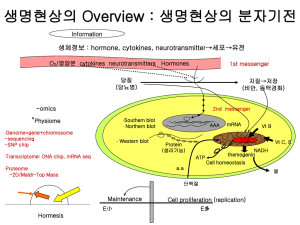
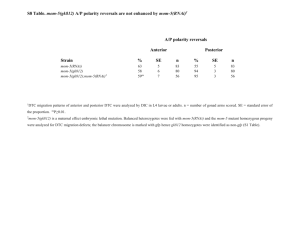
- White Rose Research Online
advertisement

Oncogene dependency and the potential of targeted, RNA interference-based anti-cancer therapy Ruiyang Yan*,†, Andrew Hallam*, Peter G. Stockley†,‡, and Joan Boyes*,‡ *Institute of Molecular and Cellular Biology, University of Leeds, Leeds LS2 9JT, United Kingdom. †Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, United Kingdom Corresponding authors: ‡Peter G. Stockley, Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK Phone: 44 (0) 113 343 3092 Email: p.g.stockley@leeds.ac.uk ‡Joan Boyes, Institute of Molecular and Cellular Biology, University of Leeds, Leeds LS2 9JT, United Kingdom. Phone: 44 (0) 113 343 3147 E-mail: j.m.boyes@leeds.ac.uk 1 Abstract Cancers arise through the progression of multiple genetic and epigenetic defects that lead to deregulation of numerous signalling networks. However, the last decade has seen the development of the concept of ‘oncogene addiction’, where tumours appear to depend on a single oncogene for survival. RNA interference (RNAi) has provided an invaluable tool in the identification of these oncogenes and oncogene-dependent cancers, and also presents great potential as a novel therapeutic strategy against them. Although RNAi therapeutics have demonstrated effective killing of oncogenedependent cancers in vitro, their efficacy in vivo is severely limited by effective delivery systems. Several virus-based RNAi delivery strategies have been explored but problems arose associated with high immunogenicity, random genome integration and nonspecific targeting. This has directed efforts towards non-viral formulations, including delivery systems based on virus-like particles, liposomes and cationic polymers, which can circumvent some of these problems by immunomasking and the use of specific tumour-targeting ligands. This review outlines the prevalence of oncogene-dependent cancers, evaluates the potential of RNAi-based therapeutics and assesses the relative strengths and weaknesses of different approaches to targeted RNAi delivery. Summary statement This review focuses on the tremendous possibilities brought about by the emerging concept of oncogene addiction and the use of RNAi technology in creating a potentially revolutionising way of combating cancer. Short title: RNAi therapeutics for oncogene-dependent cancers Keywords: Cancer, Oncogene addiction, Targeted therapy, RNAi delivery Abbreviations used: AD, adamantane; CDP, cyclodextrin-containing polymers; CML, chronic myeloid (or myelogenous) leukemia; ECE-1, endothelin converting enzyme 1; EGFR, epidermal growth factor receptor; EPR, enhanced permeability and retention; HCC, hepatocellular carcinoma; IKBKE, I Kappa B Kinase ε; IRF4, interferon regulatory factor 4; miRNA, microRNA; MM, multiple myeloma; NSCLC, non-small cell lung carcinoma; PEG, polyethylene glycol; precursor miRNA, pre-miRNA; pri-miRNA, primary miRNA; PSMA, prostate-specific membrane antigen; RISC, RNA-induced silencing complex; RNAi, RNA interference; RRM2, ribonucleotide reductase subunit M2; RSV, Rous Sarcoma virus; SNALPs, stable nucleic acid lipid particles; Tf, transferrin. 2 1. Introduction Cancer cells are remarkable in that they are able to display an extraordinary array of properties to promote their survival and growth. Being able to proliferate unchecked by the immune system, not responding to apoptotic or anti-growth signals and their ability to harness support from other cells, such as macrophages are just a few examples [1, 2]. To be able to maintain these properties, a complex collection of signalling pathways must be affected, both intra- and extracellularly. Exactly how cancerous cells are able to achieve this was unknown until the discovery of oncogenes and the subsequent emergence of evidence for oncogene dependency. 1.1 Discovery of Oncogenes The detection of the first oncogene, SRC, took decades to achieve. Rous’ preliminary experiments showed that cell filtrates from chicken sarcomas could cause cancer in healthy chickens, suggesting the presence of cancerinducing agents within the tumour, which were found to be the virus that became known as the Rous Sarcoma Virus (RSV) [3]. Such tumour viruses provided the first window in the genetics of cancer biology. Successive experiments by Baltimore and Temin revealed that RSV had an RNA genome that could convert back to DNA and integrate itself within the host’s genome [4, 5]. This discovery identified that a genetic element of the viral genome was responsible for transforming cells, which was subsequently narrowed down to a single gene, the SRC oncogene [6]. Homologues of SRC were then found in several species of bird and later across all vertebrates [7], suggesting that the viral oncogene had been acquired from host cells during evolution. This was a crucial development in cancer research as it implied the presence of oncogenes within the host genome that may cause cancer if mutated. This realisation led to the term ‘proto-oncogene’, describing a normal gene present within the genome of the organism, which, when mutated or overexpressed, can induce tumorigenesis. Since then, more than 40 protooncogenes have been identified within the human genome. 1.2 The Two-hit Hypothesis The two-hit hypothesis was proposed following the observation in retinoblastoma patients that two mutations, one in each allele of a tumour suppressor gene, are needed to trigger retinoblastoma [8]. A tumour suppressor gene encodes a protein which inhibits cell division when the cell is under unfavourable stress [9]. Knudson postulated that one faulty copy of a gene could be inherited but a further somatic mutation in the other allele was needed to produce the disease; those individuals with no inherited faulty copy required two somatic mutations [8]. 3 The two hit hypothesis was further developed to include the first hit being the activation of a proto-oncogene, which may not be enough to induce cancer as tumour suppressor genes will counteract its effect; therefore, the second hit is the loss-of-function of a tumour suppressor [10]. It is now known that multiple genetic alterations occur to trigger tumorigenesis; indeed, it has been shown that as many as 7-10 (epi)genetic events are required to bring about the cancer phenotypes [1, 11, 12]. Originally, this notion of a complex network of interacting pathways in cancers led to the idea that they may be too complicated for simple effective treatments [13, 14]. However, it has since been shown that knocking down just one specific gene can be sufficient to destroy a cancer. This phenomenon can be classed as oncogene or non-oncogene dependency. 1.3 Oncogene Dependency First coined by Bernard Weinstein in 2000, the phenomenon of oncogene addiction has now been identified in numerous cancers [2, 15-17]. This theory proposes that, despite the plethora of genetic alterations that occur in cancer progression, some cancers can become ‘addicted’ to a single oncogene on which they depend for proliferation and survival. Mouse models were initially used to identify and demonstrate oncogene dependency; for instance, the inactivation of oncogenic Myc in mice with osteosarcomas or lymphomas led to significant tumour regression [18, 19]. However, the state of oncogene addiction is not always permanent. Oncogenic escape has been observed in some tumours, which utilise their genomic instability to mutate around their addiction, by activating other oncogenes rendering the original oncogene redundant [20, 21]. In many cases the new oncoprotein influences the same molecular pathway as its predecessor, implying that it may be a particular signalling pathway to which the cancer cells are addicted rather than a specific oncogene per se. This was demonstrated in c-Myc-addicted mammary adenocarcinomas in mice [22]: following c-Myc downregulation, full regression was observed in many tumours. However, a subset of tumours continued to proliferate, in which activating mutations in KRAS2, an upstream effector of c-MYC in the MAPK pathway, were identified. 1.4 Non-Oncogene Dependency Non-oncogene addiction is related to oncogene addiction, but in this case tumours depend on normal genes for their survival [23]. One of the best studied examples of this is the dependency of some classes of multiple myelomas on interferon regulatory factor 4 (IRF4) [2], where IRF4 is absolutely required for proliferation and survival despite the fact it is not mutated or amplified [17]. 4 2. Prevalence of (Non-)Oncogene-dependent Cancers A diverse range of cancers exhibiting (non-)oncogene dependency have now been identified, these are summarised in Table 1. Some well characterised examples, including the oncogene-addicted, non-small cell lung carcinoma (NSCLC) and chronic myelogenous leukemia (CML), and nononcogene-addicted multiple myelomas (MM), are described in more detail below. 2.1 Oncogene-Dependent Cancers 2.1.1 EGFR in various cancers Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase found on the plasma membrane of epithelial cells that is involved in initiating growth and proliferation signalling pathways [24]. It is an important protooncogene due to its prevalence in several cancers, including NSCLC, breast, prostate and ovarian cancers. NSCLC is of particular significance because of the poor patient survival rates. It has been reported that NSCLC represents 85% of lung cancers [25], which account for one fifth of all cancer-related deaths [26]. Aberrant EGFR activation in these cancers can arise from mutation or overexpression. Nearly 90% of oncogenic EGFR mutations occur in the cytoplasmic kinase domain, and result in its continuous autophosphorylation [27]. The consequence of this is a constitutively active EGFR signalling cascade, through which downstream transcription factors are activated and confer various properties to the cancer phenotype, including angiogenesis, migration, proliferation, stromal invasion and resistance to apoptosis [28]. These EGFR mutations are mostly restricted to NSCLC, and are rare in other cancers. Nevertheless, there is still a high incidence of EGFR overexpression in glioblastomas [29], NSCLC, head and neck squamous cell carcinomas, and colorectal cancer [27]. Yamazaki and coworkers provided initial evidence for the therapeutic potential of targeting EGFR in cancers. They demonstrated that a ribozyme, targeted against an aberrant EGFR, could suppress its expression in the ERM5-1 cell line and significantly reduce their tumorigenic capacity in nude mice [30]. This provided evidence of tumour dependence on EGFR, which was reinforced by the subsequent emergence and clinical success of EGFRtargeted therapies in various cancers [27]. Examples include the small molecule drugs, gefitinib and erlotinib, which inhibit EGFR signalling by competing for ATP-binding sites within its intracellular kinase domain, and initially exhibited great efficacy in NSCLC patients [31, 32]. However, the success of these drugs was short-lived due to the emergence of drug resistance, often acquired by cancers via mutations in the drug-binding site of EGFR [33], thus highlighting the need for new therapeutic strategies against EGFR-dependent cancers. 