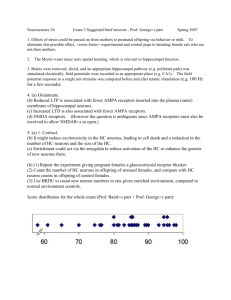
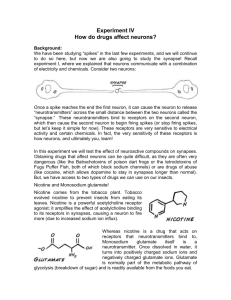
All of these
advertisement

PSYC 413 LECTURE NOTES CH.18 III-17 a. The classic antipsychotics are the phenothiazines (such as chlorpromazine, or Thorazine) and the butyropherones (such as haloperidol, or Haldol). The secondgeneration antipsychotics include clozapine (Clozaril) and risperidone (Risperdal). b. Antipsychotics are only slowly and incompletely absorbed from the gastrointestinal tract. Still, they tend to be administered orally because they are used over extended periods of time. The drugs are administered via intramuscular injection, they are released into the system so slowly that administration is needed only every 4-6 weeks. Metabolism is slow and the half-life of these drugs ranges from 11 to 58 hours. c. Classic antipsychotics (or neuroleptics) tend to antagonize dopamine transmission by competitively blocking DA receptors or by inhibiting DA release. They show a high affinity for D2 receptors, which serve as both normal postsynaptic receptors and autoreceptors in the basal ganglia, nucleus accumbens, amygdala, hippocampus, and cerebral cortex. Some of the second-generation antipsychotics can also act as serotonin antagonists. d. Homovanillic acid (HVA) is a principal DA metabolite. Clinical response to antipsychotic treatment produces an increase in HVA, indicating an initial increase in dopamine metabolism. This is followed by a gradual decrease in HVA with chronic treatment. e. 1. The mesolimbic pathway projects from the ventral tegmental area to the nucleus accumbens and other limbic areas. 2. The mesocortical pathway also projects from the VTA but sends axons to the limbic cortex. 3. The nigrostriatal pathway begins in the substantia nigra and projects to the striatum. 4. The tuberohypophyseal pathway begins in the hypothalamus and projects to he pituitary gland, where it regulates pituitary hormone secretion. f. The most serious side effect of the classic antipsychotics are the movement disorders that resemble the symptoms of Parkinson’s disease. These include tremors, akinesia (a slowing or loss of voluntary movements), muscle rigidity, akathesia (a strong feeling of discomfort in the legs and an inability to sit still), and a loss of facial expression. A second type of motor side effect is tardive dyskinesia, which is characterized by stereotyped, involuntary movements, particularly of the face and jaw but also of the arms, legs, trunk, and neck. There can be neuroendocrine effects such as breast enlargement and tenderness, decreased sex drive, lack of menstruation, increased release of prolactin, and inhibition of growth hormone release, which can be a serious issue when medicating children and adolescents. A relatively lethal condition called neuroleptic malignant syndrome may also occur, characterized by rigidity, altered consciousness, and autonomic nervous system instability (including rapid heart rate and fluctuations in blood pressure). Antipsychotic drugs can also have anti-cholinergic (dry mouth, blurred vision, constipation, difficulty in urination, and decreased gastric secretion and motility) and anti-adrenergic (sedation and low blood pressure that can lead to dizziness, faintness, and blackouts) effects. g. Unlike classical antipsychotic, second-generation antipsychotics: 1. can reduce symptoms of schizophrenia without causing significant extrapyramidal side effects 2. do not produce the predicted results in behavioral screening tests 3. can reduce negative as well as positive symptoms and are effective in treatmentresistant patients h. 1. Selective D2 receptor antagonists: Their selectivity for DA receptors minimizes their effects on the autonomic nervous system and cardiovascular system. However, hormonal side effects tend to be common, and the risk of fatal blood disorders reduces the utility of the drugs. 2. Broad-spectrum antipsychotics: This class of drugs blocks other types of receptors in addition to D2 receptors. Clozapine and risperidone act as serotonin antagonists. Although clozapine is no more effective than classic antipsychotics in treating the positive symptoms of schizophrenia, it is often effective in patients who do not respond to classic antipsychotic treatment. It can also reduce negative symptoms as well as anxiety and tension. Unfortunately, it has a wide variety of side effects, increasing the risk of seizures and producing hypersalivation, weight gain, cardiovascular problems, and a blood abnormality called agranulocytosis. Risperidone has many of the benefits of clozapine without the risk of blood disorders. The most common side effects with risperidone are insomnia, anxiety, agitation, sedation, dizziness, hypotension, weight gain, and menstrual disturbances. It also increase the risk of diabetes. 3. Dopamine system stabilizers: Some of these drugs are dopamine agonistsantagonists, which means that they bind readily to DA receptors but produce less of an effect than DA itself. They reduce both the positive and negative symptoms of schizophrenia with minimal side effects. III-18 a. 1. Dopamine hypothesis: Amphetamines can produce a psychotic reaction in healthy individuals that can be reversed by DA antagonists. Also, schizophrenics report that cocaine and amphetamine make their symptoms worse. There is a correlation between D2 receptor blockade and reduction of symptoms, and antipsychotic treatment induces changes in DA turnover. 2. Neurodevelopmental model: The negative symptoms of schizophrenia resemble the characteristics of patients with lesions of the frontal lobe. Also, the severity of these symptoms is correlated with reduced prefrontal cell metabolism. There is a correlation between poor neuropsychological test performance on tasks requiring frontal lobe function, reduced cerebral blood flow in the prefrontal cortex, and decreased DA function. When the inhibitory cortical feedback is lost, mesolimbic cells increase their activity. Psychotic experiences, hallucinations, perceptual distortions, and irrational fears are associated with electrical discharge in the limbic regions of epileptic patients. 3. Glutamate-dopamine hypothesis: Loss of glutamate input to sub-cortical DA centers leads to excess DA activity and the positive symptoms of schizophrenia. There is evidence of low glutamate levels and reduced glutamate release in schizophrenics, as well as changes in glutamate receptors in several brain areas. Some antipsychotics such as clozapine and haloperidol modify glutamate function. CH.17 III-15 a. All of these drugs reduce neuron excitability by enhancing the inhibitory effects of GABA. On a subjective level, they relieve the feelings of tension, worry, and stress that are typical of anxious individuals, producing a calm and relaxed state, with side effects that include drowsiness and mental clouding, incoordination, and prolonged reaction time. At higher doses, these drugs can also induce sleep, coma, or death. b. 1. ultrashort-acting: highly lipid soluble, can penetrate brain to put an individual to sleep within 10-20 seconds when administered intravenously, with consciousness returning in 20-30 minutes; includes thiopental (Pentothal) and hexobarbital (Evipal) 2. short/intermediate-acting: moderately lipid soluble, producing relaxation and sleep in about 20-40 minutes and lasting 5-8 hours; includes amobarbital (Amytal) and secobarbital (Seconal) 3. long-lasting: have poor lipid solubility so that their onset takes an hour or more and their action lasts 10-12 hours; used to treat seizure disorders; includes Phenobarbital (Luminal) c. The side effects of barbiturates include: 1. a reduction in REM sleep 2. cognitive side effects that include mental clouding, loss of judgment, and slowed reflexes 3. an increase in the number of liver microsomal enzymes, resulting in metabolic tolerance and reduced effectiveness 4. physical dependence and potential for abuse d. Oral ingestion of high doses in a binge fashion may be a substitute for alcohol or may occur in combination with alcohol to enhance the effect. Other users inject the barbiturate intravenously to produce a “high” similar to that achieved with heroin. Injected barbiturates may then replace heroin if the preferred drug is not available, or the two may be used in combination. Simultaneous use of a CNS stimulant like amphetamine and a barbiturate may produce an unusual mood elevation that is considered “smoother” than with the stimulant alone. e. BDZs are useful for presurgical anesthesia during which the patient is conscious but less aware of his/her surroundings and quite relaxed. They are also commonly used before major dental work as well as for a wide range of stressful diagnostic procedures. But the most common use is for anxiety relief. Several of the longer-acting BDZ are also used as hypnotics, shortening the time needed to fall asleep and increasing the duration of sleep time. Some BDZs are useful muscle relaxants and other are anticonvulsants. They can also be used to prevent acute alcohol or barbiturate withdrawal symtoms. f. BDZs relieve the sense of worry and fearfulness as well as the physical symptoms associated with anxiety with less mental clouding, loss of judgment, and motor incoordination than barbiturates. Their anti-anxiety effects show little or no tolerance. BDZs have a higher therapeutic index than other sedative-hypnotics, they rarely produce lethal overdose because they have almost no effect on the respiratory center in the medulla, and they do not increase the number of liver microsomal enzymes that normally lead to metabolic tolerance. g. The abstinence syndrome is relatively mild and develops gradually over several weeks. Symptoms include insomnia, restlessness, headache, anxiety, mild depression, subtle perceptual distortions, muscle pain, and muscle twitches. The most severe symptoms, which resemble those of other CNS depressants, include panic, delirium, and seizures. Those occur in individuals who are abusing the drugs at high doses for prolonged periods, often in combination with other drugs. h. Buspirone is much less effective in reducing the physical symptoms of anxiety than the cognitive aspects of worry and poor concentration. It has several advantages over BDZs, including its usefulness in treating depression that often accompanies anxiety. In addition, its anxiety reduction is not accompanied by sedation, confusion, or mental clouding. Buspirone does not enhance the CNS-depressant effects of alcohol or other depressants. It also has a minimum of severe side effects, and fatalities have not been reported. It has little potential for recreational use or dependence and no rebound withdrawal syndrome. One downside of buspirone is that its onset is quite long, requiring several weeks of daily use. Because it has no cross-tolerance with other sedative-hypnotics, it cannot be used as a replacement drug for those experiencing alcohol or barbiturate withdrawal. It lacks the hypnotic effects needed to treat insomnia, does not control seizures, and has no muscle-relaxant effect. i. The effectiveness of the SSRIs in treating OCD is believed to be related to their ability to enhance serotonin function. III-16 a. GABA is the major inhibitory neurotransmitter in the nervous system. It has receptors on most cells in the CNS. The GABAA receptor complex opens chloride channels and thus causes hyperpolarization of the cell. b. When BDZs bind to their receptor on the GABAA complex, they enhance the effect of GABA by increasing the number of times the chloride channel opens. However, in the absence of GABA, BDZs have no effect. Barbiturates also increase the affinity of the GABAA receptor complex for GABA, but they increase the duration of the opening of GABA-activated chloride channels rather than the number of openings. In addition, they also directly open the chloride channels without GABA. c. The central nucleus of the amygdala orchestrates the components of anxiety, including: autonomic activation, defensive behavior, enhanced reflexes, and activation of the HPA axis. It is associated with the identification of the emotional significance of events and aids in the formation of emotional memories. The prefrontal cortex exerts inhibitory control over the more primitive responses of the limbic system. Without this “cognitive” control, the anxiety response produces more limited patterns of behavior that may not be suitable for coping with modern stressors that are not resolved by a fight-or-flight response. d. Local administration of GABA or a GABA agonist into the amygdala reduces anxiety. e. BDZs can still have anxiety-reducing effects following destruction of the amygdala, suggesting that other brain areas are also involved. f. Inverse agonists can bind to the BDZ site and produce the opposite actions of the drugs, namely increased anxiety, arousal, and seizures. These inverse agonists are presumed to uncouple the GABA receptors from the chloride channels so that GABA is less effective in causing entry of chloride into the cell. Other ligands called endozepines may represent natural anxiety-reducing agents that act at the BDZ site. When individuals lean to cope with stress, anxiety may be reduced by the action of these ligands. g. In patients with panic disorder, PET scans show less BDZ binding in the CNS, particularly in portions of the frontal lobes. These patients are also less sensitive to BDZs on several psychophysiological measures. h. Norepinephrine—The locus coeruleus, where noradrenergic neurons are located, forms reciprocal connections with the amygdala. The LC shows increased firing rates when animals are presented with novel stimuli that signal either threat or reward. Anxiety disorders have been linked to abnormal autonomic response, including higherthan-normal circulating NE. NE has a significant role in the formation of emotional memories, and some anxieolytic drugs work by modulating the firing of cells in the LC. Corticotrophin-releasing factor (CRF)—This hormone controls the HPA axis and behavioral responses to stress and anxiety. Injecting CRF into the ventricles stimulates sympathetic nervous system activity. There are large numbers of CRF receptors in the amygdala, and stressful stimuli produce a release of CRF in the amygdala. Veterans with PTSD have higher-than-normal CRF levels in their cerebrospinal fluid. Injection of CRF into the cranium increases the firing rate of cells in the LC and increased NE turnover. Dopamine—Stress can increase the firing rate of mesocortical dopaminergic cells and increases DA turnover in the prefrontal cortex. Anxiety-producing drugs can increase DA metabolism in the prefrontal cortex without causing changes in DA terminals within the mesolimbic or nigrostriatal tracts. Serotonin—SSRIs desensitize terminal 5-HT autoreceptors in the orbitofrontal cortex, causing an increased release of 5-HT into synapses. CH.16 III-14 a. 1. monoamine oxidase inhibitors 2. tricyclic antidepressants 3. second-generation antidepressants (selective serotonin reuptake inhibitors and atypical antidepressants) b. With appropriate dietary restrictions, MAO-Is can be used safely and often work well for patients who are treatment resistant and who reject the idea of electroconvulsive therapy. In addition, they are also used in the treatment of anxiety and have positive effects in the treatment of bulimia and anorexia nervosa. c. By inhibiting MAO, these drugs increase the amount of monoamine neurotransmitters (NE, DA, and 5-HT) available for release into the synapse. The more common side effects include changes in blood pressure, sleep disturbances (including insomnia), and overeating—especially of carbohydrates—which may lead to excessive weight gain). MAO-Is also enhance the effect of any drug that enhances NE function, which can produce symptoms such as sweating, elevated blood pressure, and increased body temperature. Some serious side effects are due to the inhibition of MAO in the liver, which is responsible for deaminating tyramine—a naturally occurring amine formed as a by-product of fermentation in many foods, including cheeses, certain meats, and pickled foods. These foods must be avoided by individuals taking MAO-Is. Elevated tyramine levels release the higher-than-normal stores of NE at nerve endings, causing a dramatic increase in blood pressure. MAO-Is also inhibit other liver enzymes such as cytochrome P450, which normally degrade such drugs as barbiturates, alcohol, opiates, and aspirin. The effects of these drugs can be prolonged and intensified in the presence of MAO-Is. d. Tricyclic antidepressants inhibit the reuptake of NE and/or 5-HT. These drugs can also block histamine, acetylcholine and alpha-adrenergic receptors. The blockade of histamine receptors produces the sedation and fatigue that are frequent side effects. Anticholinergic side effects can include dry mouth, constipation, urinary retention, dizziness, confusion, impaired memory, and blurred vision. And alpha-adrenergic blockade in combination with the NE reuptake-blocking effects