the director or director's designee.
CAP Guidelines for Quality Control
In the general requirements, CAP states in Checklist #1 1997.1 that the overall QC program for the
entire laboratory must be clearly defined and documented. It must include general policies and
delegation of responsibilities. The QC records should be well-organized with a defined system to
permit regular review by appropriate supervisory personnel.
01.3000) Does the QC program clearly define goals for monitoring analytic performance,
procedures, policies, tolerance limits, corrective action and related information?
Hematology QC Requirements (Checklist 2, 1996.2)
(02.2005) For numeric data generated by the hematology laboratory, are Gaussian or other
longitudinal process control statistics (S.D. and C.V.) calculated at least at monthly intervals
to define analytic precision?
(02.2010) Are tolerance limits (numeric and/or non-numeric) fully defined and documented
for all hematology and coagulation control procedures?
(02.2580) Does the laboratory use preserved or stabilized whole blood preparations for
longitudinal process control?
(02.2582) Does the laboratory use retained, previously analyzed patient whole blood
samples for longitudinal process control?
(02.2587) If assayed controls are used for CBC instruments, do control values correspond to
the methodology and have target values been verified by the laboratory for quantitative
tests?
(02.2588) If unassayed controls are used, has a statistically valid target range been
established for each lot by repetitive analysis in runs that include previously tested control
materials?
Automated/General Chemistry QC Requirements (Checklist 3, 1996.2)
(03.2600) For quantitative tests, are control specimens (with a matrix appropriate for the
method) at more than once concentration (when available) used for all tests with each
analytical run?
(03.2605) If assayed controls are used, do control values correspond to the methodology and
have target values been verified by the laboratory for quantitative tests?
(03.2610) If unassayed controls are used, has a statistically valid target range been
established for each lot by repetitive analysis in runs that include previously tested control
materials?
(03.2625) Are tolerance limits defined for control procedures? Note: For tests with numeric
results, recovery ranges supplied by manufacturers of assayed controls must not be
substituted for QC range limits determined the laboratory on its own equipment.
References
1. U.S. Department of Health and Human Services. Medicare, Medicaid and CLIA programs:
Regulations implementing the Clinical Laboratory Improvement Amendments of 1988
(CLIA). Final rule. Fed Regist 1992; 57:7002-186.
2. Laessig RH, Ehrmeyer SS: New Poor Man’s (Person’) Guide to Meeting the Regulations. R
& S Consultants, Madison WI. 1997.
3. Accreditation Manual. Commission on Office Accreditation (COLA). Columbia, MD, 1996.
4. U.S. Department of Health and Human Services. Medicare, Medicaid and CLIA programs:
Regulations implementing the Clinical Laboratory Improvement Amendments of 1988
(CLIA) and Clinical Laboratory Act program fee collection. Fed Regist 1993:58:5215-37.
5. U.S. Department of Health and Human Services. Medicare, Medicaid and CLIA Programs:
Extension of Certain Effective Dates for Clinical Laboratory Requirements Under CLIA.
Fed Regist 1997;62:25855-58.
6. Accreditation Manual for Pathology and Clinical laboratory Services. Joint Commission on
Accreditation of Healthcare Organizations (JCAHO). Oakbrook Terrace, IL, 1996.
7. Laboratory Accreditation Program. College of American Pathologists (CAP). Northfield,
IL, 1997.
Biography: Sharon S. Ehrmeyer, Ph.D.
Sharon Ehrmeyer, Ph.D., MT(ASCP) is Professor of Pathology and Laboratory Medicine and
Director of the Medical Technology Program at the University of Wisconsin in Madison,
Wisconsin. Dr. Ehrmeyer is active in the American Association for Clinical Chemistry, the
American Society for Clinical Laboratory Science and the National Committee Clinical Laboratory
Standards where she serves on the Board of Directors and chairs its pH/Blood Gas Committee. Dr.
Ehrmeyer gives numerous presentations on laboratory regulations (CLIA, JCAHO, CAP and
COLA), point of care testing and various quality issues. Her research interests focus on clinical
laboratory quality and the impact of government regulations on laboratory practices.
Copyright © 2000. All rights reserved
Westgard QC, 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's new? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
Lesson of the Month:
QC - THE PLANNING PROCESS
James O. Westgard, Ph.D.
What information is needed for QC planning?
What are some practical QC planning approaches?
1. Computer Supported Planning Process
2. Manual QC planning with a workbook of OPSpecs charts
3. Manual QC planning with normalized OPSpecs charts
4. Manual QC planning with QC Selection Grids
Which approach should you use?
Education and Training in QC Planning
References
PLEASE NOTE: An updated version of this lesson is now available in Basic QC Practices, 2nd Edition.
Many of you know about my Scandinavian heritage from my frequent references to one of my
ancestors, Hagar the Horrible, who is a source of both inspiration and practical advice. In one of my
favorite Hagar cartoons, Helga asks why girls mature faster than boys. This is a good example of a
process problem that all of us have experienced as teenagers and many of us also as parents. Hagar's
answer is "poor planning." That certainly rings true. And those of us who are parents of more than
one child have been repeatedly frustrated by this process, even though we supposedly gain
experience that should make it easier the next time. The difficulty is that we really aren't able to
change the process and just have to live through it again and again. Everyone in the future will also
have to endure this problem and live through a similar experience. What is really needed is to replan
the process to eliminate the problem, but that's something that only top management has the power
to do.
In many ways, this problem is analogous to the QC problems experienced in many laboratories.
