Supplementary Methods, Figures and Tables
advertisement

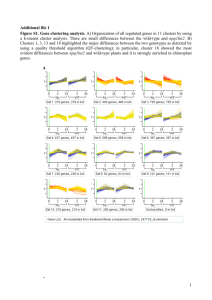
Supplementary Methods, Results, and Figures Development and Validation of a qRT-PCR Classifier for Lung Cancer Prognosis Guoan Chen, MD, PhD,* Sinae Kim, PhD,† Jeremy MG Taylor, PhD,† Zhuwen Wang, MD,* Oliver Lee, MS,† Nithya Ramnath, MD,‡ Rishindra M Reddy, MD,* Jules Lin, MD,* Andrew C Chang, MD,* Mark B Orringer, MD,* David G Beer, PhD,* † Department of Surgery, † Biostatistics, ‡ Internal Medicine, University of Michigan Medical School, Ann Arbor, Michigan, United States of America The first two authors contributed equally to this study. Address for correspondence: David G. Beer, PhD, Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan 48109. E-mail: dgbeer@umich.edu Disclosure: The authors declare no conflicts of interest. Runing title: A 91-Gene Classifier for Lung Cancer Prognosis Supplementary Methods Supplementary Results and Discussion Supplementary Figs S1-S10 1 Supplementary Methods RNA isolation and cDNA synthesis RNA was extracted using miRNeasy Mini Kit (Qiagen, Cat. no. 217004, Valencia, CA). Frozen dissected tumor tissue was placed in 700 µl of QIAzol lysis reagent (Qiagen) and disrupted with a Teflon-glass homogenizer to facilitate dissolution. An on-column DNA digestion with the RNase-Free DNase (Qiagen, cat. no. 79254) was performed. The RNA yield and OD260/280 was analyzed by NanoDrop 3300 Fluorospectrometer (Thermo Scientific, Wilmington, DE) and quality was analyzed by 2100 Bioanalyzer (Agilent Santa Clara, CA). For cDNA synthesis, 2 µg of total RNA was converted to cDNA in a 20 µl volume using the random-primed high-capacity cDNA Reverse Transcription Kit with RNase inhibitor (Applied Biosystems Ins, (ABI), PN 4374966, Foster City, CA). Custom TaqMan low density arrays and quantitative RT-PCR Custom TaqMan Low Density Arrays (384-well micro fluidic cards) were obtained from ABI (PN 4342265 Format 384 was used for 384 genes set qRT-PCR, and PN 4342259 Format 96a was used for 96 genes set qRT-PCR). The primers of survival-related genes including an endogenous loading control gene (18s RNA) and blank controls pre-coated on the cards. The preparation and running of the micro fluidic cards (qRT-PCR) followed the guidelines of product protocols (Applied Biosystems 7900HT Micro Fluidic Card Getting Started Guide, PN 4319399). Briefly, each 100-µl PCR mix for each fill reservoir of the card contained 5 µl cDNA (100 ng of total RNA converted to cDNA), 50 µl TaqMan Universal PCR Master Mix (2x) (ABI, PN 4304437) and 45 µl RNase/DNase-free water. After loading a sample-specific 100 µl PCR mix to each reservoir, the card was centrifuged at 1200 rpm twice and then sealed. The sample containing fluidic cards were then run on the ABI Prism 7900HT Sequence Detection System using a two-temperature cycling protocol: 95o C for 10 min, then 40 cycles of 97o C for 30 sec and 60o C for 1 min. Cycle threshold (Ct) values were generated for each card by automatic selection of a threshold. 2 We have tested the technical performance and the repeatability of the qRT-PCR cardbased for 10 tumor samples and 10 normal samples using different qRT-PCR cards that were examined with the same cDNA sample and also examined different tissue sections of the same tumor. Excellent correlation and reliable values were found (Supplementary Figure S1, S2 and S3). Statistical analysis Initial microarray data processing and filtering The preprocessing and filtering steps were identical to those described in Shedden et al.1 Microarray probe sets from the 439 tumor training set (dChip processed) were obtained and those showing fewer than five samples with greater than a 50 raw expression units were removed. The missing expression measurements were imputed using nearest neighbor averaging. All values were log-2 transformed. Standard deviations were calculated for each probe set and 25% of the probe sets with the smallest standard deviations were removed. In order to select genes consistently measured across the four centers (U of Michigan, Moffitt, Memorial Sloan Kettering and Dana Farber), we calculated the integrative correlation coefficients (R-package: metaArray developed by Choi, H, in extension of LM Cope et al.). 