lead to several potentially dangerous cardiovascular side effects, including orthostatic hypotension, tachycardia, and arrhythmias. Toxicity following overdose causes cardiovascular depression, delirium, convulsions, respiratory depression and coma. Heart arrhythmias may produce cardiac arrest and fatalities. e. SSRIs block the reuptake of 5-HT almost exclusively, with very little effect on NE reuptake. The side effects of SSRIs are different than those of the TCAs because they have no effect on NE, histamine, or acetylcholine receptors. Side effects include: anxiety, restlessness, movement disorders, muscle rigidity, nausea, headache, insomnia, and sexual dysfunction, which occurs in 40-70% of patients. They can have life-threatening effects when combined with other serotonergic agonists or drugs that interfere with normal metabolism of the SSRIs. These symptoms, referred to as the serotonin syndrome, are characterized by severe agitation, disorientation and confusion, ataxia, muscle spasms and exaggerated autonomic nervous system functions including fever, shivering, chills, diarrhea, elevated blood pressure, and increased heart rate. f. ECT enhances the function of several neurotransmitter systems, including NE, 5-HT, DA, and GABA, and these changes are probably responsible for its antidepressant effects. The most significant side effect is cognitive impairment, taking the form of confusion and memory loss. ECT may disrupt the ability to retain new information (anterograde amnesia) for several days or weeks after treatment. In addition, significant retrograde amnesia for events preceding the treatment may also occur. g. Lithium enhances 5-HT activity, elevating brain tryptophan, 5-HT, and 5-HIAA levels, as well as increasing 5-HT release, which ultimately alters receptor response in several brain areas. Lithium reduces catecholamine activity by enhancing reuptake and reducing release. Because it is not metabolized but is excreted by the kidneys in its intact form at a rate inversely related to sodium levels, sodium depletion (due to extreme sweating, diarrhea, vomiting, dehydration, etc) may lead to potentially toxic levels of lithium. Side effects are generally mild but may include increased thirst and urination, impaired concentration and memory, fatigue, tremor, and weight gain. Toxic effects are more severe and include camps, vomiting, diarrhea, kidney dysfunction, coarse tremor, confusion, and irritability. CH.14 III-12 a. Mescaline—The peyote cactus, which is found in the Southwestern United States and Mexico, is the source of mescaline. The crown of the peyote cactus, known as a peyote button, is cut off and dried. It is then chewed raw or cooked and then eaten. Or mescaline can be extracted from the catctus and consumed as a relatively pure powder. Psilocybin—Certain types of mushrooms, such as the genus Psilocybe, are either eaten raw, boiled in water to make tea, or cooked with other foods to cover the bitter flavor. Dimethyltryptamine (DMT)—This substance and its analog 5-MeO-DMT, are found in a number of plants that are indigenous to South America. Indigenous tribes there make hallucinogenic snuffs or drinks such as ayahuasca. In the U.S., DMT is sold in powdered form and taken by smoking. b. Albert Hofmann first synthesized LSD (d-lysergic acid diethylamide) from ergot, a substance produced by a parasitic fungus, believing it would be useful as a respiratory stimulant. Instead, he accidentally discovered its hallucinogenic properties. At first, LSD was used as a psychotherapeutic tool to uncover repressed thoughts and feelings. It was also used in the U.S. and Canada as a form of “psychedelic therapy,” intended to help patients gain insights into their problems. The US government considered using it as a mind control agent. LSD’s popularity exploded because of the experimentation of Harvard University psychologist Timothy Leary and the “acid tests” conducted by author Ken Kesey and his band of “merry pranksters.” Recreational use of LSD was banned in 1967. III-13 a. The psychedelic effects of most hallucinogens generally begin within 30 to 90 minutes of ingestion. An LSD or mescaline “trip” typically lasts 6-12 hours or even longer, whereas psilocybin’s effects may dissipate a bit sooner. The effects of smoked DMT are felt within seconds, reach a peak by 5-20 minutes, and are over within an hour or less. b. 1. Onset—occurs about 30-60 minutes after ingestion; visual effects include intensification of colors and appearance of geometric patterns that can be seen with one’s eyes closed. 2. Plateau—the next two hours; subjective sense of time slows down, visual effects become more intense. 3. Peak—generally occurs after three hours and lasts 2-3 hours; characterized by feelings of being in another world, time standing still, bizarre images, possibly synesthesia. 4. Come-down—a phase lasting for two or more hours, in which most of the effects dissipate. c. Most hallucinogenic drugs have either a serotonin-like or catecholamine-like structure. The serotonin-like (or indoleamine) hallucinogens include LSD, psilocybin, psilocin, DMT, 5-MeO-DMT. Mescaline is a catecholamine-like (phenethylamine) hallucinogen. d. LSD is a serotonin agonist that binds to at least eight different serotonergic receptor subtypes. e. Volenweider et al (1998) showed that the visual illusions and hallucinations produced by psilocybin can be blocked by 5-HT2A antagonists (such as risperidone). f. Most hallucinogens, with the exception of DMT, produce rapid tolerance, probably due to down-regulation of 5-HT2A receptors. g. One hypothesis is that hallucinogens decrease the spontaneous firing of cells in the locus coeruleus (which receives and integrates input from all of the major sensory systems and sends information to all areas of the cortex) and at the same time enhances the excitability of these cells by sensory stimulation. An alternative hypothesis is that hallucinogens disrupt normal information processing in a circuit that includes the frontal cortex, striatum, and thalamus. This hypothesis argues that hallucinogens interfere with the normal screening or “gating” of sensory information passing through this circuit, resulting in information overload at the cortical level. h. Hallucinogens are not considered addictive in the standard sense. Users do not binge on them, and it is rare for people to experience the kind of cravings seen with addictive drugs such as cocaine and opiates. They also do not produce physical dependence or withdrawal and are not effective reinforcers in animal tests. i. The most common problem associated with hallucinogens is the occurrence of “bad trips,” which are probably an interaction between the drug, the individual’s emotional state at the start of the drug experience, and the external environment. Another problem is flashbacks, which is defined in the DSM-IV as “the re-experiencing, following cessation of use of a hallucinogen, of one or more of the perceptual symptoms that were experienced while intoxicated.” With LSD there is also the potential for a psychotic breakdown, which generally occurs in individuals who had already been diagnosed with a psychotic disorder or who had experienced pre-psychotic symptoms before taking the drug. CH.13III-9 a. Marijuana—the crude mixture of dried and crumbled leaves, small stems, and flowering tops from the Cannabis sativa plant. Hashish—a relatively pure resin preparation with a very high cannabinoid content, or solvent extracts of leaves or resin that are more variable in their potency. b. Most studies have not found a correlation between breath-hold duration and subjective “high,” although Block et al found a small yet significant difference between durations of seven and 15 seconds. c. Smoking THC produces rapidly rising levels in the blood plasma of the smoker once the THC is absorbed through the lungs. After reaching a relatively fast peak, THC levels decline due to a combination of metabolism in the liver and accumulation of the drug in the body’s fat stores. In contrast, oral consumption leads to prolonged but poor absorption of THC from the gastrointestinal tract, thus resulting in low and variable plasma concentrations that increase much more slowly. d. Cannabinoid receptors are found in the basal ganglia (including the striatum, globus pallidus, and substantia nigra), cerebellum, hippocampus and cerebral cortex. e. CB1 receptors, which are found in the CNS, are metabotropic and tend to result in an inhibition of cyclic AMP formation, inhibition of calcium channels, and activation of potassium channels. They tend to be found on axon terminals instead of postsynaptic receptors, and thus inhibit the release of many different neurotransmitters, including acetylcholine, dopamine, norepinephrine, serotonin, glutamate, and GABA. f. The endocannabinoids are too lipid soluble to be stored in vesicles since they would just pass right through the vesicle membrane. And so they are probably just made and released when needed. g. Endocannabinoid release is triggered by an increase in intracellular calcium levels. After their release, they are taken up by a specific transport protein and then metabolized by several enzymes, such as fatty acid amide hydrolase (FAAH). h. Endocannabinoids may act as retrograde messengers at specific synapses in the hippocampus and cerebellum, carrying information from the postsynaptic to the presynaptic cell. In response to depolarization of the postsynaptic cell, they get released and cross the synaptic cleft, where they inhibit neurotransmitter release from the presynaptic neuron. For instance, in the hippocampus, endocannabinoids are released by pyramidal cells and inhibit release of GABA from interneurons that would normally suppress the firing of these pyramidal cells. The inhibition of GABA permits the pyramidal cell to temporarily fire at a faster-than-normal rate. III-10 a. 1. The “buzz”—a brief period during which the user may feel lightheaded or even dizzy, and may experience tingling in different parts of the body. 2. The “high”— characterized by feelings of euphoria and exhileration, as well as a sense of disinhibition that is often manifested as increased laughter. 3. Being “stoned”—the user feels calm, relaxed, perhaps even in a dreamlike state; sensory reactions experienced in this stage include floating sensations, enhanced visual and auditory perception, visual illusions, and a tremendous slowing of time. 4. The “come-down”— the effects of the drug wear off. b. There is increased blood flow to the skin, which leads to sensations of warmth or flushing. Heart rate and appetite may be stimulated, as well. c. Marijuana can evoke a psychopathological response in a small percentage of users, including strong feelings of anxiety or panic, feelings of paranoia, and delirium (most commonly be first-time users). Also, flashbacks have occasionally been reported. d. In a study by Kirk et al, the expectation of consuming cannabinoids not only enhanced the pleasurable effects of actual cannabinoid administration but it also elicited a more positive response from subjects given a placebo. Also, there is evidence that the maximum level of intoxication occurs when plasma THC levels are already declining. e. Either the brain has a delay reaction in processing information related to THC, or the metabolites of THC contribute in some way to the psychoactive properties of the drug. f. Cognitive—Marijuana intoxication produces illogical or disordered thinking, fragmented speech, and difficulty in remaining focused on a given topic of conversation, as well as deficits in a variety of verbal, spatial, time estimation, and reaction time tasks. Psychomotor—Marijuana can impair tasks such as driving an automobile. g. Regular marijuana users can discriminate cigarettes containing THC from ones containing placebo. Subjects preferred THC cigarettes or capsules over their placebocontaining analogs. h. Cannabinoids stimulate firing of DA neurons in the VTA and the release of DA in the nucleus accumbens. Mu-opioid receptors mediate the rewarding effects of cannabinoids whereas kappa-opioid receptors mediate the aversive effects. i. Endocannabinoid systems may play a role in the process of reinforcement, dependence and relapse for a number of other drugs, including ethanol, opioids, cocaine and nicotine. III-11 a. Prior experience with alcohol and tobacco, emotional problems in the family, heavy drug use in the household and/or by peers, dislike of school, poor school performance, and an early age of first use of marijuana are all risk factors in the development of heavy marijuana use by adolescents. b. Some investigators have observed tolerance following repeated administration of THC to subjects, whereas others have reported that the “high” produced by THC is similar in heavy and light users. c. Dependence—difficulty in stopping drug use, a craving for marijuana, and unpleasant withdrawal symptoms. Withdrawal—irritability, increased anxiety, depressed mood, sleep disturbances, heightened aggressiveness, and decreased appetite. d. Decreased DA cell firing in the VTA and increased corticotrophin-releasing factor in the amygdale by be linked to marijuana abstinence syndrome. e. Dependent THC users typically enter outpatient programs that involve cognitivebehavioral therapy, relapse prevention training, and motivational enhancement therapy. These approaches can be combined with an incentive program in which participants earn vouchers for THC-free urine samples. f. In younger users, greater THC use is associated with poorer grades, more negative attitudes about school and increased absenteeism. It can produce amotivational syndrome. When consumed by smoking, it can produce lung damage. Tar from cannabis smoke actually contains higher concentrations of certain carcinogens than tobacco. Cannabinoid use can suppress immune function and can possibly have reproductive effects (reduced sperm count in men; suppressed release of luteinizing hormone in women). CH.12III-6 a. Smoking has such a powerful reinforcing effect because it can deliver nicotine to the brain in about seven seconds, which is about twice as fast as an intravenous injection. b. Most nicotine is broken down into cotinine by the liver enzyme, cytochrome P450 2A6. Cotinine and other nicotine metabolites are excreted mainly in the urine. c. Nicotine works mainly by activating nicotinic cholinergic receptors, which are ionotropic receptors found in many parts of the brain, including the cerebral cortex, thalamus, striatum, hippocampus, substantia nigra, ventral tegmental area, locus coeruleus, and raphe nuclei. When nicotine binds to nicotinic receptors, it opens sodium and calcium channels. d. Mood: Nicotine is usually found to increase calmness and relaxation in abstinent smokers, probably due to relief from nicotine withdrawal symptoms. In non-smokers, nicotine elicits feelings of heightened tension or arousal along with light-headedness, dizziness, and nausea. Cognition: Abstinent smokers given nicotine show enhanced performance on many kinds of cognitive tasks, especially those involving attention. Much of this enhancement may be due to the alleviation of withdrawal-related deficits. Yet there is some indication that nicotine has positive effects on cognitive performance even in non-smokers, reducing reaction times on visual attention tasks. e. Nicotine receptors located in the VTA stimulate the firing of dopaminergic neurons, which causes increased DA release in the nucleus accumbens. f. Nicotine activates elements of both the sympathetic and parasympathetic nervous system, causing the adrenals to release epinephrine and norepinephrine and activating sympathetic ganglia. This activation elevates blood pressure and can increase a smoker’s risk for cardiovascular disease and strokes. The action of nicotine on parasympathetic ganglia increases hydrochloric acid secretion in the stomach and muscle contractions in the bowel. g. The symptoms of nicotine poisoning include nausea, excessive salivation, abdominal pain, vomiting, diarrhea, cold sweat, headaches, dizziness, disturbed hearing and vision, mental confusion, weakness, fainting, a drop in blood pressure, difficulty breathing, irregular heart rate, and even respiratory failure. h. Nicotine produces acute tolerance that wears off during the night, which is why smokers often report that their first cigarette of the day tends to be the most pleasurable. i. Foulds et al (1997) showed that a subcutaneous injection of nicotine produced symptoms of mild nicotine toxicity in non-smokers but not in smokers. This suggests that smokers have developed a tolerance to the toxic effects of nicotine, and also that this tolerance must occur before an individual can fully experience nicotine’s reinforcing effects. j. The nicotine abstinence syndrome is mediated by nicotinic receptors in both the central nervous system (VTA) and peripheral nervous system (autonomic ganglia). III-7 a. More than 70 million Americans were using tobacco as of 2002, which is approximately 