Bench level analysts often have to live with QC problems because the testing process has not been
properly planned, particularly the selection of an appropriate statistical QC procedure. The people at
the bench can't generally change the process because management retains that power. Many of you
who are reading this are probably QC specialists, supervisors, managers, or directors, and you do
have the power to change the process. You need to be able to select QC procedures that will assure
the desired analytical or clinical quality is achieved, taking into account the imprecision,
inaccuracy, and instability observed for your methods. That's what QC planning is all about.
What information is needed for QC planning?
QC planning requires information about the quality requirements for different tests, the imprecision
and inaccuracy observed for a specific testing process, and the probabilities of rejection expected
for the QC procedures of interest. Information on quality requirements will be found in
recommendations in scientific literature, regulatory guidelines, and proficiency testing criteria. This
website provides a discussion of different types of quality requirements, as well as summary tables
of analytical quality requirements, clinical quality requirements, and biological goals.
Reliable estimates of method performance can be obtained initially by following established method
evaluation protocols and later from on-going estimates of imprecision from measurements
accumulated on control materials and estimates of bias from proficiency testing surveys.
Performance characteristics of commonly used QC procedures are available in the clinical
chemistry literature. QC simulation programs may be employed by laboratory analysts to estimate
the power of new control rules, including patient data algorithms. Probability calculations may also
be utilized by industrial statisticians and laboratory analysts with the necessary mathematical skills.
What are some practical QC planning approaches?
A systematic QC planning process is needed to consider all this critical performance information in
an orderly manner. The key to practicality is being able to do QC planning in a matter of a few
minutes, rather than a few hours or days. This means supporting data calculations, preparing
graphical tools and charts, and making it easy to document the QC recommendations.
Based on my experience with QC planning over the last several years, both in the laboratory and in
classrooms and workshops, there are four approaches that are practical today. These approaches
take advantage of tools and technology that make it simpler and faster to perform QC planning. It is
essential to recognize that new tools and technology will be needed if you are to perform QC
planning quickly and efficiently. As with any new tool or technology, you need to initially spend
some time understanding the principles and theory, satisfy yourself that the approach is
scientifically sound, and then implement the process steps and operations. The planning process can
be quick and easy to perform with the new tools and technology, even though the principles and
theory may be complicated.
1. Computer supported QC planning process
The most quantitative, comprehensive, and flexible approach is to provide computer support for the
QC planning process. For several years, I used electronic spreadsheets to prepare power function
graphs and OPSpecs® charts, but this still took hours and sometimes days to assess QC
performance, select new QC procedures, and document QC recommendations. To reduce the time
required, specialized software ( our QC Validator® and EZ Rules® programs) has been developed to
perform the calculations, prepare the necessary graphs and charts, and provide documentation in a
matter of a few minutes. Use of the QC Validator program has been described in the literature [1,2]
and elsewhere on this website, where a demo and tutorials are also available to illustrate how it
works. Computer support permits both analytical and clinical quality requirements to be considered
and makes available the power functions for as many as 100 combinations of control rules and N's.
The program's HELP facility can be used to provide quick access to information, such as the CLIA
PT requirements. QC planning applications on this website illustrate the use of power function
graphs, critical-error graphs, OPSpecs charts, and QC Validation Reports that have been prepared
by the QC Validator program. For a cholesterol example, see our QC application using Cholesterol
with a clinical requirement.
2. Manual QC planning with a workbook of OPSpecs charts
A manual process can be supported using a collection of preprinted OPSpecs charts (our OPSpecs
Manual) [3] that cover a wide range of analytical quality requirements (0.5% to 50%), commonly
used control rules (12s, 12.5s, 13s, 13.5s, 13s/22s/R4s/41s), and commonly used numbers of control
measurements (2,3,4,6). This manual QC planning process is limited to analytical quality
requirements that are stated in the form of allowable total errors, such as the CLIA proficiency
testing criteria for acceptable performance. To use the OPSpecs Manual, you first define the quality
required for the test of interest and then lookup the corresponding OPSpecs charts in the manual.
You plot your observed imprecision and inaccuracy as the "operating point" and then inspect the
charts to select QC procedures whose operating limits (for the imprecision and accuracy that are
allowable) are above your operating point. Both 90% AQA and 50% AQA OPSpecs charts are
provided to plan QC procedures that will achieve 90% error detection (generally preferred) or 50%
error detection (okay for very stable methods that have few problems). Application of this approach
has been illustrated in a recent paper [4] that provides an error budget perspective and explanation
of the quality- planning models and the OPSpecs chart. For a cholesterol example, see our QC
application using Cholesterol with an analytical requirement.
3. Manual QC planning with normalized OPSpecs® charts
Standardized or normalized OPSpecs charts can be prepared that apply to any total error
requirement and a limited menu of control rules and limited numbers of control measurements.
Manual calculations are necessary to present method imprecision and inaccuracy as a percent of the
total error requirement. The normalized operating point is then plotted and its location used to
determine which control rules and N's are appropriate. Normalized charts are available in the
literature [5] and incorporate either 90% or 50% error detection for the 13.5s, 13s, 12.5s, 12s, and
13s/22s/R4s/41s control rules with N's of 2 and 4. A normalized OPSpecs calculator is available on
this website, as well as a Lesson on Normalized OPSpecs Charts.