5 2.1.2 BCR-ABL in chronic myeloid leukaemia BCR-ABL is an oncogene resulting from a chromosomal translocation found in 95% of CML [34], and in some acute myeloid and lymphoblastic leukaemias. The translocation between chromosomes 9 and 22 generates the Philadelphia chromosome, giving rise to a BCR-ABL fusion gene composed of exons 1-3 from BCR and all except the first exon from ABL [35]. The identification of this translocation in CML cells provided the first example of a chromosomal translocation in cancer [36], though it was not proven until later that it could induce tumorigenesis. When BCR-ABL was forcibly expressed in mouse models, development of several hematologic malignancies, particularly CML, was observed [37]. Wild-type ABL is a highly regulated tyrosine kinase with roles in the cell cycle, genotoxic stress response and integrin signalling [38]. The fusion of BCR-ABL results in the loss of the ABL autoinhibition domain, and ultimately leads to its deregulation [39]. This constitutively active tyrosine kinase hyperphosphorylates a vast range of substrates involved in growth, cell adhesion and inhibition of apoptosis, which results in the induction of tumorigenesis. The dependence of CML on BCR-ABL for survival was demonstrated through the clinical success of BCR-ABL inhibitors, most notably imatinib. Like gefitinib and erlotinib, imatinib is a small molecule tyrosine kinase inhibitor but has high specificity for BCR-ABL; it inhibits BCR-ABL by binding in the active site, locking the kinase in its autoinhibited conformation. The remarkable efficacy of imatinib in CML patients during clinical trials led to its rapid FDA approval to treat the disease in the USA [40]. However, it has since emerged that drug resistance can develop in CML patients that renders them unresponsive to imatinib treatment, typically through gene amplification of BCR-ABL or mutations in its catalytic domain [41]. Such drug resistance is especially prevalent during the later stages of CML, including the accelerated phase or blast crisis [42]. 2.2 Non-oncogene Dependent Cancers 2.2.1 IRF4 in multiple myeloma IRF4 is a transcription factor that is required at different stages of B cell development and, in particular, in the differentiation of B cells into plasma cells. In many MM, the malignant plasma cells are dependent on IRF4 for maintenance and survival despite the fact that the gene is not always mutated to an oncogenic form [17]. Hence this dependency is termed non-oncogene dependency. IRF4 is of notable significance because current treatments for MM are ineffective and the median survival time is only 3-4 years following initial treatment [43]. IRF4 is expressed in acutely activated B cells, and directs them to terminally differentiate into plasma cells by acting as a positive transcriptional regulator of genes involved in differentiation and proliferation. As many as 308 genes are direct or indirect targets of IRF4, of which 101 have been shown to 6 be upregulated in MM cell lines [17]. This broad regulation stems from the fact that IRF4 is at the apex in the hierarchy of gene regulators in that it regulates expression of other transcription factors which then further regulate gene expression. Moreover MYC, one transcription factor upregulated by IRF4, is a direct positive regulator of IRF4 itself [17]. Thus, IRF4 may act in a positive feedback loop to maintain its own expression whilst driving cancer progression. Dependence of MM on non-oncogenic IRF4 was demonstrated using short hairpin RNA (shRNA) screens with retroviral expression vectors [17]. Retroviruses carrying different shRNAs were transfected into MM cell lines and shRNA expression was induced by addition of doxycycline. Several shRNAs targeting IRF4 were identified that were able to kill ten different cell lines, each with a distinct molecular manifestation of MM. One particular shRNA targeting the 3’ untranslated region of IRF4 mRNA reduced its expression by 50-75%, and killed MM cells within 3 days. IRF4 is only expressed in lymphoid and myeloid cells [44]. This tissue specificity makes IRF4 an attractive therapeutic target for MM as (1) the uptake of IRF4-targeting drugs by other tissues will be unlikely to induce significant adverse effects, and (2) healthy blood cells lost due to the action of these drugs can be regenerated by natural haematopoiesis. 3. RNAi and RNAi-based Therapeutics Since its discovery in the mid-1990s [45, 46], RNAi has rapidly transformed from a curious phenomenon in worms to an invaluable tool in the study of functional genomics. The significance of RNAi technology was underlined by the award of the 2006 Nobel Prize in Physiology or Medicine to Fire and Mello [47]. RNAi describes the cellular process that occurs in various organisms, including mammals, plants and nematodes, whereby double stranded (ds) RNAs mediate specific and potent gene silencing [48]. RNAi is believed to have evolved from an early immune mechanism against viruses and transposable elements [49]. Foreign dsRNAs are recognised and processed by Dicer RNases into 21-24 nucleotide fragments, known as small interfering RNA (siRNA), then loaded onto an Argonaute-containing RNA-induced silencing complex (RISC). One strand of the siRNA (passenger strand) is degraded, whilst the other (guide strand) in complex with RISC searches cytoplasmic RNA for complementary sequences. Once located, Argonaute triggers cleavage of the targeted RNA, thereby silencing expression of the foreign gene. This RNAi pathway forms an important component of innate antiviral immunity in plants, nematodes, fungi and arthropods [49]. Another RNAi pathway, the endogenous microRNA (miRNA) pathway, enables post-transcriptional regulation of gene expression in animals and plants [50]. This pathway commences with the transcription of primary miRNAs (pri-miRNA) from the host genome. These transcripts are typically 7 excised by a microprocessor complex into 65-70 nucleotide precursor miRNAs (pre-miRNA), which are then exported to the cytoplasm by exportin-5 and Ran GTP [51]. Similar to the siRNA pathway, precursor miRNAs are processed by Dicer to form 21-26 nucleotide mature miRNAs, which can be loaded onto RISC, termed miRISC. Compared to siRNAs, miRNAs only partially base pair with their target sequences in the 3’-untranslated regions (3’-UTRs) of mRNA, mainly via 7-8 consecutive base pairs of the so-called seed region. Binding of miRISCs to 3’-UTRs inhibits 5’-cap dependent translational initiation and can trigger mRNA degradation. Individual miRNAs usually have several different mRNAs as targets. To date, 1,872 miRNA sequences have been identified within the human genome (http://microrna.sanger.ac.uk; accessed March 6, 2014) which regulate almost a third of protein-encoding genes [52]. Unsurprisingly, endogenous RNAi plays a critical role in regulating numerous vital processes, including cell growth, cell proliferation, apoptosis and tissue differentiation [53]. Cellular RNAi can be exploited to silence a gene of interest by introducing exogenous sxRNA analogues, either siRNA or shRNA, that target its mRNA. SiRNAs are competent for RISC loading, and may directly enter the RNAi pathway once delivered to the cytoplasm (see ‘Non-viral RNAi Delivery’). For shRNAs, viral vectors are typically used to deliver shRNAencoding genes into cells for expression (see ‘Viral RNAi Delivery’). Expressed shRNA undergoes processing to form siRNA, which can then be loaded onto RISC complexes. Many different sxRNA libraries now exist that cover the entire genomes of both mice and humans, enabling high-throughput loss of function analyses and the identification of essential genes for virtually any cellular process [54]. RNAi technology has played a key role in the identification of many (non-)oncogene-dependent cancers, and the oncogenes on which they rely. One example of this is the identification of the oncogene, I Kappa B Kinase ε (IKBKE), in breast cancers. A large shRNA library targeting 1,200 genes was used to screen the breast cancer cell line, MCF-7, in which three shRNAs targeting IKBKE were able to reduce proliferation and viability of MCF-7 cells, indicating their dependence on IKBKE for maintenance and survival [55]. These findings not only added to the accumulating body of evidence for oncogene dependence in cancers, but also highlighted the therapeutic potential of sxRNA-mediated RNAi against them. SxRNAs have several advantages as novel therapeutic agents. They are synthetic and relatively easy to produce compared to protein-based therapeutics, can be rationally designed to target any (non-)oncogene, and can achieve a level of specificity far higher than that of traditional cancer therapeutics. Furthermore, as sxRNAs target (non-)oncogenes at the mRNA level, they may exhibit synergistic effects when used with drugs that target at the protein level. For instance, siRNA-mediated silencing of BCR-ABL and the gene encoding multidrug resistance protein 1 has been shown to sensitise 8 CML cells to imatinib treatment [56, 57], highlighting the potential of such combinatorial therapies. However, several barriers have severely hindered the progress of RNAi therapeutics towards clinical approval, including the short plasma half-lives of RNAs, their poor cellular uptake, and the lack of tumour-specific targeting. To overcome these barriers, various sxRNA delivery systems have been developed, which aim to (1) protect the RNA from nuclease degradation, (2) evade immune surveillance, (3) prevent rapid renal clearance, (4) promote its accumulation in tumour tissues, (5) deliver the RNA to cancer cells in a highly specific manner, and (6) interact with cellular trafficking pathways to deliver the RNA to appropriate locations within the cell. A diverse range of RNAi delivery systems have been explored, these can generally be categorised as viral or non-viral. Selected examples that have shown the most promise in the laboratory or clinical trials are discussed in detail below, but several others exist [58]. 