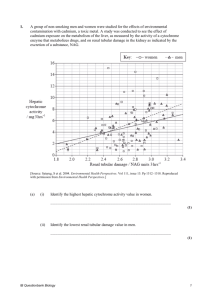
Mol
advertisement

Mol. Nutr. Food Res. 2013, 00, 1–25 DOI 10.1002/mnfr.201300522 1 REVIEW Chemoprevention of nonalcoholic fatty liver disease by dietary natural compounds Min-Hsiung Pan1,2, Ching-Shu Lai1, Mei-Ling Tsai3 and Chi-Tang Ho4∗ 1 Institute of Food Science and Technology, National Taiwan University, Taipei, Taiwan 2 Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan 3 Department of Seafood Science, National Kaohsiung Marine University, Kaohsiung, Taiwan 4 Department of Food Science, Rutgers University, New Brunswick, NJ, USA Nonalcoholic fatty liver disease (NAFLD) refers to a wide spectrum of liver disease that is not from excess alcohol consumption, but is often associated with obesity, type 2 diabetes, and metabolic syndrome. NAFLD pathogenesis is complicated and involves oxidative stress, lipotoxicity, mitochondrial damage, insulin resistance, inflammation, and excessive dietary fat intake,which increase hepatic lipid influx and de novo lipogenesis and impair insulin signaling, thus promoting hepatic triglyceride accumulation and ultimately NAFLD. Overproduction of proinflammatory adipokines from adipose tissue also affects hepatic metabolic function. Current NAFLD therapies are limited; thus,much attention has been focused on identification of potential dietary substances from fruits, vegetables, and edible plants to provide a new strategy for NAFLD treatment. Dietary natural compounds, such as carotenoids, omega-3PUFAs, flavonoids, isothiocyanates, terpenoids, curcumin, and resveratrol, act through a variety of mechanisms to prevent and improve NAFLD. Here, we summarize and briefly discuss the currently known targets and signaling pathways as well as the role of dietary natural compounds that interfere with NAFLD pathogenesis. Keywords: Chemoprevention / Nonalcoholic fatty liver disease / Dietary natural compounds Received: July 19, 2013 Revised: September 25, 2013 Accepted: October 9, 2013 Correspondence: Dr. Min-Hsiung Pan, Institute of Food Science and Technology,National TaiwanUniversity,No.1, Section 4, Roosevelt Road, Taipei 10617, Taiwan E-mail: mhpan@ntu.edu.tw Fax: +886-2-33661771 Abbreviations: _-SMA, _-smooth muscle actin; ACC, acetylcoenzyme A carboxylase; ACO, acyl-coenzyme A oxidase; ALT, alanine aminotransferase; AMPK, AMP-activated protein kinase; AST, aspartate aminotransferase; CAT, catalase; CPT-1, carnitine palmitoyl transferase 1; CYP, cytochrome P450; DHA, docosahexaenoic acid; DR5, death receptor 5; EGCG, epigallocatechin3-gallate; EPA, eicosapentaenoic acid; FA, fatty acid; FAS, FA synthase; FFA, free FA; FOXO, forkhead box protein O; G6Pase, glucose 6-phosphatase; GPx, GSH peroxidase; GSH, glutathione; GST, GSH S-transferase; HFD, high-fat diet; HMG-CoA, 3hydroxy-3-methylglutaryl-coenzyme A; HSC, hepatic stellate cell; HSL, hormone-sensitive lipase; IRS-1, insulin receptor substrate 1; JNK, c-Jun N-terminal kinase; LDL, low-density lipoprotein; MCP-1, monocyte chemotactic protein 1; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NF-_B, nuclear factor-_B; Nrf2, NF-E2-related factor 2; PEPCK, phosphoenolpyruvate carboxykinase; PI3K, phosphatidylinositol-3 kinase; PPAR, peroxisome proliferator-activated receptor; Pten, phosphatase and tensin homolog; ROS, reactive oxygen species; SCD1, stearoyl-CoA desaturase 1; SFAs, saturated FAs; SIRT, sirtuin; 1 Introduction Nonalcoholic fatty liver disease (NAFLD) has emerged as a common liver disorder that is characterized by abnormal hepatic triglyceride (TG) accumulation in the absence of excessive alcohol consumption. This disease represents a histological spectrum ranging from simple hepatic steatosis (defined as hepatic TG >5% by liver weight) that can progress to inflammatory nonalcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and ultimately end-stage liver failure or hepatocellular carcinoma [1]. Simple hepatic steatosis is a benign process without inflammation, whereas lobular inflammation and hepatocellular injury followed by fibrosis are common in NASH and are believed to drive progression to cirrhosis [2]. Liver biopsies are currently the gold standard for NASH clinical diagnosis and staging because there are no specific symptoms to distinguish this disease. Other clinical diagnostic SOCS, suppressor of cytokine signaling; SOD, superoxide dismutase; SREBP-1, sterol regulatory element binding protein 1; TF-1, theaflavin; TG, triglyceride; TGF-_1, transforming growth factor _1; TNF-_, tumor necrosis factor _; UCP, uncoupling protein; VLDL, very-low-density lipoprotein ∗Additional corresponding author: Dr. Chi-Tang Ho, E-mail: ho@aesop.rutgers.edu C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 2 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 indices include increased serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT), elevated BMI, and metabolic syndrome [1, 3]. NAFLD prevalence has increased worldwide in the past 20 years, reflecting its emergence as a major public health problem. Epidemiological studies demonstrated that NAFLD prevalence is different and varies wildly depending on population and diagnostic methods or definition [4]. Population studies estimate that approximately 25–30% of the general population in the United States [5,6] and 13–60% of the population from Japan, Italy,China, andKorea haveNAFLD[7–10]. The increasing NAFLD prevalence is not only in adults, but also in children (3–10%) and is rising up to 40–70% among obese children [11]. Although NASH is the most serious form of NAFLD, it is difficult to diagnose without a liver biopsy, which results in less population-based prevalence studies. Previous epidemiological research demonstrated that approximately 30% of simple steatosis cases progress to NASH in NAFLD patients, and approximately 20% of NASH patients develop cirrhosis. Once developed, 30–40% of cirrhosis patients succumb to liver-related death over a 10-year period [12].NASHpatients had increased cardiovascular disease and liver-related disease-induced mortality compared to simple steatosis patients. Most of these patients were diagnosed with diabetes or impaired glucose tolerance and had increased weight gain [13]. Previously, researchers suggested a number of risk factors implicated in NAFLD, including age, race, genetics, and chronic infection, and NAFLD was also strongly associated with metabolic conditions, such as obesity, type 2 diabetes, hypertension, and dyslipidemia, which are regarded as hepatic manifestations of metabolic syndrome [3, 4]. 2 NAFLD pathogenesis The liver is a necessary and important organ for whole body metabolism and energy homeostasis. Hepatic TG accumulation is a hallmark of NAFLD that results from several sources, including increased free fatty acid (FFA) delivery from adipose tissue (as lipolysis), dietary fatty acids (FAs), elevated hepatic de novo lipogenesis, reduced very-low-density lipoprotein (VLDL) export, and decreased FA _-oxidation. Except for hepatic steatosis, other histological and biological changes associated with NAFLD include lobular and portal inflammation, hepatocyte ballooning and apoptosis, increased AST levels, collagen deposition, and hepatic fibrosis [6]. This process also involves hepatocyte lipotoxicity, increased oxidative stress from mitochondrial _-oxidation, inflammatory cytokine release, and immune cell and hepatic stellate cell (HSC) activation. A classic two-hit model has been proposed to explain NAFLD pathogenesis by Day and James in 1998 [14]. The first hit refers to hepatic TG accumulation (steatosis) that increases liver sensitivity to a variety of second hits, such as inflammatory cytokines and oxidative stress that cause hepatic injury, necroinflammation, and fibrosis. However, this hypothesis has been challenged because some steatosis patients develop NASH without implicating secondhit but other external injury. A number of in vitro and in vivo epidemiological studies strongly suggest that obesity and hepatic insulin resistance are critical pathogenic factors in NAFLD. Dysfunctions in these signaling and regulation mechanisms impair hepatocellular functions and predispose patients to NAFLD pathogenesis. Although the precisemechanisms for NAFLD development and progression remain incompletely understood, NAFLD is currently recognized as a consequence of a multihit hypothesis, involving obesity, insulin resistance, oxidative stress, and proinflammatory processes [15]. 2.1 Obesity and adipokines Obesity is defined as excessive fat accumulation in adipose tissue and it is associated with numerous metabolic diseases such as cardiovascular disease, type 2 diabetes, and NAFLD [10,16]. Several population studies demonstrated that the prevalence of simple steatosis,NASH, andNAFLD was increased in overweight and obese individuals [10,17–20]. High NASH and NAFLD prevalence also occurred in severely and morbidly obese individuals, indicating that its incidence is highly associated with the degree of obesity [21–23]. Moreover, NAFLD was also found in obese insulin-resistant children and those with elevated serum ALT levels [24, 25]. As caloric intake increases, adipocytes store energy in the form of TGs that result in enhanced adipogenesis, increased adipose tissue mass, and consequently obesity [26]. Insulin inhibits adipose tissue lipolysis by lowering cyclic adenosine monophosphate levels and activating phosphodiesterase 3b, thereby attenuating protein kinase A activity and decreasing protein kinase A dependent hormone-sensitive lipase (HSL) activation, or through dephosphorylation of HSL at regulatory and basal phosphorylation sites by protein phosphatase [27, 28]. FFAs released from adipose tissue by enhanced TG hydrolysis via insulin resistance-mediated HSL increases resulted in elevated plasma FFAs. Subsequently, these FFAs are transported to the liver via the hepatic artery and portal vein and thus increase hepatic FFA influx. Increased FFAs delivered to the liver from visceral adipose tissue induce hepatic insulin resistance via reduced hepatic insulin clearance and increased circulating insulin levels, which decreases insulinstimulated glucose uptake through IRS-1-associated (where IRS-1 is insulin receptor substrate 1) phosphatidylinositol3 kinase (PI3K) signaling impairment and reduced insulinmediated hepatic glucose output suppression and endogenous glucose production [29–31]. FFAs also stimulate hepatic gluconeogenesis and TG synthesis and directly cause hepatic lipotoxicity, which promote NAFLD pathogenesis. One study demonstrated the contribution of the FFA source in NAFLD patients using multiple stable isotope techniques. Of the hepatic TG, 59% was from serum nonesterified FAs, 26% was derived from hepatic de novo lipogenesis, and only 15% was from dietary sources [32]. This study suggested C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 3 that although increased FFAs from other pathways also account for hepatic lipid accumulation, elevated FAs from peripherally expanded adipose tissue and de novo lipogenesis are the major sources of hepatic and lipoprotein fat accumulation in NAFLD. Fat distribution may also be more important than total adipose tissuemass in obesity [33]. Visceral adipose tissue is strongly associated with metabolic complications including hepatic steatosis [34, 35] and contributes to hepatic inflammation and fibrosis inNAFLD patients [36]. Compared with subcutaneous adipose tissue, visceral adipose tissue increases insulin resistance, metabolic activity, lipogenesis, and lipolysis [37, 38]. Deregulated adipokine secretion from adipose tissue is another mechanism by which obesity is involved in NAFLD via impaired insulin signaling and proinflammatory properties. Most adipokines are increased in obese adipose tissue, and some of these have been linked to insulin resistance, such as tumor necrosis factor-_ (TNF-_), leptin, and IL-6 [39]. Visceral adipose tissue-generated adipokines directly transported to the liver and cause harmful effects on hepatocytes. Obesity is significantly associated with chronic low-grade inflammation and insulin resistance, which is first evidenced by TNF-_ release from adipocytes [40]; in addition, using TNF-_ neutralizing antibodies improves insulin resistance. TNF-_ knockout mice or its receptor have ameliorated insulin resistance in both diet-induced obese and leptin-deficient (ob/ob) mice that develop severe type 2 diabetes and hypercholesterolemia [41]. Clinical studies demonstrated that both serum and hepatic TNF-_ is increased in NASH patients compared with simple steatosis patients, and the increased TNF-_ levels are correlatedwith the severity of histological changes [42–44]. Inhibition of endogenous TNF-_ production improved steatosis, steatohepatitis, and insulin resistance in NASH patients as well as both HFD-fed (where HFD is high-fat diet) and leptin-deficient (ob/ob) mice [45–47]. These studies reveal the role of TNF-_ in insulin signaling impairment in NAFLD. At the molecular level, increased TNF-_ is observed by FFA treatment of mouse hepatocytes, which downregulates insulin receptor phosphorylation and thus blocks insulin signaling [48, 49]. FFA exposure also caused hepatocyte lipotoxicity by promoting TNF-_ production and, thus, I_B kinase _/nuclear factor-_B (NF-_B) activation, resulting in abundant proinflammatory cytokine expression [48]. TNF-_ also stimulated hepatic FA synthesis and increased serum TGlevels [50]. IL-6 is another proinflammatory cytokine that is produced from visceral adipose tissue and with systemic effects on the immune response. Increased IL-6 is found in obese patients, is decreased by weight loss [51, 52], and is a predictor of insulin resistance [53]. Secreted IL-6 levels from abdominal adipose tissue are higher than that from subcutaneous adipose tissue [54]. In severely obese patients, IL-6 and TNF-_ mRNA expression in both subcutaneous and visceral adipose tissue ismore than 100-fold greater than liver tissue [51]. Visceral adipose tissue-derived IL-6 enters the liver; therefore, the liver might be a major IL-6 target organ. Several studies suggested that IL-6 caused hepatic insulin resistance by inhibiting receptor autophosphorylation, IRS-1 phosphorylation, and downstream PI3K/Akt signaling via suppressor of cytokine signaling (SOCS) 3 activation [51, 55]. An HFD animal study also demonstrated that adipose tissue-derived IL-6 increased hepatic SOCS3 expression followed by reduced insulin-stimulated AKT activation and consequent hepatic insulin resistance [56]. Leptin is mainly produced by adipocytes and is an important adipokine in the regulation of energy expenditure, food intake, energy balance, and the immune system [57, 58]. Increased serum leptin levels are found in NAFLD patients [59, 60] and are correlated with hepatic steatosis severity, but not with inflammation or fibrosis [61, 62]. Comparatively, a 6-month follow-up study revealed no association between serum leptin levels and NAFLD severity [63]. Leptin-deficient (ob/ob) mice develop obesity, fatty liver, and insulin resistance [64]. Although there is no correction of leptin and hepatic fibrosis in human studies, much evidence suggests that leptin is a fibrogenic factor that is implicated in hepatic fibrosis. Leptin is essential for HSC activation and transforming growth factor _1 (TGF-_1) production that promotes collagen synthesis and is involved in hepatic fibrosis [65,66]. However, an in vitro study demonstrated that the fibrogenic effect of leptin andHSC activation is because of interactions with hepatic Kupffer cells but not direct effects on HSC [67]. Adiponectin is an anti-inflammatory adipokine that is also produced by adipocytes and is suggested to inhibit NAFLD [43, 68]. Decreased adiponectin is observed in NASH patients compared with simple steatosis and is associated with more extensive hepatic necroinflammation [43]. Increased levels of TNF-_ and IL-6, two major proinflammatory adipokines, inhibit adiponectin expression [69]. Adiponectin inhibits NAFLD and exerts a hepatoprotective effect through multiple mechanisms, including suppression of steatosis, fibrosis, inflammation, lipotoxicity, and an increase in insulin sensitivity and as has been described in recent reviews [70, 71]. Recombinant adiponectin significantly attenuated hepatomegaly, steatosis, and hepatic inflammation in leptin-deficient (ob/ob) mice and cultured hepatocytes via enhancement of hepatic FA oxidation and reduced FA synthesis as well as TNF-_ production [72, 73]. 