30% of the population age 12 and older. The highest incidence of tobacco use is in the 18-25-year age range. Males are more likely to smoke than females, and the highest rate of smoking is among Native Americans, followed by whites, Hispanics, African Americans and then Asians. There is an inverse correlation between smoking and education. b. Teenagers may take up smoking for any of the following reasons: establishing feelings of independence and maturity; improving self-image and enhancing social acceptance; counteracting stress and boredom; curiosity. c. The fact that cigarettes devoid of nicotine have never been popular is evidence that nicotine is a big part of the reason why people smoke. Also, people smoke more when they switch from regular cigarettes to ones low in tar and nicotine. d. Nicotine abstinence syndrome is characterized by tobacco craving, irritability, impatience, restlessness, anxiety, insomnia, difficulty concentrating, hunger and weight gain. e. Sensory stimuli associated with smoking, such as the taste and smell of inhaling cigarette smoke, can become conditioned to the reinforcing effects of nicotine and are thus able to function as secondary reinforcers themselves. Also, brain imaging studies have shown that smokers have less MAO activity compared to non-smokers, suggesting that some ingredient in cigarettes acts as an MAO inhibitor. f. Cigarette smoking increases the risk of many life-threatening illnesses, including cancer, cardiovascular disease, respiratory diseases (e.g., emphysema and chronic bronchitis). Smokers are at an increased risk for heart attack, stroke, and atherosclerosis. Smoking during pregnancy is the leading cause of low birth weight, which can lead to other complications. g. Behavioral interventions are often focused on smoking prevention, through media campaigns, warnings on cigarette packages, and high taxes. There are various self-help strategies that are limited in their effectiveness, although individual and group counseling programs can be more successful—especially those that provide social support and coping-skills training to their clients. The most common pharmacological intervention for smoking cessation is nicotine replacement, such as nicotine gum, transdermal patches, nicotine spray, and lozenges. When combined with buproprion (Zyban), an antidepressant that inhibits DA and norepinephrine reuptake, the effectiveness of nicotine replacement increases considerably. III-8 a. Caffeine is completely absorbed from the gastrointestinal tract within 30-60 minutes. Caffeine absorption begins in the stomach but takes place mainly within the small intestine. About 1-2% of an administered dose is excreted unchanged. In humans, virtually all caffeine is broken down into its metabolites and then eliminated through the urine (95%), feces, and bodily fluids such as saliva. b. In animals, low doses have a stimulant effect, producing increased locomotor activity; at high doses, this effect is reversed and animals actually show reduced activity. In humans, low doses can have positive subjective effects (feelings of well-being, enhanced energy, increased alertness and self-confidence, and enhanced sociability), whereas high doses produce feelings of tension and anxiety. c. Some of caffeine’s subjective effects may be related to its peripheral physiological actions. These include increased blood pressure and respiration rate, enhanced excretion, and stimulation of catecholamine release (especially epinephrine). d. Two therapeutic uses of caffeine are: It is found in some over-the-counter analgesic agents because it has been shown to be mildly effective in the treatment of nonmigraine headache. Caffeine is also used to regularize breathing in newborn infants with breathing irregularities. e. Heavy coffee drinkers can consume caffeine shortly before bedtime and still fall asleep, whereas a late-night cup of coffee is likely to cause insomnia in someone who does not normally consume much caffeine. Chronic caffeine use also produces tolerance to the cardiovascular and respiratory effects of the drug. f. Caffeine withdrawal symptoms include headache, drowsiness, fatigue, impaired concentration and psychomotor performance, and in some cases, mild anxiety and depression. Intense coffee cravings may also occur. g. Heavy coffee drinking has been linked with increased blood pressure and a heightened risk of coronary heart disease. Caffeine consumption by pregnant women can also lead to low infant birth weight. h. Caffeine has several mechanisms of action. It blocks the enzyme that breaks down cAMP, as well as GABA and adenosine receptors. It can also stimulate calcium release in cells. The blockade of adenosine receptors may be the most important in terms of producing the subjective effects associated with caffeine consumption. i. Adenosine is a neurotransmitter that is believed to be responsible for feelings of drowsiness following periods of prolonged wakefulness. CH.11III-1 a. Source of cocaine: the coca shrub, native to South America. Method of production: After extraction, it is converted to a hydrochloride salt that can be crystallized. Cocaine hydrochloride is water soluble and breaks down when exposed to heat. To avoid this breakdown, the salt is transformed back into cocaine freebase by one of two different methods. The first involves treating the hydrochloride salt with an alkaline solution and then dissolving it in an organic base such as ether. Smoking cocaine obtained in this manner is called freebasing. The second method involves mixing dissolved cocaine HCl with baking soda, heating the mixture, and then drying it to form crack. b. IV injection and smoking produce very rapid absorption of cocaine into the bloodstream. Absorption is somewhat slower with oral administration or snorting. c. Once cocaine is in the bloodstream, it gets broken down quickly by enzymes in the bloodstream and liver. It is also eliminated rapidly, with a half life of 0.5-1.5 hours. That is why the subjective high associated with cocaine tends to last only about 30 minutes. d. When taken together, cocaine and alcohol produce a metabolite called cocaethylene, which acts like cocaine but has a longer half-life. This can exacerbate the toxic effects of cocaine on the heart and other organs. e. Cocaine blocks the reuptake of three monoamine neurotransmitters: dopamine (DA), norepinephrine (NE), and serotonin (5-HT). It binds to the transporters and thus inhibits their function. The order of affinity of cocaine for transporters is: 5-HT > DA > NE. The blockade of DA transporters seems to be the most important for predicting cocaine’s stimulating, reinforcing, and addictive properties. f. At higher concentrations, cocaine can act as a local anesthetic by blocking sodium channels. Two synthetic local anesthetics used commonly in medical and dental practice—procaine (Novocain) and lidocaine (Xylocaine)—were developed from the structure of cocaine. II-2 a. Typical aspects of the cocaine “high” are feelings of exhileration and euphoria, a sense of well-being, enhanced alertness, heightened energy, diminished fatigue, and increased self-confidence. Taken by IV injection or smoking, it also produces a brief “rush” described by some as orgasm-like. At lower doses, it increases sociability and sexual arousal, as well as aggressive behavior. b. Cocaine is sympathomimetic, which means that it produces symptoms of sympathetic nervous system arousal. The physiological consequences of acute administration of cocaine include: increased heart rate, vasoconstriction, increased blood pressure, and increased body temperature. High does can be toxic and even fatal. Some of the potential conseuqnces of heavy cocaine use are seizures, heart falure, stroke, or intracranial hemorrhaging. c. Cocaine blocks DA uptake whereas amphetamine also stimulates DA release. Cocaine produces local anesthetic effects whereas amphetamine does not. d. The rewarding and reinforcing effects of cocaine depend not only on the blockade of dopamine transporters (DAT) by cocaine but also on the interaction of the drug with other molecular targets. e. Volkow et al have used PET imaging to estimate DAT occupancy by cocaine or methylphenidate (Ritalin). When DAT occupancy reaches a minimum level (40-60%), the subject tends to experience a drug-induced “high.” However, the intensity and likelihood of the “high” depend not just on DAT occupancy but also on the rate at which the drug binds to the transporter and also the baseline level of DA activity in the mesolimbic pathway. Higher baseline levels and faster occupancy rates tend to produce a more intense “high.” f. D1 receptors are linked to the locomotor-stimulating effects of cocaine, whereas D2 receptors are linked to the reinforcing effects. III-3 a. The long-term abuse potential of the drug depends on early use of other substances, the availability of the drug, cost, the social and legal consequences of drug use, and the fear of losing control over one’s drug use. b. Psychomotor stimulants can produce either tolerance or sensitization, depending on the pattern of drug administration. Continuous administration of the drug produces tolerance, whereas once-daily use produces sensitization. c. There are two phases of sensitization: In the induction phase, sensitization is established, and in the expression phase, the sensitized response is manifested. Activation of glutamate NMDA receptors and D1 receptors is necessary for the induction of sensitization, whereas expression is dependent at least partly on enhanced reactivity of dopaminergic nerve terminals in the nucleus accumbens so that a given dose of a psychostimulant produces a greater increase in DA levels in the nucleus accumbens in sensitized compared to non-sensitized individuals. d. The three phases of cocaine abstinence are: 1. Crash—the user feels exhausted and suffers from a depressed mood 2. Withdrawal—lack of energy, anhedonia (inability to experience normal pleasures), anxiety, and a growing craving for cocaine that increases the risk of relapse 3. Extinction—symptoms subside, although relapse may still occur e. The negative mood state and craving associated with cocaine stem from temporary decreases in synaptic levels of DA. f. A single dose of cocaine may trigger a stroke or seizure. Chronic use can lead to heart complications, including chest pain, cardiac arrhythmias, cardiac myopathy (damaged heart muscle), and heart attack. Other organs, such as the lungs, gastrointestinal tract, and kidneys may be damaged by cocaine. Frequent snorting may lead to perforation of the nasal septum. Cocaine ingestion by a pregnant woman can have profound effects on the unborn child, leading to attention deficit and other cognitive-behavioral abnormalities. High doses can lead to panic attacks, delusions, and hallucinations (e.g., “cocaine bugs” which create the sensation of something crawling over the surface of one’s skin). Cocaine psychosis may occur. g. Desipramine is a tricyclic antidepressant that inhibits NE uptake. It is used to treat depression symptoms in patients recovering from cocaine abuse or dependence. h. Cocaine abuse is associated with significant impairments in verbal memory, attention, and motor function. i. Psychosocial treatment programs involved individual, group or family counseling designed to educate the user, promote behavioral change, and alleviate some of the problems caused by the cocaine abuse. Cognitive behavior therapies are aimed at restructuring cognitive processes and training the user either to avoid high-risk situations or to employ appropriate coping mechanisms in such situations (relapse prevention). Also available are 12-step programs such as Narcotics Anonymous or Cocaine Anonymous. j. Higgins et al developed a 24-week outpatient multi-component behavioral treatment program based on the premise that drug taking is an operant response that persistes mainly due to the reinforcing properties of the drug. By altering reinforcement contingencies and increasing the availability of non-drug reinforcers, the program promotes abstinence and helps the patient shift to a drug-free lifestyle. Each negative urine test is reinforced by a voucher that can be redeemed for money, YMCA passes, and educational materials. The program has community reinforcement elements that are designed to help with social support, relationships, recreational activities and job opportunities. III-4 a. Common amphetamines include 1-amphetamine (Benzedrine), d-amphetamine (Dexedrine), methamphetamine, MDMA, MDA, and MDE. The two naturally occurring plant compounds similar in structure to amphetamine are cathinone (primary active ingredient in khat) and ephedrine, found in the herb Ephedra vulgaris used in Chinese medicine. b. Ephedra sharply elevates blood pressure and increases the risk of heart attack or stroke. c. Methamphetamine is more potent than amphetamine in its effects on the central nervous system. d. Amphetamine and methamphetamine block the reuptake of catecholamines and also trigger the release of catecholamines from nerve terminals. At very high doses, they can also act as MAO inhibitors. e. Amphetamine causes heightened alertness, increased confidence, feelings of exhileration, reduced fatigue, and a generalized sense of well-being. It can improve performance on simple, repetitive psychomotor tasks, as well as a delay in sleep onset, a reduction in sleep time and in REM period duration. Amphetamine permits sustained physical effort without rest or sleep and can enhance athletic performance. It is highly reinforcing and has a few medical uses, such as in the treatment of narcolepsy and ADHD. In high doses, it can produce a psychotic reaction consisting of hallucinations, paranoia, delusions, and behavioral disorganization. f. Administration of multiple doses of methamphetamine to animals causes long-lasting reductions in the levels of DA, tyrosine hydroxylase, and DAT in the striatum, suggesting damage to DA axons and terminals. It also produces damage to serotonergic fibers in several parts of the brain, including the neocortex, hippocampus, and striatum. III-5 a. MDMA is considered an entactogen because of its ability to elicit a “touching within,” or in more direct terms, and enhanced ability to introspect and to confront disturbing or painful emotions. b.MDMA produces mild euphoria, enhanced sensory perception, increased energy, feelings of well-being and self-confidence, a desire to be with and interact with other people, and sometimes sexual arousal. It can also increase heart rate, blood pressure, body temperature, seating and salivation; and lead to tremors, and bruxism (tightening of the jaw muscles). c. MDMA differs from other amphetamines in that its primary mode of action is to enhance the release of 5-HT and inhibit 5-HT reuptake. It also stimulates DA release, but not as much as 5-HT. d. Methylphenidate and other psychostimulants in low doses can have a calming effect on individuals with ADHD. Striatal DAT density may be higher in individual with ADHD, resulting in faster clearance of DA from the synaptic cleft. NE dysfunction has also been implicated in ADHD. e. Chronic MDMA exposure may cause pruning of serotonergic axons and terminals in various forebrain areas such as the cortex and hippocampus. Although MDMA initially stimulates serotonergic activity, it seems to damage serotonergic fibers in the long-term. MDMA users have lower 5-HIAA levels in the CSF, a decreased density of the 5-HT transporter, and diminished hormonal responses to pharmacological challenge of the serotonergic system. There is some evidence of an association between heavy MDMA use and cognitive deficits on neuropsychological tests—especially on memory tasks. 