4. Manual QC planning with QC Selection Grids
Another approach is to simplify the QC planning process with the aid of QC Selection Grids
(QCSG) [6]. One QCSG has been constructed for single-rule QC procedures that can be
implemented on Levey-Jennings control charts. Another provides adaptations of the Westgard
multi-rule procedure. These are tables for looking up QC recommendations based on the expected
process capability and the process stability of a method. A quantitative estimate of process
capability is obtained by manually calculating the size of the systematic error that is medically
important; a qualitative estimate of process stability is assessed from the expected frequency of
problems or errors, classifying method stability as excellent, moderate, or poor. After the candidate
QC procedures have been selected from the tables, the actual QC performance must be assessed by
interpolation of power function graphs to make a final selection. A QCSG calculator is available on
this website.
Which approach should you use?
The easiest approach to get started with is manual QC planning using the OPSpecs Manual. The
approach can be learned by following the example applications that are included in the manual,
from information available in the laboratory management literature [4], or by participating in one of
our workshops or training programs.
Computer support is essential to use clinical quality requirements. You may be able to implement
the quality-planning models if you are skilled in using electronic spreadsheets, or you can consider
specialized software such as the QC Validator program that implements both the analytical and
clinical models. Validator manual features plus an Automatic QC Selection feature that is initiated
with the click of a button. Operation of the program can be learned from the tutorials that are
supplied with the program.
Normalized OPSpecs charts and QC Selection Grids require manual calculations, thus they are not
as simple and reliable as the OPSpecs® Manual and the QC Validator program. However, they are
documented in the scientific literature [5,6] and are readily available alternatives that should be
practical in the initial learning phase. Once the importance and usefulness of QC planning has been
established, you will want to progress to the OPSpecs Manual or the QC Validator
program.
Education and training in QC Planning
Extensive materials are available on this website. See the Archives for a complete list of available
materials.
References
1. Westgard JO, Stein B. Automated selection of statistical quality control procedures to assure
meeting clinical or analytical quality requirements. Clin Chem 1997;43:400-403.
2. Westgard JO, Stein B, Westgard SA, Kennedy R. QC Validator 2.0: a computer program for
automatic selection of statistical QC procedures for applications in healthcare laboratories.
Comput Methods Programs Biomed 1997;53:175-186.
3. Westgard JO. OPSpecs Manual - Expanded Edition. Ogunquit, ME, Westgard QC, 1996,
distributed by Dade International and also available through the AACC, ASCLS, and
CLMA.
4. Westgard JO. Error budgets for quality management: Practical tools for planning and
assuring the analytical quality of laboratory testing processes. Clin Lab Manag Review
1996;10:377-403.
5. Westgard JO. Charts of operational process specifications ("OPSpecs Charts") for assessing
the precision, accuracy, and quality control needed to satisfy proficiency testing
performance criteria. Clin Chem 1992;38:1226- 1233.
6. Westgard JO, Quam EF, Barry PL. Selection grids for planning quality control procedures.
Clin Lab Sci 1990;3:273-280.
Copyright © 2003. All rights reserved.
Westgard QC, 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's New? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
QC - THE LEVEY-JENNINGS CONTROL CHART
Patricia L. Barry, BS, MT(ASCP)
Example application
QC procedure(s) to be implemented
Calculation of control limits
Preparation of control charts
Use of control charts
Answers for this exercise
Interpretation of example test results
Please Note: an updated version of this article is now available in Basic QC Practices, 2nd Edition
This exercise is intended to show, in step-wise fashion, how to construct a Levey-Jennings control
chart, plot control values, and interpret those results. This assumes you already have (a) selected
appropriate control materials, (b) analyzed those materials to characterize method performance by
collecting a minimum of 20 measurements over at least 10 days, (c) calculated the mean and
standard deviation of those data, and (d) selected the number of control measurements to be used
per run and (e) selected the control rules to be applied.
See QC - The Materials for more information about selecting appropriate control materials. See QC
- The Calculations for detailed information about calculating the mean and standard deviation. See
QC - The Planning Process for a description of the approach, tools, and technology available to
select QC procedures on the basis of the quality required for a test and the performance observed
for a method.
Example application
For a cholesterol method, two different commercial control products have been selected that have
concentrations near the important medical decision levels of 200 mg/dL and 240 mg/dL identified
by the National Cholesterol Education Program (NCEP) guidelines for test interpretation. The
materials were analyzed once per day for a period of twenty days. From these data, the means and
standard deviations were calculated to be:
Control 1
Mean=200
smeas= 4.0 mg/dL, or 2.0% CV
Control 2
Mean=250
smeas=5.0 mg/dL, or 2.0% CV
QC procedure(s) to be implemented
Each of the two control materials will be analyzed once per run, providing a total of two control
measurements per run. Control status will be judged by either the 12s or 13s rule. These rules are
defined as follows:
12s refers to the control rule that is commonly used with a Levey-Jennings chart when the
control limits are set as the mean plus/minus 2s. In many laboratories, this rule is used to
reject a run when a single control measurement exceeds a 2s control limit.
13s corresponds to a Levey-Jennings chart having control limits set as the mean plus/minus
3s. An analytical run is rejected when a single control measurement exceeds a 3s control
limit.
The 12s rule is very commonly used today, and while it provides high error detection, the use of 2s
control limits gives an expected high level of false rejections. The 13s rule provides an alternative
QC procedure that has lower false rejections, but also lower error detection. In this exercise, you
will see how to apply both QC procedures and also get a feel for the difference in their
performance.