2.2 Insulin resistance Insulin signaling is essential for carbohydrate and lipid metabolism in various organs and tissues and is crucial for homeostatic regulation of blood glucose levels by the liver [74]. In physiological states, pancreatic _ cells secrete insulin in response to increased blood glucose levels after feeding. Insulin stimulates hepatic glucose uptake and conversion of glucose into glycogen, TG synthesis, and export to adipose tissue as VLDL. This biological function of insulin is triggered by insulin binding to the insulin receptor and activation of the intracellular signaling cascade. Once bound, the insulin receptor _-subunit with tyrosine kinase C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 4 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 activity phosphorylates IRS family members, further activating various signaling pathways that are involved in the metabolic effects of insulin [75], which involves downstream targeted insulin/IRS and PI3K/Akt pathway activation. PI3Kmediated Akt activation is important for glucose transport, glycogen and protein synthesis as well as hepatic gluconeogenesis suppression. Akt-dependent forkhead box protein O (FOXO) transcription factor phosphorylation results in their exclusion from the nucleus to the cytoplasm, thus blocking the DNA binding ability of FOXOs and subsequent downstream gluconeogenic gene transcription including genes, such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase) [75, 76]. Akt also promotes translocation of glucose transporter 4 to cell membranes together with the Cbl/Tc10 pathway that facilitates glucose uptake [77] and subsequently reduces hepatic gluconeogenesis and blood glucose levels. In addition to regulating hepatic glucose metabolism, insulin signaling stimulates hepatic de novo lipogenesis through sterol regulatory element binding protein 1 (SREBP-1) transcription factor activation that upregulates acetyl-coenzyme A carboxylase (ACC) and FA synthase (FAS) [78]. Insulin resistance is a physiological condition that is described as decreased target cell sensitivity to the normal insulin concentrations and, hence, insulin-mediated uptake and glucose utilization in insulin-sensitive organs and tissue including the liver, adipose, and muscle tissue. Insulin resistance has been linked to metabolic syndrome and is a major cause of type 2 diabetes because of pancreatic _ cell dysfunction [79]. Extensive research also has highlighted the implication of insulin resistance in NAFLD. Increased NAFLD prevalence is found in patients with impaired glucose tolerance or diagnosed diabetes [10, 25, 80]. A study of type 2 diabetic patients demonstrated thatNAFLD is extremely common in type 2 diabetes patients with 69.5% prevalence [81]. A population-based matched retrospective cohort study also demonstrated that a higher risk of advanced liver disease is found in newly diagnosed diabetic individuals compared with those who do not have type 2 diabetes [82]. Moreover, insulin resistance is associated with the degree of NAFLD, advanced fibrosis, and mortality [83, 84]. The mechanism underlying hepatic insulin resistance is poorly understood, but may involve FFA overflow, proinflammatory adipokines, hyperinsulinemia, hyperglycemia, and adipose tissue insulin resistance. Insulin signaling inactivates adipose tissue HSL to suppress lipolysis, whereas adipose tissue insulin resistance results in increased hepatic FFA influx and further lipid accumulation [27, 28]. It has been demonstrated that in HSL knockout mice, hepatic insulin sensitivity is increased by reducing TG concentrations [85]. Increased FFAs also contribute to hepatic insulin resistance as described above (Section 2.1). FFAs and proinflammatory adipokines, such as IL-6 and TNF-_, from adipose tissue impaired hepatic insulin signaling via c-Jun N-terminal kinase (JNK), I_B kinase/NF-_B, and SOCS protein activation, resulting in IRS inactivation or degradation [86, 87]. High glucose-induced hyperinsulinemia in blood caused hepatic SREBP-1 activation and promoted hepatic de novo lipogenesis [88]. This hyperinsulinemia also inhibited FFA oxidation by upregulating malonyl-CoA levels, resulting in carnitine palmitoyl transferase 1 (CPT-1) inhibition, thus decreasing FA shuttling intomitochondria and reduced hepatic lipid utilization [89, 90]. The pathogenic role of insulin resistance in NAFLD is complicated by the involvement of multiple factors and molecules between organs that amplify deregulated signaling cascades and thus alter hepatic gene expression as well as glucose and lipid metabolism. 2.3 Lipotoxicity and lipoapoptosis Accumulation of FFAs and their metabolites causes cell damage and death known as lipotoxicity and lipoapoptosis. Indeed, apoptosis is a prominent feature of NASH that correlates with histological hepatic inflammation and fibrosis [91]. As mentioned before, hepatic TG accumulation per se does not directly cause hepatotoxicity, whereas FFAs and a wide range of lipid metabolites potently cause lipotoxicity and lipoapoptosis. FFAs cause lipotoxic effects through various mechanisms, including lysosomal destabilization, mitochondrial pathways, death receptor signaling, and ER stress. FFA exposure in mouse hepatocytes causes Bax translocation to lysosomes and lysosomal destabilization with lysosomal cysteine protease (cathepsin B) release that contributes to loss of lysosomal integrity. Downregulating Bax and inhibiting lysosomal permeabilization reduced FFA-induced toxicity [92]. FFA treatment decreased Bcl-xL, which is an anti-apoptotic BCl-2 family protein, while overexpression blocked lysosomal permeabilization and apoptosis [92]. FFAs also caused NF_B-dependent TNF-_ expression that may further promote insulin resistance and hepatic lipogenesis [48, 93]. A recent study found that saturated FAs (SFAs) induced cytotoxicity and apoptosis both in human and mouse hepatocytes, but MUFAs only resulted in lipid accumulation [94]. SFAs such as palmitate and stearic acid trigger hepatic lipoapoptosis via Bim activation, which is a BH3 domain-only protein that further binds to Bax and triggers mitochondrial apoptosis pathways [95]. Pharmacological and genetic JNK inhibition or Bim knockdown by siRNA both attenuated SFA-induced cell death [95]. A further study demonstrated that FFAs mediated protein phosphatase 2A-dependent FOXO3a dephosphorylation that in turn stimulated FoxO3a-dependent Bim expression and hepatocyte apoptosis [96]. Elevated Fas receptor (CD95) expression occurred in liver specimens from NASH patients and correlated with disease severity [97]. In carbohydrate-fed mice, hepatocyte Fas expression was increased and accompanied by hepatic steatosis [98, 99]. Fas agonist administration increased hepatocyte apoptosis and liver injury in diet-induced obese mice [98]. Upregulated Fas expression was noted in FFA-treated HepG2 cells and increased the sensitivity to Fas agonist-induced apoptosis [98]. FFA treatment of hepatocytes caused JNK-dependent TNF-related C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 5 apoptosis-inducing ligand receptor death receptor 5 (DR5) upregulation and sensitization to TNF-related apoptosisinducing ligand mediated apoptosis [100]. Increased DR4 and DR5 expression was also demonstrated in livers from human NASH patients [100,101]. A recent study suggested that FFAinducedDR5 expression was transcribed by C/EBP homology protein (CHOP), an ER stress-mediated transcription factor. This study also suggested that FFAs induced lipoapoptosis by promoting DR5 clustering and lipid raft redistribution within the plasma membrane [102]. These studies indicated that DR upregulation during steatosis increased hepatocyte susceptibility to apoptosis through other mechanisms. Cellular lipid accumulation also induces ER stress. Hepatocyte FFA exposure disrupts ER homeostasis, induces ER stress, and promotes apoptosis [103]. Mice fed a high saturated fat diet had hepatic steatosis and increased ER stress via spliced X-box binding protein 1, upregulated glucose-regulated protein 78 levels and increased hepatic apoptosis [104]. Eukaryotic initiation factor-2_ phosphorylation and increased levels of both glucose-regulated protein 78 and X-box binding protein 1 were observed in NAFLD and diet-induced NASH patients [105,106]. Upregulation of these signalingmolecules is linked to lipid accumulation, insulin resistance, and hepatic inflammation via JNK, NF-_B, and reactive oxygen species (ROS) production [107]. 2.4 Oxidative stress Oxidative stress is a redox imbalance that results from excessive ROS or free radicals and decreased antioxidant defense. Elevated free radicals, increased DNA damage, and lipid peroxidation as well as reduced antioxidants have been observed in NAFLD patients [108, 109]. In addition to the direct hepatotoxic effect, FFA overload induces ROS production via mitochondrial dependent _-oxidation or microsomal enzymes. Increased hepatic microsomal FA oxidizing enzyme cytochrome P450 (CYP) 2E1 was found in mice with diet-induced hepatic steatosis and NASH patients and is considered to be a source of ROS [110, 111]. Mitochondrial _-oxidation is regulated by CPT-1, while FFAs induce peroxisome proliferator-activated receptor (PPAR) _ to up-regulate CPT-1 expression [112]. ROS overproduction causes an attack on DNA, protein, and cellular membranes, which induces lipid peroxidation and results in mitochondrial dysfunction that contributes to hepatocellular damage. Mitochondrial abnormalities include ultrastructural lesions, mitochondrial DNA depletion, impaired ATP synthesis, and decreased respiratory chain complex activity are associated with NAFLD and might result in further ROS production [113, 114]. TNF_ is also involved in mitochondrial dysfunction through induction of mitochondrial swelling and interference among mitochondrial respiratory chain complexes [115, 116]. PUFA peroxidation induced apolipoprotein B100 degradation, a critical protein component of VLDL; thus, decreased VLDL secretion may be relevant to hepatic lipid accumulation [117]. Moreover, Cu/Zn superoxide dismutase (SOD), glutathione (GSH) peroxidase (GPx), and catalase (CAT) activities are increased in NAFLD patients compared with simple steatosis, reflecting the state of oxidative stress [118]. 2.5 Hepatic inflammation NASH is an extreme form of NAFLD and has gotten more attention recently because it can progress to fibrosis and cirrhosis. NASH is characterized by steatosis with mixed lobular inflammation and hepatocyte ballooning with or without fibrosis [2,119]. Other features are also included such as portal inflammation or panacinar steatosis, which are associated with advanced liver fibrosis [119, 120]. Hepatic inflammation is a complicated condition that is caused by various factors, including FFAs, cytokines, adipokines, and oxidative stress that results in hepatocellular injury, further inflammatory cell recruitment, which releases various oxidants and proinflammatory molecules, andHSC activation, which is involved in hepatic fibrosis [2]. Themajor source of hepatic TNF-_ and IL-6 is derived from injured hepatocytes, immune cells, and activated Kupffer cells. Increased TNF-_ activates JNK signaling and results in hepatocyte apoptosis [121]. Studies have revealed that NF-_B and JNK activation are essential for inflammatory cell recruitment in NASH [122, 123]. A recent study demonstrated that hepatocytes release danger signals leading to activation of mononuclear cells and production of IL-1_ and TNF-_ after FFA exposure [124]. The role of IL-6 in liver pathology is very complicated because it is considered to have hepatoprotective effect and promote liver regeneration. Although the way IL-6 participates in NAFLD is still unclear, studies have suggested a positive correlation between hepatic IL-6 and degree of disease. Increased IL-6 expression in hepatocytes is found in patients with NASH and correlated to degree of inflammation, stage of fibrosis, and systemic insulin resistance [125]. Blockade of IL-6 signaling by neutralizing antibody against the IL-6 receptor (MR16–1) enhanced hepatic steatosis but improved hepatic injury in mice fed with an methionine choline-deficient diet [126]. Overproduction of cytokines, such as TGF-_ by Kupffer cells, infiltrating inflammatory cells, and fibroblasts triggers HSC activation and differentiation into myofibroblast-like cells, promotes collagen synthesis and blocks extracellular matrix degradation by enhancing tissue inhibitors of matrix metalloprotease expression [127]. Furthermore, adipose tissue-generated leptin increased the collagen I and III production inHSCs via a JAK- and PI3K-mediated pathway [128]. In rats with diet-induced steatohepatitis, hepatocytes are the source of lipid peroxidation at early stages followed by hepatocellular injury, further inflammatory cell recruitment, and HSC activation [129]. Activation of Kupffer cells and other inflammatory cells also generates ROS through NADPH oxidase [130, 131]. Although oxidative stress may not initiate hepatic inflammation, ROS overproduction could cause hepatocyte damage or death and, in turn, cytokine release that C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 6 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 provides positive feedback on inflammatory signaling and promotes NASH pathogenesis. 