Immunoprecipitation (Adapted from eBioscience)
Introduction
Immunoprecipitation involves the interaction between a protein and its specific antibody, the
separation of these immune complexes with Protein G or Protein A, and the subsequent
analysis by SDS-PAGE.
The protocol below offers a general guideline for immunoprecipitation for native protein at
endogenous level. Optimization may be required for each specific antigen and antibody. The
abundance of a given protein in a sample is variable and a critical factor for obtaining desired
results
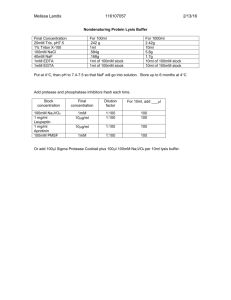
The choice of lysis buffer is critical and dependent on the nature of the protein to be studied.
NP-40, a non-ionic detergent, is the most commonly used detergent in cell lysis buffers.
Increasing the salt concentration, decreasing the detergent concentration, or changing the
detergent to Triton X-100, Saponin, Digitonin, CHAPS etc. are steps that can be taken to
optimize conditions for immunoprecipitation.
Table 1: Binding Characteristics of Some Immunoglobulins Binding Characteristics of Some
Immunoglobulins
Immunoglobulin
Protein A
Protein G
Mouse IgG1
+
++
Mouse IgG2a +++
+++
Mouse IgG2b ++
++
Mouse IgG3
+
+++
Mouse IgM
Mouse IgA
Mouse IgE
Rat IgG1
+
+
Rat IgG2a
+++
Rat IgG2b
++
Rat IgG2c
+
++
Human IgG1
+++
+++
Human IgG2
+++
+++
Human IgG3
+++
Human IgG4
+++
+++
Materials
* Cell lysate or Tissue Lysate
* Protein A or Protein G slurry
* Primary antibody
Buffers
* Lysis buffer
* Proteases Inhibitors (various brands, Roth turns out to be the cheapest and is effective)
* PBS
* SDS-PAGE sample buffer
Instruments
* Centrifuge
* Rocking platform or rotator
Method
Step Ia: Cell Lysate Preparation
1. Harvest approximately 107 cells. Note: The total number of cells per ml and the cell
equivalent loaded per lane of gel should be optimized specifically for each protein and
antibody.
2. . Wash cells with ~10 ml of PBS in a conical tube and spin at 400xg for 10 minutes.
3. Discard supernatant and repeat step 2. Note: If using tissue culture dish adherent
monolayer cells, wash cells in the flask two times with room temperature PBS and
aspirate without disturbing the monolayer.
4. After the second wash, remove supernatant completely and resuspend the cell pellet
in 1ml of cold Lysis Buffer containing 1X Protease Inhibitor Cocktail (final
concentration of 107 cells/ml). Gently vortex the tube. Note: To prevent protease
action effectively, the Lysis Buffer should be pre-chilled and all the following steps
should be kept in the cold. The Protease Inhibitor Cocktail should be added to the
required volume of the cold Lysis Buffer just before use. For adherent monolayers,
add 1ml of cold Lysis Buffer containing 1X Protease Inhibitor Cocktail per 100mm
culture dish.
5. Place the tube or the dish on ice for 30 minutes, with occasional mixing.
6. Spin cell lysate at 10,000xg for 15 minutes at 4°C.
7. Carefully collect supernatant, without disturbing the pellet and transfer to a clean
tube. The cell lysate can be frozen at this point for long-term storage at minus 80°C.
Discard pellet.
Step Ia: Tissue Lysate Preparation
1. Harvest 2 cortices from 2 mouse brains or 6 hippocampi from 3 mouse brains
2. Add lysis buffer and triturate using mini-pestels, until the tissue is completely
homogenized.
3. If there are still chunks of tissue, sonicate 3 x for 2” at medium frequency.
4. Read concentration we typically get a yield of 8-10mg of total protein.
5.
Step II: Cell Lysate Preclearing
1. Transfer 50μl of the Magna_beads Protein G beads slurry (commercially available
from several vendors, we have tried also Roth but this I would definitely not
recommend!!!) to an eppendorf tube and add 450μl cold Lysis Buffer. Spin at 10,000xg
for 30 seconds and remove the Lysis Buffer. Wash one more time with 500μl of cold
Lysis Buffer. Resuspend the beads in 50 μl of cold Lysis Buffer.