Calculation of control limits
Two sets of control limits will be needed to implement the rules described above. The first set uses
2s control limits (for implementation of the 12s rule) calculated as the mean plus or minus 2 times
the standard deviation. The second set uses 3s control limits (for implementation of the 13s rule)
calculated as the mean plus or minus 3 times the standard deviation.
For this example, Control 1 has a mean of 200 and a standard deviation of 4 mg/dL.
The upper control limit would be:
200 + 2*4, which is 208 mg/dL.
The lower control limit would be:
200 - 2*4, or 192 mg/dL.
What are the 3s control limits for Control 1?
What are the 2s control limits for Control 2?
What are the 3s control limits for Control 2?
NOTE: This Javascript Control Limit Calculator only works on browsers that support Javascript!
Enter the control mean:
Enter the control standard deviation:
Enter the control limit you wish to
evaluate (number only i.e. 2, 3, 3.5,
etc.):
Once you've entered these three
values, click this button to calculate
your limits
Your Upper Limit will appear here:
Your Lower Limit will appear here:
Results for the Cholesterol Example Control Limits
Control Material
mean +/- 2s limits
mean +/- 3s limits
Control 1
-
-
Control 2
-
-
You should end up with 3s control limits of 188 and 212 for Control 1. For Control 2, you should
have 2s control limits of 240 and 260 and 3s control limits of 235 and 265.
Preparation of control charts
This exercise shows how to construct control charts manually using standard graph paper. For this
exercise, graph paper having 10x10 or 20x20 lines per inch works well. You will need two sheets,
one for each chart of the two control materials. While it is possible to prepare both charts on a
single sheet, this may reduce the readability of the control charts. If you do not have graph paper
available at this time, print out the lower resolution grids below.
Click here if you want to print a larger version of this chart separately.
Click here if you want to print a larger version of this chart separately.
Label charts. Include the name of the test and the name of the control material in a
prominent place so that this information is quickly and easily discerned when viewing the
chart. The measurement unit, in this case mg/dL, can be included in the label or included in
the label for the y-axis. Other information typically included on the chart are the name of the
analytical system, the lot number of the control material, the current mean and standard
deviation, and the time period covered by the chart.
Scale and label x-axis. The horizontal or x-axis represents time and you will typically set
the scale to accomodate 30 days per month or 30 runs per month. For this example, divide
the x-axis into evenly sized increments and number them sequentially from 1 to 30. Label
the x-axis "Day."
Scale and label y-axis. The vertical or y-axis represents the observed control value and you
need to set the scale to accomodate the lowest and highest results expected. A generally
useful scale is to allow for a value as low as the mean - 4 standard deviations and a value as
high as the mean + 4 standard deviations. For this example, the chart for Control 1 should be
scaled to accomodate a range from 200 - 4*4, which is 184, to 200 + 4*4, which is 216. This
can be rounded to 180 to 220 to fit the 10x10 or 20x20 grids of the graph paper. Mark off
and identify appropriate concentrations on the y-axis. Label the y-axis "Control value."
What is the range for scaling the chart for Control 2?
Draw lines for mean and control limits. On the y-axis, locate the values that correspond to
the mean and draw a green horizontal line (at 200 mg/dL for Control 1). Locate the values
that correspond to the mean +2s and the mean -2s and draw yellow horizontal lines (at 192
and 208 for Control 1). Locate the values that correspond to the mean +3s and the mean -3s
and draw red horizontal lines (at 188 and 212 for control 1). What are the mean and control
limit lines for Control 2?
Click here if you want to print a larger version of this chart separately.
Click here if you want to print a larger version of this chart separately.
Use of Control Charts
Once the control charts have been set up, you start plotting the new control values that are being
collected as part of your routine work. The idea is that, for a stable testing process, the new control
measurements should show the same distribution as the past control measurements. That means it
will be somewhat unusual to see a control value that exceeds a 2s control limit and very rare to see a
control value that exceeds a 3s control limit. If the method is unstable and has some kind of
problem, then there should be a higher chance of seeing control values that exceed the control
limits. Therefore, when the control values fall within the expected distribution, you classify the run
to be "in-control," accept the results, and report patient test results. When the control values fall
outside the expected distribution, you classify the run as "out-of-control," reject the test values,
and do not report patient test results.
Plot control results. For practice, the accompanying table provides some control results for
our example cholesterol method. Plot these results, one from Control 1 and one from
Control 2, for each day. You can print the Levey-Jennings QC Practice Exercise (below) to
obtain a worksheet that shows all these control results. For day 1, the value for Control 1 is
200 and Control 2 is 247. On the chart for Control 1, find the value of 1 on the x-axis and
the value of 200 on the y-axis, follow the gridlines to where they intersect, and place a mark;
it should fall on the mean line. On the chart for Control 2, find the value of 1 on the x-axis
and the value of 247 on the y-axis, then mark that point; it should fall a little below the mean
line. In plotting control values, it is common practice to draw lines connecting the data
points on the control chart to provide a stronger visual impression and make it easier to see
patterns and shifts.
Levey-Jennings QC Practice Exercise:
Cholesterol example where:
Control 1 has a mean of 200 mg/dL and standard deviation of 4.0 mg/dL.
Control 2 has a mean of 250 mg/dL and standard deviation of 5.0 mg/dL.
Prepare appropriate control charts and interpret the results.
Day
Control 1
Value
Control 2
Value
1
200
247
2
205
250
3
195
255
4
202
243
5
186
254
6
207
263
7
194
251
8
209
264
9
200
253
10
196
244
11
190
261
12s Rule
Violation
13s Rule
Violation
Accept(A),
Warning (W),
or Reject(R)?