3 Molecular mechanism of dietary natural compounds used to treat NAFLD Development and progression of NAFLD andNASHis amultifactorial process. Despite understanding the process and mechanism, there is no established treatment or therapy for NAFLD. Epidemiological studies suggested a combination of lifestyle interventions, such as decreased caloric intake, altered dietary composition, weight loss and physical exercise, is safe and effective for improving obesity-mediated insulin resistance and NAFLD [132]. Current pharmaceutical drugs, such as insulin-sensitizers, thiazolidinediones, statins, antioxidants, and Omega-3 PUFAs, which targeting the mechanisms involved in metabolic syndrome have been evaluated in animal and clinical studies [132,133]. However, some of these therapeutic agents tested in patients with limited findings and inconsistent outcomes, and because of short durations and randomized trails, some of these have safety concerns [134, 135]. Evidence has supported the concept that NAFLD is associated with diet-associated obesity and insulin resistance. This evidence also offers novel targets for NAFLD intervention and treatment by dietary and nutritional components. Currently, researchers have become increasingly interested in searching for natural products from dietary and herbal plants that can both prevent and control NAFLD via a chemopreventive strategy. Many dietary natural compounds isolated from fruits, vegetables, and edible plants reportedly possess health-promoting properties, such as anti-inflammation, antioxidation, antiobesity, and increased insulin sensitivity. Furthermore, in vivo and in vitro studies demonstrated thatmany herbal plants have been used for management of fatty liver conditions and improvement of NAFLD by their hypoglycemic, antihyperlipidemic, and hepatoprotective effect, and without major side effect [133]. Convincing scientific evidence in animal and human studies displays the potential of these dietary natural compounds for NAFLD treatment.Understanding the regulatory role and mechanism of these dietary natural compounds may help to prevent and treat NAFLD. The chemopreventive effects and molecular targets of selected dietary natural compounds in NAFLD are highlighted below (Table 1). 3.1 Carotenoids Carotenoids are fat-soluble pigments and potent antioxidants that are rich in many plants, fruits, and flowers. Lycopene is a member of the carotenoid family, with a highly unsaturated 40-carbon molecule that contains 11 conjugated and two unconjugated double bonds. The main sources of lycopene are tomato, watermelon, papaya, and orange grapefruit. Rats administered 2 and 4 mg/kg lycopene for 6 wk displayed reduced serum ALT and AST activities and TG and cholesterol levels. Hepatic steatosis and inflammation were also reduced, followed by decreased lipid peroxidation and increased GSH as well as lowered serum TNF-_ [136]. In a NASH-promoted hepatocarcinogenesis animal study, dietary lycopene reduced HFD- and diethylnitrosamine-induced hepatic precancerous lesions. This inhibitory effect is associated with decreased hepatic lipid peroxidation, NF-_B levels, and upregulation of both nuclear NF-E2-related factor 2 (Nrf2) and its target gene heme oxygenase 1. Increased lycopene levels are found in HFD-fed but not control diet-fed rat livers, indicating that lycopene is incorporated into micelles along with dietary fat and that it has efficacy in target organs [137]. Dietary lycopene feeding in gerbils also restored HFD-induced decreased liver antioxidant enzyme defenses, including CAT, GSH reductase, and GSH S-transferase (GST) activities [138]. These studies suggested that lycopene has potent antioxidative activity by alleviating oxidative stress and upregulating antioxidants to prevent HFD-induced NAFLD. In addition to the antioxidative property, a recent study demonstrated that lycopene decreased HFD-induced hepatic steatosis and FFAinduced lipid accumulation in hepatocytes viamicroRNA-21dependent FA-binding protein 7 inhibition [139]. _-Carotene is one of the most abundant carotenoids and antioxidants in vegetables and fruits. _-Carotene consumption is distributed in a variety of tissues including liver and adipose tissues [140]. 3T3-L1 adipocytes treated with _-carotene have reduced TNF_-mediated ROS production and restored adiponectin and glucose transporter 4 production thatmay improve insulin resistance [141]. _-carotene supplementation decreased retinol deficiency-induced hepatic lipid peroxidation and enhanced CAT and GST activities, thus reducing hepatic oxidative stress [142]. Similar to lycopene, _-carotene acts as an antioxidant in liver or modulates adipokine production from adipose tissue to display indirect effects against NAFLD. Lutein is also a common carotenoid that is found in most fruits and vegetables that reduces hepatic free cholesterol, lipid peroxidation, and TNF-_ because of decreased NF-_B p65 DNA binding activity in high cholesterol diet-fed Hartley guinea pigs [143]. Fucoxanthin is a carotenoid from edible brown algae that is characterized by its unique structure including an allenic bond and 5, 6-monoepoxide that differs from common carotenoids and has exhibited antiobesity and antidiabetic effects [144, 145]. Mice fed a fucoxanthinsupplemented HFD had reduced proinflammatory cytokine levels, including leptin, TNF-_, monocyte chemotactic protein 1 (MCP-1), and IL-6, and increased adiponectin in plasma and adipose tissue [146, 147]. Fucoxanthin also decreased hepatic TG and cholesterol levels by suppressing activities of malic enzyme, FAS, glucose-6-phosphate dehydrogenase, 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, and acyl coenzyme A: cholesterol acyltransferase, thus decreasing hepatic lipogenesis and increasing _-oxidation [146]. Fucoxanthin reduces hyperglycemia and hyperinsulinemia both in HFD-fed C57BL/6N mice and diabetic/obese KK-A(y) mice [146, 147]. Fucoxanthin also decreased hepatic C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 7 Table 1. Chemopreventive activities and mechanisms of dietary natural compounds on NAFLD Class Compound Structure Dietary source Molecular mechanisms and targets References Upregulation Downregulation Carotenoids Lycopene Tomatoes, watermelon, papaya and orange _ Antioxidants (GSH, CAT, GR, and GST; SD rats, gerbils) _ Nrf2-dependent HO-1 (SD rats) _ Hepatic inflammation (SD rats) _ Lipid peroxidation (SD rats) _ FABP7 (C57BL6/J mice) [136–139] _-Carotene Red palm oil, pumpkin, and leafy green vegetables _ Adiponectin (3T3-L1 cells) _ Glut4 (3T3-L1 cells) _ Antioxidants (CAT and GST; RD rats) _ Lipid peroxidation (RD rats) _ ROS production (3T3-L1 cells) [140–142] Lutein Spinach and kale - _ Lipid peroxidation (Guinea pigs) _ NF-_B-mediated TNF-_ (Guinea pigs) [143] Fucoxanthin Brown algae _ Plasma adiponectin (C57BL6/N mice) _ Proinflammatory adipokines (C57BL6/N mice, KK-A(y) mice) _ Hepatic lipogenesis (FAS, G6PD, and HMG-CoA reductase) (C57BL6/N mice) _ SCD-1 (KK-A(y) mice) [146–148] Omega-3 PUFAs EPA Fish oils and golden algae oil _ Plasma adiponectin (Balb/cA mice) _ CPT-1 (Medaka) _ AMPK_ and PPAR_ (Pten-deficient mice) _ Hepatocytes necrosis (Balb/cA mice) _ Inflammatory cells infiltration (Balb/cA mice) _ Oxidative stress (Balb/cA mice) _ SREBP-1 transcription factor (Medaka) _ Lipogenic and fibrogenic genes (HepG2 cells, Medaka, Wistar rats) [156–159, 161–163] DHA _ SOD activity (Balb/c mice) _ Plasma adiponectin (C57BL6/N mice) _ Genes involved in lipolysis and _-oxidation (Kunming mice) _ Insulin sensitivity (C57BL6/N mice) _ Inflammation and fibrosis (Ldlr(−/−), ob/ob mice) _ Hepatic lipogenesis (FAS and SREBP-1; ob/ob, Kunming mice) _ Kupffer cells activation (Ldlr (−/−) mice) _ IL-1_ and TNF-_ (Ldlr (−/−) mice) [169–176] C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 8 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 Table 1. Continued Class Compound Structure Dietary source Molecular mechanisms and targets References Upregulation Downregulation Flavones Apigenin Parsley and celery _ AMPK signaling in adipocytes (3T3-L1 cells) _ HSL and lipolysis in adipocytes (3T3-L1 cells) [178] Luteolin Parsley and celery _ CPT-1 (HepG2 cells) _ AMPK-dependent ACC phosphorylation (HepG2 cells) _ SREBP-1 and FAS (HepG2 cells) [179] Nobiletin Citrus peels _ Adiponectin secretion (ST-13 preadipocytes, 3T3-L1 cells) _ Insulin sensitivity (ob/ob, C57BL/6J mice) _ Proinflammatory adipokines (ob/ob, C57BL/6J mice) [180–183] Baicalein Baical Skullcap _ FA oxidation (C57BL/6J mice) _ AMPK_ and PPAR_ (C57BL/6J mice) _ Hepatic inflammation (C57BL/6J mice) _ SREBP-1-dependent lipogenic genes (C57BL/6J mice) [184] Flavonols Quercetin Onion and broccoli and GPx) (C57BL/6, db/db mice) _ Nrf2-targeted genes (Wistar rats) _ _-oxidation (C57BL/6J mice) _ Glucose uptake (HepG2 cells) _ Proinflammatory cytokines (C57BL/6 mice, HepG2 cells) _ Antioxidants (SOD, CAT, _ Lipogenic and fibrogenic genes (C57BL/6J, C57BL/6 mice) _ NF-_B and JNK signaling (Wistar rats, C57BL/6 mice) _ Lipoperoxidation and DNA damage (C57BL/6 mice) [185–192] Kaempferol Broccoli and tea - _ ROS production (Chang liver cells) [193] Flavanols (catechins) Epigallocatechin3-gallate (EGCG) Tea _ PI3K/Akt signaling _ FA oxidation (UCP-2, SCD-1; New Zealand black mice) _ Dietary lipid oxidation (New Zealand black mice) _ Oxidative stress (SD rats) _ Hepatic inflammation, necrosis and fibrosis (TGF/SMAD signaling; SD rats) _ Plasma adipokines (C57BL/6 mice) C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 9 Table 1. Continued Class Compound Structure Dietary source Molecular mechanisms and targets References Upregulation Downregulation _ Inflammatory molecules (iNOS, COX-2, and TNF-_; SD rats) _ FOXO1 and NF-_B (SD rats) _ Dietary lipids incorporate to liver (C57BL/6 mice) [194–199] Theaflavin (TF-1) Black tea _ AMPK signaling (HepG2 cells) _ FA oxidation (HepG2 cells) _ ACC activity (HepG2 cells) _ Oxidative stress and hepatocytes apoptosis (I/R injury mice) _ Inflammatory cytokines (I/R injury mice) [200, 201] Flavanones Naringenin Citrus _ FA oxidation (CYP450 IVA1, PPAR_, and PGC-1_-targeted aCPT-1, ACO; ICR, Ldlr (−/−) mice) _ Insulin sensitivity (Ldlr (−/−) mice) _ VLDL-TG and VLDL-apoB secretion (Ldlr (−/−) mice) _ Lipogenic genes (Ldlr (−/−) mice) [202–204] Hesperetin Citrus - _ PAP activity and TG synthesis (SD rats) [205] Anthocyanidins Cyanidin-3-O-_glucoside Cherries and strawberries _ GSH synthesis (HepG2 cells, db/db mice) _ AMPK signaling (HepG2 cells) _ FA oxidation (CPT-1) (HepG2 cells) _ TG synthesis (mtGPAT1; HepG2 cells, KK-A(y) mice) _ Neutrophil infiltration (db/db mice) _ Plasma adipokines (db/db mice) _ ROS production (HepG2 cells, db/db mice) _ JNK signaling (db/db mice) _ ACC activity (HepG2 cells) [207–210] Isoflavones Daidzein Soybean _ Antioxidants (SOD-2 and GST; Wistar rats) _ De novo lipogenesis (C57BL/6J mice) _ SCD-1(Wistar rats, C57BL/6J mice) [211, 212] C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 10 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 Table 1. Continued Class Compound Structure Dietary source Molecular mechanisms and targets References Upregulation Downregulation _ Blood insulin and adipokines (C57BL/6J mice) Genistein _ FA oxidation (PGC-1, PPAR_ and CPT-1; HepG2 cells, C57BL/6J mice) _ Anti-oxidants (GPx, GR, and GSH; Wistar rats) _ IRS-1/PI3K/Akt signaling and Glut1 (HepG2 cells) _ Glucokinase activity (db/db mice) _ Hepatic inflammation and apoptosis (C57BL/6J mice) _ SREBP-1 and lipogenic genes (SD rats) _ Proinflammatory cytokines (C57BL/6J mice, SD rats) _ JNK and NF-_B signaling (SD rats, HepG2 cells) _ PEPCK and G6Pase (db/db mice) [213–225] Other phenolic compounds Resveratrol Grapes, red wine _ FA oxidation (CPT-1 and ACO; Zucker, SD rats) _ Nrf2-targeted genes and antioxidants (SD rats) _ UCP-2 (Wistar rats) _ IRS-1/PI3K/Akt signaling and insulin sensitivity (C57BL/6J, KK-A(y) mice) _ SIRT1 activity and AMPK signaling (HepG2 cells, SD rats) _ FOXO deacetylation (HepG2 cells) _ Lipogenic genes (SREBP-1, FAS, ACC, G6PD, and HMG-CoA reductase; HepG2 cells, C57BL/6J mice, hamsters) _ Lipid peroxidation and oxidative stress (Wistar rats) _ Proinflammatory cytokines (Wistar rats) _ ER stress (HepG2 cells) [226–229, 231–240] Curcumin Turmeric _ Mitochondria biogenesis and function (ob/ob mice, New Zealand rabbits, primary hepatocytes) _ Adiponectin of adipocytes (ob/ob mice) _ Insulin sensitivity (Akt signaling; HSCs) _ GCL activity (HSCs) _ AMPK signaling (HSCs) _ SREBP-1 and HMG-CoA reductase (ob/ob mice, SD rats) _ ROS production (New Zealand rabbits, primary hepatocytes, AML-12 hepatocytes) _ Inflammatory cells infiltration (NMRI mice) _ HSC activation and fibrogenic genes expression (HSCs) [242–255] C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 11 Table 1. Continued Class Compound Structure Dietary source Molecular mechanisms and targets References Upregulation Downregulation _ Proinflammatory cytokines (ob/ob, NMRI mice, SD rats) _ JNK, NF-_B, and SOCS3 (primary hepatocytes, ob/ob, NMRI mice) _ LDLR and LOX-1 (HSCs) [242–255] FABP7, FA-binding protein 7; G6PD, glucose-6-phosphate dehydrogenase; GCL, glutamate-cysteine ligase GR, GSH reductase; ICR, institute for cancer research; iNOS, inducible nitric oxide synthase; RD, retinol deficiency. stearoyl-CoA desaturase 1 (SCD-1) expression,which is an enzyme that catalyzes MUFA biosynthesis from SFAs through leptin signaling regulation that suppresses 18:0 desaturation into 18:1n-9 in the liver [148]. 3.2 Omega-3 PUFAs Growing evidence clearly demonstrates that an increased intake of marine omega-3 PUFAs, such as eicosapentaenoic acid (20:5 n-3, EPA) and docosahexaenoic acid (22:6 N-3, DHA), is beneficial to diverse physiological functions and human health, including the improvement of metabolic syndrome. Omega-3 PUFA supplementation has been shown to ameliorate hepatic steatosis in human studies and in different animal models [149–152]. Basically, the beneficial function of omega-3 PUFAs to NAFLD most likely is contributed by their incorporation into plasma phospholipids that modulate membrane fluidity and intracellular signaling or that alter the lipid composition of the liver [153]. In patients who have been diagnosed with a NASH, daily dietary intake of 2700 mg of EPA for 12 months resulted in decreasing the serum ALT, FFA, and thioredoxin levels, which contribute to hepatic oxidative stress. Among the 23 patients, seven of them showed improvement in hepatic steatosis and fibrosis. Decreased hepatocyte ballooning and lobular inflammation were found in six patients. The most safety concern of EPA is bleeding tendency, whereas there is no adverse event that has occurred during EPA treatment in all patients of this study [154]. A cross-sectional observational study showed the positive effect of dietary EPA in Japanese men with NAFLD but not in women that with unidentified reason [155]. In HFD-fed mice, dietary intake of EPA is effective in reducing fatty droplets by decreasing the hepatic cholesterol, TG, and nonesterified FAs [156, 157]. When mice were fed high sucrose/HFD supplemented with EPA, reduced D-galactosamine-induced hepatic injury occurred, as evidenced by a decreased hepatocyte necrosis and inflammatory cell infiltration. This result was also accompanied by lowered hepatic TG levels via the reduction of FAS and SCD-1 gene expression, decreased ROS production, and increased plasma adiponectin [158]. These effects could contribute to suppressing the progression of hepatitis. Moreover, EPA intake is found to abrogate HFD-induced SREBP-1, FAS, and ACC1 mRNAs, while increasing the CPT-1 expression; thus, it could decrease FA synthesis and promote mitochondrial _-oxidation [159]. A diet that is deficient in methyl groups, such as methionine and choline, is another animal model of NAFLD that is caused by impairedmitochondrial _oxidation [160]. Several studies have demonstrated that EPA decreased hepatic steatosis and the progression of fibrosis by suppressing TG synthesis and the expression of fibrogenic genes, such as TGF-_1, _-smoothmuscle actin (_-SMA), and collagen [161, 162]. In Pten-deficient (liver-specific deletion of phosphatase and tensin homolog) mice, an NAFLD animal model characterized by increased hepatic lipogenesis, C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 12 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 EPA improved steatohepatitis by reducing steatosis, lobular inflammation, and hepatocytes ballooning. Additionally, supplementation