Supplementary Methods and Appendix A Data extraction For each
advertisement

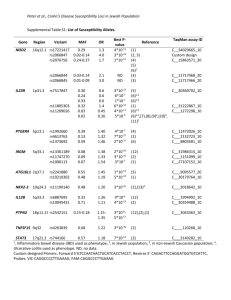
Supplementary Methods and Appendix A Data extraction For each eligible association and each eligible article thereof we recorded: the PMID of each pertinent article, the name of the first author, the journal and the year of publication, the SNP or other genetic variant analyzed, the chromosomal region, the implicated gene(s) loci (as judged by the GWAS investigators), the genetic model selected, the phenotype of interest, and the ancestral group(s) of the included subjects. Data extraction was performed by two investigators with a third investigator arbitrating in discrepancies and ambiguous information. Selection of associations - clarifications Whenever two or more eligible GWAS existed on the same association and both were eligible, we merged all the data from these investigations. The same applied for GWAS that did not fulfill the GWS criterion, but one or more other GWAS on the same association existed that fulfilled the GWS criterion. For eligible associations appearing in back-to-back publications (same issue of a journal) and no cross-publication validation was performed, we synthesized by meta-analysis the final replication samples from both publications. We considered both outcomes selected upfront in the discovery stage as well as additional outcomes assessed in the final replication stage; e.g. bipolar datasets could have been included in the replication of schizophrenia-related genes on the grounds of defining a broader severe mental disease phenotype, or SNPs associated with eosinophil count could have been tested subsequently for asthma on the grounds of a common biological pathway. We excluded secondary clinical outcomes that were a subset of the primary clinical outcome (e.g. primary=stroke and secondary=ischemic stroke) or vice versa. If the primary clinical outcome was excluded, secondary clinical outcomes were evaluated for inclusion and, should more than one have qualified for inclusion, we chose to assess only those reported first by the GWAS investigators in the text of the article. When several genetic models of inheritance were eligible (e.g. log-additive, additive, dominant, recessive, model free), we selected the one that was presented by the original investigators as the main analysis. If several models were presented in the main analysis, the allelic (log-additive) model was chosen. Whenever a meta-analysis of populations of the same ancestry was not performed in the original article, we performed a fixed-effects meta-analysis per ancestry group to document eligibility. Selection of replication datasets to avoid the winner’s curse The effect sizes for genetic associations are expected to be inflated when these are selected among those that pass a threshold based on statistical significance or some other selection metric (e.g. p-value, Bayes factor, or false-discovery threshold). When only those SNPs that pass the threshold are carried to the next stage, there can be inflation due to the winner’s curse. The inflation is greater when the power of the data is more limited to show an association at the desired threshold. This inflation could affect the comparison of effect sizes between ancestry groups, if datasets from different ancestry groups come from different stages in the discovery and replication process and these stages have different selection thresholds. Therefore, we grouped datasets in the following three groups. The first group (“agnostic discovery”) included datasets subjected to agnostic testing in a genome-wide association platform with the aim to discover new variants. The second group (“trimming down”) included datasets from intermediate stage(s) where selection of the genetic markers to move forward to the next stage was based upon passing a threshold (p-value or other) in the data in the specific stage or in the combination of the data in the specific stage and previous stage(s). The third group (“replication”) included datasets where variants surviving the previous stage(s) were assessed in one or multiple independent datasets and there was no other subsequent stage where only some best selected SNPs were tested. This third group was split to subgroup 1, where combination of the previous data had not yet reached GWS; and subgroup 2 where combination of the previous data had already reached GWS . Of note, when a new GWAS reported its results on specific SNPs that had been previously found to be GWS in another ancestry group(s), this testing represents replication (third group) even though these SNPs may have been tested as part of the agnostic genome-wide platform. For analyses other than assessing GWS, we excluded the ‘discovery’ and the ‘trimming-down’ datasets, in order to minimize bias arising from inflation of effects. Effect metrics For each population dataset included in an eligible association, we extracted the allele or genotype counts to generate the natural logarithm of the odds ratio (OR) and its standard error for the strength of the