Comments
12
204
254
13
196
239
14
207
236
15
200
250
16
205
259
17
209
257
18
197
256
19
196
249
20
198
257
21
197
241
22
195
255
23
198
250
24
199
259
25
191
247
26
197
242
27
190
256
28
202
246
Interpret control results. Apply the 12s and 13s control rules and make a decision whether
you should accept or reject the run for each day. The control values for the first day are incontrol and the patient results can be reported. Continue plotting the 2 control values per day
and interpreting those results. Circle those points that correspond to runs that should be
rejected. Keep track of the control rules that are violated on the worksheet for the LeveyJennings QC Practice Exercise. Patient results obtained in runs where the 13s rule is violated
are most likely incorrect. Results obtained in runs where the 12s rule is violated may or may
not be incorrect because there is about a 10% chance of this occurring even when the
method is working perfectly. This is a "false alarm" problem that is inherent with the use of
2s control limits with an N of 2. In spite of this serious limitation, many laboratories
continue to use 2s control limits and just routinely repeat the run and the controls, or
sometimes repeat only the controls by themselves. Note that if a control is out a second time,
the actual control rule that is being used to reject a run is a 22s rule rather than the stated 12s
rule. Unfortunately, the 22s rule by itself is not very sensitive, therefore, it is better to use the
13s and 22s rules together in a multirule procedure to improve error detection while, at the
same time, maintaining a low false rejection rate. We'll provide more discussion of multirule
QC procedures in a later lesson.
Click here to view the answers to the exercise.
Copyright © 2000. All rights reserved.
Westgard QC, Inc., 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's new? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
QC Application:
QC - THE PRACTICE
James O . Westgard, Ph.D., Elsa F. Quam, BS, MT(ASCP), and Patricia L. Barry, BS, MT(ASCP)
Purpose of QC
QC Planning
QC Implementation
QC Operation
QC Documentation and Review
Who's Responsible?
PLEASE NOTE: An updated version of this article is available in Basic QC Practices, 2nd Edition
This lesson provides a summary of the overall process of establishing and maintaining a statistical
QC procedure. The objective of this lesson is to outline all the activities that are necessary without
getting bogged down in the details of each of these activities. More detailed information is provided
by links to other materials on this website.
Purpose of QC
QC is intended to help people do good work by giving them a way to check that their work process
is functioning properly. People need tools, such as statistical QC, to help them do this. Statistical
QC provides a way of looking at the results of a work process and identifying when they exceed the
variation expected under stable routine operation, in which case, it is likely that something has gone
wrong and that the process needs to be fixed.
In a healthcare laboratory, known samples from a large number of bottles of stable control materials
are analyzed to monitor the variation of a testing process. Results on these known samples are
expected to fall within certain statistical limits, e.g., 95% within the mean plus or minus 2 standard
deviations, 99.7% within the mean plus or minus 3 standard deviations. The means and standard
deviations are calculated from laboratory measurements on these control materials, then control
charts are constructed to display the variation of these known samples over time. Control limits are
drawn to identify results that are unexpected and need to be investigated. [See QC - The Idea.]
QC Planning
The purpose of QC planning is to select a QC procedure that will assure the required quality at the
minimum cost. Optimum cost-effectiveness depends on designing QC procedures on a test-by-test
basis on each analytical system, taking into account the particular quality required for an individual
test and the particular performance achieved with the analytical method on that system.
Define the quality required for the test on the basis of clinical quality needed for proper use
and interpretation of test results, or on the basis of the analytical quality needed to satisfy
regulatory requirements, such as the CLIA proficiency testing criteria for acceptable
performance. Quality requirements in the form of medically important changes (or clinical
decision intervals) and total analytical errors (allowable total errors) can readily be used
with available QC planning tools. [See QC - The Planning Process].
Assess method performance from initial evaluation studies or from on-going QC results and
proficiency testing surverys. Initially estimate bias from the comparison of methods
evaluation experiment; estimate imprecision from a replication experiment over 10-20 days.
Utilize QC planning tools, such as OPSpecs charts, to identify the appropriate control rules
and the number of control measurements (N). [See Mapping the Road to Analytical Quality
with Charts of Operating Specifications]. In the absence of a quantitative planning process,
be aware that the use of 2s control limits (i.e., control limits set as the mean plus or minus 2
standard deviations, or the 12s control rule) will cause a high false alarm or false rejection
rate - approximately 5% for N=1, 9% for N=2, 14% for N=3, and 18% for N=4. Also be
aware that the use of 3s control limits (or the 13s control rule) may not give sufficient error
detection. Use of a 12.5s control rule or the 13s/22s/R4s or 13s/2of32s/R4s multirule procedures
would be better to reduce the false rejections compared to a 12s control procedure and
provide better error detection than a 13s control procedure. [See QC - The Chances of
Rejection.]
Identify a Total QC strategy that places appropriate emphasis on statistical QC, nonstatistical components (such as preventive maintenance, instrument function checks,
performance validation tests, patient data QC, and quality improvement). When medically
important error can be readily detected by statistical QC, rely on statistical QC and perform
the minimum other QC as required by the manufacturer's instructions, government and
accreditation requirements, and good laboratory practice. If errors cannot be readily
detected, they must be prevented by frequent maintenance, instrument function checks,
thorough operator training, operator experience, etc. [See Total Quality Control Strategies.]