with dietary EPA for 76 wk reduced the amount of hepatocellular carcinoma. The suppressed effect of EPA is due to a decrease in hepatic ROS production and upregulation of AMP-activated protein kinase (AMPK) _ and PPAR_ expression, which may repress the expression of lipogenic genes. Analyzing the lipid composition of the liver showed that EPA causes a dramatic change in the content of arachidonic acid and that EPAserves as the anti-inflammatory mechanism that acts as an inactive lipid mediator compared to arachidonic acid [163]. A recent study revealed that EPA and the oxidized form suppressed the liver X receptor agonistinduced TG synthesis in HepG2 cells by the downregulation of SREBP-1, a transcription factor that is involved in the expression of lipogenic genes, including FAS, ACC, and SCD1 [164]. Two controlled trial studies showed that children with NAFLD supplemented with 250 and 500 mg DHA per day for 6–24 months had improved liver steatosis and insulin sensitivity accompanied by lowered TG and ALT levels [165, 166]. Moreover, no adverse effect was found in 60 children with biopsy-proven NAFLD when supplemented with DHA supplementation at dosage of 250 and 500 mg for 6 months [161]. In animal models of diet-induced NAFLD, which included a high carbohydrate diet, HFD, and choline-deficient diet, the dietary intake of DHA reduced hepatic steatosis, inflammation, and fibrosis and decreased hepatic and serum total cholesterol levels and lipid peroxidation [167–169]. Increased SOD activity and decreased activity of SREBP-1 account for the inhibitory effect of DHA [167, 168]. Furthermore, alteration of the lipid and FA compositions and increases in omega-3 PUFAs in the liver provide a possible mechanism for the protective effect of DHA against NAFLD and NASH [169]. DHA was also found to ameliorate hepatic steatosis and inflammation by the downregulation of the expression of genes that are involved in TG synthesis (FAS, SREBP-1c, and PPAR_). The suppression of Kupffer cells and macrophage activation, inflammatory cytokine production (IL-1_ and TNF-_), and nuclear NF-_B accumulation occurred in dietary DHA-treated Ldlr−/− (deficient in the low-density lipoprotein [LDL] receptor) and leptin-deficient (ob/ob) mice [170, 171]. Mice treated with trans-10, cis-12conjugated linoleic acid developed NAFLD and insulin resistance, while supplementation with DHA reduced fatty liver, TG, and insulin levels and improved insulin resistance [172]. Compared to EPA, DHA is effective at restoring serum adiponectin levels, which contributes to improving hepatic insulin function [173]. An in vitro study showed that DHA decreased palmitate-induced lipid accumulation and inflammatory cytokine production through suppressing the activation of nucleotide-binding oligomerization domain (NOD) like receptor protein 3 inflammasomes, thus, in turn, blocking caspase-1-mediated IL-1_ and IL-18 release [174]. There are a number of studies that have demonstrated that both EPA and DHA regulate a variety of genes that are implicated in lipogenesis, lipolysis, and _-oxidation in liver and adipose tissue, which suggests that they function on hepatic lipid metabolism and NAFLD [175, 176]. Although dietary intake of omega-3 PUFAs is exerting a beneficial effect on NAFLD, studies exhibited that increased hepatic lipid peroxidation also occurred. With regard to this finding, the addition of antioxidants is suggested for omega-3 PUFAs in the intervention of diseases [177]. 3.3 Polyphenols Previous studies showed thatmany polyphenolic compounds in nature exert health-promoting effects when consumed in food. 3.3.1 Flavonoids Flavonoids are plant secondary metabolites that appear ubiquitously in fruits, vegetables, nuts, and seeds and can be classified into seven subgroups as follows: flavones, flavanones, flavonols, flavanonols, isoflavones, flavanols (catechins), and anthocyanidins, based on differences in the structure of the aglycone C ring. The diversity of the functional groups (by hydroxylation, methoxylation, or glycosylation) provides the structural variation and different biological properties of the flavonoids. More than 1000 natural flavonoids have been identified, and some of them exhibit a broad spectrum of biological properties and widespread beneficial effects for human health. 3.3.1.1 Flavones Apigenin (4_,5,7-trihydroxyflavone) belongs to the flavone class and is most prevalent in parsley and celery. It has been reported that apigenin inhibited lipolysis of 3T3-L1 adipocytes by decreasing HSL gene expression and upregulating AMPK signaling, which can attenuate adipogenesis and FFA release from adipocytes [178]. Luteolin is another flavone and ismost often present in thyme and other plants, including Brussels sprouts, cabbage, onion, broccoli, and cauliflower. Luteolin is also found to reduce palmitate-stimulated lipid accumulation in HepG2 cells by decreasing SREBP-1 and FAS as well as increasing CPT-1 gene expression. In addition, luteolin treatment induced AMPK signaling and, in turn, phosphorylated ACC (thus inhibiting the ACC activity) and reduced the production of malonyl-CoA, an allosteric inhibitor of CPT-1 that contributes to increased _-oxidation [179]. Nobiletin is a polymethoxyflavone that is rich in citrus peel and is reported to suppress proinflammatory adipokine production, such as the production ofMCP-1 and TNF-_, and to increase adiponectin secretion in adipocytes, both in vitro and in vivo [180–183]. In HFD-fed mice and leptin-deficient (ob/ob) mice, dietary intake of nobiletin improved plasma glucose tolerance and insulin sensitivity [181, 182], which suggests that insulin is acting on the improvement of adipose tissue-mediated C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 13 insulin resistance, which plays a central role in the pathogenesis ofNAFLD. Baicalein naturally occurs in the roots of Scutellaria baicalensis and has been found to reduce HFD-induced hepatic inflammation and lipid ectopic deposition by inhibiting SREBP-1-dependent lipogenic gene expression. Baicalein also enhances hepatic phosphorylated AMPK, PPAR_, and its targeted gene expression and, thus, represses FA and cholesterol synthesis and promotes FA oxidation [184]. 3.3.1.2 Flavonols Quercetin is a natural flavonol that is typically present in onions, broccoli, and leafy green vegetables. Many in vivo studies have supported the preventive and therapeutic efficacy of quercetin against NAFLD. In HFD-fed animals, the dietary feeding of quercetin reduced hepatic lipid accumulation, the infiltration of inflammatory cells, and portal fibrosis, and it improved insulin resistance [185–187]. The mechanisms include the reduction of lipogenic gene expression, induction of _-oxidation, upregulation of Nrf2-targeted antioxidative enzyme expression, and a decrease in NF-_B and serum IL-18 levels. Dietary quercetin also reduced hepatocellular fibrosis in HFD-fed gerbils through the regulation of Sirtuin (SIRT) 1, an NAD+-dependent protein deacetylase that is known to impair the activity of PPAR_,which results in a decrease in FA oxidation [188]. Feedingmice a methionineand choline-deficient diet supplemented with quercetin decreased liver steatosis and inflammatory cell accumulation through attenuating NF-_B and JNK as well as reducing the fibrogenic gene expression, such as the expression of _-SMA, TGF-_1, Col1_1, and Col3_1 [189]. The reduction of hepatic lipoperoxidation, DNA damage, and increased SOD, CAT, and GPx activities also contributes to the preventive action of quercetin against NAFLD [189,190]. In addition, quercetin has been shown to ameliorate hyperglycemia and to increase glucose uptake in diabetic db/db mice and oleic acid-treated HepG2 cells via increasing intracellular antioxidants and decreasing TNF-_ and IL-8 expression [191, 192]. Kaempferol is another typical flavonol that is present in broccoli, tea, and various vegetables. Both kaempferol and quercetin prevented the production of peroxides, superoxide anion, and nitric oxide induced by proinflammatory cytokines in Chang Liver cells, which could reduce hepatic oxidative stress [193]. 3.3.1.3 Flavanols (catechins) Catechins and theaflavins (TFs) are bioactive compounds in green tea and black tea that have been shown to possess wide health benefits. Epigallocatechin-3-gallate (EGCG) is the most abundant polyphenolic compound in green tea, and the dietary feeding of EGCG reduced HFD-induced hepatic steatosis, inflammation, and fibrosis, which can be attributed to decreased lipid peroxidation and _-SMA expression and increased GSH levels in liver [194,195]. EGCG prevented HFDinduced oxidative stress, and toxicity could be associated with reduced hepatic CYP2E1 that is overexpressed inNASH[194]. Long-term feeding of EGCG is effective in the reduction of HFD-induced hyperglycemia and plasma insulin levels and the improvement of insulin resistance, which could be the result of decreasing serum MCP-1, C-reactive protein, and IL-6 [195, 196]. Rats that received intraperitoneal injections of EGCG also reduced their HFD-induced hepatic fatty score, necrosis, inflammatory foci, and fibrosis, followed by decreasing TGF-_1, _-SMA, TNF-_, inducible nitric oxide synthase, and cyclooxygenase 2 gene expression through the modulation of the TGF/SMAD, PI3K/Akt/FOXO1, and NF-_B signaling pathways [197]. Male New Zealand black mice that received orally administered EGCG decreased gene expressions of malic enzyme, SCD-1, and glucokinase in the liver, whereas uncoupling protein 2 (UCP-2) was increased, which could possibly be the cause of increased FA oxidation [198]. Another study used 13C-labeled palmitate and a diet that was supplemented with corn oil as a natural source of 13Cenriched lipids; this study showed that EGCG increased the oxidation of dietary lipids and decreased the incorporation of dietary lipids in the liver, thus reducing HFD-induced lipid accumulation in the liver [199]. TFs are major polyphenols in black tea that include theaflavin (TF-1), theaflavin-3-gallate, and theaflavin-3,3_-digallate. In human HepG2 cells, TF-1, theaflavin-3-gallate, and theaflavin-3,3_-digallate are more effective than (-)-epicatechin (EC), (-)-epicatechin gallate (ECG), (-)-epigallocatechin (EGC), and EGCG on reduction of mixture of long-chain FA-induced lipid accumulation. These theaflavins are potent at inducing the activation of AMPK and the inactivation of ACC, both in HepG2 cells and in the livers of HFD-fed mice [200]. By measuring the rates of incorporation of [14C]acetate into the hepatic FAs, theaflavins were found to reduce FA synthesis while increasing FA oxidation [200]. Dietary TF-1 reduced hepatic steatosis, oxidative stress, hepatocyte apoptosis, andmacrophage infiltration in methionine- and choline-deficient diet-fed and ischemiareperfusion (I/R) injuredmice. Reduced ROS production and decreased TNF-_, IL-6, and inducible nitric oxide synthase expression could be the major mechanisms of TF-1 [201]. 3.3.1.4 Flavanones Citrus peels are a rich source of naringenin and hesperetin, both belong to the flavanones subgroup. An animal study showed that a normal diet supplemented with naringenin can increase gene and activity of various enzymes that are involved in hepatic FA oxidation, including carnitine octanoyltransferase, acyl-coenzyme A oxidase (ACO), bifunctional enzyme, and 3-ketoacyl-coenzyme A thiolase, as well as their gene expression, while they are not found in hesperetintreated mice.Naringenin also significantly increased the gene expression of microsomal CYP IV A1, which is involved in the _-oxidation of FAs [202]. Similar effects also occurred in HFD-fed Ldlr−/− mice. Dietary feeding of naringenin ameliorated hepatic steatosis, which is evidenced by a reduction in hepatic TG and VLDL–TG and VLDL–apolipoprotein B secretion. This effect resulted from naringenin increasing PPAR_ and PPAR_ peroxisome proliferator-activated receptor _ coactivator-1_, which is mediated by CPT-1 and C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 14 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 acyl-CoA oxidase gene expression (which are enzymes that are involved in mitochondrial and peroxisomal FA oxidation) [203]. In high cholesterol-fed mice, in addition to increased FA oxidation, naringenin also reduced hyperlipidemia and hepatic steatosis by the reduction of cholesterol and FA synthesis via downregulation of the expression of various genes in liver [204]. Moreover, improved glucose utilization and insulin sensitivity were found in naringenin-supplemented mice [203,204]. Another citrus flavanone, hesperetin, is found to decrease orotic acid induced hepatic TG accumulation and cholesterol levels, which contributes to reducing hepatic microsomal phosphatidate phosphohydrolaseactivity, which is the rate-limiting enzyme for TG synthesis [205]. 3.3.1.5 Anthocyanidins Anthocyanidins are plant pigments that have red and blue colors and that commonly occur in fruits and vegetables, such as blueberries and grapes. Several in vitro and in vivo studies revealed the function of cyanidin-3-O-_-glucoside on insulin-resistance-associated NAFLD. Diabetic/obese KK-A(y) mice are an animal model of type 2 diabetes that exhibit a phenotype with severe obesity, hyperlipidemia, and insulin resistance [206]. Feeding cyanidin-3-O-_-glucoside ameliorated hepatic steatosis by the reduction of TG synthesis via the downregulation of mitochondrial acyl-CoA:glycerolsn3-phosphate acyltransferase 1, an enzyme that is involved in converting glycerol-3-phosphate and acyl-CoA into phosphatidic acid, which is a precursor of TG and glycerophospholipids [207]. In both HFD-fed and diabetic db/db mice, the oral administration of cyanidin-3-O-_-glucoside reduced hepatic steatosis and neutrophil infiltration [208]. Cyanidin-3-O-_-glucoside also attenuated obesity-associated insulin resistance by lowering the fasting glucose levels. Additionally, cyanidin-3-O-_-glucoside decreased proinflammatory adipokine expression in adipose tissue and in plasma through the suppression of JNK signaling [208]. Another study showed the effect of cyanidin-3-O-_-glucoside on the reduction of hepatic steatosis, neutrophil infiltration, and hepatocyte apoptosis through preventing oxidative injury that is evidenced by the inhibition of ROS production and the increase of GSH synthesis, both in high glucose-treated HepG2 cells and in diabetic db/db mice [209]. In addition to the antioxidative property, cyanidin-3-O-_-glucoside increases cellular AMPK activity and suppresses ACC activity, thus causing decreased malonyl-CoA levels and further stimulation of CPT-1, which leads to enhanced FA _-oxidation and finally inhibits lipid accumulation in HepG2 cells [210]. 