4/1/08- Review Session II-9e Give a possible reason why reinforcement may occur. The mesolimbic DA pathway has been shown to be linked to reinforcement for psychomotor stimulants like cocaine and amphetamines. Attention to novel stimuli tends to produce activation of this pathway. So, we might theorize that the pathway may have to do with novelty-seeking behavior, which is rewarding. II-9d In progressive-ratio, first subjects are trained in an operant conditioning paradigm using continuous reinforcement. Then the reinforcement schedule shifts from a continuous to a fixed ratio schedule (e.g., a reward for every five responses). Then, the ratio is increased (the number of responses to reward) until the subject stops engaging in the behavior. That is the breaking point. The larger the ratio of the breaking point, the greater the reinforcing properties of a drug. II-2a The preganglionic neurons of both the sympathetic and parasympathetic nervous system ar cholinergic, as are the ganglionic neurons of the parasympathetic. II-2 The regulation of movement in the nigrostriatal tract involves a balance betwen DA and ACh. Too little DA results in too much ACh, and so anticholinergic drugs (orphenadrine, benztropine, trihexylphenydyl) can be helpful in the early stages of Parkinson's, bringing down ACh activity in order to boost DA. II-1a The rate of ACh synthesis is dependent on levels of choline and acetyl coA, which are the precursors, as well as the firing rate of cholinergic neruons. II-13a Non-specific effects of alcohol are related to alcohol's ability to penetrate a membrane and to change the structure of the phospholipids that make up the membrane, thus affecting the membrane's function. Specific effects: binding of ethanol to neurotransmitters, receptors, changing ion channels, stimulating the release of G proteins (second messengers) NEUROTRANSMITTER RECEPTOR AGONIST ANTAGONIST OTHER Acetylcholine Nicotinicionotropic, open Na & Ca channels, located in symp. & parasymp ns succinylcholine (muscle relaxant) D-tubocurarine (curare) blocks transmission at neuromuscular junction AChE inhibitorsreversible: neostigmine, pyridostigmine; irreversible: Sarin and Soman muscarinic-metabotropic, located in hippocampus, thalamus, striatum, smooth muscle muscarine, pilocarbine, arecoline parasympathomimetic (increase in salivation, sweating, abdominal pain) atropine, scopolamine parasympatholytic-- inhibit para ns activity used for dilating pupils; reduces salivation during surgery, produce drowsiness __________________________________________________________________________________ both-medulla, pons (dorsal raphe), midbrain Serotonin 1A- hippocamp Buspirone: agydala, septum reduces metabotropic anxiety stimulates appetite 2A-- cerebral LSD cortex striatum, nucleus, accumbens metabotropic clozapine, risperidone antipsychotic __________________________________________________________________________________ ionotropic MSG PCP or co-agonist Glutamate opens Na & domoic acid Ca channels found in cortex, projects to striatum, hippocampus, thalamus, brainstem LEARNING/ MEMORY ketamine (non-competitive) D-serine glycine __________________________________________________________________________________ A- ionotropic A- muscimol Biculline Tiagabinecortex, hippocampus, (hallucino(produces inhibits substantia nigra, genic) convulsions) GABA GABA striatum, cerebellum, transporters globus pallidus, BDZs, antiolfactory bulb barbiturates, seizure B-metabotropic & other CNS depressants Vigabatrininhibits the enzyme that breaks down GABA _________________________________________________________________________________ Opioids metabotropic, open K and close Ca channels, found throughout the n.s. opiates (morphine, naloxone naltrexone heroin, methadone) CH.10: Opiates II-15 a) When taken orally, morphine and heroin are about equally potent, but heroin becomes much more potent when injected, because it is more fat-soluble and moves across the blood-brain barrier more readily. b) When opiates bind to opioid receptors, they produce various CNS effects, including pain relief, depressed respiration (slower and more shallow), pupil constriction, drowsiness, impaired concentration, reduced awareness of the environment, reduced anxiety, cough suppression, decreased appetite, lower body temp, reduced sex drive, hormonal changes, nausea, vomiting and euphoria (or dysphoria in some cases). The biggest effect on the gastrointestinal tract is constipation. c) There are three opioid subtypes: mu, delta, and kappa. The mu-receptor is distributed throughout the brain and spinal cord and play an important role in analgesia (medial thalamus, spinal cord), reinforcement (nucleus accombens), and cardiovascular and respiratory suppression (brain stem). The delta-receptors are less widely distributed than mu-receptors, and are found mainly in forebrain structures such as the neocortex, striatum, olfactory areas, substantia nigra and nucleus accumbens. Delta-receptors have been implicated in modulating olfaction, in reinforcement, motor integration, and cognitive function. The kappa-receptors is found in the striatum and amygdale, but also in the hypothalamus and pituitary, suggesting that they besides playing a role in regulation of pain perception and mood, they have hormonal effects that influence, among other things, water balance, feeding, and temperature control. d) The endogenous opioids (also called endorphins) include: prodynorphin, POMC, and proenkephalin. They are manufactured in the soma of neurons in the brain, spinal cord, and autonomic nervous system, concentrated in areas related to mood and pain modulation. POMC is found in high concentrations in the pituitary gland. e) Opioid receptors are metabotropic and are linked to G proteins that tend to open potassium channels and close calcium channels. By opening potassium channels, endorphins hyperpolarize the cells. And by closing calcium channels, they block the release of neurotransmitters (exocytosis). Endorphins can act by inhibiting the postsynaptic neuron, activating autoreceptors that inhibit neurotransmitter release, or inhibiting axoaxonic connections in a way that also inhibits neurotransmitter release. YOU DO NOT NEED TO KNOW THE KEY TERMS “transfection” and “receptor cloning.” II-16 a) Early pain (or “first” pain) is the immediate sensory component and late pain (or “second” pain) focuses more on the emotional component. A-delta fibers, which are myelinated, transmit early pain, whereas the unmyelinated C fibers transmit late pain. Early pain goes from the spinal cord to the thalamus and then to the somatosensory cortex, whereas late pain goes from the spinal cord and thalamus to limbic structures such as the hypothalamus and amygdale, as well as the anterior cingulate, which is important in regulating emotion, attention, and motor responses. b) Opiates regulate pain in three ways: 1. within the spinal cord by small inhibitory interneurons (that block transmission up the spinal cord); 2. by two significant descending pathways originating in the periaqueductal gray, that deliver an inhibitory signal down the spinal cord and thus prevent pain signals from travelling up the spinal cord into the brain; 3. at many higher brain sites, such as the amygdala, thalamus and hypothalamus, thus affecting somatosensory, emotional and hormonal aspects of pain response. c) Directly by inhibiting spinal cord neurons or indirectly by sending an inhibitory signal down the descending pathways. d) The PAG is the starting point of two descending pathways associated with pain regulation, one of which goes through the raphe nuclei and the other through the locus coeruleus. II-17 a) Opiates injected into the VTA increase the firing of dopaminergic neurons, probably by inhibiting GABA cells in the VTA that would otherwise inhibit dopaminergic neurons. Kappa-agonists tend to have the opposite effect on dopaminergic neurons, inhibiting their firing rate. b) 1. Tolerance—The effects of opiates diminish rapidly, especially the analgesic and euphoric effects. 2. Sensitization—In some cases, the effects of the opiates increase with repeated administration; this is true for craving. 3) Physical dependence— Abstinence from opiates after continued use produces withdrawal effects that tend to involve hyperactivity. Although withdrawal symptoms are generally not life-threatening, they can be very unpleasant c) Pain, dysphoria, hyperactivity, restlessness, anxiety, flu-like symptoms (see Table 10.2). d) It is unusual to see addictive behavior in chronic pain patients, although they do show tolerance and can also experience withdrawal symptoms. e) The PAG and locus coeruleus are implicated in opiate withdrawal. f) Himmelsbach (1943) suggested that acute morphine administration disrupts the organism’s homeostrasis, but repeated administration of the drug initiates a mechanism that compensates for the original effects and returns the organism to homeostasis. This hypothesis is precursor of opponent-process theory. g) The acute effects of opioids on mu-receptors is hyperpolarization and a reduced firing rate; chronic administration produces a gradual increase in both, as tolerance develops. During opioid withdrawal, the firing rate rises to pre-treatment levels. h) Tolerance—Environmental cues paired with drug administration produce an anticipatory physiological response, because the tolerance to the drug that occurs with repeated use becomes associated with those cues. Abuse—Drug cravings become associated with environmental cues such as needles, which increase metabolic activity in brain areas such as the amygdale and anterior cingulate. Relapse—Withdrawal symptoms such as increased body temperature and restlessness can be conditioned to environmental cues. This increases the likelihood of relapse in familiar settings. II-18 a) Biopsychosocial models of therapy address: 1. The physiological effects of the drug on the nervous system. 2. The psychological status of the individual and his/her unique neurochemical makeup and history of drug use. 3. The environmental factors that provide salient cues for drug taking and powerful secondary reinforcement. b) Methadone is an opiate that eases withdrawal symptoms. Clonidine is an adrenergic agonist that inhibits norepinephrine activity triggered by opiate withdrawals and thus reduces symptoms such as chils, sweating and muscle aches. c) Methadone maintenance is the long-term substitution of one opiate for another. The rationale is that the relief of cravings reduces dangerous drug-seeking behavior. Eventually, the patient is weaned from methadone. The percentage of patients who remain abstinent 1-3 years after methadone withdrawal is estimated to be as high as 80% (and 40% after six years), compared to 12% of patients who drop out of methadone maintenance programs. They also show lower rates of criminal activity, HIV infection, and mortality. d) Methadone was chosen for use in opiate drug treatment programs for the following reasons: 1. Because of cross-dependence with heroin, methadone can prevent withdrawal symptoms associated with abstinence from those drugs. 2. Because of cross-tolerance, taking methadone reduces the euphoric effects of heroin. 3. Oral administration of methadone produces little euphoria or craving and reduces the risks associated with needle use. 4. Methadone is relatively long-lasting, which produces fewer extremes in terms of the drug effects. 5. Methadone is considered medically safe even with long-term use and does not interfere with daily activities. e) LAAM and buprenorphine (Buprenex) are longer-lasting than methadone and produce even more stable pharmacological effects, including milder withdrawal symptoms, and require less frequent administration. f) Narcotic antagonists such as naltrexone have limited usefulness because the use of these drugs requires an addict to willingly substitute an antagonist (that blocks opiate receptors) for a drug with highly reinforcing properties. Antagonists do not elminate craving, and so addicts who are less motivated stop taking these drugs. Only 10% of addicts choose to take part in antagonist treatment programs, and only 27% of these individuals complete a 12-week program. *multidimensional approach—includes a combination of detox, pharmacological support, and group/individual counseling, as well as job training, educational counseling and family therapy, in some cases 3/13/08PLEASE NOTE: THE STRUCTURE OF OUR CLASS MEETINGS WILL BE DIFFERENT THE REST OF THE SEMESTER. INSTEAD OF FORMAL LECTURES, WE WILL BE HAVING INFORMAL DISCUSSIONS OF THE MATERIAL AND I WILL BE ANSWERING YOUR QUESTIONS. I WILL POST NOTES PRIOR TO CLASS. THE BEST STRATEGY IS TO DO THE ASSIGNED READINGS AND LOOK OVER THE LECTURE NOTES BEFORE CLASS. THAT WAY, YOU CAN DECIDE WHICH TOPICS YOU WOULD LIKE TO COVER IN MORE DETAIL. Alcohol is most commonly produced through fermentation of sugars by yeast. Once the concentration reaches 15%, the yeast die off. Distillation is used to increase the concentration, up to 95.6% (alcohol is an azeotrope). How does alcohol interact with different neurotransmitter systems: 1. Glutamate-- Alcohol acts as an antagonist at NMDA receptors; this inhibits memory and learning. One possible mechanism is that alcohol binds directly to glutamate in a way that prevents it from binding to the receptor. Chronic alcohol use is going to result in up-regulation of NMDA receptors, which can lead to excitotoxicity during abstinence. This can account for certain alcohol withdrawal effects such as tremors and seizures. 2. GABA-- Alcohol enhances GABA activity at GABA-A receptors by binding to modulatory sites on the receptor. Chronic alcohol use leads to down-regulation of GABA-A receptors. This too can explain some of the symptoms of withdrawal such as seizures, tremors, and hyperexcitability. 3. Dopamine- Alcohol does stimulate dopaminergic neurons associated with the mesolimbic pathway, which is the brain's reward pathway. It acts as an agonist, increasing the firing rate of dopaminergic neurons in the VTA. That causes increased DA levels in the nucleus accumbens (NA). NA is connected to: a) the amydala-- emotion and motivation; and b) the striatum-motor activity. Chronic alcohol use results in down-regulation of DA neurons in the mesolimbic tract. 4. Opioids- Alcohol enhances opioid actvity by increasing the release of endogenous opioids (endorphins, enkephalins) and by stimulating gene expression of these opioids. Chronic alcohol use reduces the gene expression (similar to down-regulation) and leads to lower endorphin levels. Blocking opioid receptors with antagonists such as naloxone and naltrexone reduces alcohol self-administration in animals. Naltrexone is used as a pharmacotherapeutic treatment for alcohol dependence. 3/11/081970- Comprehensive Drug Abuse Prevention and Control Act (Controlled Substance Act) established five schedules of controlled substances (Schedule I drugs have no accepted medical use and high abuse potential; Schedule V drugs have important medical uses (cough suppressants) and low abuse potential). What is addiction? The term "addiction" has many connotations and so the DSM has replaced with the terms "substance dependence" and "substance abuse"). Substance dependence is characterized by tolerance (more use has less effect), withdrawal (unpleasant symptoms associated with drug abstinence), and craving (uncontrollable desire for the drug). Substance abuse is more a pattern of behavior that has negative repercussions in the life of the user and of people in the user's life. Modern conceptions of addiction focus on these qualities: 1. Compulsive drug-seeking and drug-taking behavior 2. Its relapsing nature 3. Its persistence in spite of harmful consequences Continuum of drug use-- Patterns of drug use, abuse and dependence are individual. It is hard to come up with a consistent pattern that applies to all addicts. Five categories of abuse potential: 1. withdrawal symptoms 2. strength of the drug's reinforcing properties (how much does taking the drug increase frequency of drug-seeking and drug-taking behavior). 3. Degree of tolerance 4. Degree of dependence based on difficulty quitting, relapse rate, and percentage of users who become dependent 5. Degree of intoxication produced 1= most serious 6= least serious The most problematic drug is heroin (1.9), followed by alcohol (2.5), cocaine (2.6), and nicotine (3.35). The least problematic are caffeine (5.0) and marijuana (5.4). There is not a correlation between the abuse potential of a drug and its schedule based on the controlled substance act. Models of addiction Physical dependence model-- The addicted individual uses the drug to relieve withdrawal symptoms. So drug-taking behavior is a form of negative reinforcement (when a behavior stops an unpleasant stimulus). Punishment-- negative stimulus is applied to decrease the frequency of an undesired behavior Negative reinforcement-- negative stimulus is removed when the desired behavior occurs. Criticisms of this model: 1) Some drugs, like cocaine, don't produce strong withdrawal symptoms and yet have serious addiction potential; 2) The model does not explain how dependence happens; 3) it has problems explaining the relapse of adicts who have undergone detox. Positive reinforcement model-- Using the drug reinforces drug-seeking behavior We can measure the reinforcing nature of a drug using the progressive-ratio procedure, increasing the ratio of behaviors to drug deliver (reward) until the subject reaches the "breaking point" in which they don't engage in the behavior. The more reinforcing a drug is, the higher the breaking point in terms of the reinforcement ratio. Limitations of the the positive reinvorcement model are: 1) greater drug craving is often accompanied by greater drug tolerance; 2) it doesn't explain why the negative consequences of using the drug don't counteract the positive reinforcement (advocates of this model argue that the time-frame makes a difference--that short-term consequence are more reinforcing than long-term ones); 3) it doesn't account for individual differences. Incentive-sensitization model-- The neural systems involved in reinforcement ("liking") and craving ("wanting") are different. Repeated use of an addictive drug sensitives the wanting system but not the liking system. Opponent-process model-- Neural mechanism that produces a certain affective response (a-process) triggers an opposite response (b-process). The a-process has an earlier onset and offset than the b-process, and it does not increase with repeated drug use whereas the b-process does. Koob & LeMoal (1997)-- Proposed a variation of opponent-process theory in which the change that occurs with respect to repeated drug use is not an increase in the intensity of the b-process but a change in the "hedonic set point" (the amount of response that feels good). Disease models-- are of two types: 1. Susceptibility models-- there is an inherited susceptibility to addiction 2. Exposure models-- Chronic drug use changes the brain in a way that leads to the pattern of behavior and symptoms associated with addiction. The susceptibility model promotes complete abstinence for addicts whereas the exposure model does not. Three personality-related pathways to addiction: 1. Behavioral disinhibition-- Some personality types, that are prone to impulsive, antisocial, unconventional, and even aggressive behavior, with low levels of selfrestraint and harm avoidance, are more prone to abuse certain drugs. 