Golde-PotentialEnergy - University of Pittsburgh
advertisement

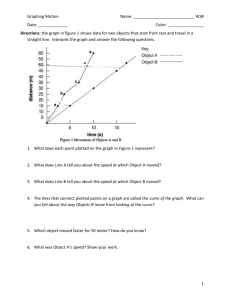
Using CAChe to explore topics associated with “potential energy versus distance” diagrams for diatomic molecules J. Noroski, M. Golde University of Pittsburgh, Pittsburgh, PA Abstract Using Property of: confirmations of a small (< ~100 atom) molecule and Property: potential energy map and Using: exhaustive search with DGauss B88-PW91/DZVP(1 label), CAChe was used to produce two side-by-side windows that allow one to easily track how the potential energy changes as the bond length changes in typical diatomic molecules. With the chosen method, however, we were largely unsuccessful in obtaining the flat regions (dissociation regions) of potential energy plots. Additionally, “locking” the bond distance to determine MO’s at non-equilibrium distances and using potential energy plots to examine qualitative ideas about force constants are briefly explored. Introduction The concept of bond formation due to a lowering of potential energy is not an easy one. This study explores ways in which CAChe might be used to elucidate this concept. CAChe software has the capability to plot a potential energy curve in one window and to show the molecule itself in another window. These small windows can be placed side-by-side, allowing one to follow an indicator (grey circle, see below) as it travels along the curve while viewing the simultaneous change in bond length. This visualization might be helpful to students. Note that for DGauss, i.e., DFT, the “energy” output is the energy required to separate the molecule into isolated nuclei and electrons. The energy is reported in atomic units (1 a.u. = 27.2114 eV = 2625.5 kJ/mol = 627.51 kcal/mol). Now, if one wants to use the potential energy plots from CAChe in a quantitative fashion to obtain information about the bond energy (enthalpy), it is probably best to focus on the energy of the flat region of the plot relative to the energy at the bottom of the potential well. In this way freshmen can determine the bond energy in a way analogous to how they calculate ΔH: products – reactants. This method allows the student and instructor to sidestep any concern over where the zero of potential energy has been set. This method assumes that the SCF calculation produces ground state atoms. That is, correct multiplicity and not ions. Concerns such as these are always relevant to computational chemistry. An exploration of the validity of this assumption is performed by comparing the energy of an isolated, ground state atom 2 to the energy of the flat region of the potential curve. These values should be equal if the molecule has been separated into two atoms with ground state multiplicities. 1 Method/Calculations Before we can perform the potential energy calculation, we must place a label on the part of the molecule in which we are interested. CAChe then performs its calculation on the labeled part of the molecule. First, we drew the diatomic molecule and beautified. Then, we clicked on one atom, held down the shift key, and clicked on the second atom. Next, one must click on “Adjust│Atom Distance”. Then, check the “Define Geometry Label” box and check the “search” button. The values that we entered for the search appear below. Note that more steps require more time, but this improves the appearance of the potential energy plot. The times are also shown below. One can easily see that the time required increases with the number of steps selected and the number of electrons present. This done, we selected Property of: confirmations of a small (< ~100 atom) molecule and Property: potential energy map and Using: exhaustive search with DGauss B88-PW91/DZVP(1 label) and performed the experiment. The multiplicity, which can be accessed in the Procedure Editor, was not changed from the default “automatic”. To determine the energy of a ground state atom we used Property of: chemical sample and Property: all molecular orbitals and Using: current geometry with B88-PW91 DFT wavefunction. When CAChe finishes the atom calculation, it displays an energy in the “Experiment status” box. For H the value was –0.50244 au. Twice this value, –1.00488 au, is what we expect at the dissociation limit. We include the values for twice the ground state energy of H, N, O, F, and Cl in the Results section, allowing us to compare the calculated and theoretical values. The calculation for Br and I would not converge. 