3.3.1.6 Isoflavones Isoflavones, such as genistein and daidzein, are abundant in soybeans; these isoflavones are a subclass of flavonoids. Isoflavones have been considered to be phytoestrogens and are recognized for improving health and aiding in the prevention of various diseases. In HFD-fed mice, daidzein reduced hepatic steatosis and de novo lipogenesis by downregulating gene expressions of ACC_, FAS, adenosine triphosphate citrate lyase, and 1-acylglycerol-3-phosphate O-acyltransferase2 [211]. Daidzein also restored HFD-induced lowered SOD-2 and GST _3 gene levels. Another study also suggested that a reduction in PPAR_ and SCD-1 could be an additional mechanism of daidzein-reduced hepatic steatosis [212]. Moreover, both studies showed that daidzein supplementation improved insulin resistance through decreasing blood insulin and adipokine (TNF-_, leptin) levels. Genistein represents a potent chemopreventive agent that acts against NAFLD through the interaction of many different mechanisms that are related to lipid metabolism, energy metabolism, insulin sensitivity, and mitochondrial function. A study compared the activity of genistein and daidzein in the modulation of lipid metabolic gene expression in liver using microarray analysis. The result demonstrated that genistein was more effective than daidzein in lowering TG levels by targetingmany genes that are involved in lipid and carbohydrate metabolism [213], although another in vitro study suggested that both genistein and daidzein upregulated CPT-1A enzyme activity [214]. Two in vitro studies also showed the ability of genistein to modulate gene expression that is involved in FA oxidation and to suppress lipogenesis via targeting the transcription factors PPAR_ and SREBP-1 [215, 216]. Dietary intake of genistein decreased hepatic steatosis, inflammatory cells infiltration, and hepatocyte ballooning in HFD-fed rats and mice. These effects could contribute to genistein alternating between adipocytemetabolism and reduced TNF_ production [217, 218]. Moreover, genistein decreased liver fat accumulation, possibly through increasing FA oxidation, as evidenced by increased PGC-1 and PPAR_-target genes, peroxisomal acyl-CoA oxidase, and mitochondrial medium chain acyl-CoA dehydrogenase, as well as UCP-2 [219]. A study that used neonatal rats fed a diet with genistein showed decreased hepatic steatosis and inflammation by reducing FAS and SREBP-1 expression, but there was no effect on PEPCK and G6Pase.Hepatocyte apoptosis and hepatic TNF-_ expression were also reduced [220]. Genistein treatment decreased HFD-induced hepatic inflammationwith lowered levels of TNF-_ and IL-6 in male sprague dawley (SD) rats. The results of molecular studies showed genistein suppression of JNK and NF-_B inflammatory signaling, which suggests that anti-inflammation is one of the mechanisms that accounts for genistein impacting the prevention ofNASH [221]. High glucose-treated rats supplemented with genistein had improved insulin resistance and liver injury through the increased activities of enzymatic (GPx, GSH reductase, and GSH) and nonenzymatic (vitamin C and E) antioxidants as well as by decreased 3-nitrotyrosine, a biomarker of inflammation that is formed by the reaction between ONOO− and the free tyrosine or tyrosine residues found in proteins [222]. Genistein also improved hepatic insulin signaling by the upregulation of IRS-1/PI3K-Akt signaling in high fructoseand HFD-fed mice [223]. Genistein supplementation elevated hepatic glucokinase activity, while suppressing the elevation of hepatic gluconeogenic G6Pase and PEPCK C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 15 activities in diabetic db/db mice, which modulates hepatic glucosemetabolism and contributes to improved insulin sensitivity [224]. In palmitate-treated HepG2 cells, genistein is found to improve glucose uptake by the upregulation of IRS1/PI3K signaling and Glut1 as well as by the inhibition of JNK signaling [225]. These studies suggested that genistein could act as an insulin sensitizer that contributes to improving insulin resistance mediated by NAFLD. 3.3.2 Resveratrol Resveratrol (3,5,4_-trihydroxystilbene), a compound found largely in the skins of red grapes, is widely accepted as a chemopreventive agent and exerts positive health effects by its multiple biological activities, such as antioxidative, antiinflammatory, anticancer, antiobesity, antidiabetic, and antiaging properties. The beneficial effect of resveratrol on metabolic syndrome has also been addressed. A number of in vivo animal models of NAFLD have exhibited the potential inhibitory effect of resveratrol. In several models of HFDinduced NAFLD, dietary intake of resveratrol efficiently suppressed hepatic steatosis; reduced lipid, cholesterol, and TG accumulation; inhibited inflammatory cell infiltration; and inhibited insulin resistance. The molecular mechanisms include decreased lipogenesis, as shown by reduced gene expression levels of SREBP-1, FAS, ACC, glucose-6-phosphate dehydrogenase, and HMG-CoA reductase, and increased FA oxidation by the upregulation of CPT-1 and ACO [226–230]. However, two studies showed that a dose–response effect was not found in resveratrol treatment [229, 231]. Male C57BL/6J mice fed 0.005% or 0.02% resveratrol reduced HFD-induced hepatic steatosis, whereas the lower dose of resveratrol (0.005%) appeared to be more beneficial than the higher dose (0.02%). Another study showed that male fa/fa Zucker rats, an animal model of obesity and liver steatosis, orally administered resveratrol at 15 and 45 mg/kg body weight reduced liver weight, TG content, and oxidative stress. This inhibitory effect of resveratrol contributes to increase enzyme activity of CPT-1 and ACO. Nevertheless, a dose–response pattern was not found of resveratrol treatment in this study. By using microarray analysis, dietary resveratrol is found to modulate the expression of various genes that are involved in hepatic lipid metabolism, including cholesterol, FA, and lipid synthesis and metabolism as well as transport [232]. Resveratrol has been shown to possess potent antioxidative activity that prevents hepatic steatosis by decreasing oxidative stress and upregulating antioxidative enzymes. Resveratrol upregulated hepatic UCP-2, an anion transporter that is located in the inner mitochondrial membrane that functions to reduce the electrochemical gradient over the membrane, and it increased the mitochondria content; thus, it could protect against HFDinduced mitochondrial dysfunction in hepatocytes [233]. The oral consumption of resveratrol is shown to suppress lipid peroxidation by the upregulation of Nrf2 and by antioxidants, including catalase, SOD, GSH, and vitamin C, which reduced fructose-induced hepatic oxidative stress [234]. Lowered hepatic TNF-_ has also occurred in resveratrol-treated rats, which suggests that there is an anti-inflammatory function of resveratrol in HFD-induced NAFLD [235]. Resveratrol attenuated insulin resistance and reduced blood glucose, serum insulin, and the hepatic glycogen content in HFD-fed mice and diabetic/ obese KK-A(y) mice, which is attributed to the upregulation of IRS/PI3K/Akt signaling in the liver and improved insulin sensitivity [236, 237]. Moreover, resveratrol is known to induce SIRT1, amember of the mammalian SIRTs, which are highly conserved protein deacetylases, and AMPK signaling that might contribute to the modulation of many transcription factors and molecules that are involved in lipogenesis and insulin signaling. In palmitate-treated HepG2 cells, treatment by resveratrol-induced SIRT1 and FOXO further downregulated SREBP-1 expression and reduced lipid accumulation [238]. When HepG2 cells were exposed to high glucose, resveratrol abrogated the impairment of the phosphorylation of AMPK and its downstream target, ACC, as well as counteracted increased expression of FAS and lipid accumulation. In addition, the activation of AMPK signaling is correlated with resveratrol-stimulated SIRT1 activity [239]. A new study showed that resveratrol ameliorated palmitate-induced deregulation of insulin signaling and ER stress through the activation of SIRT1-dependent FOXO deacetylation [240]. 3.3.3 Curcumin Curcumin (diferuloylmethane) is the major pigment from dried rhizomes of the turmeric plant (Curcuma longa Linn) and has been used as a spice and traditionalmedicine in Asia for centuries. The chemopreventive property of curcumin against various diseases and cancers has been confirmed in a number of studies. Many previous in vivo studies have also demonstrated the protective and therapeutic potential of curcumin on NASH and NAFLD using different animal models [241]. The mechanisms included anti-inflammatory and antioxidative properties, inhibition ofHSC activation, reduced lipogenesis, and improved insulin sensitivity. Dietary curcumin decreased hepatic TG levels by the downregulation of SREBP-1 and HMG-CoA reductase gene expression and by increased mitochondrial biogenesis in HFD-fed obese mice [242, 243]. Curcumin also suppressed macrophage infiltration in liver tissue with lowered NF-_B, SOCS3, MCP1, and TNF-_ in HFD-fed mice and leptin-deficient (ob/ob) mice [242–244]. In HFD-fed New Zealand rabbits, a diet supplemented with curcumin reduced hepatic steatosis, inflammation, and fibrosis, which is attributed to lower mitochondrial ROS and improved mitochondrial function [245]. Improved insulin and glucose tolerance occurs from curcumin treatment in HFD-fed C57BL/6J mice, leptin-deficient (ob/ob) mice, and cultivated human adipose tissues, and could contribute to the lowered adipose tissue-derived proinflammatory adipokines and increased adiponectin [244, 246]. TNF-_ is known to trigger the recruitment of inflammatory cells and is a pathogenic factor in NAFLD. In mice administered TNF-_ intraperitoneally, curcumin repressed the infiltration C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 16 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 Figure 1. Pathogenesis and development of NAFLD/NASH. Increased FFAs overflow from adipose tissue or diet activates JNK and NF-_B signaling where induces transcriptional expression of proinflammatory cytokines (such as TNF-_) and further cause IR. Adipokines derived from adipose tissue also cause hepatic insulin receptor (IR) via JNK and SOCS3 mediates IRS-1 serine phosphorylation, results in decreasing glucose uptake and promoting gluconeogenesis. FFAs trigger de novo lipogenesis through SREBP-1 transcription factor mediated lipogenic genes resulting TG accumulation. Upregulated PPAR_ by FFAs induce CPT-1 expression that facilitates FFAs import tomitochondria. PPAR_ also increases enzymes involved in peroxisomal and mitochondrial oxidation. Increased ROS from FAs oxidation and CYP2E1 results in lipid peroxidation and oxidative stress contributes to mitochondria dysfunction and hepatocytes injury. FFAs activated JNK, TNF-_, and oxidative stress are contributed to hepatocytes lipotoxicity and apoptosis, further induce recruitment of inflammatory cells that enhance release of cytokines and activation of HSCs. Transactivated HSCs produce fibrogenic molecules that facilitate NASH development. of Kupffer cells and neutrophils in the liver and further reducedmyeloperoxidase activity, lipid peroxidation, and nitrite content [247]. In vitro studies exhibited that curcumin treatment restored mitochondrial dysfunction and suppressed ROS production and PEPCK and G6Pase production as well as activating Akt signaling, which occurs via blocking the JNK signaling. These effects might further improve insulin sensitivity in FFA and iron overload mediated insulin resistance hepatocytes [248, 249]. Many studies suggested that the suppression of HSC activation by curcumin could be an important mechanism in preventing NASH progression. The activation of HSC occurs in response to hepatic injury and is involved in the development of hepatic fibrosis. When hepatic injury occurs, quiescent HSCs undergo enhanced cell proliferation, the loss of lipid droplets, expression of _-SMA, and excessive production of extracellular matrix. Treatment with curcumin inhibited insulin-stimulated HSC activation by increased intracellular lipid droplets and the expression of fibrogenic genes. Molecular studies demonstrated that curcumin interrupts insulin signaling and suppresses gene expression of the insulin receptor in HSCs. Moreover, curcumin eliminated insulin-induced ROS production and increased the activity of glutamate-cysteine ligase in activated HSCs, which indicates that ROS is implicated in insulin-mediatedHSC activation [250]. In high glucose- and leptin-treated HSCs, curcumin suppressed HSC activation by abrogated membrane translocation of Glut proteins. These two studies showed that curcumin interfered with hyperleptinemia-triggered p38 mitogen-activated protein kinase and IRS/PI3K/Akt signaling, which lead to inhibiting HSC activation [251, 252]. Additionally, curcumin stimulated glucokinase activity, increasing the conversion of glucose to glucose-6-phosphate in HSCs [252]. Further study revealed that curcumin abrogated leptin-induced HSC activation contributes to the upregulation of AMPK and the induction of gene expression of PPAR_, SREBP-1, and CCAAT/enhancer binding protein _, which leads to the accumulation of lipids [253]. Hypercholesterolemia is characterized by elevated levels of plasma LDL and is associated with NAFLD. Cellular uptake of oxidized LDL is mediated by binding to cell-surface LDL receptors of different cell types in the liver, which subsequently leads to cholesterol uptake and increased ROS. Curcumin treatment is found to suppress LDL and oxidized LDL induced activation of HSCs, especially through downregulated LDL receptor and lectin-like oxidized LDL receptor 1 expression, which results in decreased fibrogenic gene expression and intracellular cholesterol [254, 255]. Although curcumin is known to have various biological properties, poor absorption C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 17 and systemic bioavailability have been considered as a major limitation for its application in clinic [256]. 4 Conclusion NAFLD pathogenesis is a complicated process that is involved not only in molecular changes within the liver, but also in metabolic signaling between organs. Understanding the pathogenic factors, signaling networks, and molecular mechanisms that are implicated in NAFLD could provide biomarkers for treatment and intervention. Considerable accomplishments in NAFLD research over the past few decades have verified that increased oxidative stress, lipotoxicity, insulin resistance, ER stress, hepatic inflammation, and obesity play causal roles in the development and progression of this disease and also offer opportunities for using nutritional components as prevention or intervention. Furthermore, dietary natural compounds provide a novel strategy for obesity-associated NAFLD treatment. These dietary natural compounds have great potential to not only influence development and NAFLD progression, but also to target obesity and insulin resistance-mediated pathogenesis. This general beneficial effect of dietary natural compounds demonstrates a complex interaction of many different mechanisms, including a reduction in lipogenesis, an increase in FA oxidation, an improvement in insulin signaling, an inhibition of adipokine production, an elimination of oxidative stress, and the suppression of hepatic inflammation by targeting multiple signaling pathways, transcription factors, and enzymes. The coordination of metabolic function between liver and adipose tissue by dietary natural compounds also represents a potential mechanism to prevent hepatic steatosis, inflammation, and fibrosis (Fig. 1). Although the current knowledge suggests that dietary natural compounds could be helpful for NAFLD prevention and treatment, most of these dietary natural compounds are lack of understanding about relevance to human, such as the dosage, bioavailability, and possible adverse effects. Well-designed experiments, appropriately powered and large-scale trials are needed to examine the applicability and roles of these dietary natural compounds as chemopreventive agents of NAFLD. This study was supported by the National Science Council NSC 101–2628-B-022–001-MY4, 102-2628-B-002-053-MY3. The authors have declared no conflict of interest. 