Genetic Fingerprints of Human Embryonic Stem Cells by Copy
advertisement

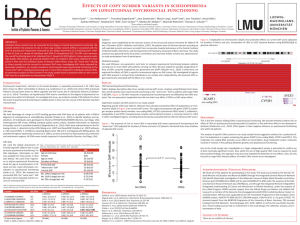
Genetic Fingerprints of Human Embryonic Stem Cells by Copy Number Variant Analysis H.Wu,a* K.J. Kim,a* K. Mehta,b S. Paxia,c A. Sundstrom,c T. Anantharaman,c A. I. Kuraishy,d T. Doan,b J. Ghosh,b A. Pyle,e,h A. Clark,f,h W. Lowry,f,h G. Fan,g,h T. Baxter,b B. Mishra,c Y. Sun,a,h M. A. Teitelld,h Departments of aPsychiatry and Biobehavioral Sciences and Molecular and Medical Pharmacology, dPathology and Laboratory Medicine, eMolecular Immunology and Medical Genetics, fMolecular, Cell and Developmental Biology, and gHuman Genetics, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA; bAgilent Laboratories, Santa Clara, CA, USA; cNYU/Courant Bioinformatics Group, Courant Institute of Mathematical Sciences, NYU, New York, USA; hMolecular Biology Institute, Jonsson Comprehensive Cancer Center, and Broad Center of Regenerative Medicine and Stem Cell Research, UCLA, Los Angeles, CA, USA Presented at The Institute for Stem Cell and Regenerative Medicine (ISCRM) Meeting, University of Washington, Seattle, November 5 2007. Abstract Microarray-based comparative genomic hybridization (array CGH) has become a powerful technique for studying DNA sequence copy number changes in a variety of cell types and tumors. The genome of human embryonic stem cells (hES) has mainly been characterized by karyotyping and comparative genomic hybridization (CGH). Here, we examined HSF1 and HSF6 hES cells using array CGH to determine the extent to which copy number variants (CNVs) account for hES cell genome variability and whether this variability is a potential source for distinct neuronal differentiation potentials. Array CGH data from 5 replicate samples for each hES cell line was analyzed using two independent analytic programs (CGH Analytics 3.4 and BuddhaCGH Pipeline). Using stringent scoring criteria, both programs identified 4 stable CNVs for HSF1 and 5 stable CNVs for HSF6 that were maintained during neuronal precursor cell differentiation or drug selection. The identified CNVs were both shared and unique between the two hES cell lines and included regions of amplifications and deletions, ranging in size from 10-Kb to 1.5Mb, involving 7 different chromosomes. The aCGH data was further used to develop a new statistical algorithm, termed “CNV Fingerprinting”, that was able to identify a distinct CNV signature for HSF1 and HSF6 hES cells. This signature also remained stable for HSF1 cells during differentiation to NPCs or with drug selection. To identify statistically significant biological process that may associate strongly with the group of amplified or deleted copy number regions identified above, a gene ontology (GO)-Stat functional genomic analysis was also performed. Nineteen GO-ID tags were shared between HSF1 and HSF6 hES cells, whereas 23 potential processes were unique to HSF1 and 12 potential processes were unique to HSF6. These associations include processes related to ectodermal and epidermal development for HSF6 CNVs and process relating to mitochondrial control of cell death and other more general developmental process for HSF1 CNVs.