2. Stress reduction-- Individuals who are high on traits such as stress reactivity, anxiety, and neuroticism, are more likely to be drawn to drug use as a way of dealing with negative emotions. 3. Reward sensitivity-- Sensation-seeking, extraverted, and who seeks drugs for their positive reinforcing qualities. 3/6/08The lecture notes below were posted prior to class because Dr. Shamas is sick today and will not be lecturing on this material. If you have any questions about the information in these notes, please ask them during the review session on April 1. II-7 a) The two major inhibitor amino acid neurotransmitters are GABA (gammaaminobutyric acid) and glycine. b) GABA is synthesized from glutamate in a one-step process catalyzed by glutamic acid decarboxylase, which removes a carbon dioxide molecule from glutamate. c) Tiagabine inhibits the GABA transporter, GAT-1. This inhibition increases GABA activity at the synapse, which in turn inhibits seizure activity. And so tiagabine is used in the treatment of epilepsy. Vigabatrin inhibits the enzyme GABA aminotransferase (GABA-T) that catalyzes one of the steps in the breakdown of GABA. This increases GABA in the brain, which has an anticonvulsant effect. And so vigabatrin has become the primary form of treatment for certain forms of epilepsy. d) GABA is widely distributed in the brain and can be found in the following areas: cerebral cortex, hippocampus, substantia nigra, cerebellum, striatum, globus pallidus, and olfactory bulb. e) GABAA receptors are ionotropic; they open up chloride channels that hyperpolarize the neuron. GABAB receptors are metabotropic. f) Muscimol, which is found in the mushroom Amanita muscaria, is a GABAA agonist. It produces hallucinations, hyperthermia, pupil dilation, elevation of mood, difficulties in concentration, loss of appetite, ataxia, and catalepsy. Bicuculline is a competitive GABAA antagonist that blocks the binding of GABA to the receptor and therefore produces convulsions. g) Benzodiazepines (BDZs) and barbiturates are agonists that bind to a site on the GABAA receptor other than the GABA binding site. They do not open up ion channels on their own but increase the efficacy of GABA (i.e., the ability of the neurotransmitter to open ion channels in the receptor). Benzodiazepines and barbiturates are two classs of CNS depressants that enhance GABAA receptor activity. Inverse agonists are substances that activate a receptor but produce the opposite effect of a typical agonist at that receptor. 3/4/08GlutamateImportant excitatory neurotransmitter, with ionotropic receptors that produce very fast postsynaptic responses. In the cortex, glutamate is used by the pyramidal cells that are the major output neurons of the cortex, projecting to the striatum, limbic system (hippocampus), thalamus, hypothalamus, and brainstem. It's important for learning, memory and synaptic plasticity. Unlike other neurotransmitters, glutamate plays a significant role in protein synthesis and cell metabolism, and so all neurons and glial cells have it, not just glutamatergic neurons. Synthesis-- glutamate is made from glucose via the intermediate, glutamine, using the enzyme glutaminase as a catalyst. Glutaminase remoses an amine group from glutamine. Release-- Three vesicular glutamate transporters (VGLUT1, VGLUT2, and VGLUT3) package glutamate into the vesicles, where it's released into the synaptic cleft. Transport- transporters EEAT1-EEAT5 (excitatory amino acid transporters) are involved in the reuptake of glutamate and aspartate from the synapse. Amyotrophic lateral sclerosis (ALS)-- Lou Gehrig's disease ("Pride of the Yankees), is a neurological disorder in which motor neurons in the spinal cord and cortex degenerate. The disease is correlated with abnormilities in EAAT2, suggesting that two much glutamate is damaging neurons in people with ALS--particularly motor neurons in the striatum. There are three subtypes of glutamate receptors, each named for a specific agonist: 1. AMPA receptors (AMPA is a synthetic amino acid) -- these receptors produce the fastest excitatory responses; they open sodium channels in the postsynaptic neuron. 2. Kainate receptors (kainic acid is derived from seaweed)-- also opens sodium channels. 3. NMDA receptors (NMDA is a synthetic amino acid)-- open sodium and calcium channels, and the calcium can act as a second messenger in the postsynaptic neuron (even the receptor is ionotropic, it has some metabotropic mechanisms). NMDA receptors are unique because they require a second neurotransmitter (either glycine or D-serine) and not just glutamate to open their ion channels. Glycine or serine are considered "co-agonists", meaning that they must bind to the receptor simultaneously to open ion channels. Four binding sites on NMDA receptors: 1. Glutamate 2. Glycine/Serine 3. Magnesium-- This site block the ion channel, and an action potential is needed to dislodge the magnesium 4. Site that binds to PCP or ketamine; both drugs are non-competitive antagonists that block the ion channels of NMDA receptors. Evidence that NMDA receptors are important in learning: 1. Treatment of animals with NMDA receptor antagonists lead to learning impairments, especially on spatial learning tasks (hippocampus has a high density of NMDA receptors) 2. NMDA receptors are involved in long-term potentiation (LTP), which is hypothesized to be involved in learning. LTP is a persistent (more than 1 hour) increase the sensitivity of postsynaptic receptors as a result of stimulating the presynaptic neuron. 3. Genetic strains of mice that have more efficient NMDA receptors show improvement on spatial learning tasks and improved rate of learning fear responses. Monosodium glutamate (MSG) produces brain damage and retinal damage in mice, as well as hormonal problems (stunted growth, obesity, and reproductive problems) due to damage to a part of the hypothalamus called the arcuate nucleus. Excitotoxicity theory-- Too much glutamate keeps the neurons depolarized in a way that damages or kills them. Evidence: injecting glutamate into a brain area of an adult animal lesions that area). NMDA receptors and excessive glutamate are implicated in: 1. necrosis-- cell death by lysis (bursting/splitting of the cell), which can happen after continuous stimulation of both NMDA and non-NMDA glutamatergic receptors for several hours. 2. apoptosis (programmed cell death)-- occurs in fetal development; it happens more slowly and doesn't involve lysis. The cell's nucleus and DNA break up, and then the whole mess is cleared by phagocytes. This is a normal development process that improves the efficiency of the nervous system. 3. ischemia-- interruption of blood flow in the brain, which can be caused by a stroke (localized) or heart attack (global). The result is a massive release of glutamate, which leads to prolonged NMDA receptor activity. In the long term, this activity is extremely damaging to the affected areas. One treatment for ischemia that has been tried successfully in animals involves using NMDA receptor antagonist tol slow down activation of NMDA receptors. Although this works great in animals, it produces symptoms of psychosis in humans. A more effective treatment is use antagonists that block the glycine/serine binding sites on the receptor. This blocks the ion channel but doesn't seem to have as many negative side effects. Domoic acid-- an excitatory amino acid that produces excitoxicity. The toxin is found in relatively small concentrations in algae, in higher concentrations in filter feeders such as clams or mussels that eat algae, and in even higher concentrations in people and animals that eat the shellfish (bioaccumulation) Victims develop the following symptoms: headache, dizziness, muscle weakness, confusion, and permanent loss of short-term memory. In some cases, it's lethal. 2/28/08One last note about ACh: The regulation of movement involves the balance between DA and ACh. Too little DA results in too much ACh, which results in overstimulation of muscle cells. Many symptoms of Parkinson's are actually related to overstimulation of muscle by ACh. In the early stages of Parkinson's, anticholinergic drugs are used to treat some of those symptoms. Orphenadrine, benztropine, trihexylphenidyl are all used to treat early Parkinson's and are all competitive ACh receptor antagonists. SEROTONIN Serotonin (5-HT; 5-hydroxytryptamine) is involved in food intake, reproductive behavior, mood, pain sensitivity, learning memory 5-HT is synthesized in a two-step process (Fig. 6-13) 5-HT levels can be affected by diet. Tryptophan competes with other amino acids to corss the blood-brain barrier, and the rate with which it crosses depends on the ratio of tryptophan to other amino acids in the bloodstream. A diet high in protein decreases the ratio of tryptophan to other amino acids because of all the amino acids contained in the protein. But a diet low in protein and high in carbohydrate increase this ratio: It stimulates insulin release, which helps in the uptake of most amino acids from the bloodstream, but NOT tryptophan. So, more tryptophan stays in the bloodstream relative to other amino acids and is more likely to cross the blood-brain barrier. There are similarities and differences between 5-HT and the catecholamines: 1. Similarities a. Serotonin is transported into the vesicles by the same vecisular transporter proteins as DA and NE. b. The storage of serotin in vesicles protects it from enzymatic breakdown in the same way that it does DA and NE, and so VMAT (vesicular transporter for monoamines) blockers like reserpine that deplete DA and NE also deplete 5-HT. c. Serotonergic autoreceptors are similar to the ones for DA and NE. The ones on the axon reduce the rate of exocytosis, and the ones on the soma and dendrites inhibit cell firing. d. The reuptake of 5-HT is similar to that of DA and NE, even though it involves a different transporter. e. Certain abused drugs such as cocaine and MDMA interact with the transporters for both 5-HT the catecholamines. f. 5-HT is broken down by MAO, as are DA and NE 2. Differences 1. Drugs like fenfluoramine that stimulate the release of 5-HT specifically but not of DA or NE. 2. It has its own transporter (5-HT transporter) 3. This transporter can be attacked specifically by a class of drugs that do not attack the transporters for DA and NE. This class of drugs is called selective serotonin reuptake inhibitors (SSRIs), which includes Prozac (fluoxetine). 4. 5-HT is not affected by the enzyme COMT, which only breaks down catecholamines. 5-hyroxyindoleacetic acid (5-HIAA) is the by-produce of 5-HT breakdown by MAO; it is measured as an indicator of serotonergic activity in the nervous system. More activity means higher levels of 5-HIAA. Where are serotonergic neurons found? Medulla, pons, and midbrain is where the vast majority of cell bodies are found. Most of the serotonergic fibers in the forebrain originate in the dorsal and median raphe nuclei. Serotonergic neurons isn the dorsal raphe are activated during movement and inhibited when an animal is attending to stimuli in the environment; these cells are also inhibited during REM sleep, when the muscles of the body are essentially paralyzed. This suggests that the 5-HT cells in the dorsal raphe facilitate motor activity and suppress sensory activity. 15 types of 5-HT receptors. The best known subtypes are 5-HT1A and 5-HT2A. 5-HT1A receptors are found in several brain areas, including the hippocampus, amygdala, septum, and raphe nuclei. They are metatropic and work by two mechanisms: 1. Reduce the synthesis of cAMP by blocking adenylyl cyclase. This in turn reduces the amount of protein kinases, which phosphorylate different kinds of proteins in the neuron. 2. Increase the opening of potassium channels, which hyperpolarize the membrane. 5-HT1A receptor agonists result in overeating (hyperphagia), which is caused by stimulating 5-HT autoreceptors, and thus inhibiting serotonin release. They can reduce anxiety (buspirone). They can reduce alcohol consumption in rats. And they can cause hypothermia. 5-HT2A receptors are also found in several brain areas, including the cerebral cortex, striatum and nucleus accumbens. 5-HT2A receptor agonists are hallucinogens in humans (example: LSD) 5-HT2A receptor antagonists are sometimes used as antipsychotic drugs. Examples: clozapine and risperidone. They block 5-HT2A receptors as well as D2 dopaminergic receptors. These drugs seem to reduce movement-related side effects produced by other antipsychotic drugs such as haloperidol, which is a dopamine antagonist. 2/26/08Announcements: Grades are now posted online under "413Grades." The grade cutoffs are: A B C D 25-30 20-24 15-19 10-14 Question #4 was thrown out and a point was awarded to EVERYONE. CH.6: Acetylcholine Acetylcholine is made in a single step from choline (derived from fats) and acetyl coenzyme A (acetyl CoA), a reaction that is catalyzed by choline acetyltransferase. This enzyme transfers an acetyl group from the coenzyme to the choline. Neurons that produce acetylcholine (ACh) as a neurotransmitter are called cholinergic neurons. These neurons make more ACh when there is more choline and acetyl coA available and when the neurons are firing at a more rapid rate. Drug Effects on Cholinergic neurons: 1. Interfering with Vesicular Transport Vesamicol blocks the vesicular ACh transporter protein that loads ACh into the vesicles. This decreases ACh levels in the vesicles but increases those levels in the cytoplasm. It also decrease the rate with which ACh is released into the synapse. 2. Interfering with the Breakdown of ACh The enzyme acetylcholinesterase (AChE) breaks down ACh into choline and acetic acid. This enzyme is found in: a. Presynaptic neurons-- where it breaks down excess ACh b. Postsynaptic neurons-- on the membrane, where it breaks down ACh after it's released into the synapse and has bound to a cholinergic receptor. c. Neuromuscular junction-- where ACH is broken down after a muscle contraction so that the muscle can relax. AChE inhibitors fall into two classes: reversible and irreversible. Reversible AChE inhibitors have a short-term effect on the enzyme by binding to it. Irreversible inhibitors can damage or neutralize the enzyme. Reversible AChE inhibitors: i. physostigmine-- naturally-occurring plant extract that was used in West Africa as a form of punishment for those accused of a crime. It crosses the blood-brain barrier and results in stimulation of cholinergic receptors in the CNS. Symptoms include slurred speech, confusion, hallucinations, loss of reflexes, convulsions, and possibly coma and death. (Other symptoms that can occur with the overstimulation of choinergic neurons include chest and abdominal pain, nausea, vomiting, sweating, and salivation) ii. Synthetic analogs of physostigmine (neostigmine and pyridostigmine) that do not cross the blood-brain barrier. They are used in treating myesthenia gravis, an autoimmune disorder that attacks cholinergic receptors at neuromuscular junctions. By inhibiting AChE, they keep ACh active longer at ther neuromuscular junction, which helps to stimulate the undamaged receptors. Irreversible AChE inhibitors: These are used as insecticides in their weak enough and as nerves gas in their strong from (Sarin, Soman). Symptoms of nerve gas poisoning include vomiting, convulsions, profuse sweating and salivation, gasping for breath. The antidote for these nerve gases are reversible AChE inhibitors, such as pyridostimine. The temporary inhibition of AChE by a reversible inhibitor protects the enzyme against permanent inactivation by the nerve gas. But the antidote must be taken BEFORE exposure to the nerve gas. Pyridostigmine when taken over a course of weeks or months may have side effects such as cognitive impairment, dizziness, and problems of balance and coordination. These are symptoms reported by veterans diagnosed with Gulf War Syndrome. Which neurons in the brain are cholinergic neurons? The preganglionic neurons of both the sympathetic and parasympathetic nervous system produce ACh. So do the ganglionic neurons of the paraysmpathetic nervous system. The parasympathetic nervous system is made up entirely of cholinergic neurons and the sympathetic nervous system is made up partially by cholinergic neurons, which are found only in the portion of those pathways lying within the CNS. The cell bodies of cholinergic neurons are found primarily in two brain areas: the midbrain and the basal forebrain. The basal forebrain cholinergic system (BFCS) has been implicated in cognitive functioning. Rats with damage to this system show impairments in memory, attention, and learning. There are two types of cholinergic receptors in the nervous system and on muscle cells: a. nicotinic receptors-- respond to nicotine as an agonist b. muscarinic receptors-- respond to muscarine as an agonist. Muscarine is an alkaloid (a nitrogen-containing compound) that is found in the fly agaric mushroom, Amanita muscaria. 