A next generation label-free quantitation strategy for
advertisement

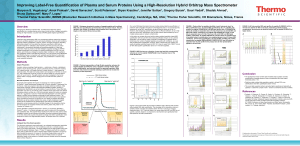
A next generation label-free quantitation strategy for analyzing complex peptide mixtures Paulo C Carvalho*1,2, Xuemei Han*1, Tao Xu1, Daniel Cociorva1, Maria da Gloria da Costa Carvalho3, Valmir C Barbosa2, and John R Yates, III1 1. Department of Chemical Physiology, The Scripps Research Institute, La Jolla, USA 2. Systems Engineering and Computer Science Program, Federal University of Rio de Janeiro, Brazil 3. Biophysics Institute, Federal University of Rio de Janeiro, Brazil Label-free approaches (e.g., spectral counting) for the quantitation of complex peptide mixtures have gained popularity because of their low cost, reasonable accuracy, and simplicity. However, they still fall behind in accuracy from those that measure abundance by comparing peptides to an internal, chemically identical standard enriched with a heavy stable isotope. Nevertheless, the latter are expensive and laborious. Here we describe a novel approach for shotgun proteomic data acquisition, termed extended data-independent acquisition (XDIA), which greatly reduces the gap between these technologies. XDIA combines high-resolution Orbitrap survey scans with multiplexed fragmentation data acquired using an ETD/CID double play. The data are then post-processed by our new software, the XDIA Processor, which enables for protein identification. A schematic of this process is shown in Figure 1. We acquired spectral counts on a yeast lysate digested with Lys-C using XDIA and compared the result to the one obtained by the widely adopted data-dependent acquisition (DDA) using ETcaD. Both identified roughly the same number of proteins; however, XDIA was more reproducible, identified ~250% additional spectra, and ~30% more unique peptides per run. Boosting the number of identified spectra significantly improves the quantitation statistics confidence; increasing identified peptides improves on coverage, making XDIA a key contribution. Figure 1 – Data-Dependent versus Extended Data-Independent Acquisition. In the DDA approach, an MS1 is acquired so precursor ions can be selected for posterior analysis by MS2. In the XDIA approach, a wide precursor window that can include multiple peptide ions is selected for fragmentation. The precursors and their charge states are detected by the XDIA processor and are included in the final MS2 file. Acknowledgments: CNPq, a FAPERJ BBP grant, and NIH P41 RR011823, R01 MH067880 for financial support.