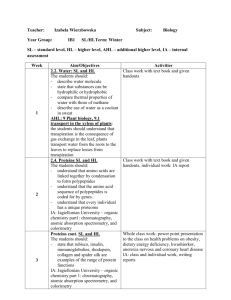
Artificial Pattern Format
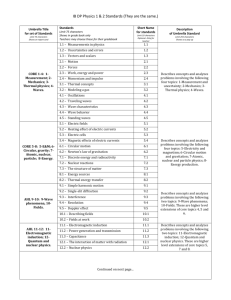
advertisement

Artificial Pattern Formation in E. Coli
David Van Valen
Oren Schaedel
Michael Amori
Abstract
Weiss et al created a band pass filter using genetically engineered building blocks and
used them to create spatial patterns of gene expression. We attempted to reconstruct this
experiment in order to investigate effects of AHL diffusion on the generation of spatial
patterns as a function of distance, location, and timing. To reconstruct this experiment,
we first transformed DH5 cells with plasmids received from the Weiss group. We then
set out to perform two sets of experiments on our transformed cells. First, we wished to
characterize the band pass filter by measuring the expression of GFP as a function of
AHL concentration. Second, we wished to examine the types of spatial patterns we could
generate by varying the location of AHL emitting cells. Unfortunately our experiments
were impeded by a number of technical dilemmas and we were only able to verify the
band-pass nature of one plasmid strand.
Plasmid Design
The design of pHD and pLD allows the concomitant transcription of CI and LacIm1 by
the transcriptional regulator LuxR (which is activated by AHL). LacIm1 inhibits the Lac
promoter on which GFP is present and CI represses the lambda promoter, onto which the
LacI is transcribed. Thus at high AHL concentrations, both CI and LacIm1 are
transcribed shutting off GFP expression. CI inhibition is stronger than LacIm1 shutting
off the native LacI, which represses GFP, therefore at medium AHL concentrations,
LacIm1 is less effective in pLac inhibition and GFP is turned on. At low AHL
concentrations, CI is transcribed at basal levels, allowing transcription of LacI (WT)
which represses GFP transcription.
When describing this as a function of distance, sender cells emit AHL which diffuses as a
gradient throughout the plate. High concentrations are close to the sender cells, and low
concentrations are present far form the cells. Plating cells with both pLD and pHD
around sender cells forms a fluorescent ring due to the gradient of AHL. 3 different
versions of pHD were constructed, high sensitivity (HD3), medium sensitivity (HD2) and
low sensitivity (HD1). HD3 cells have a low copy number of LuxR, therefore higher
AHL concentrations are required to successfully create the high detect filter. HD2 cells
have a WT LuxR, and HD1 cells have a hypersensitive mutant of LuxR, activated by
lower concentrations of AHL.
QuickTi me™ and a
TIFF (U ncompressed) decompressor
are needed to see this picture.
Figure 1 Design of a band-pass filter
Materials and Methods
We developed a number of protocols that helped streamline our experiments
o Transformation
o Transformation of the cells was done by electroporation. Frozen cells were
mixed with 1 ul of each plasmid and labeled strain BD{x}, or transformed
with one plasmid HD. Electroporation was carried out in cuivettes, cells
were transferred to LB (no antibiotics) and after 1 hour of recovery
transferred to liquid LB with appropriate antibiotics. Cells wre incubated
at 37 degrees celcius over night, and plated the next day onto LB-agar
plates with appropriate antibiotics. Glycerol stocks were prepared.
o In order to test whether transformation was successful we grew cells in a
96 well plate for AHL induction assay (discussed later).
o Cell Culture
o Pipette 5 mL of LB into a falcon tube. The antibiotics added depends on
the strain that use used. No antibiotics are added for wildtype cultures,
chloramphenicol for the single plasmid HD, and chloramphenicol plus
kanamycin for the double strain LD. 15uL of chloramphenicol and 10uL
od kanamycin are added per 5mL of media. Once the culture media is
prepared, we inoculate the culture with cells from our frozen stocks. The
cells are grown in the 37 °C room overnight
o Induction
o The cells for the induction experiment should be placed in cell culture the
previous night. We always prepare the wildtype cells in addition to the
transformed cells because measurements of their fluorescence are needed
to calculate the fold change. 30 uL of the cultured cells are placed in 8mL
of M9 media with the appropriate antibiotics several hours before the
planned experiment because M9 produces less auto-fluorescence. The
experiment also required a solution of 100mM AHL dissolved in
phosphate buffer.