association according to the selected genetic model. When allele and genotype data were not given, we used directly the provided OR and 95% CIs, or estimated this information indirectly from other published information (e.g. through the provided OR and P value). Whenever a study had used a family-based design or there was any concern about cryptic relatedness (e.g. in deCODE Icelandic populations), we preferred the relatedness-adjusted estimates over unadjusted estimates. In such cases, when we calculated estimates ourselves, we also recorded the inflation factor lambda for the study so as to adjust the standard error of the effect size (standard error is multiplied by the square root of lambda). For associations with quantitative traits rather than binary phenotypes (e.g., lipid levels, body mass index, weight, height, and waist circumference), we did the same analyses using the standardized mean difference (SMD) instead of OR. The SMD expresses the effect as a multiple of the standard deviation of the measure of interest in the population. The same considerations applied for correction for lambda. Summary odds ratio calculations used the fixed-effects model and the random-effects model methodology. Fixed-effects models assume that all studies aim at evaluating a common underlying genetic effect and results differ by chance alone. Random-effects models anticipate that the studies may have genuine differences in their results; thus, they also incorporate a between-study variance in their estimates. Random-effects models are generally more conservative (that is, they provide wider confidence intervals when there is between-study heterogeneity). APPENDIX A. Eligible genome-wide association study publications. 1-33 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. Cho, Y.S. et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet 41, 527-34 (2009). Dehghan, A. et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372, 1953-61 (2008). Easton, D.F. et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447, 1087-93 (2007). Eeles, R.A. et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet 41, 1116-21 (2009). Frayling, T.M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889-94 (2007). Graham, R.R. et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 40, 1059-61 (2008). Gudbjartsson, D.F. et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448, 353-7 (2007). Gudbjartsson, D.F. et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet 41, 342-7 (2009). Gudbjartsson, D.F. et al. Many sequence variants affecting diversity of adult human height. Nat Genet 40, 609-15 (2008). Gudmundsson, J. et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 39, 631-7 (2007). Han, J.W. et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41, 1234-7 (2009). Harley, J.B. et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40, 204-10 (2008). Hom, G. et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 358, 900-9 (2008). Ikram, M.A. et al. Genomewide association studies of stroke. N Engl J Med 360, 1718-28 (2009). Kozyrev, S.V. et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 40, 211-6 (2008). Lei, S.F. et al. Genome-wide association scan for stature in Chinese: evidence for ethnic specific loci. Hum Genet 125, 1-9 (2009). Lettre, G. et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 40, 584-91 (2008). Loos, R.J. et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet 40, 768-75 (2008). Musone, S.L. et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet 40, 1062-4 (2008). O'Donovan, M.C. et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet 40, 1053-5 (2008). Sanna, S. et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet 40, 198-203 (2008). Satake, W. et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 41, 1303-7 (2009). Simon-Sanchez, J. et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41, 1308-12 (2009). Sleiman, P.M. et al. Variants of DENND1B associated with asthma in children. N Engl J Med 362, 36-44. 25. 26. 27. 28. 29. 30. 31. 32. 33. Stacey, S.N. et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39, 865-9 (2007). Steinthorsdottir, V. et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39, 770-5 (2007). Takeuchi, F. et al. Confirmation of multiple risk Loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes 58, 1690-9 (2009). Tenesa, A. et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40, 631-7 (2008). Unoki, H. et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40, 1098-102 (2008). Weedon, M.N. et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 40, 575-83 (2008). Yasuda, K. et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 40, 1092-7 (2008). Zeggini, E. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40, 638-45 (2008). Zhou, X. et al. HLA-DPB1 and DPB2 are genetic loci for systemic sclerosis: a genome-wide association study in Koreans with replication in North Americans. Arthritis Rheum 60, 3807-14 (2009).






![[CLICK HERE AND TYPE TITLE]](http://s3.studylib.net/store/data/006920558_1-463cb03fce964e9240461fa313156bf5-300x300.png)