3.1 Viral RNAi Delivery Evolutionary fine-tuning has given viruses the ability to infiltrate specific cells and deliver genetic material for expression, making them an attractive option for delivering RNAi effectors therapeutically. Viral delivery systems typically carry shRNA expression cassettes [59]. Non-essential regions within the virus genome can be modified to incorporate a promoter and shRNA of interest, to achieve long-term shRNA expression and hence repression of targeted genes in transfected cells. Adenoviruses, adeno-associated viruses, herpesviruses and lentiviruses are among those that have been tested as shRNA delivery vectors [60]. Adenoviruses are widely used in gene therapy studies due to their ability to incorporate large genes and deliver them to the nucleus [61]. Adenoviral delivery of shRNAs has been demonstrated in several human cancer cell lines, in which delivered shRNAs targeting the mRNA of the tumour suppressor protein, p53, successfully triggered gene silencing [62]. Despite these encouraging results from in vitro studies, the inherent, high immunogenicity of adenoviruses has severely limited their use for shRNA delivery in vivo [63, 64]. Systemic administration of adenoviral vectors to human patients leads to high liver uptake and activation of innate immunity, causing severe acute inflammatory responses and even fatalities [65]. Moreover, the development of neutralising anti-adenoviral antibodies prohibits repeated drug dosing [66]. These findings seriously question the suitability of adenoviral vectors for in vivo applications. However, work is under way to address some of these issues, in particular the immunogenicity and poor targeting of adenoviruses. Various approaches, including addition of polyethylene glycol (PEG) to the virus surface [67], liposomal modification [68], and genetic incorporation of tumour-targeting ligands [61], have shown promising results. 9 Lentiviruses, of the Retroviridae family, have also been explored for therapeutic shRNA delivery. Lentiviruses are single stranded RNA viruses with the ability to reverse transcribe their RNA genome into DNA, before inserting the viral DNA into the host genome for expression [69]. Brummelkamp and coworkers successfully used lentiviruses to deliver shRNA vectors that targeted an activated K-RAS oncogene to pancreatic cancer cells. Effective knockdown of the oncogene was achieved, resulting in the elimination of the tumorigenic capacity of transfected cancer cells in mice [12]. Another lentiviral shRNA delivery system is currently under clinical evaluation for autologous cell therapy against AIDS-related non-Hodgkin’s lymphoma [70]. This involves ex vivo shRNA delivery. Haematopoietic progenitor cells are removed from patients via apheresis, transfected with lentiviral shRNA vectors and then reinfused into the patient. Early results revealed no short-term toxicity associated with the transfected haematopoietic progenitor cells, and two of four patients exhibited persistent expression of the shRNA [71]. The major drawback of retroviral shRNA delivery is that random retroviral genome insertion may cause major disruptions to the host genome, resulting in a multitude of problems, including the activation of protooncogenes [72]. This could be particularly dangerous if a large amount of virus is taken up by untargeted healthy tissues. Thus, retroviruses are not ideal candidates for shRNA delivery in vivo. Hong and coworkers demonstrated that Herpesvirus saimiri can deliver shRNA targeting the endothelin converting enzyme 1 (ECE-1) into prostate cancer cells [73]. ECE-1 was selected as the target due to its involvement in the invasion and migration of several cancers, including prostate, lung, breast, colorectal and ovarian [74]. The herpesvirus vector was tested in vitro in several cell lines and ex vivo in primary cells from three different forms of prostate cancer. Significant levels of ECE-1 knockdown were observed in all cases, which severely limited the capacity of the cancer cells to migrate and invade [73]. However, much in vivo testing is still needed to evaluate the safety of herpesviruses for therapeutic applications, especially considering the well reported tendency of vectors based on herpes simplex virus 1, another member of the herpesviridae family, to trigger severe inflammatory responses in the central nervous system [75, 76]. 3.2 Non-viral RNAi Delivery Although viral RNAi delivery has proven effective in vitro, its clinical application has largely been deterred by problems associated with its oncogenic potential, lack of tumour-specific targeting, high immunogenicity and difficulty of large scale production. As a result, non-viral RNAi delivery systems, which can potentially circumvent or minimise these problems, have attracted considerable attention in recent years [77]. 10 Several important parameters need to be considered in the design of a non-viral RNAi delivery system, including: Size: Particles should be 10-100 nm in diameter to avoid rapid renal clearance [78], and to accumulate preferentially at disease sites characterised by increased vascular permeability, such as sites of infection or tumour [79, 80]. This effect is known as enhanced permeability and retention (EPR). Zeta potential: Zeta potential is a measure of the magnitude of the electrostatic attraction or repulsion between particles, and is related to their surface charge. To minimize nonspecific electrostatic interactions with cells, which typically display a zeta potential within the range of -5 to -20 mV [81, 82], and to reduce macrophage uptake, a small negative zeta potential is generally desirable for nanoparticles [83]. Immunomasking: A highly immunogenic delivery system can trigger potentially lethal inflammatory responses in patients, as seen with adenoviruses, and also prohibits repeated dosing. This can be minimised in non-viral delivery systems via surface coverage with PEG molecules, which mask epitopes from the immune system [84]. Targeting: One common problem with many conventional cancer drugs in use today is a lack of specificity. They are readily internalised by healthy as well as cancer cells, thus producing side effects. To this end, ligands that bind cancer cell surface markers can be incorporated into non-viral RNAi delivery systems. These markers are typically receptors which are overexpressed on cancer cells, such as EGFR, and receptors for transferrin (Tf) and folate [77]. Targeting agents can be natural receptor ligands like Tf or synthetic ones like peptide or nucleic acid aptamers [85]. Aside from preferential binding to cancer cells, targeting ligands also offer a mechanism of cell entry by receptormediated endocytosis [86]. Endosomal escape: Following endocytosis, the delivery system and sxRNA must escape the endosome into the cytosol, where the sxRNA mediates its effect. This may be achieved by incorporating endosomal buffering ligands, such as histidine-rich peptides and polyethyleneimine. These ligands can absorb protons as they are pumped into the endosome, acting as ‘proton sponges’, and promoting further proton influx. This in turn is coupled to an increased influx of chloride ions and water, resulting in the osmotic swelling and eventual rupture of the endosome, which can then release its contents into the cytosol [87]. A diverse range of non-viral RNAi delivery systems have been tested and some are already in advanced stages of clinical trials. A selection of some of the most promising formulations is highlighted below. 11 3.2.1 Virus-like Particles (VLPs) Similar to viral delivery, RNAi delivery by VLPs relies on capsids for the encapsidation, nuclease protection and intracellular delivery of RNAs. However, several important distinctions exist between the two types of delivery system. Firstly, VLP delivery utilises capsids of simple viruses, typically bacteriophages, to deliver siRNAs to cells, rather than complex, geneticallymodified human viruses to deliver shRNA. Because siRNAs can directly enter the RNAi pathway and do not require expression from a vector, the virus genome can be removed, avoiding problems associated with host genome integration, as seen in retroviral shRNA delivery. Secondly, the components of a VLP delivery system are relatively simple and cheap to prepare. Coat protein subunits that constitute capsids can be produced in large quantities and purified from recombinant E. coli expression systems [88, 89]. In vitro assembly can then be performed to trigger capsid formation and package sxRNA cargo [90, 91] (Figure 1). Thirdly, VLPs can be surface-modified to incorporate a range of useful ligands for immunomasking and targeting [77, 89, 90, 92]. Thus, the resultant semi-synthetic particles, whilst retaining desirable viral attributes for RNAi delivery, exhibit enhanced functionalities. Research on VLP-mediated siRNA delivery has mostly been based on bacteriophage MS2 (Figure 1) [90, 91]. MS2 is a well characterised T=3 icosahedral virus, with a 180-subunit capsid encapsidating its single-stranded RNA genome [93]. The ease of producing MS2 coat proteins, the robust nature of its capsids, and the abundance of reactive amines on its surface for multivalent ligand display are some reasons for its selection as a model VLP delivery system. MS2 capsids are pH-sensitive, readily disassembling at acidic pH and reassembling upon mixing with a packaging signal, TR, at neutral pH (Figure 1). This not only enables simple siRNA packaging, but also provides