2 Probe sets whose consistency scores, the average of the integrative correlations over the 6 possible pair-wise comparisons across centers, less than 0.25, were removed. After pre-screening, 13,306 probes were left for further analysis. All genes in training and testing datasets are median-centered and MAD-scaled for use in subsequent analyses. Selection of survival-related clusters and genes In order to select an initial subset of genes prognostic for survival for patients with lung cancer on the Affymetrix platform, we employed two statistical methods, clustering of genes and variable selection. We hypothesized that: (a) genes that demonstrate highly-correlated expression can be separated into clusters representing potentially similar biological functional groups; and, (b) clusters and subsets of genes in each selected cluster are prognostic for survival. The selection procedure was carried out as follows: 3 Clustering of genes: Genes were separated into K groups using K-means clustering (http://projecteuclid.org/DPubS?service=UI&version=1.0&verb=Display&handle=euclid.bsmsp/ 1200512992). The number of clusters, K, was chosen to be 300. We picked K=300 since the correlations between the 300 clusters’ average gene expression values are reasonably small. A two-stage selection procedure was used; first, selection of clusters and second, selection of genes within each of the selected clusters. Backward elimination and stepwise regression on the 300 cluster mean expression values in the Cox proportional hazard model were implemented for cluster selection. Stage and age variables were included in the Cox model, but no selection was made on them. Within each of selected clusters, the second selection identified a subset of genes prognostic for survival based on a combination of various criteria: (a) the genes correlation to the cluster mean was greater than 0.5; (b) genes with smaller p-values (always less than 0.1, mostly less than 0.05) in Cox model adjusted for stage and age, within cluster; (c) the median expression and standard deviation across the four centers were similar; (d) more genes were selected from bigger sized clusters (about 15-20%) but every cluster had at least one selected gene; (e) genes with Affymetrix probes designation of _at were preferred over those with designations _s_at and _x_at which indicates gene clusters or sequences that may cross-hybridize. All 5 conditions were considered simultaneously. This approach led to a set of clusters and subsets of genes for each selected clusters considered relevant to patient survival of lung cancer. The goal was to choose approximately 370 genes, which was an appropriate number to measure on one ABI Custom TaqMan 384-well micro fluidic cards. There were some subjective choices (i.e. few important genes highly related to survival with positive beta from Cox model) about which genes to include, criteria (a) and (b) were the most important, with other criteria taken into consideration on a gene by gene basis. Selection of the 47 samples for qRT-PCR measurements Fifty of the 439 samples were selected for qRT-PCR measurement. Amongst those with tissue available to the investigator (n = 178), we identified (n = 41) patients who died before 3 years and (n = 52) who survived at least 5 years. Amongst these we identified 25 of each group who had tissue of sufficient quantity and quality for qRT-PCR measurements. Three of the 50 qRT-PCR measurements did not work satisfactorily, giving 24 who died early and 23 who lived 4 more than 5 years. We selected these 47 samples only consider the survival time and available RNA quantity and quality regardless of other clinical variables such as age, gender, stage and differentiation. There are no different when we compared these 47 samples with remaining 392 samples for these clinical variables. Normalization and imputation of qRT-PCR values Affymetrix measurements on 368 genes were obtained from 439 lung cancer patients, and of these, 47 patients were selected to have complete qRT-PCR measurements for all the 368 genes. The qRT-PCR measurements on the remaining 392 patients were then treated as missing data. A standard strategy for analyzing data with missing values is called multiple imputation.3 The method utilizes all observed data efficiently to fill in the missing qRT-PCR measurements for the 392 patients. This facilitates and simplifies subsequent analyses. It is a standard strategy 4 that has been applied in many applications, including ones with substantial fraction of missing data.5 In this paper the quality of the imputation is greatly enhanced by the high correlation between the Affymetrix and the qRT-PCR data. Furthermore, in the overall scheme of this research, the imputation is used just to help build a method for prediction, once this method is built, the imputed values do not enter into the evaluation of this method on the