QC Implementation
Because statistical QC is a quantitative technique, there are technical details that must be properly
implemented, unlike some other aspects of quality management that may be more philisophical and
less technical. Statistical QC won't work right and accomplish its intended purpose unless it is
properly implemented.
Choose appropriate control materials. This includes the number of materials necessary to
monitor the critical medical decision levels and working range of the method, as well as the
type of material that will best simulate the true patient sample. It is common practice to use
two levels of controls for many chemistry tests and three levels for hematology and
coagulation tests. [See QC - The Materials, see also Medical Decision Levels.]
Analyze the control materials to obtain a minimum of 20 measurements over at least a 10
day period. It is important to collect measurements that will characterize the actual
performance of the method under each laboratory's own operating conditions. Ten days is a
minimum period to observe factors that affect method performance; 20 days is better.
Calculate the mean and standard deviation of the control measurements. While the number
of measurements may be minimal in the beginning, the mean and standard deviation can be
updated as more measurements are accumulated. [See QC - The Calculations.]
Calculate the control limits for the control rules that are to be applied. Contrary to some
manufacturers' instructions and some laboratory practices, it is not advisable to use the
bottle values appearing on control materials to set statistical control limits. Control limits
should be calculated from the mean and standard deviation that were determined when that
control material was analyzed in the laboratory. [See QC - The Levey Jennings Control
Chart].
Prepare control charts or set the appropriate parameters in a computerized monitoring and
charting package. A sheet of graph paper is all that's needed for manual implementation,
however, computerized implementation is often necessary for high speed multi-test
automated analyzers that produce many control results in a short period of time. This
computer support may be provided by software in the analyzer, by a PC data management
station, or by a laboratory information system.
Prepare written guidelines to document the QC procedure. Written protocols are required by
accreditation and regulatory agencies, as well as for good laboratory practice. An important
part of this protocol is to describe when control samples are to be analyzed, how many
samples are to be analyzed, where they are to be located in an analytical run, how the
control results are to be interpreted, and what to do when "out-of-control."
Teach the QC procedure to ALL personnel who will be performing the test. In point- of-care
applications where some personnel perform the test infrequently, statistical QC is
particularly important for documenting operator proficiency.
Routine QC Operation
Routine operation depends on obtaining current control results and using them to determine whether
the testing process is performing as expected. It is "expected" that the current control results fall
within the established control limits if the testing process is working okay. It is unexpected for the
control results exceed a control limit or violate a control rule unless there is a problem with the
testing process.
Analyze control materials with each analytical run. According to US CLIA regulations, at
least two different control materials should be analyzed with each run Analyze control
materials with each analytical run. According to US CLIA regulations, at least two different
control materials should be analyzed with each run [See QC - The Regulations.] Note that
the run is defined by the written QC guidelines that were developed above. A run is
typically defined in terms of a length of time, a number of samples analyzed, or a physical
grouping (or batch) of samples. The definition of a run is subjective and may change as
experience is gained and more information becomes available about a method's stability or
frequency of problems.
Record the control results and plot on control charts (as necessary). According to the
government, if you didn't write it down, you didn't do it. More important, documentation
and records are an important source of information about the actual performance of the
method under the real operating conditions of a laboratory.
Review and interpret the control results to determine control status. Here's where the
decision criteria or control rules are important for achieving uniform interpretation
regardless of the experience of the person performing the test. [See QC - The Westgard
Rules.]
When control results are "in-control," i.e., none of the control rules are violated, accept the
run and report the patient test results. Our philosophy is that each analyst should have
sufficient training and experience to do this on their own, however, there may be situations
where supervisory review is necessary before patient test results can be released. [See QC The Multirule Interpretation.]
When control results are "out-of-control," i.e., one of the control rules is violated, reject the
run and do not report patient test results. Inspect the process, identify the source of
difficulty, correct the problem, then reanalyze the patients and controls. Note that the
particular control rule that is violated may be helpful in trouble-shooting; the 13s and R4s
rules tend to be more sensitive to random errors, whereas rules such as 22s and 2of32s tend to
be more sensitive to systematic errors. Random and systematic errors have different causes,
thus the problem can be narrowed down to fewer possible sources. Once a problem is fixed,
it may be best to reanalyze the controls first to determine control status, then reassay the
patient samples. [See QC - The Out-of-control Problem.]
Document any control problems and what was done to solve them. For unusual problems, it
may be useful to file a detailed trouble-shooting report for future reference and use in
training new personnel.
QC Documentation and Review
Because many factors are involved in maintaining quality, a system of records and documentation is
needed for periodic review and evaluation.
Maintain QC records for an appropriate period of time to document routine maintenance,
unscheduled maintenance, reagent lot numbers in use and expiration dates, calibration
records that include calibrator lot numbers and expiration dates, control results and summary
statistics (monthly means and standard deviations, as well as cumulative means, standard
deviations, and control limits in use), QC problems and corrective actions taken, and
trouble-shooting reports. Most records need to be kept for two years according to US
regulations, except that maintenance records must be kept for the lifetime of an instrument
system and transferred with the instrument. [See QC - The Records.]
Review monthly means for trends and small shifts that indicate systematic errors or potential
accuracy problems; review monthly standard deviations for changes in random errors or
imprecision.
Correlate these QC changes with other performance data, such as results from proficiency
testing surveys, comparison of methods results with real patient samples, changes in
reagents, and changes in calibrations. Investigate problems and recommend corrective
actions, such as recalibration, increased maintenance, and additional operator training. Make
improvements in the testing process when possible.