a mechanism of siRNA release upon entering the acidic environment of the endosome. With a diameter of 26 nm, MS2 capsids are ideal to benefit from enhanced permeability and retention (EPR). Further, MS2 capsids have a zeta potential of -25 mV [94], thus are unlikely to have strong electrostatic interactions with cells; this may also be adjusted via surface modifications if required. MS2 VLPs have recently been used to deliver siRNA targeted against the anti-apoptotic factor, BCL2, to HeLa cells [90]. Tf was conjugated to the VLP surface as a targeting ligand. VLPs successfully delivered siRNAs to the cancer cells in a Tf receptor-dependent manner, with significant levels of BCL2 knockdown and cell death observed at nanomolar siRNA concentrations. The efficiency of siRNA delivery by VLPs was comparable to that by a commercially available liposomal agent, although VLP-delivered siRNAs appeared to be more active. MS2 VLPs have also been targeted to hepatocellular carcinoma (HCC) cells for siRNA delivery [91]. A cocktail of siRNAs targeting the mRNA of several cyclins, whose overexpression is associated with 12 hepatocarcinogenesis [95], were packaged inside MS2 VLPs. The VLP surface was then decorated with (1) PEG for immunomasking, (2) SP94, a targeting peptide previously identified by affinity selection from a phage display library against HCC cell surface targets [96], and (3) a histidine-rich peptide to promote endosomal escape. Gene silencing of targeted cyclins, growth arrest and apoptosis were observed at picomolar siRNA concentrations in HCC cells, but not in normal hepatocyte cells. This demonstrated the ability of VLPs to simultaneously deliver different siRNAs to targeted cells, and to achieve multiple gene knockdowns. Such combination therapies may play a significant role in the treatment of tumours in which a diverse set of mutations are present [97]. Preliminary in vivo testing of VLP-mediated RNAi delivery has recently been carried out [98, 99]. MS2 VLPs packaging a pre-miRNA, pre-miR146a, were surface-decorated with HIV Tat peptides for cell penetration; no celltargeting ligands were used. Upon intravenous delivery to mice, these VLPs displayed widespread biodistribution in the plasma, lung, spleen and kidney, where high levels of mature miR146a were detected and knockdown of known targets of miR146a was observed. Additionally, no off-target toxicities in VLPtreated mice were reported. Despite these promising data, more is required, in particular detailed pharmacokinetic and long-term safety profiles of VLPs in vivo, to fully assess their suitability for entering clinical trials as an RNAi delivery system. 3.2.2 Cationic Liposomes Since its first report two decades ago [100], significant progress has been made towards the use of cationic liposomes for the cellular delivery of nucleic acids, particularly siRNAs. This delivery system self-assembles with siRNA via electrostatic interactions between positively charged lipids and the negatively charged phosphate backbone of siRNA, forming lipoplexes. Lipoplexes protect encapsulated siRNA from nuclease degradation, and facilitate their transport across the cell membrane. Cationic liposomes can be constituted from a variety of lipidic components, including phospholipids, cholesterols, cationic lipids and various lipid-like materials. Lipid composition can greatly influence the physical properties of liposomes [101] and their delivery efficiency [102]. For instance, cholesterols are commonly included to improve liposomal stability and uptake [103]. Some cationic derivatives of cholesterol can also facilitate endosomal escape [104]. PEGylated lipids can be incorporated to confer steric stabilisation to the nanoparticle, and prolong its blood circulation by evading the mononuclear phagocyte system [105, 106]. These PEG-displaying liposomes are termed stable nucleic acid lipid particles (SNALPs). Amino lipid derivatives containing cleavable ester bonds have recently been developed to improve biodegradability of liposomes [107]. A wide range of methods for liposome preparation are available, which can also influence important properties such as the size and dispersity of liposomes [108]. 13 Nucleic acid delivery by cationic liposomes is well established, as demonstrated by the prevalence of commercially available products such as Lipofectin, Oligofectamine, Lipofectamine and RNAifect, which are commonly used in laboratories to enhance siRNA delivery in vitro [109-111]. Furthermore, nearly half of the most promising siRNA drug candidates currently in clinical trials are formulated with cationic liposome systems for delivery [112]. For example, Alnylam Pharmaceuticals' SNALP-based drug ALN-VSP02, which is used to treat liver cancer, delivers siRNAs targeted against the mRNAs of vascular endothelial growth factor and kinesin spindle protein, both of which have important roles in tumour proliferation and survival. ALN-VSP02 has suitable physical properties for delivering siRNA in vivo, including a diameter of 80-100 nm and a small zeta potential of <6 mV at pH 7.4. Intravenous administration of ALN-VSP02 in patients with advanced cancer and liver metastases gave encouraging results, including siRNAmediated cleavage of targeted mRNAs in the liver and pronounced tumour regression [113]. One potential disadvantage of current liposomal siRNA drug candidates, including ALN-VSP02, is the lack of a targeting mechanism for cancer cells. As a result, significant levels of siRNA may be taken up by healthy cells alongside the cancer cells. It is notable that liposomes are already clinically approved to deliver other drugs, such as doxorubicin and daunorubicin, in the treatment of various cancers [114]. Whether this platform will prove to be the most effective for delivering siRNA therapeutics remains to be seen. 3.2.3 Cyclodextrin-containing Polymers Cyclodextrins are cyclic oligosaccharides that are biocompatible, exhibit low toxicity and immunogenicity in humans, and have strong resistance to degradation by human enzymes [115]. They can be incorporated into a cationic linear backbone to form cyclodextrin-containing polymers (CDPs, Figure 2A), which can package siRNA for intracellular delivery. One prominent example of CDP-based siRNA delivery is the RNAi/Oligonucleotide Nanoparticle Delivery, or RONDEL platform [116]. At the heart of RONDEL are CDPs which consist of 5-6 repeating units of βcyclodextrin coupled to amidine charge centres and terminal imidazoles (Figure 2A). These CDPs provide several functions to the delivery system: (1) positively charged amidines enable electrostatic interactions with siRNA, (2) CDPs condense siRNAs into nanoparticles and protect them from nuclease degradation, and (3) imidazole groups utilise the proton sponge effect to promote endosomal escape. CDPs also serve as a structural scaffold onto which additional components can be incorporated, including PEG for steric stabilisation [117] and immunomasking of the delivery system [118], and Tf for preferential tumour-targeting. By coupling PEG and PEG-Tf to adamantane, a cycloalkane which forms high affinity inclusion complexes with β-cyclodextrin [119], the 14 CDP-siRNA core can be non-covalently surface decorated with these ligands (Figure 2B). Assembled nanoparticles are 70 nm in diameter and thus benefit from EPR, and each contains approximately 10,000 CDPs, 2,000 siRNAs, 4,000 PEG-adamantanes and 100 Tf-PEG-adamatanes [120]. Furthermore, the zeta potential of RONDEL particles can be adjusted from +15 mV to -25 mV through incorporation of AD-PEGs modified to contain an anionic charge [121]. RONDEL demonstrated good efficacy when tested in a metastatic murine model of Ewing’s sarcoma, in which RONDEL was used to deliver siRNAs that targeted the EWS/Fli1 fusion oncogene, resulting in effective silencing of the oncogene and significant anti-tumour effects [122]. Further, good tolerability of the delivery system was shown in cynomolgus monkeys at dosages of 3 and 9 mg siRNA/kg [118]. These promising results led to the advancement of RONDEL into clinical trials. Calando Pharmaceuticals’ CALAA-01, a drug candidate currently undergoing phase 1b clinical evaluation [112], utilises RONDEL to deliver systemically siRNA targeted against the M2 subunit of ribonucleotide reductase (RRM2) to solid tumours. Ribonucleotide reductase is required for DNA synthesis and is a key component in the proliferation of cancer cells [123]. CALAA-01 has so far demonstrated promising results in human patients with metastatic melanoma refractory to standard therapies, in which delivered siRNAs localised to tumours, and levels of RRM2 mRNA decreased in a dose-dependent manner [124]. 3.2.4 Chemical Modification Whilst polymer-based delivery systems protect siRNAs from serum nucleases, they do not offer the same protection against intracellular nucleases upon cytoplasmic delivery. For this reason, chemical modifications are commonly made to siRNAs to confer resistance to these intracellular nucleases and hence increase silencing persistence [125]. Modifications typically involve the addition of phosphorothioate linkages to the phosphate backbone of siRNA, or substitution of 2′-hydroxyls with 2′-methoxy or 2′-fluoro groups on ribose sugars [126]. Importantly, these modifications do not reduce siRNA activity [127]. In fact recent studies have identified 2′-hydroxyl modifications at defined positions on the guide strand which increase siRNA potency [128]. Chemical modifications can also enhance other pharmacological properties of siRNA. For example, 2′-methoxy groups on the passenger strand, can reduce immunogenicity by preventing siRNA interaction with Tolllike receptors thus averting the type I interferon pathway [129]. Off-target effects, whereby siRNAs trigger silencing of partially complementary mRNA, can also be mitigated by 2′-methoxy substitutions at certain positions on the guide strand [130]. More recently, modifications with locked nucleic acids (LNA) and unlocked nucleic acids (UNA) have shown to improve several aspects of 15 siRNA function [131]. In LNA, a methylene bridge links the 2′-oxygen and 4′carbon of the ribose, locking the sugar ring in a C3′-endo conformation. Strategic incorporation of LNA into siRNAs increases