3. Drugs can act as nicotinic agonists or antagonists Nicotinic receptors are highly concentrated on muscle cells at neuromuscular junctions, on ganglionic cells of both the sympathetic and parasympathetic nervous system, and in certain brain areas. They are ionotropic receptors that open up sodium and calcium channels. If they open up sodium channels, they cause deparization of the membrane, and if they open up calcium channels, they stimulate release of the neurotransmitter (exocytosis). Nicotinic receptor agonist-- Succinylcholine produces continuous depolarization of the membrane that eventually results in "depolarization block," in which the resting potential of the neuron cannot be re-established and so the neuron cannot fire again. It is used as a powerful muscle relaxant during surgery. Because muscles of the diaphragm are paralyzed, the patient must be maintained on a ventilator until the drug wears off. Nicotinic receptor antagonis-- D-tubocurarine, the active ingredient in curare, is a poison that indigenous tribes of South America use on their arrowheads. It has a high affinity for nicotinic receptors and blocks the neuromuscular junction. In high enough doses, it causes respiratory paralysis that results in death. 4. Muscarinic receptor agonists and antagonists Muscarinic receptors are widely distributed in the brain (neocortex, hippocampus, thalamus, striatum, and basal forebrain). They are metabotropic receptors and have a variety of effects. In the neocortex and thalamus, they serve a cognitive function; in the striatum, a motor function. They are also linked to the reward system of the brain. These receptors are also found outside the brain in cardiac muscle and the smooth muscle of internal organs such as the stomach and intestines. They are involved in parasympathetic activation, which slows heart rate and the strength of contraction of the heart beat, and activates secretions such as saliva, sweat, and tears. Muscarinic receptor agonists-- These drugs (muscarine, arecoline) are parasympathomimetic agents, which means that they mimic the effects of parasympathetic activation. They can lead to excessive production of tears, saliva, and sweat; pupil constriction, abdominal pain, strong contractions of visceral muscles, diarrhea, and convulsions. Muscarinic receptor antagonists-- These drugs are parasympatholytic agents (atropine and scopolamine, which are both naturally occurring alkaloids) inhibit the paraysmpathetic nervous system. Atropine is used to dilate pupils and can also be used in surgery to reduce secretions such as saliva that can block airways. Also used to counteract poisoining by cholinergic agonists. Scopolamine produces drowsiness, euphoria, amnesia, fatigue, and dreamless sleep. Also used to treat seasickness by counteracting the effects of nausea. 2/19/08- Review Session Midterm 1 this Thursday. Punctuality and attendance count!!!!! Weak acids ionize better in basic (or alkaline) solutions, and weak bases ionize better in acidic solutions. I-16b. L-DOPA is a precursor to dopamine and so it's given to patients with Parkinson's disease to try to increase dopamine levels in the CNS AMPT (alpha-methyl-para-tyrosine) blocks tyrosine hydroxylase (rate-limiting enzyme in the synthesis of catecholamines), thus interfering with catecholamine synthesis; it has no therapeutic value but is used in research to simulate conditions in which there are low levels of catecholamines in the brain. I-17d DA receptor agonist-- stimulates or activates behavior DA receptor antagonist-- suppress certain kinds of behavior, can produce catalepsy (lack of spontaneous movement); D2 antagonists (e.g., haloperidol) have been used to treat schizoprhenia. I-7 key terms convergence-- several axon terminals synapse onto a single neuron divergence-- several axon terminals from the same neuron synapse onto several other neurons I-8 a) At rest, there are several forces at play in the neuron: 1. Concentration gradient-- the tendency for substances to go from areas of higher concentration to lower concentration: For sodium, the conc grad is driving the ions into the cell, and for potassium, it's driving them out. 2. Electrical gradient (electrostatic potential) -- The tendency of charged particles to go towards the opposite charge. At rest, the potential is -70mV (more negative inside the neuron), which is going to drive both sodium and potassium into the cell. 3. Potassium channels are not gated whereas sodium channels are (voltage-gated) 4. Sodium-potassium pump-- a protein in the cell membrane that exchanges sodium for potassium; for every three sodium ions that are "pumped" out of the neuron, two potassium ions are pumped in. d) Action potential: 1. The potential increases (depolarization) until it reaches -50mV, which is the threshold potential. 2. When this potential is reached, the sodium channel opens. 3. The voltage rapidly increases to +40mV, and then the sodium channel closes. 4. Meanwhile, potassium has been flowing out of the neuron because it is driven outward by both electrical and concentration gradients. 5. Sodium-potassium pump restores the potential back to -70mV by pumping out sodium and pumping in potassium in a 3/2 ratio. EPSP and IPSP-- excitatory postsynaptic potential, which is produced by opening sodium channels and depolarizing the cell; the inhibitory postsynaptic potential, which is produced by opening chloride channels and hyperpolarizing the cell. Volume transmission-- diffusion of a chemical signal through the extracellular fluid to reach target cells at a distance from the point of release. In general, neurotransmitters move across a synapse to an adjacent neuron, but they can travel longer distances to other neurons that do not synapse with the neuron producing the neurotransmitter. I-17e 6-Hydroxydopamine (6-OHDA) is a neurotoxin that kills dopaminergic cells; haloperidol is a DA receptor antagonist, which blocks the binding of DA to its receptors. The effect of 6-OHDA is to lower DA levels in the brain.The effect of haloperidol is NOT to reduce DA levels but to reduce the rate of binding of DA to receptors. BOTH result in upregulation, which is an increase in the number of receptors, although the increase happens for different reasons in each case. In the case of 6-OHDA, up-regulation happens to compensate for lower DA levels, and in the case of haloperidol, it happens to compensate for lower binding rates. I-16g tricyclic antidepressants-- inhibit the reuptake of NE and serotonin Reboxetine-- just inhibits or blocks the reuptake of NE cocaine- inhibits the reuptake of DA, NE, and serotonin The way a drug blocks reuptake is by binding to the transporter Cocaine has a high level of affinity for the transporter (it binds well to it). The term "efficacy" is used to refer to a drug or neurotransmitter's ability to exert an effect once it binds to a receptor. 2/14/08Midterm 1 Next Thursday covers Unit 1 (Lessons I-1 through I-18), which corresponds to CHs 1, 2, 3, and 5. Behavioral supersensitivity-- If an animal is given a dopamine antagonist such as haloperidol for an extended period of time and then the treatment is stopped. When the animal is then given a dopamine agonist (e.g., apomorphine), that animal is going to respond much more strongly to the agonist than a control animal. NOREPINEPHRINE Locus coeruleus-- Found in the pons; it is the primary source of norepinephrinecontaining neurons (noradranergic neurons); provides nearly all of the NE in the cortex, limbic system, thalamus, hypothalamus, cerebellum, and spinal cord. Norepinephrine is both a neurotransmitter and a hormone. NE can reach the heart by: 1. Being released from sympathetic noradrenergic neurons that are connect to the muscle tissue of the heart at a neuromuscular junction. (NE acting as a neurotransmitter) 2. Being released from the adrenal glands and traveling through the circulatory system to the heart . (NE acting as a hormone) Behavioral effects of NE: its effects can be related to triggering hunger and eating behavior; sexual behavior; fear and anxiety; pain; sleep and arousal; vigilance. Four types of adrenoreceptors: alpha1, alpha2, beta1, and beta2 Alpha receptors- act like D2 receptors, inhibiting adenylyl cyclase and thus reducing the rate of synthesis of cAMP. Beta receptors-- stimulate adenylyl cyclase and thus enhance the synthesis of cAMP Adrenergic agonistsUsed in the treatment of bronchial asthma (e.g., albuterol is a beta adrenergic agonist); these agonists constrict blood vessels in the bronchial lining and thus reduce congestion and fluid buildup in the lungs. Neosynephrine contains phenylephrine, which is an alpha-receptor agonist. It constricts blood vessels in the nose and reduces inflammation of nasal membranes; the same ingredient is found in certain eye drops because it also dilates the pupils. The simple version of the story is that NE agonists increase sympathetic response. The exception to this is alpha2 agonists such as clonidine, which actually inhibit sympathetic nervous system activity. Adrenergic antagonists"Beta blockers" are beta antagonists such as propanolol, metoprolol they're useful in treating hypertension because they dilate the blood vessels. Just as an NE agonist would, in general, result in sympathetic arousal, an NE antagonist would stimulate the parasympathetic NS or suppress sympathetic arousal. One symptom of that arousal would be constriction of blood vessels, and these antagonists do the opposite, causing dilation. They're also used to treat cardiac arrhythmia (irregular heartbeat) and angina pectoris (chest pain). They've also been used to treat the symptoms of generalized anxiety disorder such as tachycardia (racing heart). Alpha antagonists have been used in the treatment of sexual impotence, by increasing parasympathetic activity. 2/12/08Next Tues: Review session-- Bring questions if you have them. Next Thurs: First midterm. Don't be late and don't be absent. CH.5: Catecholamines Catecholamines are a class of neurotransmitters that includes dopamine (DA) and norepinephrine (NE). Although epinephrine is a catecholamine, it's not included in this discussion because it is not a neurotransmitter. The mechanism by which the brain produces catecholamines is shown in Fig 5.2. L-DOPA is used therapeutically to increase catecholamine formation. It's used in the treatment of Parkinson's alpha-methyl-para-tyrosine (AMPT)-- blocks tyrosine hydroxylase, and so it interferes with catecholamine synthesis; it doesn't have a therapeutic use but it is used in research Nomenclature Dopaminergic-- neuron or a pathway that involves dopamine (DA) Noradrenergic-- neruon or pathway that involves norepinephrine (NE) Like most neurotransmitters, catecholamines are packaged in vesicles. This packaging has two important functions: 1. It delivers a "pre-measured" amount of the neurotransmitter into the synapse. 2. It protects the neurotransmitters from getting broken down by enzymes in the axon terminal. The effects of certain drugs on catecholamines: 1. Reserpine-- Used in the treatment of high blood pressure; can act as a sedative and depressant. Blocks vesicular monoamine transporters (VMAT), which are proteins that draw the monoamine neurotransmitters into the vesicle. Monoamines include serotonin as well as the catecholamines. 2. Amphetamine and methamphetamine-- Cause exocytosis--the breaking of vesicle and the release of catecholamines into the synapse. The result is behavioral activation in animals, and symptoms such as euphoria, increased alertness and energy, and sleeplessness in humans. Autoreceptors-- found in the cell bodies, axon terminals and dendrites of dopaminergic and noradrenergic neurons. The effect of binding of the neurotransmitter to these autoreceptors is the inhibition of the release of the neurotransmitter into the synapse. a. Autoreceptors in the cell bodies and dendrites reduce the rate of firing of the neuron. b. Autoreceptors in the axon terminal reduce the rate of release of catecholamines from the vesicles by blocking calcium channels. How are catecholamines inactivated? 1. Reuptake-- DA and NE transporters that return these neurotransmitters to the axon terminal, where they are either re-packaged and re-released or broken bown by enzymes. 2. Enzymatic breakdown-- There are two enzymes that break down DA and NE into inert components: monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT). Some drugs that affect reuptake: 1. Tricyclic antidepressents-- block the transporters for NE and serotonin 2. Reboxetine-- just blocks the NE transporter 3. Cocaine- inhibits the reuptake of DA, NE, and serotonin (5-HT) Some drugs that affect the enzymes: MAO inhibitors (phenelzine, tranylcpromine) COMT inhbitors (entacapone, tolcapone) Block the enzymes that break down DA & NE (and 5-HT, in the case of MAO inhibtiors), increasing the amount of these neurotransmitters in the synapse. DOPAMINE The vast majority of dopaminergic neurons have their cell bodies in either the substantia nigra or the ventral tegmental area (VTA). Both areas are part of the tegmentum. Three major dopaminergic pathways in the brain: 1. Nigrostriatal tract-- the axons from these dopaminergic neurons extend from the substantia nigra to the striatum; this pathway is important for the regulation of movement and it is the pathway that is damaged by Parkinson's disease. 2. Mesolimbic dopaminergic pathway-- the axons start in the VTA and extend to the limbic system, including the nucleus accumbens, amygdala and hippocampus. 3. Mesocortical dopaminergic pathway-- the axons start in the VTA and extend to various cortical areas, including the prefrontal cortex, cingulate gyrus, and entorhinal cortex. Pathways 2 and 3 are considered the major reward pathways of the brain and are implicated in addiction. Less prominent is the tuberhypopheseal dopamine pathway, which starts in the hypothalamus. What happens if you damage dopaminergic pathways? Bilateral lesions in animals produce sensory neglect (they don't pay attention to stimuli in their environment), motivational deficits (e.g.,they show little interest in food or water), and motor impairments (e.g., difficulty initiating movement). Unilateral lesions (i.e., on one side only) cause the animal to lean toward the side that corresponds with the lesioned brain area. Dopamine receptors-- There are different subtypes of receptors for DA, but we will focus on two: D1 receptors-- stimulate the enzyme adenylyl cyclase, which catalyzes the synthesis of a second messenger called cAMP. This second messneger stimulates the production of a protein kinase that changes the structure of various proteins in the postsynaptic neuron. D2 receptors-- inhibit adenylyl cyclase, thus blocking the synthesis of cAMP. Some dopamine agonists such as apomorphine stimulate both D1 and D2 receptors, causing behavioral activation. Dopamine antagonists, such as haloperidol, suppress certain kinds of behavior and cause catalepsy (lack of spontaneous movement). D2 receptor antagonists have been used in the treatment of schizophrenia, although they can produce undesirable motor side effects. Let's compare the effects of two drugs: haloperidol and 6-hydroxydopamine (6-OHDA). 6-OHDA is a neurotoxin that kills dopaminergic neurons; haloperiodol is a D2 receptor antagonist.Both cause a decrease in the binding of DA to receptors, but for different reasons. In the case of 6-OHDA, there is less dopamine because the neurons that produce are DEAD. In the case of haloperidol, the neurons may be alive, but the binding of DA to the receptor is blocked. IN both cases, the postynaptic neurons will produce an increase in the number of dopamine receptors to compensate for the lack of DA binding. This is called up-regulation. 2/7/08CH.3 (Continued) Figure 3.14-- Mechanisms by which drugs can alter synaptic transmission. 