2 Results The “famous” homonuclear diatomic molecules: Molecule H2 H2 H2# N2 N2$ O2 O2^ F2* F2 Cl2 Br2 I2† Search Steps Time re (Å) Energy Energy distance (min) (au) (au) __ to __ (Å) at re at final pt. 0.4 to 2.5 16 6 0.794 –1.175 –0.967 0.4 to 6.0 24 12 0.867 –1.169 –0.92298 0.4 to 6.0 35 19 0.720 –1.175 –0.923 0.8 to 3 16 13 1.075 –109.527 –108.981 0.9 to 4 16 17 1.094 –109.530 –108.959 0.7 to 3 16 16 1.275 –150.293 –150.045 0.9 to 3 16 22 1.294 –150.291 –150.040 0.8 to 3 16 23 1.475 –199.551 –199.471 1.2 to 2.75 16 19 1.394 –199.552 –199.420 1.8 to 3.4 16 21 2.050& –920.297 –920.200 1.5 to 3.5 16 49 2.375 –5147.70 –5147.627 1.4 to 4.4 12 150 2.900 –13840.5 –13840.5 # = finished with error; correctly calculated pt. at 5.840 Å $ = finished with error; correctly calculated pt. at 3.42 Å ^ = finished with error; correctly calculated pt. at 2.475 Å * = finished with error; correctly calculated pt. at 2.83 Å & = used average of two points with the same energy † = finished with error; correctly calculated pt. at 4.15 Å Energy of two ground state atoms (au) –1.00488 –1.00488 –1.00488 –109.17018 –109.17018 –150.14032 –150.14032 –199.47464 –199.47464 –920.21104 N/A N/A The hydrogen halides: Molecule HF HCl HBr HI Search Steps Time re distance (min) (Å) __ to __ (Å) 0.5 to 2.5 16 14 1.000 1.0 to 3.65 20 25 1.265 1.0 to 3.0 12 17 1.500 1.2 to 3.2 12 18 1.700 Energy (au) at re –100.451 –460.769 –2574.45 –6920.85 Energy (au) at final pt. –100.226 –460.569 –2574.30 –6920.71 Energy of two g.s. atoms (au) –100.23976 –460.60796 N/A N/A Below, we include most of the plots of potential energy versus interatomic distance. We begin with the trial 1 H2 plot: 3 The trial 3 H2 plot is: Clearly, one can see the expected shape – the sharp rise at short distance and the asymptotic region of the plot. Also, the plot shape definitely improves when more points are included, but the time that is required increases. Note the grey indicator. The N2 plot for trial 2 has a region that is clearly flattening out: 4 O2 (trial 1) showed oscillatory behavior at larger r values. This also occurred for Cl2, but we only show the good curve for Cl2 that we obtained by running the identical Cl2 experiment again. Apparently, anomalous points occur randomly. Trial 1 for O2: 5 For trial 2 O2 gives The F2 trial 1 calculation did well up until the final few points, and then it finished with an error. It successfully plotted 12 points and, its appearance is still good: 6 For trial 2 F2 gives The Cl2 plot: 7 The Br2 plot: The I2 plot: The calculation finished with an error, but 12 points were successfully plotted. (There were 13 possible points.) We next show the hydrogen halide plots. We begin with HF, which won’t converge beyond 2.5 Å: 8 HCl won’t converge beyond 3.7 Å. The plot for HCl: 9 The HBr plot: The HI plot: Discussion There is much that can be discussed here! We begin by assessing the quality of the calculations. Including an appropriate search region (r not too small) and more steps improves the appearance of the plot. Of course, adding more steps increases the time that is required for the calculation. 10 Next, we look at the bond energies, using the “products – reactants” approach that is familiar to freshmen. Trial 3 for H2 has a definite flat region, and the final three points have the value –0.923 au, indicating a bond energy of –0.923 – (–1.175) = 0.252 au = 6.86 eV. Since the bond energy of H2 is 432.1 kJ/mol = 4.48 eV, the agreement is poor. On the other hand, if we compare the minimum energy region with two separated atoms, we obtain a second estimate of the bond energy, of -1.0049 – (-1.175) = 0.170 au = 4.62 eV, which is quite accurate. We conclude that the calculation is least successful for H2 at relatively large internuclear distances. Lacking the flat