Periodically assess whether the QC design is still appropriate based on the routine
performance being observed.
Who's responsible?
QC planning should be the responsiblity of laboratory management, usually the director, manager,
or quality specialist. The medical director has a critical role in defining the quality requirements for
the laboratory. The actual QC planning function may then be delegated to a manager or quality
specialist. Implementation is usually delegated to supervisors and technologists who are in charge
of managing specified analytical systems and testing processes. Routine operation is delegated to
everyone who performs a laboratory test.
In large laboratories, there may be several quality specialists who spend a large part of their time
dealing with quality systems. In small laboratories, the most senior technologist may inherit the
responsibilities for quality. In point-of-care situations, the central laboratory may be responsible for
the implementation, training, and maintenance of quality systems, but each healthcare worker who
performs a test should be accountable for routine QC operations.
Copyright © 2000. All rights reserved.
Westgard QC, 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-4718 or e-mail us at westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's new? | Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback
AN INDEX OF QUESTIONS
User-Submitted Questions
This page contains a list of all the User-Submitted Questions
for Westgard Web. The answers to these questions were
posted so that all users, visitors, and viewers of Westgard
Web can benefit from the responses.You can access every
public Questions page by clicking the green button. To see a
list of all the questions, click the blue preview buttons.
Go read it! Essay Title
1999 Round-up
Method Validation course questions
FAQ's about Method Validation
1998 Round-up
FAQ's about Basic QC Practices
FAQ's about CLIA'88, JCAHO & CAP
Multirules and Validator; What's a run?
FAQ's about OPSpecs charts; Coag PT & PTT
FAQ's about Power Functions and Critical-Error Graphs
FAQ's about Multirule QC; Immunoassay QC
FAQ's about QC Validator
Using Mean and Range charts
Using Mean and Range charts
How can I set up mean and range charts for a calcium method?
Read the answers
Back to Top
What is QC Validator?
How do you choose a decision level?
Preview Availability
Public
Public, BMV
Public, BMV
Public
Public, BQC
Public, BQC
Public
Public
Public
Public
Public
Public
Should the stable imprecision observed be matched to the decision level defined or should you use the average
of the imprecision observed for the different control levels?
Should the stable inaccuracy observed be matched to the decision level defined or should you use the average
over the analytical range of interest?
How is the frequency of errors determined?
Is it necessary to run Validator for both the analytical and clinical quality requirements?
Which graph is actually used to choose the control rule(s) & number of control measurements (N) ? Power
function graphs? Critical-Error graphs? OPSpecs charts?
What if you can't achieve 90% error detection with less than 5% false rejection?
Read the answers
Back to Top
FAQ's about multirule QC; Immunoassay QC
What is N?
What's the best way to chart QC for multirule applications?
Does the 12s warning rule have to be used in a computerized implementation?
Can other rules be used as warning rules rather than rejection rules?
Other than better error detection, are there any reasons to multirule procedures instead of single rules?
What rules are most sensitive for detecting systematic errors?
What causes systematic errors?
What rules are most sensitive for detecting random error?
What causes random errors?
When can a single rule QC procedure be used instead of a multirule procedure?
How do you decide if you need to apply rules across runs?
When one rule in a multirule combination is violated, do you exclude just that control value from the QC
statistics?
When should you use the 41s and 10x rules for "warning" rules and when should you use them as out-of-control
rules?
Should I have an 12s rule violation for starting evaluation of violations of 4 1s, 10x, 8x and 12x rules?
When would I use 8x and 12x rules?
Immunoassay Questions:
Are the manufacturer's specifications for acceptable control values too wide?
Should we set our own control limits based on our control data?
How do you use control charts on extremely stable immunoassay analyzers?
How do you determine the frequency with which to run controls on extremely stable analyzers?
Where can I find some example QC planning applications for immunoassay methods?
Read the answers
Back to Top
FAQ's about Power Function and Critical-Error Graphs
Why aren't actual concentration units used on the x-axis?
Why is the y-axis given as probability instead of average run length (ARL)?
Why aren't the power curves smooth?
How good are the probabilities estimated by computer simulations?
Why is a Ped of 0.90 considered to be ideal?
Read the answers
Back to Top
FAQ's about OPSpecs charts; Coag PT &PTT
Why isn't the x-axis labeled CV since imprecision is presented in percent?
Why isn't the size error on the x-axis presented in units of concentration?
Why is the y-axis labeled biastotl instead of biasmeas?
What do TEa and DInt mean in the headings of the OPSpecs charts?
How do I decide whether to use an analytical or clinical quality requirement?
What is the meaning of the SE in the %AQA(SE) heading on an OPSpecs chart?
What does %AQA mean?
Why is 80% used instead of 90% AQA for the random error OPSpecs chart?
Aren't 50% and 25% AQA too low to be useful?
What's the meaning of the "maximum limits of a stable process" line?
Is there any practical use for the "maximum limits" line?
Coagulation PT & PTT Testing
We use unassayed control materials for coagulation PT and PTT testing. Ranges must be established for each
lot. Each time ranges are established, the limits are very tight, giving way too many QC outliers. What are your
suggestions to help this?
Read the answers
Back to Top
Multirules and Validator; What's a Run?
How are the different levels of control incorporated in the power curves?
How does the number of control materials affect automatic QC selection?
How does the number of runs (R) affect the selection of control rules?
What are the implications of R in the simulation of power curves and the application of rules in a multirule
procedure?
What rules are used across runs?
Why is the R4s rule restricted to a single run?