their thermostability, and can increase nuclease resistance [132, 133], reduce immunogenicity [134], and mitigate off-target effects [135]. Conversely, incorporation of UNA, which lack a C2′-C3′ bond in their ribose groups, at certain positions in siRNA can lead to local destabilisation of the duplex [136]. Such modifications have shown to reduce off-targeting [137] and improve the biostability of siRNAs [138]. One modification which can promote tumour-targeting of siRNAs is the covalent attachment of a nucleic acid aptamer [85]. Similar to antibodies, nucleic acid aptamers can bind to a large number of biomolecules and display a high degree of specificity and affinity to their targets [139]. Aptamers offer several advantages over antibodies. They can be selected against any protein or cell targets via automated in vitro selection. They are amenable to simple chemical modifications as well as relatively cheap mass production. They elicit little immune response and their smaller size may facilitate access to epitopes that are inaccessible to antibodies [140]. The use of aptamer-siRNA chimaeras has been demonstrated by the Giangrande group [141], who used an aptamer targeting the prostate-specific membrane antigen (PSMA), an overexpressed surface marker in prostate cancers, conjugated to siRNAs against BCL2 and polo-like kinase 1. Aptamerconjugated siRNAs were specifically internalised into prostate cancer cells via PSMA, and induced knockdown of targeted genes as well as cell death. In contrast, few effects were observed with unmodified siRNAs or when aptamer-siRNAs were incubated with non-PSMA expressing cells. These aptamer-siRNA conjugates demonstrated great efficacy when tested in vivo, which led to significant regression of PSMA-expressing tumours upon systemic administration in mice [142]. Six of the fourteen most promising siRNA therapeutics in clinical trials are chemically modified, naked siRNAs [112]. It is notable that despite the advantages of these modifications, such RNA-only formulations may still encounter problems such as rapid renal clearance due to their small size, or undesired electrostatic interactions with serum proteins. Therefore, it seems likely that it will be ultimately more effective to package chemically modified siRNAs inside polymer-based delivery systems. 3.3 Problems Associated with RNAi Therapy Although RNAi holds huge therapeutic promise, it might not be as effective as it first appeared. Problems associated with off-target effects have been observed, whereby different mRNAs with siRNA seed matches in their 3-UTR' are bound and repressed by the siRNA/RISC complex [143]. Off16 target effects can also result from aberrant loading of the passenger strand into the RISC complex, possibly targeting other mRNAs and leading to their knockdown [144]. These highlight the need for careful sxRNA design to avoid sequence similarity in other genes to the target mRNA. Algorithms have been developed to facilitate specific siRNA design to reduce the likelihood of these off-target effects [145]. Modifications to the passenger strand can also be used to this end [146, 147]. Exogenous RNA is known to induce cellular stress responses and cause toxicity in vivo [59]. For sxRNAs, toxicities can arise as a result of the saturation of endogenous RNAi pathways in cells, in which important natural miRNAs are outcompeted for interaction with limiting cellular factors. Several studies have reported hepatotoxic side effects of high shRNA expression in the liver of mice, which correlated with the downregulation of liver-derived miRNAs and the upregulation of miRNA-controlled hepatic genes [148, 149]. Further experiments revealed that the silencing capacity of both shRNAs and miRNAs can be improved by overexpressing exportin-5 in mice [148]. Exportin-5 is a protein of low abundance that is essential for the nuclear export of shRNAs, miRNAs and tRNAs, hence the data suggests it might be one limiting factor over which shRNAs and miRNAs compete to exert their silencing effects. These studies underline the need for stringent control over sxRNA dosing for therapeutic applications in vivo. For viral shRNA delivery, the use of a moderate promoter and limited vector dosages should be considered to avoid saturating endogenous RNAi pathways and adverse effects in healthy cells. For non-viral siRNA delivery, as well as limiting dosages, the incorporation of PEG and tumour-targeting ligands could be considered to promote preferential siRNA uptake by cancer cells and mitigate uptake by healthy cells. 4. Conclusion RNAi technology has undoubtedly developed into a powerful laboratory tool in the last two decades, but the hypothesis that it can be translated into an effective therapeutic strategy for cancer remains to be proven. The development of RNAi-based anti-cancer therapeutics is motivated by several factors. First, the inability of some conventional cancer treatments, such as chemotherapy and radiation, to specifically and fully eradicate cancers has shifted the emphasis towards more effective, targeted therapies. Second, despite the complex array of processes associated with tumour formation and maintenance, some cancers appear to be addicted to single gene products for their survival. This (non-)oncogene dependence has been dubbed the ‘Achilles’ heel’ of cancers [15], potentially yielding effective therapies that specifically target these genes for silencing. Finally, despite the tremendous success of drugs such as imatinib and gefitinib in the treatment of various cancers, one recurring problem has been the emergence of drug resistance, typically through gene mutations. This emphasises the importance of novel, targeted therapeutic strategies against cancers. 17 Various strategies have been explored to improve the pharmacological properties of sxRNA, by enhancing their stability, prolonging their plasma halflives, promoting their cellular uptake, and facilitating tumour-specific targeting. The natural ability of viruses to deliver genetic material to cells makes them an attractive option for delivering shRNA expression vectors. However, problems with high immunogenicity, lack of tumour-specific targeting and the oncogenic potential of insertional mutagenesis have directed efforts towards non-viral siRNA delivery, which can circumvent these issues. A diverse range of promising nanoparticulate delivery systems have emerged from laboratories, some of which are already in clinical trials. Development of these platforms will be crucial to making RNAi a viable therapeutic strategy against cancer. Funding R.Y. is grateful for the financial support from the Leeds University Research Scholarship program. Conflict of interest The authors declare no conflict of interest. 18 References 1 Hanahan, D. and Weinberg, R. A. (2000) The hallmarks of cancer. Cell. 100, 57-70 2 Luo, J., Solimini, N. L. and Elledge, S. J. (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 136, 823-837 3 Rous, P. (1911) A sarcoma of the fowl transmissible by an agent from the tumor cells. Journal of Experimental Medicine. 13, 397-411 4 Baltimore.D. (1970) Viral RNA-dependent DNA Polymerase: RNAdependent DNA Polymerase in Virions of RNA Tumour Viruses. Nature. 226, 1209-1211 5 Temin, H. M. and Mizutani, S. (1970) RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 226, 1211-1213 6 Wang, L. H., Duesberg, P. H., Kawai, S. and Hanafusa, H. (1976) Location of envelope-specific and sarcoma-specific oligonucleotides on RNA of Schmidt-Ruppin Rous sarcoma virus. Proc. Natl. Acad. Sci. U. S. A. 73, 447-451 7 Stehelin, D., Varmus, H. E., Bishop, J. M. and Vogt, P. K. (1976) DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 260, 170-173 8 Knudson, A. G. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A. 68, 820-823 9 Sherr, C. J. (2004) Principles of tumor suppression. Cell. 116, 235-246 10 MacPherson, D. and Dyer, M. A. (2007) Retinoblastoma: From the twohit hypothesis to targeted chemotherapy. Cancer Research. 67, 7547-7550 11 Renan, M. J. (1993) How many mutations are required for tumorigenesis? Implications from human cancer data. Molecular Carcinogenesis. 7, 139-146 12 Brummelkamp, T. R., Bernards, R. and Agami, R. (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2, 243-247 13 Li, R. H., Yerganian, G., Duesberg, P., Kraemer, A., Willer, A., Rausch, C. and Hehlmann, R. (1997) Aneuploidy correlated 100% with chemical transformation of Chinese hamster cells. Proc. Natl. Acad. Sci. U. S. A. 94, 14506-14511 14 Hahn, W. C. and Weinberg, R. A. (2002) Modelling the molecular circuitry of cancer. Nat. Rev. Cancer. 2, 331-341 15 Weinstein, I. B. (2002) Cancer: Addiction to oncogenes - The Achilles heal of cancer. Science. 297, 63-64 16 Weinstein, I. B. and Joe, A. (2008) Oncogene addiction. Cancer Research. 68, 3077-3080 17 Shaffer, A. L., Emre, N. C. T., Lamy, L., Ngo, V. N., Wright, G., Xiao, W. M., Powell, J., Dave, S., Yu, X., Zhao, H., Zeng, Y. X., Chen, B. Z., 19 Epstein, J. and Staudt, L. M. (2008) IRF4 addiction in multiple myeloma. Nature. 454, 226-231 18 Pelengaris, S., Littlewood, T., Khan, M., Elia, G. and Evan, G. (1999) Reversible activation of c-Myc in skin: Induction of a complex neoplastic phenotype by a single oncogenic lesion. Molecular Cell. 3, 565-577 19 Jain, M., Arvanitis, C., Chu, K., Dewey, W., Leonhardt, E., Trinh, M., Sundberg, C. D., Bishop, J. M. and Felsher, D. W. (2002) Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 297, 102-104 20 Karlsson, A., Giuriato, S., Tang, F., Fung-Weier, J., Levan, G. and Felsher, D. W. (2003) Genomically complex lymphomas undergo sustained tumor regression upon MYC inactivation unless they acquire novel chromosomal translocations. Blood. 101, 2797-2803 21 Gunther, E. J., Moody, S. E., Belka, G. K., Hahn, K. T., Innocent, N., Dugan, K. D., Cardiff, R. D. and Chodosh, L. A. (2003) Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev. 17, 488-501 22 D'Cruz, C. M., Gunther, E. J., Boxer, R. B., Hartman, J. L., Sintasath, L., Moody, S. E., Cox, J. D., Ha, S. I., Belka, G. K., Golant, A., Cardiff, R. D. and Chodosh, L. A. (2001) c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nature medicine. 