The M9 solution of cells is then diluted to an OD600 value of .05. For our
experiments we induced the cells with concentrations of AHL ranging
from 100-.01. The most effective way to arrange our plate was to use the
following configuration
A
B
C
D
E
F
G
H
1
X
X
X
X
X
X
X
X
2
100
10
1
.1
0
WT
M9
X
3
90
9
.9
.09
4
80
8
.8
.08
5
70
7
.7
.07
6
60
6
.6
.06
7
50
5
.5
.05
8
40
4
.4
.04
9
30
3
.3
.03
10
20
2
.2
.02
11
10
1
.1
.01
0
WT WT WT WT WT WT WT WT WT
M9 M9 M9 M9 M9 M9 M9 M9 M9
X
X
X
X
X
12
X
X
X
X
X
X
X
X
To achieve these concentrations, we first performed a 1:10 dilution of the
100mM AHL stock. It should be noted that if we wished to explore
different orders of magnitude, all we have to do is either perform a higher
dilution of the stock, or just use the stock directly. We then pipetted 220 uL
of cells into row A and 180 uL into rows B-D. The following volumes of
the 10 mM AHL solution were added:
Cell
A2
A3
A4
A5
A6
A7
A8
Concentration
(uM)
100
90
80
70
60
50
40
Amount to add
(uL)
2.2
1.98
1.76
1.54
1.32
1.11
.88
A9
A10
A11
30
20
10
.66
.44
.22
Once the AHL is added, we then performed a serial 1:10 dilution of row A.
20 uL from row A are pipetted into row B, 20 uL of row B are pipetted into
row C, and 20 uL from row B are pipetted into row D. The space between
the wells is filled with dH2O and the plate is covered with the special
plastic top generously provided by Dhananjay Tampe to prevent
evaporation during culture. The plate is then cultured overnight in the plate
reader. The proper setup file is accessible on snowdome, as are the
MATLAB files we created to interpret the data.
The final result of the experiment is to calculate the fold change, which is
given by
where all values are taken relative the values of the M9 media.
o Cell Plating: this experiment was orchestrated in order to test whether the band
pass filter functioned as a function of distance from a point source. Cells emitting
AHL were plated in the center of the pitri dish, and BD or HD cells were plated
around. A double layer of M9-agar was plated in the following fashion: the
bottom layer consisted of M9-agar at 1.5% with appropriate antibiotics was
poured and let to dry. A second layer of M9-agar 0.7% was plated on top
premixed with the receiving cells, and chloramphenicol only (sender cells have
only Chloramphenicol resistance). After second layer dried, filter paper was cut at
the diameter of a 1 ml tube, loaded with sender cells and placed in the center of
the Pitri dish. Two plates were made per strain. Controls for this setup were a WT
dish, and a sender dish W/O receiver cells.
Results
QuickTime™ and a
TIFF (U ncompressed) decompressor
are needed to see this pi cture.
Figure 2 Fluorescence response of transformed cells to AHL
Unfortunately, we only had one successful trial of the induction experiment. While we
were unable to calculate fold-change of gene expression in this experiment, the
fluorescence per cell clearly shows a band pass behavior for the BD1 plasmid. From our
first experiment, we thought that the concentration points we used were simply to spread
out to properly observe the peak of BD2, and did not include high enough concentrations
to observe the peak for BD3. We aimed to repeat the experiment for the BD2 and BD3
strains using more a better range of concentrations.