1. Acts as a precursor for a neurotransmitter-Example: L-DOPA is a precursor for dopamine 2. Inhibits neurotransmiiter (NT) synthesis-A form of tyrosine inhibits the enzyme (tryrosine hyroxylase) that catalyzes the reaction which forms dopamine and norepinephrine. 3. Prevents NT storage in the vesicles-Example: Reserpine (a drug that reduces high bp) blocks the storage of dopamine, norepinephrine, and serotonin. 4. Stimulates release of NT-Example: amphetamine stimulates exocytosis of dopamine and norepinephrine. 5. Inhibits the release of NT-Example: botox inhibits release of acetylcholine 6. Stimulates postsynaptic receptors (acting as an agonist for the NT)-An agonist either heightens the effect of the neurotransmitter or mimics that effect. Example: Opiates stimulate opioid receptors. 7. Blocks postsynaptic receptors (acting as an antagonist)Example: Antipsychotic drugs are antagonists that block dopamine receptors. 8. Stimulates autoreceptors, which inhibits the release of the NTExample: clonidine reduces the symptoms of opiate withrrawal by stimulating the autoreceptors that inhibit the release of norepniphrine and serotonin. 9. Blocks autoreceptors, thus stimulating release of the NTExample: Yohimbine blocks autoreceptors for norepinephrine, trigger a flood of NE into the synapse, and thus producing the symptoms of a panic attack. 10. Inhibits NT degradationExample: MAO inhibitors block the enzyme monoamine oxidase (MAO), which breaks down dopamine, serotonin, and norepinephrine. 11. Blocks NT reuptake Example: Cocaine binds to dopamine transporters and thereby blocks reuptake of DA. ENDOCRINE SYSTEM- The glands of the body and the hormones they produce (secrete). Both hormones and neurotransmitters interact with receptor proteins. Drugs interact with hormone systems as well as neurotransmitter systems. The correspondence between the glands of the endocrine system and the chakra system: 1st Chakra-- Prostate gland in men (seminal fluid)/ Skene's glans (female ejaculation) 2nd Chakra-- Gonads Ovaries-- estrogens (estradiol) and progesterone Testes-- androgens (testosterone) 3rd Chakra-- Adrenals and Pancreas Pancreas-- located between the adrenals, just above the kidneys secretes insulin and glucagon, both of which regulate blood sugar Adrenals have two components: 1. Inside (adrenal medulla) secretes epinephrine and norepinephrine, which mobilize glucose and provide immediate energy for a fight-or-flight response 2. Outside (adrenal cortex) secretes glucocorticoids (e.g., cortisol), which maintain blood glucose levels and help store excess glucose for future use. 4th Chakra-- Thymus Secretes thymic hormones that regulate the immune system 5th Chakra-- Thyroid Found in the throat area; secretes thyroxine and triiodothyronine, which are important for energy metabolism Underactive thyroid (hypothyroidism)-- weakness and fatigue Overactive thyroid (hyperthyroidism)-- excessive energy and nervousness 6th Chakra-- Hypothalamus and Pituitary The pituitary gland has two components: 1. anterior pituitary-- secretes hormones that stimulate the other glands of the body: thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH) stimulates the adrenals; follicle-stimulating hormone (FSH) and luteinizing hormone (LH) stimulate the overies; growth hormone (GH); and prolactin 2. posterior pituitary-- secretes vasopressin (antidiuretic hormone) and oxytocins (stimulates uterine contractions in labor and lactation). The hypothalamus releases hormones that activate the pituitary: a) thyrotropinreleasing hormone (TRH) stimulates the release of the TSH by the anterior pituitary; b) corticotropin-releasing hormone (CRH) stimulates the release of ACTH; c) gonadotropin-releasing hormone (GnRH) stimulates the release of LH and FSH. 7th Chakra Pineal gland-- located over the brain stem in the center of the brain; secretes melatonin, which is associated with the body clock and the sleep-waking cycle. Two roles of testosterone: 1. In early development, it acts within the brain to determine gender. 2. In puberty, plays a role in stimulating sexual desire in both genders. Two types of hormones-- Peptide hormones (amino acids) and steroid and thyroid hormones (lipids) Peptide hormones-- attach to extracellular (membrane) receptors and act just like metabotropic neurotropic neurotransmitters, working through a second messenger. Steroid and thyroid hormones-- use intracellular receptors located within the cell nucleus, and act as transcription factors that turn on or off the expression of a specific gene, which in turn affects protein synthesis 2/5/08CH.3 Synapse- the fundamental unit of neurotransmission that includes the presynaptic terminal, synaptic cleft, and part of the postsynaptic cell presynaptic terminal-- axon terminal on the neuron that is sending the message postsynaptic neuron-- neuron receiving the message synaptic cleft-- gap between the two neurons (20nm) Types of synapes: 1. axodendritic synapse-- most common;; axon terminal from the presynaptic neuron communicates with a dendrite on the postsynaptic neuron. 2. axosomatic synapse-- axon terminal communicates with a neuron cell body 3. axoaxonic-- axon terminal communicates with another axon; these types of synapses generally modulate or regulate neurotransmitter release. neuromuscular junction-- connection between a neuron and a muscle; although it's not technically a synapse, it has similarities to a synapse. Neurotransmitters-- these are the chemical messengers of the nervous system. The definitional criteria for neurotransmitters are: 1. The presynaptic neuron manufactures and contains it. 2. The substance is released from an axon terminal. 3. It binds with receptors on the postsynaptic neuron. 4. The binding of the molecule (or of an agonist) with the receptor triggers an effect. 5. Applying an antagonist that blocks the receptor inhibits the molecule's effect. 6. There is a mechanism for inactivating this substance. Types of neurotransmitters: 1. Amino acids (Glutamate/GABA) 2. Monoamine (Dopamine, Serotonin, Norepinephrine) 3. Acetylcholine 4. Neuropeptides (Endorphins)-- chains of amino acids 5. Lipids (Anandamide)-- long-chain hydrocarbons, includes fats 6. Gases (nitric oxide, NO) Neuropeptides are distinct from the other neurotransmitters in that they are made in the cell body, unlike other neurotransmitter which are made in the axon terminal. As a result, neuropeptides are replenished more slowly than other neurotransmitters. The steps in neurotransmission: 1. Neurotransmitter is synthesized in the presynaptic neuron and stored in vesicles (sac-like structure that can contain and transport as many as several thousand neurotransmitter molecules). 2. The action potential moves down the membrane of the presynaptic neuron until it reaches the axon terminal. 3. Upon reaching the axon terminal, it opens up calcium channels. 4. Calcium ions flow into the terminal. 5. Calcium causes vesicles to fuse with the the membrane of the axon terminal (exocytosis). 6. Neurotransmitters get released from the terminal. 7. Neurotransmitters bind to receptors on the membrane of the postsynaptic neuron. 8. The binding results in an opening or closing of ion channels in the postynaptic neuron. 9. The net effect is either an excitatory postsynaptic potential (EPSP; depolarization) or an inhibitory postsynaptic potential (IPSP; hyperpolarization). 10. Vesicle gets retrieved from the plasma membrane. 11. The neurotransmitter is released from the receptor and becomes inactivated. Factors that regulate the rate at which neurotransmitters are released by the presynaptic neuron: 1. The rate of cell firing (how many action potentials move along the membrane per unit time). 2. Probability that the neurotransmitters will be released from the axon terminal as a result of an action potential (sometimes an action potential does NOT result in exocytosis, or the release of neurotransmitters). 3. The presence of autoreceptors. There are two types of autoreceptors: a) Terminal autoreceptors are found on the axon terminal and are specific to the neurotransmitter released by that terminal; the binding of the neurotransmitter to this receptor inhibits the release of more neurotransmitters. b) Somatodendritic autoreceptors. These are found on either the cell body or the dendrites of the presynaptic neuron; binding of the neurotransmitter to these receptors inhibits cell firing. 4. Axoaxonal synapses have heteroreceptors (receptors for a neurotransmitter that is released by another neuron) that may either enhance or reduce the amount of neurotransmitter released. Inactivation of the neurotransmitter: 1. Enzymatic breakdown. An enzyme breaks down the neurotransmitter into smaller and inert pieces. 2. Neurotransmitter is removed from the synaptic cleft by a transporter (protein) found on the membrane of the presynaptic neuron (REUPTAKE). 3. Neurotransmitter can also be removed from the synapse by a transporter on a neighboring cell (UPTAKE). Cocaine binds to transporters for dopamine, serotonin and norepinephrine, preventing those neurotransmitters from undergoing reuptake. Two key concepts for understanding receptors: 1. Almost all neurotransmitters have more than one receptor (there are different receptor subtypes for the same neurotransmitter). 2. Most receptors fall into two categories: a) Ionotropic-- fast acting; the receptor has an ion channel at its center that is opened or closed when the neurotransmitter binds to it. b) Metabotropic- slow-acting; the receptor is composed of a single large protein that triggers a chain reaction in the postsynaptic neuron. This chain reaction involves the activation of G-proteins, which in turn trigger a chain of events that cause an ion channel to open up somewhere in the cell at some point in the future. G proteins operate by two mechanisms: 1. They can stimulate or inhibit the opening of an ion channel in the cell membrane. 2. They can stimulate certain enzymes on the membrane of the postsynaptic neuron, called effector enzymes. Most of these enzymes are involved in the synthesis or breakdown of a second messenger that initiates a biochemical reaction within the cell. The second messenger activates an enzyme called a protein kinase. This enzyme adds a phosphate group to another protein (phosphorylation); the protein on which this enzyme acts can be an ion channel, an enzyme involved in neurotransmitter synthesis, a receptor, a transporter, a structural protein, or a different kind of protein. The phosphorylation reaction can change the function of a protein in some important ways (e.g., it can ause an ion channel channel to open, can make a receptor more sensitive to a neurotransmiiter, or can cause a protein to be produced that turns on or off a specific gene in that neuron). 1/31/08The Nervous System Fig. 2.15 Central nervous system-- brain and spinal cord Peripheral nervous system-- everything else PNS can be subdivided into: Autonomic nervous system- innervates the internal organs sympathetic nervous system-- energy utilization, specifically in response to environmental stressors (i.e., the "fight-or-flight" response) parasympathetic nervous system-- energy conservation and metabolism (e.g., digestion) Somatic nervous system-- innervates the skeletal muscles sensory afferent-- nerve that delivers signal from sense organs (e.g., receptors on the skin) to the spinal cord motor efferent-- nerve that delivers a signal from the spinal cord to the muscle of the body nucleus-- within the central nervous system, is a cluster of neuronal cell bodies tract-- within the CNS, is a bundle of axons ganglion-- within the peripheral nervous system, is a cluster of neuronal cell bodies nerve-- within the PNS, is a bundle of axons meninges-- three layers of membranes that surround the brain and spinal cord; they also hold the cerebrospinal fluid (CSF), which helps to protect the brain and spinal cord and also in the exchange of nutrients and waste with the blood. ORGANIZATION OF THE ADULT HUMAN BRAIN I. Spinal cord-- motor neurons send information to the muscles through the ventral horn of the spinal cord, and sensory receptors send information to the brain through the dorsal horn. II. MyelencephalonA. medulla-- regulates vital functions, including heart rate, digestion, respiration, blood pressure, coughing and vomiting 1. area postrema- it has less of a blood-brain barrier than other brain areas; this allows toxins into this area that would not enter other brain areas. If there are enough toxins and they are sufficiently noxious, they will be expelled. III. Metencephalon A. pons- At its core is the reticular formation, which is a se to nuclei that are important for arousal and attention (sleep-waking cycle), muscle tone, cardiac and respiratory reflexes. Two sets of nuclei of the reticular formation are especially pertinent to this course: 1. locus coeruleus-- where the vast majority of neurons that produce norepinephrine are found (these neurons are called "noradrenergic"); they stimulate arousal, vigilance and attention. The amphetamines trigger noradrenergic pathways in the brain, enchancing alertness and causing sleeplessness. 2. raphe nuclei (this is a set of nuclei that includ the dorsal and median raphe)-- where most of the neurons that produce serotonin (these are called "serotonergic"); they are important in regulating sleep, aggression, impulsiveness, and other emotions (serotonin is often an inhibitory neurotransmitter) B. cerebellum-- involved in muscle coordination, balance, timing of movements, and smooth muscle movements IV. Mesencephalon (midbrain) A. tectum1. superior colliculus-- reflexes of the visual system (such as pupillary reflex) 2. inferior colliculus-- the auditory system B. tegmentum 1. periaqueductal gray (PAG)-- surrounds the aqueduct that connects the third and fourth ventricle; has large concentrations of opioid receptors, which means that it is involved in the control of pain. 2. substantia nigra-- contains neurons that produce dopamine (dopaminergic); the nigrostriatal tract in initiation of movement; damage to this area is associated with Parkinson's disease 3. ventral tegmental area (VTA)-- dopaminergic neurons in this brain area have axons that become two important tracts in the brain's reward system: mesolimbic tract connects the VTA to the nucleus accumbens and the amygdala; the mesocortical tract connects to the prefrontal cortex (the orbitofrontal cortex, OFC) and the cingulate gyrus. V. Diencephalon A. Thalamus-- relay station by which sensory impulses are distributed to the appropriate parts of the cortex. Four out of the five senses go through the thalamus (NOT olfaction) B. Hypothalamus-- Regulates virtually every basic drive, including hunger, thirst, body temperature, sexual behavior, and some emotional behavior. Controls the sympathetic and parasympathetic nervous system through its connection to the pituitary gland. VI. Telencephalon A. Basal ganglia-- motor control, also play a part in the reward pathway 1. Caudate nucleus 2. Putamen 3. Globus pallidus 4. Nucleus accumbens Caudate and putamen make up the "striatum" B. 1. 2. 3. 4. Limbic system Cingulate gyrus-- involved in the emotional component of pain Olfactory bulb Hippocampus- memory, specially long-term memory and spatial memory Amygdala-- processing emotion, especially fear C. Neocortex-- Two cerebral hemispheres connected by a set of fibers called commissures, the largest of which is the corpus collosum). Each hemisphere is in turn divided into four lobes: 1. 2. 3. 4. Occipital-- vision Temporal-- hearing Parietal-- somatosensory cortex (awareness of touch, temperature and pain) Frontal-- motor cortex (where movement is initiated) a. Prefrontal cortex-- decision-making, planning, evaluation of strategies, and impulse control 1/29/08CHAPTER 2 Neurons Three types of neurons in the NS: 1. Sensory neurons-- transducers, which convert some kind of external energy (light, mechanical energy) into electrochemical impulses. 2. Motor neurons-- trigger a muscle contraction 3. Interneurons-- only connected to other neurons within the central nervous system (CNS); they tend to be connected in complex networks Structure of neurons 1. dendrites-- receive signals from either other neurons or other parts of the same neuron; they are covered with dendritic spines, which are projections that increase the receptive area of the dendrite 2. axons- each neuron has only one axon even though it can have many dendrites; an axon can be branched, meaning that it has side branches called axon collaterals. At the end of the axon is the terminal button (axon terminal) which is where neurotransmitters are released. Synaptic vesicles-- these carry neurotransmitters from the soma, or cell body, to the terminal button. Myelin-- fatty insulating coating ("Lorenzo's Oil") nodes of Ranvier-- spaces in between the myelin coating where action potentials can occur. 