7, 235-239 23 Solimini, N. L., Luo, J. and Elledge, S. J. (2007) Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 130, 986-988 24 Kolch, W. and Pitt, A. (2010) Functional proteomics to dissect tyrosine kinase signalling pathways in cancer. Nat. Rev. Cancer. 10, 618-629 25 Ettinger, D. S., Akerley, W., Bepler, G., Blum, M. G. and Yang, S. C. (2010) Non Small Cell Lung Cancer. J. Natl. Compr. Cancer Netw. 8, 740-801 26 Ferlay, J., Autier, P., Boniol, M., Heanue, M., Colombet, M. and Boyle, P. (2007) Estimates of the cancer incidence and mortality in Europe in 2006. Annals of Oncology. 18, 581-592 27 Ciardiello, F. and Tortora, G. (2008) Drug therapy: EGFR antagonists in cancer treatment. New England Journal of Medicine. 358, 1160-1174 28 Nair, P. (2005) Epidermal growth factor receptor family and its role in cancer progression. Current Science. 88, 890-898 29 Mellinghoff, I. K., Cloughesy, T. F. and Mischel, P. S. (2007) PTENmediated resistance to epidermal growth factor receptor kinase inhibitors. Clin. Cancer Res. 13, 378-381 30 Yamazaki, H., Kijima, H., Ohnishi, Y., Abe, Y., Oshika, Y., Tsuchida, T., Tokunaga, T., Tsugu, A., Ueyama, Y., Tamaoki, N. (1998) Inhibition of tumor growth by ribozymemediated suppression of aberrant epidermal growth factor receptor gene expression. J. Natl. Cancer Inst. 90, 581–587 31 Lynch, T. J., Bell, D. W., Sordella, R., Gurubhagavatula, S., Okimoto, R. A., Brannigan, B. W., Harris, P. L., Haserlat, S. M., Supko, J. G., Haluska, F. G., Louis, D. N., Christiani, D. C., Settleman, J. and Haber, D. A. (2004) 20 Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 350, 2129-2139 32 Pao, W., Miller, V., Zakowski, M., Doherty, J., Politi, K., Sarkaria, I., Singh, B., Heelan, R., Rusch, V., Fulton, L. (2004) EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA. 101, 13306–13311 33 Tartarone, A., Lazzari, C., Lerose, R., Conteduca, V., Improta, G., Zupa, A., Bulotta, A., Aieta, M. and Gregorc, V. (2013) Mechanisms of resistance to EGFR tyrosine kinase inhibitors gefitinib/erlotinib and to ALK inhibitor crizotinib. Lung Cancer. 81, 328-336 34 Wada, H., Mizutani, S., Nishimura, J., Usuki, Y., Kohsaki, M., Komai, M., Kaneko, H., Sakamoto, S., Delia, D., Kanamaru, A. and Kakishita, E. (1995) Establishment and molecular characterization of a novel leukemic cell line with Philadelphia chromosome expressing p230 BCR/ABL fusion protein. Cancer Research. 55, 3192-3196 35 Faderl, S., Talpaz, M., Estrov, Z., O'Brien, S., Kurzrock, R. and Kantarjian, H. M. (1999) Mechanisms of disease - The biology of chronic myeloid leukemia. New England Journal of Medicine. 341, 164-172 36 Nowell, P. C. and Hungerford, D. A. (1960) A Minute Chromosome in Human Chronic Granulocytic Leukemia. Science. 132, 1497-1497 37 Daley, G. Q., Vanetten, R. A. and Baltimore, D. (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 247, 824-830 38 Deininger, M. W. N., Goldman, J. M. and Melo, J. V. (2000) The molecular biology of chronic myeloid leukemia. Blood. 96, 3343-3356 39 Pluk, H., Dorey, K. and Superti-Furga, G. (2002) Autoinhibition of c-Abl. Cell. 108, 247-259 40 Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., Lydon, N. B., Kantarjian, H., Capdeville, R., Ohno-Jones, S. and Sawyers, C. L. (2001) Efficacy and safety of a specific inhibitor of the BCRABL tyrosine kinase in chronic myeloid leukemia. New England Journal of Medicine. 344, 1031-1037 41 Gambacorti-Passerini, C. B., Gunby, R. H., Piazza, R., Galietta, A., Rostagno, R. and Scapozza, L. (2003) Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 4, 75-85 42 Druker, B. J. (2008) Translation of the Philadelphia chromosome into therapy for CML. Blood. 112, 4808-4817 43 Raab, M. S., Podar, K., Breitkreutz, I., Richardson, P. G. and Anderson, K. C. (2009) Multiple myeloma. Lancet. 374, 324-339 21 44 Eisenbeis, C. F., Singh, H. and Storb, U. (1995) Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 9, 1377-1387 45 Fire, A., Xu, S. Q., Montgomery, M. K., Kostas, S. A., Driver, S. E. and Mello, C. C. (1998) Potent and specific genetic interference by doublestranded RNA in Caenorhabditis elegans. Nature. 391, 806-811 46 Baulcombe, D. C. (1996) RNA as a target and an initiator of posttranscriptional gene silencing in transgenic plants. Plant Mol Biol. 32, 79-88 47 Bernards, R. (2006) [The Nobel Prize in Physiology or Medicine for 2006 for the discovery of RNA interference]. Ned Tijdschr Geneeskd. 150, 2849-2853 48 Wilson, R. C. and Doudna, J. A. (2013) Molecular mechanisms of RNA interference. Annual review of biophysics. 42, 217-239 49 Obbard, D. J., Gordon, K. H., Buck, A. H. and Jiggins, F. M. (2009) The evolution of RNAi as a defence against viruses and transposable elements. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 364, 99-115 50 Carthew, R. W. (2006) Gene regulation by microRNAs. Current opinion in genetics & development. 16, 203-208 51 Lund, E. and Dahlberg, J. E. (2006) Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harbor symposia on quantitative biology. 71, 59-66 52 Macfarlane, L. A. and Murphy, P. R. (2010) MicroRNA: Biogenesis, Function and Role in Cancer. Current genomics. 11, 537-561 53 Esquela-Kerscher, A. and Slack, F. J. (2006) Oncomirs - microRNAs with a role in cancer. Nature reviews. Cancer. 6, 259-269 54 Latterich, M., ed. (2008) RNAi. Taylor & Francis Group, New York 55 Boehm, J. S., Zhao, J. J., Yao, J., Kim, S. Y., Firestein, R., Dunn, I. F., Sjostrom, S. K., Garraway, L. A., Weremowicz, S., Richardson, A. L., Greulich, H., Stewart, C. J., Mulvey, L. A., Shen, R. R., Ambrogio, L., Hirozane-Kishikawa, T., Hill, D. E., Vidal, M., Meyerson, M., Grenier, J. K., Hinkle, G., Root, D. E., Roberts, T. M., Lander, E. S., Polyak, K. and Hahn, W. C. (2007) Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 129, 1065-1079 56 Wohlbold, L., van der Kuip, H., Miething, C., Vornlocher, H. P., Knabbe, C., Duyster, J. and Aulitzky, W. E. (2003) Inhibition of bcr-abl gene expression by small interfering RNA sensitizes for imatinib mesylate (STI571). Blood. 102, 2236-2239 57 Vasconcelos, M. H., Lima, R. T. and Guimarães, J. E. (2007) Overcoming K562Dox resistance to STI571 (Gleevec) by downregulation of P-gp expression using siRNAs. Cancer Therapy. 5, 67-76 58 Lee, J. M., Yoon, T. J. and Cho, Y. S. (2013) Recent developments in nanoparticle-based siRNA delivery for cancer therapy. Biomed Res Int. 2013, 782041 22 59 Castanotto, D. and Rossi, J. J. (2009) The promises and pitfalls of RNA-interference-based therapeutics. Nature. 457, 426-433 60 Heilbronn, R. and Weger, S. (2010) Viral vectors for gene transfer: current status of gene therapeutics. Handb Exp Pharmacol, 143-170 61 Campos, S. K. and Barry, M. A. (2007) Current advances and future challenges in Adenoviral vector biology and targeting. Curr Gene Ther. 7, 189-204 62 Shen, C., Buck, A. K., Liu, X., Winkler, M. and Reske, S. N. (2003) Gene silencing by adenovirus-delivered siRNA. FEBS Lett. 539, 111-114 63 Liu, Q. and Muruve, D. A. (2003) Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 10, 935-940 64 Hartman, Z. C., Appledorn, D. M. and Amalfitano, A. (2008) Adenovirus vector induced innate immune responses: impact upon efficacy and toxicity in gene therapy and vaccine applications. Virus Res. 132, 1-14 65 Marshall, E. (1999) Gene therapy death prompts review of adenovirus vector. Science. 286, 2244-2245 66 Yang, Y., Greenough, K. and Wilson, J. M. (1996) Transient immune blockade prevents formation of neutralizing antibody to recombinant adenovirus and allows repeated gene transfer to mouse liver. Gene Ther. 3, 412-420 67 Wonganan, P. and Croyle, M. A. (2010) PEGylated Adenoviruses: From Mice to Monkeys. Viruses. 2, 468-502 68 Wan, Y., Han, J., Fan, G., Zhang, Z., Gong, T. and Sun, X. (2013) Enzyme-responsive liposomes modified adenoviral vectors for enhanced tumor cell transduction and reduced immunogenicity. Biomaterials. 34, 30203030 69 Sakuma, T., Barry, M. A. and Ikeda, Y. (2012) Lentiviral vectors: basic to translational. The Biochemical journal. 443, 603-618 70 Burnett, J. C., Rossi, J. J. and Tiemann, K. (2011) Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 6, 1130-1146 71 DiGiusto, D. L., Krishnan, A., Li, L., Li, H., Li, S., Rao, A., Mi, S., Yam, P., Stinson, S., Kalos, M., Alvarnas, J., Lacey, S. F., Yee, J. K., Li, M., Couture, L., Hsu, D., Forman, S. J., Rossi, J. J. and Zaia, J. A. (2010) RNAbased gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2, 36ra43 72 Yi, Y., Hahm, S. H. and Lee, K. H. (2005) Retroviral gene therapy: safety issues and possible solutions. Curr Gene Ther. 5, 25-35 73 Hong, Y., Macnab, S., Lambert, L. A., Turner, A. J., Whitehouse, A. and Usmani, B. A. (2011) Herpesvirus saimiri-based endothelin-converting enzyme-1 shRNA expression decreases prostate cancer cell invasion and migration. Int J Cancer. 129, 586-598 74 Smollich, M., Gotte, M., Yip, G. W., Yong, E. S., Kersting, C., Fischgrabe, J., Radke, I., Kiesel, L. and Wulfing, P. (2007) On the role of 23 endothelin-converting enzyme-1 (ECE-1) and neprilysin in human breast cancer. Breast Cancer Research and Treatment. 106, 361-369 75 Wood, M. J. A., Byrnes, A. P., Pfaff, D. W., Rabkin, S. D. and Charlton, H. M. (1994) Inflammatory Effects of Gene-Transfer into the Cns with Defective Hsv-1 Vectors. Gene Therapy. 1, 283-291 76 McMenamin, M. M., Byrnes, A. P., Charlton, H. M., Coffin, R. S., Latchman, D. S. and Wood, M. J. (1998) A gamma34.5 mutant of herpes simplex 1 causes severe inflammation in the brain. Neuroscience. 83, 12251237 77 Cheng, Z., Al Zaki, A., Hui, J. Z., Muzykantov, V. R. and Tsourkas, A. (2012) Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 338, 903-910 78 Choi, H. S., Liu, W., Misra, P., Tanaka, E., Zimmer, J. P., Itty Ipe, B., Bawendi, M. G. and Frangioni, J. V. (2007) Renal clearance of quantum dots. Nature biotechnology. 25, 1165-1170 79 Matsumura, Y. and Maeda, H. (1986) A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs1. Cancer Research. 46, 6387-6392 80 Tabata, T., Murakami, Y. and Ikada, Y. (1998) Tumor accumulation of poly(vinyl alcohol) of different sizes after intravenous injection. Journal of Controlled Release. 50, 123-133 81 Zhang, Y., Yang, M., Portney, N. G., Cui, D., Budak, G., Ozbay, E., Ozkan, M. and Ozkan, C. S. (2008) Zeta potential: a surface electrical characteristic to probe the interaction of nanoparticles with normal and cancer human breast epithelial cells. Biomed Microdevices. 