5 References [1] Angulo, P., Nonalcoholic fatty liver disease. N. Engl. J.Med. 2002, 346, 1221–1231. [2] Shifflet, A., Wu, G. Y., Non-alcoholic steatohepatitis: an overview. J. Formos. Med. Assoc. 2009, 108, 4–12. [3] Neuschwander-Tetri, B. A., Clark, J. M., Bass, N. M., Van Natta, M. L. et al., Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology 2010, 52, 913–924. [4] Vernon, G., Baranova, A., Younossi, Z. M., Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [5] Bhala, N., Jouness, R. I., Bugianesi, E., Epidemiology and natural history of patients with NAFLD. Curr. Pharm. Des. 2013, 19, 5169–5176. [6] Levene, A. P., Goldin, R. D., The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 2012, 61, 141–152. [7] Fan, J. G., Zhu, J., Li, X. J., Chen, L. et al., Prevalence of and risk factors for fatty liver in a general population of Shanghai, China. J. Hepatol. 2005, 43, 508–514. [8] Jimba, S., Nakagami, T., Takahashi, M.,Wakamatsu, T. et al., Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabet. Med. 2005, 22, 1141–1145. [9] Bedogni, G., Miglioli, L.,Masutti, F., Tiribelli, C. et al., Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [10] Kwon, Y. M., Oh, S. W., Hwang, S. S., Lee, C. et al., Association of nonalcoholic fatty liver disease with components of metabolic syndrome according to body mass index in Korean adults. Am. J. Gastroenterol. 2012, 107, 1852–1858. [11] Bellentani, S., Scaglioni, F., Marino, M., Bedogni, G., Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [12] McCullough, A. J., The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 521–533. [13] Ekstedt, M., Franzen, L. E., Mathiesen, U. L., Thorelius, L. et al., Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [14] Day, C. P., James, O. F., Steatohepatitis: a tale of two “hits”? Gastroenterology 1998, 114, 842–845. [15] Tilg, H., Moschen, A. R., Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [16] Reaven, G., Abbasi, F., McLaughlin, T., Obesity, insulin resistance, and cardiovascular disease. Recent Prog. Horm. Res. 2004, 59, 207–223. [17] Ong, J. P., Elariny, H., Collantes, R., Younoszai, A. et al., Predictors of nonalcoholic steatohepatitis and advanced fibrosis in morbidly obese patients. Obes. Surg. 2005, 15, 310–315. [18] Colicchio, P., Tarantino, G., del, G. F., Sorrentino, P. et al., Non-alcoholic fatty liver disease in young adult severely obese non-diabetic patients in South Italy. Ann. Nutr. Metab. 2005, 49, 289–295. [19] Liew, P. L., Lee, W. J., Wang, W., Lee, Y. C. et al., Fatty liver disease: predictors of nonalcoholic steatohepatitis and gallbladder disease in morbid obesity. Obes. Surg. 2008, 18, 847–853. [20] Huang, H. L., Lin, W. Y., Lee, L. T., Wang, H. H. et al., Metabolic syndrome is related to nonalcoholic C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 18 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 steatohepatitis in severely obese subjects. Obes. Surg. 2007, 17, 1457–1463. [21] Dolce, C. J., Russo, M., Keller, J. E., Buckingham, J. et al., Does liver appearance predict histopathologic findings: prospective analysis of routine liver biopsies during bariatric surgery. Surg. Obes. Relat. Dis. 2009, 5, 323–328. [22] Harnois, F.,Msika, S., Sabate, J.M., Mechler, C. et al., Prevalence and predictive factors of non-alcoholic steatohepatitis (NASH) in morbidly obese patients undergoing bariatric surgery. Obes. Surg. 2006, 16, 183–188. [23] Gholam, P. M., Flancbaum, L., Machan, J. T., Charney, D. A. et al., Nonalcoholic fatty liver disease in severely obese subjects. Am. J. Gastroenterol. 2007, 102, 399–408. [24] Yoo, J., Lee, S., Kim, K., Yoo, S. et al., Relationship between insulin resistance and serum alanine aminotransferase as a surrogate of NAFLD (nonalcoholic fatty liver disease) in obese Korean children. Diabetes Res. Clin. Pract. 2008, 81, 321–326. [25] Patton, H. M., Yates, K., Unalp-Arida, A., Behling, C. A. et al., Association between metabolic syndrome and liver histology among children with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2010, 105, 2093–2102. [26] Galic, S., Oakhill, J. S., Steinberg, G. R., Adipose tissue as an endocrine organ. Mol. Cell Endocrinol. 2010, 316, 129–139. [27] Choi, S. M., Tucker, D. F., Gross, D. N., Easton, R. M. et al., Insulin regulates adipocyte lipolysis via anAkt-independent signaling pathway. Mol. Cell Biol. 2010, 30, 5009–5020. [28] Stralfors, P., Honnor, R. C., Insulin-induced dephosphorylation of hormone-sensitive lipase. Correlation with lipolysis and cAMP-dependent protein kinase activity. Eur. J. Biochem. 1989, 182, 379–385. [29] Lam, T. K., Yoshii, H., Haber, C. A., Bogdanovic, E. et al., Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E682–E691. [30] Boden, G., Cheung, P., Stein, T. P., Kresge, K. et al., FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. Am. J. Physiol. Endocrinol.Metab. 2002, 283, E12–E19. [31] Boden, G., Chen, X., Ruiz, J.,White, J. V. et al., Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Invest. 1994, 93, 2438–2446. [32] Donnelly, K. L., Smith, C. I., Schwarzenberg, S. J., Jessurun, J. et al., Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005, 115, 1343–1351. [33] Kissebah, A. H., Krakower, G. R., Regional adiposity and morbidity. Physiol. Rev. 1994, 74, 761–811. [34] van der, K. K., Leenen, R., Seidell, J. C., Deurenberg, P. et al., Abdominal diameters as indicators of visceral fat: comparison between magnetic resonance imaging and anthropometry. Br. J. Nutr. 1993, 70, 47–58. [35] Nielsen, S., Guo, Z., Johnson, C. M., Hensrud, D. D. et al., Splanchnic lipolysis in human obesity. J. Clin. Invest. 2004, 113, 1582–1588. [36] van der, P. D., Milner, K. L., Hui, J., Hodge, A. et al., Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [37] Ibrahim, M. M., Subcutaneous and visceral adipose tissue: structural and functional differences. Obes. Rev. 2010, 11, 11–18. [38] Preis, S. R., Massaro, J.M., Robins, S. J., Hoffmann,U. et al., Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity (Silver Spring) 2010, 18, 2191–2198. [39] Guilherme, A., Virbasius, J. V., Puri, V., Czech, M. P., Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [40] Hotamisligil, G. S., Shargill, N. S., Spiegelman, B. M., Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [41] Uysal, K. T., Wiesbrock, S. M., Marino, M. W., Hotamisligil, G. S., Protection from obesity-induced insulin resistance inmice lacking TNF-alpha function. Nature 1997, 389, 610–614. [42] Haukeland, J. W., Damas, J. K., Konopski, Z., Loberg, E. M. et al., Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [43] Hui, J. M., Hodge, A., Farrell, G. C., Kench, J. G. et al., Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004, 40, 46–54. [44] Crespo, J., Cayon, A., Fernandez-Gil, P., Hernandez-Guerra, M. et al., Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [45] Li, Z., Yang, S., Lin, H., Huang, J. et al., Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [46] Van Wagner, L. B., Koppe, S. W., Brunt, E. M., Gottstein, J. et al., Pentoxifylline for the treatment of non-alcoholic steatohepatitis: a randomized controlled trial. Ann. Hepatol. 2011, 10, 277–286. [47] Yalniz,M., Bahcecioglu, I. H., Kuzu, N., Celebi, S. et al., Amelioration of steatohepatitis with pentoxifylline in a novel nonalcoholic steatohepatitis model induced by high-fat diet. Dig. Dis. Sci. 2007, 52, 2380–2386. [48] Feldstein, A. E., Werneburg, N. W., Canbay, A., Guicciardi, M. E. et al., Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [49] Chakraborty, C., Biochemical andmolecular basis of insulin resistance. Curr. Protein Pept. Sci. 2006, 7, 113–121. [50] Feingold, K. R., Soued, M., Serio, M. K., Adi, S. et al., The effect of diet on tumor necrosis factor stimulation of hepatic lipogenesis. Metabolism 1990, 39, 623–632. [51] Moschen, A. R., Molnar, C., Geiger, S., Graziadei, I. et al., Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut 2010, 59, 1259–1264. C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 19 [52] Fernandez-Real, J.M., Vayreda, M., Richart, C., Gutierrez, C. et al., Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J. Clin. Endocrinol. Metab. 2001, 86, 1154–1159. [53] Wang, X., Bao,W., Liu, J., Ouyang, Y. Y. et al., Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [54] Fontana, L., Eagon, J. C., Trujillo, M. E., Scherer, P. E. et al., Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010– 1013. [55] Senn, J. J., Klover, P. J., Nowak, I. A., Zimmers, T. A. et al., Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [56] Sabio, G., Das, M., Mora, A., Zhang, Z. et al., A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [57] Friedman, J. M., Halaas, J. L., Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [58] Lam, Q. L., Lu, L., Role of leptin in immunity. Cell. Mol. Immunol. 2007, 4, 1–13. [59] Huang, X. D., Fan, Y., Zhang, H., Wang, P. et al., Serum leptin and soluble leptin receptor in non-alcoholic fatty liver disease. World J. Gastroenterol. 2008, 14, 2888–2893. [60] Lemoine, M., Ratziu, V., Kim, M., Maachi, M. et al., Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int. 2009, 29, 1431–1438. [61] Chitturi, S., Farrell, G., Frost, L., Kriketos, A. et al., Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology 2002, 36, 403–409. [62] Huang, X. D., Fan, Y., Zhang, H., Wang, P. et al., Serum leptin and soluble leptin receptor in non-alcoholic fatty liver disease. World J. Gastroenterol. 2008, 14, 2888–2893. [63] Canbakan, B., Tahan, V., Balci, H., Hatemi, I. et al., Leptin in nonalcoholic fatty liver disease. Ann. Hepatol. 2008, 7, 249–254. [64] Anania, F. A., Leptin, liver, and obese mice—fibrosis in the fat lane. Hepatology 2002, 36, 246–248. [65] Ikejima, K., Takei, Y., Honda, H., Hirose, M. et al., Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellularmatrix in the rat. Gastroenterology 2002, 122, 1399–1410. [66] Leclercq, I. A., Farrell, G. C., Schriemer, R., Robertson, G. R., Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [67] Wang, J., Leclercq, I., Brymora, J. M., Xu, N. et al., Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [68] Targher, G., Bertolini, L., Scala, L., Poli, F. et al., Decreased plasma adiponectin concentrations are closely associated with nonalcoholic hepatic steatosis in obese individuals. Clin. Endocrinol. (Oxf.) 2004, 61, 700–703. [69] Louthan, M. V., Barve, S., McClain, C. J., Joshi-Barve, S., Decreased serum adiponectin: an early event in pediatric nonalcoholic fatty liver disease. J. Pediatr. 2005, 147, 835– 838. [70] Buechler, C., Wanninger, J., Neumeier, M., Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol. 2011, 17, 2801–2811. [71] Tilg, H., The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 179–185. [72] Xu, A.,Wang, Y., Keshaw, H., Xu, L. Y. et al., The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest. 2003, 112, 91–100. [73] Awazawa, M., Ueki, K., Inabe, K., Yamauchi, T. et al., Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem. Biophys. Res. Commun. 2009, 382, 51–56. [74] Saltiel, A. R., Kahn, C. R., Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [75] Taniguchi, C.M., Emanuelli, B., Kahn, C. R., Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [76] Puigserver, P., Rhee, J., Donovan, J., Walkey, C. J. et al., Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [77] Chiang, S. H., Baumann, C. A., Kanzaki, M., Thurmond, D. C. et al., Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 2001, 410, 944–948. [78] Postic, C., Girard, J., Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 2008, 118, 829–838. [79] Kahn, S. E., The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [80] Browning, J. D., Szczepaniak, L. S., Dobbins, R., Nuremberg, P. et al., Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004, 40, 1387–1395. [81] Targher, G., Bertolini, L., Padovani, R., Rodella, S. et al., Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [82] Porepa, L., Ray, J. G., Sanchez-Romeu, P., Booth, G. L., Newly diagnosed diabetes mellitus as a risk factor for serious liver disease. Can. Med. Assoc. J. 2010, 182, E526–E531. [83] Prashanth, M., Ganesh, H. K., Vima, M. V., John, M. et al., Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. J. Assoc. Physicians India 2009, 57, 205–210. [84] Adams, L. A., Harmsen, S., St Sauver, J. L., Charatcharoenwitthaya, P. et al., Nonalcoholic fatty liver disease increases risk of death among patients with diabetes: a communitybased cohort study. Am. J. Gastroenterol. 2010, 105, 1567– 1573. [85] Voshol, P. J., Haemmerle, G., Ouwens, D. M., Zimmermann, R. et al., Increased hepatic insulin sensitivity together with C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 20 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology 2003, 144, 3456–3462. [86] Smith, B. W., Adams, L. A., Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat. Rev. Endocrinol. 2011, 7, 456–465. [87] Tarantino, G., Caputi, A., JNKs, insulin resistance and inflammation: a possible link between NAFLD and coronary artery disease.World J. Gastroenterol. 2011, 17, 3785–3794. [88] Schwarz, J. M., Linfoot, P., Dare, D., Aghajanian, K., Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [89] bu-Elheiga, L., Matzuk, M. M., bo-Hashema, K. A., Wakil, S. J., Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [90] An, J., Muoio, D. M., Shiota, M., Fujimoto, Y. et al., Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat. Med. 2004, 10, 268–274. [91] Feldstein, A. E., Canbay, A., Angulo, P., Taniai, M. et al., Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [92] Feldstein, A. E., Werneburg, N. W., Li, Z., Bronk, S. F. et al., Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1339–G1346. [93] Li, Z., Berk, M., McIntyre, T. M., Gores, G. J. et al., The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [94] Li, Z. Z., Berk, M., McIntyre, T. M., Feldstein, A. E., Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [95] Malhi, H., Bronk, S. F., Werneburg, N. W., Gores, G. J., Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [96] Barreyro, F. J., Kobayashi, S., Bronk, S. F., Werneburg, N. W. et al., Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J. Biol. Chem. 2007, 282, 27141–27154. [97] Feldstein, A. E., Canbay, A., Angulo, P., Taniai, M. et al., Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [98] Feldstein, A. E., Canbay, A., Guicciardi, M. E., Higuchi, H. et al., Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J. Hepatol. 2003, 39, 978–983. [99] Inoue, Y., Asanuma, T., Smith, N., Saunders, D. et al., Modulation of Fas-FasL related apoptosis by PBN in the early phases of choline deficient diet-mediated hepatocarcinogenesis in rats. Free Radic. Res. 2007, 41, 972–980. [100] Malhi, H., Barreyro, F. J., Isomoto, H., Bronk, S. F. et al., Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 2007, 56, 1124–1131. [101] Volkmann, X., Fischer, U., Bahr, M. J., Ott, M. et al., Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 2007, 46, 1498–1508. [102] Cazanave, S. C., Mott, J. L., Bronk, S. F., Werneburg, N. W. et al., Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Biol. Chem. 2011, 286, 39336–39348. [103] Wei, Y., Wang, D., Topczewski, F., Pagliassotti, M. J., Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [104] Wang, D., Wei, Y., Pagliassotti, M. J., Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943– 951. [105] Puri, P., Mirshahi, F., Cheung, O., Natarajan, R. et al., Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [106] Rinella, M. E., Siddiqui, M. S., Gardikiotes, K., Gottstein, J. et al., Dysregulation of the unfolded protein response in db/db mice with diet-induced steatohepatitis. Hepatology 2011, 54, 1600–1609. [107] Hotamisligil, G. S., Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [108] Seki, S., Kitada, T., Sakaguchi, H., Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol. Res. 2005, 33, 132–134. [109] Madan, K., Bhardwaj, P., Thareja, S., Gupta, S. D. et al., Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD). J. Clin. Gastroenterol. 