validation dataset of 101 patients. Before imputation, we developed a method of normalization for the PCR data that resulted in the best correlation between PCR and Affymetrix measurements. First, we subtracted the mean PCR level for 18S, a ubiquitous gene, from each measurement. Next, we calculated the mean expression level for each gene across all 47 samples. These overall means are then subtracted from the individual 18S normalized measurements leaving us with the residuals for each gene and patient. Finally, the average residual for each gene was calculated and subtracted from each individual's 18S normalized PCR expression level for that gene. Through this method we were able to achieve the highest levels of Spearman's correlation between PCR and Affymetrix measurements (Table 2 and Supplementary Figure S5). The imputation was performed using IVEware (http://www.isr.umich.edu/src/smp/ive/) which uses a sequential regression imputation method. We ran the multiple imputation 5 algorithms on the normalized PCR data. The imputation approach incorporated both the Affymetrix and PCR measurements on all 368 genes as well as stage, age, and survival time. Survival time was log transformed. For censored observations survival time was regarded as missing with a lower bound at the censoring time. Ten iterations of the sequential regression scheme were run to create each imputed dataset and a total of 20 imputed sets were created. Alternative strategies of selecting training datasets for building a prediction model that can be used on the 101 validation dataset are (a) build a model just on the 47 subjects, (b) build a model directly from the normalized Affymetrix microarray data of 439 training set (mediancentered and MAD-scaled ) and apply it to the qRT-PCR validation data (similarly MAD-scaled, validation set of 101 patients). Method (a) was not used because these 47 subjects were considered too small to obtain a reliable prediction model, and it was not a random sample of the 439 training set. Method (b) was one of the standard strategies of microarray normalization or batch adjustment (MAD scale for both microarray value and qRT-PCR value) for two different platforms in which both the training set and test set were given the similar scale. We also implemented the approach of method (b) and found that it gave a similar prediction results (Supplementary Table S4 and Supplementary Figure S8) as compared to the multiple imputation strategy used in this study (Table 3 and Figure 2A), providing some reassurance that the strategy (i.e. multiple imputation) we developed was reasonable. Random Survival Forests (RSF) for survival analysis and prediction The random survival forests (RSF) method developed by Ishwaran, et al in R package (http://www.bio.ri.ccf.org/Resume/Pages/Ishwaran/ishwaran.html) (6) was used to relate the expression data to survival and to give a model for prediction. The RSF is an ensemble tree method for analysis of right-censored survival data. Each of the 1000 decision trees of the forest was grown by dividing patients at each node into two groups, where the split is chosen to maximize the survival differences via a log-rank test based on a randomly selected subset of variables at each node. The Random Survival Forests (RSF) method was used for three purposes in this study. For all uses, subject age and stage were included as covariates as well as the genes of interest. The first RSF was used to build a predictor from the Affymetrix data for the 368 genes from the 6 439 patients in the training data. The second RSF was applied to the imputed qRT-PCR data on the 439 patients in the training set and used to aid in the selection of the final 91 genes. The third RSF was built from the imputed qRT-PCR data on the 439 training patients using the 91 genes. This third predictor was then tested on the independent validation dataset of 101 subjects using qRT-PCR. Once the RSF prediction model was built, the model was applied to the test sets for prediction. The cumulative hazard function (CHF) was derived from each tree of the RSF, and an ensemble CHF, an average over 1000 survival trees was determined. Mortality (which we call the mortality risk index in this study, or MRI) was obtained as a weighted sum over ensemble CHF, weighted by the number of individuals at risk at the different time points. Higher mortality values implied higher risk. To test the significance of the mortality risk index it was used as a continuous covariate in a Cox model. For graphical representation we used the mortality risk index to