10, 321-328 82 Bondar, O. V., Saifullina, D. V., Shakhmaeva, I. I., Mavlyutova, I. I. and Abdullin, T. I. (2012) Monitoring of the Zeta Potential of Human Cells upon Reduction in Their Viability and Interaction with Polymers. Acta Naturae. 4, 78-81 83 Zahr, A. S., Davis, C. A. and Pishko, M. V. (2006) Macrophage uptake of coreshell nanoparticles surface modified with poly(ethylene glycol). Langmuir. 22, 8178-8185 84 Knop, K., Hoogenboom, R., Fischer, D. and Schubert, U. S. (2010) Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angewandte Chemie-International Edition. 49, 6288-6308 85 Bunka, D. H. J. and Stockley, P. G. (2012) CHAPTER 6 Therapeutic Applications of Nucleic Acid Aptamer Conjugates. In DNA Conjugates and Sensors. pp. 140-165, The Royal Society of Chemistry 86 Bareford, L. M. and Swaan, P. W. (2007) Endocytic mechanisms for targeted drug delivery. Advanced drug delivery reviews. 59, 748-758 87 Behr, J. P. (1997) The Proton Sponge - A Trick to Enter Cells the Viruses Did Not Exploit. Chimia. 51, 34-36 24 88 Mastico, R. A., Talbot, S. J. and Stockley, P. G. (1993) Multiple presentation of foreign peptides on the surface of an RNA-free spherical bacteriophage capsid. J Gen Virol. 74 ( Pt 4), 541-548 89 Wu, M., Brown, W. L. and Stockley, P. G. (1995) Cell-specific delivery of bacteriophage-encapsidated ricin A chain. Bioconjug Chem. 6, 587-595 90 Galaway, F. A. and Stockley, P. G. (2013) MS2 viruslike particles: a robust, semisynthetic targeted drug delivery platform. Mol Pharm. 10, 59-68 91 Ashley, C. E., Carnes, E. C., Phillips, G. K., Durfee, P. N., Buley, M. D., Lino, C. A., Padilla, D. P., Phillips, B., Carter, M. B., Willman, C. L., Brinker, C. J., Caldeira Jdo, C., Chackerian, B., Wharton, W. and Peabody, D. S. (2011) Cell-specific delivery of diverse cargos by bacteriophage MS2 virus-like particles. ACS Nano. 5, 5729-5745 92 Steinmetz, N. F. and Manchester, M. (2009) PEGylated viral nanoparticles for biomedicine: the impact of PEG chain length on VNP cell interactions in vitro and ex vivo. Biomacromolecules. 10, 784-792 93 Strauss, J. H., Jr. and Sinsheimer, R. L. (1963) Purification and properties of bacteriophage MS2 and of its ribonucleic acid. Journal of molecular biology. 7, 43-54 94 Pang, L., Nowostawska, U., Ryan, J. N., Williamson, W. M., Walshe, G. and Hunter, K. A. (2009) Modifying the surface charge of pathogen-sized microspheres for studying pathogen transport in groundwater. J Environ Qual. 38, 2210-2217 95 Masaki, T., Shiratori, Y., Rengifo, W., Igarashi, K., Yamagata, M., Kurokohchi, K., Uchida, N., Miyauchi, Y., Yoshiji, H., Watanabe, S., Omata, M. and Kuriyama, S. (2003) Cyclins and cyclin-dependent kinases: comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology. 37, 534-543 96 Lo, A., Lin, C. T. and Wu, H. C. (2008) Hepatocellular carcinoma cellspecific peptide ligand for targeted drug delivery. Molecular cancer therapeutics. 7, 579-589 97 Greaves, M. (2013) Return of the malingering mutants. Br J Cancer. 109, 1391-1393 98 Pan, Y., Jia, T., Zhang, Y., Zhang, K., Zhang, R., Li, J. and Wang, L. (2012) MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int J Nanomedicine. 7, 5957-5967 99 Pan, Y., Zhang, Y., Jia, T., Zhang, K., Li, J. and Wang, L. (2012) Development of a microRNA delivery system based on bacteriophage MS2 virus-like particles. FEBS J. 279, 1198-1208 100 Felgner, P. L., Gadek, T. R., Holm, M., Roman, R., Chan, H. W., Wenz, M., Northrop, J. P., Ringold, G. M. and Danielsen, M. (1987) Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. U. S. A. 84, 7413-7417 101 Yitbarek, E. (2011) Characterization and Analytical Applications of DyeEncapsulated Zwitterionic Liposomes. BiblioBazaar 25 102 Kim, H. K., Davaa, E., Myung, C. S. and Park, J. S. (2010) Enhanced siRNA delivery using cationic liposomes with new polyarginine-conjugated PEG-lipid. International Journal of Pharmaceutics. 392, 141-147 103 Gao, X. and Huang, L. (1995) Cationic liposome-mediated gene transfer. Gene Ther. 2, 710-722 104 Kim, H. R., Kim, I. K., Bae, K. H., Lee, S. H., Lee, Y. and Park, T. G. (2008) Cationic solid lipid nanoparticles reconstituted from low density lipoprotein components for delivery of siRNA. Mol Pharm. 5, 622-631 105 Gabizon, A. and Papahadjopoulos, D. (1988) Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl. Acad. Sci. U. S. A. 85, 6949-6953 106 Senior, J., Delgado, C., Fisher, D., Tilcock, C. and Gregoriadis, G. (1991) Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: studies with poly(ethylene glycol)-coated vesicles. Biochim Biophys Acta. 1062, 77-82 107 Maier, M. A., Jayaraman, M., Matsuda, S., Liu, J., Barros, S., Querbes, W., Tam, Y. K., Ansell, S. M., Kumar, V., Qin, J., Zhang, X., Wang, Q., Panesar, S., Hutabarat, R., Carioto, M., Hettinger, J., Kandasamy, P., Butler, D., Rajeev, K. G., Pang, B., Charisse, K., Fitzgerald, K., Mui, B. L., Du, X., Cullis, P., Madden, T. D., Hope, M. J., Manoharan, M. and Akinc, A. (2013) Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther. 21, 1570-1578 108 Laouini, A., Jaafar-Maalej, C., Limayem-Blouza, I., Sfar, S., Charcosset, C. and Fessi, H. (2012) Preparation, Characterization and Applications of Liposomes: State of the Art. Journal of Colloid Science and Biotechnology. 1, 147-168 109 Dalby, B., Cates, S., Harris, A., Ohki, E. C., Tilkins, M. L., Price, P. J. and Ciccarone, V. C. (2004) Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 33, 95-103 110 Baker, B. E., Kestler, D. P. and Ichiki, A. T. (2006) Effects of siRNAs in combination with Gleevec on K-562 cell proliferation and Bcr-Abl expression. J Biomed Sci. 13, 499-507 111 Bjorge, J. D., Pang, A. S., Funnell, M., Chen, K. Y., Diaz, R., Magliocco, A. M. and Fujita, D. J. (2011) Simultaneous siRNA targeting of Src and downstream signaling molecules inhibit tumor formation and metastasis of a human model breast cancer cell line. PloS one. 6, e19309 112 Bouchie, A. (2012) Companies in footrace to deliver RNAi. Nature biotechnology. 30, 1154-1157 113 Tabernero, J., Shapiro, G. I., LoRusso, P. M., Cervantes, A., Schwartz, G. K., Weiss, G. J., Paz-Ares, L., Cho, D. C., Infante, J. R., Alsina, M., Gounder, M. M., Falzone, R., Harrop, J., White, A. C., Toudjarska, I., Bumcrot, D., Meyers, R. E., Hinkle, G., Svrzikapa, N., Hutabarat, R. M., Clausen, V. A., Cehelsky, J., Nochur, S. V., Gamba-Vitalo, C., Vaishnaw, A. K., Sah, D. W., Gollob, J. A. and Burris, H. A., 3rd. (2013) First-in-humans trial 26 of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 3, 406-417 114 Chang, H. I. and Yeh, M. K. (2012) Clinical development of liposomebased drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 7, 49-60 115 Davis, M. E. and Brewster, M. E. (2004) Cyclodextrin-based pharmaceutics: past, present and future. Nature reviews. Drug discovery. 3, 1023-1035 116 Heidel, J. D. and Schluep, T. (2012) Cyclodextrin-containing polymers: versatile platforms of drug delivery materials. Journal of drug delivery. 2012, 262731 117 Bellocq, N. C., Davis, M. E., Engler, H., Jensen, G. S., Liu, A., Machemer, T., Maneval, D. C., Quijano, E., Pun, S. H., Schluep, T. and Wen, S. (2003) Transferrin-targeted, cyclodextrin polycation-based gene vector for systemic delivery. Mol. Ther. 7, S290 118 Heidel, J. D., Yu, Z., Liu, J. Y., Rele, S. M., Liang, Y., Zeidan, R. K., Kornbrust, D. J. and Davis, M. E. (2007) Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc. Natl. Acad. Sci. U. S. A. 104, 5715-5721 119 Pun, S. H. and Davis, M. E. (2002) Development of a Non-Viral Gene Delivery Vehicle for Systemic Application. Bioconjugate Chem. 13, 630-639 120 Bartlett, D. W. and Davis, M. E. (2007) Physicochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjug Chem. 18, 456-468 121 Davis, M. E. (2009) The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 6, 659-668 122 Hu-Lieskovan, S., Heidel, J. D., Bartlett, D. W., Davis, M. E. and Triche, T. J. (2005) Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res. 65, 8984-8992 123 Cerqueira, N. M., Fernandes, P. A. and Ramos, M. J. (2007) Ribonucleotide reductase: a critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat Anticancer Drug Discov. 2, 11-29 124 Davis, M. E., Zuckerman, J. E., Choi, C. H., Seligson, D., Tolcher, A., Alabi, C. A., Yen, Y., Heidel, J. D. and Ribas, A. (2010) Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 464, 1067-1070 125 Czauderna, F., Fechtner, M., Dames, S., Aygun, H., Klippel, A., Pronk, G. J., Giese, K. and Kaufmann, J. (2003) Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Research. 31, 2705-2716 27 126 Chiu, Y. L. and Rana, T. M. (2003) siRNA function in RNAi: a chemical modification analysis. Rna. 9, 1034-1048 127 Soutschek, J., Akinc, A., Bramlage, B., Charisse, K., Constien, R., Donoghue, M., Elbashir, S., Geick, A., Hadwiger, P., Harborth, J., John, M., Kesavan, V., Lavine, G., Pandey, R. K., Racie, T., Rajeev, K. G., Rohl, I., Toudjarska, I., Wang, G., Wuschko, S., Bumcrot, D., Koteliansky, V., Limmer, S., Manoharan, M. and Vornlocher, H. P. (2004) Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 432, 173-178 128 Kenski, D. M., Butora, G., Willingham, A. T., Cooper, A. J., Fu, W., Qi, N., Soriano, F., Davies, I. W. and Flanagan, W. M. (2012) siRNA-optimized Modifications for Enhanced In Vivo Activity. Mol Ther Nucleic Acids. 1, e5 129 Robbins, M., Judge, A., Liang, L., McClintock, K., Yaworski, E. and MacLachlan, I. (2007) 2'-O-Methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 15, 1663–1669 130 Jackson, A. L., Burchard, J., Leake, D., Reynolds, A., Schelter, J., Guo, J., Johnson, J. M., Lim, L., Karpilow, J., Nichols, K., Marshall, W., Khvorova, A. and Linsley, P. S. (2006) Position-specific chemical modification of siRNAs reduces "off-target" transcript silencing. Rna. 12, 1197-1205 131 Bramsen, J. B. and Kjems, J. (2012) Development of TherapeuticGrade Small Interfering RNAs by Chemical Engineering. Frontiers in genetics. 