2. Add this 50μl of prepared Protein G slurry and 500μl of Cell Lysate to an eppendorf
tube and incubate on ice for 30-60 minutes.
3. Spin at shortly at 4°C and then transfer to the magnetic holder on ice, transfer the
supernatant to a fresh eppendorf on ice.
Step III: Immunoprecipitation
1. Add 5-10μg of antibody to the eppendorf tube containing the cold precleared lysate.
Usually bad Abs do not work even when u put 50ul so make sure u have a good Ab to
start with.
2. Incubate at 4°C for 1 hour.
3. Add 50μl of washed Protein G slurry in prechilled Lysis Buffer (prepared as instructed in
Preclearing Step 1 above).
4. Incubate for 1 hour at 4°C on a rocking platform or a rotator.
5. Spin at shortly at 4°C and then transfer to the magnetic holder on ice.
6. Carefully remove supernatant completely and wash the beads 3-5 times with 500μl of
Lysis Buffer. To minimize background, care should be given to remove the supernatant
completely in these washes.
After the last wash, aspirate supernatant and add 50μl of 1X Laemmli sample buffer to bead
pellet. Vortex and heat to 90-100°C for 10 minutes.
7. Spin at 10,000xg for 2 minutes, then transfer to the magnetic holder, collect supernatant
and load onto the gel.
Supernatant samples can be collected and kept frozen at this point if the gel is to be run
later.
8. Follow manufacturer’s instructions for SDS-PAGE. Stain the gel for visual analysis of
the immunoprecipitated protein.
Immunoprecipitation Buffers
* NP-40 Cell Lysis Buffer:
o 50mM Tris-HCl pH 8.0
o 150mM NaCl
o 1% NP-40
* Protease Inhibitor Cocktail (100X):
o PMSF, 5mg (50μg/ml)
o Aprotinin, 100μg (1μg/ml)
o Leupeptin, 100μg (1μg/ml)
o Pepstatin, 100μg (1μg/ml)
o 100% Ethanol bring up to 1ml, aliquot and keep at minus 20
Trouble shooting
No binding
Possible causes:
Target protein is not present in sample? Make sure sample is appropriate
Target protein lost or degraded? Prepare fresh lysate und add suitable protease inhibitors,
keep sample cold at all times, avoid using frozen lysate
Antibody not suitable for IP? Check datasheet, polyclonal antibodies generally perform
better than monoclonal antibodies
Wrong beads used? Check which beads (e.g. Protein A, Protein G) binds best to the
species and subclass of primary antibody
Antibody concentration too low? Increase concentration (check datasheet for
recommended starting dilution)
Antibody incubation time too short? Increase incubation time
Protein A/G incubation time too short? Increase incubation time
Interfering substances in sample? Make sure sample is free of BME, DTT and other
reducing agents as they may destroy antibody function. Excessive detergents and
extreme pH may interfere with antibody-antigen interaction
Wash step too stringent? Decrease number of wash steps, reduce stringency (e.g. less
detergent and salt or different detergent)
Protein glycosylated? Glycosylation can mask the epitope by steric blocking preventing the
antibody binding. Denature protein by heating up the lysate for several minutes
High background/non-specific bands
Possible causes:
Non-specific binding to agarose beads or antibody? Perform a pre-clear step by incubating
the lysate with Protein A/G/L agarose conjugate without the antibody
Aggregated protein, membrane fractions etc. in lysate? Centrifugate lysate and remove
aggregated protein before adding antibody
Antibody concentration too high? Use a lower concentration
Wash step to short or not stringent enough? Increase number of wash steps, increase
stringency (e.g. higher concentration of detergent or salt)
Contaminated buffers and equipment? Use freshly prepared buffers and clean equipment
Dirty membrane? Do not touch membrane with bare hands
Many specific bands
Possible causes:
Proteolytic degradation? Multiple bands below expected molecular weight may indicate
proteolytic degradation of sample. Prepare new lysate and add fresh protease
inhibitors
55kDa and 28kDa – heavy and light chain? Run sample under non-reducing conditions so
IgG runs at 150 kDa
 0
0