What about using the R4s across materials within a run?
Does the R4s rule require consecutive control measurements?
Is the R4s rule violated if one control is +2.4 SD and another is _1.8 SD?
What's a Run?
How is a "run" defined for automated random access analyzers?
Does it make any difference whether a pair of control materials are analyzed immediately, one after the other,
or in random order, separated by time, say one in the morning and one in the afternoon?
Is it important to include patient averages to assure quality and detect preanalytical as well as analytical factors
that may not be observed with control samples?
How do QC procedures that make use of moving averages compare to multirule procedures?
Read the answers
Back to Top
FAQ's about CLIA'88, JCAHO, and CAP
How many CLIA’88 certificates should my hospital have?
Who will inspect me -- HCFA (for CLIA’88), JCAHO or CAP?
The hospital is accredited by JCAHO. Will JCAHO inspect all laboratory testing?
The hospital is accredited by JCAHO, but the central laboratory is inspected by CAP. Will CAP also inspect
POCT?
Will JCAHO accept CAP’s inspection?
Will CAP accept JCAHO’s inspection?
Why are CLIA’88 requirements important to me when my institutions testing is inspected by JCAHO or CAP?
My JCAHO laboratory is in a state that has exempt status from HCFA. Do I still need to meet specific state
requirements?
All testing in my institution is inspected by JCAHO and several CLIA numbers are involved. As part of quality
assurance, do we just need to compare results from different methodologies under the same CLIA number?
I am a doctoral scientist without board certification and I direct a laboratory that does high complexity testing.
The April 24, 1995 Federal Register extended the certification requirement to September 1, 1996. Can I still be
the laboratory director?
Read the answers
Back to Top
FAQ's about Basic QC Practices
How should I check out a new lot number of a control product?
What should I do when control products with long expiration dates are not available (such as the case for many
hematology control materials?
What should I do when a control value is outside the calculated control limits?
Do I have to record every result for a control material?
Which QC data should be excluded when calculating monthly statistics (mean, standard deviation, and
coefficient of variation)?
Which QC results should be included in updating your mean and SD statistics?
What is N?
Will multirule QC assure ideal QC performance?
How exactly do you apply the 41s control rule? the R4s? the 10x?
What control measurements can be used when applying "bias" rules?
What do I do about patient results that were reported before a problem was detected with a control rule applied
across runs?
Is there a multirule QC procedure for computerized immunoassay QC which fulfills all regulations?
What references describe how to apply and interpret multirule QC?
What training is available to help me decide what QC to use?
Read the answers
Back to Top
1998 Round-up of questions
What's a trend?
How to apply the R4s rule?
Manufacturer Ranges: Use them or lose them?
Significant Figures
Finding quality requirements for unregulated analytes
Standard deviation ratios; verifying a new lot of reagents
Where to get bias
Patient-based QC and Average of Normals
Read the answers
Back to Top
FAQ's about Method Validation
Why is it necessary to validate method performance when the manufacturer already has?
What analytical performance is needed for a laboratory test?
Who should perform the validation studies in a laboratory?
In setting up a new method for validation studies, how important is it to calibrate the method using primary
standards instead of commerical calibrators?
What performance characteristics are usually validated?
What experiments are usually performed?
Does linearity have to be validated?
Does detection limit have to be validated for all tests?
How many materials need to be analyzed in a replication experiment?
What comparison method should be used in the comparison of methods experiment?
Why is there so much emphasis placed on the comparison of methods experiment?
Why can't the correlation coefficient be used to judge the agreement between methods in a comparison of
methods study?
Why are regression statistics still recommended?
What's the proper way to use t-test statistics?
What's the proper way to use regression statistics?
What's Deming regression?
What's the alternative to more complicated regression calculations?
What tests are likely to have a narrow range of data and require more care?
Why can't acceptability be judged by tests of significance, such as t-test and F-test?
How does the "method decision chart" approach compare with the "performance criteria" approach?
Where can I find more detailed protocols and statistical guidelines for method validation?
Read the answers
Back to Top
Method Validation course questions
This group of questions came from students in Chem 555, which is a graduate course in Clinical
Chemistry at West Chester University, West Chester, PA (Fall '99). Dr. Al Caffo was the course
instructor (acaffo@wcupa.edu) and an old friend from DuPont/Dade/Behring days (the "good old
days"). A great opportunity to discuss method validation practices.
Read this essay
Back to Top
1999 Round-Up of Questions
What is N? What is a run?
How can I find quality requirements for hematology?
Can I make up my own control limits?
The z-value and defect rates?
How do you do external QC for a test where there is only a small or no peer group?
Read this essay
Back to Top
Your questions are encouraged!! Please send them in to westgard@westgard.com or mail them to
the address below. While we can't guarantee that all questions can be answered, we regularly
post a series of questions and answers here on the website so that visitors can take advantage and
benefit from previous questions.
James O. Westgard, PhD, is a professor of pathology and laboratory medicine at the
University of Wisconsin Medical School, Madison. He also is president of Westgard QC,
Inc., (Madison, Wis.) which provides tools, technology, and training for laboratory
quality management.
Copyright © 2000. All rights reserved.
Westgard QC, 7614 Gray Fox Trail, Madison WI 53717
Call 608-833-47183 or e-mail us at westgard@westgard.com
A Message from JOW
QC Lessons | QC Applications | Questions | Multirule
CLIA Requirements | What's New?| Catalog | Demo Download
Home | Glossary | ARCHIVES | Links | Feedback