separate patients into three tertiles (high, med, and low risk). Each tree provides a measure of its predictive error as described by Ishwaran 6 with smaller number indicating a better tree. The prediction error is calculated by 1 - C-index (i.e. the Harrell’s concordance index) in the out-of-bag data which were not used for building a tree each time. Variable importance scores (VIMPs) for all the variables used to grow trees were also generated. Large VIMPs indicate variables are good predictors for outcome whereas zero or negative values identify non-predictability. These scores were used to select genes relevant to survival in the final gene selection step of selecting the 91 genes from the 368 genes using the qRT-PCR data. In order to set a cut-off on VIMP for gene selection in final step, a set of 100 “noisy” and independent variables from a uniform distribution was created and added to each of 20 imputed datasets. The VIMPs for those “noisy” variables were expected to be very low. Genes with VIMPs that were larger than the averaged 20 VIMPs for “noisy” variables were selected. The number 91 for the gene selection size was chosen because it is a practical number to measure with the typical size (two 18S RNA, two blank controls and one test primer included in 7 the card) of a qRT-PCR card-based TaqMan Low Density Array (384-well micro fluidic cards) platform. With this platform one can either run four individual samples or run two samples in duplicate on each card. Calculation of area under the curve (AUC) To evaluate the discriminative ability of the predictions from the RSF’s, a receiver operating characteristic (ROC) curve was constructed for the validation dataset of 101 subjects. For this comparison we considered the vital status (dead or alive) of the person at two years, 5 subjects were removed who were censored before 2 years. The ROC curves were constructed by varying the cut-off of the MRI, and the AUC calculated. Supplementary Results and Discussion. Pre-selection and testing of 368 survival-related genes To minimize potential microarray batch affects, we used only one uniformly measured data set that included 439 lung adenocarcinomas as our training set. 1 Utilizing K-means clustering, 300 clusters were generated based on the training data set. A total of 73 clusters, whose average gene expression was found to be related to patient survival, were then selected. From these clusters, a total of 368 genes were selected by the criteria described in the methods. As an initial assessment of the survival predictability of these 368 genes, two independent Affymetrix platform-based datasets (Bild et al., 110 samples 7 and Raponi et al., 130 samples 8) were tested using RSF. The RSF prediction model was developed from the training data set, and then both test sets were applied to the model built from the training set for testing. The prediction error rates were 41.1% and 34.6%, respectively for the Bild and Raponi sets. For both test sets, low, intermediate and high-risk groups were clearly separated by MRI with Kaplan-Meier survival curves (for Bild data, HR = 1.00, 1.16, 1.87 and log-rank test p value = 0.12, low vs high p = 0.05; for Raponi data, HR = 1.00, 2.25, 3.20 and log-rank test p value = 0.005, low vs high p = 0.002) (Supplementary Table S1 and Supplementary Figure S4). This demonstrated the potential use of these 368 genes as predictors for patient survival of lung cancer. 8 Our prediction results using the Raponi test set, although comprising all squamous cell lung cancers, was better than using the Bild data set which included both adenocarcinomas and squamous cell lung cancers. Surprisingly, when we analyzed the 58 adenocarcinomas and 53 SCCs of Bild set separately, we found a better prediction results in 58 adenocarcinomas (test error rate is 33.9%, even better than Rapoin's SCC set 36.3%) as compared to 53 SCCs (test error rate is 43.9%). This indicated that our 91-gene (or 368-gene) classifier has a better prediction in adenocarcinomas than SCC, and it is possibly due to the classifier was built from all adenocarcinomas in the training set. Applying MRI to patient groups who received adjuvant therapy or didn’t in the 101 validation set In our present study, all tumors in the 439 training set and 101 validation set were adenocarcinomas with mixed stage and adjuvant therapy. We tried to develop a qRT-PCR based 91-gene classifier for lung cancer prognosis including early stage cancer and test it on 101 new samples regardless post-operative therapy which was done by most studies including breast