region at large r, the potential energy curves for the other molecules do not have the desired appearance. This is due to the inability of this method to converge at large r. When larger ranges in r are attempted, the calculation finishes with an error, stopping at the first point where convergence is not found. In some cases “large” r is not much more than only 1 Å or so from re. This is the reason for all of the footnotes for the homonuclear diatomic table in the Results section. In addition, most of the potential curves show an error at large internuclear distances, analogous to that for H2. The experimental bond energies are as follows: N2: 9.76 eV; O2: 5.12 eV; F2: 1.60 eV; Cl2: 2.51 eV; Br2: 2.00 eV; HF: 5.87 eV; HCl: 4.49 eV. For O2 and N2, there are additional problems concerned with electronic spin. For N2, for instance, the experimental ground state dissociates to two N atoms with quartet multiplicity: this is unlikely to be reproduced by the CAChe calculation. In summary, we can see that without fail the bond energy that is predicted by our chosen method is too high. For this reason it appears that quantitative analysis with this method is not feasible or fruitful – certainly for freshman. Qualitative analysis, however, is possible in terms of the curve shape, equilibrium bond lengths, and how the curve changes with changing bond length. Note the I2 curve, which has been included as an illustration of the need to choose your search distance carefully. Using an r that is too small yields an obscured well. The bond lengths, re, at the bottom of the potential agree fairly well with the accepted values of 0.741, 1.10, 1.21, 1.42, and 1.99 Å for H2, N2, O2, F2, and Cl2. For the hydrogen halides the equilibrium bond lengths are 0.917, 1.27, 1.41, and 1.60 Å (from HF to HI). The addition of more points ought to help generate even better agreement. It was one of our initial hopes that, by clicking on various points along the PE curve, we could view the MO surfaces at each point where we click. We were unsuccessful in this attempt. Perhaps, this function exists. One can, however, build a molecule, lock the bond length, and then run an experiment. In a manner similar to that described above, one must click on “Adjust│Atom Distance”, enter the desired bond length, check the “Define Geometry Label” box, check the “lock” button, click “apply”, and then “OK”. One can then run an experiment at the current geometry. As an example, we will show the MO’s that are generated for H2 at bond lengths of 2, 2.5, and 2.75 Å. We used all molecular orbitals and current geometry with B88PW91 wavefunction. The 2 Å MO is 11 The 2.5 Å MO is 12 The 2.75 Å MO: The color difference between the MO’s is without meaning. This type of work might be beneficial to freshman as well as junior level physical chemistry students. As the bond length continues to grows, the student can see the MO as it reverts back to the original 1s atomic orbitals. From the decrease in energy, the student can see that the potential energy is lowered as the electrons are allowed to enter a larger MO, where their motion is less restricted (recall the uncertainty principle) than in an atomic orbital. Lastly, the analysis that we performed allows one to focus on the steepness of the walls of the potential wells for various diatomic molecules and relate that to the different force constant that are present. We consider H2 and N2. If we change the N2 plot (trial 1, not shown above) to the same range along each axis that is used for the H2 plot (trial 1), the much steeper walls for the potential well of N2 are obvious. The revised N2 plot: 13 Of course, there might be one or two such figures in a physical chemistry textbook, but CAChe allows us to explore this concept with many more molecules. Finally, note that the times for these experiments can be a bit long. If one is designing a computer lab with CAChe for potential energy curve calculations, this must be considered. We have, perhaps, only provided a source of further discussion with this paper. If so, that’s fine! Clearly, many aspects of CAChe remain a mystery. 14