Unfortunately, we ran into a problem during the next set of experiments. We expect our
cells to be in the log phase of growth, but when we ran the experiment again, we
observed a constant OD600, implying no growth. Plotting the OD600 as a function of
AHL concentration, we saw the following for BD2:
QuickTime™ and a
TIFF (U ncompressed) decompressor
are needed to see this pi cture.
Figure 3 OD600 as a function of AHL concentration for BD2
From the graph, we can see that the OD600 decreases as the concentration of AHL
increases? Why is that? After some investigation, we were able to determine that the
acidic ethyl acetate that the AHL was dissolved in was actually dissolving polystyrene.
Almost everything that we used in the lab was made of polystyrene, including the plates
used for the plate reader. We attempted to redo the experiment using lower
concentrations of AHL, and hence less solvent but we were unsuccessful. Our solution
was to order AHL as a solid compound and dissolve it in a phosphate buffer to make a
100mM solution. Working with AHL in a different buffer solves the ethyl acetate
problem, but unfortunately we had issues with cell growth in the M9 media. As a result,
none of our most recent experiments can provide any useful data.
In parallel with our efforts to characterize the plasmids, we also plated them to see if we
would get the pattern formation described by Weiss et al. The experiment was rather
lengthy, and we were only able to attempt it once. We did not see any fluorescence on the
plates. Our main suspicion is that either the cells were unable to grow in the media, or
that the production of AHL and its diffusion through the agar was so high and so fast that
we missed the window where the concentration in the plate was within the necessary
concentration range.
Conclusions
Since we don’t have many results, we can advise of steps to improve future attempts.
1. AHL should be ordered a week in advance, solidified in PBS, pH 6.5 aliquoted
and frozen.
2. Calibrating cells to AHL sensitivity after transformation: on a plate arrange 4
orders of magnitude for strains BD2 and BD3 ranging from 0.01mM to 100 mM
AHL, and 2 orders of magnitude for BD1 from 0.001 to 0.1 mM. Controls are M9
medium, WT (untransformed) DH5 cells and relevant strain without AHL
induction.
3. Testing fluorescence as a function of distance: prepare M9 medium autoclave
with 1.5% agar, and a stirrer, let sit in heat source. Prepare a separate medium of
M9 with 0.7% agar and stirrer, and let sit on a heat source. Once it reaches a
temperature you’re comfortable to leave your hands on for a few seconds (don’t
stick a thermometer in side), add relevant antibiotics and pour 1.5% medium into
Petri dish. It takes around 15 minutes to dry. In that time, spin down sender cells,
and resuspend pellet in fresh M9 in 20 fold volume decrease. Pour 0.7% agar-M9
mixed with relevant strain onto first layer and let dry. During that time, plate
100ul of cells onto filter paper. We recommend that the filter paper used should
be received as small circles pre-cut and trimmed and sterile. Cutting filters on-site
slices filter threading which was observed after 2 days (filter fiber was seen in 10x
magnification 1 day after plating). Furthermore cutting the filter increases the risk
of contamination. According to the materials and methods section in Weiss et al, a
fluorescence response was observed after 15 hours. This is due to transformation
efficiency and should not be taken at face value.
We had several experiments which came to mind:
1. Placing filter paper at various locations and observing pattern formation.
2. Plating strains at different times with different fluorescence colors.
3. Sharpening the bands on the agar plates: since the fluorescence is a function of the
diffusion coefficient of agar (which can be altered as a function of agar %), a
relatively high agar concentration should sharpen the bands of the strains. It
would be interesting to see if there is a difference between the sharpness levels of
the different strains as a function of agar concentration.
4. If we were able to obtain an inhibitor of AHL, we may be able to engineer
symmetry breaking in our system. If you look at the diffusion equation, once you
include a term that accounts for the reaction, then you break the invariance of the
equation under actions of SO2. This means that the concentration of AHL would
no longer be invariant under rotations and you would expect the spatial pattern of
gene expression to experience the same symmetry breaking. This experiment
would be even cooler if were able to engineer the cells to be genetically identical
because then our system would be able to mimic the symmetry breaking you see
in development.