Readme fil
advertisement

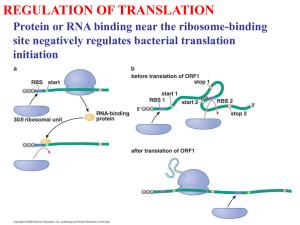
Analysis of codon pair frequencies in genomic sequence Buchan, JR, Aucott, L. and Stansfield, I Introduction Perl version 5.005_3 was used to run all programs described. The E.coli geneome sequence has been supplied for test purposes in a trimmed format (a sample of 100 ORFs only in TIGRecoli-trim.txt). The genome has been reformatted to remove all hard returns (paragraph marks) except those that separate one sequence from another. Sequences are in the format: >sequence name1 ###ATGNNNNNNNNNNNNNNNNNNNN hr >sequence name2 ###ATGNNNNNNNNNNNNNNNNNNNN hr Where: ‘ATG’ represents the first codon of each open reading frame, and ‘###’ represents marker text defining the start of the open reading frame . hr denotes a hard return. ‘N’ denotes any nucleotide. In the instructions below, bold font indicates a command to enter in a DOS window whilst italicised comments represent the name of a file (e.g. programs file, spreadsheet, sequence file). Menu commands are underlined. Generating codoncount data using E.coli as an example The program codoncount.txt counts all the codon pairs in a genome ORF by ORF and records the total observed count of each of the 3904 pair types (61 sense codons x 64 sense and stop codons). It also calculates the expected number of codon pairs as each ORF is processed, and records a cumulative expected codon pair count for the whole genome. Results are stored in the file named ‘results.txt’, and can be opened in Excel as a tabdelimited .txt file. In Excel the spreadsheet will comprise an upper block of data 64 rows X 130 columns (starts in cell A3). This is the observed codon pair data while the lower 64R X 130C block (starts in cell A68) is the expected codon pair count data. The data can be converted into a more manageable form using the spreadsheet ‘Codoncount clean.xls’ Select the entire results.txt worksheet, copy it, and paste the data into the ‘Codoncount RAW’ worksheet of Codoncount clean.xls at cell A1. This data is then referenced in the ‘Cleaned data’ worksheet of Codoncount clean.xls such that observed codon pair data (top) and expected codon pair data bottom is listed in 65R x 62C blocks. 5’ P site codons are listed in rows 3 (observed) and 71(expected) and 3’ A site codons are listed in column A. Analysing codoncount data in other spreadsheets Data in Codoncount clean.xls can now be directly pasted into the spreadsheet Residual calcualtor.xls. This normalises expected count data for amino acid pair bias and calculates residual scores for ever codon pair. Copy the observed codon pair data (cells A1:BJ67) from Codoncount clean.xls and Paste special values into the Residual calcualtor.xls ‘observed’ worksheet at cell A1. Copy the observed expected pair data (cells A69:BJ165) from Codoncount clean.xls and Paste special values into Residual calcualtor.xls ‘expected’ worksheet at cell A1. Codonpairindex The program codonpairindex.txt, calculates the mean codon pair residual score (i.e. the Codon Pair index) for each ORF in a genome. The program is first ‘loaded’ with a library of residual scores for each potential codon pair. The supplied version of the program contains the E.coli codon pair residual library. The filename for the genome being analysed must be accurately referenced in codonpairindex.txt (version supplied refers to TIGRecoli-trim.txt). Once the program is run, the mean CPI score for each ORF is written to the file results.txt. This can be opened in Excel. Random Random.txt is a program that generates a randomised genome for a given organism based on the codon usage values of that organism. Random.txt is set to generate random ORFs with an E.coli codon bias. This is achieved by selecting a random number between 0 and 1. Each codon is assigned to a discrete band of values within this range. The width of that band is proportional to that codon’s biased usage among the 61 sense codons of the genetic code. An ‘if’ statement then determines which of the 61 sense codons will be chosen in response to each random number generation. Gene size and the number of genes generated are specified by editing the program at lines 2 and 5. The text “###” is introduced prior to the gene sequence so that the output genome file is ready for analysis by the programs listed above.