2006, 40, 930–935. [110] Weltman, M. D., Farrell, G. C., Liddle, C., Increased hepatocyte CYP2E1 expression in a rat nutritionalmodel of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [111] Weltman, M. D., Farrell, G. C., Hall, P., Ingelman-Sundberg, M. et al., Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 1998, 27, 128–133. [112] Reddy, J. K., Hashimoto, T., Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [113] Caldwell, S. H., Swerdlow, R. H., Khan, E. M., Iezzoni, J. C. et al., Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [114] Perez-Carreras, M., Del, H. P., Martin, M. A., Rubio, J. C. et al., Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [115] Sanchez-Alcazar, J. A., Schneider, E., Martinez, M. A., Carmona, P. et al., Tumor necrosis factor-alpha increases the steady-state reduction of cytochrome b of the mitochondrial respiratory chain in metabolically inhibited L929 cells. J. Biol. Chem. 2000, 275, 13353–13361. C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 21 [116] Higuchi, M., Proske, R. J., Yeh, E. T., Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene 1998, 17, 2515–2524. [117] Pan, M., Cederbaum, A. I., Zhang, Y. L., Ginsberg, H. N. et al., Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J. Clin. Invest. 2004, 113, 1277–1287. [118] Perlemuter, G., vit-Spraul, A., Cosson, C., Conti, M. et al., Increase in liver antioxidant enzyme activities in nonalcoholic fatty liver disease. Liver Int. 2005, 25, 946–953. [119] Brunt, E. M., Janney, C. G., Di Bisceglie, A. M., Neuschwander-Tetri, B. A. et al., Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [120] Chalasani, N., Wilson, L., Kleiner, D. E., Cummings, O. W. et al., Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 829–834. [121] Schwabe, R. F., Uchinami, H., Qian, T., Bennett, B. L. et al., Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004, 18, 720–722. [122] Dela, P. A., Leclercq, I., Field, J., George, J. et al., NF-kappaB activation, rather than TNF, mediates hepatic inflammation in amurine dietary model of steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [123] Schattenberg, J. M., Singh, R., Wang, Y., Lefkowitch, J. H. et al., JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 2006, 43, 163–172. [124] Csak, T., Ganz, M., Pespisa, J., Kodys, K. et al., Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [125] Wieckowska, A., Papouchado, B. G., Li, Z., Lopez, R. et al., Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379. [126] Yamaguchi, K., Itoh, Y., Yokomizo, C., Nishimura, T. et al., Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine cholinedeficient diet-fed mice. Lab. Invest. 2010, 90, 1169–1178. [127] Dooley, S., ten, D. P., TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [128] Choudhury, J.,Mirshahi, F.,Murthy, K. S., Yager, D. R. et al., Physiologic concentrations of leptin increase collagen production by non-immortalized human hepatic stellate cells. Metabolism 2006, 55, 1317–1322. [129] George, J., Pera, N., Phung, N., Leclercq, I. et al., Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J. Hepatol. 2003, 39, 756–764. [130] Teufelhofer, O., Parzefall, W., Kainzbauer, E., Ferk, F. et al., Superoxide generation from Kupffer cells contributes to hepatocarcinogenesis: studies on NADPH oxidase knockout mice. Carcinogenesis 2005, 26, 319–329. [131] Gornicka, A., Morris-Stiff, G., Thapaliya, S., Papouchado, B. G. et al., Transcriptional profile of genes involved in oxidative stress and antioxidant defense in a dietary murine model of steatohepatitis. Antioxid. Redox Signal. 2011, 15, 437–445. [132] Musso, G., Cassader, M., Rosina, F., Gambino, R., Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [133] Kim, M. S., Kung, S., Grewal, T., Roufogalis, B. D., Methodologies for investigating natural medicines for the treatment of nonalcoholic fatty liver disease (NAFLD). Curr. Pharm. Biotechnol. 2012, 13, 278–291. [134] Musso, G., Gambino, R., Cassader, M., Pagano, G., A metaanalysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [135] Nissen, S. E.,Wolski, K., Rosiglitazone revisited: an updated meta-analysis of risk for myocardial infarction and cardiovascularmortality. Arch. Intern. Med. 2010, 170, 1191–1201. [136] Bahcecioglu, I. H., Kuzu, N., Metin, K., Ozercan, I. H. et al., Lycopene prevents development of steatohepatitis in experimental nonalcoholic steatohepatitis model induced by high-fat diet. Vet. Med. Int. 2010, 2010, 1–8. [137] Wang, Y., Ausman, L. M., Greenberg, A. S., Russell, R. M. et al., Dietary lycopene and tomato extract supplementations inhibit nonalcoholic steatohepatitis-promoted hepatocarcinogenesis in rats. Int. J. Cancer 2010, 126, 1788– 1796. [138] Choi, S. K., Seo, J. S., Lycopene supplementation suppresses oxidative stress induced by a high fat diet in gerbils. Nutr. Res. Pract. 2013, 7, 26–33. [139] Ahn, J., Lee, H., Jung, C. H., Ha, T., Lycopene inhibits hepatic steatosis viamicroRNA-21-induced downregulation of fatty acid-binding protein 7 in mice fed a high-fat diet. Mol. Nutr. Food Res. 2012, 56, 1665–1674. [140] Gugger, E. T., Bierer, T. L., Henze, T. M., White, W. S. et al., Beta-carotene uptake and tissue distribution in ferrets (Mustela putorius furo). J. Nutr. 1992, 122, 115–119. [141] Kameji, H., Mochizuki, K., Miyoshi, N., Goda, T., betaCarotene accumulation in 3T3-L1 adipocytes inhibits the elevation of reactive oxygen species and the suppression of genes related to insulin sensitivity induced by tumor necrosis factor-alpha. Nutrition 2010, 26, 1151–1156. [142] Sangeetha, R. K., Bhaskar, N., Baskaran, V., Comparative effects of beta-carotene and fucoxanthin on retinol deficiency induced oxidative stress in rats. Mol. Cell. Biochem. 2009, 331, 59–67. [143] Kim, J. E., Clark, R. M., Park, Y., Lee, J. et al., Lutein decreases oxidative stress and inflammation in liver and eyes of guinea pigs fed a hypercholesterolemic diet. Nutr. Res. Pract. 2012, 6, 113–119. [144] Woo, M. N., Jeon, S. M., Shin, Y. C., Lee, M. K. et al., Antiobese property of fucoxanthin is partly mediated by altering lipid-regulating enzymes and uncoupling proteins of visceral adipose tissue in mice. Mol. Nutr. Food Res. 2009, 53, 1603–1611. C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 22 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 [145] Maeda, H., Hosokawa, M., Sashima, T., Murakami- Funayama, K. et al., Anti-obesity and anti-diabetic effects of fucoxanthin on diet-induced obesity conditions in amurine model. Mol. Med. Rep. 2009, 2, 897–902. [146] Woo, M. N., Jeon, S. M., Kim, H. J., Lee, M. K. et al., Fucoxanthin supplementation improves plasma and hepatic lipid metabolism and blood glucose concentration in high-fat fed C57BL/6N mice. Chem. Biol. Interact. 2010, 186, 316– 322. [147] Hosokawa, M., Miyashita, T., Nishikawa, S., Emi, S. et al., Fucoxanthin regulates adipocytokine mRNA expression in white adipose tissue of diabetic/obese KK-Ay mice. Arch. Biochem. Biophys. 2010, 504, 17–25. [148] Beppu, F., Hosokawa, M., Yim, M. J., Shinoda, T. et al., Down-regulation of hepatic stearoyl-CoA desaturase-1 expression by fucoxanthin via leptin signaling in diabetic/ obese KK-A(y) mice. Lipids 2013, 48, 449–455. [149] Zhu, F. S., Liu, S., Chen, X.M., Huang, Z. G. et al., Effects of n3 polyunsaturated fatty acids from seal oils on nonalcoholic fatty liver disease associated with hyperlipidemia. World J. Gastroenterol. 2008, 14, 6395–6400. [150] Janczyk,W., Socha, P., Lebensztejn, D., Wierzbicka, A. et al., Omega-3 fatty acids for treatment of non-alcoholic fatty liver disease: design and rationale of randomized controlled trial. BMC Pediatr. 2013, 13, 85. [151] Masterton, G. S., Plevris, J. N., Hayes, P. C., Review article: omega-3 fatty acids—a promising novel therapy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 31, 679–692. [152] Kuda, O., Jelenik, T., Jilkova, Z., Flachs, P. et al., n-3 fatty acids and rosiglitazone improve insulin sensitivity through additive stimulatory effects on muscle glycogen synthesis in mice fed a high-fat diet. Diabetologia 2009, 52, 941–951. [153] Lamaziere, A., Wolf, C., Barbe, U., Bausero, P. et al., Lipidomics of hepatic lipogenesis inhibition by omega 3 fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 149–154. [154] Tanaka, N., Sano, K., Horiuchi, A., Tanaka, E. et al., Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2008, 42, 413–418. [155] Oya, J., Nakagami, T., Sasaki, S., Jimba, S. et al., Intake of n-3 polyunsaturated fatty acids and non-alcoholic fatty liver disease: a cross-sectional study in Japanese men and women. Eur. J. Clin. Nutr. 2010, 64, 1179–1185. [156] Nemoto, N., Suzuki, S., Kikuchi, H., Okabe, H. et al., Ethyl-eicosapentaenoic acid reduces liver lipids and lowers plasma levels of lipids in mice fed a high-fat diet. In Vivo 2009, 23, 685–689. [157] Shiba, S., Tsunoda, N., Wakutsu, M., Muraki, E. et al., Regulation of lipid metabolism by palmitoleate and eicosapentaenoic acid (EPA) in mice fed a high-fat diet. Biosci. Biotechnol. Biochem. 2011, 75, 2401–2403. [158] Kajikawa, S., Harada, T., Kawashima, A., Imada, K. et al., Suppression of hepatic fat accumulation by highly purified eicosapentaenoic acid prevents the progression of dgalactosamineinduced hepatitis in mice fed with a highfat/ high-sucrose diet. Biochim. Biophys. Acta 2009, 1791, 281–288. [159] Matsumoto, T., Terai, S., Oishi, T., Kuwashiro, S. et al., Medaka as a model for human nonalcoholic steatohepatitis. Dis. Model. Mech. 2010, 3, 431–440. [160] Varela-Rey, M., Embade, N., Ariz, U., Lu, S. C. et al., Nonalcoholic steatohepatitis and animal models: understanding the human disease. Int. J. Biochem. Cell Biol. 2009, 41, 969–976. [161] Kajikawa, S., Imada, K., Takeuchi, T., Shimizu, Y. et al., Eicosapentaenoic acid attenuates progression of hepatic fibrosis with inhibition of reactive oxygen species production in rats fed methionine- and choline-deficient diet. Dig. Dis. Sci. 2011, 56, 1065–1074. [162] Kajikawa, S., Harada, T., Kawashima, A., Imada, K. et al., Highly purified eicosapentaenoic acid ethyl ester prevents development of steatosis and hepatic fibrosis in rats. Dig. Dis. Sci. 2010, 55, 631–641. [163] Ishii, H., Horie, Y., Ohshima, S., Anezaki, Y. et al., Eicosapentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific Pten-deficient mice. J. Hepatol. 2009, 50, 562–571. [164] Nanthirudjanar, T., Furumoto, H., Hirata, T., Sugawara, T., Oxidized eicosapentaenoic acids more potently reduce LXRalpha-induced cellular triacylglycerol via suppression of SREBP-1c, PGC-1beta and GPA than its intact form. Lipids Health Dis. 2013, 12, 73. [165] Nobili, V., Bedogni, G., Alisi, A., Pietrobattista, A. et al., Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353. [166] Nobili, V., Alisi, A., Della, C. C., Rise, P. et al., Docosahexaenoic acid for the treatment of fatty liver: randomised controlled trial in children. Nutr. Metab. Cardiovasc. Dis. 2012, in press. [167] Takayama, F., Nakamoto, K., Totani, N., Yamanushi, T. et al., Effects of docosahexaenoic acid in an experimental rat model of nonalcoholic steatohepatitis. J. Oleo. Sci. 2010, 59, 407–414. [168] Poudyal, H., Panchal, S. K., Ward, L. C., Brown, L., Effects of ALA, EPA and DHA in high-carbohydrate, high-fat diet-induced metabolic syndrome in rats. J. Nutr. Biochem. 2013, 24, 1041–1052. [169] Tang, X., Li, Z. J., Xu, J., Xue, Y. et al., Short term effects of different omega-3 fatty acid formulation on lipid metabolism in mice fed high or low fat diet. Lipids Health Dis. 2012, 11, 70. [170] Depner, C. M., Philbrick, K. A., Jump, D. B., Docosahexaenoic acid attenuates hepatic inflammation, oxidative stress, and fibrosis without decreasing hepatosteatosis in a Ldlr(−/−) mouse model of western diet-induced nonalcoholic steatohepatitis. J. Nutr. 2013, 143, 315–323. [171] Kim, H. J., Lee, K. T., Park, Y. B., Jeon, S. M. et al., Dietary docosahexaenoic acid-rich diacylglycerols ameliorate hepatic steatosis and alter hepatic gene expressions in C57BL/6JLep( ob/ob) mice. Mol. Nutr. Food Res. 2008, 52, 965–973. C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 23 [172] Fedor, D. M., Adkins, Y., Mackey, B. E., Kelley, D. S., Docosahexaenoic acid prevents trans-10, cis-12-conjugated linoleic acid-induced nonalcoholic fatty liver disease in mice by altering expression of hepatic genes regulating fatty acid synthesis and oxidation. Metab. Syndr. Relat. Disord. 2012, 10, 175–180. [173] Vemuri, M., Kelley, D. S., Mackey, B. E., Rasooly, R. et al., Docosahexaenoic acid (DHA) but not eicosapentaenoic acid (EPA) prevents trans-10, cis-12 conjugated linoleic acid (CLA)-induced insulin resistance in mice. Metab. Syndr.Relat. Disord. 2007, 5, 315–322. [174] Luo, X., Yang, Y., Shen, T., Tang, X. et al., Docosahexaenoic acid ameliorates palmitate-induced lipid accumulation and inflammation through repressing NLRC4 inflammasome activation in HepG2 cells. Nutr. Metab. (Lond.) 2012, 9, 34. [175] Tsuzuki, T., Kawakami, Y., Suzuki, Y., Abe, R. et al., Intake of conjugated eicosapentaenoic acid suppresses lipid accumulation in liver and epididymal adipose tissue in rats. Lipids 2005, 40, 1117–1123. [176] Sun, C., Wei, Z. W., Li, Y., DHA regulates lipogenesis and lipolysis genes in mice adipose and liver. Mol. Biol. Rep. 2011, 38, 731–737. [177] Gladine, C., Roy, N. C., Rigaudiere, J. P., Laillet, B. et al., Increasing intake of long-chain n-3 PUFA enhances lipoperoxidation and modulates hepatic gene expression in a dosedependent manner. Br. J. Nutr. 2011, 1–20. [178] Ono, M., Fujimori, K., Antiadipogenic effect of dietary apigenin through activation of AMPK in 3T3-L1 cells. J. Agric. Food Chem. 2011, 59, 13346–13352. [179] Liu, J. F., Ma, Y., Wang, Y., Du, Z. Y. et al., Reduction of lipid accumulation in HepG2 cells by luteolin is associated with activation of AMPK and mitigation of oxidative stress. Phytother. Res. 2011, 25, 588–596. [180] Kunimasa, K., Kuranuki, S., Matsuura, N., Iwasaki, N. et al., Identification of nobiletin, a polymethoxyflavonoid, as an enhancer of adiponectin secretion. Bioorg. Med. Chem. Lett. 2009, 19, 2062–2064. [181] Lee, Y. S., Cha, B. Y., Saito, K., Yamakawa, H. et al., Nobiletin improves hyperglycemia and insulin resistance in obese diabetic ob/ob mice. Biochem. Pharmacol. 2010, 79, 1674– 1683. [182] Lee, Y. S., Cha, B. Y., Choi, S. S., Choi, B. K. et al., Nobiletin improves obesity and insulin resistance in high-fat dietinduced obese mice. J. Nutr. Biochem. 2013, 24, 156–162. [183] Miyata, Y., Tanaka, H., Shimada, A., Sato, T. et al., Regulation of adipocytokine secretion and adipocyte hypertrophy by polymethoxyflavonoids, nobiletin and tangeretin. Life Sci. 2011, 88, 613–618. [184] Pu, P., Wang, X. A., Salim, M., Zhu, L. H. et al., Baicalein, a natural product, selectively activating AMPKalpha(2) and ameliorates metabolic disorder in diet-induced mice. Mol. Cell. Endocrinol. 