3, 154 132 Bramsen, J. B., Laursen, M. B., Nielsen, A. F., Hansen, T. B., Bus, C., Langkjaer, N., Babu, B. R., Hojland, T., Abramov, M., Van Aerschot, A., Odadzic, D., Smicius, R., Haas, J., Andree, C., Barman, J., Wenska, M., Srivastava, P., Zhou, C., Honcharenko, D., Hess, S., Muller, E., Bobkov, G. V., Mikhailov, S. N., Fava, E., Meyer, T. F., Chattopadhyaya, J., Zerial, M., Engels, J. W., Herdewijn, P., Wengel, J. and Kjems, J. (2009) A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 37, 2867-2881 133 Glud, S. Z., Bramsen, J. B., Dagnaes-Hansen, F., Wengel, J., Howard, K. A., Nyengaard, J. R. and Kjems, J. (2009) Naked siLNA-mediated gene silencing of lung bronchoepithelium EGFP expression after intravenous administration. Oligonucleotides. 19, 163-168 134 Hornung, V., Guenthner-Biller, M., Bourquin, C., Ablasser, A., Schlee, M., Uematsu, S., Noronha, A., Manoharan, M., Akira, S., de Fougerolles, A., Endres, S. and Hartmann, G. (2005) Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nature medicine. 11, 263-270 135 Bramsen, J. B., Laursen, M. B., Damgaard, C. K., Lena, S. W., Babu, B. R., Wengel, J. and Kjems, J. (2007) Improved silencing properties using small internally segmented interfering RNAs. Nucleic Acids Res. 35, 58865897 136 Langkjaer, N., Pasternak, A. and Wengel, J. (2009) UNA (unlocked nucleic acid): a flexible RNA mimic that allows engineering of nucleic acid duplex stability. Bioorganic & medicinal chemistry. 17, 5420-5425 28 137 Bramsen, J. B., Pakula, M. M., Hansen, T. B., Bus, C., Langkjaer, N., Odadzic, D., Smicius, R., Wengel, S. L., Chattopadhyaya, J., Engels, J. W., Herdewijn, P., Wengel, J. and Kjems, J. (2010) A screen of chemical modifications identifies position-specific modification by UNA to most potently reduce siRNA off-target effects. Nucleic Acids Res. 38, 5761-5773 138 Laursen, M. B., Pakula, M. M., Gao, S., Fluiter, K., Mook, O. R., Baas, F., Langklaer, N., Wengel, S. L., Wengel, J., Kjems, J. and Bramsen, J. B. (2010) Utilization of unlocked nucleic acid (UNA) to enhance siRNA performance in vitro and in vivo. Molecular bioSystems. 6, 862-870 139 Ellington, A. D., Szostak, J. W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature. 346, 818 - 822 140 Dey, A. K., Griffiths, C., Lea, S. M. and James, W. (2005) Structural characterization of an anti-gp120 RNA aptamer that neutralizes R5 strains of HIV-1. Rna. 11, 873-884 141 McNamara, J. O., 2nd, Andrechek, E. R., Wang, Y., Viles, K. D., Rempel, R. E., Gilboa, E., Sullenger, B. A. and Giangrande, P. H. (2006) Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nature biotechnology. 24, 1005-1015 142 Dassie, J. P., Liu, X. Y., Thomas, G. S., Whitaker, R. M., Thiel, K. W., Stockdale, K. R., Meyerholz, D. K., McCaffrey, A. P., McNamara, J. O., 2nd and Giangrande, P. H. (2009) Systemic administration of optimized aptamersiRNA chimeras promotes regression of PSMA-expressing tumors. Nature biotechnology. 27, 839-849 143 Birmingham, A., Anderson, E. M., Reynolds, A., Ilsley-Tyree, D., Leake, D., Fedorov, Y., Baskerville, S., Maksimova, E., Robinson, K., Karpilow, J., Marshall, W. S. and Khvorova, A. (2006) 3' UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nature Methods 3, 199–204 144 Schwarz, D. S., Hutvagner, G., Du, T., Xu, Z., Aronin, N. and Zamore, P. D. (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell. 115, 199-208 145 Bartlett, D. W. and Davis, M. E. (2006) Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Research. 34, 322-333 146 Lorenz, C., Hadwiger, P., John, M., Vornlocher, H. P. and Unverzagt, C. (2004) Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg Med Chem Lett. 14, 4975-4977 147 Zhang, J., Zheng, J., Lu, C., Du, Q., Liang, Z. and Xi, Z. (2012) Modification of the siRNA passenger strand by 5-nitroindole dramatically reduces its off-target effects. Chembiochem : a European journal of chemical biology. 13, 1940-1945 148 Grimm, D., Streetz, K. L., Jopling, C. L., Storm, T. A., Pandey, K., Davis, C. R., Marion, P., Salazar, F. and Kay, M. A. (2006) Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 441, 537-541 29 149 Ahn, M., Witting, S. R., Ruiz, R., Saxena, R. and Morral, N. (2011) Constitutive expression of short hairpin RNA in vivo triggers buildup of mature hairpin molecules. Hum Gene Ther. 22, 1483-1497 150 Rolfsson, O., Toropova, K., Morton, V., Francese, S., Basnak, G., Thompson, G. S., Homans, S. W., Ashcroft, A. E., Stonehouse, N. J., Ranson, N. A. and Stockley, P. G. (2008) RNA packing specificity and folding during assembly of the bacteriophage MS2. Comput Math Method M. 9, 339-349 151 Borodavka, A., Tuma, R. and Stockley, P. G. (2012) Evidence that viral RNAs have evolved for efficient, two-stage packaging. Proc. Natl. Acad. Sci. U. S. A. 109, 15769-15774 152 Stockley, P. G., Rolfsson, O., Thompson, G. S., Basnak, G., Francese, S., Stonehouse, N. J., Homans, S. W. and Ashcroft, A. E. (2007) A simple, RNA-mediated allosteric switch controls the pathway to formation of a T=3 viral capsid. Journal of molecular biology. 369, 541-552 153 Valegard, K., Murray, J. B., Stockley, P. G., Stonehouse, N. J. and Liljas, L. (1994) Crystal structure of an RNA bacteriophage coat proteinoperator complex. Nature. 371, 623-626 154 Weinberg, R. A. (2007) The Biology of Cancer. Garland Science, New York 155 Arteaga, C. L. (2002) Epidermal growth factor receptor dependence in human tumors: More than just expression? Oncologist. 7, 31-39 156 Chen, Y. F., Wang, J. Y., Wu, C. H., Chen, F. M., Cheng, T. L. and Lin, S. R. (2005) Detection of circulating cancer cells with K-ras oncogene using membrane array. Cancer Lett. 229, 115-122 157 Guan, H. Y., Zhang, H., Cai, J. C., Wu, J. H., Yuan, J., Li, J., Huang, Z. S. and Li, M. F. (2011) IKBKE is over-expressed in glioma and contributes to resistance of glioma cells to apoptosis via activating NF-kappa B. J. Pathol. 223, 436-445 158 Lutterbach, B., Zeng, Q. W., Davis, L. J., Hatch, H., Hang, G. Z., Kohl, N. E., Gibbs, J. B. and Pan, B. S. (2007) Lung cancer cell lines harboring MET gene amplification are dependent on met for growth and survival. Cancer Research. 67, 2081-2088 159 Yoshida, T., Okamoto, I., Okamoto, W., Hatashita, E., Yamada, Y., Kuwata, K., Nishio, K., Fukuoka, M., Janne, P. A. and Nakagawa, K. (2010) Effects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib-resistant non-small cell lung cancer cells with acquired MET amplification. Cancer Sci. 101, 167-172 160 Wilda, M., Fuchs, U., Wossmann, W. and Borkhardt, A. (2002) Killing of leukemic cells with a BCR/ABL fusion gene by RNA interference (RNAi). Oncogene. 21, 5716-5724 161 Sharma, S. V. and Settleman, J. (2007) Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes & Development. 21, 3214-3231 30 162 Iida, S., Rao, P. H., Butler, M., Corradini, P., Boccadoro, M., Klein, B., Chaganti, R. S. K. and DallaFavera, R. (1997) Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nature Genet. 17, 226-230 163 Ashwell, S. and Zabludoff, S. (2008) DNA damage detection and repair pathways - Recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 14, 4032-4037 164 Muller, A. J. and Scherle, P. A. (2006) Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat. Rev. Cancer. 6, 613-625 31 Figure 1. Construction of an MS2 VLP-based siRNA delivery system. MS2 coat proteins associate as non-covalent dimers (magenta, PDB: 1MSC). Capsid assembly from these dimers can be triggered at neutral pH by addition of the MS2 translational operator (TR), a 19-nt RNA stem-loop [150, 151]. TRbinding induces a conformation change in the coat protein dimer (green, PDB: 1ZDI), which can then act as an assembly initiation complex [152, 153]. Any siRNA conjugated to TR will be packaged inside assembled capsids [90]. The capsid surface can then be chemically modified to display ligands for immunomasking, endosomal escape or tumour targeting. Examples presented include PEG, a nucleic acid aptamer, transferrin (Tf) and an antibody. These are not drawn to scale. Figure 2. The RONDEL siRNA delivery platform. (A) Chemical structure of a cyclodextrin-containing polymer (CDP) composed of repeating units of β–cyclodextrin, positively charged amidine centres and terminal imidazoles. (B) Schematic illustrating RONDEL assembly. CDPs form high affinity inclusion complexes with adamantane (AD) coupled to PEG or PEG-Tf. Addition of siRNAs triggers compaction of the components via electrostatic interactions between amidines within CDPs and the phosphate backbone of siRNA. The resulting nanoparticle, approximately 70 nm in diameter, contains condensed siRNAs in its core and multiple PEG and PEGTf ligands on its exterior. Adapted from [121]. Table 1. Summary of (non-)oncogene dependencies and their incidence in cancers. Abbreviations: NSCLC, non-small cell lung cancer; CML, chronic myeloid leukaemia, AML, acute myeloid leukaemia; ALL, acute lymphoblastic leukaemia. Dash indicates not applicable. (Table 1 is presented on p 33.) 32 CANCERS IN WHICH MUTATED CANCERS IN WHICH OVEREXPRESSED REFERENCE EGFR Cell surface receptor tyrosine kinase; induction of various proliferative pathways including the MAP kinase and PI3K pathways NSCLC Various epithelial cancers including 40% glioblastomas, 10-20% squamous cell carcinomas, NSCLC, breast, head and neck, gastric, colorectal, prostate, bladder, renal, pancreatic, ovarian [27, 154, 155] K-RAS Signal transduction intermediate for mitogenic and anti-apoptotic stimuli >50% colorectal; 75% pancreatic; 48% NSCLC; 44% adrenocortical tumours 5-10% lung, ovarian and bladder carcinomas [154, 156] IKBKE Serine/ threonine protein kinase; anti-apoptotic - 30% breast, ovarian, prostate, glioma [157] MET Cell surface receptor tyrosine kinase; induction of various proliferative pathways - 20% gastric carcinomas, NSCLC, 4% oesophageal & lung [154, 158, 159] BCR-ABL Tyrosine kinase; ABL normally a regulator of the cell cycle 95% CML, 30% ALL, 2% AML - [34, 160] c-KIT Receptor tyrosine kinase Gastrointestinal stromal tumours Gastrointestinal stromal tumours [16] HER2/ ERBB2 Cell surface receptor tyrosine kinase related to EGFR - 30% breast, ovarian, NSCLC [161] VEGF (extrinsic) Signal protein; promote growth of blood vessels - - [16] IRF4 (intrinsic) Transcription factor; B-cell differentiation and proliferation Multiple myeloma - [162] CHK1 (intrinsic) Cell cycle checkpoint kinase; DNA damage response and cell cycle arrest - - [163] COX2 (extrinsic) Roles in immune suppression, angiogenesis, invasiveness - Colorectal, breast, pancreatic, NSCLC [164] PROTEIN FUNCTION ONCOGENE DEPENDENT NONONCOGENE DEPENDENT 33