3. soma-- the cell body, which contains: nucleus-- contains genetic material (chromosomes) and also transcription factors (proteins that aid in the manufacture of proteins) transcription-- the manufacture of mRNA from the DNA in the nucleus translation-- mRNA-coded manufacture of proteins, which happens in the ribosomes mitochondria-- glucose is converted to ATP, which is the fundamental energy storage unit in the body Ion channels-- found in the membranes of neurons. Their characteristics include: 1. They are relatively specific in terms of allowing certain ions through (sodium, potassium, calcium, and chloride channels are most common in neuronal membranes). 2. Most ion channels are closed the majority of the time, and only open on a very temporary basis. Such channels are called gated channels. 3. There are two types of gated channels: a. Ligand-gated-- a ligand such as a drug, hormone or neurotransmitter binds to a receptor that opens the channel. b. Voltage-gated-- a change in voltage across the membrane caused the channel to open. negative voltage-- more negative than positive ions inside the membrane positive volltage-- more positive ions inside the membrane With voltage-gated channels, there is only a specific range of voltages in which the channel opens. 4. The flow of ions through the channel depends on: a. Concentration gradient-- Dissolved substances move from areas of greater concentration to areas of lower concentration. b. Electrical gradient (electrostatic potential)-- Ions move towards the opposite charge and away from the same charge. If there is a negative voltage, that is going to drive positive ions into the neuron. Glial cells- Support cells of the nervous system, which provide insulation, protection, and metabolic support. Four types of glial cells: 1. Oligodendroglia- Produce myelin on axons of neurons in the CNS; each glial cell is dedicated to myelinating a single neuron. 2. Schwann cells-- Produce myelin on axons of neurons in the PNS; one glial cell can serve several neurons. 3. Astrocytes-- structural support to neurons and regulate the chemical environment, helping to move nutrients from the blood into the neuron and waste from the neuron into the blood. 4. microglia-- scavengers, remove dying cells; help in the immune response of the CNS Electrical potentials in the nervous system: Resting potential-- at rest, the voltage of most neurons is -70 mV (the inside of the membrane is more negative than the outside) Forces responsible for the resting potential: 1. Concentration gradient a. drives sodium ions into the neuron b. drives potassium ions out 2. Electrical gradient a. drives sodium ions in b. drives potassium ions in 3. Potassium channels are not gated, which means that potassium ions can move relatively freely. Equilibrium potential is reached in which the numbers of potassium ions going in is equal to that going out. 4. Sodium-potassium pump At times, sodium leaks into the cell at rest, and the sodium-potassium pump moves sodium out while bringing potassium in. For every 3 sodium ions that are pumped out, 2 potassium ions are pumped in. depolarization-- When a membrane at rest becomes more positive (or less negative), moving closer to 0mV hyperpolarization-- When a membranes becomes more negative Local potentials-- These happen on dendrites and cell bodies. They have the following characteristics: 1. are graded-- the larger the electrical stimulus, the greater the potential 2. decay rapidly 3. summation-- several depolarizations and/or hyperpolarizations can be added together Action potentials-- these are potentials that are transmitted through the axon membrane, and they get regenerated at different points along the membrane Here is how they work: 1. The potential increase from a resting potential of -70mV to -50mV, which is the threshold potential. 2. Sodium channels open and sodium ions pour into the axon. 3. The voltage changes immediately from -50mV to +40mV 4. When the voltage hits +40mV, the sodium channel closes again (These channels are gated in such a way that they only open between -50 and +40mV) 5. Sodium-potassium pum-- moves two potassium ions in for every three sodium ions that it removes. Why does the resting potential start at -50mV instead of just zero? Having a negative potential at the start of the action potential speeds up that potential, increasing the rate with which the sodium ions flow across the membrane. Action potentials follow the all-or-none law. If the membrane is depolarized enough to hit its threhold potential, the action potential is going to happen. A greater depolarization doesn't result in a greater potential. In myelinated axons, the speed of conduction is 15 times faster than in unmyelinated axons. The process of transmission of the potential along the membrane of a myelinated axon is called saltatory ("jumping") conduction. Local anesthetics- Block sodium channels so that pain signals can't reach the CNS procaine (Novocaine), lidocaine (Xylocaine), benzocaine (Anesthesin) 1/24/08Announcements: Syllabus, study guide, and lecture notes are posted at: http://vas.web.arizona.edu EliminationFirst-order kinetics-- The rate of elimination of a drug from the body is an exponential function of the concentration of the drug in the blood stream, so that 50% of the free remaining drug is removed at each interval. Zero-order kinetics-- Drug is eliminated at a constant rate, purely as a function of time, and regardless of its concentration in the bloodstream. Example: alcohol. The single most important route of elimination of drugs from the body is through urine. The liver produces enzymes that help break down the drug and ionize it into a form that can be dissolved easily and passed through urine. Pharmacodynamics The mechanism of action of drugs in the body, which involves binding to receptors. With psychoactive drugs, the receptors are associated with neurons somewhere in the nervous system. Receptors are proteins that are either found on the surface of cell membranes (extracellular) or inside the cell (intracellular). Substances that bind to receptors are ligands, and the most common ligands are neurotransmitters, hormones, and drugs. The biggest difference between a neurotransmitter and a hormone has to do with how distributed its effects are on the body. Neurotransmitters tend to be localized whereas hormones are distributed widely throughout the body. Agonist vs antagonist-- An agonist is a ligand that binds to a receptor and activates it. An antagonist blocks the activity of the receptor, either directly or indirectly. Competitive vs. non-competitive antagonists-- A competitive antagonist competes with the agonist for binding sites on the receptor. Example: naloxone competes with morphine and blocks it effects on the body. A non-competitive antagonist reduces the effect of the agonist without necessarily competing with it for binding sites on the receptor. Example: chlorothiazide (CTZ), a diuretic that indirectly interferes with glutamate receptors. Characteristics of receptors: 1. They have the ability to recognize specific molecular shapes (lock-and-key model). 2. The binding of the ligand is temporary. 3. The shape of the receptor changes as a result of binding by the ligand; this change triggers a chain of intracellular events that produce a specific effect (e.g, opening up ion channels in a neuron). 4. Receptors are proteins that have a specific life cycle; they can be modified in terms of number and sensitivity. Up-regulation (increase in the number of receptors) and down-regulation (decrease in the number of receptors). The potency of a drug is a measure of the amount of the drug needed to produce a desired effect. Potency is a function of: Affinity- Attraction of the drug to the receptor (how well does it bind) Efficacy- Ability of the drug to trigger a certain action by the recepto once it does bind. ED50-- The dose that produces 50% of maximal effectiveness. Ideally, drug manufacturers want this number to be as low as possible, meaning that the drug produces a relatively large effect with a relatively small dose. TD50-- The dose that produces 50% of maximal toxicity. Ideally, drug manufacturers want this number to be as high as possible, meaning the it takes a large dose to produce toxicity. Therapeutic index-- The ratio of TD50/ED50. The larger the effective dose is, compared to the toxic dose, the better. Tolerance-- Diminished response to a drug after repeated exposure to it. Characteristics of tolerance: 1. It's reversible. If you stop taking a specific drug, the tolerance gets reduced. 2. It depends on the pattern of drug use. Higher frequency and dosage produces more tolerance. 3. It depends on the type of drug. Some drugs produce rapid tolerance (LSD), some produce slow tolerance, and some no tolerance (antipsychotics). 4. It depends on the effect-- With morphine, the nausea produced by the drug has rapid tolerance, whereas the constipating effect has no tolerance. 5. It can have different mechanisms, such as: metabolic tolerance (drug disposition)- This occurs when repeated use of the drug reduces the amount available at binding sites, probably because of enzyme induction. More drug in the system equals more enzymatic breakdown of that drug. pharmacodynamic tolerance-- This is a change in nerve cell function that compensates for continued presence of the drug (down-regulation). Homeostatic processes that compensate for an imbalance resulting from drug use. behavioral tolerance-- This occurs when drug effects diminish only in certain environments that are associated with drug administration but not in novel environments. habituation- Desensitization to familiar vs novel stimuli. Orienting response-- You hear a buzzer goes off and your brain produces an evoked potential. The amplitude of that potential decrease with repeated exposure to the same stimulus. classical conditioning-- can explain some types of tolerance. Tolerance may start out as an unconditioned response to the stimulus (i.e., the drug) but then it can become a conditioned response to environmental cues. operant conditioning-- If you're rewarded for showing less of an effect of a drug, then you will start to show less of an effect (Example: If you're rewarded for driving effectively under the influence of the drug, then you will learn to function better as a driver under the influence). state-dependent learning-- Tasks learned under the influence of a psychoactive drug are performed better under the influence of that drug. The drug effect becomes part of the environmental cues for memory retrieval. 1/22/08Study guide is at http://vas.web.arizona.edu, along with the syllabus. Lecture notes will be posted there, as well. CH.1- Principles of Pharmacology What is pharmacology? It's the study of the action of drugs and their effects on living organisms. Pharmacology can be divided into three sub-disciplines: 1. Neuropharmacology-- focuses on the effects of drugs on the nervous system. 2. Psychopharmacology- focuses on the effects of drugs on the mind (experience, thought, and action). 3. Neuropsychopharmacology- focuses on the development of drugs that counter the effects of injury, disease or other damaging environmental factors. Drug effects can be categorized as follows: therapeutic vs side effects-- therapeutic effects are intentional physical or psychological effects, whereas side effects are unintentional. specific vs non-specific effects-- specific effects are based on drug-receptor interactions that can be predicted; non-specific effects are due to personality factors (P), situational or environmental factors (E) and the interaction of the two (P x E), which are much less predictable. Included among non-specific effects are placebo effects, which are the effects produced as a result of taking an inert substance. Pharmacokinetics-- The study of the factors that affect the rate with which drug effects are initiated and terminated. These factors include: 1. Route of administration-- How and where a drug is delivered into the body. 2. Absorption and distribution-- How the drug enters the bloodstream and where it is delivered by the blood. 3. Binding-- How the drug binds to receptors. 4. Inactivation-- Breakdown of the drug through some metabolic process into an inert form. 5. Excretion-- How the drug is eliminated from the body. I. Routes of Administration a) Oral--- Taken through the mouth, usually in the form of a pill or capsule. b) Intravenous-- Injected directly into the bloodstream, through a vein. c) Intramuscular-- Injected into muscle tissue. d) Subcutaneous-- Injected just below the skin e) Inhalation-- Absorbed through the longs (smoking) f) Topical-- Application to the mucous membranes (e.g., snorting) g) Transdermal-- Through the skin (nicotine patch, scopolamine) h) Epidural-- Injection into the cerebrospinal fluid (CSF), used with drugs that would not cross the blood-brain barrier fast enough otherwise. IV use poses several hazards: 1. Drugs that are impure or of unknown quality can produce overdose or poisoning. 2. Lack of sterile needles and procedures can lead to infections such as hepatitis and HIV> 3. Filler materials in IV drugs that don't dissolve properly and create clots and other blockages in the body. II. Absorption Absorption of the drug is determined by several factors: 1. Route of administration-- affects where the drug is absorbed and the amount of the drug destroyed by metabolic processes. 2. Drug concentration-- Depends on dosage, but also on age, sex, and body size. 3. Differences in solubility and ionization of different drugs The ionization of a drug, which is usually a salt of some kind, is the breakdown of the drug molecules into positive and negative ions. The rate of ionization of a drug depends on: a. pH (measure of the acidity/alkalinity of a solution)-- Drugs that are weak acids dissolve and ionize better in alkaline solutions, and weak bases ionize better in acidic solutions. b. pKa-- The equilibrium constant for the dissociation or ionization of a drug; this is an inherent or intrinsic characteristic of the drug. The ionized form of a drug does not get absorbed by membranes as effectively as the non-ionized form. The membranes found in the lining of the stomach and intestines are made up of phospholipids. The phosphate "head" of these molecules is hydrophilic ("water loving") and therefore dissolve in water. The hydrocarbon "tail" is hydrophobic ("water fearing") and therefore not only do not dissolve in water but also repel substances that are ionic or polar. Why do so many prescribed drugs have to be taken just before a meal and with water? The stomach empties into the small intestine faster when it's empty, and the small intestine has more surface area and slower movement of material than the stomach, allowing greater absorption of the drug. III. Distribution Highest concentrations of a drug that has been ingested are in organs that have a high blood flow, such as the heart, brain, kidneys, and liver. In theory, the brain, which has a very high blood flow, should be inundated with drugs, but it's not because of the blood-brain barrier. This barrier consists of endothelial cells that make up the walls of brain capillaries. The cells have tight junctions that restrict the movement of substances out of the capillary and into the brain. The same kind of barrier can be found in the placenta. Some drugs pass through this placental barrier relatively easily, including opiates, alcohol, barbiturates, cocaine, and carbon monoxide from smoking. Many of these substances are teratogens (cause birth defects in a developing fetus). IV. BINDING Binding to receptors in the brain is the topic of pharmacodynamics (next section). Depot binding is the binding of drugs to inactive sites in the body, such as blood plasma, muscles, fat, and bone, where drugs accumulate. The effects of depot binding include: 1. Lowered concentrations of the drug at sites of action because only freely-circulating drugs can bind to these sites. This can slow the onset of a drug or reduces its effects. 2. Competition among different drugs for depot-binding sites can produce higher concentrations than expected of a drug (in the bloodstream); this is the source of certain drug interactions. 3. Bound drugs cannot be metabolized by liver enzymes because they don't make it into the liver (some drugs, such as THC, can stay in your system for weeks after you've taken it). 4. Binding to depots may inactivate the drug, at least temporarily. V. INACTIVATION Biotransformation (metabolism of a drug into an inactive form) is of two types: Type I-- The breakdown of the drug into smaller components through oxidation, reduction, and hydrolysis. Type II-- The drug can combine with other molecules to form new, larger, and also inactive substances. Factors that affect drug metabolism or inactivation: 1. enzyme induction-- many drugs cause an increase in the levels of the liver enzymes that biotransform them. 2. enzyme inhibition-- other drugs can inhibit the action of enzymes (MAO inhibitors). 3. drug competion-- a drug may block the breakdown of another drug 4. individual differences that cause some people to metabolize a drug more efficiently than others (factors such as genetics, age, and gender can affect metabolic rates).