2012, 362, 128–138. [185] Panchal, S. K., Poudyal, H., Brown, L., Quercetin ameliorates cardiovascular, hepatic, and metabolic changes in diet-induced metabolic syndrome in rats. J. Nutr. 2012, 142, 1026–1032. [186] Jung, C. H., Cho, I., Ahn, J., Jeon, T. I. et al., Quercetin reduces high-fat diet-induced fat accumulation in the liver by regulating lipid metabolism genes. Phytother. Res. 2013, 27, 139–143. [187] Hoek-van den Hil E. F., Keijer, J., Bunschoten, A., Vervoort, J. J. et al., Quercetin induces hepatic lipid omega-oxidation and lowers serum lipid levels in mice. PLoS One 2013, 8, e51588. [188] Ying, H. Z., Liu, Y. H., Yu, B., Wang, Z. Y. et al., Dietary quercetin ameliorates nonalcoholic steatohepatitis induced by a high-fat diet in gerbils. Food Chem. Toxicol. 2013, 52, 53–60. [189] Marcolin, E., San-Miguel, B., Vallejo, D., Tieppo, J. et al., Quercetin treatment ameliorates inflammation and fibrosis in mice with nonalcoholic steatohepatitis. J.Nutr. 2012, 142, 1821–1828. [190] Marcolin, E., Forgiarini, L. F., Rodrigues, G., Tieppo, J. et al., Quercetin decreases liver damage in mice with nonalcoholic steatohepatitis. Basic Clin. Pharmacol. Toxicol. 2013, 112, 385–391. [191] Jeong, S. M., Kang, M. J., Choi, H. N., Kim, J. H. et al., Quercetin ameliorates hyperglycemia and dyslipidemia and improves antioxidant status in type 2 diabetic db/db mice. Nutr. Res. Pract. 2012, 6, 201–207. [192] Vidyashankar, S., Sandeep, V. R., Patki, P. S., Quercetin ameliorate insulin resistance and up-regulates cellular antioxidants during oleic acid induced hepatic steatosis in HepG2 cells. Toxicol. In Vitro 2013, 27, 945–953. [193] Crespo, I., Garcia-Mediavilla, M. V., Almar, M., Gonzalez, P. et al., Differential effects of dietary flavonoids on reactive oxygen and nitrogen species generation and changes in antioxidant enzyme expression induced by proinflammatory cytokines in Chang Liver cells. Food Chem. Toxicol. 2008, 46, 1555–1569. [194] Kuzu, N., Bahcecioglu, I. H., Dagli, A. F., Ozercan, I. H. et al., Epigallocatechin gallate attenuates experimental nonalcoholic steatohepatitis induced by high fat diet. J. Gastroenterol. Hepatol. 2008, 23, e465–e470. [195] Bose, M., Lambert, J. D., Ju, J., Reuhl, K. R. et al., The major green tea polyphenol, (-)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice. J. Nutr. 2008, 138, 1677–1683. [196] Chen, Y. K., Cheung, C., Reuhl, K. R., Liu, A. B. et al., Effects of green tea polyphenol (-)-epigallocatechin-3-gallate on newly developed high-fat/Western-style diet-induced obesity and metabolic syndrome in mice. J. Agric. Food Chem. 2011, 59, 11862–11871. [197] Xiao, J., Ho, C. T., Liong, E. C., Nanji, A. A. et al., Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur. J. Nutr. 2013, in press. [198] Klaus, S., Pultz, S., Thone-Reineke, C., Wolfram, S., Epigallocatechin gallate attenuates diet-induced obesity in mice by decreasing energy absorption and increasing fat oxidation. Int. J. Obes. (Lond.) 2005, 29, 615–623. [199] Friedrich, M., Petzke, K. J., Raederstorff, D., Wolfram, S. et al., Acute effects of epigallocatechin gallate from green C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com 24 M.-H. Pan et al. Mol. Nutr. Food Res. 2013, 00, 1–25 tea on oxidation and tissue incorporation of dietary lipids in mice fed a high-fat diet. Int. J. Obes. (Lond.) 2012, 36, 735–743. [200] Lin, C. L., Huang, H. C., Lin, J. K., Theaflavins attenuate hepatic lipid accumulation through activating AMPK in human HepG2 cells. J. Lipid Res. 2007, 48, 2334–2343. [201] Luo, X. Y., Takahara, T., Hou, J., Kawai, K. et al., Theaflavin attenuates ischemia-reperfusion injury in a mouse fatty liver model. Biochem. Biophys. Res. Commun. 2012, 417, 287–293. [202] Huong, D. T., Takahashi, Y., Ide, T., Activity and mRNA levels of enzymes involved in hepatic fatty acid oxidation in mice fed citrus flavonoids. Nutrition 2006, 22, 546–552. [203] Mulvihill, E. E., Allister, E. M., Sutherland, B. G., Telford, D. E. et al., Naringenin prevents dyslipidemia, apolipoprotein B overproduction, and hyperinsulinemia in LDL receptornull mice with diet-induced insulin resistance. Diabetes 2009, 58, 2198–2210. [204] Assini, J.M., Mulvihill, E. E., Sutherland, B. G., Telford, D. E. et al., Naringenin prevents cholesterol-induced systemic inflammation, metabolic dysregulation, and atherosclerosis in Ldlr(−)/(−) mice. J. Lipid Res. 2013, 54, 711–724. [205] Cha, J. Y., Cho, Y. S., Kim, I., Anno, T. et al., Effect of hesperetin, a citrus flavonoid, on the liver triacylglycerol content and phosphatidate phosphohydrolase activity in orotic acid-fed rats. Plant Foods Hum. Nutr. 2001, 56, 349–358. [206] Iwatsuka, H., Shino, A., Suzuoki, Z., General survey of diabetic features of yellow KK mice. Endocrinol. Jpn. 1970, 17, 23–35. [207] Guo, H., Li, D., Ling,W., Feng, X. et al., Anthocyanin inhibits high glucose-induced hepatic mtGPAT1 activation and prevents fatty acid synthesis through PKCzeta. J. Lipid Res. 2011, 52, 908–922. [208] Guo, H., Xia,M., Zou, T., Ling,W. et al., Cyanidin 3-glucoside attenuates obesity-associated insulin resistance and hepatic steatosis in high-fat diet-fed and db/db mice via the transcription factor FoxO1. J. Nutr. Biochem. 2012, 23, 349– 360. [209] Zhu, W., Jia, Q., Wang, Y., Zhang, Y. et al., The anthocyanin cyanidin-3-O-beta-glucoside, a flavonoid, increases hepatic glutathione synthesis and protects hepatocytes against reactive oxygen species during hyperglycemia: involvement of a cAMP-PKA-dependent signaling pathway. Free Radic. Biol. Med. 2012, 52, 314–327. [210] Guo, H., Liu, G., Zhong, R., Wang, Y. et al., Cyanidin-3-Obetaglucoside regulates fatty acidmetabolism via an AMPactivated protein kinase-dependent signaling pathway in human HepG2 cells. Lipids Health Dis. 2012, 11, 10. [211] Kim, M. H., Park, J. S., Jung, J. W., Byun, K. W. et al., Daidzein supplementation prevents non-alcoholic fatty liver disease through alternation of hepatic gene expression profiles and adipocytemetabolism. Int. J. Obes.(Lond.) 2011, 35, 1019–1030. [212] Crespillo, A., Alonso, M., Vida, M., Pavon, F. J. et al., Reduction of body weight, liver steatosis and expression of stearoyl-CoA desaturase 1 by the isoflavone daidzein in diet-induced obesity. Br. J. Pharmacol. 2011, 164, 1899– 1915. [213] Takahashi, Y., Odbayar, T. O., Ide, T., A comparative analysis of genistein and daidzein in affecting lipid metabolism in rat liver. J. Clin. Biochem. Nutr. 2009, 44, 223–230. [214] Shin, E. S., Cho, S. Y., Lee, E. H., Lee, S. J. et al., Positive regulation of hepatic carnitine palmitoyl transferase 1A (CPT1A) activities by soy isoflavones and L-carnitine. Eur. J. Nutr. 2006, 45, 159–164. [215] Kim, S., Shin, H. J., Kim, S. Y., Kim, J. H. et al., Genistein enhances expression of genes involved in fatty acid catabolism through activation of PPARalpha. Mol. Cell. Endocrinol. 2004, 220, 51–58. [216] Shin, E. S., Lee, H. H., Cho, S. Y., Park, H.W. et al., Genistein downregulates SREBP-1 regulated gene expression by inhibiting site-1 protease expression in HepG2 cells. J. Nutr. 2007, 137, 1127–1131. [217] Kim, M. H., Kang, K. S., Lee, Y. S., The inhibitory effect of genistein on hepatic steatosis is linked to visceral adipocyte metabolism in mice with diet-induced non-alcoholic fatty liver disease. Br. J. Nutr. 2010, 104, 1333–1342. [218] Yalniz, M., Bahcecioglu, I. H., Kuzu, N., Poyrazoglu, O. K. et al., Preventive role of genistein in an experimental nonalcoholic steatohepatitis model. J. Gastroenterol. Hepatol. 2007, 22, 2009–2014. [219] Lee, Y. M., Choi, J. S., Kim, M. H., Jung, M. H. et al., Effects of dietary genistein on hepatic lipid metabolism and mitochondrial function in mice fed high-fat diets. Nutrition 2006, 22, 956–964. [220] Huang, C., Qiao, X., Dong, B., Neonatal exposure to genistein ameliorates high-fat diet-induced non-alcoholic steatohepatitis in rats. Br. J. Nutr. 2011, 106, 105–113. [221] Ji, G., Yang, Q., Hao, J., Guo, L. et al., Anti-inflammatory effect of genistein on non-alcoholic steatohepatitis rats induced by high fat diet and its potential mechanisms. Int. Immunopharmacol. 2011, 11, 762–768. [222] Mohamed, S. S., Nallasamy, P., Muniyandi, P., Periyasami, V. et al., Genistein improves liver function and attenuates non-alcoholic fatty liver disease in a rat model of insulin resistance. J. Diabetes 2009, 1, 278–287. [223] Arunkumar, E., Karthik, D., Anuradha, C. V., Genistein sensitizes hepatic insulin signaling and modulates lipid regulatory genes through p70 ribosomal S6 kinase-1 inhibition in high-fat-high-fructose diet-fed mice. Pharm. Biol. 2013, 51, 815–824. [224] Ae, P. S., Choi, M. S., Cho, S. Y., Seo, J. S. et al., Genistein and daidzeinmodulate hepatic glucose and lipid regulating enzyme activities in C57BL/KsJ-db/db mice. Life Sci. 2006, 79, 1207–1213. [225] Lei, H., Lu, F., Dong, H., Xu, L. et al., Genistein reverses free fatty acid-induced insulin resistance in HepG2 hepatocytes through targeting JNK. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2011, 31, 185–189. [226] Shang, J., Chen, L. L., Xiao, F. X., Sun, H. et al., Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharmacol. Sin. 2008, 29, 698–706. [227] Jeon, B. T., Jeong, E. A., Shin, H. J., Lee, Y. et al., Resveratrol attenuates obesity-associated peripheral and central C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com Mol. Nutr. Food Res. 2013, 00, 1–25 25 inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454. [228] Cho, I. J., Ahn, J. Y., Kim, S., Choi, M. S. et al., Resveratrol attenuates the expression of HMG-CoA reductase mRNA in hamsters. Biochem. Biophys. Res. Commun. 2008, 367, 190–194. [229] Gomez-Zorita, S., Fernandez-Quintela, A., Macarulla, M. T., Aguirre, L. et al., Resveratrol attenuates steatosis in obese Zucker rats by decreasing fatty acid availability and reducing oxidative stress. Br. J. Nutr. 2012, 107, 202–210. [230] Alberdi, G., Rodriguez, V. M., Macarulla, M. T., Miranda, J. et al., Hepatic lipid metabolic pathways modified by resveratrol in rats fed an obesogenic diet. Nutrition 2013, 29, 562–567. [231] Cho, S. J., Jung, U. J., Choi, M. S., Differential effects of low-dose resveratrol on adiposity and hepatic steatosis in diet-induced obese mice. Br. J. Nutr. 2012, 108, 2166–2175. [232] Ahn, J., Cho, I., Kim, S., Kwon, D. et al., Dietary resveratrol alters lipid metabolism-related gene expression of mice on an atherogenic diet. J. Hepatol. 2008, 49, 1019–1028. [233] Poulsen, M. M., Larsen, J. O., Hamilton-Dutoit, S., Clasen, B. F. et al., Resveratrol up-regulates hepatic uncoupling protein 2 and prevents development of nonalcoholic fatty liver disease in rats fed a high-fat diet. Nutr. Res. 2012, 32, 701–708. [234] Bagul, P. K., Middela, H., Matapally, S., Padiya, R. et al., Attenuation of insulin resistance, metabolic syndrome and hepatic oxidative stress by resveratrol in fructose-fed rats. Pharmacol. Res. 2012, 66, 260–268. [235] Bujanda, L., Hijona, E., Larzabal, M., Beraza, M. et al., Resveratrol inhibits nonalcoholic fatty liver disease in rats. BMC Gastroenterol. 2008, 8, 40. [236] Kang, W., Hong, H. J., Guan, J., Kim, D. G. et al., Resveratrol improves insulin signaling in a tissue-specific manner under insulin-resistant conditions only: in vitro and in vivo experiments in rodents. Metabolism 2012, 61, 424–433. [237] Chen, S., Li, J., Zhang, Z., Li, W. et al., Effects of resveratrol on the amelioration of insulin resistance in KKAymice. Can. J. Physiol. Pharmacol. 2012, 90, 237–242. [238] Wang, G. L., Fu, Y. C., Xu,W. C., Feng, Y. Q. et al., Resveratrol inhibits the expression of SREBP1 in cellmodel of steatosis via Sirt1-FOXO1 signaling pathway. Biochem. Biophys. Res. Commun. 2009, 380, 644–649. [239] Hou, X., Xu, S., Maitland-Toolan, K. A., Sato, K. et al., SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [240] Jung, T. W., Lee, K. T., Lee, M. W., Ka, K. H., SIRT1 attenuates palmitate-induced endoplasmic reticulum stress and insulin resistance in HepG2 cells via induction of oxygenregulated protein 150. Biochem. Biophys. Res. Commun. 2012, 422, 229–232. [241] Shapiro, H., Bruck, R., Therapeutic potential of curcumin in non-alcoholic steatohepatitis. Nutr. Res. Rev. 2005, 18, 212–221. [242] Kuo, J. J., Chang, H. H., Tsai, T. H., Lee, T. Y., Positive effect of curcumin on inflammation and mitochondrial dysfunction in obese mice with liver steatosis. Int. J. Mol. Med. 2012, 30, 673–679. [243] Zeng, C. H., Zeng, P., Deng, Y. H., Shen, N. et al., The effects of curcumin derivative on experimental steatohepatitis. Zhonghua Gan. Zang. Bing. Za Zhi 2011, 19, 454–459. [244] Weisberg, S. P., Leibel, R., Tortoriello, D. V., Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology 2008, 149, 3549–3558. [245] Ramirez-Tortosa, M. C., Ramirez-Tortosa, C. L., Mesa, M. D., Granados, S. et al., Curcumin ameliorates rabbits’s steatohepatitis via respiratory chain, oxidative stress, and TNFalpha. Free Radic. Biol. Med. 2009, 47, 924–931. [246] Qu, X. B., Zhao, S. P., Xu, J., Dong, L. N., Effects of curcumin on secretion of adiponectin and interleukin-6 in human adipose tissues: an in vitro study. Zhong. Xi. Yi. Jie. He. Xue. Bao. 2008, 6, 711–715. [247] Mouzaoui, S., Rahim, I., Djerdjouri, B., Aminoguanidine and curcumin attenuated tumor necrosis factor (TNF)-alphainduced oxidative stress, colitis and hepatotoxicity inmice. Int. Immunopharmacol. 2012, 12, 302–311. [248] Kuo, J. J., Chang, H. H., Tsai, T. H., Lee, T. Y., Curcumin ameliorates mitochondrial dysfunction associated with inhibition of gluconeogenesis in free fatty acid-mediated hepatic lipoapoptosis. Int. J. Mol. Med. 2012, 30, 643–649. [249] Messner, D. J., Rhieu, B. H., Kowdley, K. V., Iron overload causes oxidative stress and impaired insulin signaling in AML-12 hepatocytes. Dig. Dis. Sci. 2013, 58, 1899–1908. [250] Lin, J., Zheng, S., Chen, A., Curcumin attenuates the effects of insulin on stimulating hepatic stellate cell activation by interrupting insulin signaling and attenuating oxidative stress. Lab. Invest. 2009, 89, 1397–1409. [251] Lin, J., Chen, A., Curcumin diminishes the impacts of hyperglycemia on the activation of hepatic stellate cells by suppressing membrane translocation and gene expression of glucose transporter-2. Mol. Cell. Endocrinol. 2011, 333, 160–171. [252] Tang, Y., Chen, A., Curcumin prevents leptin raising glucose levels in hepatic stellate cells by blocking translocation of glucose transporter-4 and increasing glucokinase. Br. J. Pharmacol. 2010, 161, 1137–1149. [253] Tang, Y., Chen, A., Curcumin protects hepatic stellate cells against leptin-induced activation in vitro by accumulating intracellular lipids. Endocrinology 2010, 151, 4168–4177. [254] Kang, Q., Chen, A., Curcumin eliminates oxidized LDL roles in activating hepatic stellate cells by suppressing gene expression of lectin-like oxidized LDL receptor-1. Lab. Invest. 2009, 89, 1275–1290. [255] Kang, Q., Chen, A., Curcumin suppresses expression of low-density lipoprotein (LDL) receptor, leading to the inhibition of LDL-induced activation of hepatic stellate cells. Br. J. Pharmacol. 2009, 157, 1354–1367. [256] Garcea, G., Jones, D. J., Singh, R., Dennison, A. R. et al., Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br. J. Cancer 2004, 90, 1011–1015. C_ 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mnf-journal.com