cancer.9 Next, we applied the MRI to patient groups who received adjuvant therapy and who didn’t in the 101 validation set. This classifier could significantly separate high- and low-risk groups (median MRI as cutoff, HR = 2.47, 1.04-5.9, p = 0.035) among 58 patients without adjuvant therapy (Supplementary Figure S9A). Unfortunately, like Zhu’s study, 13 it couldn’t separate high- and low-risk groups (HR = 1.14, 0.49 – 2.6, p = 0.77) among 43 patients who received adjuvant therapy (Supplementary Figure S9B). One of the reasons for our 91-gene classifier couldn’t predict (or separate) high- or lowrisk groups of patients who received adjuvant therapy could be the number of patients receiving adjuvant therapy in the 439 training set was too small (only 108 received adjuvant therapy and most of them were stage 1b and above 1b patients). Zhu’s 15-gene classifier didn’t work for adjuvant therapy as well and may due to the lack of patients receiving adjuvant therapy in the training set. We believe the balance of population of the training set is crucial if one want a prediction model works for different subtypes of patients. 9 Effect of chemotherapy for patient groups defined by MRI The ideal classifier should not only predict patient survival but also predict the effect of adjuvant therapy (i.e. Identify a high risk group of patients who become low-risk when given some sort of postoperative therapy), and importantly this kind of classifier should be verified by other different cohorts or clinical trials such as 21-gene or 70-gene classifier for breast cancer. 912 We didn’t find any classifier that identifies a high risk group of patients who become low-risk when given some sort of postoperative therapy in these 20 more published papers listed in Supplementary Table S5 and Table S6. Most studies including this manuscript are trying to find a prognosis signature for patient survival rather than a predictive signature for benefit of adjuvant therapy because the adjuvant therapy was not randomly assigned to patients or the therapy information not clear in the published data sets except for the Zhu et al. study.13 In our data sets, no benefit was observed for patient survival from adjuvant therapy for both the 101 validation set (Supplementary Fig. S10A) and 439 training set (data not show) were observed although there was a 5.4% benefit from adjuvant chemotherapy in another randomized clinical trial study. 14 In order to determine the potential effect of adjuvant therapy for the 3 risk groups defined by the MRI in the 101 validation set shown in Fig. 2A, we compared the survival difference in each risk group regarded whether adjuvant therapy was given. A significantly worse survival for low-risk patients was seen if adjuvant therapy was given (p = 0.01) (Supplementary Fig. S10B). There was also no benefit from adjuvant therapy in high and medium-risk groups (p = 0.8 and 0.5, respectively) (Supplementary Fig. S10C and D). It is difficult to make firm conclusions from this because: (a) the 101 validation set was not a clinical trial study and there wasn’t standard criteria for the type of adjuvant therapy (chemo-/radio) given; (b) the number of samples in each subgroup was too small (from 11 to 22); (c) since the MRI didn’t separate highrisk patients from 43 adjuvant therapy shown in Fig. S9B, it is difficult to examine the benefit of adjuvant therapy; (d) further clinical trials are needed to test if the MRI could predict the benefit from additional post-operative therapy in each risk group such as done in a recent breast cancer study.10 10 References 1. Shedden K, Taylor JM, Enkemann SA, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med 2008;14:822-827. 2. Choi H, Shen R, Chinnaiyan AM, et al. A latent variable approach for meta-analysis of gene expression data from multiple microarray experiments. BMC Bioinformatics 2007;8:364. 3. Raghunathan TE, Lepkowski JM, Van Hoewyk J, et al. A multivariate technique for multiply imputing missing values using a sequence of regression models. . Surv Methodol 2001;27:11. 4. Marshall A, Altman DG, Holder RL, et al. Combining estimates of interest in prognostic modelling studies after multiple imputation: current practice and guidelines. BMC Med Res Methodol 2009;9:57. 5. Taylor JM, Cooper KL, Wei JT, et al. Use of multiple imputation to correct for nonresponse bias in a survey of urologic symptoms among African-American men. Am J Epidemiol 2002;156:774-782. 6. Ishwaran H, Kogalur UB, Blackstone EH, et al. Random Survival Forests. Ann Appl Stat 2008;2:841-860. 7. Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006;439:353-357. 8. Raponi M, Zhang Y, Yu J, et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res 2006;66:7466-7472. 9. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, nodenegative breast cancer. N Engl J Med 2004;351:2817-2826. 10. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with nodenegative, estrogen receptor-positive breast cancer. J Clin Oncol 2006;24:3726-3734. 11. Cardoso F, Van't Veer L, Rutgers E, et al. Clinical application of the 70-gene profile: the MINDACT trial. J Clin Oncol 2008;26:729-735. 12. Sparano JA, Paik S. Development of the 21-gene assay and its application in clinical practice and clinical trials. J Clin Oncol 2008;26:721-728. 13. Zhu CQ, Ding K, Strumpf D, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol 2010;28:4417-4424. 14. Pignon JP, Tribodet H, Scagliotti GV, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol 2008;26:3552-3559. 11 Supplementary Figs S1-S10 Fig. S1A. Image of qRT-PCR results for 18S-RNA control gene for all samples used in this study. The amplification plot for cycle vs delta Rn showing that small variance (all samples were in one cycle range) of 18S-RNA expression among all samples. Fig. S1B. Representative image of qRT-PCR amplification results using ABI 384 card platform with 368 survival related genes, positive and negative control genes for sample A01. 12 Fig. S2A. Representative image of qRT-PCR amplification results using ABI 384 card platform with 91 survival related genes, positive and negative control genes for samples C002-C005. Fig. S2B. Representative image of qRT-PCR results using ABI 384 card platform with 91 survival related genes, positive and negative control genes for samples C002-C005 showing the well position vs CT value for each gene in four samples. 13 Fig. S3A. Repeatability of qRT-PCR for the same RNA sample C023 using all 91 survivalrelated genes (r = 0.98). Fig. S3B. Repeatability of qRT-PCR for the correlation of genes within the same sample but between different portion of the same tumor for sample C023 using all 91 survival-related genes (r = 0.98). 14 Fig. S4. Prediction results on two test sets by Kaplan-Meier survival curve using RSF (mortality risk index separated patient to low, medium, high-risk groups, 1/3rd in each group) built from training set using 368 genes with stage and age. A, HR = 1.00, 1.16, 1.87 and log-rank test pvalue = 0.12, low vs high p = 0.05; B, HR = 1.00, 2.25, 3.20 and log-rank test p-value = 0.005, low vs high p = 0.002. Fig. S5. Scatter plot of the correlation between microarray value and qRT-PCR values for one gene with 47 samples (r = 0.95). 15 Fig. S6. Major biological process of 91 survival-related genes. Fig. S7. ROC curve of 91-gene classifier on qRT-PCR validation set (2 year survival, censored patients removed) for all patients (A) and stage 1 patients (B). 16 Fig. S8. Prediction results of the 91-gene qRT-PCR signature in the 101 sample validation set. Kaplan-Meier survival curve using patient mortality risk index (MRI) from the RSF prediction model built from training set (Microarray data in the training set was normalized using MADscaled method described in Supplementary method) including 91 genes, stage and age. This predictor could significantly separate high, medium and low-risk groups (1/3rd in each group, HR = 1.00, 3.15, 6.12, p = 0.00006) among all 101 patients. 17 Fig. S9. Kaplan-Meier survival curve on 101 validation set using MRI from the RSF prediction model built from training set. This predictor could significantly separate high- and low-risk groups (median of MRI as cutoff value) among 58 patients without adjuvant therapy (A), but couldn’t separate high- and low-risk groups among 43 patients received adjuvant therapy (B). No = no adjuvant therapy, Th = adjuvant therapy, L = low-risk, H = high-risk. 18 Fig. S10. Kaplan-Meier survival curve showing the difference in all patients (A) and different risk groups (B, C and D) identified by mortality risk index (MRI) in 101 validation set presented in Figure 2A. (A), No benefit from adjuvant therapy as compared to patients without adjuvant therapy in 101 validation set (p = 0.05); (B), No benefit, even worse survival from adjuvant therapy as compared to patients without adjuvant therapy in low-risk group (p = 0.015); (C and D), No benefit for survival from adjuvant therapy as compared to patients without adjuvant therapy in medium- or high-risk groups (p = 0.46, and 0.8, respectively). noTh = no adjuvant therapy, Th = adjuvant therapy, L = low-risk, M = medium-risk, H = high-risk. 19